


A high capacity V$$_4$$ $$_3$$ $$_z$$ $$_2$$
 0
0 
Abstract
The use of highly concentrated electrolytes to enlarge the operational electrochemical window of MXenes is a strategy to enhance their energy density. Here, we demonstrate that V
Keywords
INTRODUCTION
The development of more efficient, sustainable and cost-effective energy storage devices is one of the top priorities in our society enabling the migration to renewable energies and supplying for the energy storage needs of a growing market of a range of applications including wearable electronics. The greatest challenge is to develop energy storage devices with high energy density and power density. Supercapacitors are high power density devices, as compared to batteries, but supply a comparatively lower energy density. Hence, enhancing energy density of supercapacitors is a current focus of intensive research.
MXenes have emerged as excellent material for supercapacitor applications[1]. MXenes, M
MXenes have demonstrated a high-rate performance in aqueous electrolytes of typically 1 M concentration. However, an area of improvement is the enlarging of typically negative electrochemical windows (EWs) vs. open circuit potential (OCP) to improve energy density. Particularly, Ti
Recently, water-in-salt electrolytes (WISEs) have emerged as a suitable alternative to enlarge the electrochemically stable windows, hence energy density, of MXene-based supercapacitors, overcoming the limitations imposed by standard aqueous electrolytes. WISEs are highly concentrated electrolytes (HCEs) where the salt outnumbers the water in weight and volume[15,16], and very few water molecules are free to form hydrogen bonds[17]. Unlike their salt-in-water counterpart, in WISEs, cation-anion interactions such as contact ion pairs (CIPs) and aggregated cation-anion pairs (ACAPs) dominate over ion-solvent interactions leading to unusual physicochemical properties[15,18]. The advantages of WISEs in energy storage were first visualized and utilized in battery applications to enlarge the electrochemically stable windows, where both HER and OER shifted to lower and higher potentials, respectively[15]. HER shifted to lower potentials as a result of a preferential reduction reaction of anions in cation-anion adducts (formed as a result of the very high concentration of salt) over water molecules[15]. This reduction reaction resulted in the formation of a passivation layer that eventually suppressed HER[15]. The shift of OER to positive potentials has been explained by several works. When coordinated with cations such as Li
Another category of electrolytes of even higher salt concentration is hydrate melts, in which all the water molecules are coordinated with cations or CIPs and no water molecules are free to form hydrogen bonds[17]. Especially designed hydrate melts favor water solvation of cations disabling the formation of CIPs[17]. A disadvantage of WISEs and some hydrate melts is a substantial reduction of the reaction kinetics of energy storage process due to low ionic conductivity and high viscosity of such HCEs.
In the case of MXenes, WISEs have been used to enlarge the negative limit of electrochemically stable windows, while being paired with electrodes operating at positive potentials for the achievement of full cells. Ti
Herein, we investigate the charge storage processes of V
EXPERIMENTAL METHODS
Chemicals
Hydrofluoric acid (HF, 48 wt.
Synthesis of V$$ _4 $$ $$ _3 $$
As reported previously in[13]. Vanadium powder (99.5%, -325 mesh, Alfa Aesar), aluminum powder (99.5%, -325 mesh, Alfa Aesar), and graphite (99
Synthesis of etched V$$ _4 $$ $$ _3 $$
As reported previously in[13]. Prior to etching procedures, particle size selection was performed. Powder of particle size
After etching, the reaction mixture was placed in conical centrifuge tubes (150 mL); DI water was added and centrifugation was done at 5, 000 rpm for 10 min. The acidic supernatant was collected with a pipette and discarded. This step was repeated until the supernatant reached pH 5-6. The etched powder was then further washed and collected by vacuum-assisted filtration, using polyvinylidene fluoride (PVDF) membrane filters (0.22
Delamination of etched V$$ _4 $$ $$ _3 $$
As reported previously in[13]. For delamination, the etched powder was added to 10 wt.
V$$ _4 $$ $$ _3 $$ $$ _z $$
As reported previously in[13]. Delaminated films were manufactured using vacuum-assisted filtration. A standard glass vacuum filtration system and a Celgard 3501 filter membrane (64 nm pore size) were used. To determine the suspension volume to filter to achieve a determined film thickness, calibration was necessary according to the concentration of the suspension. In general, to reduce concentration and facilitate faster filtration, as-produced suspensions were purified by a centrifugation step at 5, 000 rpm for 30 min and collection of the top 70
Electrolyte preparation and property measurements
LiBr/water electrolytes of 5, 7.5, 10, 12.5, 15, 17.5 and 19 molal concentration were prepared at room temperature by adding the LiBr salt in small portions to DI water (25 mL) while magnetically stirring (100 rpm). Complete dissolution, especially in the most concentrated electrolytes, was ensured by stirring overnight. The recipients containing the electrolyte were maintained closed to avoid water evaporation caused by the highly exothermic dissolution.
The following observations are worth noting. In this work, we found the limit of solubility of LiBr in DI water to be 19.0 m and not 19.2 m, as previously reported[24,25]. Previous studies in LiBr aqueous solubility as a function of temperature state a theoretical equilibrium solubility of a 0.625 wt.
