

Advances in high-temperature shock technology for structural engineering of electrode materials: a review
 0
0 Abstract
Precise control of structure is critical for improving electrochemical performance of electrode materials. Developing effective strategies for structural engineering has therefore become a central pursuit in advancing energy storage and conversion systems. In this aspect, high-temperature shock (HTS) technology has emerged as a powerful tool for engineering electrode materials with precise structural control. By utilizing ultrafast Joule heating to create extreme thermal gradients, HTS enables the rapid synthesis of materials in non-equilibrium states, providing new possibilities for their design. This review focuses on how HTS can be used to achieve designed structural features such as size modulation, defect engineering, phase transformation, and interfacial optimization. Specifically, the unique HTS strategy promotes the creation of nanoscale materials with superior electrochemical properties, while also facilitating the formation and healing of defects that enhance charge storage and transport. HTS not only induces phase transformations but can also stabilize complex phases, such as high-entropy alloys. Moreover, HTS facilitates sophisticated interface engineering, which plays a key role in improving electrochemical performance and durability. The structural regulation enabled by HTS not only provides a distinct perspective on materials design but also exhibits great potential in practical applications, such as advancing lithium- and sodium-ion batteries, optimizing electrocatalysts in fuel cells, and improving efficiency in water splitting. Overall, this review highlights the transformative potential of HTS in the design of next-generation electrode materials for energy storage and conversion applications.
Keywords
INTRODUCTION
The fast-growing demand for efficient energy technologies has stimulated intensive research into the design of functional electrode materials for both energy storage and conversion[1-4]. Recent studies have revealed that rather than solely relying on chemical composition, the structural characteristics at multiple length scales of the electrode material play a decisive role in determining its performance, including defect concentration, phase distribution, interface stability and grain size[5-8]. For example, the introduction of atomic-scale point defects can modulate the local electronic structure, introduce abundant active sites, and accelerate ion/electron transport kinetics[9]. Meanwhile, the coexistence of different crystalline phases within a single material is designed to form heterogeneous but functionally synergistic regions for optimizing performance[10]. At larger length scales, constructing hierarchical nanostructures and reducing grain size enhance electrochemically active surface area, shorten diffusion distance and maintain interface stability[11-13]. In this regard, structural engineering from the atomic to the mesoscale is considered a key strategy to improve the electrochemical performance of electrode materials.
Recent years have witnessed the successful employment of various traditional synthesis strategies, including sol-gel, hydro/solvothermal methods, and solid-state reactions, to construct special-structure electrode materials[14-18]. For example, the sol-gel route enables homogeneous mixing of metallic species at the molecular level and thus facilitates the formation of coated materials, while hydrothermal processes are effective in directing the growth of well-defined nanostructures[19-22]. However, these techniques still rely on relatively slow heat transfer and long-duration thermal treatment under near-equilibrium conditions. From a physico-chemical point of view, such quasi-equilibrium processing provides sufficient time for thermodynamic relaxation, leading to the gradual elimination of high-energy defects and the transformation of metastable phases into thermodynamically stable structures[23-25]. In addition, extended dwelling at high temperature promotes continuous grain growth and local strain relaxation, thereby reducing the structural imperfections that often enhance ion transport kinetics. These kinetic limitations inevitably restrict the degree of structural tunability that can be achieved, making it challenging to precisely maintain defect-rich or strain-coupled lattice states during synthesis.
High-temperature shock (HTS) synthesis technology - also referred to as Joule heating or carbon thermal shock (CTS) - is an emerging materials synthesis strategy that relies on the rapid conversion of electrical energy into heat by passing current through a resistive substance. HTS offers a fundamentally different approach by driving chemical reactions under non-equilibrium conditions[26-30]. In HTS processes, the temperature of the precursor material is rapidly raised to several hundred or thousand degrees Celsius within a short time (typically < 1 min), followed by rapid cooling. The extreme heating and cooling rates give rise to localized stress fields inside the material, which can dramatically lower the kinetic barrier for defect formation and lattice reconstruction. In addition, the abrupt heat input can promote partial phase reorganization, providing access to metastable structures that cannot be obtained via conventional thermal processes. Meanwhile, the fast precursor decomposition promotes instantaneous nucleation, which can suppress grain growing and favor the formation of nanoscale architectures with good uniformity[24,31-33].
These unique non-equilibrium features endow HTS with great potential to manipulate electrode materials at multiple structural levels, including the generation of non-stoichiometric defects, local lattice reordering, nanoscale phase redistribution, and size regulation of particles[34-38]. They allow HTS not only to reveal dynamic processes of material evolution under extreme conditions but also to create structures difficult to achieve through conventional synthesis. Such advances have direct implications for practical applications, including enhancing the energy density and stability of lithium-ion and sodium-ion batteries, improving catalytic efficiency and durability in fuel cells, and accelerating electrochemical reactions in water splitting. Consequently, HTS is emerging as a powerful strategy to bridge structural engineering with the pressing demands of materials for future energy storage and conversion devices. Several reviews have previously discussed Joule heating or HTS techniques from different views[34,35,39]. An extensive survey of the literature reveals that most existing reviews primarily emphasize the broad applications of HTS in energy storage, catalysis, and material recycling, while systematic discussions of its structural modulation mechanisms remain limited. With growing interest in the structure-property relationships of electrochemical materials, it is highly desirable to systematically summarize recent progress in HTS-assisted synthesis, particularly the underlying physico-chemical principles and emerging strategies for constructing specialized structures to meet the demanding requirements of next-generation electrode materials for energy storage and conversion.
HIGH-TEMPERATURE SHOCK
HTS is a non-equilibrium synthesis method that relies on electrical current to generate intense heat within a conductive element. HTS synthesis can typically be categorized into two modes, depending on the heat source [Figure 1]. In the first mode, electric current directly passes through a conductive precursor, enabling rapid Joule heating and triggering ultrafast reactions within the material itself. In the second mode, the precursor is placed on a conductive substrate, such as a metal foil or carbon cloth, where the current flows through the substrate and induces resistive heating. The generated heat is then transferred to the precursor, driving its transformation[39-41]. Because heat is generated internally and directly transferred to the precursor, temperatures exceeding 3,000 K, with heating and cooling rates of 104 ~ 105 K/s, can be achieved within milliseconds to seconds, far surpassing the capabilities of traditional furnace systems [Figure 2A and B].
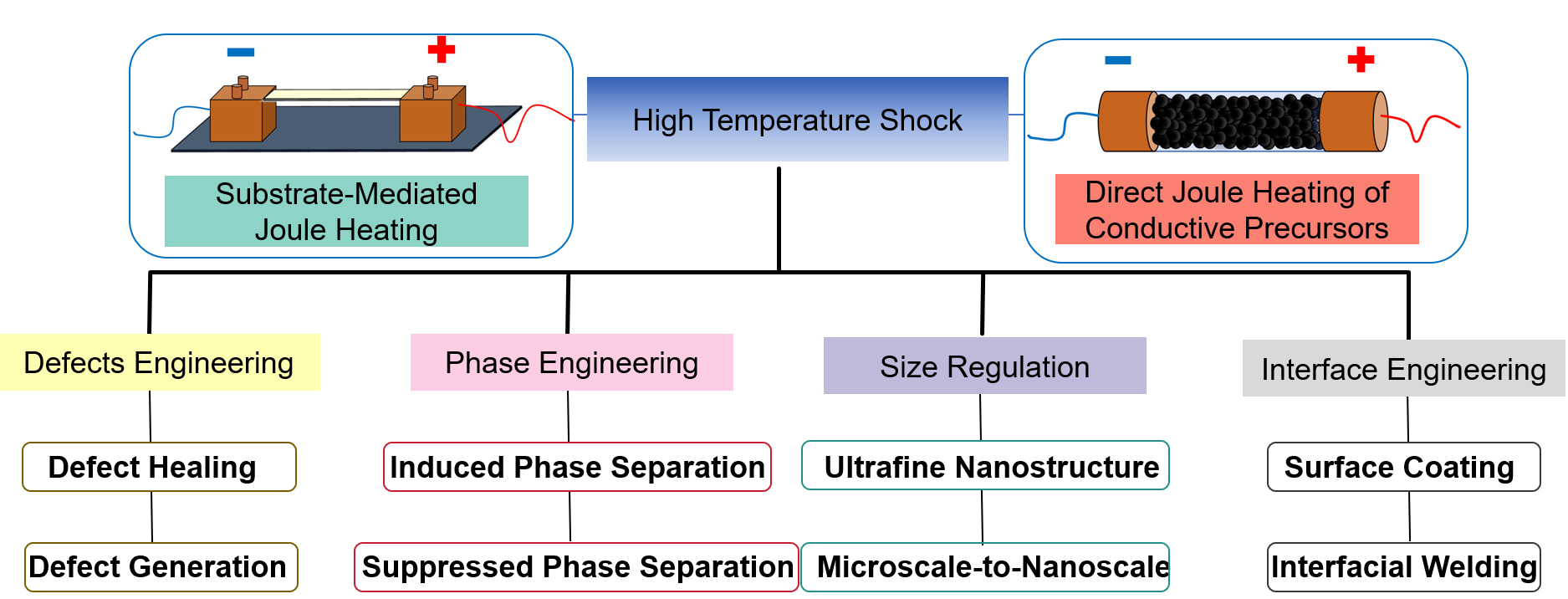
Figure 1. Two representative heating modes of HTS and its structural regulation in electrode materials. HTS: High-temperature shock.
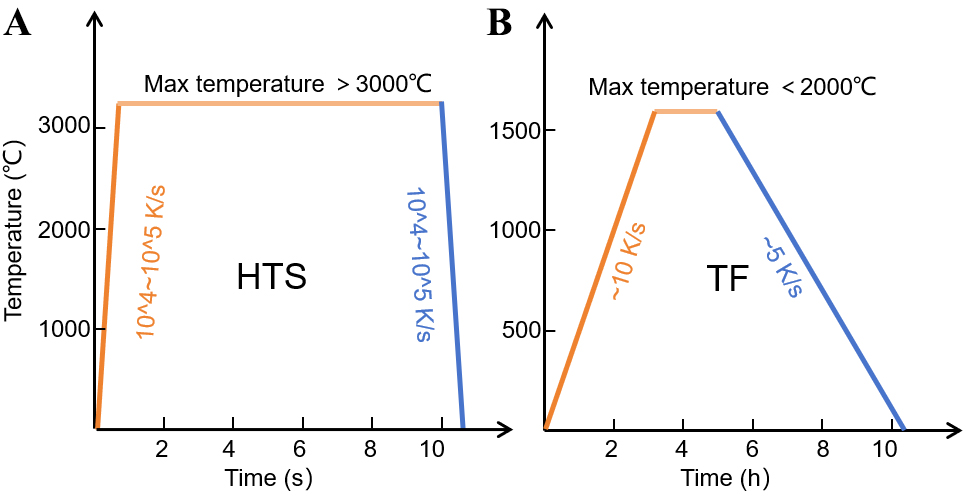
Figure 2. Comparison of two heating methods: (A) high temperature shock and (B) tube furnace. HTS: High-temperature shock; TF: tube furnace.
The design of HTS devices is crucial for achieving precise control of material structure and reproducible material synthesis. The device typically includes a high current power supply capable of delivering controlled current, a sample stage (such as a fixture or electrode) that ensures minimum resistance and uniform current distribution on the sample, a high-sensitivity temperature measurement device, and a reaction chamber that allows for flexible control of the atmosphere (inert, reducing, or oxidizing). Parameters such as current density, pulse duration, voltage, and heating/cooling rate are key to controlling the intensity of thermal shock and the time scale of structural transformation. The sample stage and the contact between the sample stage and the sample determine the current path, heating uniformity, and rate, which are important for inducing specific structural features. The reaction atmosphere - inert, reducing, or oxidizing - can also significantly affect phase stability and chemical reactions. In addition, the cooling rate can affect defect healing and phase equilibrium. Moreover, accurate temperature measurement and careful insulation to prevent energy loss are crucial for reproducibility.
The ultrafast heating and cooling rates inherent to HTS (often > 105 K s-1) drive materials far from equilibrium, creating unique thermodynamic and kinetic environments for structural evolution [Figure 1][35,39]. The rapid thermal ramp allows phase transitions to occur on extremely short time scales, which effectively minimizes thermal diffusion and inhibits phase separation or elemental segregation that would normally occur under slow heating. Equally important, the subsequent rapid quenching prevents the structural relaxation and grain growth commonly seen in conventional heat treatments. As a result, HTS is particularly effective at stabilizing non-equilibrium structures, retaining metastable crystal phases, and preserving local lattice distortions generated during the heating stage. Another distinct feature of HTS is that the thermal field is highly localized around the resistive element, producing steep thermal gradients and transient stress fields within a short time frame. These conditions can reduce the formation energy of structural defects, facilitate lattice reconstruction, and even promote partial redistribution of atomic species at the nanoscale. In addition, the violent thermal shock also promotes particle fragmentation and restrains grain growth, enabling the formation of nanosized or even ultrafine domains that shorten ion and electron transport pathways. These structural effects greatly reduce ion diffusion length and create more accessible active sites, ultimately improving the electrochemical reactivity and rate capability of the resulting material.
Benefiting from its fast kinetics, simple setup, and energy efficiency, HTS has demonstrated strong potential in the synthesis of advanced electrode materials, and has also been applied in electrocatalysis, metal alloying, and functional ceramic fabrication[35,42-45]. The flexible control over current density, resistance, and duration allows fine adjustment of heating intensity and energy delivery, making HTS well-suited for both laboratory studies and scalable production of high-performance materials with special structures.
DEFECT ENGINEERING ENABLED BY HTS
Defects are a critical structural element governing the electrochemical behavior of functional materials. Depending on the type and concentration, defects can either enhance ion/electron transport and surface activity, or, in contrast, lead to structural instability and degradation during long-term operation[46-48]. Unlike conventional thermodynamic synthesis, HTS provides an extremely rapid non-equilibrium pathway that can either generate useful defects within pristine materials or eliminate detrimental defects accumulated during electrochemical cycling under appropriate conditions. This dual capability makes HTS a versatile tool not only for the synthesis of defect-engineered electrode and catalyst materials, but also for the rapid regeneration of structurally deteriorated electrodes.
Defect generation
Defects are ubiquitous in crystalline solids and play an essential role in determining their electrochemical properties. On the one hand, defects could effectively increase the number of available ion migration sites and lower the activation barrier for ionic hopping between adjacent lattice positions. For example, twin boundaries can serve as preferential transport pathways, thereby enhancing kinetic performance and potentially improving structural stability by accommodating lattice strain[49-51]. On the other hand, defects often act as catalytically active centers by altering the electronic structure of surface sites and modifying the adsorption energy of reaction intermediates. For example, vacancies can enhance active sites to promote redox reactions and modify the electronic structure to enhance conductivity[52-54]. Consequently, defect engineering has emerged as an effective strategy for simultaneously improving performance and reaction kinetics.
