




Beyond preclinical promise: can mesenchymal stromal cell-derived extracellular vesicles reliably target tubular epithelial cells?
 0
0 
Abstract
Kidney disease, encompassing both acute kidney injury (AKI) and chronic kidney disease (CKD), represents a major global health challenge. A pivotal aspect of the pathogenesis of these conditions is damage to renal tubular epithelial cells (TECs), which contributes to maladaptive repair mechanisms and fibrosis. Due to their essential role, TECs are regarded as a promising target for innovative therapeutic strategies. Mesenchymal stromal cell-derived extracellular vesicles (MSC-EVs) have attracted increasing attention for their therapeutic potential in kidney disease, with extensive literature documenting their beneficial effects on TEC damage through targeted mechanisms. In this review, we critically examine the existing literature on the targeting of TECs by MSC-EVs in both in vitro and in vivo settings. Furthermore, we highlight the limitations and potential of MSC-EV-based strategies for TEC targeting, aiming to provide insights for future clinical trials and therapeutic applications.
Keywords
INTRODUCTION
Kidney disease and tubular epithelial cell damage
Kidney disease is broadly categorized into two primary forms: acute kidney injury (AKI) and chronic kidney disease (CKD). AKI, marked by damage to renal tubular epithelial cells (TECs), is a common and potentially life-threatening condition among hospitalized patients, affecting 10%-15% of all admissions and approximately 50% of individuals in intensive care units. Severe, recurrent, and inadequately managed AKI can progress to CKD, a process known as the AKI-to-CKD transition[1]. CKD is characterized by the gradual loss of kidney function and poses a major global public health challenge[2]. According to estimates from the World Health Organization (WHO), CKD accounted for 1.5% of global deaths in 2012 and is projected to become the fifth leading cause of mortality worldwide by 2040[3].
Renal fibrosis, particularly tubulointerstitial fibrosis (TIF), which is marked by tubular atrophy and extracellular matrix (ECM) accumulation[4], is an inevitable outcome of all progressive CKD. Currently, no effective treatment exists to reverse kidney fibrosis and prevent progression to kidney replacement therapy. Among the various renal cell types, TECs are the primary targets of injury. They undergo cell death followed by regeneration and repair[5]. However, incomplete recovery can lead to maladaptive repair, fibrosis, and ultimately kidney dysfunction[6]. Recent studies have increasingly focused on the mechanisms underlying tubular damage, highlighting the role of TECs in the pathogenesis of renal disease[6,7]. Accordingly, TECs are actively involved in both the acute and chronic phases of kidney disease, making them a promising therapeutic target.
Pathophysiology of TECs
TECs, particularly those in the proximal tubule, constitute more than 50% of the renal parenchyma[8]. These are essential for renal function, playing a pivotal role in the selective reabsorption of water, electrolytes such as sodium and potassium, and nutrients including glucose and amino acids from the glomerular filtrate. Concurrently, TECs actively secrete metabolic waste products, such as urea and creatinine, along with various toxins, into the urine. They also contribute to acid-base homeostasis by reabsorbing bicarbonate and excreting hydrogen ions[9]. Owing to their high metabolic activity and energy demands, TECs are particularly susceptible to damage from hypoxia, proteinuria, and toxins.
In response to acute injury, TECs facilitate the regeneration and repair of renal tubules, a process vital for the restoration of kidney function. However, maladaptive repair can result in the AKI-to-CKD transition, culminating in kidney fibrosis[10]. Emerging research indicates that TECs are not merely passive targets of injury but also active initiators of the fibrotic response to diverse insults[11]. Thus, TECs have been identified as critical therapeutic targets due to their significant involvement in kidney injury and repair mechanisms.
MSC-EVs
Mesenchymal stromal cells (MSCs), also known as mesenchymal stem cells, are multipotent stromal cells that can be isolated from various adult tissues. They have the capacity to differentiate into multiple cell types, including adipocytes, osteocytes, and chondrocytes[12]. Since their first clinical application as a therapeutic agent in 1995[13], MSCs have been considered a promising tool in regenerative and immunomodulatory medicine. Numerous preclinical and clinical studies have demonstrated their potential in kidney protection and repair[14]. MSCs exhibit complex and potent endocrine and paracrine functions, with their therapeutic potential largely attributed to their secretome, particularly extracellular vesicles (EVs). Their functions, including growth factor secretion, regulation of inflammation, promotion of cell proliferation, anti-apoptotic and anti-inflammatory effects, reduction of fibrosis, and stimulation of angiogenesis, have been extensively documented in the literature[15,16].
EVs are nanoparticles with a bilayer phospholipid membrane, released by nearly all cell types and commonly detected in various body fluids or cell culture supernatants[17]. Based on their biogenesis and size, EVs are generally classified into three subtypes: exosomes (exos), microvesicles (MVs), and apoptotic bodies (ABs), with respective diameters of 30-150 nm, 100-1,000 nm, and 800-5,000 nm[15]. Exos (30-150 nm) are generated through inward budding of the cell membrane, forming endocytic vesicles that develop into multivesicular bodies (MVBs). Fusion of MVBs with the plasma membrane leads to the release of intraluminal vesicles into the ECM. Exosomal membranes are enriched with protein markers such as tetraspanins (CD9, CD63, CD81) and heat shock proteins (HSP60, HSP70, HSP90). MVs (150-1,000 nm) are produced by outward budding of the plasma membrane, releasing vesicles that typically contain ADP-ribosylation factor 6 (ARF-6) and mirror the membrane composition of their cells of origin[18]. ABs (500 to 2,000 nm) are formed during apoptosis[19]. EVs carry a diverse molecular cargo - including nucleic acids, proteins, and lipids - that reflects the physiological state of their parental cells, such as MSCs. This cargo plays a key role in intercellular communication. Owing to their stability and unique biological properties, MSC-derived extracellular vesicles (MSC-EVs) exhibit significant therapeutic potential in kidney disease, while addressing several limitations associated with direct stem cell therapy[14].
Nonetheless, the therapeutic application of MSC-EVs is complicated by their inhernt heterogeneity and the challenges of precise characterization. The biological properties and molecular cargo of MSC-EVs vary considerably, influenced by multiple factors including the tissue origin of the parental MSCs, donor-specific characteristics (such as age and health status), and culture conditions. Variables such as passage number, two-dimensional vs. three-dimensional culture systems, oxygen levels, and exposure to inflammatory cytokines all contribute to this variability. Such heterogeneity presents a significant obstacle to the development of standardized therapeutic products[20].
A growing body of evidence from both in vivo and in vitro studies suggests that MSC-EVs exert protective effects against TEC damage through targeted mechanisms. However, inconsistencies in EV isolation methods and dosing regimens remain a challenge, and it is still unclear whether MSC-EVs act mainly through direct interactions with TECs or indirectly via neighboring cells. In this paper, we critically review the existing literature concerning the targeting of TECs by MSC-EVs in both in vitro and in vivo settings. We also discuss the limitations and therapeutic potential of MSC-EVs in TEC targeting, with the aim of informing future clinical trials and advancing the clinical application of MSC-EV-based therapies.