Measurements of electrolyte viscosity and conductivity were done at 25
Equipment
Centrifugation was performed using a centrifuge (Thermo Scientific Heraeus Multifuge X1R) equipped with a TX-400 rotor (16.8 cm radius). The relative centrifugal force (RCF), measured in gravity acceleration (g) units, is defined as
where S is the centrifugal rotational speed (rpm), and r is the radius of the rotor (cm). This formula should be used to convert the
The mass of the powders was measured using a balance (Sartorius, Germany) with a 0.0001
Materials characterization methods
Materials characterization was performed in our previous work[13]. Here, we describe briefly instrumentation. Scanning electron microscopy (SEM) was performed in a ZEISS Merlin (ZEISS, Germany) microscope equipped with a GEMINI II column and using the In Lens detector. Standard and high-resolution high angle annular dark field scanning transmission electron microscopy (HAADF-STEM) and electron-energy loss spectroscopy (EELS) were conducted using a double C
X-ray diffraction (XRD) was performed in a STOE STADI P diffractometer with a Mo K
Electrochemical methods
Electrochemical cell. Electrochemical tests were performed in 1/4 inch 3-electrode perfluoroalkoxy alkane (PFA) Swagelok cells using 6 mm diameter glassy carbon rods as current collectors. Cells were assembled using disc electrodes of V
Electrochemical tests. Electrochemical tests were performed in a VMP300 (BioLogic) potentiostat using EC-Lab software. Cyclic voltammetry (CV) was the main technique used. The capacitance C (F) was calculated as:
where anodic (
Galvanostatic cycling with potential limitation (GCPL) was performed according to the potential limits established by CV and according to the electrolyte concentration. GCPL rate tests were performed at 0.25 to 10 A g-1 current rates. The CE from GCPL studies was calculated as (Q
Staircase potentio-EIS (SPEIS), a technique suitable for performing successive EIS measurements during a potential scan, was used to study ion transport processes[13]. SPEIS was performed in half-cell electrode setups using electrolytes of selected concentrations. The measurements were conducted along a CV scan following an OCP to V
Capacitance was calculated from EIS studies according to[30]:
where
where
Results and discussion
Materials characterization
The MXene V
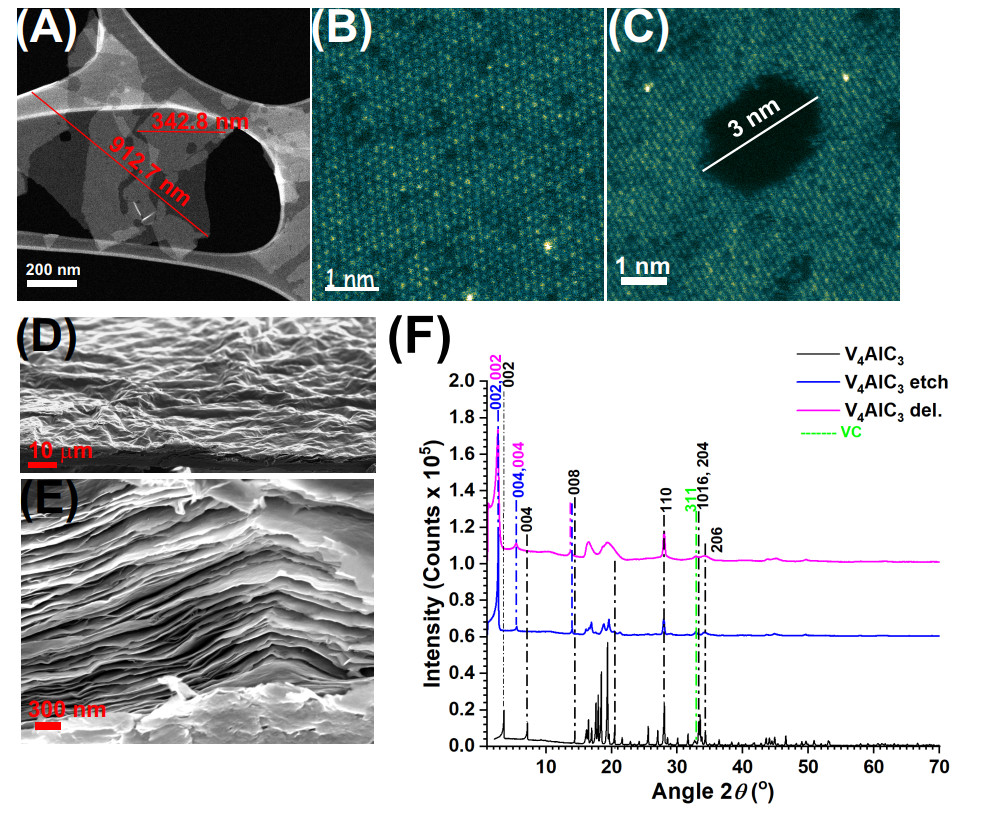
Figure 1. (A) HAADF-STEM image of V4C3Tz flakes with lateral size indicated, (B and C) HAADF-STEM images of a V4C3Tz monolayers showing vacancies and a (C) pore, (D and E) SEM images of a V4C3Tz film obtained by vacuum filtration, (F) XRD patterns of V4AlC3 and corresponding etched and delaminated samples. Reprinted from[13], Copyright (2024), with permission from Elsevier.
In our previous work, XRD studies and Rietveld refinement confirmed the crystal structure of V
Electrolyte formulations
The electrochemical activity of V
LiBr/H
| LiBr/H | |||||||
| Molality | 5 | 7.5 | 10 | 12.5 | 15 | 17.5 | 19 |
| wt. LiBr/wt. H | 0.43 | 0.65 | 0.87 | 1.09 | 1.30 | 1.52 | 1.65 |
| vol. LiBr/vol. H | 0.13 | 0.20 | 0.27 | 0.34 | 0.41 | 0.48 | 0.54 |
| mol LiBr/mol H | 0.09 | 0.14 | 0.18 | 0.23 | 0.27 | 0.32 | 0.34 |
| mol H | 11.11 | 7.40 | 5.55 | 4.44 | 3.70 | 3.17 | 2.92 |
Based on these arguments, we consider the 12.5 m, 15 m, 17.5 m and 19 m electrolytes as high-concentration electrolytes, where the 19 m is a HCE but not WISE, whereas the other 5 m, 7.5 m, 10 m electrolytes are considered low-concentration or salt-in-water electrolytes.
Electrochemical windows
The stability of EWs was studied. CVs were performed at 0.5 mV s-1 from OCP to negative potentials vs. Ag (all the potentials in this work are referred to as an Ag pseudoreference electrode) in steps of 0.05 V. The same was done for positive potentials using a different electrode. In addition, CAs were performed from OCP to the same potentials for 15-30 min ensuring current stabilization and using different electrodes for positive and negative potentials vs. OCP. The OCP of the electrodes varied with electrolyte concentration.
For the test using the 10 m electrolyte and negative EW, the OCP was at 0.37 V [Supplementary Figure S3A]. In this electrolyte, significant CV cathodic currents were observed at potentials
The EWs for the other low-concentration electrolytes, 5 m and 7.5 m followed a similar behavior at the negative limit, having and onset of HER at -0.7 V [Supplementary Figure S2]. However, at the positive limit of the EW, the onset of high anodic currents was at a lower potential of 0.6 V for the electrode tested in the 5 m electrolyte [Supplementary Figure S2B]. For the electrode tested in the 7.5 m electrolyte, the electrochemically stable positive EW limit was 0.7 V [Supplementary Figure S2D].
For the 15 m electrolyte and positive EW, the OCP was 0.54 V; significant cathodic CV currents were observed at potentials
For the 19 m electrolyte and negative EW, the OCP was 0.5 V; significant CV cathodic currents were observed at potentials
A suitable observation on all these tests is that the evolving currents beyond the set negative EW limits are larger than those evolving beyond the set EW positive limits, describing a larger electrochemical activity due to HER than the activity at the positive limits.
Based on these studies, EW potential limits were set for further studies. A negative EW potential limit of -0.7 V was set for tests in all electrolytes. Positive EW potential limits were set as 0.8 V for tests in 12.5 m, 15 m, 17.5 m and 19 m electrolytes; and 0.7 V for tests in 5 m, 7.5 m and 10 m electrolytes. An EW positive potential limit of 0.6 V applies to tests in the 5 m electrolyte. However, a setting at 0.7 V was preferred for the sake of comparison of charge storage in a common EW with tests in 7.5 m and 10 m electrolytes. Cycling stability studies indeed showed that a positive EW limit of 0.7 V leads to poor cycling stability and poor CE in these electrolytes (Sections "Cyclability tests", "Analysis of CVs of cycling tests"), thus, confirming that a lower positive EW limit of a maximum of 0.6 V should be used.
