


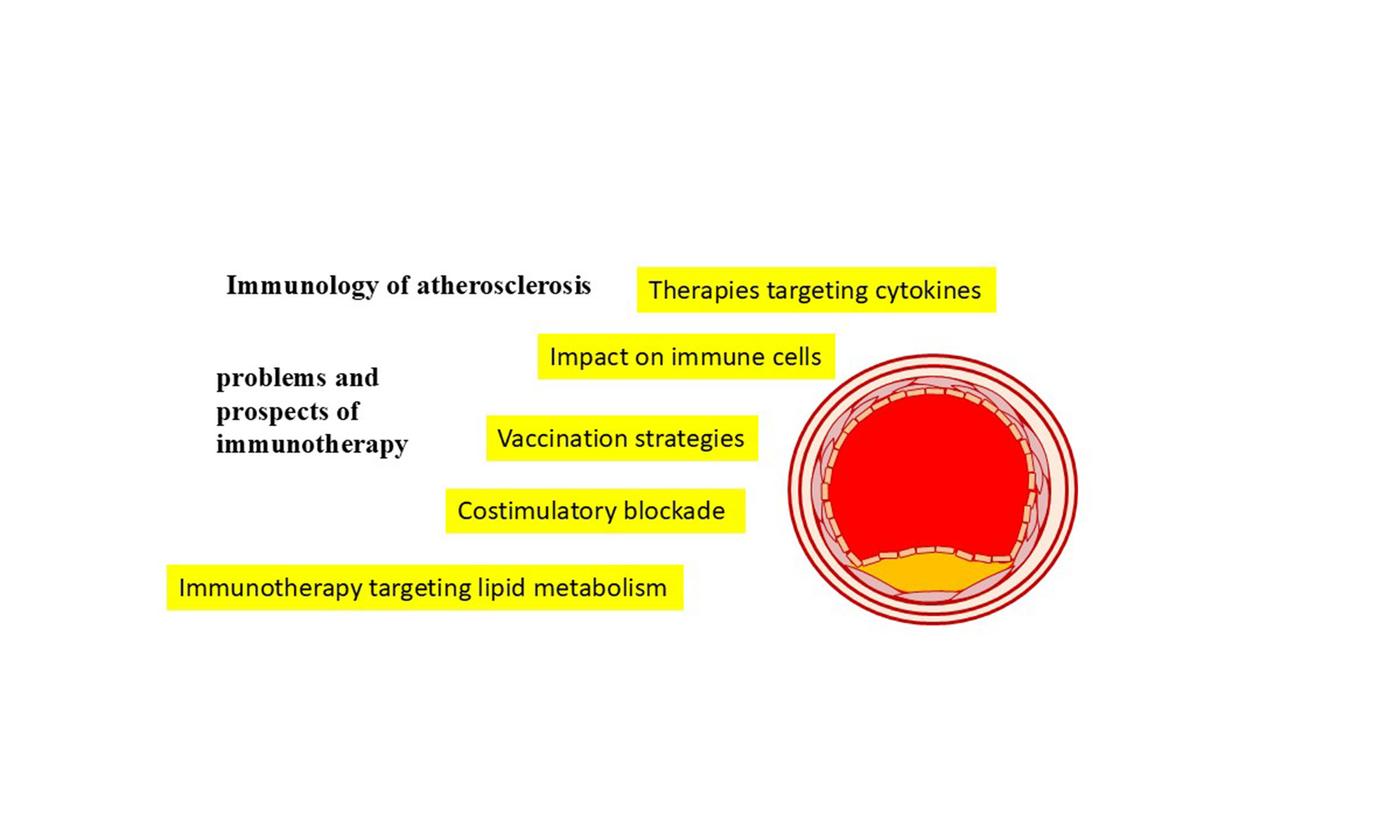
Immunology of atherosclerosis: problems and prospects of immunotherapy
 0
0 
Abstract
Atherosclerosis is one of the key problems of modern society. A growing body of evidence suggests that atherosclerosis is not only a metabolic but also an immune disease with local and systemic mechanisms. Dyslipidemia, local disturbances in vascular hemodynamics, and inflammation in the vascular wall involving various immune and non-immune cells such as endothelial and smooth muscle cells, are thought to be important for the development of atherosclerosis. In this context, there is a growing clinical and research interest in the possibility of therapeutically targeting different immune mechanisms. The aim of the current review is to discuss recent advances in understanding the immune mechanisms of atherogenesis and the challenges and prospects for immunotherapy of atherosclerosis.
Keywords
INTRODUCTION
Atherosclerosis is one of the key problems of modern society due to its high prevalence and social significance[1-3]. Indeed, atherosclerosis is the cause of such widespread diseases as ischemic heart disease and cerebral stroke, as well as peripheral arterial disease. These diseases contribute significantly to hospitalizations, disability, and premature mortality in many countries. In addition, they carry a heavy economic burden because of the direct costs of diagnosis and treatment, as well as the indirect costs associated with temporary and permanent disability. In this regard, atherosclerosis is an actively researched topic. A substantial body of landmark studies has significantly advanced the understanding of atherogenesis, directly contributing to enhanced diagnostic capabilities and therapeutic strategies. The contemporary paradigm of atherosclerosis reflects a complex evolution in scientific thought regarding its pathogenesis. Originally conceptualized as a disease driven chiefly by lipid (particularly cholesterol) infiltration of the vascular wall, atherosclerosis is now recognized as fundamentally an inflammatory disorder. Its initiation and progression involve the interplay of diverse immune and non-immune cell populations and the activation of distinct immune pathways[4,5]. Furthermore, it has become clear that the function of lipids is more complex than just being the morphological basis of atherosclerosis: lipids and lipid mediators are involved in many immune processes[6,7]. Lipid mediators are involved in the regulation of inflammation and are therefore of growing interest and considered as a potential therapeutic strategy. Despite the potential promise of immunotherapy, the key focus for the prevention and treatment of atherosclerosis has, for many years, been on blood lipid levels rather than immune mechanisms. Despite this, the key focus for the prevention and treatment of atherosclerosis has remained for many years on targeting blood lipid levels rather than immune mechanisms.
An important development in the immunotherapy of atherosclerosis is the recent inclusion of colchicine in the European Society of Cardiology clinical guidelines for the treatment of chronic coronary heart disease. Colchicine is a known anti-inflammatory agent that inhibits NLR family pyrin domain-containing 3 (NLRP3) activation and interleukin-1β (IL-1β) release. In addition, the use of monoclonal antibodies, another important area of immunotherapy, is increasing in clinical practice. In this regard, identification of new immune targets for pharmacotherapy of atherosclerosis is a promising area for research[8]. Thus, the aim of the current review is to discuss recent advances in understanding the immune mechanisms of atherogenesis, as well as the challenges and prospects of immunotherapy of atherosclerosis.
IMMUNOLOGY OF ATHEROSCLEROSIS AND PROMISING DIRECTIONS FOR CELL-TARGETED THERAPIES
Atherosclerosis arises from a complex cascade of events within the vascular wall, involving diverse cell types originating both from the artery itself and the bloodstream [Figure 1]. Research has established that its development results from a combination of systemic and local factors. Previously considered mainly a metabolic disease associated with lipid accumulation, atherosclerosis is now recognized as a chronic inflammatory disease in which innate immunity plays a leading role at all stages - from onset to progression and plaque rupture. Moreover, not only specialized immune cells such as macrophages or neutrophils, but also many other cells, including epithelial cells or endothelial cells, have an innate immune function, as the innate immune system utilizes universal non-selective mechanisms that are implemented upon initial contact with the pathogen. The mechanisms that innate immune cells utilize include pathogen uptake by phagocytosis, cytokine production, and production of antimicrobial peptides. Atherosclerosis is characterized by chronic non-infectious inflammation in the vascular wall, although there are a number of studies that have demonstrated the association of some pathogens with atherosclerosis[9,10].
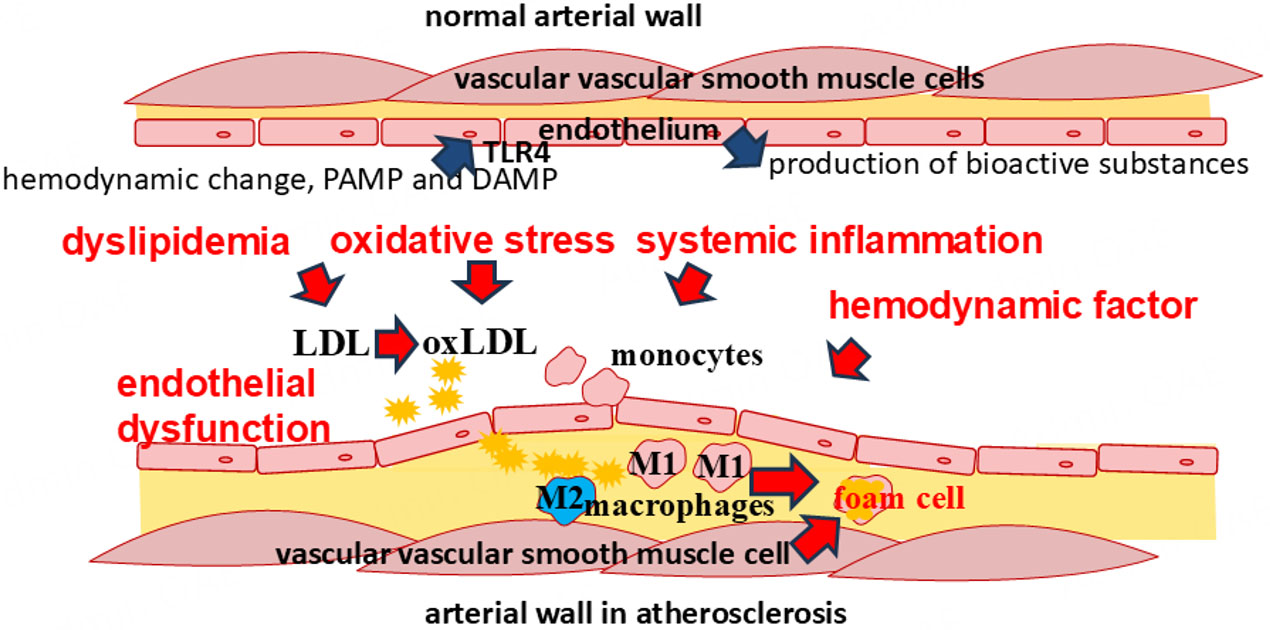
Figure 1. Mechanisms of atherogenesis. Atherosclerosis develops as a result of a chain of events including impaired vascular hemodynamics, endothelial dysfunction, dyslipidemia, systemic inflammation, and oxidative stress. These factors promote inflammation involving various cells in both the vascular wall and the peripheral blood flow.