DO MSC-EVs SHOW THERAPEUTIC EFFECTS ON KIDNEY TECs?
In vitro evidence
In vitro research has become a widely adopted approach among researchers due to its cost-effectiveness and relative simplicity. TECs, key mediators of kidney fibrosis in response to injury, are increasingly used in
The principal mechanisms through which MSC-EVs confer renoprotective effects include the promotion of cellular proliferation, regulation of apoptosis, autophagy, necroptosis, and inflammatory responses, as well as the mitigation of oxidative stress and cell cycle arrest[23].
These effects are mediated via various signaling pathways and molecular targets. For example, the p53 signaling pathway and caspase activation represent key regulatory mechanisms of apoptosis and cell cycle control[24]. In the HK-2 hypoxia model, MSC-EVs were observed to attenuate cell cycle arrest and apoptosis in TECs by downregulating p53 expression and modulating Bcl-2 and Bax, thereby inhibiting apoptosis[25]. In a cisplatin-induced HK-2 damage model, EVs derived from MSC-like cells reprogrammed from induced pluripotent stem cells (iMSC-EVs) enhanced cell proliferation and suppressed apoptosis by reversing ERK1/2 pathway activation[26]. Similarly, Yin et al. reported that bone marrow MSC-derived exosomes (BM-MSC-Ex) inhibit TGF-β1-induced epithelial-mesenchymal transition (EMT) in HK-2 cells through the activation of autophagy[27].
A large body of research has shown that MSC-EVs primarily exert their effects by transferring molecular cargos such as microRNAs (miRNAs), proteins, messenger RNAs (mRNAs), and lipids. Among these, miRNAs have been studied most extensively, with increasing evidence documented in the literature[25,28,29]. MiRNAs are small non-coding RNAs, 19-24 nucleotides in length, that regulate up to 30% of mammalian protein-coding genes. Several miRNAs, including miR-125b-5p, miR-20a-5p, and miR-30, have been reviewed for their roles in apoptosis, antioxidation, inflammation, and proliferation[30]. In a study by Lindoso et al., MSC-EV mechanisms were investigated in an ischemia-reperfusion (I/R) injury model, demonstrating that EV-mediated miRNA transfer and the transcriptional regulation of miRNAs in TECs contributed to protective effects[31]. Predicted targets of these miRNAs include genes associated with apoptosis, cytoskeletal reorganization, and hypoxia.
In addition, MSC-EVs can directly deliver various pro-angiogenic transcription factors, including vascular endothelial growth factor (VEGF), insulin-like growth factor 1 (IGF-1), and basic fibroblast growth factor (bFGF), to damaged TECs, thereby promoting angiogenesis - a critical process in tissue regeneration[32]. MSC-EVs, particularly MVs, also demonstrate potential in transferring mitochondria and related components in various injury settings. However, their primary role appears to be the modulation of mitochondrial function rather than direct transfer[33].
It is widely accepted that MSC-EVs exert their effects through internalization by TECs, which is typically confirmed using uptake assays. However, comparisons across studies are complicated by differences in experimental design, including incubation times (4-24 h), readout methods (e.g., uptake efficiency or fluorescence intensity), and cell conditions (injured vs. normal)[28,29,34]. Additionally, critical methodological details, including EV labeling strategies, dye concentrations, removal of unbound dye, and the use of appropriate controls to rule out false positives (e.g., dye aggregation or nonspecific binding), are often inconsistently reported, hindering cross-study comparability. Moreover, uptake assays conducted over 24 h may yield nonspecific signals, potentially resulting in an overestimation of internalization. Importantly, the functional relevance of uptake remains unclear in many studies, as they often do not assess whether internalization translates into the anticipated therapeutic effect in functional assays.
The specific uptake of MSC-EVs by TECs is mediated by ligand-receptor interactions. Lindoso et al. showed that MSC-EVs can be internalized by proximal tubular epithelial cells (PTECs) following ATP depletion, mediated via integrins CD29 and CD44 present on the EV membrane. This uptake was inhibited when both integrins were blocked[31]. However, the receptors or molecules on PTECs responsible for this interaction were not fully identified. Kidney injury molecule-1 (KIM-1), a marker of tubular damage expressed on injured proximal tubules, also facilitates EV internalization by recognizing phosphatidylserine (PS). Engineered EVs modified with the peptide LTHVVWL, which has a strong affinity for KIM-1, enhanced targeted delivery to injured PTECs[35]. Additionally, Cao et al. observed that downregulation of ICAM-1 and VCAM-1 in hypoxia/reoxygenation (H/R)-injured HK-2 cells significantly diminished the uptake of Dil-labeled MSC exos[25].
Taken together, current in vitro evidence indicates that MSC-EVs exert direct protective effects on damaged TECs, with certain studies showing specific ligand-receptor interactions. However, most in vitro studies apply MSC-EVs either as a pre-treatment[34,36] or in co-culture[24], implying that MSC-EVs are introduced prior to the occurrence of damage, which may not accurately reflect in vivo conditions. Therefore, it is crucial to further evaluate these effects in animal models and clarify the mechanisms by which MSC-EVs reach the site of injury in vivo.
In vivo evidence
In vivo animal models provide valuable tools for investigating the functionality of MSC-EVs under pathophysiological conditions, as they preserve intact organ systems and physiological parameters including blood flow and systemic immune responses. Because TECs play crucial roles in both the acute stage (AKI) and the late stage (TIF) of kidney disease, they are commonly used to evaluate the effects of MSC-EVs
Among the various models of kidney damage, cisplatin-induced damage, I/R, and LPS-induced sepsis are frequently employed to investigate AKI, often in conjunction with in vitro TEC assays[38,39]. By contrast, models such as unilateral ureteral obstruction (UUO), diabetic nephropathy (DN), doxorubicin-induced injury, and adenine-induced injury are more closely related to chronic kidney damage and fibrosis[40]. Although animal models cannot fully replicate the complexity of human disease, the careful selection of appropriate models and outcome measures is critical for accurately assessing the effects of MSC-EVs in different contexts. Currently, the anti-apoptotic, pro-angiogenic, anti-inflammatory, and anti-fibrotic properties of MSC-EVs are recognized as key mechanisms underlying their ability to ameliorate kidney damage induced by diverse etiologies[14,22]. Furthermore, considering that TECs are rich in mitochondria and highly vulnerable to energy depletion, endpoints such as different types of cell death (apoptosis, autophagy, ferroptosis, pyroptosis), mitochondrial damage, oxidative stress, and inflammation are widely used in
In one study, rats with cisplatin-induced kidney injury were pretreated with human umbilical cord mesenchymal stem cell-derived extracellular vesicles (hucMSC-EVs) via renal capsule injection, which reduced the number of renal tubules exhibiting edema and structural damage. The study further suggested that activation of autophagy is essential for this protective effect, as corroborated by in vitro experiments using the rat tubular epithelial cell line (NRK-52E)[41]. In another study, Zhao et al. demonstrated that in the
Macrophages, as pivotal inflammatory mediators, have also been shown to play a central role in AKI progression[44]. In a porcine model of diet-induced metabolic syndrome and renal artery stenosis, Eirin et al. demonstrated that MSC-EVs injected into the stenotic renal artery colocalized with tubular cells and macrophages four weeks later. This treatment reduced renal inflammation and stenosis, increased the population of reparative M2 macrophages, and upregulated interleukin-10 (IL-10). Notably, these protective effects were absent when IL-10-depleted MSC-EVs were administered[45]. Similarly, in a UUO model, hucMSC-Exos ameliorated renal tubular injury by suppressing macrophage-to-myofibroblast transition (MMT). HucMSC-Exos reduced collagen deposition, lowered tubular injury scores, and downregulated fibrosis markers such as collagen I, vimentin, and α-SMA. Mechanistically, they inhibited MMT by downregulating the circadian gene ARNTL (BMAL1), as evidenced by reduced α-SMA+/F4/80+ dual-positive cells in vivo and in TGF-β-induced MMT in vitro[46].