Cyclic voltammetry tests
Next, the electrochemical activity of V
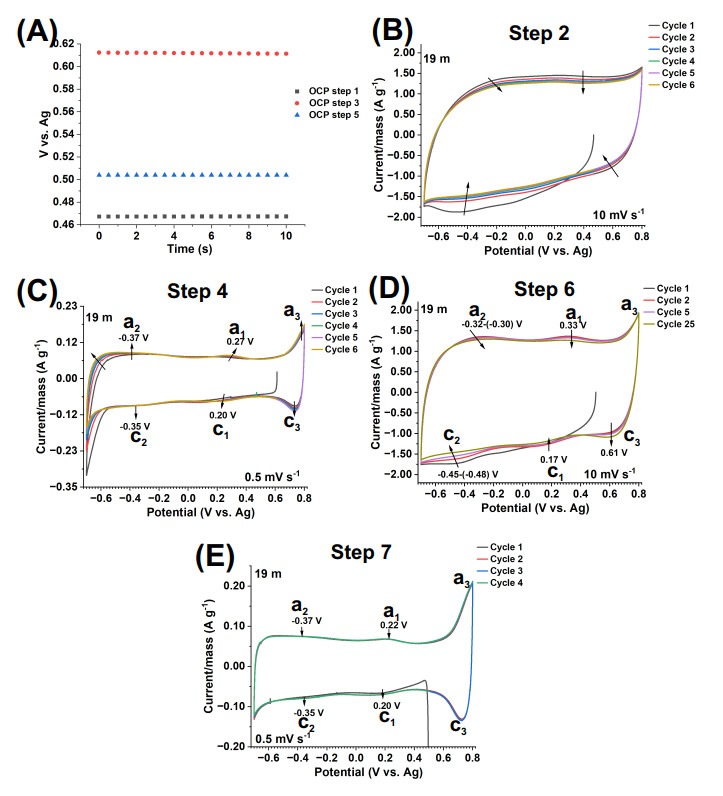
Figure 2. CV tests of V
The electrochemical processes undergone by the V
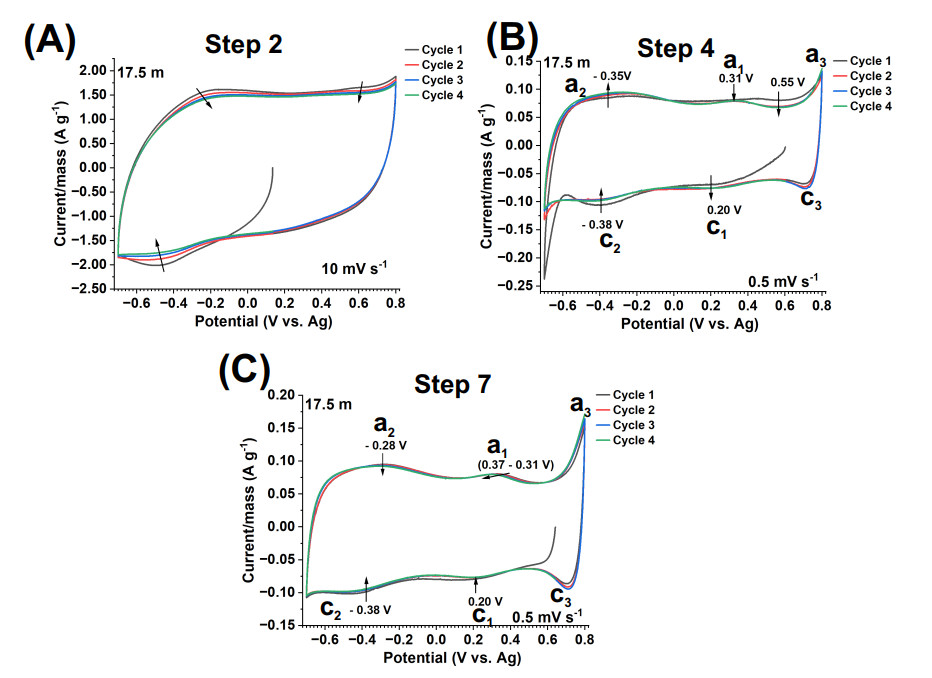
Figure 3. CV tests of V
The electrochemical processes of the V
Similar electrochemical processes were undergone by the V
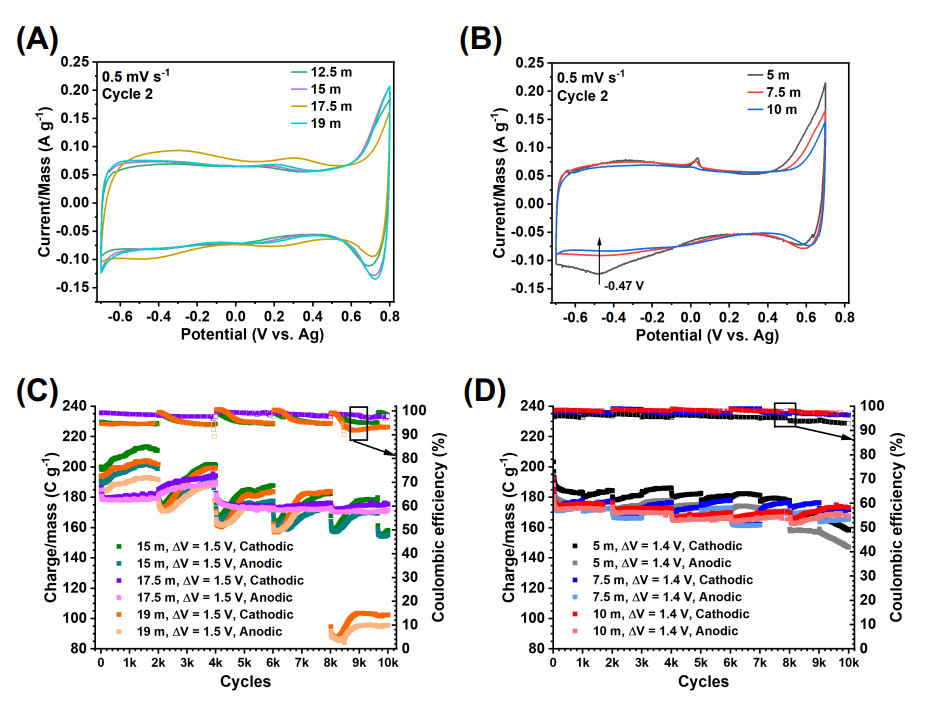
Figure 4. Charge storage performance of V
The CV tests in electrodes tested in
An anodic peak at 0.0 V was proved to be related to the glassy carbon current collector [Supporting Information, Supplementary Section S5, Supplementary Figure S10]. The cathodic/anodic currents at c
In order to evaluate the nature of the charge storage contribution of the V
In summary, the electrodes tested in 19 m, 17.5 m, 15 m, and 12.5 m showed a similar electrochemical activity consisting of (c
Cyclability tests
Cyclability tests revealed further insights into charge storage mechanisms and EW stability. Electrodes were tested by CV at 10 mV s-1 up to 10, 000 cycles in 7.5 m, 10 m, 15 m, 17.5 m and 19 m LiBr [Figure 4C and D]. In order to understand the cycling trends, the following note is relevant. Preliminary tests showed that the Celgard separator is not stable in LiBr and tends to degrade over cycling. For this reason, the separator and electrolyte were changed every 1, 000 or 2, 000 cycles. Other separators, including glass fiber, were tested with no better performance.