The adaptive immune system plays a complex and dual role in atherosclerosis pathogenesis: it can promote plaque progression and instability (primarily through proinflammatory actions) or exert protective, stabilizing effects. Adaptive immunity is activated following antigen presentation by innate immune cells, chiefly dendritic cells, and contributes significantly to all phases of the disease[11,12].
Role of endothelial cells
Research results show that the localization of atherosclerosis is not random, but is associated with local hemodynamic disturbances. It is believed that under normal conditions, blood flow in arteries is laminar, with blood moving in uniform layers, the speed of which varies from the highest in the center of the vessel to the lowest at its wall. The physical force that acts on the vascular wall by this blood flow is called shear stress and is an important determinant of endothelial function. Arteries are characterized by high shear stress, reaching 20-40 dyne/cm2. On the other hand, arterial curvatures and bifurcations disrupt the normal laminar blood flow, promoting turbulence, which is characterized by low values of shear stress that are detected by endothelial cells. Areas with turbulent flow and low shear stress are more prone to atherosclerosis[13-16]. One reason is that endothelial cells can detect shear stress and respond by changing their polarization, which involves cell shape and function[17-19]. Endothelial cells have extensive functions in the regulation of hemodynamics through the production of various bioactive substances that can influence vascular tone. In addition, research in recent decades has greatly increased the understanding of endothelial cell functions in the immunology of the vascular wall. The vasoregulatory and immune functions of endothelial cells have been found to overlap closely. Endothelial cells are involved in the production of biologically active substances that fulfil many different functions. Nitric oxide, a known hemodynamically important agent, also acts as an important messenger in other processes and is a chemically active substance that performs a number of regulatory and immune functions. Disruption of nitric oxide production is characteristic of endothelial dysfunction, one of the important steps in the development of atherosclerosis[20-22]. In addition to nitric oxide, endothelial cells are involved in the production of lipid mediators, which participate in both hemodynamic and inflammatory regulation. Endothelial cells also perform functions of cytokine secretion, phagocytosis, antigen presentation, recognition of pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs), and generally perform proinflammatory and anti-inflammatory, immunostimulatory, and immunosuppressive functions[23,24]. To perform immune functions, endothelial cells express pattern recognition receptors such as Toll-like receptors (TLRs) [Figure 1][25,26]. The disruption of all these immune functions in endothelial dysfunction is part of the mechanisms of atherogenesis, many of which are still being researched.
The participation of endothelial cells in atherogenesis is also associated with the fact that they regulate the penetration of lipids and immune cells into the subendothelial space. It is believed that low-density lipoproteins (LDL) penetrate under the endothelium by transcytosis, which is facilitated by high levels of LDL in the bloodstream, as well as impaired polarization of endothelial cells in atherosclerosis[27,28]. In addition, endothelial cells promote the recruitment of monocytes from the bloodstream and their infiltration beneath the endothelium, where these cells participate in inflammation[29].
Given the extensive data on the role of the endothelium in atherogenesis, endothelial dysfunction is an important and promising target for treatment. Proinflammatory cytokine antagonists and immunosuppressive/anti-inflammatory cytokines (such as interleukin (IL)-10 and IL-37) are considered as promising agents for immunotherapy of endothelial dysfunction[30,31]. Micro ribonucleic acids (MicroRNAs) represent another promising area for research in the treatment of endothelial dysfunction[32,33]. For example, endothelial microRNA-125a/b-5p has been shown to downregulate the expression of endothelin-1, an important vasoactive substance that is also involved in the regulation of the production of bioactive lipid mediators of inflammation, in vascular endothelial cells[34-36]. It should be noted that microRNAs are also not a simple object for clinical application, as many microRNAs have several targets and a complete understanding of their function is still an object of research.
Thus, endothelial cells are important participants in the vascular immune system, and endothelial dysfunction is considered an initiating step in atherogenesis. Thus, endothelial cells are important participants in the vascular immune system, and endothelial dysfunction is considered the initial stage of atherogenesis. Participation in the production of bioactive lipid mediators of inflammation, mechanisms of immune cell recruitment, and their penetration through the endothelium represent interesting potential targets for therapy.
Involvement of macrophages in atherogenesis
Macrophages are a key participant in the body's innate immune defence and an important player in atherogenesis[9,37]. These cells in the vascular wall originate from two sources: residual tissue macrophages that maintain local populations and macrophages derived from circulating peripheral blood monocytes. Monocyte recruitment is an important part of atherogenesis[38-40]. Endothelial cells and macrophages already present in the vascular wall play an important role in this process. It is important to note that macrophages perform different roles in inflammation. They can be proinflammatory, producing proinflammatory cytokines and nitric oxide via inducible nitric oxide synthase (iNOS), or conversely, they can perform anti-inflammatory functions, producing ornithine and proline, which are necessary for tissue repair during inflammation. This is due to the polarization of macrophages, i.e., switching their metabolism and function. Several subtypes of macrophages are known, among which the most described are proinflammatory or M1 macrophages and anti-inflammatory M2 macrophages[41,42]. Proinflammatory macrophages may contribute to nitrosative stress, progression of atherosclerosis, and instability of atherosclerotic plaques.
Given the important role of macrophages, there is growing clinical interest in the potential ability to regulate their polarization. The dipeptidyl peptidase 4 (DPP-4) inhibitor linagliptin enhances the polarization of M2 macrophages, which helps to reduce the size of atherosclerotic lesions and slow their progression[43]. Blocking the KCa3.1 channel reduces the expression of proinflammatory genes and prevents macrophages from differentiating toward the M1 phenotype, thereby stabilizing plaques[44]. Glucagon-like peptide-1 (GLP-1) receptor agonist liraglutide attenuates already formed atherosclerosis in apolipoprotein E-deficient mice through regulation of immune cell phenotypes and proinflammatory mediators[45]. Effects on foam cells are also of interest, such as experiments using a foam cell vaccine. Immunization with this vaccine decreased the level of proinflammatory cytokines (interferon-γ (IFN-γ), monocyte chemoattractant protein-1 (MCP-1), IL-6) and increased transforming growth factor beta 1 (TGF-β1) levels and collagen deposition, contributing to lesion stability[46]. Studies to find therapeutic agents aimed at restoring efferocytosis due to decreased Mer tyrosine kinase (MerTK) expression in macrophages are of interest. Removal of apoptotic cells through efferocytosis is critical to prevent the progression of atherosclerosis, and impaired efferocytosis contributes to the worsening of atherosclerosis. Targeted delivery of MerTK protein using nanoparticles embedded in the cell membrane enhances efferocytosis and slows the development of atherosclerosis in ApoE-/- mice with diabetes[47]. However, this strategy is experimental and the prospects for its clinical application are unclear. Thus, despite the importance of macrophages in the pathogenesis of atherosclerosis, therapeutic strategies aimed at these cells are mainly experimental in nature, due to the difficulty of targeted drug delivery to macrophages in the vascular wall without disrupting their immune function in other tissues.
Lymphocyte involvement in atherogenesis
A growing body of evidence suggests the involvement of cells of the adaptive immune system in atherogenesis. The mechanisms of the adaptive immune system in atherosclerosis are predominantly associated with T cells, which are consistently present at all stages of atherogenesis in experimental studies. T lymphocytes play a significant role in the development and progression of atherosclerosis. They regulate the severity of the atherogenic proinflammatory response, influencing the stability of atherosclerotic lesions and the tendency to thrombus formation. They promote lesion progression and remodeling by increasing inflammation in atherosclerotic plaques[48,49].
Atherosclerotic lesions are dominated by CD4+ T cells, which include T helper (Th)1, Th2, Th17, and regulatory T cells (Treg). Th1 cells are pro-atherogenic, Th2 and Th17 cells can be both pro- and anti-atherogenic depending on the cytokines they secrete, and Treg cells have anti-inflammatory effects and may attenuate atherogenesis[11,50-52]. Th1, which secrete IFNγ, are the most common T cells found in atherosclerotic plaques[11]. Type 2 T cells secrete anti-inflammatory cytokines such as IL-10 and TGF-β. T cells are activated by antigen-presenting cells (APCs) through costimulatory and co-inhibitory signals. This activation is crucial for their role in atherogenesis.
Similarly, cytotoxic CD8+ T cells exert a proatherogenic effect by secreting interferon-gamma and stimulating monocytopoiesis[11]. CD8+ T cells may play a protective role in progressive atherosclerosis by limiting the accumulation of Th1 and macrophages, thereby contributing to a more stable disease course[53]. CD8+ T cell deficiency in advanced atherosclerosis resulted in less stable lesions, increased macrophage content, and increased necrotic core area compared with controls[53].
B lymphocytes play a multifaceted role in the development and progression of atherosclerosis. B1 cells, especially the B1a subtype, are generally considered atheroprotective. They produce natural immunoglobulin M (IgM) antibodies that help to eliminate modified LDL and apoptotic cells, reducing inflammation and plaque formation[54-57]. B1 cells also secrete the anti-inflammatory interleukin-10 (IL-10), which can suppress the proinflammatory responses of other immune cells[54,58]. The number of IL-10+ B cells is inversely proportional to the severity of atherosclerosis. The protective effect of IL-10+ B cells can be offset by elevated cholesterol levels[58].
