

Design of efficient dual-atom catalysts for energy conversion
 0
0  ,
, Abstract
Dual-atom catalysts (DACs) have triggered the burgeoning interest in the field of catalysis. The identification of coordination structures and the understanding of the catalytic role of dual-atom centers are of great significance to designing highly efficient DACs. This review summarizes the current synthesis methods for the construction of DACs. Then, we highlight the differences in geometric and electronic structures of DACs in terms of modulation strategies and engineering dual-metal interaction. In particular, the combination of various advanced characterizations and density functional theory (DFT) insights disclose the state of the dual-atom active center microenvironment. Moreover, the catalytic role of DACs in heterogeneous catalysis is discussed, involving the electronic effect of one metal as the main active center and the synergistic effect between dual metals. The structure-activity relationship and reaction mechanism based on experimental studies and theoretical exploration are discussed. Finally, the challenges and opportunities of DACs for the efficient valorization of energy-related small molecules are proposed. This review can offer new inspiration on developing DACs for energy conversion.
Keywords
INTRODUCTION
With the growing energy crisis and global population caused by the excessive consumption of fossil fuels, significant efforts have been devoted to developing clean energy conversion technologies[1-3]. Most of them are related to the catalytic processes, particularly electrocatalysis showing a promising platform to reduce fossil fuel dependence[4-7]. Designing effective catalysts with remarkable reactivity is important for catalytic reactions and industrial applications. Taking advantage of robust and easy separation, heterogeneous catalysts especially supported metal catalysts are widely investigated[8-12]. To improve the performance, a simple method is to decrease the particle size of metals to the nanoscale. In this regard, the finding that Au nanoparticles are active for carbon monoxide oxidation at cryogenic temperatures has ignited excitements in nanocatalysis[13]. The ultimate goal is to achieve single-atom dispersion of metal species.
Single-atom catalysts (SACs) have been a new frontier in catalysis during the past decade, which have been widely investigated owing to their high atom utilization and distinct structure[14-23]. In 2011, Qiao et al. proposed the concept of “single-atom catalysis” for the first time[24]. The intrinsic activity of SACs can be enhanced by further tailoring the electronic state and coordination structure[25-29]. Despite achieving significant progress, the challenges still lie in the development of SACs. To keep the single-atom dispersion, the metal loading of SACs is usually low (< 1.5 wt%) to avoid aggregation; thus, the total activity is not satisfactory[30-35]. In addition, since the active center of SACs is single, it is difficult to break the limitation by a linear scaling relationship. Moreover, SACs may not be desired for reactions that need different metal sites to activate reactants or multiple-step catalytic processes.
Dual-atom catalysts (DACs), as an extension of SACs, have attracted much attention in recent years[36-40]. The timeline of the representative breakthroughs in DACs is presented in Figure 1[24,41-47]. The addition of the other metal atoms into SACs can improve the metal loadings, tune the electronic structures, and break the linear relationship. Generally speaking, the d-band center will be adjusted owing to the coordination of the adjacent two atoms. Furthermore, DACs provide more active sites to adsorb and activate the reactants, giving priority to realizing multiple reaction pathways[48-54]. Benefiting from the synergistic effect, DACs are promising candidates for catalyzing tandem reactions via rational design. At present, several reviews on preparation, characterization, and electrochemical application have been reported[55-60]. However, these papers mainly focus on homonuclear and heteronuclear DACs and relevant applications in electrocatalysis. So far, identifying the active structure and understanding the catalytic role of DACs in heterogeneous catalysis are still lacking.
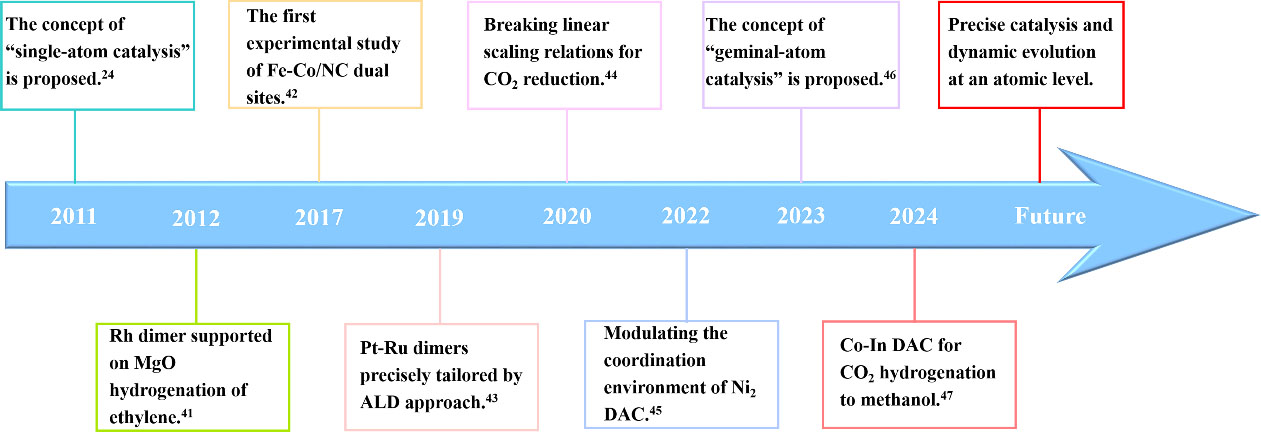
Figure 1. Timeline of breakthroughs during the last decade of DACs. DACs: Dual-atom catalysts; ALD: atomic layer deposition.
In this review, we first present an overview of synthetic approaches for DACs, involving atomic layer deposition (ALD), wet chemical approach, high-temperature pyrolysis, and atom trapping. We introduce and discuss the different principles to guide the dual-atom design on diverse substrates. Next, we highlight the identification of active center structures of DACs, with a focus on elucidating local environments and dual-atom interactions. Moreover, we illustrate the catalytic role of DACs in boosting performance and disclose the reaction mechanisms, involving electronic effect and synergistic effect. Finally, the current challenges and future opportunities of DACs are put forward to point out directions for the development of DACs.
SYNTHESIS STRATEGIES OF DACs
The fabrication of DACs is more rigorous than that of SACs due to diverse requirements. Developing facile preparation methods becomes an important issue to be solved. First, the interaction between metal precursors and substrates is critical to stabilize the metal dimers. Another feasible approach is via spatial confinement utilizing porous materials, such as metal - organic frameworks (MOFs) and covalent organic frameworks (COFs). In this part, we summarize the preparation methods of DACs, including ALD, wet chemical approach, high-temperature pyrolysis, and atom trapping. It is worth mentioning that the synthetic approaches reported up to now are still in their infancy, with expensive reagents and high-temperature conditions. The effective and controllable synthesis needs to be explored in the future.
Atomic-layer deposition
The ALD method is a powerful tool for the precise control of catalysts from clusters to single atoms. The ALD process involves two sequential self-limiting surface reactions separated by inert gas sweeping[61,62]. This unique character makes the ALD technique possible to construct single or few atoms on high-surface-area support controllably and precisely. Besides, dimers prepared by ALD may be different from the reported dual-metal sites. As a typical example, Pt-Ru dimers on nitrogen (N)-doped carbon nanotubes (NCNT) were synthesized by the ALD process[43]. As shown in Figure 2A, the single Pt atoms were obtained by using MeCpPtMe3 as the precursor and nitrogen as the purge gas. The Pt atoms tend to bond with N sites on NCNT by forming strong interactions. Then, Ru atoms were selectively dispersed on Pt atoms to form Pt-Ru dimers. The atomic resolution electron microscopy images showed dimers dispersed on the substrate [Figure 2B]. The two atoms exhibited distinct contrasts, indicating that the dimer structure contained two different elements, Pt and Ru [Figure 2C]. The detailed characterization and theoretical calculation indicated that the electronic state of Ru was strongly affected by the Pt atom, resulting in good hydrogen evolution activity. In the synthesis of Pt2 dimers, nucleation sites were firstly constructed on graphene nanosheets via acid oxidation followed by thermal reaction[63]. The Pt1 single atoms were first generated by alternately exposing MeCpPtMe3 and O2. Then, these atoms were further used as nucleation sites for binding the second MeCpPtMe3 to form Pt2 dimers. Furthermore, to regulate the interspace of Pt1 and Ni1 atoms, Chen et al.[64] fabricated Pt1Ni1 oxo dimers and Pt1+Ni1/C3N4 DAC by ALD [Figure 2D]. The aberration-corrected high-angle annular dark-field scanning transmission electron microscopy
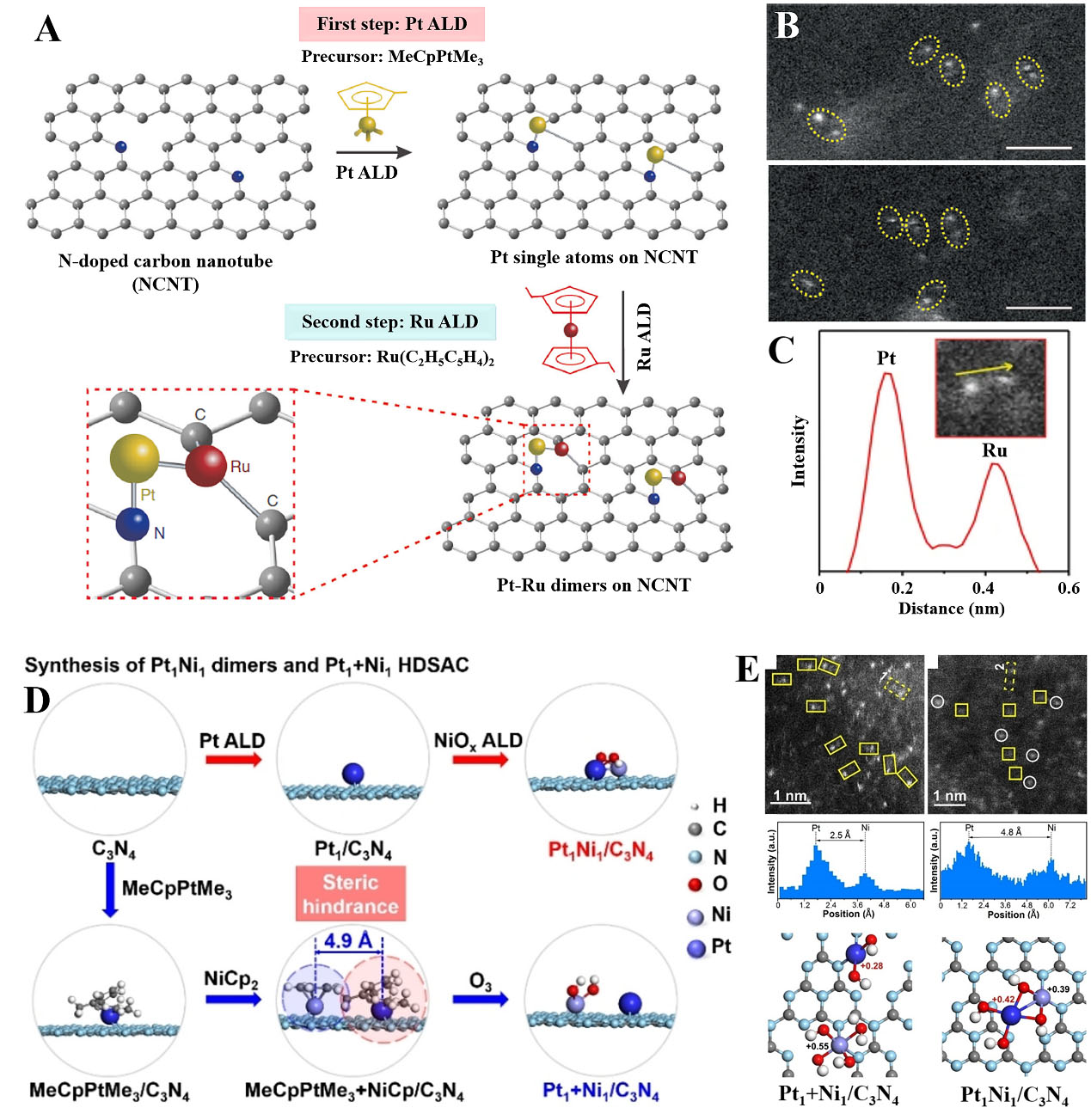
Figure 2. (A-C) The preparation of DACs by ALD and AC HAADF-STEM images of Pt-Ru pairs. Reproduced with permission from Ref.[43]. Copyright 2019, Nature Publishing Group; (D) The schematic synthesis of Pt1Ni1/C3N4 and Pt1+Ni1/C3N4 dimers; (E) AC HAADF-STEM images and structural models; (D-E) Reproduced with permission from Ref.[64]. Copyright 2022, Wiley-VCH. DACs: Dual-atom catalysts; ALD: atomic layer deposition; AC HAADF-STEM: aberration-corrected high-angle annular dark-field scanning transmission electron microscopy.
Wet chemical method
The wet chemical method is the most intensively used approach for synthesizing supported metal catalysts. It has been illustrated as an effective strategy to construct well-dispersed atomic pairs on substrates with suitable anchoring sites. Li et al. prepared an Ag2/graphene catalyst with an AgN3-AgN3 site via the wet chemical method[65]. Typically, the binuclear Ag complex containing aromatic molecules was prepared and used as the metal precursor. Then, the precursor was adsorbed on the graphene surface via π-π interaction, and the ligands were removed by following the pyrolysis process. Besides, Cu2 dimers anchored on carbon nitride were fabricated for efficient partial oxidation of methane[66]. Similarly, the Cu dimer precursor was first synthesized by complexation reaction, followed by self-assembly of Cu dimer complex on g-C3N4. Subsequently, the mixture was treated at 250 °C in air to anchor Cu species via Cu-N bonding. As shown in Figure 3A, the Ni2 site comprising two N atoms bridged Ni-N4 structures was prepared toward electrocatalytic CO2 reduction[67]. The ligand-protected dinuclear Ni2(dppm)2Cl3 precursor was first prepared by a controllable sluggish reaction of NiCl2 and dppm complex. The precursor was then impregnated with zeolite imidazolate framework 8 (ZIF-8)-derived carbon. Finally, the mixture was pyrolyzed at 400 °C in Ar to remove the organic ligands, resulting in the formation of a diatomic Ni2/N-doped carbon (NC) catalyst. The corresponding AC HAADF-STEM image demonstrated Ni2 species uniformly dispersed on the NC substrate [Figure 3A]. Interestingly, it was found that the coordinated unsaturated Ni could adsorb oxygen species in the electrolyte, producing an O-Ni2-N6 configuration, which decreased the energy barrier for the activation of CO2 and enhanced CO2 reduction to CO performance. Using a dinuclear Ni complex as the precursor, dual-atom Ni2 supported on NCNT was developed[68]. It was indicated that the directly bonded NiN3-NiN3 structure decreased the reaction energy for *COOH generation and promoted CO2 electroreduction. By changing the type of metal precursors, Tian et al. prepared diatomic Fe2 dispersed mesoporous carbon nitride, which exhibited superior activity toward alkene epoxidation[69]. Furthermore, they anchored the dinuclear Pd2 precursor on acetylene black as displayed in Figure 3B, and KPF6 was added to ensure the Pd deposited on the surface of the carbon substrate[70]. Yu et al. synthesized dinuclear Ir2 sites bridged by oxygen on NC, which exhibited higher activity than Ir SACs for electrochemical chlorine evolution reaction[71]. In particular, Guan and Yardimci
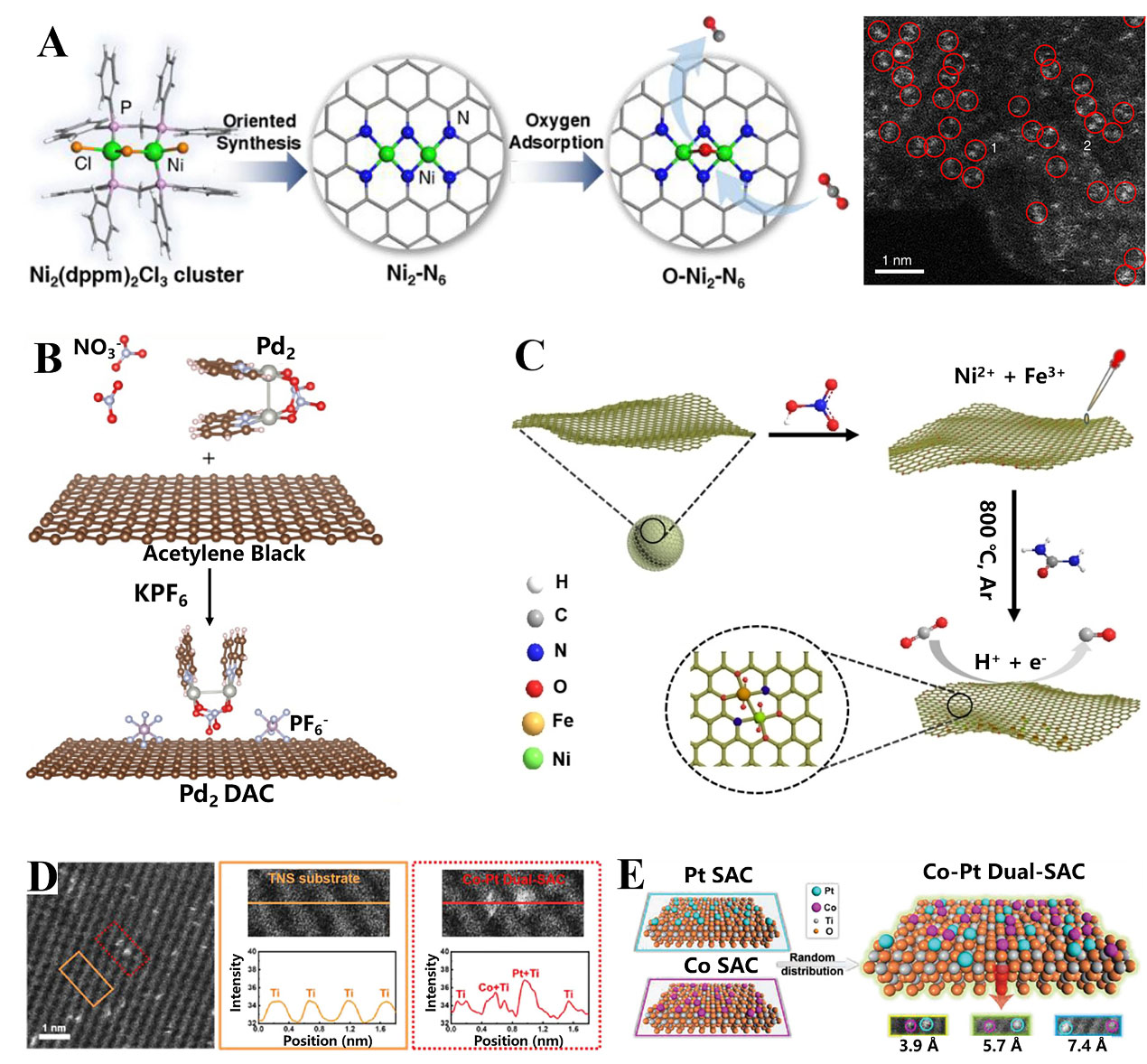
Figure 3. (A) The preparation strategy for dinuclear Ni2/NC and atomically dispersed Ni2 species highlighted by red circles. Reproduced with permission from Ref.[67]. Copyright 2021, American Chemical Society; (B) The synthesis strategy for Pd2 DAC. Reproduced with permission from Ref.[70]. Copyright 2021, Wiley-VCH; (C) Schematic of synthesis of dual-atom Ni/Fe on carbon blacks. Reproduced with permission from Ref.[73]. Copyright 2021, Wiley-VCH; (D) Atomic scale identified the brightness of Pt and Co dual atoms in the selected area; (E) Schematically illustration of random atomic sites; (D-E) Reproduced with permission from Ref.[75]. Copyright 2021, Wiley-VCH. DACs: Dual-atom catalysts; SACs: single-atom catalysts.