Taken together, these studies clearly indicate that MSC-EVs possess therapeutic properties in vivo across various models of tubular injury. These effects involve both direct actions on TECs and indirect mechanisms mediated by other cell types, such as macrophages. The kidney, being a complex organ composed of multiple cell types, presents an environment where intercellular interactions can influence the efficacy of MSC-EVs. However, data on the uptake and interaction of MSC-EVs with different nephron cell types remain limited. For instance, damage in one region can exacerbate injury in another by releasing vesicles, as evidenced by injured podocyte-derived EVs inducing apoptosis in TECs[47]. Within the nephron system, MSC-EVs can engage with a variety of cell types, including podocytes, TECs, fibroblasts, and macrophages, thereby contributing to renal protection. Consequently, simple in vitro co-culture systems involving only MSC-EVs and TECs may not fully capture the complexity of these interactions. More advanced in vitro models incorporating multiple cell types will be needed to better elucidate the therapeutic potential of MSC-EVs in the kidney.
BIODISTRIBUTION AND KIDNEY TARGETING
Despite considerable evidence supporting both the direct and indirect therapeutic effects of MSC-EVs on TECs, it is crucial to investigate their biodistribution in order to clarify the actual fate of MSC-EVs in vivo. Such knowledge is fundamental for developing more precise targeting strategies in the future.
Influence of EV purification, dosage and administration route
Several administration routes have been employed for delivering MSC-EVs in animal studies, including intravenous, intraperitoneal, and in situ administration. Regardless of the method used, the in vivo biodistribution of injected EVs is most commonly observed in the liver, spleen, lungs, and kidneys[48]. This indicates that the injected dose does not necessarily correspond to the amount of EVs reaching the injured site. Increasingly, studies on kidney damage models are incorporating biodistribution analyses or in vivo uptake assays. However, significant variability exists across studies with respect to animal species, EV isolation methods, dosages, observation time points, and tracking techniques (e.g., fluorescent dyes, bioluminescence, or radiolabeling).
In studies investigating MSC-EVs for the treatment of kidney disease, rats and mice are the most widely used animal models. The most common administration route is intravenous injection via the tail vein, although some studies employ subcapsular or intrarenal injections to achieve higher kidney retention[49]. The isolation method also significantly influences the biodistribution and effective dose of MSC-EVs. Techniques include differential ultracentrifugation (dUC), density gradient separation, size exclusion chromatography (SEC), flow-based separation, and charge- or molecular recognition-based methods[50]. These approaches can result in substantial variability in EV purity and cargo composition[51,52]. Among them, dUC is the most frequently reported in literature, typically followed by EV quantification based on total protein content measured by standard colorimetric assays. Reported injection doses in rodent models vary widely, from 20 to 200 µg[53,28]. Of note, quantification based on total protein may overestimate vesicle dose because of co-isolated protein contaminants, particularly when less specific isolation methods are used[54]. Another dosing strategy reported in literature is based on particle number, typically measured by light scattering technologies (such as nanoparticle tracking analysis), with doses ranging from 109 to 1011 particles per administration[43,55]. Some studies also report doses in terms of the number of producer cells[56]. Table 1 summarizes recent research on the biodistribution of intravenously administered MSC-EVs in kidney disease models characterized by tubular damage.
In vivo biodistribution studies of intravenously administered MSC-EVs
| Isolation | Injury model | EV source | Dose | Labeling | Biodistribution | Effects observed | Time points | Ref. |
| dUC | UUO | BM-MSCs | 100 μg (mice) | PKH67 | Liver, lung, kidneys, brain, spleen, heart; Peak accumulation in obstructed kidney at 72 h | Interstitial fibrosis ↓ | 6, 12, 24, 72, 168, 240 h | [57] |
| hucMSCs | 10 mg/kg (rats) | DiR | Liver, lung, kidneys; Enriched in injured kidney | Interstitial fibrosis ↓ | 48 h | [58] | ||
| AMSCs | 1 × 103 μg/mL (mice) | Cy5 | Liver, lung, kidneys | Interstitial fibrosis ↓ Peritubular capillaries loss ↓ | 12 h | [59] | ||
| I/R | hPMSCs | 100 μg (mice) | Dil | Injured kidney | Interstitial fibrosis ↓ Mitochondrial FAO ↑ Mitochondrial homeostasis ↑ | Day 1, 3, 5, 7 | [60] | |
| BM-MSCs | 6.96 × 1010 particles (mice) | Sulfo-Cy7-NHS ester | Liver, lung, kidney, spleen; TECs at 24 h | Mitochondrial damage ↓ Inflammation ↓ | 1, 4, 6, 24 h | [42] | ||
| hucMSCs | 100 μg (rats) | PKH-26 | Kidney tissue, TECs | Interstitial fibrosis ↓ Angiogenesis ↑ VEGF↑ | 24 h | [56] | ||
| hPMSCs | 80 μg (mice) | AIEgen (DPA-SCP) | Liver, lung, spleen, kidneys; Enriched in injured kidney and TECs | Mitochondrial homeostasis ↑ | 2, 24, 72 h | [61] | ||
| Diabetic | hucMSCs | 10 mg/kg (mice) | Cy7/PKH67 | Liver, healthy kidneys, TECs | Apoptosis ↓ EMT ↓ | 24 h | [62] | |
| FA-induced CKD | iPSC-MSCs | 400 μg (mice) | CellTracker orange CMTMR tetramethylrhodamine | Kidney tissue | Interstitial fibrosis ↓ Inflammation ↓ Cell death ↓ | 66 h | [63] | |
| TFF | Cisplatin-induced AKI | AMSCs | 2 × 108 particles (mice) | Cy5.5 | Liver, kidneys; Enriched in injured kidney | Inflammation ↓ Proliferation ↑ Apoptosis ↓ | 48 h | [64] |
| AEC | Rhabdomyolysis-induced AKI | iPSC-MSCs | 150 μg (mice) | NanoLuc luciferase protein (NanoLuc-sEV) | Brain, lung, heart, liver, spleen, kidneys; Enriched in injured kidney | Inflammation ↓ Proliferation ↑ Klotho loss ↓ | 1 h | [65] |
| Commercial kit | UUO | Human MSCs | 1 mg/kg (mice) | PKH67 | Injured kidney | Interstitial fibrosis ↓ | 24, 48 h | [66] |
This table highlights a critical barrier: the considerable variability in EV dosage and assessment time points across studies. Even when the same isolation methods are employed, the lack of standardization poses a major obstacle for translating preclinical findings into clinical applications. Another inherent limitation is that most biodistribution studies rely on a single injection, whereas therapeutic studies typically involve multiple administrations. Furthermore, some reports have used the same EV dose for both in vivo animal studies and in vitro cell experiments, despite these contexts being fundamentally different and not directly comparable[43,56,61]. Although a standardized dosing protocol has yet to be established, significant efforts are required to bridge the gap between in vitro and in vivo research.