B2 B cells are the most common subtype in vessel walls. They can activate proinflammatory T cells, contributing to the progression of atherosclerosis[56,57,59]. Deletion of B2 cells has been shown to reduce the manifestations of atherosclerosis in animal models, emphasizing their role in the development of the disease[59]. B cells produce antibodies, including those against oxidized low-density lipoprotein (oxLDL), which can have both protective and deleterious effects. Antibodies against oxLDL may promote the excretion of harmful lipids, but their exact role in the development of atherosclerosis is still under investigation[60,61].
It should be noted that B2 B cells are subdivided into follicular B cells and marginal zone B cells, which have different effects on the development of atherosclerotic plaques. Follicular B cells are thought to contribute to the development of atherosclerosis. They contribute to the disease by differentiating into plasma cells that produce IgG antibodies, which can accumulate in atherosclerotic lesions and increase inflammation and plaque formation[62,63]. On the other hand, marginal zone B cells protect against atherosclerosis. They are involved in the production of natural IgM antibodies, which play a protective role by acting in a Tfh-dependent manner and binding to oxLDL and apoptotic cells, promoting their elimination[63-65]. Marginal zone B cells control the response of follicular T helper cells to a high cholesterol diet[66].
It is known that IgM binds to oxidized LDL, oxidized phospholipids, inflammatory microvesicles, and apoptotic cells, thereby helping to reduce inflammation mainly by neutralizing molecular patterns associated with damage and stimulating complement-mediated opsonization and phagocytosis of apoptotic cells[67]. Thus, lymphocytes are attracting growing interest due to their multifaceted and complex role in atherogenesis and atheroprotection and are considered potential targets for therapy. Lymphocyte activation gene 3 (LAG3) is a promising target for blocking immune checkpoints. Monoclonal antibodies targeting LAG3 can modulate T cell activity and inflammation in mice, although their effect on plaque size and stability requires further study[68].
Lymphocyte-specific tyrosine kinase (Lck) plays an important role in T cell receptor (TCR) signaling, which is necessary for the development, activation, and function of T cells. Lck is involved in T cell activation, which may contribute to the inflammatory processes underlying atherosclerosis. In atherosclerosis, T cells can destabilize plaques through their effector functions, and abnormal TCR signaling can lead to T cell hyperactivity, exacerbating the disease[69,70]. Lck inhibitors are of interest for the treatment of atherosclerosis because they stabilize plaques, improve cholesterol metabolism, and modulate immune responses. The LCK inhibitor attenuates atherosclerosis in ApoE-/- mice by regulating T cell differentiation and reverse cholesterol transport[71]. The LCK inhibitor suppressed CD4+ T cell infiltration into plaques, secretion of proatherosclerotic cytokines INF-γ and TNF-α, synthesized mainly by Th1 cells, and activation of the PI3K/AKT/mTOR signaling pathway[72]. LCK inhibition also affects T cell differentiation, increasing the number of regulatory T cells (Treg) and decreasing the number of type 1 T helper cells (Th1), which leads to a decrease in the secretion of proatherogenic cytokines such as INF-γ and TNF-α[72].
Strategies aimed at improving the function of marginal zone B cells or increasing the production of atheroprotective IgM antibodies may be useful in the treatment of atherosclerosis. For example, TLR7 stimulation has been shown to increase the number of MZ B cells and IgM production, leading to a reduction in atherosclerotic changes in experimental models[73]. Monoclonal antibodies against BAFFR selectively deplete atherogenic B2 cells, slowing the progression of atherosclerosis but not affecting other B cell subtypes in hyperlipidemic ApoE-/- mice[74].
Preclinical studies in mice have shown that depletion of B2 cells with antibodies against CD20 can reduce the risk of atherosclerosis and complications of myocardial infarction without affecting protective IgM-producing B1 cells[75]. Rituximab is a monoclonal antibody against CD20. The efficacy and safety of rituximab have been confirmed in patients with myocardial infarction. Its effect on cardiac remodeling after myocardial infarction is currently being studied (NCT05211401)[76].
It is known that IgM, produced in response to the modification of biomolecules by reactive oxygen species, plays an atheroprotective role[77-79]. In this regard, B cell engineering, a technology that can be used to develop cell therapies, is of potential interest. This is a promising therapeutic strategy that could change the situation and increase the production of atheroprotective IgM[67]. B cells reprogrammed using CRISPR/Cas9 to produce pathogen-specific antibodies have been studied previously[80]. However, this strategy of modifying B cells with atheroprotective IgM has not yet been implemented. Thus, the effects on immune cells are of clinical interest and require further research.
PROBLEMS AND PROSPECTS OF OTHER DIRECTIONS FOR IMMUNOTHERAPY OF ATHEROSCLEROSIS
Given the complexity of the processes occurring in the vascular wall in atherosclerosis and the fact that they occur over many years, the treatment of atherosclerosis is a difficult task. Current research in the field of immunotherapy for atherosclerosis focuses on many areas related to the mechanisms of both innate and adaptive immunity [Figure 2].
Figure 2. Potential targets for immunotherapy of atherosclerosis.
Therapies targeting cytokines
Considering the success of anti-cytokine antibodies in a bunch of medical fields, cytokine-targeted therapy is one of the top priorities in treating atherosclerosis. This is especially true since the treatment of certain rheumatic diseases with these drugs has also led to positive results in patients with concomitant atherosclerotic cardiovascular diseases.
Interleukin-1β
Interleukin-1β (IL-1β) is a key proinflammatory cytokine that plays a central role in the initiation and progression of atherosclerosis. It contributes to endothelial dysfunction, inflammation, and plaque destabilization, making it a crucial therapeutic target[81,82]. IL-1β is mainly produced by macrophages, endothelial cells, and vascular smooth muscle cells (VSMCs) via activation of the NLRP3 inflammasome (e.g., by the action of oxidized LDL, cholesterol crystals, or metabolic stress)[82,83]. IL-1β has multiple effects at all stages of atherosclerosis[82,84,85]. IL-1β promotes endothelial expression of adhesion molecules (VCAM-1, ICAM-1), facilitating monocyte recruitment. It stimulates the release of chemokines (e.g., MCP-1), enhancing leukocyte infiltration into the arterial wall. It also activates macrophages and VSMCs to produce other cytokines (IL-6, TNF-α) and matrix metalloproteinases (MMPs) that promote plaque inflammation and loosen the fibrotic plug.
IL-1β promotes foam cell formation by enhancing macrophage uptake of oxidized LDL through regulation of scavenger receptors (e.g., CD36). In advanced stages of atherosclerosis, IL-1β is involved in plaque destabilization. IL-1β induces MMPs (e.g., MMP-9) that degrade collagen in the fibrous cap, increasing the risk of rupture. It also promotes apoptosis of VSMCs and endothelial cells, reducing plaque stability[81,83,86-89]. Given these and other data on the role of IL-1β in the pathogenesis of atherosclerosis, the therapeutic use of inhibition of this cytokine has been suggested. The CANTOS study used canakinumab, a monoclonal antibody targeting IL-1β. It was shown to reduce the incidence of recurrent cardiovascular events[90]. However, the increased risk of fatal infections due to immunosuppression offset the beneficial effects of canakinumab.
Anakinra is a recombinant IL-1 receptor antagonist (IL-1Ra). IL-1 blockade with anakinra significantly reduced the formation and progression of atherosclerotic plaques in the aortic arch in ApoE-/- mice fed an atherogenic diet. Furthermore, anakinra suppressed the expression of inflammatory biomarkers such as IL-6, MMP-9, and monocyte chemoattractant protein-1 (MCP-1) in human umbilical vein endothelial cells (HUVECs), rat aortic smooth muscle cells (RAOSMC), and 3T3-L1 adipocytes, suggesting that anakinra treatment may be a useful strategy to block inflammatory signals mediating the process of atherosclerosis and systemic inflammation. Anakinra induced a significant dose-dependent decrease in MMP-9 mRNA expression and also reduced the migration of rat aortic smooth muscle cells, which is a model of vascular remodeling[91]. Anakinra reduced plaque size in the aortic arch (by 30.6% and 25.2% at doses of 25 and
Gevokizumab is a monoclonal antibody targeting interleukin-1β (IL-1β). Studies in mouse models of myocardial infarction and atherosclerosis demonstrate that Gevokizumab preserves left ventricular function and reduces markers of atherosclerosis. Administration of gevokizumab reduced the size of myocardial infarction. Immediate and late administration of gevokizumab improves left ventricular remodelling associated with heart failure. Gevokizumab improves coronary vascular status in heart failure[92]. Additionally, single-dose Gevokizumab (1 mg/kg) enhances reendothelialization and reduces neointima formation in rats following carotid artery injury[93].
NLRP3 inflammasome inhibitors (e.g., MCC950, DFV890) suppress IL-1β production. MCC950, a potent and selective NLRP3 inflammasome inhibitor, has shown efficacy in reducing inflammation and atherosclerotic plaque formation in preclinical models. It inhibits the NLRP3/ASC/Caspase-1 axis, reducing macrophage pyroptosis and IL-1β and IL-18 production[94-96]. MCC950 has also been shown to prevent the development of atherosclerosis by inhibiting inflammation and pyroptosis in macrophages[95]. However, its clinical use was suspended due to hepatotoxicity, leading to the development of new-generation inhibitors[97]. In an ongoing Phase IIa clinical trial (ClinicalTrials.gov NCT06097663), the efficacy, safety, and tolerability of DFV890-another NLRP3 inhibitor-and MAS825, a bispecific monoclonal antibody targeting IL-1β/IL-18, are being assessed[98,99].