Although conventional thermal processes can induce certain types of defects through high-temperature annealing or controlled doping, their effectiveness is limited due to long reaction times and near-equilibrium growth conditions, which tend to eliminate high-energy defects and restore long-range structural order. HTS, in contrast, provides a powerful approach for producing and stabilizing such structural imperfections. HTS instantly increases the energy of the precursor material to a high level, far exceeding the thermal equilibrium conditions achievable in traditional furnaces. The rapid temperature increase forces the lattice into a highly non-equilibrium state, which could significantly accelerate cation migration and exchange between sites, resulting in the formation of antisite defects and cation/anion vacancies. At the same time, the sudden cooling process prevents atomic motion, and these non-equilibrium defect structures are effectively “frozen” in the lattice, with no time for relaxation or migration back to equilibrium positions[34,35,39].
HTS has been widely employed in constructing functional materials with controlled defects. Extensive efforts have been devoted to demonstrating the application of high-temperature thermal shock in preparing defect-rich materials. For example, LiFePO4 cathodes synthesized through HTS show a high concentration of Li-Fe antisite defects (5.37%) and tensile lattice strain, which together create isotropic Li+ diffusion pathways and markedly enhance ionic conductivity [Figure 3A]. As a result, the HTS-treated LiFePO4 delivers superior rate performance, excellent low-temperature capability, and long-term cycling stability (126 mAh g-1 after 550 cycles at 1 C)[55]. Similarly, in layered oxides such as Li1.2Ni0.13Co0.13Mn0.54O2, HTS induces coherent twin boundaries that stabilize the layered structure, suppress oxygen loss, and mitigate phase transitions [Figure 3B], yielding a high discharge capacity of 278 mAh g-1 and robust cycling stability[56]. In the case of spinel LiMn2O4, HTS not only produces twin boundaries enhancing Li+ diffusion but also enables rapid dopant incorporation [Figure 3C], suppressing Jahn-Teller distortion and Mn dissolution; the resulting Ni-doped spinel exhibits excellent long-term stability with 86.5% capacity retention after 500 cycles[57].
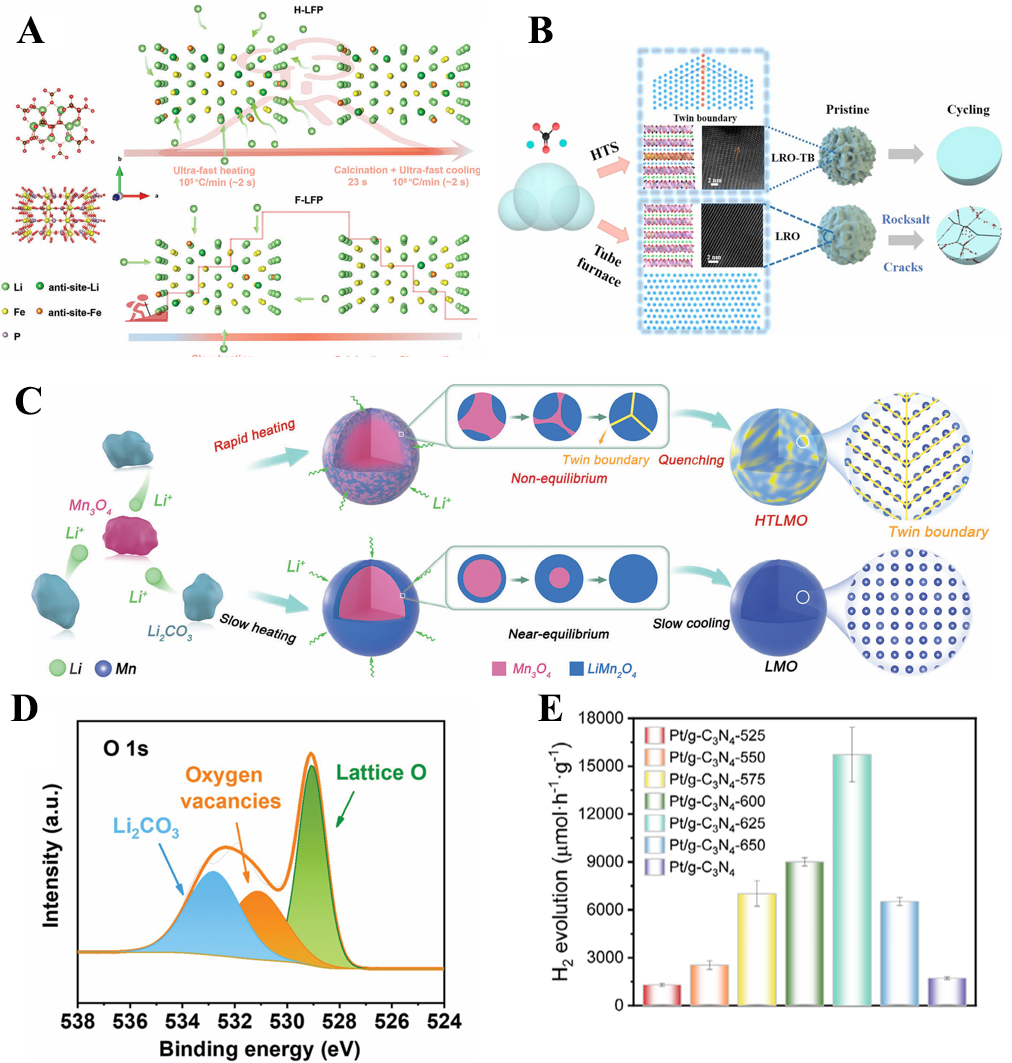
Figure 3. Schematic illustration of (A) Li+ diffusion in HTS calcination and furnace calcination process[55]; (B) synthesis of Li1.2Ni0.13Co0.13Mn0.54O2 with and without twin boundaries[56] and (C) preparation of LiMn2O4 through HTS and tube furnace[57]; (D) XPS spectrum of O 1s in LiMn2O4[58]; (E) Hydrogen production rate of Pt/g-C3N4[59]. HTS: High-temperature shock; XPS: X-ray photoelectron spectroscopy; g-C3N4: graphitic carbon nitride. LRO: Li1.2Ni0.13Co0.13Mn0.54O2; LRO-TB: Li1.2Ni0.13Co0.13Mn0.54O2 with twin boundaries; HTLMO: LiMn2O4 synthesized through HTS.
Beyond cation/anion defects, HTS has also been applied to deliberately introduce oxygen vacancies. For instance, in LiCoO2, LiMn2O4 [Figure 3D] and Li-rich oxides, HTS-driven oxygen deficiency improves electronic conductivity and simultaneously stabilizes the structure at high voltages, leading to improved rate capability and suppressed structural degradation. In spinel LiMn2O4, oxygen vacancies suppress Mn dissolution and reduce charge-transfer resistance[58]. Moreover, in non-oxide systems such as graphitic carbon nitride (g-C3N4), flash Joule heating generates abundant nitrogen vacancies and cyano groups, which act as electron traps and catalytic sites. The resulting defective Pt/g-C3N4 exhibits dramatically enhanced photocatalytic hydrogen evolution, greatly surpassing its defect-free counterpart [Figure 3E][59].
Taken together, these studies establish HTS as a universal and efficient strategy for defect engineering across diverse classes of electrode and catalytic materials. By leveraging ultrafast heating and cooling under non-equilibrium conditions, HTS enables the controllable introduction of various lattice imperfections - antisite defects, twin boundaries, and oxygen/nitrogen vacancies - that significantly boost ionic conductivity, charge-transfer kinetics, and structural stability. This capability makes HTS a transformative tool for developing next-generation energy materials with superior electrochemical and catalytic performance.
Defect healing and structural regeneration
During long‐term electrochemical cycling or catalytic operation, electrode materials inevitably undergo structural deterioration, including dislocation accumulation, cation/anion vacancy formation, antisite defects, and phase decomposition[60-62]. These degradation phenomena destroy long‐range order and impede ion transport pathways, leading to rapid capacity fading and performance degradation. Traditional recycling routes - such as calcination and hydrothermal treatment - are typically destructive, resource‐intensive and time-consuming[63-65]. By contrast, HTS has emerged as a highly energy-efficient regeneration strategy, enabling rapid elimination of structural defects within a few minutes. During the HTS process, the ultrafast heating triggers spontaneous thermodynamic reordering of the lattice, which facilitates the annihilation of dislocations and vacancies, relieves accumulated strain, and promotes the phase transformation of impurity phases back to their original crystalline forms. Importantly, the extremely short heating duration prevents unwanted phenomena such as lithium volatilization, surface overgrowth, or secondary phase nucleation, allowing the bulk lattice to regain long-range order, which not only repairs defects but also enhances the overall structural integrity of the material, making it more suitable for electrochemical applications[35,39,66].
This unique “rapid healing” capability has been demonstrated in various degraded electrode systems. For example, Yin et al. reported that rapid Joule heating of spent LiCoO2 at 1,440 K for 8 s not only repairs spinel Co3O4 impurities but also replenishes lithium vacancies via in situ relithiation [Figure 4A], successfully restoring the layered structure[67]. Remarkably, the regenerated cathode delivers an initial discharge capacity of 133 mAh g-1 and maintains ~ 100 mAh g-1 after 300 cycles at 0.1 C, which is almost equivalent to commercial fresh LiCoO2. The energy consumption and treatment time are greatly reduced compared with those of traditional electrode material regeneration schemes[67]. Similarly, Cheng et al. reported an electrothermal rejuvenation process performed at 750 °C for 30 s together with Mg/Al doping [Figure 4B], which restores Li/Co stoichiometry and eliminates oxygen vacancies in degraded LiCoO2[68]. The regenerated cathode exhibits a high discharge capacity of 203 mAh g-1 and shows 84% capacity retention over 200 cycles, outperforming even commercial LiCoO2 (only 4% retention under identical conditions)[68].
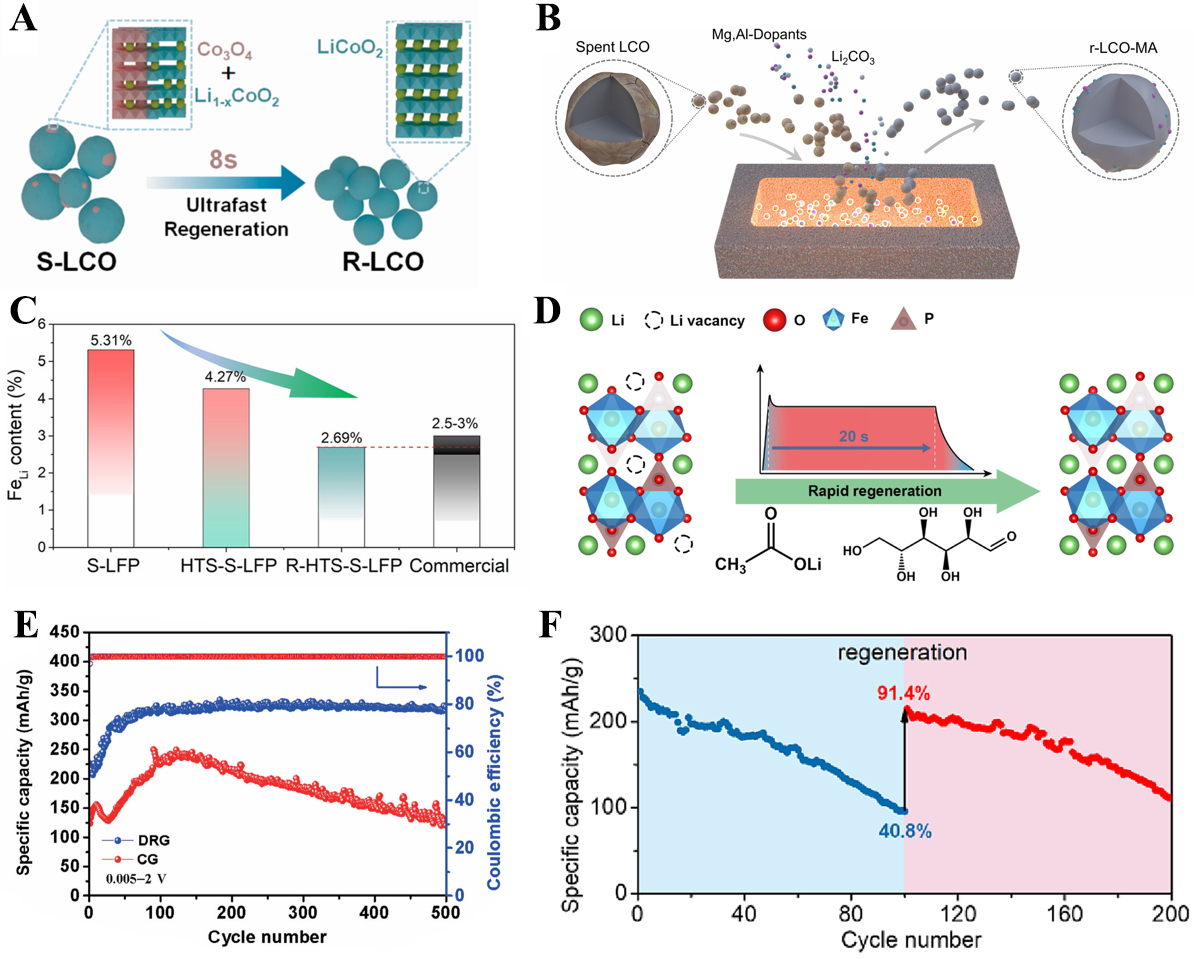
Figure 4. Schematic illustration of (A) regeneration of spent LiCoO2[67] and (B) spent LiCoO2 regeneration with doping[68]; (C) Decrease of Li/Fe antisite contents through HTS treatment[69]; (D) Schematic illustration of regeneration process for spent LiFePO4[70]; (E) Cycling stability of DRG and CG[71]; (F) Cycling performance of Li1.2Co0.4Nb0.4O2 before and after regeneration[72]. HTS: High-temperature shock; DRG: defect-rich regenerated graphite; CG: commercial graphite. S-LCO: R-LCO: LFP: r-LCO-MA:
The regeneration effect of HTS is not limited to layered cathode materials. In LiFePO4 (LFP) systems, Guo et al. showed that the regeneration treatment through HTS dramatically reduces Li/Fe antisite defects from 5.31% to 2.69% [Figure 4C], enabling efficient Li-ion transport across the olivine framework[69]. The regenerated cathodes recover a high discharge capacity of 156.7 mAh g-1 at 0.1 C and maintain 94.1 mAh g-1 at 5 C, demonstrating both excellent capacity retention and enhanced high-rate capability[69]. Zheng et al. further demonstrated that a 20-s HTS treatment combined with Li-acetate and sucrose efficiently compensates for Li loss and restores the olivine lattice [Figure 4D], enabling a significant improvement in the Li+ diffusion coefficient and achieving stable cycling over 400 cycles at 2 C without decay[70].