The cycling performance of the electrodes tested in the low-concentration electrolytes 5 m and 7.5 m was relatively stable but highly irreversible [Figure 4D]. Namely, cathodic capacity retention of 77.8
The cycling performance of the electrodes tested in the high-concentration electrolytes 15 m, 17.5 m and 19 m electrolytes was different as compared to those tested in the low-concentration electrolytes [Figure 4C]. For the tests in the 15 m and 19 m electrolytes, there was an increasing capacity trend for each set of 2, 000 cycles with a poor CE. For instance, in the 15 m LiBr, the CE was 94.4-95.4
On the other hand, the electrode tested in the 17.5 m electrolyte, although with a slightly lower capacity than the electrode tested in the 15 m electrolyte (195.5 C g-1 (maximum), 174.5 C g-1 (at cycle 5, 000) cathodic capacity), had a superior CE (CE of 99.9 to 96.5
In general, the electrodes tested in the high-concentration electrolytes have a higher capacity but poorer cycling stability and CE than the low-concentration electrolytes. This is correlated to electrolyte properties (Section "Electrolyte properties"). However, electrodes tested in the 7.5 m and 5 m low-concentration electrolytes have a different source of instability linked to an irreversible electrochemical activity at the positive EW potential limit, as demonstrated in the next section ("Analysis of CVs of cycling tests"), which indicates that the EW positive limit should be lowered to at least 0.6 V. This instability is improved for the test in the 10 m electrolyte. The 17.5 m electrolyte offers the best performance balance among all the electrolytes.
Analysis of CVs of cycling tests
A close examination of the electrochemical activity revealed by CVs of the cyclability tests gave insights into the cycling trends. The test sequence followed here was: (Step 1) 10 mV s-1 for five cycles, (Step 2) a CV at 0.5 mV s-1 for five cycles and (Step 3) a CV at 10 mV s-1 for 25 cycles. Due to an analysis of CVs at 10 mV s-1, instead of 0.5 mV s-1 analyzed in the previous section "Cyclic voltammetry tests", a different notation is used to describe the CV electrochemical activity.
The CVs of the tests in the 10 m electrolyte showed fairly stable activity over cycling [Supplementary Figure S12]. The electrochemical activity at 0.045 V (A, anodic), 0.7 V (B, anodic), 0.53 V (C, cathodic), -0.52 V (D, cathodic) and -0.53 V (E, anodic) potentials was analyzed. The anodic current peak A has already been identified as correlated to a reaction with the glassy carbon current collector. The current peaks B and C (c
The CVs of the tests in the 15 m LiBr electrolyte showed a different electrochemical activity [Supplementary Figure S13]. The same current peaks as in the case of the 10 m electrolyte were found at 0.045 V (A, anodic), 0.8 V (B, anodic), 0.60 V (C, cathodic), -0.52 V (D, cathodic) and -0.53 V (E, anodic). In addition, an anodic current at 0.28 V (A') emerged at cycle 2 and moved to 0.16 V on the following cycles [Supplementary Figure S13A]. Other new cathodic currents emerged at 0.08 V, which moved to -0.02 V over cycling (C'), and at -0.40 V (C"). The latter most likely is current D but displaced over cycling. A current at -0.33 V (F) grew over cycling. At cycle 2, 000, the current A was enhanced, along with the currents B and C [Supplementary Figure S13A]. At the same time, C' and C" vanished while a current at -0.20 V (C"') emerged. Upon change of separator and electrolyte, at cycle 2002, the activity in B, C and C"' decreased drastically. From cycles 2, 002 to
The CVs of the tests in the 19 m LiBr electrolyte showed similar electrochemical activity and evolution over cycling than the 15 m [Supplementary Figure S14].
In summary, the electrochemical activity in A is related to the current collector-electrolyte interface. The electrochemical activity in B and C is definitely linked to electrolyte reaction and explains the poor cycling stability and poor CE of the electrodes tested in 5 m and 7.5 m electrolytes.
Electrode rate performance
The charge storage evolution over CV scan rate and as a function of the LiBr electrolyte concentration was studied. Electrodes were tested by CV (at 0.5 to 500 mV s-1 scan rates) in 5 m to 19 m LiBr electrolytes [Supplementary Figure S15]. The cathodic and anodic charge per each scan rate and the CE [Figure 5 and Supplementary Figure S16] were calculated. The trends of the cathodic and anodic charge vs. scan rates were similar [Supplementary Figure S16A-C]. At scan rates
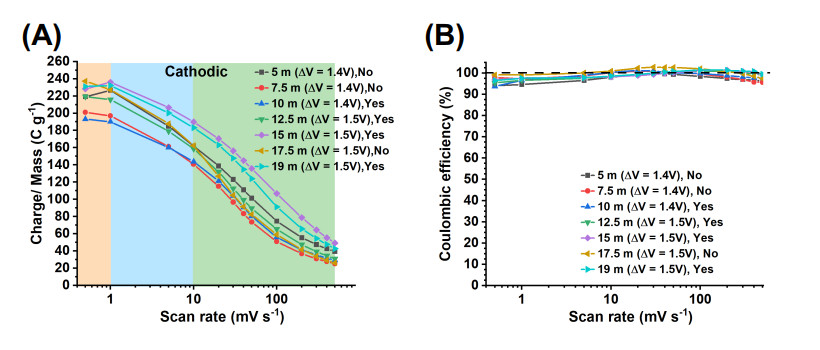
Figure 5. (A) Cathodic charge storage vs. scan rate curves for electrodes tested in 5 to 19 m LiBr electrolytes, and (B) the corresponding Coulombic efficiency curves. The notes "Yes" or "No" indicate whether the electrode underwent an activation procedure (step 6).