Alternative approaches to NLRP3 inhibition are being explored. Emodin, an anthraquinone derived from certain plants, was found to suppress atherosclerotic plaque development by reducing NLRP3/GSDMD-driven inflammation through inhibition of the TLR4/MyD88/NF-κB signaling pathway in macrophages[100]. Emodin was shown to attenuate atherosclerotic lesions in apoE-/- mice fed a high-fat diet[100]. Tranilast acts as a direct NLRP3 inhibitor, consequently suppressing NLRP3 inflammasome expression and activation within atherosclerotic lesions[101]. In experimental models, tranilast slowed atherosclerosis progression in low-density lipoprotein receptor-deficient and apolipoprotein E-deficient mice[102].
As noted, colchicine, which is involved in reducing IL-1β production, has recently been included in clinical guidelines for the management of chronic coronary syndromes[103]. Studies have shown that colchicine at low doses reduces the risk of cardiovascular and cerebrovascular events, as well as myocardial infarction, stroke and the need for coronary revascularization in a wide range of patients with coronary heart disease (CHD)[104]. The LoDoCo2 study investigated the use of low-dose colchicine to prevent cardiovascular events. The results of the study showed that low-dose colchicine significantly reduced the incidence of cardiovascular events, including myocardial infarction and stroke, compared to placebo[105,106]. In patients with recent myocardial infarction, colchicine at a dose of 0.5 mg daily significantly reduced the risk of ischemic cardiovascular events compared with placebo[107].
Thus, IL-1β remains a cornerstone of the inflammation hypothesis in atherosclerosis and is a promising target for therapy. It should be noted, however, that this cytokine is involved in many inflammatory diseases and is also part of the normal immune response to infections. In this regard, the therapeutic possibilities of long-term inhibition of IL-1β for the treatment of atherosclerosis are still limited.
Interleukin-6
Interleukin-6 (IL-6) plays a multifaceted role in the development of atherosclerosis, affecting both inflammatory and metabolic pathways. IL-6 is produced by various cells involved in the development of atherosclerosis, including smooth muscle and endothelial cells, monocytes, and T cells. IL-6 controls several aspects of vascular and hematopoietic cell function during the chronic inflammatory process that leads to atherogenesis[108]. IL-6 plays a key role in the inflammatory cascade by stimulating the production of acute phase proteins such as C-reactive protein (CRP) and fibrinogen, which are involved in the pathogenesis of atherosclerosis[109,110]. High plasma IL-6 levels are associated with an increased risk of adverse cardiovascular events, making it a biomarker of residual inflammatory risk in coronary heart disease. IL-6 affects various pathophysiological processes including endothelial dysfunction, lipid metabolism, and plaque stability[111,112]. IL-6 regulates extracellular matrix metabolism, increasing collagen degradation by activating MMPs, which can lead to plaque instability and rupture[113].
Some clinical trials have shown that lowering IL-6 levels can reduce cardiovascular events, suggesting that targeting IL-6 may be an effective strategy to reduce residual inflammatory risk in atherosclerosis[114,115]. IL-6 receptor (IL-6R) blockade is considered a promising treatment for various cardiovascular diseases including coronary heart disease and myocardial infarction[116].
Tocilizumab is a monoclonal antibody targeting the IL-6 receptor. Tocilizumab has shown promising results in improving endothelial function and reducing arterial stiffness in patients with rheumatoid arthritis, which may be useful in atherosclerosis[117,118]. However, it increases total cholesterol and LDL levels, which may have uncertain long-term effects[119].
Zevacizumab (ziltivekimab) is a human monoclonal narrow-spectrum antibody against IL-6 ligands. Inhibition of IL-6 with ziltivekimab in patients at high risk for atherosclerosis (RESCUE) reduced levels of biomarkers of inflammation and thrombosis associated with atherosclerosis[120]. The results of the RESCUE-2 study support the possibility of using ziltivekimab for secondary prevention and treatment of patients at high risk of atherosclerosis[121].
Tumor necrosis factor-alpha
Tumor necrosis factor-alpha (TNF-α) is a crucial proinflammatory cytokine that plays a significant role in the development and progression of atherosclerosis[122]. It promotes vascular inflammation, endothelial dysfunction, and macrophage foam cell formation, which are key steps in the development of atherosclerotic plaques[123-125]. Studies in TNF-α-deficient mice demonstrated a significant reduction in atherosclerotic lesion size, indicating TNF-α involvement in the formation and progression of atherosclerotic plaques[124,126]. TNF-α deficiency results in fewer advanced lesions and less intraplaque necrosis, suggesting its role in lesion maturation[126]. TNF-α induces endothelial cell apoptosis, thereby promoting endothelial dysfunction, a key early step in atherogenesis[127]. This dysfunction facilitates the adhesion and infiltration of inflammatory cells into the vascular wall[128]. TNF-α stimulates macrophages to produce more inflammatory mediators, amplifying the inflammatory response and promoting foam cell formation[129,130]. Foam cells play a key role in the development of fatty streaks, the initial stage of atherosclerosis[129]. TNF-α increases the expression of adhesion molecules such as intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) on endothelial cells, which are essential for attracting leukocytes to the focus of inflammation[128].
In this regard, TNF blockade with monoclonal antibodies is a promising therapeutic target. Patients with psoriatic arthritis taking TNF-α inhibitors were less likely to have plaques in carotid arteries compared with patients taking conventional anti-rheumatic drugs (15.8% and 40.4% of patients, respectively)[131]. In another study, patients with severe psoriasis taking biologic drugs (most patients (> 80%) received TNF-α inhibitors) had age- and sex-adjusted risk ratios (HR) of 0.28 (95%CI: 0.12-0.64) compared with other treatments in the control group[132].
Patients with rheumatoid arthritis taking TNF-α blockers had a lower risk of cardiovascular events [0.46 (95%CI: 0.25-0.85, P = 0.013)] compared with untreated patients[133]. In a study including 10,156 patients with rheumatoid arthritis, 4,684 of whom were receiving TNF-α inhibitors, the risk of the primary combined cardiovascular endpoint was lower (OR 0.39, 95%CI: 0.19 to 0.82) in those receiving TNF-α inhibitors compared with those taking non-biologic agents[134].
Soluble TNF receptors, including soluble TNFRII (sTNFRII), participate in modulating the inflammatory response[135]. sTNFRII can enhance inflammation by preventing the degradation and clearance of TNF-α, thereby prolonging its activity. Consequently, sTNFRII may exacerbate inflammatory responses and promote atherosclerosis development[136-138]. The peptide vaccine (AtheroVax™) targets a specific splicing variant of sTNFRII [sTNFRII(Δ7)], which is associated with prolonged TNFα activity and inflammation. By targeting this variant, the vaccine aims to reduce the inflammatory response without affecting the beneficial membrane-bound TNFRII[138]. In mouse models, the sTNFRII-targeting peptide vaccine demonstrated potential to reduce inflammation and stabilize atherosclerotic plaques, suggesting it may be an effective therapeutic strategy for treating atherosclerosis[138].
Thus, TNF-α represents a promising target for pharmacotherapy of atherosclerosis. It should be noted that TNF-α plays a multifaceted role in inflammation and its long-term inhibition should be evaluated in future studies.
Interleukin-2
Interleukin-2 (IL-2) is a proinflammatory cytokine that plays an important role in the development and progression of atherosclerosis. IL-2 is mainly produced by activated T cells and is crucial for the proliferation and activation of these cells[139]. In atherosclerosis, IL-2 promotes the activation of T helper cells (Th1), which produce other proinflammatory cytokines such as IFN-γ and tumor necrosis factor-β (TNF-β)[140]. This activation leads to an enhanced inflammatory response in atherosclerotic plaques[141]. Elevated serum IL-2 levels have been observed in patients with CHD. Moreover, higher IL-2 levels correlate with disease severity, indicating its role in disease progression[142,143]. Targeting IL-2 signalling pathways could be a potential therapeutic strategy for atherosclerosis. Modulation of IL-2 activity may help to reduce inflammation and slow disease progression[144,145]. IL-2 plays a crucial role in immune homeostasis by determining the balance between T effectors (Teffs) and regulatory T cells (Tregs)[146,147]. IL-2/anti-IL-2 monoclonal antibody complexes have been shown to increase the number of Treg and reduce the size of atherosclerotic lesions, which makes them a promising tool for immunomodulation in the treatment of atherosclerosis[144,145].
Low-dose IL-2 therapy is a novel approach in immunotherapy for the treatment of a wide range of autoimmune and inflammatory diseases[148]. It has been shown that low doses of IL-2 can selectively increase the number of anti-inflammatory regulatory T cells (Treg)[149]. The LILACS (Low Dose Interleukin-2 in Patients with Stable Coronary Heart Disease and Acute Coronary Syndromes) study primarily investigated the safety and biological efficacy of low-dose IL-2 in patients with CHD and acute coronary syndromes (ACS)[147,150]. The LILACS study established the safety of low-dose IL-2 and its biological efficacy in CHD. The IVORY study (NCT04241601) is designed to evaluate the effects of low-dose IL-2 on vascular inflammation in patients with ACS[147]. Its results have not yet been published. Thus, IL-2 is a promising target for immunotherapy of atherosclerosis and requires new studies.
Immunotherapy targeting lipid metabolism
Given the leading role of dyslipidemia in atherogenesis, various links in lipid metabolism are promising targets for treatment, such as mechanisms related to LDL metabolism. It is important to note that lipids are a chemically diverse group of substances and have various biological functions. Lipid mediators are involved in the activation, maintenance, and resolution of inflammation. The role of leukotrienes, thromboxane, and various prostaglandins in these processes is well known[151-154]. In recent years, there has been a significant increase in therapeutic interest in specialized pro-resolving mediators, which are derivatives of polyunsaturated fatty acids and include such classes of lipid mediators as Lipoxins, Resolvins (classes E and D), Maresins, and Protectins. They have powerful pro-resolving and anti-inflammatory effects, making them of great clinical interest in various inflammatory diseases, including atherosclerosis[155-157].