Apart from homonuclear dual atoms, the precursor-preselected wet-chemistry strategy is also effective for the construction of heteronuclear dual atoms. As presented in Figure 3C, the Ni/Fe-NC dimers were prepared by a two-step procedure[73]. In brief, carbon black was activated by nitric acid to trap and adsorb
The above advances suggest several critical factors for the controlled preparation of dual atoms by the wet chemical method: (1) selecting appropriate binuclear metal complexes and moderate interaction with substrates. Particularly, a strong interaction between binuclear metal and the substrate is significant for obtaining high dispersion and stable DACs; (2) constructing porous substrate to take full advantage of the pore confinement effect to stabilize diatomic metals; (3) taking advantage of functional groups or defects on the surface of substrates to stabilize metal complexes; (4) using the π–π electrostatic interplay to anchor binuclear metal complexes. The corresponding approaches have been verified to be effective in obtaining highly dispersed DACs.
High-temperature pyrolysis
The high-temperature pyrolysis is a commonly used method for fabricating SACs and DACs, especially in NC-supported metal catalyst systems. MOFs are extensively considered as the precursors for pyrolysis. On the one hand, MOFs can be facilely tuned by regulation of metal nodes or organic linkers with some functions[76]. On the other hand, they possess abundant pores that confine different metal species, making them a promising candidate to stabilize single and few atoms[77,78]. In recent work, Lu et al. reported a competitive complexation strategy to synthesize isolated Zn-Co atomic pairs[79]. As presented in Figure 4A, chitosan was chosen as C and N sources, and zinc chloride and cobalt acetate were used as metal precursors. The 4s and 4p electrons of Co2+ and Zn2+ could be bound with the -NH2 and -OH on chitosan chains, thus obtaining the uniform dispersion of Co and Zn atoms after pyrolysis at 750 °C. The characterization and DFT calculations revealed ZnCoN6 as an active site; such architecture enhanced the activation ability of O2, thus leading to outstanding oxygen reduction performance. In another work, the monomer melamine,
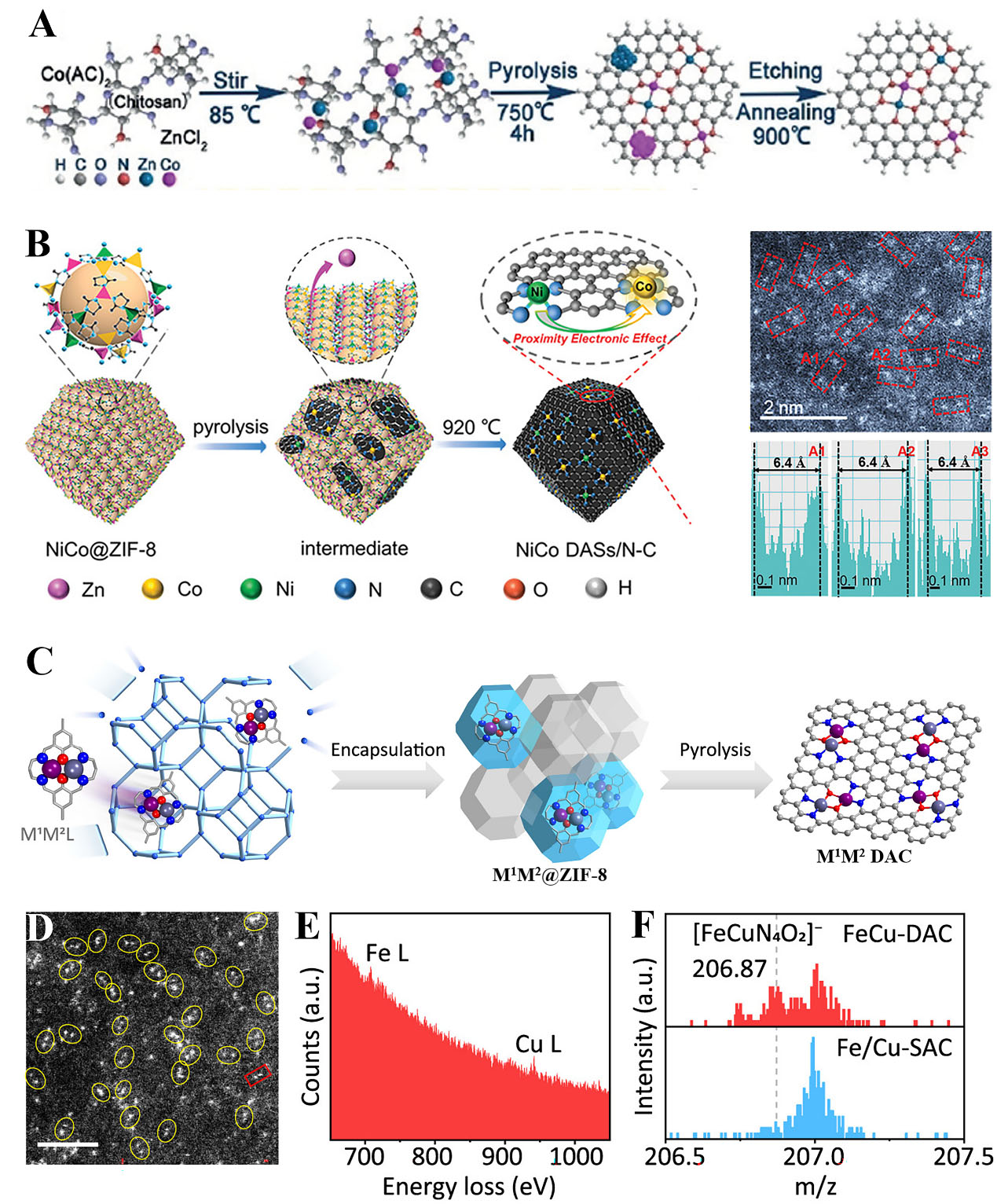
Figure 4. (A) The preparation process for Zn/CoN-C. Reproduced with permission from Ref.[79]. Copyright 2019, Wiley-VCH; (B) Scheme of the synthesis process of NiCo DASs/N-C. Reproduced with permission from Ref.[81]. Copyright 2022, Wiley-VCH; (C) Macrocyclic precursor-mediated encapsulation-pyrolysis to prepare DACs; (D) AC HAADF-STEM image of FeCu-DAC and the scale bar is 2 nm; (E) EELS of a FeCu dual atoms as labeled in the red rectangle in Figure 4D; (F) TOF-SIMS spectra of FeCu-DAC and Fe/Cu-SAC; (C-F) Reproduced with permission from Ref.[87]. Copyright 2023, American Chemical Society. DACs: Dual-atom catalysts; AC HAADF-STEM: aberration-corrected high-angle annular dark-field scanning transmission electron microscopy; TOF-SIMS: time-of-flight secondary ion mass spectrometry.
In addition, the preparation of DACs via pyrolysis of MOF precursors is an epidemic method. Wang et al. designed Fe-Co atomic pairs based on control over binding between Co nodes and adsorbed Fe within the confined space of MOFs[42]. The Fe-Co/NC comprising Fe-Co direct bonding structure was formed after heating at 900 °C. Benefiting from the MOF-assisted host-gust strategy, isolated Ni/Co dual atoms anchored on the NC substrate were fabricated [Figure 4B][81]. First, Ni and Fe atoms were encapsulated in the ZIF-8 owing to the atom size matching. Then it was pyrolyzed under N2 at 920 °C to evaporate Zn atoms, resulting in the Ni-N4 and Co-N4 moieties uniformly dispersed on carbon substrate. The bright spots in AC HAADF-STEM indicated the presence of NiCo dual atoms, and the distance of atomic pairs was about 6.4 Å [Figure 4B]. It was demonstrated that Co-N4 was the dominant active center, and the adjacent Ni-N4 was a modulator to tune the electronic localization of Co-N4 for synergistically enhancing oxygen reduction reaction (ORR) and hydrogen evolution reaction (HER) processes. Under the two-solvent route, the
Apart from heteronuclear DACs, homonuclear DACs can also be obtained based on an encapsulation-pyrolysis strategy. For instance, Zhang et al. developed a general approach for developing a DAC library in a wide range of homonuclear (Fe2, Co2, Ni2, Cu2, Mn2, and Pd2) and heteronuclear (Fe-Cu, Fe-Ni, Cu-Mn, and Cu-Co) bimetal centers[87]. As displayed in Figure 4C, a binuclear complex M1M2L was used as a precursor, which was confined by the pores of ZIF-8. The pyrolysis was at 900 °C to carbonize the M1M2L@ZIF-8 and yield DACs. The AC HAADF-STEM image revealed the atomically dispersed metal sites [Figure 4D], which were further confirmed to be Fe and Cu atoms by electron energy-loss spectroscopy (EELS) in Figure 4E. In other evidence, they performed time-of-flight secondary ion mass spectrometry (TOF-SIMS) to analyze the mass fragments. The diatomic fragment of [FeCuN4O2]- held a higher intensity in FeCu-DAC compared to Fe/Cu-SAC [Figure 4F], suggesting a similar structure between FeCu-DAC and [FeCuN4O2] complex. This work opens a new way of using polynuclear complexes to construct a large variety of DACs. Besides, Leng et al. constructed dual atoms (Fe, Cu, or Ir) derived from metal dimer functionalized MOFs[88]. First, the metal dimer was immobilized on the surface of CTAB-functionalized ZIF-8. Second, the sample was coated with polydopamine to inhibit thermal atomic migration. The Fe pairs were anchored on a cubic nanocage with the formation of well-defined N-coordinated Fe2 atoms, which exhibited superior activity for ORR. Moreover, the coordination environments of Ni2 DACs were modulated by the control of pyrolysis temperature. Three Ni2-NC catalysts including Ni2-N7, Ni2-N5C2, and Ni2-N3C4 were synthesized by pyrolysis at 700, 800, and 900 °C[45]. As a result, Ni2-N3C4 showed the highest electrochemical CO2 reduction activity, which originated from the electronic state change to the Ni center and the appropriate adsorption strength of key intermediates. However, a considerable amount of single atoms was yielded along with DACs. Therefore, great attention should be given to ensuring that pyrolysis proceeds in a more controlled way. Additionally, the pyrolysis will cause a part of active metals embedded in the bulk of the matrix materials. The construction of ultrathin substrates is a promising strategy to enable dual-metal site exposure and boost catalysis.
Atom trapping
The atom trapping approach, originally designed for the synthesis of thermally stable single-atom Pt on ceria, has recently been illustrated as effective for fabricating DACs as well. This method makes use of the abundant oxygen vacancies in reducible substrates, such as CeO2. By aging in the air at 800 °C, the Pt group metals emitted as metal oxides migrated and then were trapped by substrate with defect sites[89]. As presented in Figure 5A, Pt and Pd dual atoms were anchored on vacancy-rich CeO2, which exhibited outstanding three-way catalysis performance[90]. The AC HAADF-STEM image revealed Pt and Pd in pairs dispersed on the surface of CeO2 [Figure 5B]. Furthermore, the extended X-ray absorption fine structure (EXAFS) spectra showed peaks including Pt-O, Pd-Ce, and Pd-O-M coordination, indicating the existence of atomic pairs [Figure 5C]. The charge density showed that the abundant delocalized charges gathered along with the Pt-O-Pd bond efficiently lowered the reaction energy barrier [Figure 5D]. The atom trapping has also been verified to be versatile. Pd1 and Rh1 dual atoms were developed by calcining the PdPh@CeO2 precursor in air at 700 °C[91]. The synergistic interaction between adjacent Pd and Rh demonstrated a remarkable improvement in methane combustion. Besides, metal dimers could be generated dynamically under the reaction condition. Meier et al. investigated CO oxidation by a model Pt/Fe3O4 and found that (PtCO)2 dimers produced via the agglomeration of mobile Pt-carbonyl species[92]. Each Pt atom was bonded to two oxygen atoms on adjacent rows of the substrate structure [Figure 5E]. Noncontact atomic force microscopy (AFM) displayed the Pt2 species as faint dark protrusions [Figure 5F]. When the tip was closer, two bright protrusions appeared above each Pt2 dimer [Figure 5G]. This study manifests that metastable active species can be formed under a reaction environment and the introduction of just one atom can make a significant difference to SACs.