Given the similar trends of cathodic and anodic charge vs. scan rate [Supplementary Figure S16], an analysis of the trends across electrolytes is based on the cathodic charge [Figure 5A]. The charge storage vs. concentration trends varied according to three different scan rate regimes: very low (0.5 mV s-1), medium (1-10 mV s-1) and high (20-500 mV s-1). The charge vs. electrolyte concentration trends are as follows. At the very low scan rate of 0.5 mV s-1, the charge storage decreased as: 17.5 m
These charge storage vs. scan rate trends are determined by several variables. First, electrolyte properties, which are a function of concentration, play a key role (Section "Electrolyte properties"). Second, different EWs were considered, i.e., 1.4 V for tests in 5 m, 7.5 m and 10 m electrolytes and 1.5 V for 12.5 m, 15 m, 17.5 m, 19 m electrolytes. A higher charge storage is expected for larger EWs. However, this is not true for the test in 5 m electrolyte (1.4 V EW), which showed higher charge storage than for tests in, e.g., 12.5 m (1.5 V EW) at 1-500 mV s-1 scan rates. A third variable was the performance or not of the activation process (25 cycles at 10 mV s-1, step 6), which was done for tests in 19 m, 15 m, 12.5 m and 10 m electrolytes but not for tests in 17.5 m, 7.5 m and 5 m electrolytes. Electrodes for which activation was not performed had a more drastic decreasing charge storage at high scan rates (20-500 mV s-1). This is the case for the electrode tested in the 17.5 m electrolyte, which had a marked charge storage decrease from 20 to 500 mV s-1, falling below the charge storage of electrodes tested in 5 m (non-activated) and 12.5 m (activated). The same can be said about the non-activated electrode tested in 7.5 m electrolyte, which had a marked charge storage decrease from 20 to 500 mV s-1, which fell below the charge storage of the electrode tested in the 10 m electrolyte (activated). In contrast, electrodes tested in 15 m and 19 m, which underwent activation, did not show a drastic decrease of capacity at 20 to 500 mV s-1 scan rates. Thus, activation procedures are important in keeping a high storage capacity at high scan rates and this becomes important for HCEs.
A clear picture of the advantages of the 17.5 m electrolyte over the 5 m electrolyte was provided by GCPL studies performed on electrodes without activation and at a range of rates from 0.25 to 10 A g-1 [Figure 6A]. At the very low current rate of 0.25 A g-1, the electrodes performed poorly in both electrolytes, especially in the 17.5 m, attributed to the high viscosity and low ionic conductivity at this high concentration. However, at 0.5 to 2 A g-1, the electrode tested in the 17.5 m electrolyte had a higher capacity than the electrode tested in the 5 m electrolyte. The CE was below 100
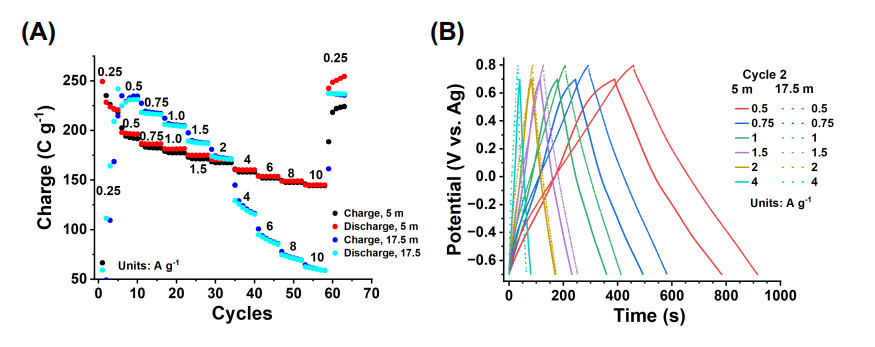
Figure 6. GCPL rate studies of electrodes tested at 0.25 to 10 A g-1 current rates in 5 m and 17.5 m LiBr electrolytes. (A) Charge vs. cycles curves, (B) GCPL curves.
Charge storage performance in context
The maximum charge storage achieved by the electrode tested in the 17.5 m electrolyte at 0.5 mV s-1 was 237.1 C g-1 (65.8 mAh g-1)/158 F g-1/745.5 C cm-3, which delivers and energy density of 49.4 Wh kg-1/155.3 mWh cm-3. The electrode tested in the 15 m electrolyte had superior performance at higher scan rates from 1-500 mV s-1, i.e., 235.9 C g-1 (65.5 mAh g-1)/157.2 F g-1/759.6 C cm-3 and 49.1 Wh kg-1/158.2 mWh cm-3. This performance is superior to the 120 F g-1 reported for Ti
Equally, the performance of the V
Electrolyte properties
The electrolyte properties were studied. The charge storage vs. concentration trends are summarized in Figure 7A (the same data as in Figure 5A). The ionic conductivity and dynamic viscosity were measured as a function of concentration [Figure 7B]. As the electrolyte concentration increased, the ionic conductivity first increased (from 5 m to 7.5 m concentration) and then decreased (for concentrations
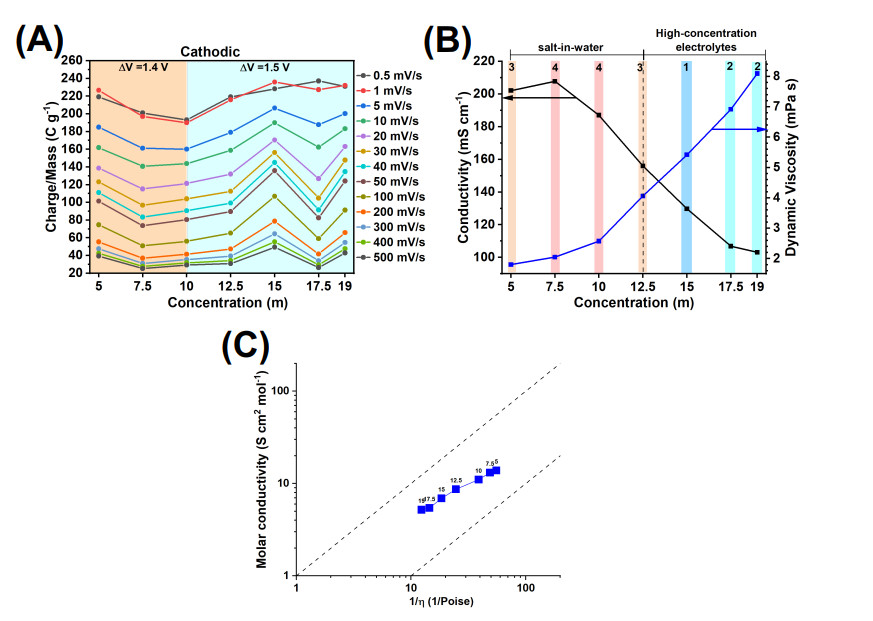
Figure 7. Charge storage performance and electrolyte properties. (A) Charge storage obtained from CVs at a range of scan rates vs. LiBr electrolyte concentration curves, (B) electrolyte conductivity and dynamic viscosity vs. LiBr electrolyte concentration curves, and (C) Walden plot of the LiBr electrolytes at the range of concentration considered in this study (indicated with labels). In (B), the following standard deviations apply to dynamic viscosity values: (5 m)
Using the two pieces of evidence, charge storage trends and electrolyte properties, the following conclusions can be drawn. Considering the charge vs. concentration curves at medium to high scan rates (1-500 mV s-1), it is clear that the highest charge storage in the 15 m LiBr electrolyte has place at intermediate conductivity and viscosity values of 129.7 mS cm-1 and 5.42 mPa s [Figure 7B, region 1]. On the other hand, according to the condition wt. LiBr/wt. H
At the lowest testing scan rate of 0.5 mV s-1, transport processes controlled by diffusion become important. The 17.5 m
In conclusion, these measured electrolyte properties provided guidance to explain charge storage performance trends as a function of electrolyte concentration. Importantly, these experiments showed that the highest charge storage performance was achieved in the 17.5 m (0.5-1 mV s-1) and 15 m (5-500 mV s-1) electrolytes. Therefore, from the capacity point of view, and considering operation at room temperature, the use of extremely high concentrated electrolytes of 19 m (solubility limit at room temperature) is unnecessary, precluding associated problems of electrolyte crystallization (imminent upon slight variation of temperature or concentration). Further details are provided in the Experimental section. In addition, the results from cyclability tests [Figure 4C], reporting the best performance for the electrode tested in the 17.5 m electrolyte, and despite the high viscosity (but lower than for the 19 m electrolyte), further support that this electrolyte best performs. Performance over scan rates could be further improved with suitable electrode activation processes and further engineering of the electrode and/or utilization at applications at temperatures higher than room temperature.