Immunotherapy targeting lipoprotein metabolism
PCSK9 inhibitors
PCSK9 inhibitors are a new class of drugs designed to lower LDL cholesterol. Proprotein convertase subtilisin/kexin type 9 (PCSK9) is an enzyme that disrupts LDL receptors (LDLR) in the liver, reducing the removal of LDL from the bloodstream[158-160]. PCSK9 has also been shown to be involved in the production of proinflammatory cytokines in macrophages, to regulate TLR4 expression and NF-κB activation, and to be involved in the regulation of apoptosis and autophagy[161]. PCSK9 inhibitors prevent LDL receptor degradation, thereby increasing the number of receptors available for LDL removal. This leads to a significant reduction in LDL levels[158-160].
PCSK9 inhibitors have been studied as adjunctive therapy to statins or as monotherapy in high-risk cardiovascular patients[159]. They have shown promising results in reducing the incidence of serious cardiovascular events, making them a potential therapy for high-risk patients, including those with familial hypercholesterolemia and those who cannot tolerate statins[159].
The PCSK9 inhibitors evolocumab and alirocumab are monoclonal antibodies to PCSK9. Evolocumab and alirocumab have been shown to reduce LDL-C levels by approximately 50%-60% when used as monotherapy or in combination with other lipid-lowering drugs[159,162]. Evolocumab significantly reduces LDL cholesterol levels. Studies have shown a reduction in levels of up to 58.5% when combined with statins[163,164]. This reduction in LDL levels is associated with a reduction in atheroma volume in the coronary arteries, indicating regression of atherosclerotic plaques[165-167]. In patients with peripheral arterial disease, everolocumab in combination with statins reduced arterial stenosis and claudication, improving overall patient outcomes[162,168]. In addition, PCSK9 inhibitors were found to significantly reduce lipoprotein (a) levels, which may be beneficial for patients with elevated lipoprotein (a) levels. In the FOURIER study, the use of Evolocumab resulted in a 59% reduction in LDL cholesterol levels, which corresponded to a 15%-20% reduction in the incidence of major cardiovascular events. In the SPIRE-2 study, there was a 21% reduction in the risk of the primary composite endpoint with the use of Bococizumab, another PCSK9 inhibitor, although the study was terminated early. In the ODYSSEY Outcomes study, LDL cholesterol levels decreased by 57% with Alirocumab. There was also a 15% reduction in the incidence of adverse events[159].
Combining monoclonal antibodies with other lipid-lowering drugs such as statins has shown increased efficacy in slowing the progression of atherosclerosis and improving plaque stability. Triple treatment with alirocumab, evinacumab, and atorvastatin reduced lesion size from baseline in the thoracic aorta by 50% and in the aortic orifice by 36% (both P < 0.05 compared with baseline), reduced macrophage accumulation by decreasing their proliferation, and reduced lesion severity[169]. In addition to regulating LDL metabolism, PCSK9 inhibitors have been shown to modulate inflammation. In patients taking PCSK9 inhibitors, carotid artery plaques contain less inflammatory proteins NLRP3 and caspase-1, as well as reduced levels of IL-1β, TNF-α, and NF-κB[170].
Thus, monoclonal antibodies against PCSK9 are a promising class of drugs for the treatment of atherosclerosis. It should be noted that in addition to antibodies, small interfering RNAs (siRNAs) targeting PCSK9 iRNAs (the drug Inclisiran) are also of interest as other PCSK9 inhibitors[160,171]. Antisense oligonucleotides (ASO) that reduce PCSK9 expression are also being studied[172,173]. However, a detailed description of this class of drugs is beyond the scope of the current review. Thus, PCSK9 inhibitors represent a significant breakthrough in lipid-lowering therapy by providing significant reductions in LDL cholesterol levels.
Cholesterol ester transfer protein
Cholesterol transport disorders play an important role in atherogenesis. In this regard, studies on the inhibition of cholesterol ester transfer protein (CETP), which transfers cholesterol from HDL to the atherogenic LDL fraction, are of interest[174]. Inhibition of CETP results in an increase in HDL cholesterol concentration and a decrease in LDL cholesterol concentration. Despite the potential benefit of such inhibition in rabbits, the ILLUMINATE clinical trial showed increased mortality with the CETP inhibitor torcetrapib[175], which may be related to the role of CETP in the innate immune system and involvement in LPS removal and the impairment of this function when CETP is inhibited[176]. The ILLUMINATE clinical trial was prematurely terminated due to other unexpected pharmacological effects of torcetrapib, which resulted in an increased risk of cardiovascular events and death[177].
Angiopoietin-like protein 3
Angiopoietin-like protein 3 (ANGPTL3) is a liver-derived protein that plays an important role in lipid metabolism by inhibiting lipoprotein lipase and endothelial lipase, resulting in increased plasma triglyceride and LDL cholesterol levels[178-180]. Studies have shown a positive correlation between plasma ANGPTL3 levels and carotid artery intima-media thickness (CA-IMT) and femoral artery intima-media thickness (FA-IMT) in healthy subjects, indicating that high ANGPTL3 levels are associated with increased arterial wall thickness, which is a marker of atherosclerosis[181]. ANGPTL3 is also involved in endothelial dysfunction and inflammation, which are key processes in the development of atherosclerosis[182].
ANGPTL3 also has pro-angiogenic properties that are important for recovery after myocardial infarction (MI) by stimulating endothelial progenitor cell (EPC) function and angiogenesis[183]. This dual role suggests that although inhibition of ANGPTL3 may reduce lipid levels and the risk of atherosclerosis, patients with CHD should be careful not to impair angiogenesis and recovery from myocardial infarction[183].
Targeting ANGPTL3 with monoclonal antibodies, antisense oligonucleotides or RNA interference techniques has shown promising results in reducing plasma triglyceride and LDL cholesterol levels, potentially offering a new strategy for the treatment of atherogenic dyslipidaemia[178,184,185]. Early clinical trials have demonstrated the safety and efficacy of these inhibitors in significantly lowering lipid levels[178,185].
Thus, ANGPTL3 plays a multifaceted role in lipid metabolism, inflammation, and endothelial function, making it an important factor in the development of atherosclerosis and its potential treatment. Although ANGPTL3 inhibition is a promising means of reducing atherogenic lipids, its pro-angiogenic properties require a balanced approach to therapeutic application, especially in patients with CHD[178,181,185].
Lectin-like oxidized low-density lipoprotein (LDL) receptor-1
LOX-1 is a transmembrane glycoprotein that serves as a receptor for oxidized LDL and is involved in endothelial dysfunction, monocyte adhesion, smooth muscle cell proliferation, foam cell formation, inflammation, and plaque instability[186,187]. LOX-1 is also involved in the activation of NF-κB, which induces the expression of IL-6, thereby promoting the progression of atherosclerosis[188]. LOX-1 binds and uptakes oxidized LDL, leading to endothelial dysfunction, smooth muscle cell proliferation, and foam cell formation[189-191]. LOX-1 is involved in various proatherogenic processes, including endothelial activation, monocyte adhesion, and apoptosis[190-192].
Exposure to LOX-1 is anticipated using a DNA vaccine. The vaccine is designed to produce an immune response against the LOX-1 protein, potentially preventing atherosclerosis[193]. LOX-1 vaccination may slow the development of atherosclerosis by reducing foam cell formation, reducing aortic wall thickness, and modulating inflammation[194]. The LOX-1 vaccine may potentially slow the progression of atherosclerosis by reducing NF-κB activation, increasing endothelial nitric-oxide synthase (eNOS) expression, and attenuating the development of atherosclerotic plaques in a rat study[194]. However, further studies are needed to assess the efficacy of the vaccine, its safety and potential for clinical use in the prevention of atherosclerosis.
Oxidized LDL
Oxidized LDL causes damage to vascular endothelial cells, leading to endothelial dysfunction, which is a critical early event in atherosclerosis[195-197]. Oxidized LDL is taken up by macrophages, which is one of the mechanisms of foam cell formation. The foam cells accumulate in arterial walls, contributing to plaque formation[195-198]. In addition, oxidized LDL stimulates the recruitment and proliferation of SMCs in the arterial intima, promoting plaque stability and growth[198,199].
Vaccines targeting oxidized LDL aim to modulate the immune system's response to these lipoproteins. These vaccines are designed to produce protective antibodies that promote the elimination of oxLDL and antigen-specific regulatory T cells that counteract the Th1 autoimmune response against oxLDL[200,201]. Studies have shown that immunization with oxLDL induces antibody formation (both IgG and IgM) and protects against the development of atherosclerosis[202,203]. Immunization with oxLDL has been shown to induce the production of protective antibodies and reduce the risk of atherosclerosis in animal models[204,205]. Specifically, Tregs activated through the induction of oral tolerance to oxidized LDL can counteract oxLDL-specific T cells, thereby reducing plaque formation[206,207].
Thus, vaccines targeting oxidized LDL represent a promising strategy to modulate the immune response in atherosclerosis. Although animal studies have shown a protective effect, further studies are needed to optimize vaccine formulations, ensure specificity, and evaluate long-term safety and efficacy in humans.
Therapies targeting inflammatory mediators
Specialized pro-resolving mediators
As already mentioned, polyunsaturated fatty acids (PUFAs) serve as substrates for the formation of various proinflammatory and anti-inflammatory lipid mediators. Specialized pro-resolving mediators (SPMs) are a group of substances derived from PUFAs that play an important role in resolving inflammation and restoring tissue homeostasis. These mediators are divided into several classes depending on their biosynthetic origin and functions[208].