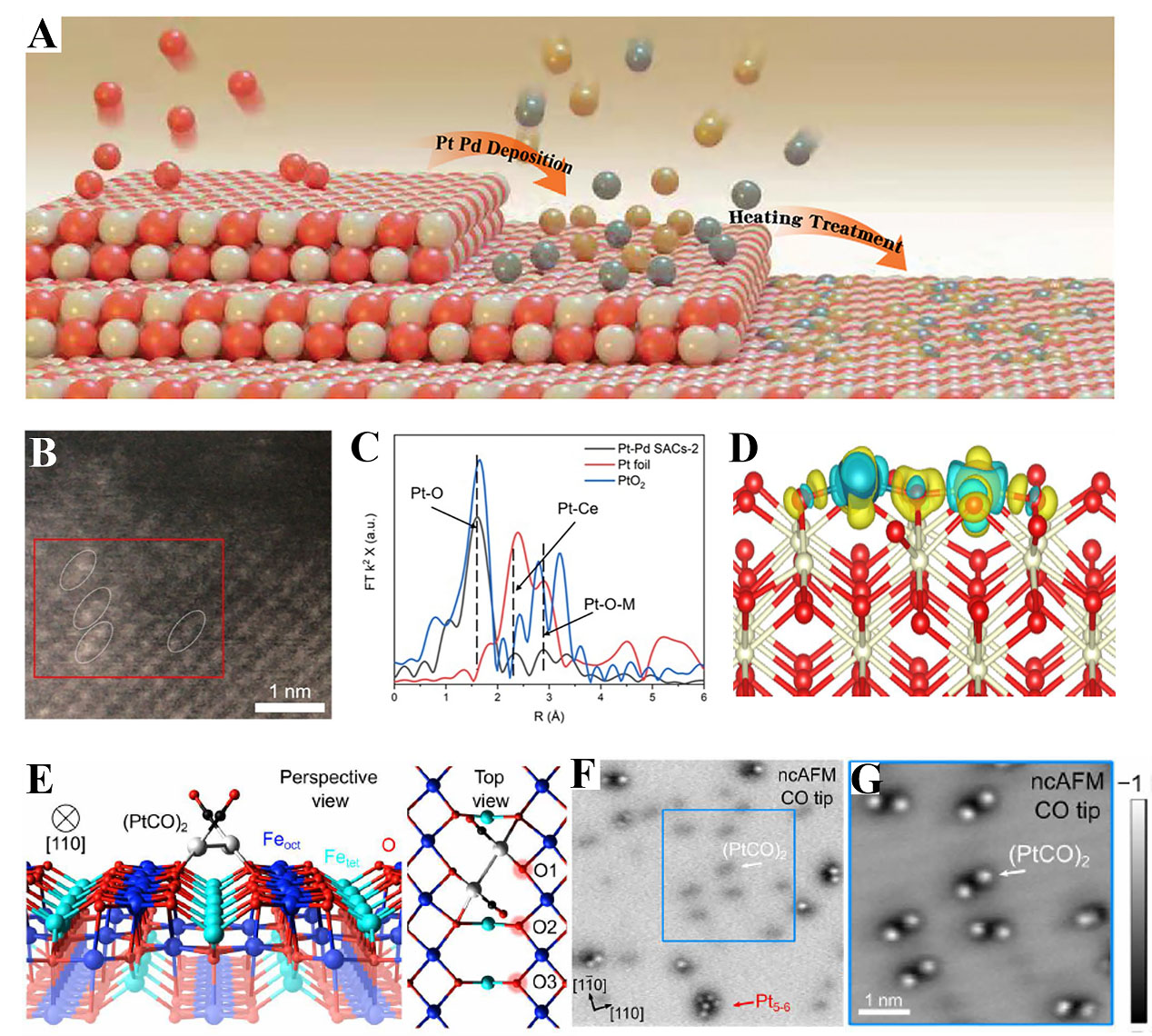
Figure 5. (A) The method for preparing Pt-Pd DAC; (B) HAADF-STEM image of Pt-Pd DAC; (C) EXAFS spectra in the R space of Pd K-edge; (D) Charge density of Pt-Pd DAC; (A-D) Reproduced with permission from Ref.[90]. Copyright 2022, Wiley-VCH; (E) Perspective and top view of the (PtCO)2 dimer on Fe3O4(001); (F) Constant-height ncAFM image taken with a CO-functionalized tip; (G) Constant-height ncAFM image of the (PtCO)2 dimers; (E-G) Reproduced with permission from Ref.[92]. Copyright 2022, American Association for the Advancement of Science. DACs: Dual-atom catalysts; HAADF-STEM: high-angle annular dark-field scanning transmission electron microscopy; EXAFS: X-ray absorption fine structure; AFM: atomic force microscopy.
In short, we summarize the preparation approaches for achieving dual-atom dispersion, including ALD, wet-chemistry method, high-temperature pyrolysis, and atom trapping. Each strategy has its superiority and drawbacks and may be suitable for particular cases. Among the various methods, the ALD technique shows a high level of precision in controlling the catalysts. Although there is an advantage in uniformity and atomic-scale regulation, the high cost limits its large-scale fabrication. Besides, the high-temperature atmosphere can lead to atom sintering, and deposition conditions usually depend on experience. For the precursor-preselected wet-chemistry method, the preparation of dinuclear precursors is complicated and the type of metal complexes is limited. High-temperature pyrolysis is usually used to synthesize dual atoms on NC materials. It is worth noting that the uniformity of samples is difficult to control, and such an approach can also result in a part of the active metals embedded in the bulk of the substrate matrix, making it hardly accessible during the reaction process. The atom trapping technique has a requirement for support, usually being appropriate for reducible oxides with abundant vacancies. Therefore, the selection of proper metallic precursors, reaction atmosphere, and temperature is critical to obtaining uniformly dispersed dual atoms.
THE ACTIVE CENTER STRUCTURE OF DACs
Precise identification of active center structures of DACs is difficult but significant for the study of the structure-activity relationship. To verify the successful synthesis of DACs, characterization at the atomic scale is urgently needed. Due to the flexible coordination structure of DACs, it brings more difficulties in the identification of the local environment and electronic configurations. In recent years, advanced techniques have been developed to enable atomic-level interrogation of the nature of dual-atom structures. Here, we focus on several techniques that are sensitive to the dual-atom structures, such as AC HAADF-STEM, X-ray absorption spectroscopy, in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTs), and other modern sophisticated techniques. The AC HAADF-STEM is a critical tool to provide the direct observation of dual-atom dispersion. XAFS is used to illustrate the electronic structure and coordination environment of metal atoms. Moreover, infrared spectroscopy is also important in characterizing single or dual atoms through special probe molecular adsorption. It is significant to combine various characterization techniques to clarify the structure of dual metal atoms and explore their unique performance. The synthesis strategies, active center structures, and catalytic applications for DACs are summarized in Table 1.
Summary of synthesis strategies, active center structures, and catalytic applications for DACs
| Catalysts | Active center structures | Synthesis strategies | Applications | Ref. (year) |
| PdCu-NC | Pd-N4, Cu-N4 | electrochemical reduction method | ethylene glycol oxidation | 2022[93] |
| PtFe-NC | Pt-N4, Fe-N4 | high-temperature pyrolysis | ORR | 2021[94] |
| NiFe-NC | Ni-N4, Fe-N4 | high-temperature pyrolysis | CO2RR | 2021[95] |
| CuCo-NC | Cu-C4, Co-N4 | misplaced deposition strategy | oxidative esterification | 2021[96] |
| NiCo-NC | Ni-N4, Co-N4 | two-step pyrolysis | CO2RR | 2023[97] |
| FeMn-NC | N2Fe-N2-MnN2 | molten salt assisted pyrolysis | ORR and OER | 2022[98] |
| NiFe-NC | N2Ni-N2-FeN2 | adsorption and pyrolysis | CO2RR | 2024[99] |
| NiCo-NC | N2Ni-N2-CoN2 | assembly and carbonization | polysulfide conversion | 2024[100] |
| ZnCo-NC | N2Zn-N2-CoN2 | competitive complexation strategy | propane dehydrogenation | 2023[101] |
| CuFe-NC | N2Cu-ON2-FeN2 | assembly and pyrolysis | electrochemical NO reduction | 2023[102] |
| CuNi-NC | N2Cu-S-NiN2 | co-impregnation method | CO2RR | 2024[103] |
| CoNi-NC | N3Co-NiN3 | coating and pyrolysis | ORR and OER | 2019[104] |
| CuSn-NC | N2Cu-SnN2 | dissolution and carbonization method | CO2RR | 2023[105] |
| CuZn-NC | N3Cu-ZnN3 | high-temperature pyrolysis | acetylene semihydrogenation | 2024[106] |
| NiCu-NC | N3Ni-CuN3 | host-guest strategy | CO2RR | 2022[107] |
| FeSn-NC | N2Fe-SnN2 | two-step pyrolysis | ORR | 2024[108] |
DACs with non-bonding interaction
Identification and understanding of the long-range interaction between neighboring single-metal atoms is of great significance to catalysis. Moges et al. prepared Pd-N4/Cu-N4 sites by an electrochemical reduction method[93]. The coordination structure was confirmed by EXAFS fitting results. The synergetic properties of the Pd-N4 acted as an active center and Cu-N4 served as a structural site, which showed outstanding reactivity for ethylene glycol electrooxidation. Besides, the adjacent Pt-N4 moiety was considered to tune the 3d electronic orbitals of the Fe-N4 center to improve ORR activity[94]. Based on the EXAFS spectra, the isolated Pt-N4 and Fe-N4 structure was confirmed. Moreover, they found that this modulation effect was not effective in optimizing the ORR performance of Co-N4/Pt-N4 and Mn-N4/Pt-N4. In addition, Jiao et al. constructed a new Fe1-Ni1-N-C with a non-bonding interaction of adjacent Fe and Ni dimers for CO2 electroreduction[95]. As presented in Figure 6A, NC embedded by neighboring Fe-N4 and Ni-N4 was synthesized via pyrolysis. The atomic-resolution EELS line scan showed an atomic pair containing one Fe atom and one Ni atom, strongly verifying the formation of Ni-Fe dual atoms [Figure 6B]. To further distinguish the microenvironment, the EXAFS fitting showed isolated Fe-N4 and Ni-N4 configurations with an interatomic distance of about 4.1 Å [Figure 6C]. Wang et al. exploited a simple method to fabricate isolated FeCo-NC catalysts[109]. Specifically, they used ZIF-8 to adsorb Fe(acac)3 and Co(acac)3 solutions and the obtained precursors were subsequently pyrolyzed at 900 °C under 5% H2/Ar atmosphere. It was confirmed to be Fe-N4 and Co-N4 configurations from EXAFS fitting. Based on a typical M-N4 structure, Yang et al. synthesized FeCu DACs on commercial carbon black[110]. The EELS validated the co-existence of Fe and Cu atoms. The EXAFS fitting further indicated that both Fe and Cu atoms were bonded to four N atoms as
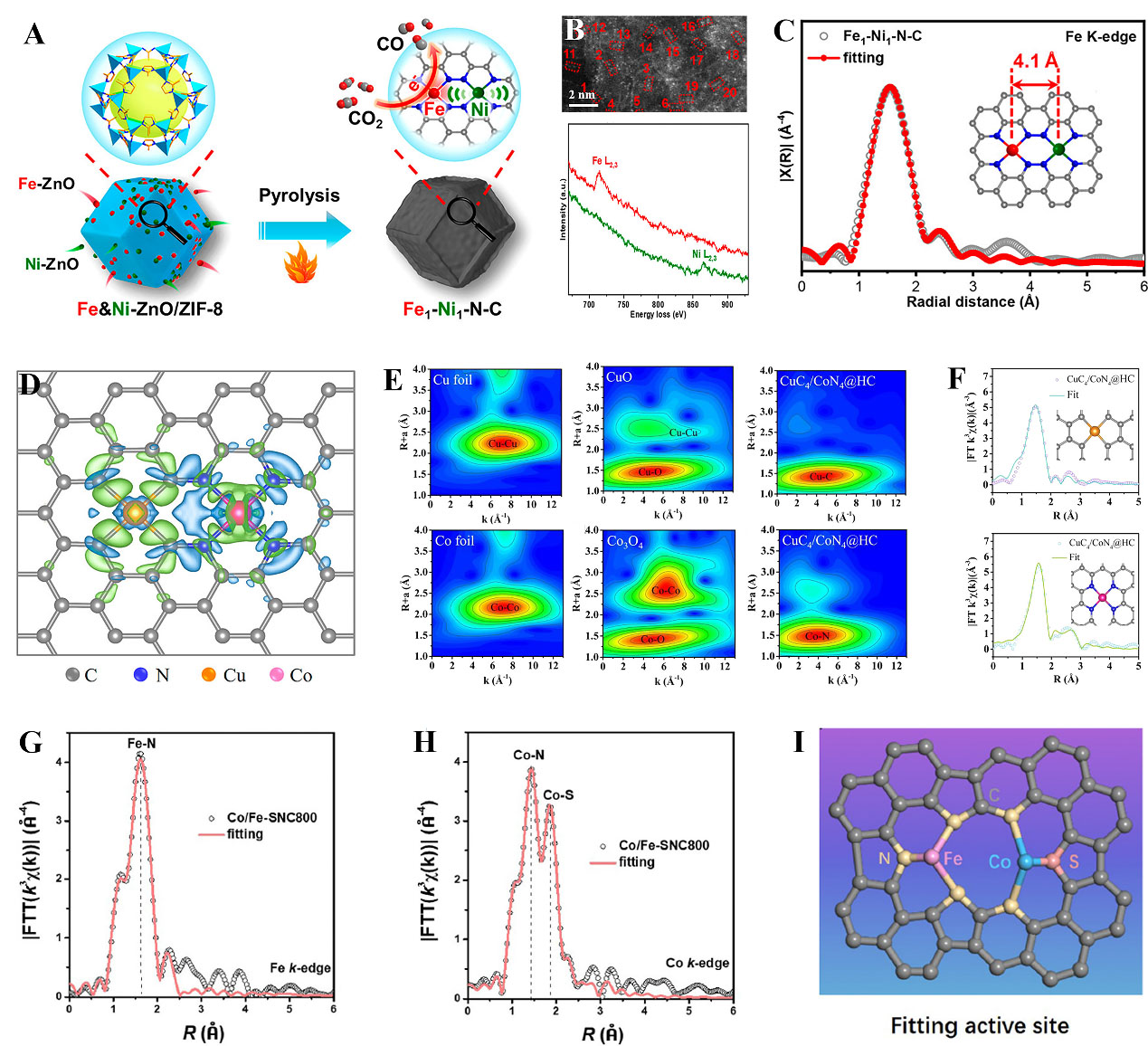
Figure 6. (A) Preparation of Fe1-Ni1-N-C on ZIF-8; (B) AC HAADF-STEM image and EELS spectra; (C) Fe K-edge EXAFS spectrum fitting and (inset) optimized configuration; (A-C) Reproduced with permission from Ref.[95]. Copyright 2021, American Chemical Society; (D) Top view of the differential charge densities of CuC4/CoN4@HC; (E) Wavelet-transformed k3-weighted EXAFS spectra of Cu/Co foil, CuO, Co3O4, and CuC4/CoN4@HC; (F) Cu/Co K-edge EXAFS fitting curves; (D-F) Reproduced with permission from Ref.[96]. Copyright 2021, American Chemical Society; (G) Fe k-edge EXAFS fitting; (H) Co k-edge EXAFS fitting; (I) Model of fitting active site; (G-I) Reproduced with permission from Ref.[114]. Copyright 2023, The Royal Society of Chemistry. AC HAADF-STEM: Aberration-corrected high-angle annular dark-field scanning transmission electron microscopy; EELS: electron energy-loss spectroscopy; EXAFS: extended X-ray absorption fine structure.
Inspired by theoretical calculations, Tamtaji et al. designed graphene-supported DACs in FeMN6 and FeMN8 configurations (M:Fe, Co, Ni, Cu, and Zn)[111]. Importantly, the non-bonding interaction of these counterpart metals could change the ORR activity of Fe sites. The FeNi-N8 structure demonstrated a superior activity comparable to the benchmark Pt/C. This work suggests a DFT-guided strategy for designing FeM-NC DACs for electrochemical reactions. Apart from Fe-based DACs, Cu and Zn dual-metal atoms were observed to promote ORR. For instance, Tong et al. constructed an atomically dispersed Cu-N4 and Zn-N4 on NC substrate by pyrolyzing a CuZn-ZIF precursor[112]. Operando X-ray absorption near edge structure (XANES) showed that the Cu-N4 as an active site underwent the change from atomic dispersion to cluster with the synergy of Zn-N4 during the reaction process, and then turned to single atoms again after the reaction. The low electronegativity Zn atom could transfer electrons to Cu, facilitating the cleavage of O-O on Cu-N4 sites. In addition, Bi/Zn DACs featuring Bi-N4 and Zn-N4 moiety were effective for syngas production with controlling CO/H2 ratios from 0.20 to 2.92[113]. Zhao et al. reported a misplaced deposition strategy for the construction of differently coordinated DACs[96]. As proof of the concept, they fabricated the Cu and Co dual atoms. The charge distribution pattern [Figure 6D] was simulated to reveal the electrical properties of Cu and Co sites. The as-prepared catalyst with the asymmetric deployment of CuC4 and CoN4 sites led to a greatly polarized surface charge distribution including both electron-rich CuC4 and electron-deficient CoN4 sites. The EXAFS fitting and wavelet transform results confirmed the CuC4 and CoN4 structure [Figure 6E-F]. This finding may open new avenues for designing DACs with coexistent electron-rich and electron-deficient sites for efficient synergistic catalysis. Except for Co and Cu dual atoms, other metals such as Ni can also modulate selectivity and optimize the electrochemical CO2 reduction to syngas. It was reported that the ratio of CO/H2 could be controlled by tuning the Co/Ni ratio in individual NiN4 and CoN4 dual sites[97]. This work offers a new way to achieve industrial-level syngas production by CO2 electroreduction.