SPEIS studies
Next, details of ion transport processes were investigated by SPEIS. SPEIS experiments were performed in half-cell setups for electrodes tested in 5 m, 15 m, 17.5 m and 19 m LiBr electrolytes. PEIS was performed every 25 mV during the first cycle of a CV following an OCP to V
The Nyquist plots of the electrodes tested at OCP and in the 17.5 m and 19 m LiBr electrolytes showed a semicircle at high frequencies, whereas the others tested in 5 m and 15 m LiBr electrolytes did not [Figure 8A]. Such semicircle persisted (and varied) across polarization potentials over the entire EW and all the cathodic and anodic scans (e.g., for tests in the 17.5 m electrolyte shown in Figure 8B, C and Supplementary Figure S17B, D, F). Since the semicircle is present at OCP, its origin cannot be ascribed to a charge transfer resistance but rather an interfacial resistance experienced by the Li
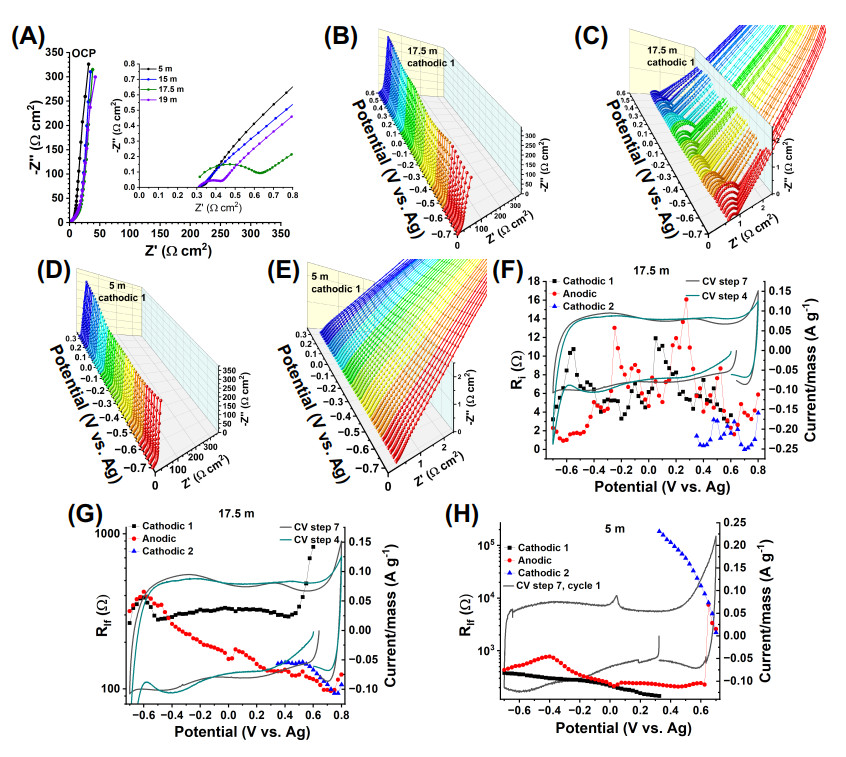
Figure 8. (A) Nyquist plots of electrodes polarized at OCP for electrodes tested in 5 m, 15 m, 17.5 m and 19 m LiBr electrolytes, (B), (C) 3D Nyquist plots of electrodes tested in the 17.5 m LiBr electrolyte (cathodic scan 1), (D), (E) 3D Nyquist plots of electrodes tested in the 5 m LiBr electrolyte (cathodic scan 1), (F) R
We focus first on the analysis of the EIS data of electrodes tested in the 17.5 m electrolyte. An EIS model 1 was proposed to describe the data [Figure 9].
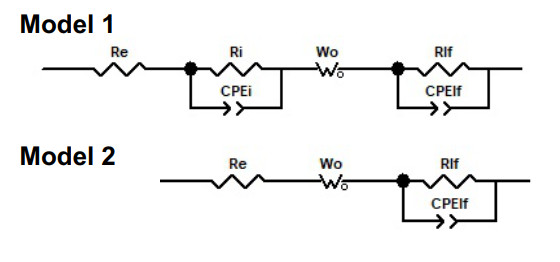
Figure 9. Equivalent circuits of model 1 and model 2.
The circuit R
At medium and low frequencies, the 17.5 m EIS data set showed two main contrasting EIS spectra. In the first case, the Nyquist data showed a semi-infinite linear diffusion behavior at medium frequencies (Z'
The EIS data of the electrode tested in the 17.5 m LiBr electrolyte was adequately modeled using model 1 for the cathodic 1, anodic and cathodic 2 scans [Supplementary Figure S19]. Next, we analyze the evolution of physical parameters with polarization potential.
During the cathodic scan 1, R
During the cathodic scan 1, R
The EIS data of the electrode tested in the 5 m LiBr electrolyte had no semicircle at high frequencies in the cathodic scan 1, and most of the anodic scan from -0.7 to 0.625 V [Figures 8D and E, Supplementary Figure S18A-D]. In this case, a Model 2 was utilized [Figure 9]. The fit data is reported in Supplementary Figure S20. The absence of such a semicircle indicated the absence of an interfacial film. During the cathodic scan 1 from OCP to -0.7 V, R
It is clear, thus, that a key difference between tests in the 5 m and the 17.5 m electrolytes is the high current and irreversible process at the positive EW potential limit, confirmed by CV (Section "Cyclic voltammetry tests"), analysis of CVs of cycling tests (Section "Analysis of CVs of cycling tests"), GCPL (Section "Electrode rate performance") and EIS studies. The electrode tested in the 17.5 m electrolyte was more stable at the positive EW potential limit allowing the EW to be enlarged up to 0.8 V.