Lipoxins (LXs) are a class of biologically active lipid mediators derived from arachidonic acid. Lipoxin A4 (LXA4) is known for its powerful anti-inflammatory, pro-resolving, and anti-atherogenic properties. LXA4 helps protect the endothelium and reduces leukocyte adhesion by inhibiting the expression of VCAM-1, ICAM-1, and E-selectin on the endothelium. In addition, lipoxins block leukocyte adhesion and their transmigration into the vascular intima, and also participate in the control of inflammation by promoting the transition of macrophages from a proinflammatory (M1) phenotype to an anti-inflammatory/pro-resolving (M2) phenotype[209-211]. In this regard, lipoxins are considered a potential target for the treatment of atherosclerosis[212-214]. However, they are chemically unstable, so work is underway to search for their synthetic analogs[215]. Since lipoxins act primarily through the ALX/FPR2 receptor, specific agonists of this receptor are being developed, which are not necessarily structural analogues of lipoxins.
E-series resolvins and D-series resolvins (RvD) are families of anti-inflammatory and anti-inflammatory lipid mediators derived from eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), respectively. Resolvin is a large group of substances. Resolvin E series includes four representatives (RvE1-RvE4) and their epimer analogues formed by aspirin. Among the resolvin D series, six representatives are known (RvD1-RvD6). Resolvin has numerous anti-inflammatory effects. They participate in the control of neutrophils, limiting chemotaxis, adhesion, and transendothelial migration of neutrophils to the site of inflammation, and also promote apoptosis of dying neutrophils[216-219]. Experimental studies suggest the ability of Resolvins and their synthetic analogs to reduce the adverse effects of ischemia and decrease the size of myocardial infarction[220]. Given these and other data, Resolvins and their synthetic analogues may be used as potentially useful agents in the treatment of atherosclerotic lesions[216,220,221].
Docosahexaenoic acid is a substrate for the formation of another class of SPMs, such as Maresins. Several representatives of this class are known: MaR1, MaR2, MaR1-d5, MaR2-d5, MCTR1, MCTR2, and MCTR3. Maresins are produced mainly by macrophages and are powerful factors that resolve inflammation with a wide range of biological actions. Maresins act through several different mechanisms and are interesting subjects for research[222-225]. However, to date, all studies on the use of Maresins for the treatment of atherosclerosis are at the preclinical stage. Omega-3 docosahexaenoic acid (DHA) is a substrate for the formation of Protectin, one of the classes of pro-resolving lipid mediators. Protectin D1 (PD1) is formed with the participation of various cells and plays an important role in actively stopping inflammation, protecting tissues, and restoring homeostasis. Due to their unique ability to actively eliminate inflammation, protect vascular wall cells, and stabilize atherosclerotic plaques, Protectins are promising candidates for a new class of therapeutic agents against atherosclerosis[226-228]. Despite the promising results of preclinical studies of SPM analogues, these drugs are not yet used in the treatment of atherosclerosis due to the complexity of their biosynthesis and the difficulty of targeted delivery to the site of atherosclerotic lesions.
Vaccination strategies
In this context, numerous experimental studies have contributed to the understanding of vaccine development by exploring different routes of exposure. Key strategies include passive immunization using natural immunoglobulin G (IgG) antibodies and active immunization targeting LDL cholesterol and apolipoproteins such as apolipoprotein B100 (ApoB100) and apolipoprotein CIII (ApoCIII). Other approaches include vaccines targeting proteins that regulate lipoprotein metabolism, including PCSK9, CETP, and ANGPTL3[76]. Two AFFITOPE® peptide vaccines have been proposed for the treatment of hypercholesterolemia by producing PCSK9-specific antibodies, of which AT04A has demonstrated significant activity in lowering LDL levels[229]. Another experimental trend involves vaccination using dendritic cells stimulated with oxLDL. Transfer of oxLDL-induced mature dendritic cells (mDCs) to LDL receptor-deficient mice resulted in a significant reduction in the size of atherosclerotic plaques and an increase in their stability. In addition, vaccination led to the induction of oxLDL-specific T cells with a reduced Th1 profile and increased levels of oxLDL-specific IgG, which contributed to a reduction in foam cell formation[202]. Immunotherapeutic strategies based on dendritic cells, including vaccines and innovative combinations with nanotechnology-based drug delivery platforms, are promising for the treatment of atherosclerosis[230]. For example, IL-37-treated dendritic cells have been shown to be a novel therapeutic agent for the treatment of acute coronary syndrome (ACS)[231].
Studies of passive immunization in atherosclerosis
Passive immunization involves the administration of ready-made antibodies to provide immediate protection or therapeutic effect. Studies have shown that recombinant human IgG1 antibodies against specific epitopes of oxLDL can significantly reduce atherosclerotic lesions and inflammation in mice[232,233].
The GLACIER study investigated immunotherapy using injectable immunoglobulins. The aim of the study was to evaluate the efficacy of an injectable human recombinant monoclonal IgG1 antibody (MLDL1278A) against oxidized LDL-C (ApoB epitope) in patients with stable plaques in the carotid artery or aorta[234]. It was assumed that this antibody could attenuate inflammation in atherosclerosis by forming immune complexes with oxLDL and thereby prevent the activation of inflammatory macrophages in plaques. However, the study showed a lack of efficacy, which could be due to several methodological limitations[76]. Passive immunization with natural IgM antibodies specific for hypochlorite-oxLDL (HOCl-oxLDL) has been shown to reduce plaque volume and cholesterol levels in LDL receptor-deficient mice[235]. Passive immunization with natural IgG antibodies to ox-LDL-C shows promising results in the control of atherosclerosis by targeting specific epitopes of ox-LDL. IgG and IgM antibodies have shown potential in preclinical studies, which emphasizes the therapeutic potential of immunomodulation in cardiovascular disease[236].
Phosphorylcholine is one of the major epitopes of oxLDL, playing a central role in the development of atherosclerosis due to its atherogenic and proinflammatory properties. ATH3G10 is a fully human IgG1 monoclonal antibody designed to target phosphorylcholine[237,238]. ATH3G10 treatment has been shown to reduce vascular inflammation by reducing the expression of endoplasmic reticulum stress markers and CCL2 production, which play a key role in the inflammatory process in atherosclerosis[238]. The antibody inhibits the uptake of oxidized LDL by macrophages, thereby reducing foam cell formation and subsequent plaque development[238]. Thus, this direction of immunotherapy still needs further experimental and clinical studies.
Vaccines targeting apolipoprotein B-100
Apo-B-100 is a major LDL protein that plays an important role in the development of atherosclerosis. Vaccines targeting ApoB-100 aim to modulate the immune response, thereby reducing inflammation and atherosclerotic plaque formation. This is achieved through the induction of regulatory T cells (Treg) and specific antibodies that attenuate the autoimmune response[239-241]. Immunization with ApoB-100 peptides has been shown to significantly reduce the risk of atherosclerotic plaque development in various animal models. For example, vaccination with p210-CTB resulted in a 35% reduction in aortic lesion size, while other studies have reported a 66% reduction[239,240]. These vaccines not only reduce plaque size, but also modulate the immune system by increasing the production of protective antibodies and regulatory T cells that help inhibit proinflammatory responses associated with atherosclerosis[239-242]. Despite promising preclinical results, translating these discoveries into safe and effective therapies for humans remains a challenge. Issues such as long-term safety, optimal dosing, and potential side effects need to be addressed[76,243].
Costimulatory blockade in the treatment of atherosclerosis
The development of an immune response begins with the recognition, via the T cell receptor (TCR), of an antigenic peptide within the major histocompatibility complex (MHC). However, antigen recognition is necessary but not sufficient for T lymphocyte activation. The involvement of costimulatory molecules is necessary for cell activation. The best known costimulatory system is signal transduction via the CD28 molecule on the T lymphocyte surface as a result of its interaction with ligands B7-1 (CD80) and B7-2 (CD86) on the surface of the antigen-presenting cell[244].
Costimulatory molecules play an important role in the pathogenesis of atherosclerosis by modulating immune responses. These molecules, members of the tumor necrosis factor receptor (TNFR) and CD28 immunoglobulin superfamily, influence T and B cell responses and the behavior of antigen-presenting and non-immune cells. Inhibition of the CD30-CD30L pathway reduces atherosclerotic plaque formation by modulating T cell responses. In particular, treatment of mice with antibodies against CD30L resulted in a 35% reduction in atherosclerotic plaques and a significant decrease in the number of T cells[245]. CD137 and its ligand CD137L are involved in atherogenesis. CD137 activation increases inflammation, T cell infiltration, and expression of proinflammatory cytokines, contributing to the progression of atherosclerosis[246,247].
The CD40-CD40L dyad plays a key role in the inflammatory response in atherosclerosis. It regulates the interaction between immune cells, as well as between immune and non-immune cells, which makes it a potential therapeutic target[248]. Disruption of CD40-CD40L interaction can reduce the manifestations of atherosclerosis but may lead to immune suppression. However, selective inhibition with TRAF-STOPs, which block the CD40-TRAF6 interaction while preserving other CD40-mediated immune functions, has shown promising results in reducing the manifestations of atherosclerosis without compromising overall immunity[249].
Increased expression of CTLA-4 (cytotoxic T lymphocyte-associated protein 4; CD152), a negative regulatory molecule, reduces atherosclerotic plaque formation and decreases the accumulation of macrophages and CD4+ T cells in plaques. This suggests that CTLA-4 suppresses proatherogenic immune responses[250].
The costimulatory molecule B7-1 has been identified as an important factor affecting atherosclerotic inflammation. Inhibition of B7-1 with RhuDex® has been shown to reduce T cell activation and inflammation in atherosclerotic plaques[251].
TRAF-STOPs are small-molecule inhibitors that have demonstrated efficacy against atherosclerosis by acting on macrophages and reducing the influx of inflammatory monocytes. This approach has been shown to halt plaque progression and promote plaque stability in mouse models[249].
Costimulation blockade offers a novel and targeted approach to treat atherosclerosis by modulating immune responses. Despite promising results, this method requires careful study of potential side effects and a balanced approach to immunomodulation. Future studies and clinical trials will be crucial to optimizing these therapies for safe and effective use in patients with atherosclerosis.