Recently, some studies have also focused on S doping, and results showed the charge redistribution of central metal atoms owing to the different electronegativity compared to N species. In this case, Chen et al. design Co-Fe dual toms to optimize OER performance[114]. As presented in Figure 6G-H, the R-space fitting results of Fe and Co K-edge indicated atomically dispersed Fe and Co species, where the configuration was revealed to FeN3 and CoN2S coordination structure [Figure 6I]. The interplay between Fe and Co sites was a key factor for improved oxygen evolution reaction (OER) activity, where the Co atom bound with S and N enabled efficient site-to-site electron transfer, while the Fe atom bound with N facilitated stability of the Co site. In the other study, the reversed electron transfer mechanism on Au and Co DACs was reported[115]. The coordination number of Au-S and Co-S was about 2 and 4, respectively. It was revealed that the localized electrons around Co and delocalized electrons from Au created a polarized region to boost CO2 reduction.
DACs with N/O/S/Cl-bridged interaction
The geometry configurations and electronic states are the most critical factors to optimize reaction performance. At present, some attention has been paid to different active structures of DACs, especially nonmetallic atoms (e.g., N, O, S, Cl, etc.) bridged dual-metal atoms. Yang et al. implanted Mn-N moiety in Fe-N-C to prepare FeMn-N-C atomic pairs, where dicyandiamide was used as C and N sources, and FePc and manganese nitrate were used as metal precursors[116]. On the basis of the simulated Fe K-edge spectra of XANES and the calculated structural model, the FeMn-N6 structure was suggested as the most possible configuration for an as-prepared catalyst. The introduction of the Mn-N site caused FeIII electron delocalization and made the spin state of FeIII transition from low spin (t2g5eg0) to intermediate spin (t2g4eg1), readily penetrating the antibonding π-orbital of oxygen and achieving the outstanding ORR activity. Most previously reported NC supports for anchoring DACs show three-dimensional (3D) bulk structures, making lots of active sites buried inside and insufficient mass transfer. To address this, Cui et al. designed N-bridged Fe-N4 and Mn-N4 sites dispersed on ultrathin two-dimensional NC nanosheets with porous architecture, which exhibited excellent bifunctional activities for ORR and OER[98]. In a similar case, the FeNi-N6 configuration was found effective in acidic ORR than its single-atom counterpart[117]. Most recently, Chen et al. reported the effect of Ni and Fe atomic distance on the performance of CO2 electroreduction
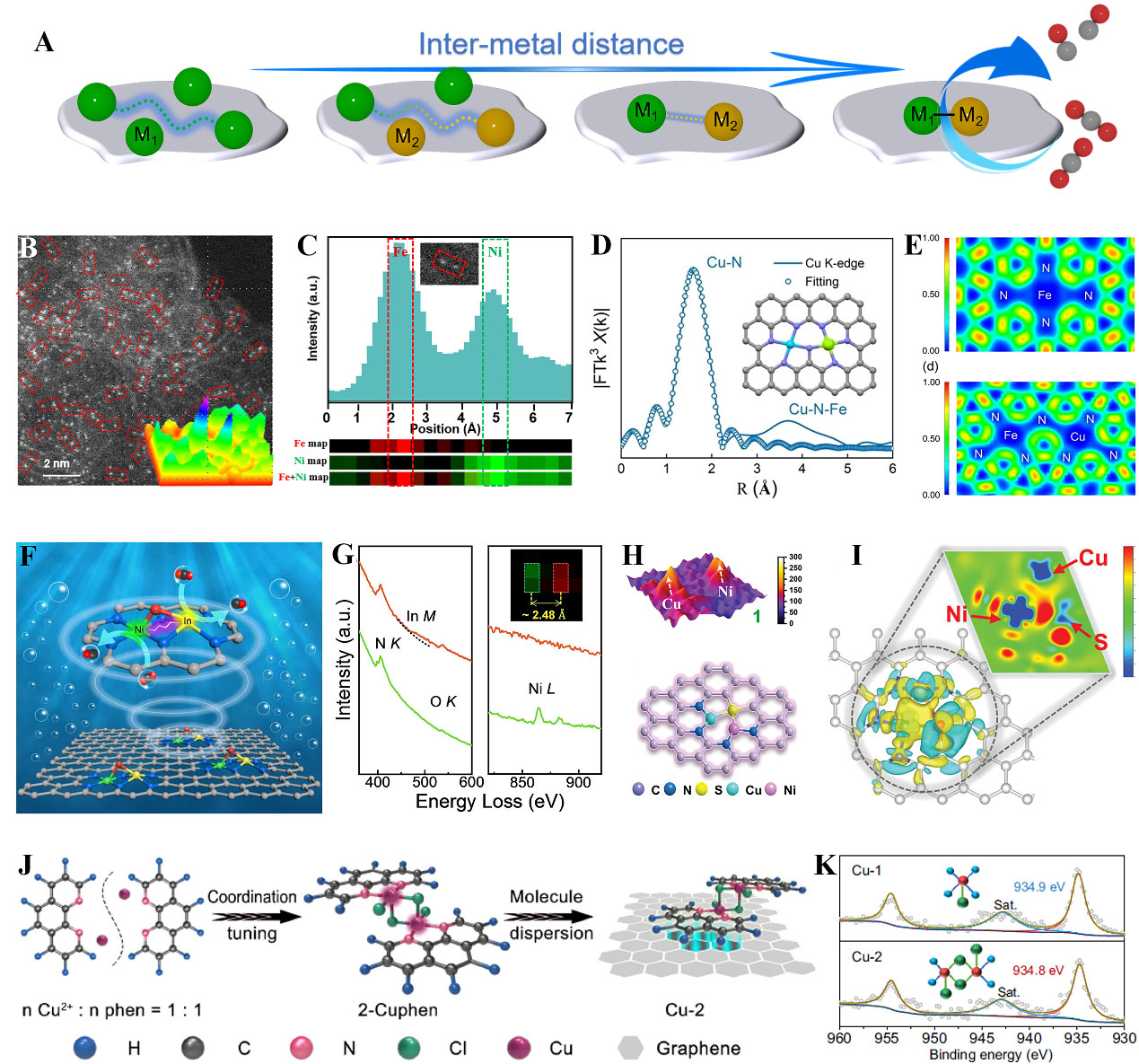
Figure 7. (A) Schematic of DACs showing the distance change of inter-NiFe on different models with SAC as a comparison; (B) AC HAADF-STEM image of NiFe-N bridge and 3D intensity surface plot; (C) Atomic resolution EELS mapping of NiFe-N bridge; (A-C) Reproduced with permission from Ref.[99]. Copyright 2024, Wiley-VCH; (D) R-space curve-fitting and corresponding fitted structure of the Fe and Cu atoms; (E) ELF analysis for FeN4 and FeCuN7; (D-E) Reproduced with permission from Ref.[123]. Copyright 2024, Wiley-VCH; (F) Illustration of axial oxygen bridged In-N6-Ni structure for electrochemical CO2 reduction; (G) The EELS spectra of In-Ni atomic pair; (F-G) Reproduced with permission from Ref.[124]. Copyright 2023, Wiley-VCH; (H) 3D model of Cu-S-Ni sites and structural model of Cu-S-Ni/SNC; (I) Electron density difference plot of Cu-S-Ni site; (H-I) Reproduced with permission from Ref.[103]. Copyright 2024, Wiley-VCH; (J) The preparation for Cu-2; (K) Cu 2p XPS spectra; (J-K) Reproduced with permission from Ref.[128]. Copyright 2024, Springer Nature. DACs: Dual-atom catalysts; AC HAADF-STEM: aberration-corrected high-angle annular dark-field scanning transmission electron microscopy; 3D: three-dimensional; EELS: electron energy-loss spectroscopy.
In the NiCo-NC system, the presence of Ni and Co atom pairs alongside N-bridged Ni-N4 and Co-N4 coordination environments was observed[100]. The NiCo-NC DAC demonstrated exceptional activity in the conversion of lithium polysulfide, indicating promising prospects for lithium-sulfur batteries. Very recently, two N-bridged Fe-N4 and Co-N4 diatomic sites were developed for Zn-air/iodide hybrid batteries[121]. The strong coupling between 3d orbitals of Fe and Co weakened the binding strength between active sites and intermediates, promoting the activities for both ORR/OER and iodide/iodate redox. The N-colligated Zn1Co1 dual atoms were found as the main active sites for high-temperature propane dehydrogenation (PDH)[101]. The role of N-colligated Zn1Co1 species was further disclosed, which originated not only for the coordination of N atoms for C-H bond scission but also for the feasible channel for C3H6 desorption via the retrofitted Co1 site neighboring with Zn1 site. An important work reported that the activity of CuCo DACs for ORR, OER, and HER processes was determined by coordination shells. By controlling the amount of thiomalic acid in the precursors, three DACs with unique coordination structures were constructed as S atoms that directly bond to CuCo-N6 in the lower coordination shell, indirectly bond in the neighboring coordination shell, and doped in the higher coordination shell, respectively[122]. Interestingly, the direct bonding of S made Cu with a comparatively low chemical state of + 1.6, promoting ORR. The indirect bonding of S resulted in a moderate chemical state (e.g., Cu + 1.8), improving OER. When S atoms were located at greater distances, the metal atoms showed a higher chemical state (e.g., Cu + 2.0), promoting HER. These findings offer a new strategy to design highly efficient DACs by tuning the coordination environments beyond the first shell for diverse applications.
In addition to dual-metal atoms sharing two N atoms, some studies demonstrated that one N-bridged atomic pair was effective in improving reactivity. Bi et al. developed a FeCu DAC for direct nitrite reduction to ammonia[123]. As depicted in Figure 7D, the EXAFS fitting analysis revealed the existence of N3Fe-N-CuN3 configuration. In light of this evidence, the electron localization function (ELF) was performed to further reveal the degree of electronic delocalization of N3Fe-N-CuN3. As plotted in Figure 7E, the N3Fe-N-CuN3 configuration showed a larger region area around Fe-N atoms than that of Fe-N4 in ELF, suggesting that the N3Fe-N-CuN3 possessed richer delocalized electrons. The introduction of Cu atoms changed the energy of the Fe 3d orbital and decreased the reaction barriers, promoting the nitrite reduction reaction. Besides, it was found that one N-bridged CoNi DAC (N3Co-N-NiN3) achieved 95% polystyrene conversion and 92% ethylbenzene yield, greatly higher than the corresponding SACs[80]. The experimental results and theoretical calculations revealed that the Co site could activate the C=C bond easily, while the Ni site spatially optimized the adsorption configuration of the styrene owing to the electronic interplay. To further regulate the electronic properties and local environment of DACs, Gong et al. regulated the coordination structure around Ni2 centers by changing pyrolysis temperatures[45]. It was disclosed that the C2NNi-N-NiNC2 structure significantly boosted activity for CO2 electroreduction to CO. This finding provides a way to boost catalytic performance via tailoring the local environments of DACs.
Except for N-bridged configuration, O-bridged dual atoms are also efficient for enhanced catalysis. For instance, Fan et al. designed an axial oxygen-bridged indium-nickel DAC with an O-In-N6-Ni configuration [Figure 7F][124]. The EELS spectra extracted from the two labeled regions confirmed the atomically dispersed In and Ni species [Figure 7G]. In situ attenuated total reflection surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS) combined with DFT results indicated that the synergy of In-Ni dual sites and bridged O atoms not only lowered the reaction barrier for *COOH formation but also inhibited HER. Similarly, an axial oxygen coordinated Cu-Fe DAC as O-Fe-N6-Cu configuration was efficient for NO electroreduction to NH3 in neutral electrolyte[102]. In another work, the O-bridged W-Mo atoms were deposited on N-doped graphene defects featuring W-O-Mo-O-C[125]. The electron delocalization of this configuration optimized the adsorption strength of H and improved the HER kinetics. Apart from an oxygen-bridged structure, a novel sulfur-bridged Cu-S-Ni DAC was reported with high efficiency for CO2 reduction[103]. The 3D intensity map outlined the locations of Cu-S-Ni dual sites and the structural model was depicted as N2Cu-S-Ni-N2 [Figure 7H]. As shown in Figure 7I, the charge asymmetry in Cu-S-Ni sites was observed via the electron density difference map. Charge transfer between Cu, S, and Ni atoms emphasized the important regulating role of the S-bridged in electron transfer and charge distribution. It was revealed that Cu as a major active site promoted *COOH formation, thereby achieving superior CO2 reduction performance. Apart from the S-bridged interaction, the electronic structure of the metal active center can be regulated by the remote interactions of the heteroatoms. The activity of the single Fe-N4 site could be tuned by incorporating sulfur functionalities into the carbon plane[126]. The introduction of oxidized sulfur functionality decreased the d-band center of irons by withdrawing electrons, thereby lowering the adsorption energy of the intermediate at the Fe-N4 site and improving the activity. Besides, adjusting the electronic structure of Cu-N4 sites by heteroatom was investigated[127]. Experimental and theoretical results indicated that the long-range interaction with B atoms decreased the electronic density of Cu active sites, and optimized the adsorption energy for reactant activation. Impressively, Yang et al. developed chlorine-bridged binuclear Cu atoms for lithium-sulfur batteries[128]. As illustrated in Figure 7J, the 2-Cuphen complex was first prepared by tailoring the mole ratio of Cu2+ and 1,10-phenanthroline ligand into 1:1. Subsequently, the obtained Cu complex was supported on a graphene substrate for fabricating
DACs with direct metal bonding interaction
Recent studies of DACs with metal-metal interaction have achieved great progress in tailoring the configuration and local coordination structure, enabling multiple reaction adsorption sites and outstanding reactivity. For instance, Wang et al. designed a new Zn-porphyrin/RuCu-pincer for selective CO2 photoreduction[129]. As presented in Figure 8A, the heteronuclear Ru-Cu sites easily promoted C1 intermediate coupling owing to the strong gradient orbital coupling of Ru4d-Cu3d resonance. The two *CO intermediates of Ru-Cu showed a weaker electrostatic repulsion for an asymmetric charge distribution, which resulted in side-to-side adsorption and lowered the reaction barrier for C-C coupling. In contrast, the homoatomic counterparts exhibited the best selectivity for CO. Besides, covalent triazine frameworks supported the Pd1-Co1 catalyst, which was efficient for ambient CO2 conversion to formate[130]. It was revealed that the Pd atom was conducive to the dissociation of hydrogen, and the Co atom facilitated the adsorption and activation of the captured CO2, leading to a low-energy barrier for CO2 conversion. Xiao
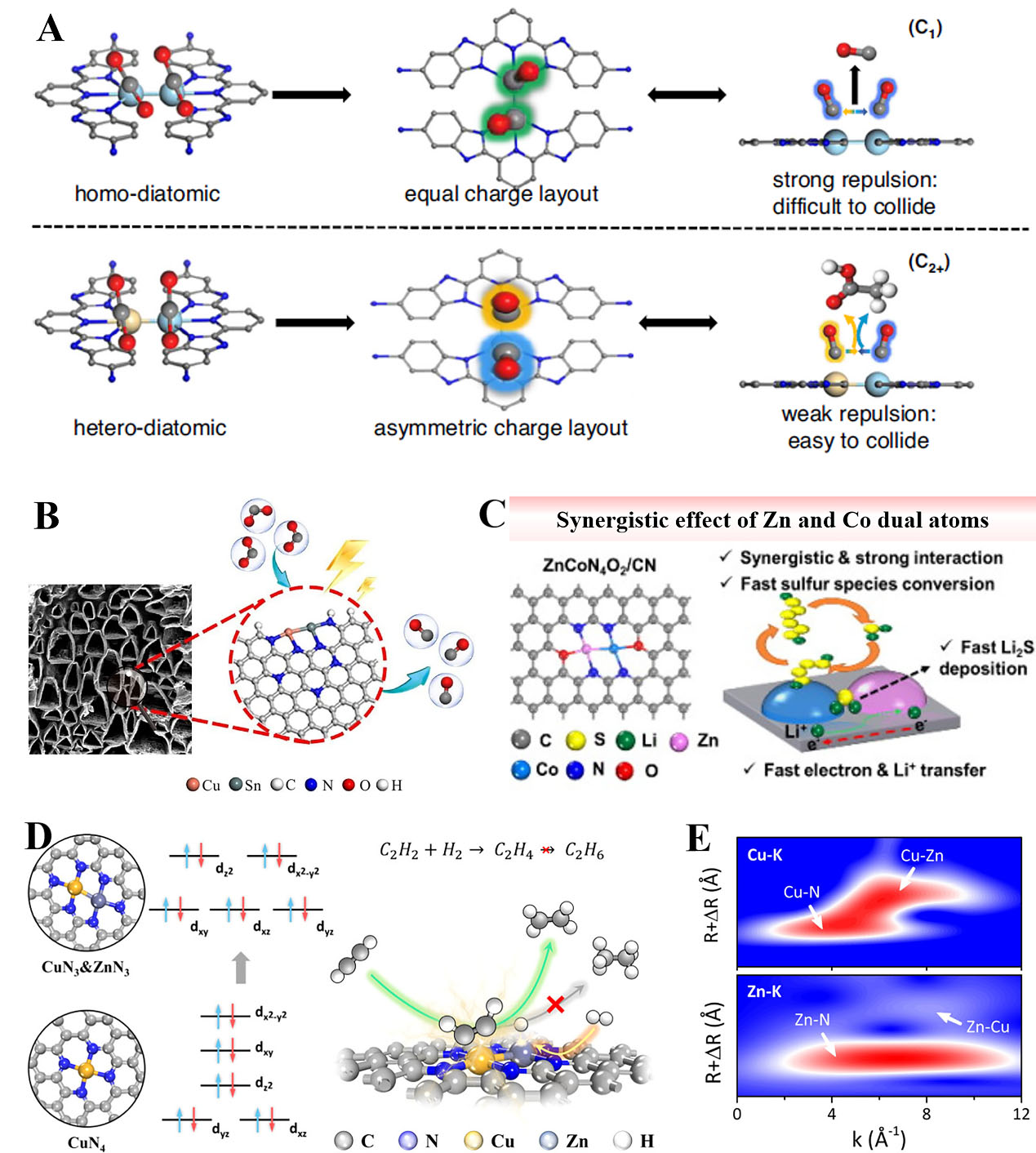
Figure 8. (A) CO2 reduction mechanism of C1 and C2+ product formation by homo- and hetero-DACs, respectively. Reproduced with permission from Ref.[129]. Copyright 2023, Springer Nature; (B) Schematic of the Cu-Sn DACs for CO2RR. Reproduced with permission from Ref.[105]. Copyright 2023, Springer; (C) Operational principles of ZnCo DAC in Li-S batteries. Reproduced with permission from Ref.[135]. Copyright 2023, Elsevier; (D) Schematic structures and d-orbital splitting manner for CuZnN6 and CuN4; (E) Wavelet transform of Cu and Zn K-edge spectra in CuZn/NC; (D-E) Reproduced with permission from Ref.[106]. Copyright 2024, American Chemical Society. DACs: Dual-atom catalysts.