Evidence of MSC-EVs targeting TECs
Existing biodistribution data indicate that MSC-EVs exhibit therapeutic effects on tubular damage. Current research on EV-cell interactions mainly focuses on direct engagement, in which EVs are thought to be internalized by cells or interact with cell surface receptors. To substantiate the hypothesis of direct targeting to TECs, comprehensive in situ analyses are required to confirm the presence of MSC-EV within renal tubules. However, their rapid clearance from circulation to organs such as the lungs, spleen, and kidneys within minutes, coupled with challenges in accurate labeling and tracking, poses a significant challenge. Consequently, many studies investigating TECs provide limited evidence of direct targeting, reducing the strength of their conclusions regarding the targeting efficacy of MSC-EVs. Cao et al. conducted a real-time in vivo tracking study of MSC-EVs in a murine model of I/R injury[61]. To facilitate tracking, the researchers labeled MSC-EVs with aggregation-induced emission luminogens (AIEgens), specifically DPA-SCP. A dose of 80 µg of MSC-EVs was administered intravenously, and fluorescence imaging was performed at multiple time points between 2 and 72 h post-injection. The results demonstrated that MSC-EV signals peaked at
Challenges in targeting: passive vs active approaches
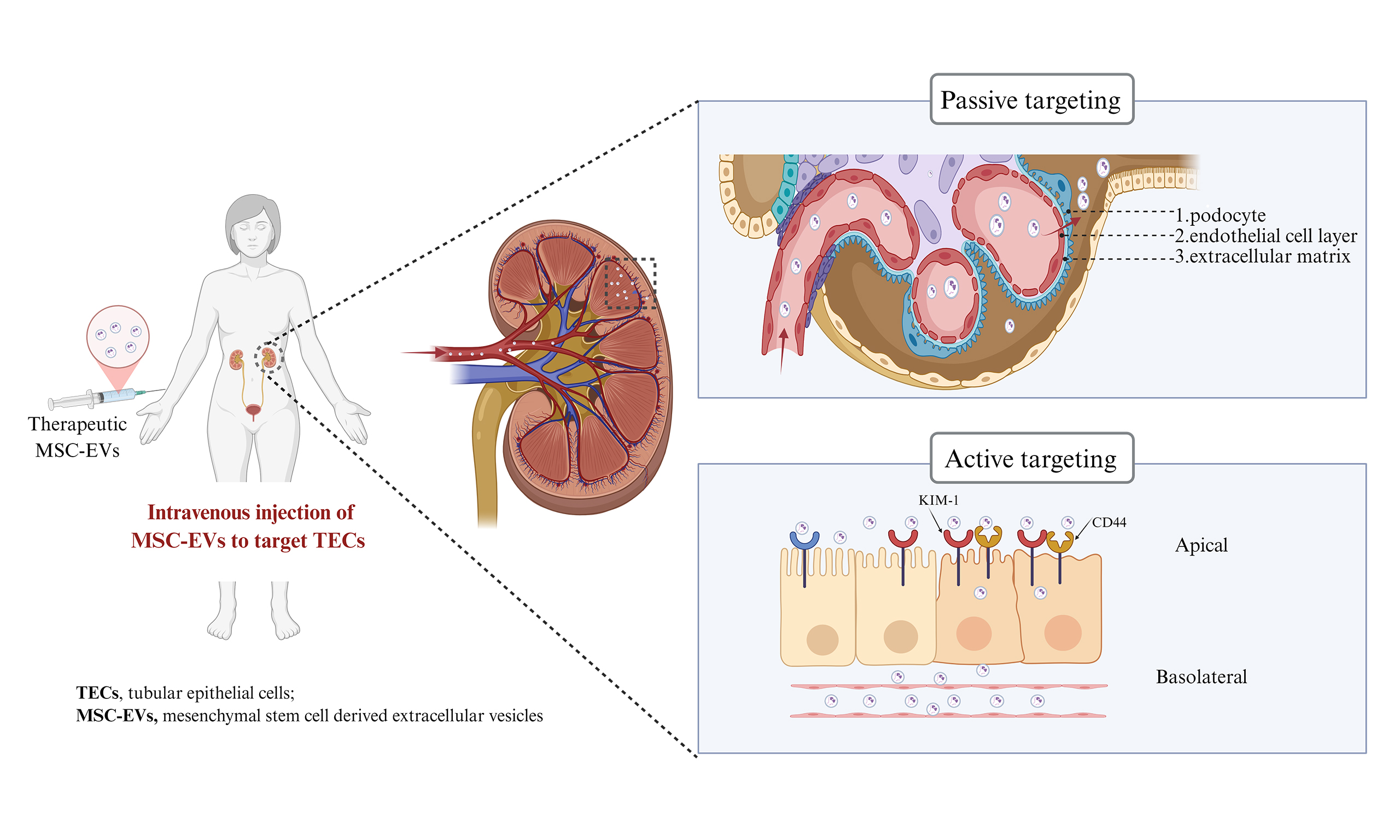
Nanoparticles in circulation, including MSC-EVs, are being investigated for their ability to selectively target TECs through both passive and active mechanisms [Figure 1]. Active targeting relies on specific ligand-receptor interactions between molecules expressed on TECs and MSC-EVs, facilitating therapeutic delivery via receptor-mediated internalization, as discussed in the “in vitro effects” section. For instance, the uptake of these vesicles by TECs has been demonstrated in vitro, and this process can be inhibited by antibodies against CD44 and CD29 on the MSC-EV membrane[31]. Notably, CD44, a ubiquitously expressed type I transmembrane glycoprotein with high affinity for hyaluronic acid (HA), is overexpressed in damaged TECs[67]. Engineered EVs derived from PEGylated, HA-modified MSCs selectively accumulate in injured kidneys through HA-mediated binding to CD44 on TECs[64]. Similarly, engineering EVs with specific ligands, such as peptides that bind to KIM-1, has been shown to enhance TEC targeting[35].