CONCLUSION
The data accumulated to date suggest that atherosclerosis is the result of a disruption in the complex relationships between metabolic and immune mechanisms. Cells of the innate and adaptive immune systems are involved in the development of atherosclerosis. In this regard, a promising strategy is to target the immune mechanisms underlying atherosclerosis. Currently, many strategies for immunotherapy of atherosclerosis are being developed, targeting cellular and humoral mechanisms. The genetic basis of these disorders is of interest, as it is important for the diagnosis of risk groups[252,253]. In addition, there is growing interest in the regulation of epigenetic mechanisms using microRNAs[254-256].
Despite significant progress in the study of the immunology of atherosclerosis, the therapeutic potential of this knowledge remains underutilized [Figure 3]. Currently, only a few groups of drugs are used in the immunotherapy of atherosclerosis. This is due to the fact that inflammation in the vascular wall in atherosclerosis is low-intensity and lasts for many years. In addition, atherogenic immune mechanisms are most often universal for inflammation regardless of its location, as a result of which blocking inflammation in the vascular wall leads to similar disturbances of inflammation in other organs, which can lead to a disruption of normal immunity and response to infection. Many promising anti-inflammatory drugs, although effective in preclinical models, have failed to demonstrate safety in clinical settings because they disrupt the normal immune response, leading to the risk of infectious complications such as pneumonia. In this regard, new methods of delivering anti-inflammatory agents to the site of inflammation are being developed that do not disrupt the normal protective mechanisms of the body as a whole. It is possible that new methods of delivering these agents will be able to solve their safety issues.
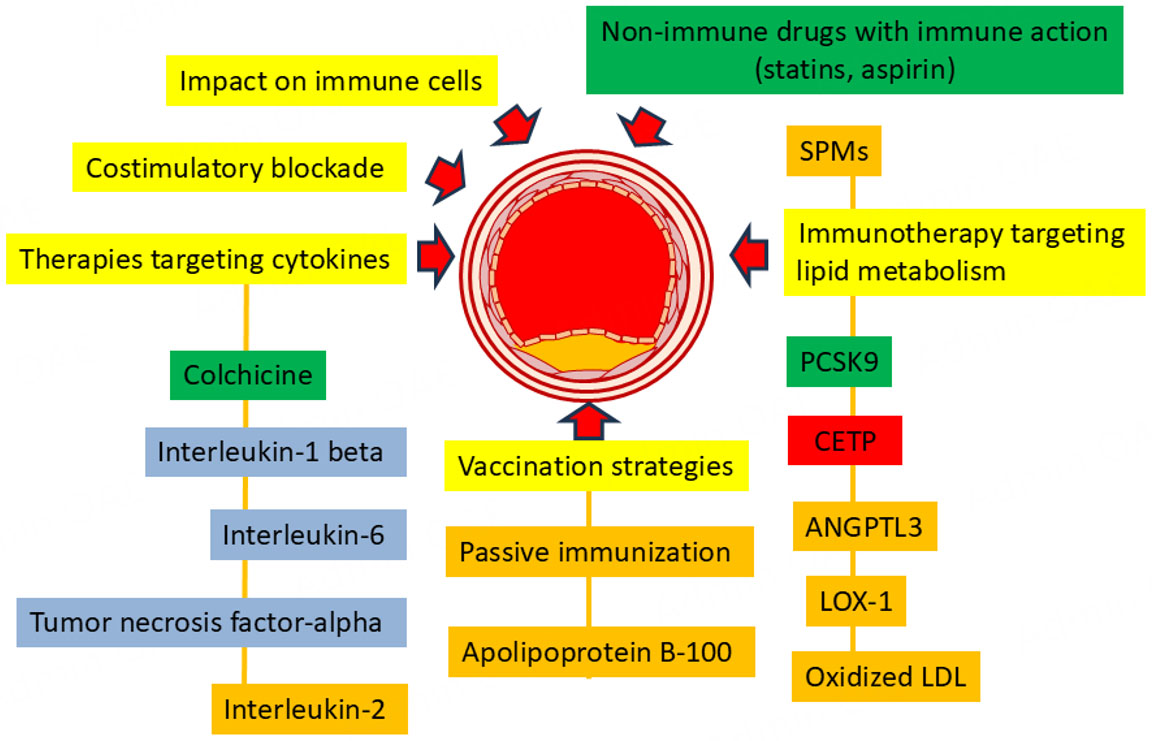
Figure 3. Schematic representation of immunotherapy approaches to atherosclerosis. green indicates approaches currently used to treat atherosclerosis, blue indicates treatments for other diseases that have a positive effect on atherosclerosis, promising immunotherapy approaches that have demonstrated efficacy in preclinical studies are marked in orange, and approaches that have not demonstrated safe and effective action are marked in red.
On the other hand, there is growing evidence that some drugs whose main action is not related to immunity also have an immunomodulatory effect. Aspirin, a drug widely used in cardiology, has the important ability to acetylate platelet cyclooxygenase (COX)-1, blocking the formation of thromboxane A2 (TXA2) and platelet aggregation, but it can also acetylate COX-2 in endothelial cells, leukocytes, and other inflammatory cells. Acetylated COX-2 acquires a new enzymatic activity - it converts arachidonic acid not into PGG2, but into 15(R)-hydroxyeicosatetraenoic acid (15 (R)-HETE) with the subsequent formation of 15-epi-lipoxin A4 (known as aspirin-triggered (AT) lipoxin A4 (ATL)). Aspirin also promotes the formation of the “aspirin-triggered” 18(R)-form of Resolvins E from eicosapentaenoic acid and D-series Resolvins from docosahexaenoic acid. Aspirin-triggered specialized pro-resolving mediators (AT-SPMs) act like other SPMs by inhibiting NF-κB activation, the production of proinflammatory cytokines (TNF-α, IL-1β, IL-6), chemokines, and the expression of adhesive molecules[157,257-259]. Thus, aspirin is an important anti-inflammatory drug that modulates the activity of inflammation in the vascular wall.
Statins, a widely used group of drugs that have become a reliable part of the therapeutic arsenal for treating atherosclerosis, have a diverse effect on the immune system. By regulating cholesterol biosynthesis, statins influence cellular cholesterol levels, which are critical for macrophage function. Cholesterol is an essential component of the plasma membrane, contributing to its biophysical properties and serving as a platform for transmembrane proteins that perform important transport and receptor functions. The cholesterol content of plasma membranes determines the stability of lipid rafts, which serve as platforms for the placement of membrane proteins[260]. The TLR4 receptor, which is actively involved in the mechanisms of the innate immune system in macrophages, is localized in lipid rafts, and its activity depends on the cholesterol content in them[261]. In addition, statins have been shown to prevent NLRP3 inflammasome activation through various mechanisms[262-264]. Another anti-inflammatory mechanism of statins is their involvement in the production of specialized pro-resolving mediators. Statins induce the same metabolic switching of COX-2 as aspirin, which promotes the production of epimeric forms of lipoxins[265-267]. In this regard, the use of statins in the treatment of atherosclerosis has several goals, including the modulation of immune mechanisms. Thus, immunotherapy of atherosclerosis represents a promising strategy. In this regard, the study of immune mechanisms involved in atherogenesis represents diagnostic and therapeutic prospects.
DECLARATIONS
Authors’ contributions
The author contributed solely to the article.
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
The author declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
1. Ceasovschih A, Mantzouranis E, Dimitriadis K, et al. Coronary artery thromboembolism as a cause of myocardial infarction with non-obstructive coronary arteries (MINOCA). Hellenic J Cardiol. 2024;79:70-83.
2. Nedkoff L, Briffa T, Zemedikun D, Herrington S, Wright FL. Global trends in atherosclerotic cardiovascular disease. Clin Ther. 2023;45:1087-91.
3. Chen W, Li Z, Zhao Y, Chen Y, Huang R. Global and national burden of atherosclerosis from 1990 to 2019: trend analysis based on the Global Burden of Disease Study 2019. Chin Med J. 2023;136:2442-50.
4. Roy P, Orecchioni M, Ley K. How the immune system shapes atherosclerosis: roles of innate and adaptive immunity. Nat Rev Immunol. 2022;22:251-65.
6. Fredman G, Hellmann J, Proto JD, et al. An imbalance between specialized pro-resolving lipid mediators and pro-inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat Commun. 2016;7:12859.
7. Lubrano V, Ndreu R, Balzan S. Classes of lipid mediators and their effects on vascular inflammation in atherosclerosis. Int J Mol Sci. 2023;24:1637.
8. Kong P, Cui ZY, Huang XF, Zhang DD, Guo RJ, Han M. Inflammation and atherosclerosis: signaling pathways and therapeutic intervention. Signal Transduct Target Ther. 2022;7:131.
9. Hou P, Fang J, Liu Z, et al. Macrophage polarization and metabolism in atherosclerosis. Cell Death Dis. 2023;14:691.
10. Gusev E, Sarapultsev A. Atherosclerosis and inflammation: insights from the theory of general pathological processes. Int J Mol Sci. 2023;24:7910.
11. Snijckers RPM, Foks AC. Adaptive immunity and atherosclerosis: aging at its crossroads. Front Immunol. 2024;15:1350471.
12. Ley K. Role of the adaptive immune system in atherosclerosis. Biochem Soc Trans. 2020;48:2273-81.
13. Yoshizumi M, Abe JI, Tsuchiya K, Berk BC, Tamaki T. Stress and vascular responses: atheroprotective effect of laminar fluid shear stress in endothelial cells: possible role of mitogen-activated protein kinases. J Pharmacol Sci. 2003;91:172-6.
14. Traub O, Berk BC. Laminar shear stress: mechanisms by which endothelial cells transduce an atheroprotective force. Arterioscler Thromb Vasc Biol. 1998;18:677-85.
15. Meng Q, Pu L, Qi M, et al. Laminar shear stress inhibits inflammation by activating autophagy in human aortic endothelial cells through HMGB1 nuclear translocation. Commun Biol. 2022;5:425.