In addition to noble metal DACs, NC-anchored Fe-Co dual sites were active for ORR under acidic conditions[42]. The preparation was based on controlling the formation of the N3Fe-CoN3 structure. DFT calculations indicated the dual sites were favored for O-O activation, which was important for four-electron ORR. A similar structure was also found to show good ORR activity and durability in alkaline media, promising in practical zinc-air batteries and flexible zinc-zir batteries[132]. From the viewpoint of theoretical calculation, a variety of heterogeneous DACs implanted in the graphitic carbon nitride were investigated as the potential candidates for NO3RR by high-throughput in silico first-principles calculations based on DFT[133]. By testing 21 catalysts, FeMo and CrMo showed a high performance with low limiting potentials for electrochemical NO3RR. In addition, Co-Ni dual atoms stabilized by N-doped hollow carbon nanocubes with N3Co-NiN3 coordination exhibited superior reversibility for ORR/OER[104]. Atomically dispersed Fe-Ni dual atoms were developed with extraordinary performance for ORR/OER[134]. DFT calculation revealed that Fe as an active center facilitated four-electron reduction, while the Ni site regulated the electronic properties of Fe and decreased the energy barrier of the rate-limiting step. Recently, asymmetrical FeSn dual atom sites on C2N nanosheets were developed for ORR[108]. The p-d orbital hybridization between p-block Sn and
Compared to heteronuclear DACs, homonuclear DACs with uniform dispersion are more easily obtained, which offers great potential for promoting reactions. For instance, Zhang et al. constructed dual-atom Fe on the surface of N-doped graphene for boosting conversion kinetics of Lithium-sulfur batteries[136]. The
UNDERSTANDING THE CATALYTIC ROLE OF DACs
With dual metal centers close to each other, DACs possess the advantages of SACs and also present opportunities to tune the interplay between active centers and reactants. The electronic effect is an essential factor affecting the catalytic reaction. Previous studies have stressed the importance of electronic structures of metal active centers on performance[139,140]. Although a few reviews introduced the spin state change of metal active centers and its effect on electrocatalytic performance[53]. The nature of spin state change is to influence the electronic properties or charge distribution of metal centers. Generally, dual metal atoms play two types of roles in reactions. On the one hand, one of the metals serves as the active center, and the other metal as a promoter tailors the electronic properties of the active center. On the other hand, dual-metal atoms behave cooperatively to play a bifunctional role, and linear scaling relations may be broken. In this section, we sketch out the current landscape of DACs. In particular, we concentrate on understanding the role of DACs, including electronic effects and synergistic effects, discussing the structure-activity relationship, and providing possible guidelines for the design of efficient catalysts. These effects are typical and significant for DACs.
Electronic effects
With the promising development of DACs, the chemical interaction between isolated dual atoms has been revealed as a key factor in changing electronic structures and catalytic performance. According to Sabatier’s principle, ideal heterogeneous catalysts should have optimized affinity strength between the key intermediates and the surface of a catalyst, which should be neither too weak nor too strong. Constructing an atomic pair structure is regarded as a powerful approach to alter the catalytic behaviors due to the electronic modulation effect. As shown in Figure 9A, the incorporation of Cu positively shifted the Ni 3d orbital energy to the Fermi level, implying an enhanced binding strength of intermediates on the NiCuN6 sites[107]. Additionally, the introduction of Cu eliminated the band gap of Ni 3d [Figure 9B], which was considered to lower the energy barrier for intermediate adsorption. As illustrated in Figure 9C, the NiMN6 dual atoms showed decreased Gibbs free energy for the *COOH formation than the NiN4 site, while NiCuN6 promoted *CO desorption, achieving better overall kinetics. Hao et al. found the strong electron interactions of Cu and Ni induced by the electronegativity offset[141]. The averaged and compensated electronic structures optimized the binding strength of *COOH and enhanced CO2 reduction. Furthermore, a distance threshold of about 5.3 Å between neighboring Ni-N4 and Cu-N4 was found to trigger effective electronic modulation and improved activity and selectivity for CO2 electroreduction[142]. Moreover, Zhang et al. designed a Ni-Cu DAC with a direct bonding structure for CO2 electroreduction to CO in a wide pH range[143]. To explore the origin of the enhanced activity, ATR-SEIRAS was used to monitor reaction intermediates. As shown in Figure 9D, the peaks at 1,215 cm-1 were attributed to *COOH intermediate, indicating that CO2 was easily protonated to generate *COOH on Cu/Ni-NC. The Cu atoms induced the d-band center of Ni closer to the Fermi level than that of Ni in the NiN3 site, promoting the adsorption of reactants [Figure 9E]. As shown in Figure 9F, the Ni site acted as an active center to adsorb and convert CO2, and Cu atoms tuned the electronic state of Ni to boost CO2 reduction. Recently, an asymmetric coordination Fe-Se DAC was designed for CO2 electroreduction to CO[144]. It was demonstrated that the electronic hybridization effect induced by asymmetrically coordinated Fe-Se dual sites enhanced the adsorption of *COOH and promoted the desorption of *CO, leading to excellent CO2 reduction performance. This work provides valuable insights into the design of highly efficient DACs through the electronic hybridization effect.
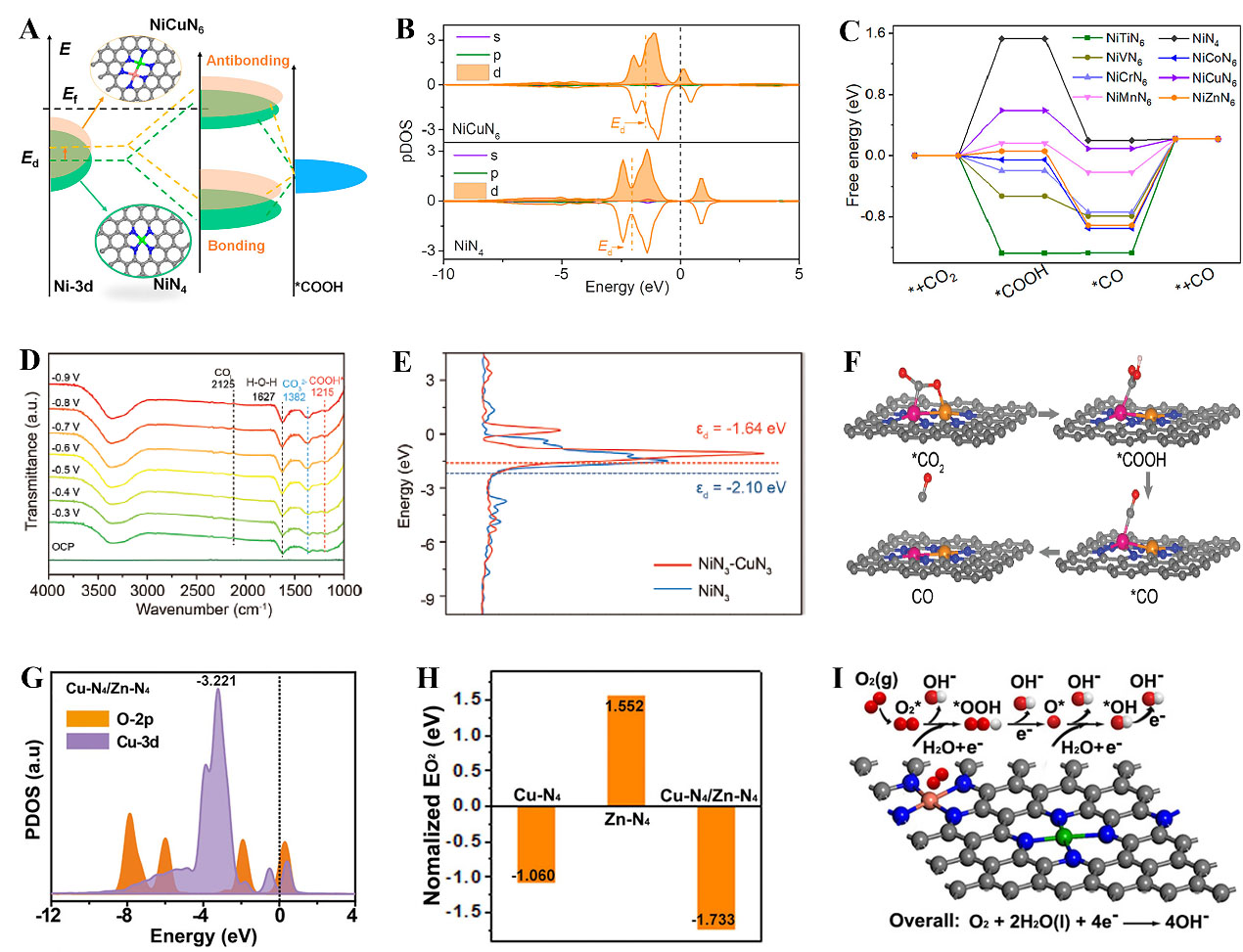
Figure 9. (A) Change of metal-adsorbate interaction by changing the metal d band center; (B) pDOS of Ni in NiN4 and NiCuN6; (C) Gibbs free energy profiles for CO2 electroreduction on NiMN6 and NiN4; (A-C) Reproduced with permission from Ref.[107]. Copyright 2022, American Chemical Society; (D) In situ ATR-SEIRAS test on Cu/Ni-NC in neutral medium; (E) The density of state of Ni d-orbitals in NiN3-CuN3 and NiN3; (F) The CO2 reduction pathway on NiN3-CuN3; (D-F) Reproduced with permission from Ref.[143]. Copyright 2023, Wiley-VCH; (G) Cu 3d PDOS and O 2p PDOS of Cu-N4/Zn-N4; (H) Free energy for ORR on the three catalysts; (I) Proposed ORR mechanism on the Cu-N4/Zn-N4 site; (G-I) Reproduced with permission from Ref.[112]. Copyright 2021, Wiley-VCH. ORR: Oxygen reduction reaction.
In addition to NiCu DACs, Xie et al. developed a NiSn atomic pair for CO2 reduction to formate[145]. Different from the NiCu system, it was verified that Sn served as the intrinsic active center and the neighboring Ni site imposed the electron redistribution of Sn. This facilitated the formation of *OCHO intermediate and made the potential-determining step thermodynamically spontaneous. Besides, main-group metal In could tune the electronic property of the neighboring Cu site to boost CO2 reduction to CO without the HER side reaction[146]. The electronic donation effect of electron-rich Cu sites strengthened the adsorption of *COOH on Cu sites and enhanced activity. Except for experimental explorations, significant efforts have been devoted to catalyst screening via theoretical calculations. Taking Fe SAC as an example, it was demonstrated that the design of a dual-atom center by the introduction of Cu allowed for CO2 reduction to produce CH3OH and CH4[147]. Theoretical results suggested that Fe served as the active site and Cu acted as the promoter to enhance activity, which promoted the formation of multi-electron products. The theoretical prediction will simulate experiments to fabricate excellent DACs for CO2 reduction to high-value chemicals. Although some progress has been made, the choice of metal of dual atoms as the main active center is still controversial. It is worth making great efforts to explore more DACs to understand the origin of differences in electronic structures.
Constructing dual-metal atoms may open a new avenue for ORR catalyst design. For instance, dual-metal atoms consisting of Cu-N4 and Zn-N4 were constructed[112]. As presented in Figure 9G, an energy-negative shift of Cu dual-atom structure for O2 adsorption could be observed, indicating strong adsorption of O2 on Cu-N4/Zn-N4. Moreover, the O2 adsorption energy of the dual-metal site was -1.733 eV, smaller than that of the single-atom site [Figure 9H]. After adsorption on the Cu site, the O-O bond was easy to break due to the low electronegativity of Zn that could transfer electrons to Cu. The proposed mechanism of ORR was depicted in Figure 9I. Besides, it was reported that the strain effect caused by CuN4 replacing the adjacent carbon environment of the FeN4 could adjust the electronic state and reduce the *OH adsorption on FeN4 sites, boosting the ORR process[148]. Similarly, Li et al. reported that the neighboring Ni-N4 could tune the electronic localization of the Co-N4 center, facilitating the *OH desorption and *H adsorption, thus enhancing the ORR and HER process[81]. As for NiMn-NC DAC, it was revealed that the MnN4 acted as the active center for ORR with the assistance of CoN4 as electron transfer from Co to Mn[149]. Besides, the addition of the Co-N4 site into Ru/Co-NC could increase Ru electron density, which then optimized the bonding strength between adsorption species and Ru sites, thus promoting OER and HER[150]. These works provide new insights into the regulation of the electronic nature of dual-atom centers by choosing suitable metal atoms for energy conversion.
The modulation of electronic structure is closely linked to the intrinsic activity of reactions. In one case, one of the metals acts as an electron donator to give electrons to the active metal site. The electron transfer between them may facilitate maintaining the chemical state of active metal and avoiding over-oxidation that would cause catalyst deactivation. In the other case, the introduction of another metal can make the active metal d orbital energy shift to the Fermi level, which promotes the adsorption of key intermediates and accelerates the rate-limiting step, thus boosting performance. Nevertheless, although DACs possess many advantages, the control of electron transfer direction is still a great challenge. It is difficult to predict dual-atom interaction and the distribution of active sites is not uniform as well. Moreover, electronic regulation is complicated when it involves factors beyond the direct coordination atom, such as the influence of the second shell and further. Therefore, more efforts should be devoted to tailoring dual-atom structures and catalytic properties for heterogeneous catalysis.