HTS has also shown great potential for the regeneration of anode and cation‐disordered rocksalt (DRX) materials. Luo et al. regenerated spent graphite anodes using a 1,500 °C shock treatment, selectively preserving beneficial atomic-level defects while restoring the layered graphite structure[71]. The defect-rich regenerated graphite (DRG) shows a remarkably high specific capacity of 323 mAh g-1 at 2 C and retains 323 mAh g-1 after 500 cycles - much higher than commercial graphite (CG) (~ 120 mAh g-1) [Figure 4E][71]. In addition, Luan et al. utilized HTS to repair lattice collapse in spent Li1.2Co0.4Nb0.4O2 DRX cathodes, fully restoring lattice fringes and recovering 91.4% of the original specific capacity [Figure 4F], with the regenerated materials exhibiting similar cycling stability to pristine electrode materials[72].
Collectively, these examples clearly demonstrate that HTS treatment provides a rapid, energy-efficient, and non-destructive route for defect elimination and structural regeneration across a broad spectrum of energy materials. By enabling the annihilation of bulk defects, recovery of long-range order, and preservation (or even optimization) of surface-active structures, HTS not only restores the original electrochemical performance but, in many cases, delivers enhanced rate capability and extended cycling stability. Therefore, HTS represents a highly promising “waste-to-value” strategy for sustainable regeneration and long-term utilization of electrochemical materials.
PHASE ENGINEERING VIA HTS
Phase engineering is another important aspect enabled by HTS. The extreme heating-cooling dynamics of HTS can drastically alter phase stability and transformation pathways, providing opportunities to manipulate phase structures difficult to achieve under conventional conditions. On the one hand, the non-equilibrium environment can induce phase separation or promote the formation of metastable phases with unique functionalities. On the other hand, the ultrafast quenching can suppress phase segregation and stabilize otherwise unstable solid solutions, such as in high-entropy alloys (HEAs). These dual capabilities highlight the versatility of HTS in tailoring phase structures for advanced electrode materials.
Induced phase separation
Heterogeneous phases, consisting of two or more structurally different domains such as crystalline/amorphous, layered/spinel and multiphase carbide interfaces, are highly desirable in functional materials. The coexistence of different phases often gives rise to synergistic properties, such as fast ion transport pathways, abundant active sites, and improved structural robustness during repeated cycling[73-75]. However, traditional solid-state or solvothermal synthesis routes typically rely on long-term heat treatment under equilibrium conditions, making it difficult to stabilize metastable phase combinations or precisely control the spatial distribution of multiple phases. Moreover, conventional processes often require complicated multi-step procedures, such as doping, pre-crystallization and post-annealing, to obtain the desired heterostructure[10,76].
The HTS process causes the precursor to instantly reach a high-energy non-equilibrium state, which induces rapid bond breaking, local lattice rearrangement, and selective nucleation of specific phases. As the material reaches high temperatures, these processes occur much faster than in conventional heating, preventing thermodynamic equilibrium from being achieved. The subsequent fast quenching “freezes” these transient phases, resulting in phase separation or heterostructure formation[77-79]. For example, Huang et al. reported a spatiotemporally controlled electrothermal strategy via Joule heating that produces hard carbon containing graphitic microcrystals with enlarged interlayer spacing coexisting with amorphous, pore-rich regions [Figure 5A][80]. The resulting heterostructure delivered a high sodium-storage capacity of 306.83 mAh g-1, a high initial Coulombic efficiency of ~ 92%, and long-term stability (79.45% capacity retention after 1,000 cycles)[80]. Similarly, Duan et al. achieved a crystalline-amorphous vanadium oxide heterostructure (V2O3/a-NVO) using flash Joule heating [Figure 5B], which provided complementary functions of fast charge transfer and rich Zn2+ storage sites, achieving a high specific capacity of 138 mAh g-1 after 5,000 cycles in aqueous zinc-ion batteries [Figure 5C][81].
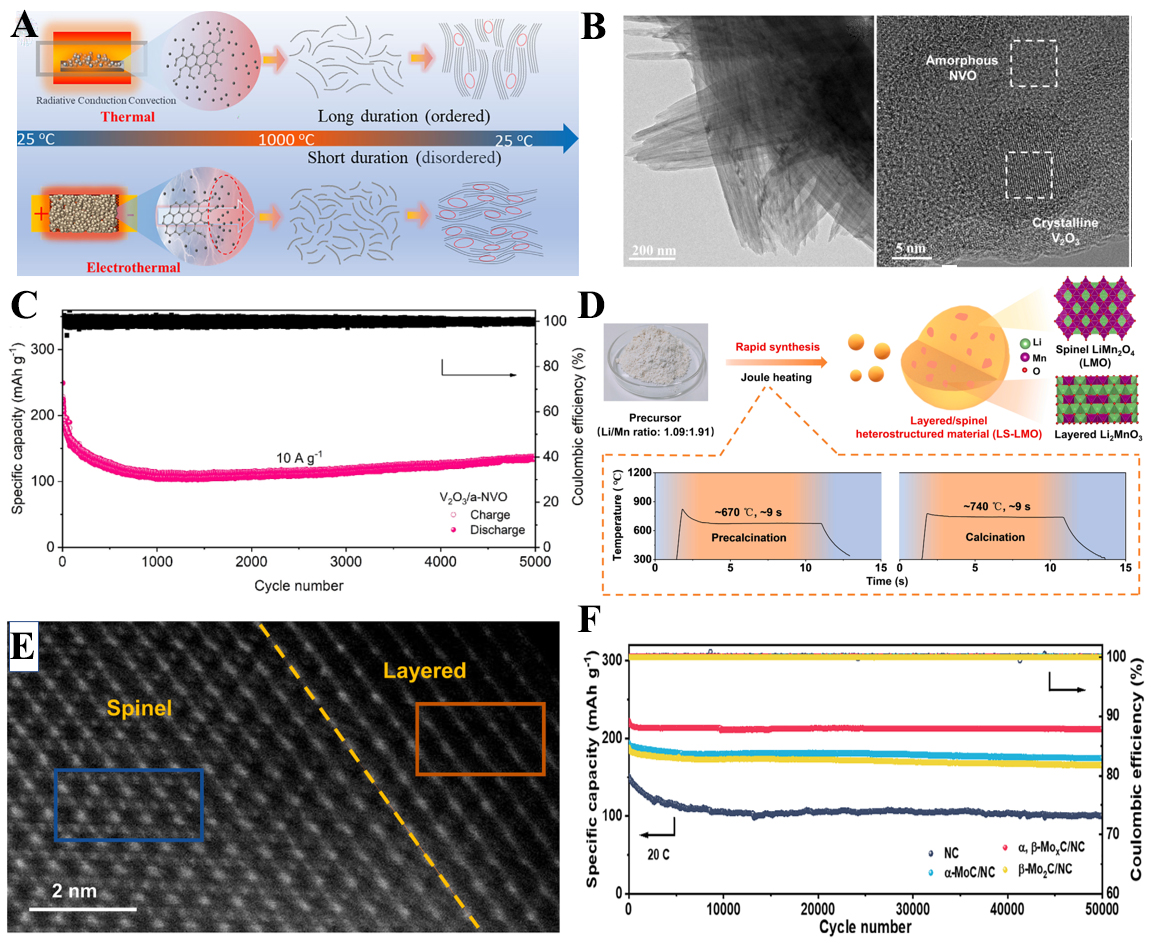
Figure 5. (A) Schematic illustration of preparing hard carbon[80]; (B) Transmission electron microscopy image of V2O3 /a-NVO[81]; (C) Cycling stability of V2O3/a-NVO[81]; (D) Schematic diagram for synthesis of layered/spinel heterostructured LS-LMO[82]; (E) High-angle annular dark-field scanning transmission electron microscopy image of LS-LMO[82]; (F) Long-term cycling stability of Zn-I2 batteries[83]; LS-LMO: a layered Li2MnO3 and spinel LiMn2O4 heterostructured cathode material.
Beyond crystalline-amorphous hybrids, HTS also enables precise modulation of phase composition to engineer interfacial phase junctions with built-in electric fields. Zhu et al. developed a layered/spinel heterostructured cathode LS-LMO (a layered Li2MnO3 and spinel LiMn2O4 heterostructured cathode material) consisting of layered Li2MnO3 and spinel LiMn2O4 by Joule heating [Figure 5D and E], which efficiently suppressed phase transition and Mn dissolution, resulting in excellent cycling stability (83% capacity retention after 800 cycles at 5 C)[82]. Chen et al. further demonstrated that HTS can tune Mo/C ratios to form α-MoC/β-Mo2C phase junctions, where the built-in electric field accelerated iodine redox kinetics and enabled zinc-iodine batteries with a remarkable lifespan of 50,000 cycles and 95.2% capacity retention [Figure 5F][83]. In addition, Dong et al. introduced a one-step HTS approach to synthesize W-W2C heterostructures anchored on graphene for Li-S batteries[84]. The abundant nanoscale interfaces and internal electric fields enhanced polysulfide conversion and effectively mitigated shuttle behavior, providing high-rate performance (665 mAh g-1 at 5 C) and ultralong cycling life (0.06% capacity decay per cycle over 1,000 cycles)[84].
Overall, these representative studies highlight that HTS delivers an unrivaled kinetic platform for generating heterogeneous phases with tailored interfacial properties. By taking advantage of non-equilibrium reaction dynamics and field-assisted bond engineering, HTS provides a powerful route to design multifunctional heterostructures that simultaneously optimize ion diffusion, electrical conductivity, and structural integrity.
Suppressed phase separation
High-entropy materials (HEMs), consisting of multiple principal elements incorporated into a single phase, offer vast compositional freedom and enhanced configurational entropy that can stabilize metastable structures. These features endow HEMs with unique electronic structures, abundant active sites, and robust stability, making them promising for next-generation batteries and electrocatalysis[85-87]. Nevertheless, a fundamental challenge is the strong thermodynamic driving force toward phase separation, especially during high-temperature synthesis, where atomic diffusion promotes segregation into element-rich domains. Such instability deteriorates structural integrity, hinders ion transport, and results in rapid capacity fading or catalytic deactivation.
During the HTS process, ultrafast heating rapidly raises the system temperature, promoting atomic diffusion and lattice mixing among multiple elements. However, subsequent ultrafast quenching leaves insufficient time for elemental segregation or long-range diffusion. As a result, diverse atoms become kinetically trapped within a single homogeneous lattice, stabilizing a metastable solid solution instead of allowing equilibrium phase separation. This dynamic balance between accelerated diffusion and rapid quenching effectively suppresses demixing, enabling the formation of high-entropy or compositionally complex phases that exhibit enhanced conductivity, ion transport, and catalytic stability[34,35,39].
Recent studies exemplify these benefits. Wu et al. employed HTS method to synthesize a high-entropy Na super ionic conductors (NASICON) cathode [Na3.45V0.4Fe0.4Ti0.4Mn0.45Cr0.35(PO4)3][88]. The rapid thermal kinetics homogenized five transition metals within a single crystallographic site, suppressing phase separation and raising configurational entropy (1.61R). This stabilized heterophase enabled reversible multi-electron redox, delivering excellent rate performance, long-term cycling stability (83.7% retention after 2,000 cycles at 5 C) [Figure 6A], and remarkable wide-temperature operability (retaining 73.7 mAh g-1 even at -50 °C)[88]. Yuanet al. employed an ultrafast Joule heating technique to synthesize a novel rocksalt structure high-entropy oxide (HEO) Fe0.2Co0.2Ni0.2Cu0.2Zn0.2O within just 3 s [Figure 6B][89]. The synergistic effects of multiple cations, including reversible Fe/Co/Ni redox contributions, enabled exceptional lithium storage reversibility. As a result, the HEO anode delivered outstanding electrochemical performance, achieving a reversible capacity of 1,310 mAh g-1 over 200 cycles at 0.1 A g-1, highlighting the critical role of entropy engineering in inhibiting phase separation for stable cycling[89].
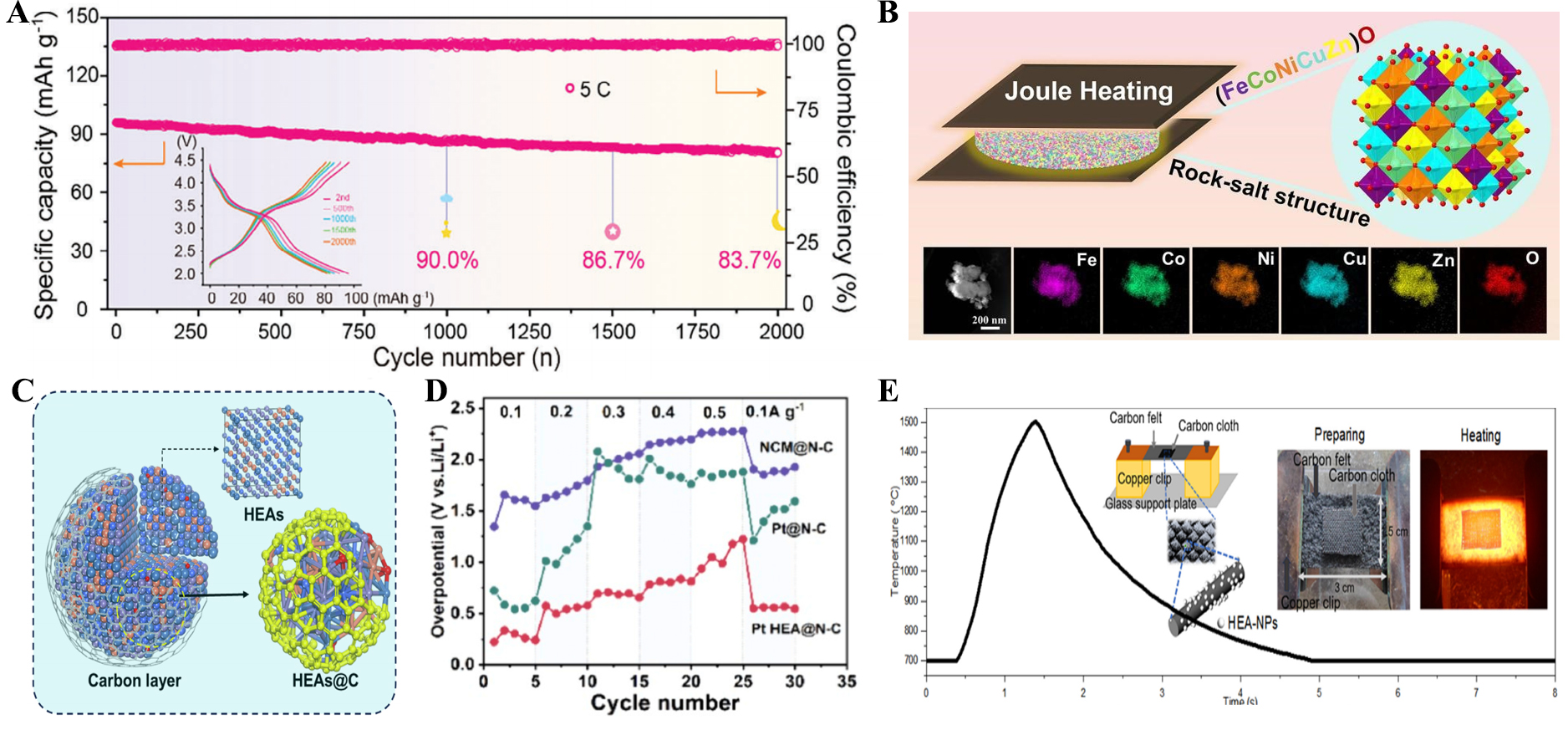
Figure 6. (A) Cycling performance of Na3.45V0.4Fe0.4Ti0.4Mn0.45Cr0.35(PO4)3[88]; (B) Preparation of Fe0.2Co0.2Ni0.2Cu0.2Zn0.2O and the element mapping[89]; (C) Model structure of MnFeCoNiCu@C[90]; (D) Overpotentials of samples under different currents[91]; (E) Process for synthesizing FeCoMnCuAl HEA electrocatalyst[92]. HEA: High-entropy alloy.