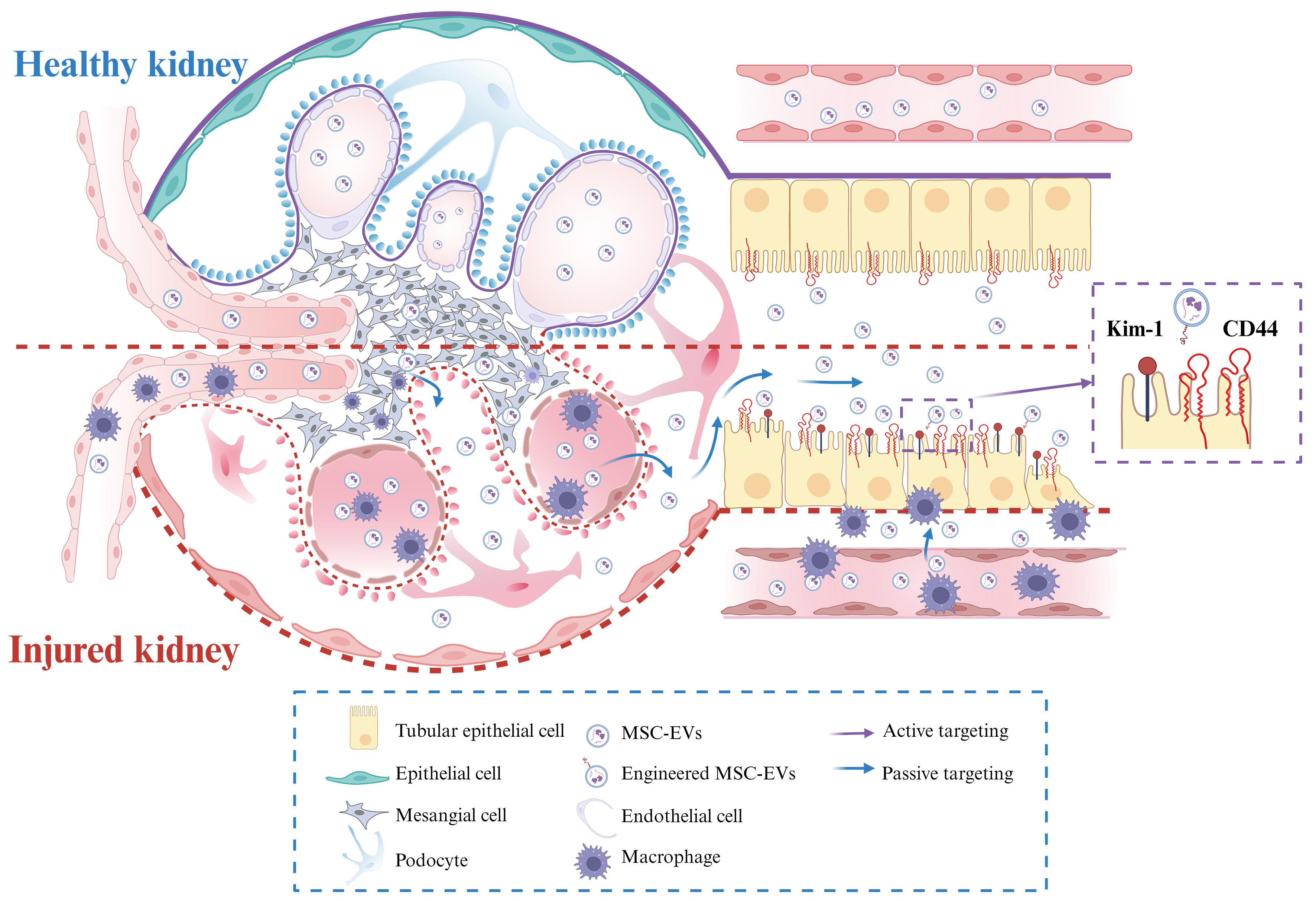
Figure 1. Schematic illustration of MSC-EV delivery and targeting to TECs. Kidney injury can involve disruption of the glomerular filtration barrier, inflammation, immune cell infiltration, and tubular epithelial cell injury. Passive targeting: MSC-EVs can extravasate through a compromised glomerular barrier to reach the tubular lumen or access the interstitial space via injury-induced increases in peritubular capillary permeability. Active targeting: MSC-EVs can be surface-modified for homing to injured TECs using ligands such as CD44-specific antibodies or peptides binding to KIM-1. Created in BioRender. pan, L. (2025) https://BioRender.com/zg9iyea. MSC-EV: Mesenchymal stem cell-derived extracellular vesicle; TEC: tubular epithelial cell; CD44: cluster of differentiation 44; KIM-1: kidney injury molecule-1.
Passive targeting, conversely, exploits the intrinsic physiological features of the kidney to promote preferential accumulation. Given the complex kidney architecture, which encompasses glomerular filtration and peritubular capillary circulation, nanoparticle size is a key determinant of distribution to TECs. Studies on renal nanomedicine delivery have identified two main theoretical passive pathways: (1) glomerular filtration followed by luminal uptake; and (2) transport through peritubular capillaries followed by basolateral uptake[68].
To reach TECs via glomerular filtration, MSC-EVs must first cross the endothelial cell layer, which is lined with a dense glycocalyx and fenestrations measuring 60-80 nm. Beyond this lies the glomerular basement membrane (GBM), a non-cellular meshwork of negatively charged ECM macromolecules with pores of approximately 10 nm. The GBM is externally covered by podocytes, whose interdigitating foot processes are separated by slit diaphragms averaging 12 nm. These structural constraints suggest that passive diffusion of intact EVs across a healthy GBM is unlikely[48,69]. However, under pathological conditions, an impaired GBM with enlarged pores may permit MSC-EV passage, often coinciding with proteinuria. Exploiting such altered barriers, MSC-EVs could potentially traverse the GBM and reach the tubular lumen. Still, whether natural MSC-EVs might also be captured by other injured cells, such as endothelial cells or podocytes, remains unclear. Because glomerular barrier damage is typically accompanied by injury to glomerular cells that can also capture EVs, direct TEC targeting under these conditions becomes more complex. The exact upper size limit for passive passage through a diseased GBM remains to be determined.
Alternatively, MSC-EVs may reach TECs via peritubular capillaries, which lie adjacent to the TEC basal membrane. This anatomical arrangement enables direct transport from the bloodstream into tubular compartments, bypassing glomerular filtration. Although peritubular capillary fenestrae are only ~5 nm, studies using synthetic nanoparticles (e.g., gold or silicon) have shown that particles of 100-200 nm and even 300-400 nm can reach peritubular capillaries and enter TECs through the basolateral membrane via endocytosis and exocytosis[70]. Kidney injury, which often occurs in clinical contexts, may further facilitate this process by increasing capillary permeability. Nonetheless, nanoparticle delivery to the tubules is influenced not only by size but also by charge, shape, and nanostructure - critical factors when designing engineered vesicles.
Taken together, future precision medicine strategies employing MSC-EVs to target TECs will likely require an integrated approach, combining passive mechanisms (leveraging nanoparticle size and pathological alterations in diseased kidneys) with active targeting strategies (using TEC-specific ligands) to enhance delivery efficiency while minimizing off-target effects.