Next, diffusion coefficients were calculated for tests in the 5 m, 15 m, 17.5 m and 19 m LiBr electrolytes at 0 V polarization potential [Supplementary Figure S21D]. This polarization potential was considered an intermediate value within the EW where the secondary processes, other than the intrinsic storage processes of the electrode, observed at the limits of the EW could be avoided. The anomalous diffusion coefficients were calculated according to the parameters derived from the fit using model 2 (5 m and 15 m LiBr EIS data) and model 1 (17.5 m and 19 m LiBr EIS data) and Supplementary Equation S4. Supplementary Figure S21A-C shows the experimental and calculated EIS data. The
The diffusion coefficients were calculated at all polarization potentials for the tests in the 5 m and 17.5 m electrolytes. There were variations with potential, which is expected from the basis of diffusion theory upon which, the open Warburg element is derived [Supplementary Equation S1)[42]. The Faradaic current due to an overpotential equals the diffusion flux (Fick's first law) and steady-state conditions are applied[42]. However, the calculation of T
A final point of interest is an insight of the influence of electrolyte properties such as viscosity on electrode rate performance that can be given by EIS studies (complementing studies in Sections "Electrode rate performance", "Electrolyte properties"). A measure of the response rate of a capacitor is the so-called "dielectric relaxation time constant' (
In order to study the effect of viscosity on capacitor rate response, we considered it tests in two electrolytes having viscosity values in the low and high limit: 5 m and 17.5 m LiBr with a viscosity of 1.8 and 6.92 mPa
In conclusion, the EIS data showed that in the HCE of 17.5 m, an interfacial film formation takes place, which may involve faradaic processes that upon cycling may derive into an SEI. This is in agreement with the (c
CONCLUSIONS
The electrochemical processes undergone by V
The use of the 17.5 m electrolyte suppressed HER activity and an irreversible process at the positive limit of the EW, which might consist of OER or Br-1 oxidation reactions. Cycling tests confirmed the irreversibility of these processes, enhanced in the low-concentration electrolytes (5 m to 7.5 m) and EIS confirmed a correlated high resistance at low frequencies. Further studies would be necessary to confirm the precise nature of this process. CV and EIS confirmed evidence of an interfacial film at the electrode-electrolyte interface enabled by 17.5 m and 19 m electrolytes that may lead to the formation of a stabilizing SEI.
Studies of the electrolyte properties showed that an optimized charge storage performance requires a careful balance of ionic conductivity and viscosity, which was achieved for the electrolytes at a concentration (17.5 m) well below the solubility limit of LiBr (19.0 m). The higher the concentration, the greater the viscosity and lower the conductivity, which brings disadvantages to rate performance and cycling stability. Key to the improvement of rate performance is the electrode architecture to facilitate electrolyte access, evidenced here for activated electrodes. Further engineering of the electrode is necessary to improve further the rate performance.
DECLARATIONS
Acknowledgments
Yury Gogotsi (Drexel University) is acknowledged for his kind gesture of providing MAX materials. Dr. Alessandro Mariani (Elettra Sincrotrone Trieste) is acknowledged for the preliminary MD simulations. Dr. Christopher Klein is acknowledged for access to rheology instrumentation and for his kind support with measurements. Prof. Juan Bisquert (UPV-CSIC) and Assoc. Prof. Mathijs Janssen (NMBU) are acknowledged for useful EIS discussions.
Authors' contributions
Beatriz Mendoza-Sánchez (Corresponding and Senior author): Conceptualization, Methodology, Software, Validation, Formal analysis, Investigation, Resources, Data Curation, Writing - Original Draft, Writing - Review
Availability of data and materials
The data that support the findings of this work are available upon request to the corresponding author.
Financial support and sponsorship
This work was supported by the Helmholtz Association with a Helmholtz ERC Recognition Award (ERC-RA-0028) granted to Beatriz Mendoza-Sánchez. Camille Douard and Thierry Brousse acknowledge Labex STORE-EX (ANR-10-LABX-7601) for financial support. This work contributes to the research performed at the Centre for Electrochemical Energy Storage Ulm-Karlsruhe (CELEST).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Authors 2025.
REFERENCES
1. Mendoza-Sánchez, B.; Gogotsi, Y. Synthesis of two-dimensional materials for capacitive energy storage. Adv. Mater. 2016, 28, 6104-35.
2. Anasori, B.; Lukatskaya, M. R.; Gogotsi, Y. 2D metal carbides and nitrides (MXenes) for energy storage. Nat. Rev. Mater. 2017, 2, 16098.
3. VahidMohammadi, A.; Rosen, J.; Gogotsi, Y. The world of two-dimensional carbides and nitrides (MXenes). Science 2021, 372, eabf1581.
4. Lukatskaya, M. R.; Mashtalir, O.; Ren, C. E.; et al. Cation intercalation and high volumetric capacitance of two-dimensional titanium carbide. Science 2013, 341, 1502-5.
5. Ghidiu, M.; Lukatskaya, M. R.; Zhao, M. Q.; Gogotsi, Y.; Barsoum, M. W. Conductive two-dimensional titanium carbide 'clay' with high volumetric capacitance. Nature 2014, 516, 78-81.
6. Yoon, Y.; Lee, M.; Kim, S. K.; et al. A strategy for synthesis of carbon nitride induced chemically doped 2D MXene for high-performance supercapacitor electrodes. Adv. Energy. Mater. 2018, 8, 1703173.
7. Shan, Q.; Mu, X.; Alhabeb, M.; et al. Two-dimensional vanadium carbide (V
8. VahidMohammadi, A.; Mojtabavi, M.; Caffrey, N. M.; Wanunu, M.; Beidaghi, M. Assembling 2D MXenes into highly stable pseudocapacitive electrodes with high power and energy densities. Adv. Mater. 2019, 31, 1806931.
9. Halim, J.; Kota, S.; Lukatskaya, M. R.; et al. Synthesis and characterization of 2D molybdenum carbide (MXene). Adv. Funct. Mater. 2016, 26, 3118-27.
10. Anasori, B.; Xie, Y.; Beidaghi, M.; et al. Two-dimensional, ordered, double transition metals carbides (MXenes). ACS. Nano. 2015, 9, 9507-16.
11. Pinto, D.; Anasori, B.; Avireddy, H.; et al. Synthesis and electrochemical properties of 2D molybdenum vanadium carbides - solid solution MXenes. J. Mater. Chem. A. 2020, 8, 8957-68.
12. Zhao, S.; Chen, C.; Zhao, X.; et al. Flexible Nb
13. Mendoza-Sánchez, B.; Ladole, A. H.; Samperio-Niembro, E.; et al. On the atomic structure of monolayer V
14. Avireddy, H.; Byles, B. W.; Pinto, D.; et al. Stable high-voltage aqueous pseudocapacitive energy storage device with slow self-discharge. Nano. Energy. 2019, 64, 103961.