HTS has also enabled the design of HEAs for electrocatalysis. Zou et al. developed MnFeCoNiCu@C HEAs via Joule heating [Figure 6C], where oxygen doping and carbon coating acted as dual confinements, preventing atomic-scale demetalization and suppressing surface degeneration[90]. This structure sustained stable zinc-air battery operation for over 1,600 h under high current densities, outperforming conventional catalysts[90]. Similarly, Wang et al. upcycled spent Li-ion cathodes into PtNiCoMn HEA catalysts through a one-step HTS route[91]. The ultrafast process not only inhibited elemental segregation but also induced reverse electron transfer from Pt to surrounding 3d metals, optimizing the d-band center and enabling ultralow polarization (0.27 V) with a prolonged cycling life (240 cycles) [Figure 6D][91]. Zhu et al. synthesized FeCoMnCuAl and nanoparticles via carbothermal shock (CTS) [Figure 6E], achieving uniform elemental distribution and lattice distortion that tuned electronic states[92]. The high entropy alloy delivered low oxygen evolution reaction (OER) overpotentials (≈ 280 mV at 10 mA cm-2) and high long-term stability[92].
Collectively, these works demonstrate that HTS provides a unique synthetic window to stabilize high-entropy heterophases against phase separation. By kinetically trapping multielement distributions, HTS not only preserves structural integrity but also enhances electrochemical performance, enabling fast ion transport, reversible multi-electron redox, low polarization, and exceptional stability under extreme conditions. This synergy between entropy stabilization and thermal shock kinetics highlights HTS as a critical route for designing durable, high-performance HEM-based electrodes and electrocatalysts.
PARTICLE SIZE REGULATION THROUGH HTS
Size engineering has long been recognized as a powerful strategy to enhance the electrochemical performance of electrode materials. Reducing particle dimensions to the nanoscale not only shortens ion and electron transport pathways but also improves strain accommodation during cycling and increases the density of electrochemically active sites[93-95]. However, conventional high-temperature synthesis or annealing approaches often lead to excessive grain coarsening, agglomeration, or particle sintering, making it extremely difficult to precisely tailor material dimensions at the nanoscale. HTS, enabled by ultrafast Joule heating, has recently emerged as a transformative approach to regulate particle size and morphology. By generating extreme heating and cooling rates within milliseconds to seconds, HTS decouples crystallization from particle growth, kinetically suppressing aggregation and enabling precise size control[35,79]. This unique capability allows the fabrication of nanostructures that are inaccessible through traditional furnace-based methods, thereby unlocking new opportunities for advanced electrode materials.
Microscale-to-nanoscale transformation
A key advantage of HTS lies in its unique capability to induce rapid size modulation from the microscale to the nanoscale, which cannot be easily achieved by conventional thermal processing routes. During HTS treatment, the ultrafast Joule heating generates extremely high local temperatures that far exceed the melting or sublimation points of the precursor, leading to instantaneous melting or partial vaporization of bulk or micron-sized particles. Subsequently, the ultrafast quenching arrests atomic diffusion and suppresses grain growing, allowing the resolidified material to reassemble into uniformly distributed nanosized domains. Such processes not only downscale materials into well-dispersed nanoparticles but also allow for anisotropic dimensional engineering, leading to the formation of nanowires or confined nanostructures.
A representative demonstration of HTS-enabled size modulation is the dramatic transformation of silicon microparticles (SiMPs, ~ 1.5 μm) into uniform silicon nanoparticles (SiNPs, 10-15 nm) within a conductive reduced graphene oxide (RGO) matrix [Figure 7A]. Under radiative thermal shock reaching ~ 1,800 K, SiMPs rapidly melted and redistributed across defect-rich RGO nanosheets, followed by ultrafast quenching to nucleate well-dispersed SiNPs in only 30 s. This non-equilibrium pathway effectively prevented particle agglomeration and enabled the synthesis of nanosized silicon that is otherwise difficult to obtain from bulk precursors. The resulting RGO-SiNP composite demonstrated outstanding Li-ion battery anode performance, delivering 1,165 mAh g-1 after 100 cycles at 200 mA g-1. The superior cycling stability originates from the nanoscale particle size, which better accommodates the drastic ~ 300% volume expansion of silicon during lithiation, while the conductive RGO framework ensures efficient electron transport. Importantly, this strategy has also been extended to metals such as Sn and Al, further confirming the universality of HTS in downscaling bulk precursors into high-performance nanosized anodes[96].
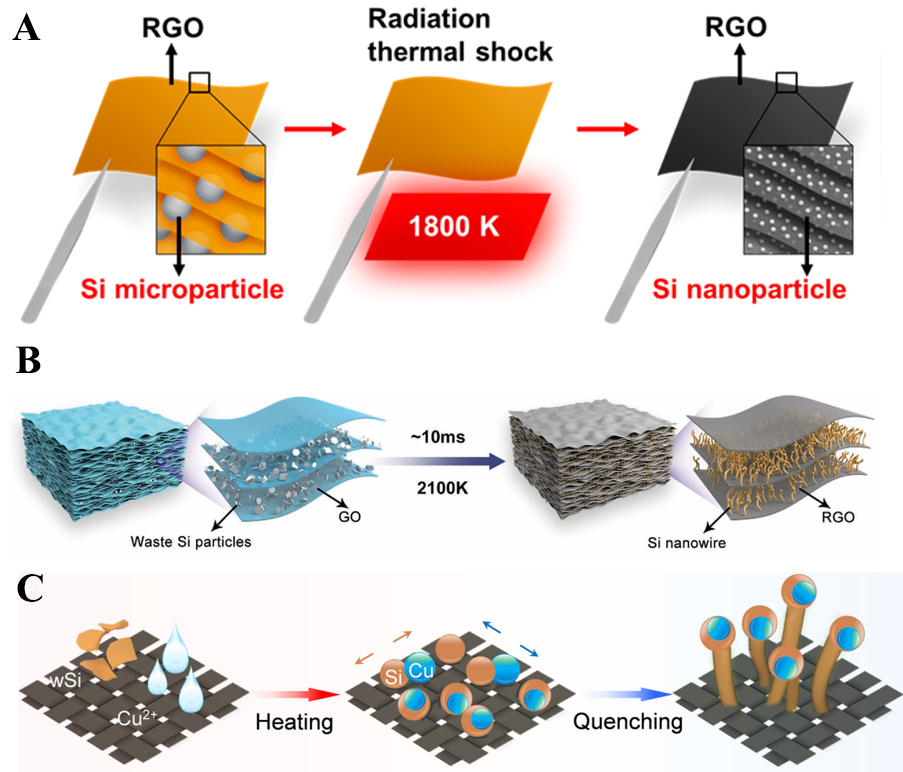
Beyond spherical nanoparticles, HTS also enables anisotropic dimensional control, such as transforming micron-sized silicon waste particles into vertically aligned nanowires (SiNWs) with diameters around 50 nm. Using electrothermal shock at ~ 2,100 K within just 10 ms, molten silicon atoms were driven by extreme thermal gradients to diffuse directionally and solidify anisotropically between confined graphene oxide layers [Figure 7B]. The physical confinement of RGO sheets enforced one-dimensional growth, while native oxide layers on waste silicon (wSi) acted as nucleation templates to restrict radial expansion. Rapid quenching locked the anisotropic morphology, simultaneously generating 30-50 nm SiNPs on the film surfaces. The resulting SiNWs@RGO composite exhibited an ultrahigh capacity of 2,381.7 mAh g-1 at 1 A g-1 for 500 cycles, with minimal electrode swelling (~ 20%). Such dimensional engineering effectively overcame the pulverization problem of bulk silicon and illustrated how HTS can valorize industrial waste into advanced nanostructured anodes[97]. Furthermore, Shen et al. developed a novel value-added recycling strategy to convert photovoltaic wSi into high-performance amorphous silicon nanowires (a-SiNWs) using a CTS technique[98]. Crucially, copper nanoparticles were introduced to modulate the surface energy of Si atoms, catalyzing the formation of a-SiNWs with controlled dimensions. The resulting nanowires exhibited a uniform diameter of approximately 15 nm and a high length-to-diameter ratio, growing in situ on a carbon cloth substrate to form a self-supporting electrode (a-SiNWs@CC) [Figure 7C]. This structural conversion from micron-sized wSi particles (D50 ~ 380 nm) to nanoscale amorphous wires was pivotal in achieving exceptional electrochemical performance as a lithium-ion battery anode. The a-SiNWs@CC demonstrated a remarkably high initial coulombic efficiency (91.35%) and outstanding cycling stability, maintaining a capacity of 2,150 mA h g-1 after 250 cycles at 2 A g-1. The success of this CTS approach underscores its effectiveness in overcoming traditional kinetic limitations for rapid, scalable nanomaterial synthesis with precise size control[98].
Ultrafine nanostructure formation
While microscale-to-nanoscale transformations are crucial, HTS also offers unprecedented control at the ultrafine and atomic scales. For example, ultrafast radiative heating was used to synthesize a library of BiSb, SnSb, and CoSb alloy nanoparticles with sizes restricted to 10-20 nm and with high metal loadings [Figure 8A]. By leveraging heating/cooling rates of ~ 104 K s-1 and introducing heteroatom-doped carbon anchors, particle coarsening, evaporation, and agglomeration were successfully suppressed. The BiSb alloy produced via HTS showed remarkable potassium-ion battery performance, maintaining 324.8 mAh g-1 after 800 cycles. The ultrafine alloy size accelerated K+ diffusion and buffered large volume changes during alloying, features unattainable in coarse-grained analogs[99].
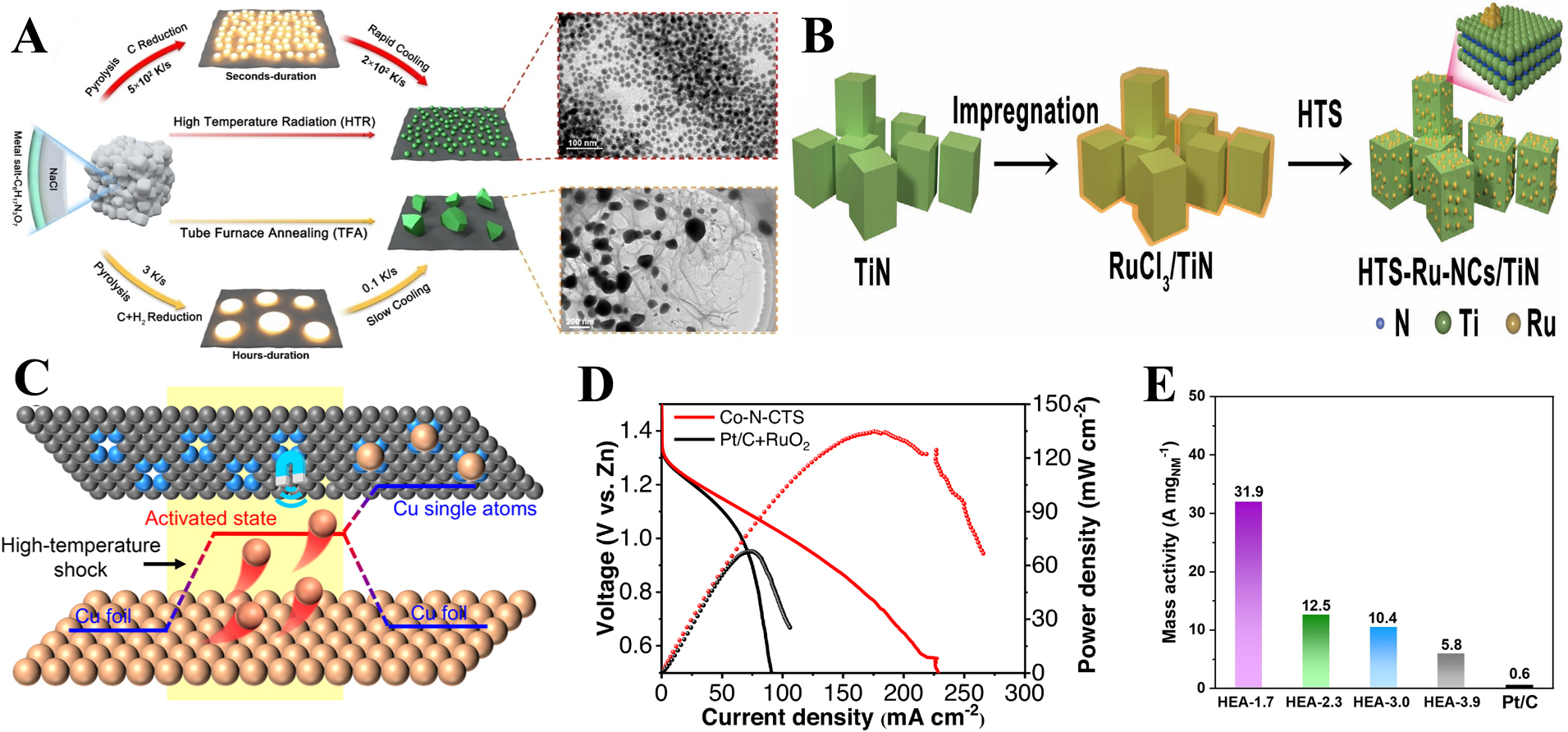
Figure 8. Schematic diagram for (A) preparation of bimetallic nanoparticles[99]; (B) Preparation of HTS-Ru-NCs/TiN[100] and (C) Proposed mechanism for the formation Cu single atom catalyst[101]; (D) Power density curves of Co-N-CTS and Pt/C + RuO2[102]; (E) Comparison of mass activity for different samples at an overpotential of 50 mV[103]. HTS: High-temperature shock; CTS: carbon thermal shock.