DISCUSSION
Kidney diseases, including AKI and CKD, pose significant global health challenges. TECs play a pivotal role in both the initial injury and the maladaptive repair processes that lead to fibrosis, making them critical targets for therapeutic intervention. MSC-EVs have shown promising renoprotective effects, largely attributable to the bioactive molecules contained within their cargos. This review consolidates current evidence on the mechanisms by which MSC-EVs target and influence TECs, emphasizing both recent advancements and ongoing challenges.
In vitro studies provide compelling evidence that MSC-EVs exert direct therapeutic effects on damaged TECs by reducing apoptosis, inflammation, and oxidative stress. These effects are mediated through the transfer of molecules, such as miRNAs and proteins, which modulate key signaling pathways. Importantly, in vivo studies indicate that MSC-EVs can actively target TECs through specific ligand-receptor interactions. For example, integrins such as CD29 and CD44 have been implicated in MSC-EV uptake by TECs[31]. Additionally, engineering EVs with peptides that bind to markers upregulated on injured TECs, such as KIM-1, enhances targeted delivery[35].
Despite these encouraging results, data remain limited on whether and how MSC-EVs effectively reach TECs in vivo - a crucial consideration for designing precise targeting strategies. Biodistribution studies consistently show that systemically administered MSC-EVs predominantly accumulate in organs such as the liver, spleen, lungs, and kidneys, regardless of the administration route. Consequently, only a small proportion of the administered dose reaches the kidneys, and specifically TECs. Some in vivo tracking studies demonstrate co-localization of labeled MSC-EVs with renal tubules, suggesting uptake by TECs; however, they also reveal uptake by other cell types, such as macrophages, which contribute to kidney repair. This suggests that the renoprotective effects of MSC-EVs are likely mediated by a coordinated “network-level” modulation involving multiple cell parts and intercellular communication within the injured kidney.
The precise mechanisms by which MSC-EVs reach TECs in vivo are not yet fully elucidated. Both passive and active targeting processes are likely involved. Passive targeting is influenced by the physiological and pathological state of the kidney. Two plausible pathways include: (1) glomerular filtration followed by luminal uptake; and (2) access via peritubular capillaries followed by basolateral uptake. Under normal conditions, the glomerular filtration barrier largely restricts EV passage due to their size. While kidney injury may increase pore size, the degree to which intact EVs can traverse this barrier and be selectively internalized by TECs remains unclear. In contrast, peritubular capillary access circumvents the glomerular filtration process, providing direct entry to the basal membrane of TECs. Research involving synthetic nanoparticles indicates that particles of similar size to EVs can reach peritubular capillaries and enter TECs, a process potentially facilitated by increased capillary permeability after injury. In addition to size, EV characteristics such as charge and shape can modulate passive targeting.
A notable limitation in the current literature is the inconsistency in experimental methodologies, especially regarding MSC-EV isolation, characterization, and dosing. Different isolation techniques yield heterogeneous EV populations that are often inadequately defined, with many studies failing to specify the EV subtypes analyzed. Although particle size distributions frequently indicate a predominance of small vesicles, the lack of precise classification complicates dose quantification based on protein content. The wide variation in doses, coupled with the inappropriate application of identical doses for both in vitro and in vivo studies, impedes meaningful comparisons. Additionally, comprehensive in vivo biodistribution studies and high-resolution tracking are often lacking, limiting the ability to definitively confirm TEC-specific uptake and clarify underlying mechanisms. Real-time in vivo EV tracking remains a significant technical challenge.
Future research efforts should prioritize enhancing the targeted delivery of MSC-EVs to TECs. This involves optimizing EV characteristics to facilitate passive targeting and, importantly, engineering EVs with ligands that bind to markers upregulated on injured TECs, such as KIM-1 or CD44. Studies must rigorously report methodological details, including EV subtypes, isolation and purification techniques, and comprehensive characterization data. Establishing standardized protocols for EV isolation and quantification is essential to ensure reproducibility. Moreover, advanced in vivo biodistribution studies employing high-resolution tracking methods are necessary to verify targeted delivery, quantify the actual dose reaching TECs, and elucidate uptake mechanisms.
CONCLUSION
MSC-EVs exhibit considerable potential for the treatment of kidney diseases by mitigating TEC damage. This potential is substantiated by robust in vitro evidence demonstrating direct protective effects and specific cellular interactions. However, clinical translation is challenged by nonspecific biodistribution and methodological inconsistencies in EV production and characterization. To overcome these obstacles, future research should focus on: (1) standardizing reporting metrics, including particle number, producing cell number, protein content for dosing, and isolation methods; and (2) developing engineered EVs functionalized with ligands targeting markers of injured TECs, such as KIM-1. Combined with advanced
DECLARATIONS
Acknowledgments
The graphic abstract were created in Biorender [pan, L. (2025) https://BioRender.com/g9vqetp]
Authors’ contributions
Writing original draft: Pan L, Franquesa M, Garcia SG
Provided intellectual input and contributed substantially to the editing of the manuscript: Garcia SG, Franquesa M
Review and editing: Garcia SG, Franquesa M, Clos-Sansalvador M, Font-Morón M, Sanroque-Muñoz M, Borràs FE, de Miguel Garcia G
Table and graph: Pan L, Font-Morón M, Franquesa M
Availability of data and materials
Not applicable.
Financial support and sponsorship
This study was supported by the Instituto de Salud Carlos III (ISCIII) through the project “PI20/00097” and the RICORS networks RD21/0005/0009 and RD24/0004/0005, all co‐funded by the European Union. Pan L is supported by a CSC-UAB fellowship. Garcia SG is supported by a grant from the Catalan Health department (“Departament de Salut”) PERIS‐PIF‐Salut (SLT017/20/000158). de Miguel Garcia G is funded by Ministerio de Ciencia, Innovación y Universidades/Agencia Estatal de Investigación (MICIU/AEI) (DIN2024-01340). Borràs FE and Franquesa M are researchers from Germans Trias i Pujol Health Science Research Institute, supported by the Health Department of the Catalan Government (Generalitat de Catalunya).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
1. Kosanović M, Milutinovic B, Glamočlija S, Morlans IM, Ortiz A, Bozic M. Extracellular vesicles and acute kidney injury: potential therapeutic avenue for renal repair and regeneration. IJMS. 2022;23:3792.
2. Reiss AB, Jacob B, Zubair A, Srivastava A, Johnson M, De Leon J. Fibrosis in chronic kidney disease: pathophysiology and therapeutic targets. JCM. 2024;13:1881.
3. Foreman KJ, Marquez N, Dolgert A, et al. Forecasting life expectancy, years of life lost, and all-cause and cause-specific mortality for 250 causes of death: reference and alternative scenarios for 2016-40 for 195 countries and territories. Lancet. 2018;392:2052-90.
4. Qi R, Yang C. Renal tubular epithelial cells: the neglected mediator of tubulointerstitial fibrosis after injury. Cell Death Dis. 2018;9:1126.