In-situ characterizations and theoretical calculations are essential to reveal the active center structures and reaction mechanisms. For instance, ATR-SEIRAS is a critical technique to detect reaction intermediates in electrochemical reactions. Operando 57Fe
Synergistic effects
For DACs, the adjacent atomic metal species can exert a synergistic effect for boosting catalytic performance. For example, Li et al. observed synergistic effects of Ni-Zn DAC for the electrochemical CO2 conversion[156]. The band gap of Ni 3d orbitals and Fermi level was narrowed and the electronic interaction between the intermediate and active center was enhanced, leading to superior activity for CO2 electroreduction. Furthermore, Zeng et al. reported that the orbital coupling between Fe and Ni resulted in the higher valance state of Fe and weakened adsorption of intermediates, achieving extraordinary activity for both CO2 reduction and oxygen evolution[157]. A Ni-Ag DAC was featured as N3-Ni-Ag-N3 with Faradic efficiency of CO surpassing 90% in a wide range of applied potentials[158]. It was revealed that the energy barrier of *COOH formation was decreased by the Ni atom, while the Ag atom promoted the *CO desorption. This synergy of DACs offers a promising way for the design of heteronuclear dual atoms for enhanced performance. To further modulate the spatial structure and coordination environment, an S,
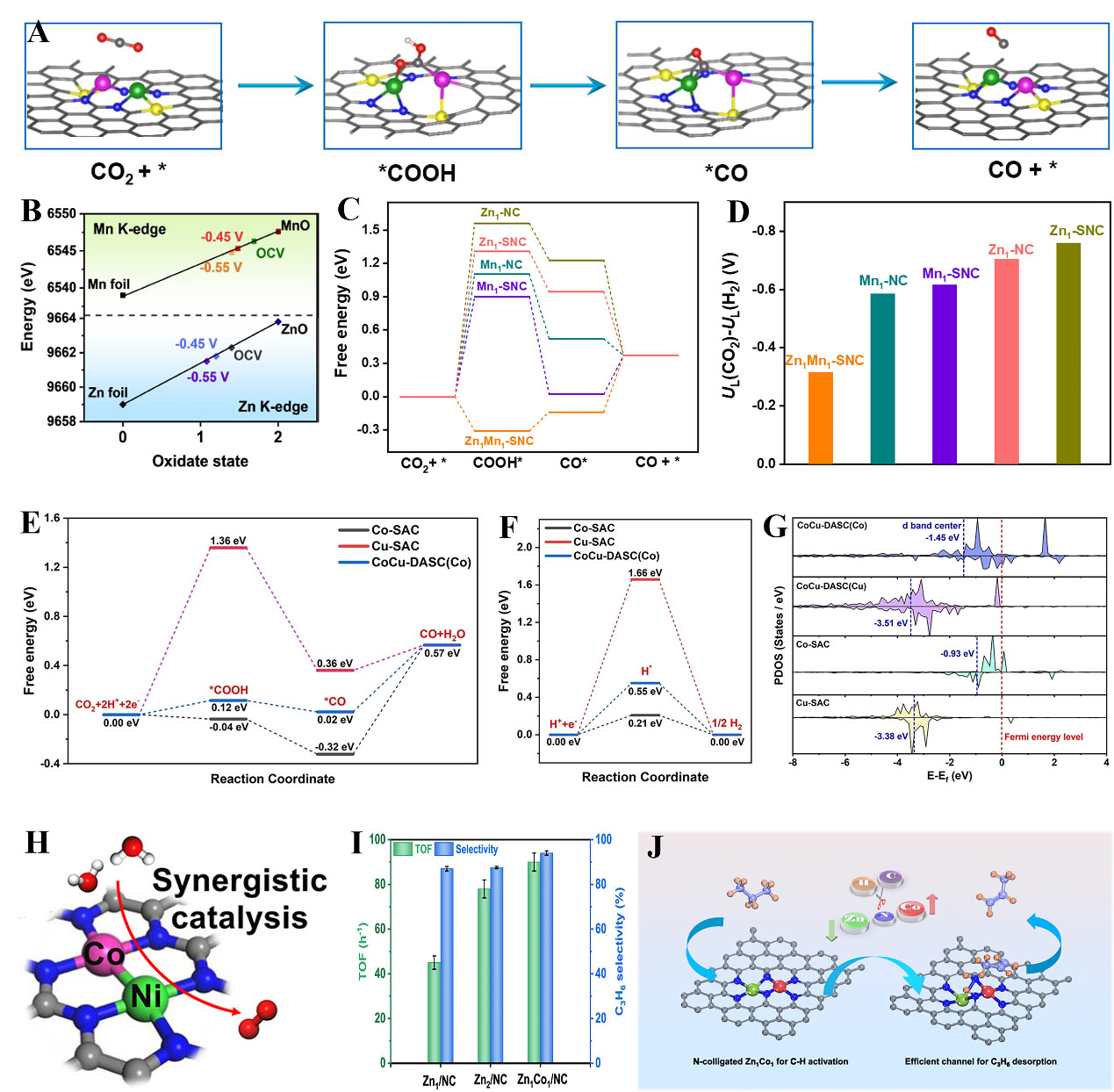
Figure 10. (A) Illustration of CO2 reduction on Zn-Mn DAC; (B) In situ ATR infrared spectra; (C) Free-energy profiles for CO2RR to CO; (D) The differences in limiting potential for CO2RR and HER; (A-D) Reproduced with permission from Ref.[159]. Copyright 2024, Wiley-VCH; (E) Free energy profiles of CO2 reduction; (F) Free energy profiles of HER; (G) The pDOS for the CoCu-DASC, Co-SAC, and Cu-SAC; (E-G) Reproduced with permission from Ref.[160]. Copyright 2022, Wiley-VCH; (H) Schematics of the NiCo synergy for OER. Reproduced with permission from Ref.[163]. Copyright 2022, Wiley-VCH; (I) TOF values and C3H6 selectivity during PDH reaction on Zn1/NC, Zn2/NC, and Zn1Co1/NC; (J) Schematic diagram of the enhanced activation of C3H8 and selectivity of C3H6 for the PDH reaction; (I-J) Reproduced with permission from Ref.[101]. Copyright 2024, American Chemical Society. DACs: Dual-atom catalysts; HER: hydrogen evolution reaction; SACs: single-atom catalysts; ATR: attenuated total reflection; OER: oxygen evolution reaction; TOF: turnover frequency; PDH: propane dehydrogenation.
Except for the synergy of DACs in electrochemical CO2 reduction, the cooperative role between dual-metal atoms makes it a promising candidate for the OER and ORR processes. Through strong metal-support interactions (SMSI) offered by nickel-vanadium layered double hydroxides (LDH), the significant interplay could anchor the Ru site to reach a fluctuation in oxidation state for cathodic HER, while the Ni site contributed to superior OER performance[161]. Similarly, IrFe dual atoms with N2Ir-N2-FeN2 configuration exhibited favorable bifunctional oxygen conversion. The Fe site facilitated the ORR, while the Ir site played a crucial role in the OER[162]. Pei et al. designed atomically dispersed NiCo DAC towards OER[163]. The synergistic interaction from the NiCo atomic pair optimized the electronic state and lowered the energy barrier; thus, the most favorable OER pathway was proposed [Figure 10H]. Additionally, ZnCu-NC DAC with atomic Cu-C2N and Zn-N4 sites was constructed by dynamic synthetic strategy[164]. Importantly, the Zn and Cu sites could effectively adsorb O2 via the bridged-adsorption model, enhancing the orbital hybridization effect between metallic 3d and O 2p, thus improving ORR activity. Similarly, Zhao et al. found that the Fe-N2-Fe structure could tune the adsorption configuration of O2 and lower the cleavage barrier, thus accelerating the catalytic kinetics[165]. Other than the intrinsic activity, structural and shape engineering of supports to expose more active sites are also of great importance in realizing excellent reactivity. A
In short, the cooperative effect of dual metal centers can largely enhance reactivity. The synergy is manifested in the following aspects. Firstly, compared to single atoms, the synergistic effect can break the restriction of scaling relations on activity. The adsorption-desorption behaviors of reactions can be optimized simultaneously on two different metal sites. Secondly, the DAC plays a bifunctional role in some reactions, such as when one metal is active for hydrogen production, and the other metal leads to superior performance for oxygen evolution. The dual-site bifunctional catalyst shows its great potential for practical application. Thirdly, the unique geometric configuration makes the structure diverse and flexible. The different configurations may result in different metal atomic distances, which determine the inter-metal interaction and adsorption behaviors of reactants. The specific configuration of DACs will influence the adsorption mode and strength of reactants, intermediates, and products, thus affecting the activity and selectivity. Particularly for the critical intermediates of C-C coupling, the synergy of dual metals can optimize adsorption configuration and promote the formation of C2+ products. Therefore, a universal macro-descriptor rigorously correlating the dual-atom configuration and intrinsic activity is critical to guide the design and preparation of advanced DACs. This opens a new avenue for future studies on DACs.
CONCLUSION AND OUTLOOK
DACs have been regarded as the new frontier in the field of energy conversion. In this review, we overview the recent progress in the controlled preparation of DACs. Then, the electronic structures and configurations of active centers are discussed. We focus on the microenvironment manipulation of active sites, including dual atoms with non-bonding interaction, N/O/S/Cl-bridged interaction, and direct metal bonding interaction. The merits in geometric and electronic configurations render the active moiety proper adsorption behavior towards intermediates. To better understand the structure-activity relationship and reaction mechanism, the catalytic role of DACs in the energy conversion application is clarified. Particularly, a fundamental understanding of the electronic effect and synergistic effect is demonstrated to guide the development of highly efficient DACs for practical applications.
So far, great progress has been achieved in DACs, but some important concerns and challenges are still worth considering and addressing.
Precise construction of uniform DACs
Current preparation approaches inevitably introduce single atoms, as well as a mixture of homonuclear and heteronuclear dual atoms. Developing new strategies to prepare uniform DACs is of great importance to establishing clear and definite structure-activity relationships.
From dual-center to multi-center design
DACs play a crucial role in bifunctional catalysts by the merits of dual metal centers. Except for DACs, some attention needs to be focused on the atomic clusters which include three or few atoms due to their unique structure and high efficiency. In addition, the well-regulated anchoring of diverse metal atoms into a single matrix shows good potential, such as high-entropy multi-atoms. This broadens the family of SACs and DACs, opening a new avenue for developing specific-atom catalysts with multi-centers.
In situ/operando characterization techniques
The characterizations of DACs are mainly by means of HAADF-STEM and X-ray adsorption spectroscopy. These techniques have difficulty differentiating the dual-metal atoms and identifying the specific metal species. In some situations, the scatting of small clusters is overlooked in the XAS analysis. Infrared spectroscopy with special probe molecules would be another powerful technique to provide additional structural information on dual atoms.
DACs library for universal design principles
Theoretical calculations not only aid in experimental design but also play a key role in elucidating the reaction mechanism. While traditional DFT calculations are usually performed, they are not feasible for high-throughput screening. The machine learning technique presents promise in the construction of predictive models. Pioneering works have already built up the library of databases. These remarkable examples offer a database for the next generation of highly efficient catalyst exploration with artificial intelligence.
Practical application in emerging fields of catalysis
Compared to classic thermocatalysis, electrocatalysis, and photocatalysis, plasms-catalysis and microwave-assisted catalysis remain at a nascent stage. The couple of multi-field catalysis of DACs is expected to be promising in the future. Another future direction is to bridge the gap between heterogeneous catalysis and bio-catalysis. Dual-metal centers are extensively present in metalloprotease. The structural similarity opens a new avenue for dual-atom structure regulation and their application in enzymatic and biomimetic catalysis.
DECLARATIONS
Authors’ contributions
Prepared the manuscript: Chen, Y.
Revised the manuscript: Lin, J.
Availability of data and materials
Not applicable.
Financial support and sponsorship
This work was supported by the National Natural Science Foundation of China (22378379, 22022814), the China Postdoctoral Science Foundation (2023M743430), the NSFC Basic Science Center Program for Single-Atom Catalysis (22388102), and the Young Top-notch Talents of Liaoning Province (XLYC2203140).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Zhang, Q.; Uchaker, E.; Candelaria, S. L.; Cao, G. Nanomaterials for energy conversion and storage. Chem. Soc. Rev. 2013, 42, 3127-71.
2. Falcone, P. M. Sustainable energy policies in developing countries: a review of challenges and opportunities. Energies 2023, 16, 6682.
3. Chu, S.; Majumdar, A. Opportunities and challenges for a sustainable energy future. Nature 2012, 488, 294-303.
4. Zhang, F.; Zhu, Y.; Lin, Q.; Zhang, L.; Zhang, X.; Wang, H. Noble-metal single-atoms in thermocatalysis, electrocatalysis, and photocatalysis. Energy. Environ. Sci. 2021, 14, 2954-3009.
5. Ding, S.; Hülsey, M. J.; Pérez-ramírez, J.; Yan, N. Transforming energy with single-atom catalysts. Joule 2019, 3, 2897-929.
6. Liang, J.; Ma, F.; Hwang, S.; et al. Atomic arrangement engineering of metallic nanocrystals for energy-conversion electrocatalysis. Joule 2019, 3, 956-91.
7. Luna P, Hahn C, Higgins D, Jaffer SA, Jaramillo TF, Sargent EH. What would it take for renewably powered electrosynthesis to displace petrochemical processes? Science 2019, 364, eaav3506.
8. Dong, C.; Li, Y.; Cheng, D.; et al. Supported metal clusters: fabrication and application in heterogeneous catalysis. ACS. Catal. 2020, 10, 11011-45.
9. Chen, Y.; Lin, J.; Jia, B.; Wang, X.; Jiang, S.; Ma, T. Isolating single and few atoms for enhanced catalysis. Adv. Mater. 2022, 34, e2201796.
10. Liu, L.; Corma, A. Metal catalysts for heterogeneous catalysis: from single atoms to nanoclusters and nanoparticles. Chem. Rev. 2018, 118, 4981-5079.
11. Wang, Y.; Mao, J.; Meng, X.; Yu, L.; Deng, D.; Bao, X. Catalysis with two-dimensional materials confining single atoms: concept, design, and applications. Chem. Rev. 2019, 119, 1806-54.
12. Rong, H.; Ji, S.; Zhang, J.; Wang, D.; Li, Y. Synthetic strategies of supported atomic clusters for heterogeneous catalysis. Nat. Commun. 2020, 11, 5884.
13. Haruta, M.; Tsubota, S.; Kobayashi, T.; Kageyama, H.; Genet, M.; Delmon, B. Low-temperature oxidation of CO over gold supported on TiO2, α-Fe2O3, and Co3O4. J. Catal. 1993, 144, 175-92.
14. Yang, X. F.; Wang, A.; Qiao, B.; Li, J.; Liu, J.; Zhang, T. Single-atom catalysts: a new frontier in heterogeneous catalysis. Acc. Chem. Res. 2013, 46, 1740-8.
16. Zhang, H.; Liu, G.; Shi, L.; Ye, J. Single-atom catalysts: emerging multifunctional materials in heterogeneous catalysis. Adv. Energy. Mater. 2018, 8, 1701343.
17. Mitchell, S.; Vorobyeva, E.; Pérez-Ramírez, J. The multifaceted reactivity of single-atom heterogeneous catalysts. Angew. Chem. Int. Ed. 2018, 57, 15316-29.
18. Kaiser, S. K.; Chen, Z.; Faust, A. D.; Mitchell, S.; Pérez-Ramírez, J. Single-atom catalysts across the periodic table. Chem. Rev. 2020, 120, 11703-809.
19. Lang, R.; Du, X.; Huang, Y.; et al. Single-atom catalysts based on the metal-oxide interaction. Chem. Rev. 2020, 120, 11986-2043.
20. Chen, Y.; Ji, S.; Chen, C.; Peng, Q.; Wang, D.; Li, Y. Single-atom catalysts: synthetic strategies and electrochemical applications. Joule 2018, 2, 1242-64.
21. Mitchell, S.; Pérez-Ramírez, J. Single atom catalysis: a decade of stunning progress and the promise for a bright future. Nat. Commun. 2020, 11, 4302.
22. Wang, A.; Li, J.; Zhang, T. Heterogeneous single-atom catalysis. Nat. Rev. Chem. 2018, 2, 65-81.
23. Liu, P.; Zhao, Y.; Qin, R.; et al. Photochemical route for synthesizing atomically dispersed palladium catalysts. Science 2016, 352, 797-801.