Even more striking, HTS has enabled the stabilization of nanoclusters and single atoms. For instance, sub-2 nm Ru nanoclusters anchored on TiN supports were fabricated within milliseconds via Joule heating of RuCl3 precursors [Figure 8B]. Rapid nucleation and immobilization at Ru-Ti interfacial sites prevented sintering, yielding uniform 1.24 nm clusters with remarkable hydrogen evolution activity. The electrocatalyst required only 16.3 mV overpotential to achieve 10 mA cm-2[100]. Similarly, Fang et al. demonstrated that bulk Cu foil could be atomized into isolated Cu atoms under 0.5-s HTS pulses [Figure 8C][101]. These single Cu atoms were trapped by nitrogen defects in carbon to form Cu-N4 sites, achieving > 90% Faradaic efficiency for nitrite-to-ammonia electroreduction with record-high ammonia production rates. Such atomic-scale engineering underscores the ability of HTS to overcome kinetic barriers to aggregation, stabilizing metastable species that are otherwise inaccessible via equilibrium processing[101].
Another advancement is coordinated CTS, which combines metal-ligand coordination chemistry with ultrafast Joule heating. This approach produced high-density nanoparticles, 1-5 nm in size, uniformly dispersed on porous carbons. Ligand coordination confined metal atoms, while millisecond-scale pyrolysis suppressed diffusion, leading to monodisperse nanoparticles with significantly enhanced electrochemical surface areas. In Zn-air batteries, CTS-derived catalysts Co-N-CTS outperformed commercial Pt/C and RuO2 due to superior oxygen redox activity and structural stability [Figure 8D][102]. Extending further, HTS combined with spatial confinement has even enabled the synthesis of sub-2-nm HEA nanoparticles with finely tuned electronic states. These ultrasmall HEAs demonstrated high mass activities and long-term durability for both hydrogen evolution reaction (HER) and oxygen reduction reaction (ORR) [Figure 8E], emphasizing the size-dependent modulation of catalytic properties achievable only through HTS[103].
INTERFACIAL ENGINEERING WITH HTS
Interfacial engineering plays a decisive role in determining the performance and reliability of electrochemical energy-storage systems. In batteries, the interfaces between electrodes and electrolytes are often sites of parasitic reactions, ion transport bottlenecks, and mechanical instability, all of which lead to capacity fading and safety concerns[104-106]. Similarly, in electrocatalytic systems, poorly designed interfaces limit charge transfer, active-site accessibility, and structural durability. To overcome these challenges, researchers have long pursued strategies such as atomic layer deposition, sol-gel coatings, and conventional furnace sintering. However, these methods often suffer from slow kinetics, limited scalability, and difficulty in simultaneously achieving conformal coverage, high crystallinity, and strong bonding without inducing undesirable side reactions.
HTS provides a transformative solution for interfacial design. Distinguished by heating and cooling rates of 103-104 K s-1, HTS allows non-equilibrium processing that kinetically suppresses detrimental interdiffusion while promoting localized fusion, crystallization, and controlled phase transformations. As a result, HTS can achieve outcomes that are inaccessible under conventional thermal conditions, such as ultrafast densification of ceramic electrolytes, defect-controlled interfacial bonding, and precise construction of core-shell architectures. Importantly, the short duration of HTS minimizes bulk degradation while enabling nanoscale structural tailoring at interfaces.
Interfacial welding
The interfacial stability between solid electrolytes and electrodes remains one of the most critical challenges for all-solid-state batteries (ASSBs)[107-109]. Conventional high-temperature sintering often fails to deliver satisfactory results, as sluggish diffusion kinetics and extended thermal exposure induce parasitic reactions, interdiffusion, and void formation, ultimately leading to high interfacial resistance and poor electrochemical stability. HTS techniques offer a radically different approach that leverages ultrafast Joule heating with extreme heating/cooling rates. During HTS treatment, the ultrafast Joule heating generates a transient high-temperature gradient at the material interface, leading to localized atomic diffusion, lattice softening, and partial melting at contact points. This extreme but short-lived thermal excitation promotes atomic intermixing and grain boundary fusion between adjacent particles or layers without causing bulk degradation, which results in robust interfacial welding with high mechanical and electrical continuity.
Kong et al. demonstrated the effectiveness of this approach in garnet-type solid electrolytes[110]. Through ultrafast high-temperature sintering (UHS), they synthesized high-entropy disordered rocksalt cathodes (HE-DRXs) and directly sintered them onto dense Li7La3Zr2O12 (LLZTO) substrates. The process, conducted at 1,100 °C for only 3 s [Figure 9A], exploited entropy stabilization to suppress transition-metal migration while forming a chemically stable, conformal interface [Figure 9B]. This innovation achieved an exceptionally low area-specific resistance of 31.6 Ω·cm2 at room temperature, nearly three orders of magnitude lower than conventional LiCoO2|LLZTO contacts. The resulting ASSBs delivered 239.7 mAh g-1 at 25 mA g-1 and retained 95% of their capacity after 100 cycles at 150 °C [Figure 9C], highlighting the potential of HTS for stabilizing ceramic-ceramic interfaces under extreme conditions[110].
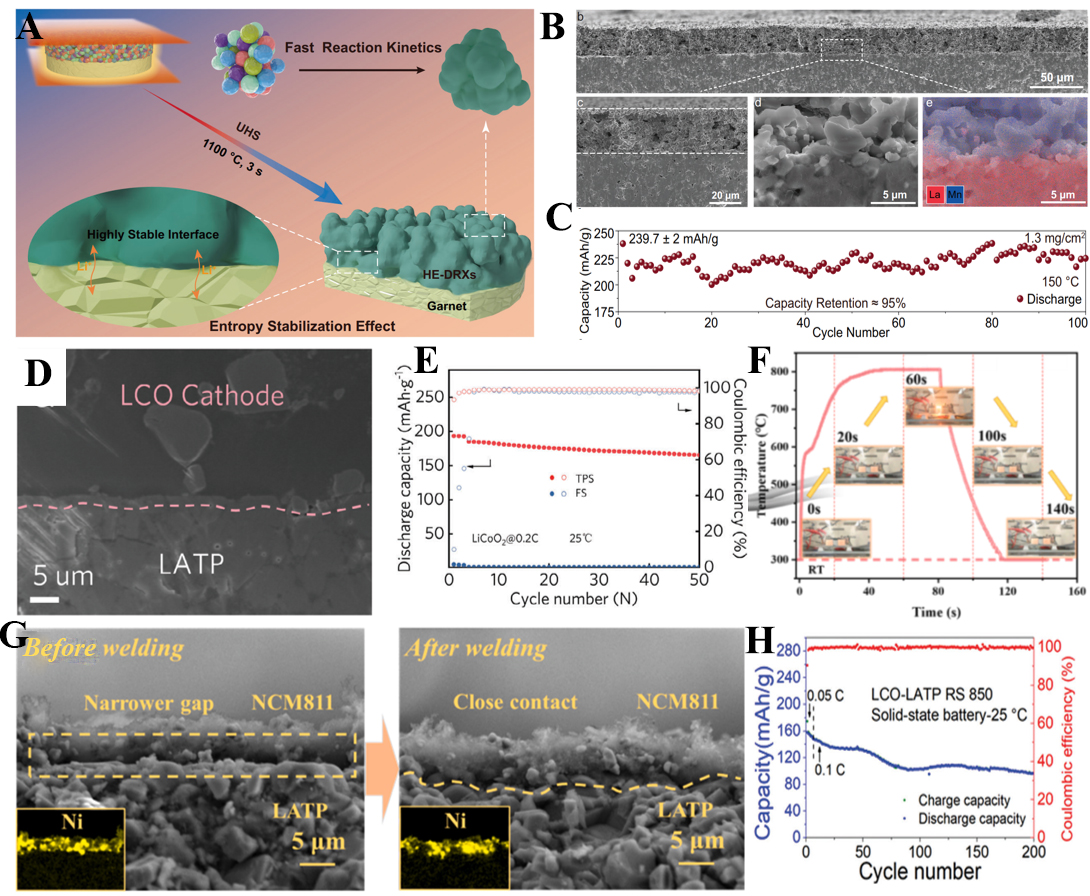
Figure 9. (A) Schematic diagram of the preparation of the HE-DRX | LLZTO interface[110]; (B) SEM image and element mapping of the HE-DRX | LLZTO interface[110]; (C) Cycling performance of the solid battery based on HE-DRX at 25 mA/g[110]; (D) SEM image of LiCoO2| LATP interface[111]; (E) Cycling stability of LCO solid battery[111]; (F) Preparation process via Joule heating[112]; (G) SEM image of the interface between NCM811 and LATP before and after treatment[112]; (H) Cycling performance of LCO-LATP pellets[113]. HE-DRX: High-entropy disordered rocksalt cathode; LLZTO: Li7La3Zr2O12; SEM: scanning electron microscope; LATP: Li1.3Al0.3Ti1.7(PO4)3; NCM811: LiNi0.8Co0.1Mn0.1O2.
Expanding on this concept, Yao et al. introduced thermal pulse sintering (TPS) for NASICON-type Li1.3Al0.3Ti1.7(PO4)3 (LATP) electrolytes[111]. Unlike continuous sintering, TPS applies segmented Joule-heating pulses (~ 10 s in total), enabling simultaneous optimization of cathode, electrolyte, and anode interfaces. At the cathode-electrolyte interface, localized Joule heating induced instantaneous physical welding with residual void fractions below 1.8% [Figure 9D], while suppressing elemental diffusion and parasitic phase formation. For bulk LATP, the thermal pulses promoted nanowire growth along the (113) plane, resulting in 98.1% relative density and enhanced ionic conductivity (8.20 × 10-4 S cm-1). At the anode interface, a graphene oxide-carbon nanotube (CNT)-MXene composite buffer was transformed into a conductive, graphitized interlayer, homogenizing lithium flux and mitigating reductive decomposition of LATP. Electrochemically, LiCoO2-based cells demonstrated stable cycling at 4.6 V with 185 mAh g-1 capacity [Figure 9E], while LFP cells retained 90.1% of capacity after 500 cycles[111].
Cui et al. further developed a nanoparticle-assisted ultrafast sintering (NUHS) strategy combined with thermal pulse welding (TPW) [Figure 9F][112]. The dual treatment densified LATP electrolytes to nearly defect-free levels, while TPW eliminated ~ 8 μm interfacial gaps in full-cell assemblies [Figure 9G]. This reduced interfacial resistance by 43.5% and enabled Li||LATP||LiNi0.8Co0.1Mn0.1O2 (NCM811) cells with 185.9 mAh g-1 capacity and 90.9% retention after 100 cycles[112]. Complementary to this, Chen et al. applied Joule-heating pulses to generate ultrathin amorphous Li+-conductive interphases between LATP and layered cathodes[113]. These amorphous layers suppressed interdiffusion, reduced resistance by more than 50%, and enabled long-term cycling (> 200 cycles) of thick composite cathodes [Figure 9H][113].
Surface coating
In addition to welding solid-solid interfaces, HTS provides a powerful pathway for constructing core-shell electrode architectures. Such structures are highly desirable, as they combine the advantages of bulk phases with protective or conductive shells, thereby improving ionic transport, suppressing surface degradation, and enhancing structural integrity[114,115]. However, conventional coating techniques, including sol-gel and furnace sintering, often yield non-uniform or thick shells due to slow diffusion and uncontrolled thermal exposure. HTS, by contrast, leverages ultrafast thermal kinetics to rapidly form thin, conformal shells with minimal side reactions, enabling the scalable synthesis of high-performance core-shell materials.
Yin et al. utilized HTS to fabricate Na3V2(PO4)3 (NVP) cathodes with carbon shells in just 15 s at ~ 850 °C [Figure 10A][116]. This process generated large, crystalline NVP cores encapsulated by 3-5 nm conductive carbon layers [Figure 10B], while simultaneously preserving a carbon framework that interconnected the particles. The resulting materials exhibited high reversible capacity (110 mAh g-1 at 0.1 C), outstanding rate capability (83.5 mAh g-1 at 50 C), and remarkable cycling stability (89.9% retention after 1,600 cycles). Importantly, the ultrafast kinetics resolved the trade-off between tap density and conductivity, a long-standing issue in polyanionic cathodes[116].
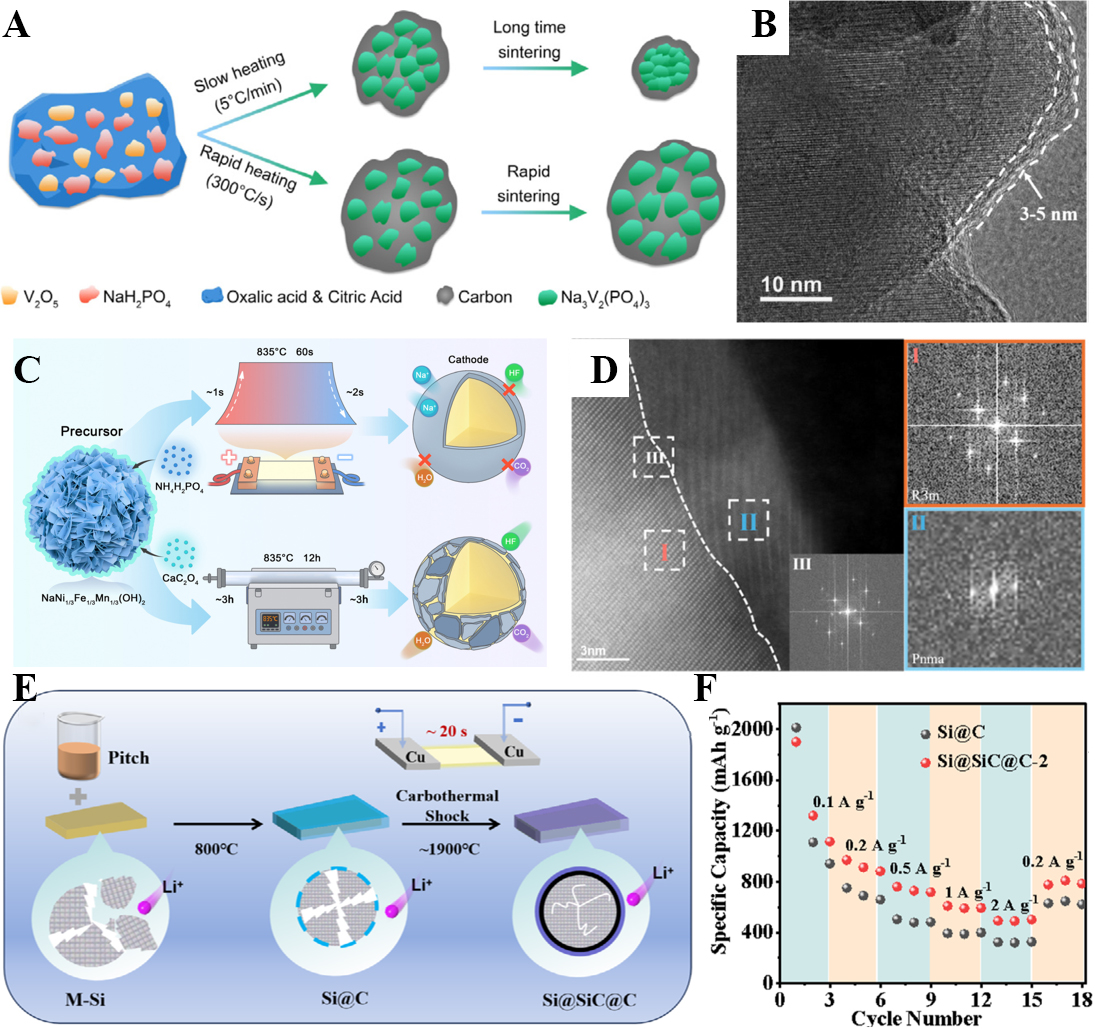
Figure 10. (A) Schematic diagram for synthesizing NVP@C[116]; (B) TEM image of NVP@C[116]; (C) Schematic diagram for preparing NFM333@NCP[117]; (D) High-angle annular dark field scanning transmission electron microscopy of NFM333@NCP[117]; (E) Schematic illustration for synthesizing Si@SiC@C[118]; (F) Rate performance of samples[118]. NVP@C: Na3V2(PO4)3@C; TEM: transmission electron microscope; NFM333@NCP: NaNi1/3Fe1/3Mn1/3O3@NaCaPO4.