5. Li ZL, Li XY, Zhou Y, Wang B, Lv LL, Liu BC. Renal tubular epithelial cells response to injury in acute kidney injury. EBioMedicine. 2024;107:105294.
6. Zou Y, Yiu WH, Lok SWY, et al. Tubular FoxP2 and kidney fibrosis. J Am Soc Nephrol. 2025;36:544-58.
7. Lake BB, Menon R, Winfree S, et al; KPMP Consortium. An atlas of healthy and injured cell states and niches in the human kidney. Nature. 2023;619:585-94.
8. Clark JZ, Chen L, Chou CL, Jung HJ, Lee JW, Knepper MA. Representation and relative abundance of cell-type selective markers in whole-kidney RNA-Seq data. Kidney Int. 2019;95:787-96.
9. Balzer MS, Rohacs T, Susztak K. How many cell types are in the kidney and what do they do? Annu Rev Physiol. 2022;84:507-31.
10. Koh ES, Chung S. Recent update on acute kidney injury-to-chronic kidney disease transition. Yonsei Med J. 2024;65:247-56.
11. Liu BC, Tang TT, Lv LL, Lan HY. Renal tubule injury: a driving force toward chronic kidney disease. Kidney Int. 2018;93:568-79.
12. Zhou J, Shi Y. Mesenchymal stem/stromal cells (MSCs): origin, immune regulation, and clinical applications. Cell Mol Immunol. 2023;20:555-7.
13. Lazarus HM, Haynesworth SE, Gerson SL, Rosenthal NS, Caplan AI. Ex vivo expansion and subsequent infusion of human bone marrow-derived stromal progenitor cells (mesenchymal progenitor cells): implications for therapeutic use. Bone Marrow Transplant. 1995;16:557-64.
14. Ceccotti E, Quaglia M, Camussi G, Bruno S. Mesenchymal stem cells derived extracellular vesicles for chronic kidney disease: pleiotropic mechanisms of actions of a versatile therapy. Front Bioeng Biotechnol. 2025;13:1612193.
15. Zheng Y, Wang H, Li X, Xie J, Fan J, Ren S. Extracellular vesicles in chronic kidney disease: diagnostic and therapeutic roles. Front Pharmacol. 2024;15:1371874.
16. Kosanović M, Milutinović B, Kutzner TJ, Mouloud Y, Bozic M. Clinical prospect of mesenchymal stromal/stem cell-derived extracellular vesicles in kidney disease: challenges and the way forward. Pharmaceutics. 2023;15:1911.
17. Welsh JA, Goberdhan DCI, O'Driscoll L, et al; MISEV Consortium. Minimal information for studies of extracellular vesicles (MISEV2023): from basic to advanced approaches. J Extracell Vesicles. 2024;13:e12404.
18. Barr SI, Abd El-Azeem EM, Bessa SS, Mohamed TM. Role of exosomes in pathogenesis, diagnosis, and treatment of diabetic nephropathy. BMC Nephrol. 2025;26:230.
19. Battistelli M, Falcieri E. Apoptotic bodies: particular extracellular vesicles involved in intercellular communication. Biology. 2020;9:21.
20. Witwer KW, Van Balkom BWM, Bruno S, et al. Defining mesenchymal stromal cell (MSC)-derived small extracellular vesicles for therapeutic applications. J Extracell Vesicles. 2019;8:1609206.
21. Tang T, Zhang Y, Crowley SD, Lv L, Liu B. Shedding light on the role of extracellular vesicles in renal fibrosis. Fundam Res. 2024;Epub ahead of print.
22. Huang Y, Yang L. Mesenchymal stem cells and extracellular vesicles in therapy against kidney diseases. Stem Cell Res Ther. 2021;12:219.
23. Birtwistle L, Chen XM, Pollock C. Mesenchymal stem cell-derived extracellular vesicles to the rescue of renal injury. Int J Mol Sci. 2021;22:6596.
24. Convento MB, de Oliveira AS, Boim MA, Borges FT. Umbilical cord mesenchymal stem cell-derived extracellular vesicles as natural nanocarriers in the treatment of nephrotoxic injury in vitro. Cells. 2024;13:1658.
25. Cao JY, Wang B, Tang TT, et al. Exosomal miR-125b-5p deriving from mesenchymal stem cells promotes tubular repair by suppression of p53 in ischemic acute kidney injury. Theranostics. 2021;11:5248-66.
26. Hong S, Kim H, Kim J, Kim S, Park TS, Kim TM. Extracellular vesicles from induced pluripotent stem cell-derived mesenchymal stem cells enhance the recovery of acute kidney injury. Cytotherapy. 2024;26:51-62.
27. Yin S, Zhou S, Ren D, et al. Mesenchymal stem cell-derived exosomes attenuate epithelial-mesenchymal transition of HK-2 cells. Tissue Eng Part A. 2022;28:651-9.
28. Bian Z, Wang X, Zhu R, Chen S. miR-21-5p in extracellular vesicles obtained from adipose tissue-derived stromal cells facilitates tubular epithelial cell repair in acute kidney injury. Cytotherapy. 2023;25:310-22.
29. Liang M, Zhang D, Zheng D, He W, Jin J. Exosomes from miR-374a-5p-modified mesenchymal stem cells inhibit the progression of renal fibrosis by regulating MAPK6/MK5/YAP axis. Bioengineered. 2022;13:4517-27.
30. Wang W, Wang J, Liao D. Effects and mechanisms of extracellular vesicles in different models of acute kidney injury. Stem Cells Int. 2025;2025:1075016.
31. Lindoso RS, Collino F, Bruno S, et al. Extracellular vesicles released from mesenchymal stromal cells modulate miRNA in renal tubular cells and inhibit ATP depletion injury. Stem Cells Dev. 2014;23:1809-19.
32. Tomasoni S, Longaretti L, Rota C, et al. Transfer of growth factor receptor mRNA via exosomes unravels the regenerative effect of mesenchymal stem cells. Stem Cells Dev. 2013;22:772-80.
33. Mao J, Li C, Wu F, et al. MSC-EVs transferring mitochondria and related components: a new hope for the treatment of kidney disease. Front Immunol. 2022;13:978571.
34. Yu Y, Chen M, Guo Q, et al. Human umbilical cord mesenchymal stem cell exosome-derived miR-874-3p targeting RIPK1/PGAM5 attenuates kidney tubular epithelial cell damage. Cell Mol Biol Lett. 2023;28:12.
35. Tang TT, Wang B, Li ZL, et al. Kim-1 targeted extracellular vesicles: a new therapeutic platform for RNAi to Treat AKI. J Am Soc Nephrol. 2021;32:2467-83.
36. Huang TY, Chien MS, Su WT. Therapeutic potential of pretreatment with exosomes derived from stem cells from the apical papilla against cisplatin-induced acute kidney injury. Int J Mol Sci. 2022;23:5721.
37. Li JK, Yang C, Su Y, et al. Mesenchymal stem cell-derived extracellular vesicles: a potential therapeutic strategy for acute kidney injury. Front Immunol. 2021;12:684496.