15. Suo, L.; Borodin, O.; Gao, T.; et al. "Water-in-salt" electrolyte enables high-voltage aqueous lithium-ion chemistries. Science 2015, 350, 938-43.
16. Han, J.; Mariani, A.; Passerini, S.; Varzi, A. A perspective on the role of anions in highly concentrated aqueous electrolyte. Energy. Environ. Sci. 2023, 16, 1480-501.
17. Yamada, Y.; Usui, K.; Sodeyama, K.; Ko, S.; Tateyama, Y.; Yamada, A. Hydrate-melt electrolytes for high-energy-density aqueous batteries. Nat. Energy. 2016, 1, 16129.
18. Zhang, H.; Liu, X.; Li, H.; Hasa, I.; Passerini, S. Challenges and strategies for high-energy aqueous electrolyte rechargeable batteries. Angew. Chem. Int. Ed. 2021, 60, 598-616.
19. Yoshida, K.; Nakamura, M.; Kazue, Y.; et al. Oxidative-stability enhancement and charge transport mechanism in Glyme lithium salt equimolar complexes. J. Am. Chem. Soc. 2011, 133, 13121-9.
20. Yokoyama, Y.; Fukutsuka, T.; Miyazaki, K.; Abe, T. Origin of the electrochemical stability of aqueous concentrated electrolyte solutions. J. Electrochem. Soc. 2018, 165, A3299.
21. Tian, Z.; Deng, W.; Wang, X.; et al. Superconcentrated aqueous electrolyte to enhance energy density for advanced supercapacitors. Funct. Mater. Lett. 2017, 10, 1750081.
22. McOwen, D. W.; Seo, D. M.; Borodin, O.; Vatamanu, J.; Boyle, P. D.; Henderson, W. A. Concentrated electrolytes: decrypting electrolyte properties and reassessing Al corrosion mechanisms. Energy. Environ. Sci. 2014, 7, 416-26.
23. Han, J.; Mariani, A.; Zarrabeitia, M.; et al. Zinc-ion hybrid supercapacitors employing acetate-based water-in-salt electrolytes. Small 2022, 18, 2201563.
24. Wang, X.; Mathis, T. S.; Sun, Y.; T; et al. Titanium carbide MXene shows an electrochemical anomaly in water-in-salt electrolytes. ACS. Nano. 2021, 15, 15274-84.
25. Wang, X.; Bak, S. M.; Han, M.; et al. Surface redox pseudocapacitance of partially oxidized titanium carbide MXene in water-in-salt electrolyte. ACS. Energy. Lett. 2022, 7, 30-5.
26. Kim, K.; Ando, Y.; Sugahara, A.; et al. Dense charge accumulation in MXene with a hydrate-melt electrolyte. Chem. Mater. 2019, 31, 5190-6.
27. Kim, K.; Okubo, M.; Yamada, A. Interfacial dissociation of contact-ion-pair on MXene electrodes in concentrated aqueous electrolytes. J. Electrochem. Soc. 2019, 166, A3739.
28. Duvall, K. N.; Dirksen, J. A.; Ring, T. A. Ostwald-meyers metastable region in LiBr Crystallization - comparison of measurements with predictions. J. Colloid. Interface. Sci. 2001, 239, 391-8.
29. Boryta, D. A. Solubility of lithium bromide in water between -50.deg. and +100.deg. (45 to 70% lithium bromide). J. Chem. Eng. Data. 1970, 15, 142-4.
30. Taberna, P. L.; Simon, P.; Fauvarque, J. F. Electrochemical characteristics and impedance spectroscopy studies of carbon-carbon supercapacitors. J. Electrochem. Soc. 2003, 150, A292-300.
31. Mendoza-Sanchez, B.; Samperio-Niembro, E.; Dolotko, O.; et al. Systematic study of the multiple variables involved in V
32. CV Classifier; 2024. Available from: http://supercapacitor-battery-artificialintelligence.vistec.ac.th/CV[Last accessed on 22 Mar 2025].
33. Deebansok, S.; Deng, J.; Le Calvez, E.; et al. Capacitive tendency concept alongside supervised machine-learning toward classifying electrochemical behavior of battery and pseudocapacitor materials. Nat. Commun. 2024, 15, 1133.
34. Yim, C. H.; Tam, J.; Soboleski, H.; Abu-Lebdeh, Y. On the correlation between free volume, phase diagram and ionic conductivity of aqueous and non-aqueous lithium battery electrolyte solutions over a wide concentration range. J. Electrochem. Soc. 2017, 164, A1002.
35. Yoshizawa, M.; Xu, W.; Angell, C. A. Ionic liquids by proton transfer: vapor pressure, conductivity, and the relevance of
36. Videa, M.; Angell, C. A. Glass formation, ionic conductivity, and conductivity/viscosity decoupling, in LiAlCl
37. Bisquert, J.; Garcia-Belmonte, G.; Bueno, P.; Longo, E.; Bulhoes, L. O. S. Impedance of constant phase element (CPE)-blocked diffusion in film electrodes. J. Electroanal. Chem. 1998, 452, 229-34.
38. Brug, G. J.; van den Eeden, A. L. G.; Sluyters-Rehbach, M.; Sluyters, J. H. The analysis of electrode impedances complicated by the presence of a constant phase element. . J. Electroanal. Chem. Inter. Electrochem. 1984, 176, 275-95.
39. Cruz-Manzo, S.; Greenwood, P. Frequency transition from diffusion to capacitive response in the blocked-diffusion warburg impedance for EIS analysis in modern batteries. J. Electrochem. Soc. 2020, 167, 140507.
40. Ho, C.; Raistrick, I. D.; Huggins, R. A. Application of A-C techniques to the study of lithium diffusion in tungsten trioxide thin films. J. Electrochem. Soc. 1980, 127, 343-50.
41. Cruz-Manzo, S.; Greenwood, P. Blocked-diffusion with frequency dispersion for study of EIS in NiMH batteries. J. Electrochem. Soc. 2019, 166, A1176.
42. Cruz-Manzo, S.; Greenwood, P.; Chen, R. An impedance model for EIS analysis of nickel metal hydride batteries. J. Electrochem. Soc. 2017, 164, A1446.
43. Peng, W.; Aranda, C.; Bakr, O. M.; Garcia-Belmonte, G.; Bisquert, J.; Guerrero, A. Quantification of ionic diffusion in lead halide perovskite single crystals. ACS. Energy. Lett. 2018, 3, 1477-81.
44. Cruz-Manzo, S.; Chen, R.; Greenwood, P. Analysis of the performance of an open-cathode polymer electrolyte fuel cell stack using simultaneous electrochemical impedance spectroscopy measurements. ECS. Trans. 2014, 48, 47.
Cite This Article
 , ... Christopher E. Shuck
, ... Christopher E. ShuckHow to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].