Similarly, Li et al. employed HTS to coat NaNi1/3Fe1/3Mn1/3O3 (NFM333) cores with ultra-uniform NaCaPO4 (NCP) shells [Figure 10C][117]. Within 2 s at ~ 835 °C, a crystalline ~ 8 nm NCP layer was formed [Figure 10D], preventing Ni segregation into electrochemically inactive phases. The conformal coating enhanced Na+ transport (10-9-10-10 cm2 s-1), suppressed electrolyte corrosion, and stabilized the O3-phase core against structural degradation. Electrochemically, the core-shell cathodes retained 80% capacity after 1,000 cycles in half cells and 70% after 700 cycles in pouch cells, outperforming conventional counterparts by more than 27%[117].
For alloy anodes, Li and Yan developed a Si@SiC@C triple-layer architecture using carbothermal shock at ~ 1,900 °C [Figure 10E][118]. The process transformed a Si@C intermediate into a silicon core, a rigid SiC buffer layer, and a conductive carbon shell. This design effectively mitigated ~ 400% volume expansion of silicon during cycling while providing fast electron and ion transport. The optimized anodes delivered 900.1 mAh g-1 after 1,000 cycles with 89.3% retention and exhibited superior rate performance (492.7 mAh g-1 at 2 A g-1) [Figure 10F], demonstrating the feasibility of HTS for producing durable silicon-based anodes[118].
Beyond batteries, HTS has also been extended to electrocatalysis. Shiet al. reported a roll-to-roll carbothermal shock process to continuously fabricate multielement heterostructured catalysts with precise core-shell morphologies[119]. This scalable method produced PtCo@La-TiO2 catalysts with HER overpotentials as low as 15 mV at 10 mA cm-2, while enabling large-area electrode fabrication at industrially relevant throughput[119]. In a related approach, Zeng et al. employed sequential thermal shocks to create Pd-decorated HEA (NHEA@NHEA-Pd) catalysts[120]. The atomic-scale Pd dispersion on the HEA surface achieved record ethanol oxidation activity (7.34 A mg-1 Pd) and > 91.8% stability over 2,000 cycles, while reducing noble-metal usage[120].
CONCLUSION AND OUTLOOK
Although HTS technology has demonstrated remarkable capability in enabling structural engineering of electrode materials [Table 1], several challenges remain that must be addressed to further advance its application. A key issue lies in the controllability and reproducibility of the ultrafast heating and quenching processes. The dynamic nature of HTS often leads to non-uniform temperature distribution across bulk samples, which in turn results in heterogeneous defect density, inconsistent phase states, or uncontrolled grain-size variations. From a mechanistic perspective, the interplay between ultrafast energy input, defect kinetics, and non-equilibrium phase evolution remains poorly understood, hindering the rational design of HTS protocols tailored for specific material systems. Bridging this knowledge gap will require advanced in situ characterization tools capable of capturing transient phenomena on sub-millisecond timescales, complemented by multiscale simulations that link atomic-scale processes with mesoscale structural evolution.
Summary of some materials synthesized by HTS and their performance
| Products | Synthesis method | HTS parameters | Medium | Performance | Application | Reference |
| LiFePO4 with Li-Fe antisite defects and tensile strain | Ball milling, HTS | 750 °C, 25 s, argon atmosphere | Carbon cloth | 126 mAh g-1 after 550 cycles at 1 C, 84.4% capacity retention after 2,000 cycles | Lithium-ion batteries (LIBs) | [55] |
| Li1.2Ni0.13Co0.13Mn0.54O2 with twin boundaries | Ball milling, HTS | 850 °C, 45 s, Air | Nickel foil | 278 mA h g-1 at 0.1 C, a capacity retention of 89.4% after 100 cycles at 0.5 C | LIBs | [56] |
| Spinel LiMn2O4 with twin boundaries | Ball milling, HTS | 750 °C, 60 s, Air | Nickel foil | 132 mA h g-1 at 1 C, 81 mAh g-1 at 5 C | LIBs | [57] |
| LiCoO2 with ultrasmall particle sizes and abundant oxygen vacancies | Combustion method, HTS | 860 °C, 16 s, Air | Carbon cloth | 139.5 mAh g-1 at 0.1C, a high capacity retention of 84.6% after 300 cycles at 1 C | LIBs | [58] |
| LiMn2O4 with small size and oxygen vacancies | Combustion method, HTS | 970 °C, 8 s, Air | Carbon cloth | 116.9 mAh g-1 at 0.1 C within 3.5-4.3 V, 82.5% after 100 cycles at 1 C | LIBs | [58] |
| Pt/nitrogen-rich defective graphitic carbon nitride | HTS | 625 °C, heating of 5 s and cooling of 5 s for 6 cycles, environment of the pressure of 10e-7 Pa | Carbon paper | Hydrogen evolution rate of 16,936.5 µmol h-1 g-1 | Photocatalytic hydrogen evolution | [59] |
| Regenerated LiCoO2 | Ball-mixing with Li2CO3, pressing into pellets, joule heating | 1,440 K, 8 s | Carbon paper | 133.0 mAh g-1 at 0.1 C | LIBs | [67] |
| Mg/Al doped regenerated LiCoO2 | Mixing with Li2CO3 and dopants, HTS | 750 °C, 30 s | Carbon felt | 203 mAh g-1 at 0.2 C,84% capacity retention after 200 cycles at 0.2 C | LIBs | [68] |
| Regenerated LiFePO4 | Mixing with LiOH, electric heating | 720 °C, 20 s, Ar | Carbon paper | 152.8 mAh g-1 at 0.1 C, 97.56% capacity retention after 300 cycles at 1 C (pouch cell) | LIBs | [69] |
| Regenerated LiFePO4 | Mixing with lithium acetate, ball milling, HTS | 800 °C, 20 s, Ar | / | 152 mAh g-1 at 0.1 C, 400 cycles at 2 C without capacity recession | LIBs | [70] |
| Defect-rich recycled graphite | HTS | 1,500 °C, 60 s, Ar | Carbon paper | 323 mAh/g at 2 C | LIBs | [71] |
| Hard carbon maintaining abundant micropores and expanded interlayer spacing | Pre-carbonization, Joule heating | 1,000 °C, 30 s, nitrogen | Hard carbon | 306.83 mAh g-1, initial Coulombic efficiency of 91.99%, 79.45% capacity retention after 1000 cycles | Sodium-ion batteries (SIBs) | [80] |
| Crystalline V2O3 and amorphous sodium vanadate heterostructure | Dissolution & recrystallization, flash Joule heating | 1,600 °C, 3 s | Graphite paper | 414.6 mAh g-1 at 0.2 A g-1 and a capacity of 329.2 mAh g-1 at a current density of 1 A g-1 after 1,000 cycles. | Zinc-ion batteries (ZIBs) | [81] |
| Heterostructure cathode material consisting of Li2MnO3 and LiMn2O4 | Two-step HTS | 670 °C, 10 s; 740 °C, 9 s | Carbon cloth | 109 mAh g-1 at 0.1 C, capacity retention of 83% after 800 cycles at 5 C | LIBs | [82] |
| Na3.45V0.4Fe0.4Ti0.4Mn0.45Cr0.35(PO4)3 | Sol-gel method, pre-calcination, HTS | 800 °C for 20 s | / | 92.8% after 400 cycles at -40 °C and a capacity of 73.7 mAh g-1 even at -50 °C | SIBs | [88] |
| Fe0.2Co0.2Ni0.2Cu0.2Zn0.2O | Ball milling, compression, Joule heating | 1,350 K, 3 s | Graphite flakes | 1,310 mAh g-1 for 200 cycles at 0.1 A g-1 705 mAh g-1 for 3,000 cycles at 5 A g-1 | LIBs | [89] |
| FeCoMnCuAl | Impregnation, HTS | 1,500 °C, 0 s | Carbon felt | overpotential of 280 mV at 10 mA cm2 and a low Tafel slope of 76.13 mV dec1 in a 1.0 M KOH solution | Oxygen evolution reaction | [92] |
| RGO composite with silicon nanoparticles | Mixing, HTS | 1,800 K, 30 s | RGO | 1,957 mA h g-1 at 50 mA g-1, 1,165 mA h g-1 over 100 cycles | LIBs | [96] |
| Si nanowires@RGO | Mixing, HTS | 2,100 K, 10 ms | RGO | 2,381.7 mAh g-1 at 1 A g-1 for more than 500 cycles, 89.5% initial Coulombic efficiency | LIBs | [97] |
| BiSb with narrow size distribution (10-20 nm) | Liquid phase mixing, freeze-drying, HTS | 1,300 K, 15 s | Carbon cloth | 322.4 mAh/g at the 200th cycles at 0.5 A/g, negligible degradation after 800 cycles | Potassium-ion batteries | [99] |
| Sub-2 nm Ru nanocrystals (NCs) on titanium nitride | Gel method, HTS | 2,200 °C, Ar | / | Overpotentials of 16.3 and 86.6 mV to achieve 10 and 100 mA cm-2 | HER | [100] |
| Cu-N-C single atom catalysts | HTS | 1,700 K, 80 cycles, holding for 0.5 s | Carbon cloth | 90% Faradaic efficiency across the entire working potential range and an ammonia production rate of up to 11.12 mg cm-2 h-1 at -1.2 V | Nitrite reduction | [101] |
| Li1.3Mn2+0.1Co2+0.1Mn3+0.1Cr3+0.1Ti0.1Nb0.2O1.7F0.3|Li6.4La3Zr1.4Ta0.6O12 interfaces | HTS | 1,100 °C, 3 s | Carbon strips | 31.6 Ω cm2 of interface resistance, 239.7 ± 2 mAh g-1 at 25 mA g-1, with a capacity retention of 95% after 100 cycles (150 °C, all-solid-state Li batteries) | Solid batteries | [110] |
| LFP/LATP interfaces | Mixing, HTS | three 800 °C thermal pulse, Ar | / | 168 mAh g-1 at 0.2 C, 99.1% after 120 cycles | Solid batteries | [111] |
| LCO/LATP interfaces | Mixing, HTS | three 800 °C thermal pulse, Ar | / | 185 mAh g-1 with a coulombic efficiency of 98.0% at 0.2 C | Solid batteries | [111] |
| Na3V2(PO4)3@C | Ball milling, HTS | 850 °C, Ar | Carbon paper | 110 mAh/g at 0.1 C with 89.9% capacity retention after 1,600 cycles at 1 C and specific capacity of 83.5 mAh g-1 at 50 C rate | SIBs | [116] |
| NaNi1/3Fe1/3Mn1/3O2 @NaCaPO4 | Coating, HTS | 835 °C for 60 s, oxygen | nickel foil | 147 mAh g-1 at 0.1 C, 80% capacity retention after 1,000 cycles at a 1 C | SIBs | [117] |
| FeCoNiSn@ FeCoNiSn-Pd | Two-step HTS | 1,750 K for 1 s,1,118 K for 0.3 s | Carbon cloth | mass activity of 7.34 A mg1Pd, > 91.8% retention after 2,000 cycles | Ethanol oxidation reaction | [120] |
While HTS has shown great potential in laboratory-scale material synthesis, scaling the technique for industrial production presents several challenges. The primary issues involve ensuring uniform heating across larger volumes of material, managing heat dissipation in continuous systems, and maintaining process efficiency. To address these challenges, ongoing research is focused on developing continuous HTS reactors that can efficiently heat and quench larger batches of materials. For example, a roll-to-roll production equipment relies on carbon cloth impregnated with metal precursors passing through two graphite rollers that are loaded with high current to generate Joule heat. Based on this device, continuous production of various catalysts can be achieved at 1,000 °C and a carbon cloth moving speed of 7 m/min[119]. Furthermore, future work should explore strategies to minimize equipment wear from frequent thermal cycles and to improve material stability during industrial processing. As the HTS technique moves towards industrial implementation, these advancements will be key to realizing its potential in large-scale manufacturing of high-performance energy materials.
The integration of HTS with artificial intelligence (AI) represents a promising direction for accelerating materials design and optimization. Firstly, AI models require large datasets to effectively learn structure-property-performance relationships. HTS, with its ultrafast synthesis capability, can generate numerous samples with systematically varied conditions in a short time, providing abundant high-quality data for training machine learning algorithms[121]. Secondly, combined with in situ diagnostics and real-time feedback, AI-guided HTS can predict optimal synthesis parameters, enabling precise control over defects, phases, particle sizes, and interfaces. This synergy offers a pathway toward autonomous, high-throughput fabrication of electrode materials with tailored structures for next-generation energy storage and conversion technologies.