38. Nho JH, Jung HK, Lee MJ, et al. Beneficial effects of cynaroside on cisplatin-induced kidney injury in vitro and in vivo. Toxicol Res. 2018;34:133-41.
39. Wang SY, Xu Y, Hong Q, Chen XM, Cai GY. Mesenchymal stem cells ameliorate cisplatin-induced acute kidney injury via let-7b-5p. Cell Tissue Res. 2023;392:517-33.
40. Liang J, Liu Y. Animal models of kidney disease: challenges and perspectives. Kidney360. 2023;4:1479-93.
41. Wang B, Jia H, Zhang B, et al. Pre-incubation with hucMSC-exosomes prevents cisplatin-induced nephrotoxicity by activating autophagy. Stem Cell Res Ther. 2017;8:75.
42. Zhao M, Liu S, Wang C, et al. Mesenchymal stem cell-derived extracellular vesicles attenuate mitochondrial damage and inflammation by stabilizing mitochondrial DNA. ACS Nano. 2021;15:1519-38.
43. Lopes JA, Collino F, Rodrigues-Ferreira C, et al. Early effects of extracellular vesicles secreted by adipose tissue mesenchymal cells in renal ischemia followed by reperfusion: mechanisms rely on a decrease in mitochondrial anion superoxide production. Int J Mol Sci. 2022;23:2906.
44. Mu YF, Mao ZH, Pan SK, et al. Macrophage-driven inflammation in acute kidney injury: therapeutic opportunities and challenges. Transl Res. 2025;278:1-9.
45. Eirin A, Zhu XY, Puranik AS, et al. Mesenchymal stem cell-derived extracellular vesicles attenuate kidney inflammation. Kidney Int. 2017;92:114-24.
46. Guo Q, Li P, Chen M, et al. Exosomes from human umbilical cord stem cells suppress macrophage-to-myofibroblast transition, alleviating renal fibrosis. Inflammation. 2024;47:2094-107.
47. Jeon JS, Kim E, Bae YU, et al. microRNA in extracellular vesicles released by damaged podocytes promote apoptosis of renal tubular epithelial cells. Cells. 2020;9:1409.
48. Kang M, Jordan V, Blenkiron C, Chamley LW. Biodistribution of extracellular vesicles following administration into animals: a systematic review. J Extracell Vesicles. 2021;10:e12085.
49. Liu Y, Cui J, Wang H, et al. Enhanced therapeutic effects of MSC-derived extracellular vesicles with an injectable collagen matrix for experimental acute kidney injury treatment. Stem Cell Res Ther. 2020;11:161.
50. Malvicini R, De Lazzari G, Tolomeo AM, et al. Influence of the isolation method on characteristics and functional activity of mesenchymal stromal cell-derived extracellular vesicles. Cytotherapy. 2024;26:157-70.
51. Llorens-Revull M, Martínez-González B, Quer J, et al. Comparison of extracellular vesicle isolation methods for miRNA sequencing. Int J Mol Sci. 2023;24:12183.
52. Clos-Sansalvador M, Monguió-Tortajada M, Roura S, Franquesa M, Borràs FE. Commonly used methods for extracellular vesicles’ enrichment: implications in downstream analyses and use. Eur J Cell Biol. 2022;101:151227.
53. Wang SJ, Qiu ZZ, Chen FW, et al. Bone marrow mesenchymal stem cell-derived extracellular vesicles containing miR-181d protect rats against renal fibrosis by inhibiting KLF6 and the NF-κB signaling pathway. Cell Death Dis. 2022;13:535.
54. Gupta D, Zickler AM, El Andaloussi S. Dosing extracellular vesicles. Adv Drug Deliv Rev. 2021;178:113961.
55. Noda P, Francini ALR, Teles F, et al. Extracellular vesicles (EVs) derived from mesenchymal stem cells (MSCs) as adjuvants in the treatment of chronic kidney disease (CKD). Cells. 2025;14:434.
56. Zou X, Gu D, Xing X, Cheng Z, Gong D, Zhang G. Human mesenchymal stromal cell-derived extracellular vesicles alleviate renal ischemic reperfusion injury and enhance angiogenesis in rats. Am J Transl Res. 2016;8:4289-99.
57. Hu X, Shen N, Liu A, et al. Bone marrow mesenchymal stem cell-derived exosomal miR-34c-5p ameliorates RIF by inhibiting the core fucosylation of multiple proteins. Mol Ther. 2022;30:763-81.
58. Shi L, Hu Y, Zeng H, et al. Mesenchymal stem cell-derived extracellular vesicles ameliorate renal interstitial fibrosis via the miR-13474/ADAM17 axis. Sci Rep. 2024;14:17703.
59. Zhang X, Zhao J, Ge R, et al. Arg-Gly-Asp engineered mesenchymal stem cells as targeted nanotherapeutics against kidney fibrosis by modulating m6A. Acta Biomater. 2025;198:85-101.
60. Gao Z, Zhang C, Peng F, et al. Hypoxic mesenchymal stem cell-derived extracellular vesicles ameliorate renal fibrosis after ischemia-reperfusion injure by restoring CPT1A mediated fatty acid oxidation. Stem Cell Res Ther. 2022;13:191.
61. Cao H, Cheng Y, Gao H, et al.
62. Cui C, Zang N, Song J, et al. Exosomes derived from mesenchymal stem cells attenuate diabetic kidney disease by inhibiting cell apoptosis and epithelial-to-mesenchymal transition via miR-424-5p. FASEB J. 2022;36:e22517.
63. Kim H, Hong S, Kim S, Kim TM. Extracellular vesicles from induced mesenchymal stem cells inhibit acute kidney injury to chronic kidney disease transition. Int J Stem Cells. 2025;18:286-300.
64. Kim SH, Kim CH, Lee CH, et al. Glycoengineered stem cell-derived extracellular vesicles for targeted therapy of acute kidney injury. Biomaterials. 2025;318:123165.
65. Deng XH, Wu ZC, Sun Q, et al. The effects of Klotho delivering mesenchymal stem cell-derived small extracellular vesicles on acute kidney injury. J Nanobiotechnology. 2025;23:427.
66. Yang Y, Wang J, Zhang Y, Hu X, Li L, Chen P. Exosomes derived from mesenchymal stem cells ameliorate renal fibrosis via delivery of miR-186-5p. Hum Cell. 2022;35:83-97.
67. Matsushita K, Toyoda T, Akane H, Morikawa T, Ogawa K. Role of CD44 expressed in renal tubules during maladaptive repair in renal fibrogenesis in an allopurinol-induced rat model of chronic kidney disease. J Appl Toxicol. 2024;44:455-69.
68. Huang Y, Wang J, Jiang K, Chung EJ. Improving kidney targeting: the influence of nanoparticle physicochemical properties on kidney interactions. J Control Release. 2021;334:127-37.
69. Huang Y, Ning X, Ahrari S, et al. Physiological principles underlying the kidney targeting of renal nanomedicines. Nat Rev Nephrol. 2024;20:354-70.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].