24. Qiao, B.; Wang, A.; Yang, X.; et al. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 2011, 3, 634-41.
25. Zhang, J.; Yang, H.; Liu, B. Coordination engineering of single-atom catalysts for the oxygen reduction reaction: a review. Adv. Energy. Mater. 2021, 11, 2002473.
26. Zhu, Y.; Sokolowski, J.; Song, X.; He, Y.; Mei, Y.; Wu, G. Single-atom catalysts: engineering local coordination environments of atomically dispersed and heteroatom-coordinated single metal site electrocatalysts for clean energy-conversion. Adv. Energy. Mater. 2020, 10, 1902844.
27. Guo, W.; Wang, Z.; Wang, X.; Wu, Y. General design concept for single-atom catalysts toward heterogeneous catalysis. Adv. Mater. 2021, 33, e2004287.
28. Sun, T.; Mitchell, S.; Li, J.; et al. Design of local atomic environments in single-atom electrocatalysts for renewable energy conversions. Adv. Mater. 2021, 33, e2003075.
29. Li, X.; Rong, H.; Zhang, J.; Wang, D.; Li, Y. Modulating the local coordination environment of single-atom catalysts for enhanced catalytic performance. Nano. Res. 2020, 13, 1842-55.
30. Lin, J.; Wang, A.; Qiao, B.; et al. Remarkable performance of Ir1/FeOx single-atom catalyst in water gas shift reaction. J. Am. Chem. Soc. 2013, 135, 15314-7.
31. Resasco, J.; DeRita, L.; Dai, S.; et al. Uniformity is key in defining structure-function relationships for atomically dispersed metal catalysts: the case of Pt/CeO2. J. Am. Chem. Soc. 2020, 142, 169-84.
32. Muravev, V.; Spezzati, G.; Su, Y.; et al. Interface dynamics of Pd-CeO2 single-atom catalysts during CO oxidation. Nat. Catal. 2021, 4, 469-78.
33. Peterson, E. J.; DeLaRiva, A. T.; Lin, S.; et al. Low-temperature carbon monoxide oxidation catalysed by regenerable atomically dispersed palladium on alumina. Nat. Commun. 2014, 5, 4885.
34. Wei, H.; Liu, X.; Wang, A.; et al. FeOx-supported platinum single-atom and pseudo-single-atom catalysts for chemoselective hydrogenation of functionalized nitroarenes. Nat. Commun. 2014, 5, 5634.
35. DeRita, L.; Resasco, J.; Dai, S.; et al. Structural evolution of atomically dispersed Pt catalysts dictates reactivity. Nat. Mater. 2019, 18, 746-51.
36. Li, R.; Wang, D. Superiority of dual-atom catalysts in electrocatalysis: one step further than single-atom catalysts. Adv. Energy. Mater. 2022, 12, 2103564.
37. Zhu, P.; Xiong, X.; Wang, D.; Li, Y. Advances and regulation strategies of the active moiety in dual-atom site catalysts for efficient electrocatalysis. Adv. Energy. Mater. 2023, 13, 2300884.
38. Lei, L.; Guo, X.; Han, X.; Fei, L.; Guo, X.; Wang, D. G. From synthesis to mechanisms: in-depth exploration of the dual-atom catalytic mechanisms toward oxygen electrocatalysis. Adv. Mater. 2024, 36, e2311434.
39. Zhang, W.; Chao, Y.; Zhang, W.; et al. Emerging dual-atomic-site catalysts for efficient energy catalysis. Adv. Mater. 2021, 33, e2102576.
40. Chen, Y.; Lin, J.; Pan, Q.; Liu, X.; Ma, T.; Wang, X. Inter-metal interaction of dual-atom catalysts in heterogeneous catalysis. Angew. Chem. Int. Ed. 2023, 62, e202306469.
41. Yardimci, D.; Serna, P.; Gates, B. C. Surface-mediated synthesis of dimeric rhodium catalysts on MgO: tracking changes in the nuclearity and ligand environment of the catalytically active sites by X-ray absorption and infrared spectroscopies. Chemistry 2013, 19, 1235-45.
42. Wang, J.; Huang, Z.; Liu, W.; et al. Design of N-coordinated dual-metal sites: a stable and active Pt-free catalyst for acidic oxygen reduction reaction. J. Am. Chem. Soc. 2017, 139, 17281-4.
43. Zhang, L.; Si, R.; Liu, H.; et al. Atomic layer deposited Pt-Ru dual-metal dimers and identifying their active sites for hydrogen evolution reaction. Nat. Commun. 2019, 10, 4936.
44. Ouyang, Y.; Shi, L.; Bai, X.; Li, Q.; Wang, J. Breaking scaling relations for efficient CO2 electrochemical reduction through dual-atom catalysts. Chem. Sci. 2020, 11, 1807-13.
45. Gong, Y. N.; Cao, C. Y.; Shi, W. J.; et al. Modulating the electronic structures of dual-atom catalysts via coordination environment engineering for boosting CO2 electroreduction. Angew. Chem. Int. Ed. 2022, 61, e202215187.
46. Hai, X.; Zheng, Y.; Yu, Q.; et al. Geminal-atom catalysis for cross-coupling. Nature 2023, 622, 754-60.
47. Dostagir, N. H. M.; Tomuschat, C. R.; Oshiro, K.; et al. Mitigating the poisoning effect of formate during CO2 hydrogenation to methanol over Co-containing dual-atom oxide catalysts. JACS. Au. 2024, 4, 1048-58.
48. Zhang, H.; Jin, X.; Lee, J. M.; Wang, X. Tailoring of active sites from single to dual atom sites for highly efficient electrocatalysis. ACS. Nano. 2022, 16, 17572-92.
49. Wong, M.; Foo, J. J.; Loh, J. Y.; Ong, W. Leveraging dual-atom catalysts for electrocatalysis revitalization: exploring the structure-performance correlation. Adv. Energy. Mater. 2024, 14, 2303281.
50. Gao, Y.; Liu, B.; Wang, D. Microenvironment engineering of single/dual-atom catalysts for electrocatalytic application. Adv. Mater. 2023, 35, e2209654.
51. Li, Y.; Wang, H.; Yang, X.; O'Carroll, T.; Wu, G. Designing and engineering atomically dispersed metal catalysts for CO2 to CO conversion: from single to dual metal sites. Angew. Chem. Int. Ed. 2024, 63, e202317884.
52. Pu, T.; Ding, J.; Zhang, F.; et al. Dual atom catalysts for energy and environmental applications. Angew. Chem. Int. Ed. 2023, 62, e202305964.
53. Tang, T.; Bai, X.; Wang, Z.; Guan, J. Structural engineering of atomic catalysts for electrocatalysis. Chem. Sci. 2024, 15, 5082-112.
54. Li, Y.; Li, Y.; Sun, H.; et al. Current status and perspectives of dual-atom catalysts towards sustainable energy utilization. Nano-Micro. Lett. 2024, 16, 139.
55. Li, H.; Li, R.; Liu, G.; Zhai, M.; Yu, J. Noble-metal-free single - and dual-atom catalysts for artificial photosynthesis. Adv. Mater. 2024, 36, e2301307.
56. Liu, H.; Rong, H.; Zhang, J. Synergetic dual-atom catalysts: the next boom of atomic catalysts. ChemSusChem 2022, 15, e202200498.
57. Zhang, J.; Huang, Q. A.; Wang, J.; Wang, J.; Zhang, J. J.; Zhao, Y. F. Supported dual-atom catalysts: preparation, characterization, and potential applications. Chin. J. Catal. 2020, 41, 783-98.
58. Tang, T.; Wang, Z.; Guan, J. Structural optimization of carbon-based diatomic catalysts towards advanced electrocatalysis. Coor. Chem. Rev. 2023, 492, 215288.
59. Zhang, T.; Liu, Y.; Xue, L.; Sun, J.; Xiong, P.; Zhu, J. Engineering of geometrical configurations in dual-atom catalysts for electrocatalytic applications. J. Energy. Chem. 2024, 94, 273-87.
60. Zhang, S.; Wu, Y.; Zhang, Y.; Niu, Z. Dual-atom catalysts: controllable synthesis and electrocatalytic applications. Sci. China. Chem. 2021, 64, 1908-22.
62. O’neill, B. J.; Jackson, D. H. K.; Lee, J.; et al. Catalyst design with atomic layer deposition. ACS. Catal. 2015, 5, 1804-25.
63. Yan, H.; Lin, Y.; Wu, H.; et al. Bottom-up precise synthesis of stable platinum dimers on graphene. Nat. Commun. 2017, 8, 1070.
64. Chen, S.; Gong, B.; Gu, J.; et al. Dehydrogenation of ammonia borane by platinum-nickel dimers: regulation of heteroatom interspace boosts bifunctional synergetic catalysis. Angew. Chem. Int. Ed. 2022, 61, e202211919.
65. Li, Y.; Chen, C.; Cao, R.; Pan, Z.; He, H.; Zhou, K. Dual-atom Ag2/graphene catalyst for efficient electroreduction of CO2 to CO. Appl. Catal. B:. Environ. 2020, 268, 118747.
66. Xie, P.; Ding, J.; Yao, Z.; et al. Oxo dicopper anchored on carbon nitride for selective oxidation of methane. Nat. Commun. 2022, 13, 1375.
67. Ding, T.; Liu, X.; Tao, Z.; et al. Atomically precise dinuclear site active toward electrocatalytic CO2 reduction. J. Am. Chem. Soc. 2021, 143, 11317-24.
68. Liang, X.; Wang, H.; Zhang, C.; Zhong, D.; Lu, T. Controlled synthesis of a Ni2 dual-atom catalyst for synergistic CO2 electroreduction. Appl. Catal. B:. Environ. 2023, 322, 122073.
69. Tian, S.; Fu, Q.; Chen, W.; et al. Carbon nitride supported Fe2 cluster catalysts with superior performance for alkene epoxidation. Nat. Commun. 2018, 9, 2353.
70. Zhang, N.; Zhang, X.; Kang, Y.; et al. A supported Pd2 dual-atom site catalyst for efficient electrochemical CO2 reduction. Angew. Chem. Int. Ed. 2021, 60, 13388-93.
71. Yu, Z.; Xia, G.; Diaconescu, V. M.; et al. Atomically dispersed dinuclear iridium active sites for efficient and stable electrocatalytic chlorine evolution reaction. Chem. Sci. 2024, 15, 9216-23.
72. Guan, E.; Debefve, L.; Vasiliu, M.; Zhang, S.; Dixon, D. A.; Gates, B. C. MgO-supported iridium metal pair-site catalysts are more active and resistant to CO poisoning than analogous single-site catalysts for ethylene hydrogenation and hydrogen-deuterium exchange. ACS. Catal. 2019, 9, 9545-53.
73. Gong, Q.; Wang, Y.; Ren, X.; He, C.; Liu, J.; Zhang, Q. Ultra-low-loaded Ni-Fe dimer anchored to nitrogen/oxygen sites for boosting electroreduction of carbon dioxide. ChemSusChem 2021, 14, 4499-506.
74. Wang, Y.; Wan, X.; Liu, J.; et al. Catalysis stability enhancement of Fe/Co dual-atom site via phosphorus coordination for proton exchange membrane fuel cell. Nano. Res. 2022, 15, 3082-9.
75. Wang, C.; Wang, K.; Feng, Y.; et al. Co and Pt dual-single-atoms with oxygen-coordinated Co-O-Pt dimer sites for ultrahigh photocatalytic hydrogen evolution efficiency. Adv. Mater. 2021, 33, e2003327.
76. Hou, C.; Wang, H.; Li, C.; Xu, Q. From metal-organic frameworks to single/dual-atom and cluster metal catalysts for energy applications. Energy. Environ. Sci. 2020, 13, 1658-93.
77. Liu, J.; Zhu, D.; Guo, C.; Vasileff, A.; Qiao, S. Design strategies toward advanced MOF-derived electrocatalysts for energy-conversion reactions. Adv. Energy. Mater. 2017, 7, 1700518.
78. Zhu, Z.; Duan, J.; Chen, S. Metal-organic framework (MOF)-based clean energy conversion: recent advances in unlocking its underlying mechanisms. Small 2024, 20, e2309119.
79. Lu, Z.; Wang, B.; Hu, Y.; et al. An isolated zinc-cobalt atomic pair for highly active and durable oxygen reduction. Angew. Chem. Int. Ed. 2019, 58, 2622-6.
80. Li, R.; Zhang, Z.; Liang, X.; et al. Polystyrene waste thermochemical hydrogenation to ethylbenzene by a N-bridged Co, Ni dual-atom catalyst. J. Am. Chem. Soc. 2023, 145, 16218-27.
81. Li, M.; Zhu, H.; Yuan, Q.; et al. Proximity electronic effect of Ni/Co diatomic sites for synergistic promotion of electrocatalytic oxygen reduction and hydrogen evolution. Adv. Funct. Mater. 2023, 33, 2210867.
82. Ren, W.; Tan, X.; Yang, W.; et al. Isolated diatomic Ni-Fe metal-nitrogen sites for synergistic electroreduction of CO2. Angew. Chem. Int. Ed. 2019, 58, 6972-6.
83. Li, Y.; Shan, W.; Zachman, M. J.; et al. Atomically dispersed dual-metal site catalysts for enhanced CO2 reduction: mechanistic insight into active site structures. Angew. Chem. Int. Ed. 2022, 61, e202205632.
84. Liu, M.; Li, N.; Cao, S.; et al. A “pre-constrained metal twins” strategy to prepare efficient dual-metal-atom catalysts for cooperative oxygen electrocatalysis. Adv. Mater. 2022, 34, e2107421.
85. Yang, X.; Li, X.; Liu, M.; Yang, S.; Xu, Q.; Zeng, G. Confined synthesis of dual-atoms within pores of covalent organic frameworks for oxygen reduction reaction. Small 2024, 20, e2306295.
86. Yao, Y.; Wang, C.; Yang, Y.; et al. Mn-Co dual sites relay activation of peroxymonosulfate for accelerated decontamination. Appl. Catal. B:. Environ. 2023, 330, 122656.
87. Zhang, Y. X.; Zhang, S.; Huang, H.; et al. General synthesis of a diatomic catalyst library via a macrocyclic precursor-mediated approach. J. Am. Chem. Soc. 2023, 145, 4819-27.
88. Leng, K.; Zhang, J.; Wang, Y.; et al. Interfacial cladding engineering suppresses atomic thermal migration to fabricate well-defined dual-atom electrocatalysts. Adv. Funct. Mater. 2022, 32, 2205637.
89. Jones, J.; Xiong, H.; DeLaRiva, A. T.; et al. Thermally stable single-atom platinum-on-ceria catalysts via atom trapping. Science 2016, 353, 150-4.
90. Zhou, X.; Han, K.; Li, K.; et al. Dual-site single-atom catalysts with high performance for three-way catalysis. Adv. Mater. 2022, 34, e2201859.
91. Pu, T.; Ding, J.; Tang, X.; et al. Rational design of precious-metal single-atom catalysts for methane combustion. ACS. Appl. Mater. Interfaces. 2022, 14, 43141-50.
92. Meier, M.; Hulva, J.; Jakub, Z.; et al. CO oxidation by Pt2/Fe3O4: metastable dimer and support configurations facilitate lattice oxygen extraction. Sci. Adv. 2022, 8, eabn4580.
93. Moges, E. A.; Chang, C.; Huang, W.; et al. Sustainable synthesis of dual single-atom catalyst of Pd-N4/Cu-N4 for partial oxidation of ethylene glycol. Adv. Funct. Mater. 2022, 32, 2206887.
94. Han, A.; Wang, X.; Tang, K.; et al. An adjacent atomic platinum site enables single-atom iron with high oxygen reduction reaction performance. Angew. Chem. Int. Ed. 2021, 60, 19262-71.
95. Jiao, L.; Zhu, J.; Zhang, Y.; et al. Non-bonding interaction of neighboring Fe and Ni single-atom pairs on MOF-derived N-doped carbon for enhanced CO2 electroreduction. J. Am. Chem. Soc. 2021, 143, 19417-24.
96. Zhao, X.; Wang, F.; Kong, X. P.; Fang, R.; Li, Y. Dual-metal hetero-single-atoms with different coordination for efficient synergistic catalysis. J. Am. Chem. Soc. 2021, 143, 16068-77.
97. Chen, Y.; Xia, M.; Zhou, C.; et al. Hierarchical dual single-atom catalysts with coupled CoN4 and NiN4 moieties for industrial-level CO2 electroreduction to syngas. ACS. Nano. 2023, 17, 22095-105.