Looking ahead, HTS has the potential to evolve from a materials-processing technique into a general platform for discovering non-equilibrium structures and novel functionalities. Several promising directions may accelerate this transition. Future research may focus on scalable, continuous processes, such as roll-to-roll or laser-based HTS, enabling industrial-scale fabrication of functional materials. Additionally, the combination of HTS with in-situ transmission electron microscopy and ultrafast spectroscopy could provide real-time insights into shock-induced transformations. More importantly, the integration of HTS with AI and machine learning will open new possibilities for data-driven discovery, as HTS can rapidly generate large libraries of materials with tunable structures, providing abundant datasets for predictive modeling and optimization. Furthermore, future developments should emphasize energy efficiency and environmental sustainability, extending HTS applications across energy storage, catalysis, and flexible electronics.
In summary, HTS synthesis has emerged as a transformative strategy for engineering the structural complexity of functional materials. Unlike conventional thermal treatments that rely on slow, equilibrium-driven processes, HTS exploits ultrafast, non-equilibrium dynamics to manipulate particle size, defect chemistry, phase distribution, and interfacial architectures with remarkable precision and speed. These structural interventions enable access to metastable configurations, unconventional defect states, and nanoscale architectures that are otherwise inaccessible, thereby creating unique opportunities for performance enhancement in energy storage and conversion devices. Beyond its synthetic utility, HTS provides a powerful platform for uncovering fundamental design principles that link metastability and dynamic structural evolution with electrochemical functionality. Its ability to engineer defects, redistribute phases, and even regenerate degraded structures highlights a new paradigm in materials design - where rapid transformation and dynamic healing can be harnessed as functional advantages. Nevertheless, significant challenges remain. A deeper mechanistic understanding of structural evolution under extreme conditions is needed, supported by advanced in situ characterization and theoretical modeling. Scalability, energy efficiency, and reproducibility must be resolved before HTS can be adopted on an industrial scale. Moreover, extending HTS to more complex chemistries and establishing robust correlations between HTS-induced architectures and long-term device performance remain critical. Overcoming these challenges will not only refine HTS as a methodology but also unlock its broad potential in lithium- and sodium-ion batteries, fuel cells, and water splitting. In conclusion, HTS enriches the materials chemistry toolkit and sets the stage for more efficient, durable, and sustainable energy technologies.
DECLARATIONS
Authors’ contributions
Made substantial contributions to the conception and design of the review: Tao, X. S.; Chen, Y.
Performed literature collection, data analysis, and interpretation: Chu, F.; Tao, X. S.; Meng, J.
Drafted the manuscript and prepared figures: Tao, X. S.; Wang, S.; Fang, X.; Zhang, J.
Provided critical revision of the manuscript for important intellectual content: Sha, J.; Chen, Y.
Availability of data and materials
Not applicable.
Financial support and sponsorship
This work was supported by the National Natural Science Foundation of China (Grant Nos. 52402257, 52403307), the Shandong Provincial Natural Science Foundation (ZR2024QE142), the China Postdoctoral Science Foundation under Grant Number 2024M751105, Young Talent of Lifting Engineering for Science and Technology in Shandong China (SDAST2025QTA095), Research and Innovation Team of Jining University (24KCTD03) and the Youth Innovation Team Project of Shandong Province (2024KJH108, 2024KJG027).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
1. Quilty, C. D.; Wu, D.; Li, W.; et al. Electron and ion transport in lithium and lithium-ion battery negative and positive composite electrodes. Chem. Rev. 2023, 123, 1327-63.
2. Deng, L.; Liu, Y.; Qi, H.; et al. A nanoengineered lithium-hosting carbon/zinc oxide composite electrode material for efficient non-aqueous lithium metal batteries. Nat. Nanotechnol. 2025, 20, 1439-48.
3. Huang, J.; Yang, Q.; Hu, A.; et al. Enhanced specific energy in fast-charging lithium-ion batteries negative electrodes via Ti-O covalency-mediated low potential. Nat. Commun. 2025, 16, 6243.
4. Lin, F.; Luo, H.; Li, L.; et al. Synthesis of isolated Ru-O3 sites on hexagonal close-packed intermetallic penta-metallene for hydrogen oxidation electrocatalysis. Nat. Synth. 2024, 4, 399-409.
5. Jain, R.; Lakhnot, A. S.; Bhimani, K.; et al. Nanostructuring versus microstructuring in battery electrodes. Nat. Rev. Mater. 2022, 7, 736-46.
6. Singhvi, C.; Sharma, G.; Verma, R.; et al. Tuning the electronic structure and SMSI by integrating trimetallic sites with defective ceria for the CO2 reduction reaction. Proc. Natl. Acad. Sci. U. S. A. 2025, 122, e2411406122.
7. Song, N.; Ma, J.; Liang, Y.; et al. Phase and orbital engineering effectuating efficient adsorption and catalysis toward high-energy lithium-sulfur batteries. Adv. Mater. 2025, 37, 2420588.
8. Bai, L.; Xu, Y.; Liu, Y.; et al. Metal-organic framework glass stabilizes high-voltage cathodes for efficient lithium-metal batteries. Nat. Commun. 2025, 16, 3484.
9. Zhang, Y.; Zhang, Y.; Zhang, H.; et al. Defect engineering in metal sulfides for energy conversion and storage. Coord. Chem. Rev. 2021, 448, 214147.
10. Yun, Q.; Ge, Y.; Shi, Z.; et al. Recent progress on phase engineering of nanomaterials. Chem. Rev. 2023, 123, 13489-692.
11. Sung, J.; Kim, N.; Ma, J.; et al. Subnano-sized silicon anode via crystal growth inhibition mechanism and its application in a prototype battery pack. Nat. Energy. 2021, 6, 1164-75.
12. Jin, H.; Zhang, Y.; Cao, Z.; Liu, J.; Ye, S. Atomically dispersed Sn on core‐shell MoS2 nanoreactors as Mott‐Schottky phase junctions for efficient electrocatalytic hydrogen evolution. Adv. Mater. 2025, 37, 2502977.
13. Kang, J.; Yang, X.; Hu, Q.; Cai, Z.; Liu, L. M.; Guo, L. Recent progress of amorphous nanomaterials. Chem. Rev. 2023, 123, 8859-941.
14. Liu, S.; Yan, L.; Huang, J.; Zhang, Q.; Zhou, B. Controlling upconversion in emerging multilayer core-shell nanostructures: from fundamentals to frontier applications. Chem. Soc. Rev. 2022, 51, 1729-65.
15. Hong, S.; Jin, S.; Deng, Y.; et al. Efficient scalable hydrothermal synthesis of MnO2 with controlled polymorphs and morphologies for enhanced battery cathodes. ACS. Energy. Lett. 2023, 8, 1744-51.
16. Zhu, K.; Yang, H.; Guo, G.; et al. Pd Single atoms/clusters at the oxygen defect-rich WOxC nanowire structure facilitate H* adsorption and desorption for efficient and stable hydrogen evolution reaction. ACS. Catal. 2025, 15, 9563-73.
17. Song, Y.; Zhou, Z.; Cui, B.; et al. High‐temperature long‐term cycling capability of lithium batteries enabled by releasing local constriction. Angew. Chem. Int. Ed. Engl. 2025, 64, e202510172.
18. Liu, F.; Fan, Z. Defect engineering of two-dimensional materials for advanced energy conversion and storage. Chem. Soc. Rev. 2023, 52, 1723-72.
19. Kang, S.; Wang, C.; Chen, J.; Meng, T.; E, J. Progress on solvo/hydrothermal synthesis and optimization of the cathode materials of lithium-ion battery. J. Energy. Storage. 2023, 67, 107515.
20. Jiang, Y.; Zou, Q.; Liu, S.; et al. The Li3V2(PO4)3@C materials prepared by freeze-drying assisted sol-gel method for an aqueous zinc ion hybrid battery. J. Electroanal. Chem. 2021, 900, 115685.
21. Li, Q.; Jiao, Q.; Li, Z.; et al. Sandwich‐like MXene@Mn3O4@PPy hollow microspheres synergistically enabled ultra-long cycling life in aqueous zinc ion batteries. Small. 2024, 21, 2409217.
22. Choi, S.; Kim, S.; Hwang, C.; et al. Plasma assisted hydrothermal synthesis of 2D & 3D Water Intercalated V2O5 nanosheet clusters for high performing aqueous zinc ion battery. Small. Struct. 2025, 6, 2500269.
23. Lei, Y.; Li, S.; Du, M.; et al. Preparation of double‐shell Si@SnO2@C nanocomposite as anode for lithium‐ion batteries by hydrothermal method. Rare. Metals. 2023, 42, 2972-81.
24. Liu, Y.; Tian, X.; Han, Y. C.; Chen, Y.; Hu, W. High-temperature shock synthesis of high-entropy-alloy nanoparticles for catalysis. Chin. J. Catal. 2023, 48, 66-89.
25. Huang, W.; Zhu, X.; Zhu, H.; et al. High temperature shock (HTS) synthesis of carbon-based nanomaterials for electrochemical applications. Carbon. Neutral. 2025, 4, e189.
26. Cui, X.; Liu, Y.; Chen, Y. Ultrafast micro/nano-manufacturing of metastable materials for energy. Natl. Sci. Rev. 2024, 11, nwae033.
27. Zeng, C.; Duan, C.; Guo, Z.; et al. Ultrafastly activated needle coke as electrode material for supercapacitors. Prog. Nat. Sci:. Mater. Int. 2022, 32, 786-92.
28. Hua, Y.; Li, X.; Li, J.; et al. Fast fabrication of a hierarchical nanostructured multifunctional ferromagnet. Science. 2024, 385, 634-41.
29. Choi, C. H. â.; Shin, J.; Eddy, L.; et al. Flash-within-flash synthesis of gram-scale solid-state materials. Nature. Chem. 2024, 16, 1831-7.
30. Silva, K. J.; Wyss, K. M.; Teng, C. H.; Cheng, Y.; Eddy, L. J.; Tour, J. M. Graphene derived from municipal solid waste. Small. 2024, 21, 2311021.
31. Liu, S.; Shen, Y.; Zhang, Y.; et al. Extreme environmental thermal shock induced dislocation‐rich Pt nanoparticles boosting hydrogen evolution reaction. Adv. Mater. 2021, 34, 2106973.
32. Chen, W.; Cheng, Y.; Chen, J.; et al. Nondestructive flash cathode recycling. Nat. Commun. 2024, 15, 6250.
33. Wang, J.; Xiao, G.; Gao, N.; et al. Highly selective CO2 electrocatalytic reduction on nickel single-atom catalyst in a high-temperature shockwave method. Fuel. 2023, 338, 127312.
34. Hu, X.; Zuo, D.; Cheng, S.; et al. Ultrafast materials synthesis and manufacturing techniques for emerging energy and environmental applications. Chem. Soc. Rev. 2023, 52, 1103-28.
35. Guo, Z.; Liu, Z.; Zhang, J.; et al. Joule heating ultrafast synthesis. ACS. Appl. Energy. Mater. 2025, 8, 9926-37.
36. Zou, J.; Tang, L.; Kang, L. Innovative heating for the nano age: exploring the potentials of carbothermal shock. ACS. Nano. 2025, 19, 152-86.
37. Qiu, Y.; Hu, Z.; Li, H.; Ren, Q.; Chen, Y.; Hu, S. Hybrid electrocatalyst Ag/Co/C via flash Joule heating for oxygen reduction reaction in alkaline media. Chem. Eng. J. 2022, 430, 132769.
38. Dou, S.; Xu, J.; Cui, X.; et al. High-temperature shock enabled nanomanufacturing for energy-related applications. Adv. Energy. Mater. 2020, 10, 2001331.
39. Zhao, P.; Wu, X.; Zhang, Y.; et al. Ultrafast thermal engineering in energy materials: design, recycling, and future directions. ACS. Nano. 2025, 19, 17199-227.
40. Dong, Y.; Rao, Y.; Liu, H.; et al. Highly efficient chemical production via electrified, transient high-temperature synthesis. eScience. 2024, 4, 100253.
41. Wang, C.; Li, Z.; Miao, Z.; et al. A soluble precursor facilitates ultra-fast synthesis of O3 layered oxides for sodium-ion batteries. Sci. China. Mater. 2025, 68, 1967-73.
42. Liu, Y.; Xu, Y.; Tian, Y.; et al. Frank partial dislocation pinning effect engineered IrNi alloy nanoparticles for water splitting. ACS. Catal. 2025, 15, 3378-90.
43. Zhang, L.; Ma, M.; Hu, Z.; et al. Coupling joule heating with vibration ball milling for synthesizing carbon-supported NixFex nanoparticles achieving efficient oxygen evolution and alkaline water electrolysis. ACS. Appl. Eng. Mater. 2024, 2, 2919-32.
44. Mazo, I.; Palmieri, B.; Martone, A.; Giordano, M.; Sglavo, V. M. Flash sintering in metallic ceramics: finite element analysis of thermal runaway in tungsten carbide green bodies. J. Mater. Res. Technol. 2023, 23, 5993-6004.
45. Shan, Y.; Li, X.; Zhao, W.; et al. Programmable and rapid fabrication of complex-shape ceramics. Nat. Commun. 2024, 15, 9973.
46. Lang, C.; Xu, Y.; Yao, X. Perfecting HER catalysts via defects: recent advances and perspectives. Chin. J. Catal. 2024, 64, 4-31.
47. Reynaud, M.; Serrano-Sevillano, J.; Casas-Cabanas, M. Imperfect battery materials: a closer look at the role of defects in electrochemical performance. Chem. Mater. 2023, 35, 3345-63.
48. Zhao, Y.; Du, H.; Kang, Y.; et al. Spent battery regeneration for better recycling. Nat. Rev. Mater. 2025, 10, 722-4.
49. Wang, R.; Chen, X.; Huang, Z.; et al. Twin boundary defect engineering improves lithium-ion diffusion for fast-charging spinel cathode materials. Nat. Commun. 2021, 12, 3085.
50. Zong, J.; Liang, Y.; Liu, F.; et al. Engineering twin structures and substitutional dopants in ZnSe0.7Te0.3 anode material for enhanced sodium storage performance. Nat. Commun. 2025, 16, 4406.
51. Zou, G.; Wang, J.; Sun, Y.; et al. A nanotwinned-alloy strategy enables fast sodium deposition dynamics. Nat. Commun. 2025, 16, 1795.
52. Zhong, H.; Zeng, C.; Lai, J.; et al. Enhanced oxygen evolution by activating vacancy defects on metal-organic framework-derived Co3O4/NC. Carbon. Neutralization. 2025, 4, e70030.
53. Zhao, M.; Li, S.; Wu, X.; Sun, L. Regulating oxygen vacancies in ammonium vanadate electrode materials for advanced aqueous zinc ion batteries. iScience. 2024, 27, 110926.
54. Liang, J.; Jiang, L.; Liu, H.; et al. Engineering interfacial oxygen vacancies of Zn-Cr sites for CO2 activation and hydrogenation. ACS. Catal. 2025, 15, 7340-50.