98. Cui, T.; Wang, Y. P.; Ye, T.; et al. Engineering dual single-atom sites on 2D ultrathin N-doped carbon nanosheets attaining ultra-low-temperature zinc-air battery. Angew. Chem. Int. Ed. 2022, 61, e202115219.
99. Chen, Y.; Zhao, J.; Pan, X.; et al. Tuning the inter-metal interaction between Ni and Fe atoms in dual-atom catalysts to boost CO2 electroreduction. Angew. Chem. Int. Ed. 2024, 63, e202411543.
100. Zhao, C.; Huo, F.; Yang, Y.; et al. Development of synergistically efficient Ni-Co pair catalytic sites for enhanced polysulfide conversion in lithium-sulfur batteries. Adv. Funct. Mater. 2024, 34, 2402175.
101. Chai, Y.; Chen, S.; Chen, Y.; et al. Dual-atom catalyst with N-colligated Zn1Co1 species as dominant active sites for propane dehydrogenation. J. Am. Chem. Soc. 2024, 146, 263-73.
102. Wang, D.; Zhu, X.; Tu, X.; et al. Oxygen-bridged copper-iron atomic pair as dual-metal active sites for boosting electrocatalytic NO reduction. Adv. Mater. 2023, 35, e2304646.
103. Sun, Z.; Li, C.; Wei, Z.; et al. Sulfur-bridged asymmetric CuNi bimetallic atom sites for CO2 reduction with high efficiency. Adv. Mater. 2024, 36, e2404665.
104. Han, X.; Ling, X.; Yu, D.; et al. Atomically dispersed binary Co-Ni sites in nitrogen-doped hollow carbon nanocubes for reversible oxygen reduction and evolution. Adv. Mater. 2019, 31, e1905622.
105. Liu, W.; Li, H.; Ou, P.; et al. Isolated Cu-Sn diatomic sites for enhanced electroreduction of CO2 to CO. Nano. Res. 2023, 16, 8729-36.
106. Yue, Y.; Wang, B.; Jin, C.; et al. Tailoring Cu-Zn dual-atom sites with reordering d-orbital splitting manner for highly efficient acetylene semihydrogenation. ACS. Catal. 2024, 14, 3900-11.
107. Zhu, J.; Xiao, M.; Ren, D.; et al. Quasi-covalently coupled Ni-Cu atomic pair for synergistic electroreduction of CO2. J. Am. Chem. Soc. 2022, 144, 9661-71.
108. Wang, X.; Zhang, N.; Guo, S.; et al. p-d Orbital hybridization induced by asymmetrical FeSn dual atom sites promotes the oxygen reduction reaction. J. Am. Chem. Soc. 2024, 146, 21357-66.
109. Wang, D.; Xu, H.; Yang, P.; et al. Fe-N4 and Co-N4 dual sites for boosting oxygen electroreduction in Zn-air batteries. J. Mater. Chem. A. 2021, 9, 13678-87.
110. Yang, H.; Huang, H.; Wang, Q.; Shang, L.; Zhang, T.; Wang, S. Fe, Cu dual-metal single atom catalyst on commercial carbon black for efficient oxygen reduction reaction. J. Mater. Chem. A. 2023, 11, 6191-7.
111. Tamtaji, M.; Peng, Q.; Liu, T.; et al. Non-bonding interaction of dual atom catalysts for enhanced oxygen reduction reaction. Nano. Energy. 2023, 108, 108218.
112. Tong, M.; Sun, F.; Xie, Y.; et al. Operando cooperated catalytic mechanism of atomically dispersed Cu-N4 and Zn-N4 for promoting oxygen reduction reaction. Angew. Chem. Int. Ed. 2021, 60, 14005-12.
113. Meng, L.; Zhang, E.; Peng, H.; et al. Bi/Zn dual single-atom catalysts for electroreduction of CO2 to syngas. ChemCatChem 2022, 14, e202101801.
114. Chen, C.; Sun, M.; Zhang, F.; et al. Adjacent Fe Site boosts electrocatalytic oxygen evolution at Co site in single-atom-catalyst through a dual-metal-site design. Energy. Environ. Sci. 2023, 16, 1685-96.
115. Zhang, Y.; Johannessen, B.; Zhang, P.; Gong, J.; Ran, J.; Qiao, S. Z. Reversed electron transfer in dual single atom catalyst for boosted photoreduction of CO2. Adv. Mater. 2023, 35, e2306923.
116. Yang, G.; Zhu, J.; Yuan, P.; et al. Regulating Fe-spin state by atomically dispersed Mn-N in Fe-N-C catalysts with high oxygen reduction activity. Nat. Commun. 2021, 12, 1734.
117. Zhou, Y.; Yang, W.; Utetiwabo, W.; et al. Revealing of active sites and catalytic mechanism in N-coordinated Fe, Ni dual-doped carbon with superior acidic oxygen reduction than single-atom catalyst. J. Phys. Chem. Lett. 2020, 11, 1404-10.
118. Wang, Y.; Wang, Y.; Yan, Y.; et al. Tailoring the d-band center of Ni on a dual-single-atom Ni-ZnNC catalyst for efficient H2O2 production. Mater. Today. Energy. 2023, 38, 101459.
119. Cheng, H.; Wu, X.; Feng, M.; et al. Atomically dispersed Ni/Cu dual sites for boosting the CO2 reduction reaction. ACS. Catal. 2021, 11, 12673-81.
120. Wang, Y.; Yin, H.; Dong, F.; et al. N-coordinated Cu-Ni dual-single-atom catalyst for highly selective electrocatalytic reduction of nitrate to ammonia. Small 2023, 19, e2207695.
121. Qiao, J.; You, Y.; Kong, L.; et al. Precisely constructing orbital-coupled Fe-Co dual-atom sites for high-energy-efficiency Zn-Air/iodide hybrid batteries. Adv. Mater. 2024, 36, e2405533.
122. Chen, Y.; Mao, J.; Zhou, H.; et al. Coordination shell dependent activity of CuCo diatomic catalysts for oxygen reduction, oxygen evolution, and hydrogen evolution reaction. Adv. Funct. Mater. 2024, 34, 2311664.
123. Bi, Z.; Hu, J.; Xu, M.; Zhang, H.; Zhou, Y.; Hu, G. Nitrogen-bridged Fe-Cu atomic pair sites for efficient electrochemical ammonia production and electricity generation with Zn-NO2 batteries. Angew. Chem. Int. Ed. 2024, 63, e202313434.
124. Fan, Z.; Luo, R.; Zhang, Y.; et al. Oxygen-bridged indium-nickel atomic pair as dual-metal active sites enabling synergistic electrocatalytic CO2 reduction. Angew. Chem. Int. Ed. 2023, 62, e202216326.
125. Yang, Y.; Qian, Y.; Li, H.; et al. O-coordinated W-Mo dual-atom catalyst for pH-universal electrocatalytic hydrogen evolution. Sci. Adv. 2020, 6, eaba6586.
126. Mun, Y.; Lee, S.; Kim, K.; et al. Versatile strategy for tuning ORR activity of a single Fe-N4 site by controlling electron-withdrawing/donating properties of a carbon plane. J. Am. Chem. Soc. 2019, 141, 6254-62.
127. Zhou, X.; Ke, M. K.; Huang, G. X.; et al. Identification of fenton-like active Cu sites by heteroatom modulation of electronic density. Proc. Natl. Acad. Sci. USA. 2022, 119.
128. Yang, Q.; Cai, J.; Li, G.; et al. Chlorine bridge bond-enabled binuclear copper complex for electrocatalyzing lithium-sulfur reactions. Nat. Commun. 2024, 15, 3231.
129. Wang, J. M.; Zhu, Q. Y.; Lee, J. H.; et al. Asymmetric gradient orbital interaction of hetero-diatomic active sites for promoting C - C coupling. Nat. Commun. 2023, 14, 3808.
130. Zhai, S.; Sun, J.; Sun, L.; et al. Heteronuclear dual single-atom catalysts for ambient conversion of CO2 from air to formate. ACS. Catal. 2023, 13, 3915-24.
131. Xiao, M.; Zhu, J.; Li, S.; et al. 3d-Orbital occupancy regulated Ir-Co atomic pair toward superior bifunctional oxygen electrocatalysis. ACS. Catal. 2021, 11, 8837-46.
132. Zhang, L.; Dong, Y.; Li, L.; Wei, L.; Su, J.; Guo, L. Enhanced oxygen reduction activity and stability of double-layer nitrogen-doped carbon catalyst with abundant Fe-Co dual-atom sites. Nano. Energy. 2023, 117, 108854.
133. Shu, Z.; Chen, H.; Liu, X.; Jia, H.; Yan, H.; Cai, Y. High-throughput screening of heterogeneous transition metal dual-atom catalysts by synergistic effect for nitrate reduction to ammonia. Adv. Funct. Mater. 2023, 33, 2301493.
134. Yu, D.; Ma, Y.; Hu, F.; et al. Dual-sites coordination engineering of single atom catalysts for flexible metal-air batteries. Adv. Energy. Mater. 2021, 11, 2101242.
135. Song, C.; He, Q.; Zeng, Z.; et al. Isolated diatomic Zn-Co metal-nitrogen/oxygen sites with synergistic effect on fast catalytic kinetics of sulfur species in Li-S battery. J. Energy. Chem. 2023, 79, 505-14.
136. Zhang, Y.; Qiu, Y.; Fan, L.; et al. Dual-atoms iron sites boost the kinetics of reversible conversion of polysulfide for high-performance lithium-sulfur batteries. Energy. Storage. Mater. 2023, 63, 103026.
137. Zhang, S.; Zhang, Y.; Ma, L.; et al. Constructing orbital coupling-modulated homogeneous dual-atom Fe-Fe sites for boosting bidirectional conversion of polysulfides. ACS. Appl. Mater. Inter. 2024, 16, 33527-38.
138. Cao, X.; Zhao, L.; Wulan, B.; et al. Atomic bridging structure of nickel-nitrogen-carbon for highly efficient electrocatalytic reduction of CO2. Angew. Chem. Int. Ed. 2022, 61, e202113918.
139. Xu, W.; Wang, Y.; Zhang, C.; et al. Insights into the electronic structure coupling effect of dual-metal atomic electrocatalytic platform for efficient clean energy conversion. Chem. Eng. J. 2023, 461, 141911.
141. Hao, J.; Zhuang, Z.; Hao, J.; et al. Interatomic electronegativity offset dictates selectivity when catalyzing the CO2 reduction reaction. Adv. Energy. Mater. 2022, 12, 2200579.
142. Yao, D.; Tang, C.; Zhi, X.; et al. Inter-metal interaction with a threshold effect in NiCu dual-atom catalysts for CO2 electroreduction. Adv. Mater. 2023, 35, e2209386.
143. Zhang, L.; Feng, J.; Liu, S.; et al. Atomically dispersed Ni-Cu catalysts for pH-universal CO2 electroreduction. Adv. Mater. 2023, 35, e2209590.
144. Li, Z.; Zhu, Z.; Wang, J.; et al. Asymmetric coordination of heterogeneous Fe-Se dual-atom sites boosts CO2 electroreduction. Adv. Funct. Mater. . DOI: 10.1002/adfm.202410552.
145. Xie, W.; Li, H.; Cui, G.; et al. NiSn atomic pair on an integrated electrode for synergistic electrocatalytic CO2 reduction. Angew. Chem. Int. Ed. 2021, 60, 7382-8.
146. Hu, C.; Wang, Y.; Chen, J.; et al. Main-group metal single-atomic regulators in dual-metal catalysts for enhanced electrochemical CO2 reduction. Small 2022, 18, e2201391.
147. Wei, X.; Wei, S.; Cao, S.; et al. Cu acting as Fe activity promoter in dual-atom Cu/Fe-NC catalyst in CO2RR to C1 products. Appl. Surf. Sci. 2021, 564, 150423.
148. Xiao, Z.; Sun, P.; Qiao, Z.; et al. Atomically dispersed Fe-Cu dual-site catalysts synergistically boosting oxygen reduction for hydrogen fuel cells. Chem. Eng. J. 2022, 446, 137112.
149. Wang, J.; Zhong, H.; Yang, J.; et al. Tuning the atomic configuration environment of MnN4 sites by Co cooperation for efficient oxygen reduction. J. Energy. Chem. 2023, 82, 547-59.
150. Rong, C.; Shen, X.; Wang, Y.; et al. Electronic structure engineering of single-atom Ru sites via Co-N4 sites for bifunctional pH-universal water splitting. Adv. Mater. 2022, 34, e2110103.
151. Zeng, Y.; Zhao, J.; Wang, S.; et al. Unraveling the electronic structure and dynamics of the atomically dispersed iron sites in electrochemical CO2 reduction. J. Am. Chem. Soc. 2023, 145, 15600-10.
152. Yang, J.; Liu, W.; Xu, M.; et al. Dynamic behavior of single-atom catalysts in electrocatalysis: identification of Cu-N3 as an active site for the oxygen reduction reaction. J. Am. Chem. Soc. 2021, 143, 14530-9.
153. Xu, S.; Carter, E. A. Theoretical insights into heterogeneous photo.electrochemical CO2 reduction. Chem. Rev. 2019, 119, 6631-69.
154. Li, L.; Chang, X.; Lin, X.; Zhao, Z. J.; Gong, J. Theoretical insights into single-atom catalysts. Chem. Soc. Rev. 2020, 49, 8156-78.
155. Tian, D.; Denny, S. R.; Li, K.; Wang, H.; Kattel, S.; Chen, J. G. Density functional theory studies of transition metal carbides and nitrides as electrocatalysts. Chem. Soc. Rev. 2021, 50, 12338-76.
156. Li, Y.; Wei, B.; Zhu, M.; et al. Synergistic effect of atomically dispersed Ni-Zn pair sites for enhanced CO2 electroreduction. Adv. Mater. 2021, 33, e2102212.
157. Zeng, Z.; Gan, L. Y.; Bin, Y. H.; et al. Orbital coupling of hetero-diatomic nickel-iron site for bifunctional electrocatalysis of CO2 reduction and oxygen evolution. Nat. Commun. 2021, 12, 4088.
158. Guo, Z.; Zhu, H.; Yang, G.; et al. Synergistic engineering of heteronuclear Ni-Ag dual-atom catalysts for high-efficiency CO2 electroreduction with nearly 100% CO selectivity. Chem. Eng. J. 2023, 476, 146556.
159. Pei, J.; Yang, L.; Lin, J.; et al. Integrating host design and tailored electronic effects of yolk-shell Zn-Mn diatomic sites for efficient CO2 electroreduction. Angew. Chem. Int. Ed. 2024, 63, e202316123.
160. Yi, J. D.; Gao, X.; Zhou, H.; Chen, W.; Wu, Y. Design of Co-Cu diatomic site catalysts for high-efficiency synergistic CO2 electroreduction at industrial-level current density. Angew. Chem. Int. Ed. 2022, 61, e202212329.
161. Sun, H.; Tung, C. W.; Qiu, Y.; et al. Atomic metal-support interaction enables reconstruction-free dual-site electrocatalyst. J. Am. Chem. Soc. 2022, 144, 1174-86.
162. Yu, Z.; Si, C.; Lagrow, A. P.; et al. Iridium-iron diatomic active sites for efficient bifunctional oxygen electrocatalysis. ACS. Catal. 2022, 12, 9397-409.
163. Pei, Z.; Lu, X. F.; Zhang, H.; Li, Y.; Luan, D.; Lou, X. W. D. Highly efficient electrocatalytic oxygen evolution over atomically dispersed synergistic Ni/Co dual sites. Angew. Chem. Int. Ed. 2022, 61, e202207537.
164. Deng, D.; Qian, J.; Liu, X.; et al. Non-covalent interaction of atomically dispersed Cu and Zn pair sites for efficient oxygen reduction reaction. Adv. Funct. Mater. 2022, 32, 2203471.
165. Zhao, S.; Liu, M.; Qu, Z.; et al. Cascade synthesis of Fe-N2-Fe dual-atom catalysts for superior oxygen catalysis. Angew. Chem. Int. Ed. 2024, 63, e202408914.
166. Huang, S.; Qiao, Z.; Sun, P.; et al. The strain induced synergistic catalysis of FeN4 and MnN3 dual-site catalysts for oxygen reduction in proton-/anion-exchange membrane fuel cells. Appl. Catal. B:. Environ. 2022, 317, 121770.
Cite This Article
 , Jian Lin
, Jian LinHow to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].