55. Luo, J.; Zhang, J.; Guo, Z.; et al. Coupling antisite defect and lattice tensile stimulates facile isotropic Li‐ion diffusion. Adv. Mater. 2024, 36, 2405956.
56. Liu, Z.; Zeng, C.; Zhang, J.; et al. Twin boundaries induced by high-temperature shock boost the structural stability of Li-rich layered-oxide. J. Mater. Chem. A. 2024, 12, 23712-23722.
57. Guo, Z.; Jiang, H.; Sun, X.; et al. Ultrafast non-equilibrium phase transition induced twin boundaries of spinel lithium manganate. Adv. Energy. Mater. 2023, 14, 2302484.
58. Zhu, W.; Zhang, J.; Luo, J.; et al. Ultrafast non-equilibrium synthesis of cathode materials for Li-ion batteries. Adv. Mater. 2022, 35, 2208974.
59. Xiao, J.; Chen, Y.; Cai, C.; et al. Flash joule heating synthesis of nitrogen-rich defective g‐C3N4 for highly efficient photocatalytic hydrogen evolution. Small. 2025, 21, 2503335.
60. Tian, L.; Hu, F.; Dong, E.; et al. Defect-mediated heteroepitaxial Co-regeneration strategy for direct regeneration of spent cathodes materials. Adv. Energy. Mater. 2025, 15, e02546.
61. Lv, X.; Lin, J.; Sun, X.; et al. Direct recycling of spent LiFePO4 cathodes through photocatalytic correction of anti-site defects. Adv. Mater. 2025, 37, 2503398.
62. Li, Y.; Cai, J.; Wang, J.; et al. A comprehensive review on reductive recycling of cathode materials of spent lithium-ion batteries. Chemistry. 2024, 30, e202400566.
63. Bin Abu Sofian, A. D. A.; Majid, S.; Kang, K.; Kim, J. K.; Show, P. Upcycling and recycling of spent battery waste for a sustainable future: progress and perspectives. Prog. Mater. Sci. 2025, 153, 101478.
64. Biswal, B. K.; Zhang, B.; Thi Minh Tran, P.; Zhang, J.; Balasubramanian, R. Recycling of spent lithium-ion batteries for a sustainable future: recent advancements. Chem. Soc. Rev. 2024, 53, 5552-92.
65. Fan, M.; Chang, X.; Meng, Q.; Wan, L.; Guo, Y. Progress in the sustainable recycling of spent lithium-ion batteries. SusMat. 2021, 1, 241-54.
66. Tang, Y.; Yang, Y.; Pan, M.; et al. Critical review of thermal reduction processes for sustainable recovery of valuable metals from cathode materials in spent lithium-ion batteries. Adv. Funct. Mater. 2025, e13322.
67. Yin, Y. C.; Li, C.; Hu, X.; et al. Rapid, direct regeneration of spent LiCoO2 cathodes for Li-ion batteries. ACS. Energy. Lett. 2023, 8, 3005-12.
68. Cheng, Y.; Chen, J.; Chen, W.; et al. Rapid electrothermal rejuvenation of spent lithium cobalt oxide cathode. Energy. Environ. Sci. 2025, 18, 6085-93.
69. Guo, Y.; Yao, Y.; Guo, C.; et al. Atomistic observation and transient reordering of antisite Li/Fe defects toward sustainable LiFePO4. Energy. Environ. Sci. 2024, 17, 7749-61.
70. Zheng, S. H.; Wang, X. T.; Gu, Z. Y.; et al. Direct and rapid regeneration of spent LiFePO4 cathodes via a high-temperature shock strategy. J. Power. Sources. 2023, 587, 233697.
71. Luo, J.; Zhang, J.; Guo, Z.; et al. Recycle spent graphite to defect-engineered, high-power graphite anode. Nano. Res. 2022, 16, 4240-5.
72. Luan, C.; Jiang, L.; Zheng, X.; et al. Direct observation of the ultrafast formation of cation-disordered rocksalt oxides as regenerable cathodes for lithium-ion batteries. Chem. Eng. J. 2023, 462, 142180.
73. Yang, M.; Lin, Y.; Chen, P.; et al. Unlocking ultrafast-kinetics asymmetric heterojunction with multi-anionic redox chemistry enables high energy/power density and low-temperature zinc-ion batteries. Angew. Chem. Int. Ed. Engl. 2025, 64, e202510907.
74. Ni, J.; Shi, Z.; Bai, J.; et al. Heterointerface anchored Ir with localized strong orbital coupling for durable proton exchange membrane water electrolysis. Angew. Chem. Int. Ed. Engl. 2025, 64, e202509985.
75. Dong, X.; Chen, X.; Tong, X.; et al. Red/black phosphorus heterostructure anchored on graphene for lithium-sulfur batteries with accelerated conversion kinetics and improved long-term stability. Chem. Eng. J. 2025, 519, 164704.
76. Tatarinov, D. A.; Schleusener, A.; Krahne, R. Controlling energy flow in perovskite heterostructures through dimensionality and phase engineering. Adv. Sci. (Weinh). 2025, 12, e05971.
78. Zhao, Z.; Sun, J.; Li, X.; Zhang, Z.; Meng, X. Joule heating synthesis of NiFe alloy/MoO2 and in-situ transformed (Ni,Fe)OOH/MoO2 heterostructure as effective complementary electrocatalysts for overall splitting in alkaline seawater. Appl. Catal. B:. Environ. 2024, 340, 123277.
79. Qin, X.; Yu, C.; Zhou, W.; Xu, Z.; Chang, J.; Wang, X. Joule heating: a versatile and sustainable heating strategy with diverse applications in materials science and waste management. J. Mater. Chem. A. 2025, 13, 12828-54.
80. Huang, P.; Guo, Z.; Li, Z.; et al. Spatiotemporal evolution in hard carbon synthesis via electrothermal coupling strategy for high-performance sodium-ion batteries. Adv. Mater. 2025, 37, 2507521.
81. Yan, D.; Li, H.; Yang, A.; et al. Ultrafast synthesis of vanadium-based oxides with crystalline-amorphous heterostructure for advanced aqueous zinc-ion batteries. Chem. Eng. J. 2025, 504, 158966.
82. Zhu, W.; Su, H.; Bai, P.; et al. A layered/spinel heterostructured cathode for Li-ion batteries prepared by ultrafast Joule heating. Chem. Eng. J. 2024, 480, 148045.
83. Chen, S.; Ma, J.; Chen, Q.; et al. Phase junction induction through atomic ratio tuning of molybdenum carbides for enhanced stepwise iodine conversion. Adv. Funct. Mater. 2025, 35, 2505201.
84. Dong, H.; Wang, L.; Cheng, Y.; et al. Flash joule heating: a promising method for preparing heterostructure catalysts to inhibit polysulfide shuttling in Li-S batteries. Adv. Sci. (Weinh). 2024, 11, 2405351.
85. Kar, N.; Skrabalak, S. E. Synthetic methods for high-entropy nanomaterials. Nat. Rev. Mater. 2025, 10, 638-53.
86. Yu, L.; Zeng, K.; Li, C.; et al. High‐entropy alloy catalysts: from bulk to nano toward highly efficient carbon and nitrogen catalysis. Carbon. Energy. 2022, 4, 731-61.
87. Shi, W.; Liu, H.; Liu, S.; et al. Heterostructure engineering in high-entropy alloy catalysts. SusMat. 2024, 5, e261.
88. Du, M.; Li, K.; Yu, N.; et al. Ultrafast preparation of high-entropy NASICON cathode enables stabilized multielectron redox and wide-temperature (-50-60 °C) workability in sodium-ion batteries. Adv. Mater. 2025, 37, 2418219.
89. He, X.; Zeng, X.; Wang, W.; et al. A novel rock-salt structure high-entropy oxide Fe0.2Co0.2Ni0.2Cu0.2Zn0.2O as a highly reversible lithium storage material. Nano. Res. 2025, 18, 94907784.
90. Zou, X.; Zhi, S.; Pang, B.; et al. Dual-confinement strategy improves the stability of high-entropy alloys in ultra-large current zinc-air batteries. Energy. Environ. Mater. 2025, 8, e70057.
91. Wang, P.; Guo, S.; Xu, Y.; et al. Upcycling spent cathodes from Li-ion batteries into a high-entropy alloy catalyst with reverse electron transfer for Li-O2 batteries. ACS. Nano. 2025, 19, 17589-605.
92. Zhu, X.; Huang, W.; Lou, Y.; et al. Ultrafast joule-heating synthesis of FeCoMnCuAl high-entropy-alloy nanoparticles as efficient OER electrocatalysts. Prog. Nat. Sci.:. Mater. Int. 2024, 34, 880-7.
93. Hassanzadeh-Tabrizi, S. Precise calculation of crystallite size of nanomaterials: a review. J. Alloys. Compd. 2023, 968, 171914.
94. Shah, S. S.; Niaz, F.; Ehsan, M. A.; et al. Advanced strategies in electrode engineering and nanomaterial modifications for supercapacitor performance enhancement: a comprehensive review. J. Energy. Storage. 2024, 79, 110152.
95. Abdelhamid, A. A.; Mendoza-Garcia, A.; Ying, J. Y. Advances in and prospects of nanomaterials’ morphological control for lithium rechargeable batteries. Nano. Energy. 2022, 93, 106860.
96. Chen, Y.; Li, Y.; Wang, Y.; et al. Rapid, in situ synthesis of high capacity battery anodes through high temperature radiation-based thermal shock. Nano. Lett. 2016, 16, 5553-8.
97. Lu, J.; Liu, S.; Liu, J.; et al. Millisecond conversion of photovoltaic silicon waste to binder-free high silicon content nanowires electrodes. Adv. Energy. Mater. 2021, 11, 2102103.
98. Shen, L.; Sun, K.; Xi, F.; et al. Conversion of photovoltaic waste silicon into amorphous silicon nanowire anodes. Energy. Environ. Sci. 2025, 18, 4348-61.
99. Dou, S.; Xu, J.; Zhang, D.; et al. Ultrarapid nanomanufacturing of high-quality bimetallic anode library toward stable potassium-ion storage. Angew. Chem. Int. Ed. Engl. 2023, 62, e202303600.
100. Zong, L.; Lu, F.; Li, P.; et al. Thermal shock synthesis for loading sub‐2 nm Ru nanoclusters on titanium nitride as a remarkable electrocatalyst toward hydrogen evolution reaction. Adv. Mater. 2024, 36, 2403525.
101. Fang, R.; Yang, J.; Song, W. S.; et al. High-temperature shock-induced transformation of bulk copper into single-atom catalyst. Nano. Res. 2025, 18, 94907300.
102. Shi, W.; Li, Z.; Gong, Z.; et al. Transient and general synthesis of high-density and ultrasmall nanoparticles on two-dimensional porous carbon via coordinated carbothermal shock. Nat. Commun. 2023, 14, 2294.
103. Cai, H.; Yang, H.; He, S.; et al. Size-adjustable high-entropy alloy nanoparticles as an efficient platform for electrocatalysis. Angew. Chem. Int. Ed. Engl. 2025, 64, e202423765.
104. Zhang, Y.; Lin, Y.; Duan, T.; Song, L. Interfacial engineering of heterogeneous catalysts for electrocatalysis. Mater. Today. 2021, 48, 115-34.
105. Chen, B.; Sui, S.; He, F.; et al. Interfacial engineering of transition metal dichalcogenide/carbon heterostructures for electrochemical energy applications. Chem. Soc. Rev. 2023, 52, 7802-47.
106. Du, Y.; Li, B.; Xu, G.; Wang, L. Recent advances in interface engineering strategy for highly‐efficient electrocatalytic water splitting. InfoMat. 2022, 5, e12377.
107. Chen, C.; Jiang, M.; Zhou, T.; et al. Interface aspects in all-solid-state Li-based batteries reviewed. Adv. Energy. Mater. 2021, 11, 2003939.
108. Wan, H.; Wang, Z.; Zhang, W.; He, X.; Wang, C. Interface design for all-solid-state lithium batteries. Nature. 2023, 623, 739-44.
109. Zahiri, B.; Patra, A.; Kiggins, C.; et al. Revealing the role of the cathode-electrolyte interface on solid-state batteries. Nat. Mater. 2021, 20, 1392-1400.
110. Kong, X.; Gu, R.; Jin, Z.; et al. Maximizing interface stability in all-solid-state lithium batteries through entropy stabilization and fast kinetics. Nat. Commun. 2024, 15, 7247.
111. Yao, X.; Chen, S.; Wang, C.; et al. Interface welding via thermal pulse sintering to enable 4.6 V solid-state batteries. Adv. Energy. Mater. 2023, 14, 2303422.
112. Cui, X.; Chen, X.; Lin, C.; et al. An integrated optimization strategy by Joule heating technique enabling rapid fabrication of robust Li1.3Al0.3Ti1.7(PO4)3 solid-state electrolyte for all-solid-state lithium metal batteries. J. Colloid. Interface. Sci. 2025, 686, 660-671.
113. Chen, J.; Chen, W.; Deng, B.; Li, B.; Kittrell, C.; Tour, J. M. Cathode interface construction by rapid sintering in solid-state batteries. Small. 2023, 20, 2307342.
114. Tubtimkuna, S.; Danilov, D. L.; Sawangphruk, M.; Notten, P. H. L. Review of the scalable core-shell synthesis methods: the improvements of Li‐ion battery electrochemistry and cycling stability. Small. Methods. 2023, 7, 2300345.
115. Wang, F.; Zhang, W.; Wan, H.; et al. Recent progress in advanced core-shell metal-based catalysts for electrochemical carbon dioxide reduction. Chin. Chem. Lett. 2022, 33, 2259-69.
116. Yin, R.; Guo, Z.; Liu, R.; Tao, X. S. Ultrafast synthesis of Na3V2(PO4)3 cathode for high performance sodium-ion batteries. Chin. Chem. Lett. 2025, 36, 109643.
117. Li, Z.; Huang, P.; Zhang, J.; et al. Ultra-uniform interfacial matrix via high-temperature thermal shock for long-cycle stability cathodes of sodium-ion batteries. Energy. Environ. Sci. 2025, 18, 2962-72.
118. He, W.; Cai, J.; Li, Y.; Wang, Z.; Li, Y.; Yan, X. Transforming of rigid-flexible micro-sized silicon anodes: carbothermal shock method yields durable, high-capacity electrodes. ChemistrySelect. 2025, 10, e202501453.
119. Shi, W.; Liu, H.; Zhang, J.; et al. Roll-to-roll synthesis of multielement heterostructured catalysts. Nat. Synth. 2025, 4, 836-847.
120. Zeng, K.; Zhang, J.; Gao, W.; et al. Surface-decorated high-entropy alloy catalysts with significantly boosted activity and stability. Adv. Funct. Mater. 2022, 32, 2204643.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0









Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].