16. Nigro P, Abe J, Berk BC. Flow shear stress and atherosclerosis: a matter of site specificity. Antioxid Redox Signal. 2011;15:1405-14.
17. Roux E, Bougaran P, Dufourcq P, Couffinhal T. Fluid shear stress sensing by the endothelial layer. Front Physiol. 2020;11:861.
18. Annink ME, Kraaijenhof JM, Stroes ESG, Kroon J. Moving from lipids to leukocytes: inflammation and immune cells in atherosclerosis. Front Cell Dev Biol. 2024;12:1446758.
19. Wojciak-Stothard B, Ridley AJ. Shear stress-induced endothelial cell polarization is mediated by Rho and Rac but not Cdc42 or PI 3-kinases. J Cell Biol. 2003;161:429-39.
20. Janaszak-Jasiecka A, Płoska A, Wierońska JM, Dobrucki LW, Kalinowski L. Endothelial dysfunction due to eNOS uncoupling: molecular mechanisms as potential therapeutic targets. Cell Mol Biol Lett. 2023;28:21.
21. Tran N, Garcia T, Aniqa M, Ali S, Ally A, Nauli S. Endothelial nitric oxide synthase (eNOS) and the cardiovascular system: in physiology and in disease states. Am J Biomed Sci Res. 2022;15:153-77.
22. Cyr AR, Huckaby LV, Shiva SS, Zuckerbraun BS. Nitric oxide and endothelial dysfunction. Crit Care Clin. 2020;36:307-21.
23. Shao Y, Saredy J, Yang WY, et al. Vascular endothelial cells and innate immunity. Arterioscler Thromb Vasc Biol. 2020;40:e138-52.
24. Mai J, Virtue A, Shen J, Wang H, Yang XF. An evolving new paradigm: endothelial cells-conditional innate immune cells. J Hematol Oncol. 2013;6:61.
25. Salvador B, Arranz A, Francisco S, et al. Modulation of endothelial function by Toll like receptors. Pharmacol Res. 2016;108:46-56.
26. Deravi N, Poudineh M, Pirzadeh M, et al. The Yin and Yang of toll-like receptors in endothelial dysfunction. Int Immunopharmacol. 2022;108:108768.
27. Bolanle IO, de Liedekerke Beaufort GC, Weinberg PD. Transcytosis of LDL across arterial endothelium: mechanisms and therapeutic targets. Arterioscler Thromb Vasc Biol. 2025;45:468-80.
28. Zhang X, Fernández-Hernando C. Transport of LDLs into the arterial wall: impact in atherosclerosis. Curr Opin Lipidol. 2020;31:279-85.
29. Mestas J, Ley K. Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends Cardiovasc Med. 2008;18:228-32.
30. Giachini FR, Zemse SM, Carneiro FS, et al. Interleukin-10 attenuates vascular responses to endothelin-1 via effects on ERK1/2-dependent pathway. Am J Physiol Heart Circ Physiol. 2009;296:H489-96.
31. Shao Y, Cheng Z, Li X, Chernaya V, Wang H, Yang XF. Immunosuppressive/anti-inflammatory cytokines directly and indirectly inhibit endothelial dysfunction--a novel mechanism for maintaining vascular function. J Hematol Oncol. 2014;7:80.
32. Desantis V, Potenza MA, Sgarra L, et al. microRNAs as biomarkers of endothelial dysfunction and therapeutic target in the pathogenesis of atrial fibrillation. Int J Mol Sci. 2023;24:5307.
33. Nemecz M, Alexandru N, Tanko G, Georgescu A. Role of MicroRNA in endothelial dysfunction and hypertension. Curr Hypertens Rep. 2016;18:87.
34. Bickford JS, Ali NF, Nick JA, et al. Endothelin-1-mediated vasoconstriction alters cerebral gene expression in iron homeostasis and eicosanoid metabolism. Brain Res. 2014;1588:25-36.
35. Li D, Yang P, Xiong Q, et al. MicroRNA-125a/b-5p inhibits endothelin-1 expression in vascular endothelial cells. J Hypertens. 2010;28:1646-54.
36. Rae GA, Trybulec M, de Nucci G, Vane JR. Endothelin-1 releases eicosanoids from rabbit isolated perfused kidney and spleen. J Cardiovasc Pharmacol. 1989;13 Suppl 5:S89-92.
37. Bobryshev YV, Ivanova EA, Chistiakov DA, Nikiforov NG, Orekhov AN. Macrophages and their role in atherosclerosis: pathophysiology and transcriptome analysis. Biomed Res Int. 2016;2016:9582430.
38. Chen S, Saeed AFUH, Liu Q, et al. Macrophages in immunoregulation and therapeutics. Signal Transduct Target Ther. 2023;8:207.
39. Strizova Z, Benesova I, Bartolini R, et al. M1/M2 macrophages and their overlaps - myth or reality? Clin Sci. 2023;137:1067-93.
40. den Bossche J, O'Neill LA, Menon D. Macrophage immunometabolism: where are we (going)? Trends Immunol. 2017;38:395-406.
41. Liu Y, Xu R, Gu H, et al. Metabolic reprogramming in macrophage responses. Biomark Res. 2021;9:1.
42. Liu N, Zhang B, Sun Y, Song W, Guo S. Macrophage origin, phenotypic diversity, and modulatory signaling pathways in the atherosclerotic plaque microenvironment. Vessel Plus. 2021;5:43.
43. Nishida S, Matsumura T, Senokuchi T, et al. Inhibition of inflammation-mediated DPP-4 expression by linagliptin increases M2 macrophages in atherosclerotic lesions. Biochem Biophys Res Commun. 2020;524:8-15.
44. Xu R, Li C, Wu Y, et al. Role of KCa3.1 channels in macrophage polarization and its relevance in atherosclerotic plaque instability. Arterioscler Thromb Vasc Biol. 2017;37:226-36.
45. Bruen R, Curley S, Kajani S, et al. Liraglutide attenuates preestablished atherosclerosis in apolipoprotein E-deficient mice via regulation of immune cell phenotypes and proinflammatory mediators. J Pharmacol Exp Ther. 2019;370:447-58.
46. Wang F, Zhang Z, Fang A, et al. Macrophage foam cell-targeting immunization attenuates atherosclerosis. Front Immunol. 2018;9:3127.
47. Qiu S, Liu J, Chen J, et al. Targeted delivery of MerTK protein via cell membrane engineered nanoparticle enhances efferocytosis and attenuates atherosclerosis in diabetic ApoE-/- mice. J Nanobiotechnol. 2024;22:178.
48. Sun L, Zhang W, Zhao Y, et al. Dendritic cells and T cells, partners in atherogenesis and the translating road ahead. Front Immunol. 2020;11:1456.
49. Gotsman I, Sharpe AH, Lichtman AH. T-cell costimulation and coinhibition in atherosclerosis. Circ Res. 2008;103:1220-31.
50. Aukrust P, Otterdal K, Yndestad A, et al. The complex role of T-cell-based immunity in atherosclerosis. Curr Atheroscler Rep. 2008;10:236-43.
51. Li N. CD4+ T cells in atherosclerosis: regulation by platelets. Thromb Haemost. 2013;109:980-90.
52. Sasaki N, Yamashita T, Takeda M, Hirata K. Regulatory T cells in atherogenesis. J Atheroscler Thromb. 2012;19:503-15.
53. van Duijn J, Kritikou E, Benne N, et al. CD8+ T-cells contribute to lesion stabilization in advanced atherosclerosis by limiting macrophage content and CD4+ T-cell responses. Cardiovasc Res. 2019;115:729-38.
54. Kyaw T, Tipping P, Bobik A, Toh BH. Protective role of natural IgM-producing B1a cells in atherosclerosis. Trends Cardiovasc Med. 2012;22:48-53.
55. Nus M, Tsiantoulas D, Mallat Z. Plan B (-cell) in atherosclerosis. Eur J Pharmacol. 2017;816:76-81.
56. Hamze M, Desmetz C, Guglielmi P. [B lymphocytes: a promising target to treat atherosclerosis?]. Med Sci. 2014;30:874-81.
58. Douna H, Amersfoort J, Schaftenaar FH, et al. Bidirectional effects of IL-10+ regulatory B cells in Ldlr-/- mice. Atherosclerosis. 2019;280:118-25.
59. Sage AP, Mallat Z. Multiple potential roles for B cells in atherosclerosis. Ann Med. 2014;46:297-303.
60. Tay C, Kanellakis P, Hosseini H, et al. B cell and CD4 T cell interactions promote development of atherosclerosis. Front Immunol. 2019;10:3046.
61. Leeuwen M, Damoiseaux J, Duijvestijn A, Tervaert JW. The therapeutic potential of targeting B cells and anti-oxLDL antibodies in atherosclerosis. Autoimmun Rev. 2009;9:53-7.
62. Tay C, Liu YH, Kanellakis P, et al. Follicular B cells promote atherosclerosis via T cell-mediated differentiation into plasma cells and secreting pathogenic immunoglobulin G. Arterioscler Thromb Vasc Biol. 2018;38:e71-84.
63. Ma SD, Mussbacher M, Galkina EV. Functional role of B cells in atherosclerosis. Cells. 2021;10:270.
64. Tsiantoulas D, Sage AP, Mallat Z, Binder CJ. Targeting B cells in atherosclerosis: closing the gap from bench to bedside. Arterioscler Thromb Vasc Biol. 2015;35:296-302.
65. Harrison J, Newland SA, Jiang W, et al. Marginal zone B cells produce 'natural' atheroprotective IgM antibodies in a T cell-dependent manner. Cardiovasc Res. 2024;120:318-28.
66. Nus M, Sage AP, Lu Y, et al. Marginal zone B cells control the response of follicular helper T cells to a high-cholesterol diet. Nat Med. 2017;23:601-10.
67. Ransegnola BP, Pattarabanjird T, McNamara CA. Tipping the scale: atheroprotective IgM-producing B cells in atherosclerosis. Arterioscler Thromb Vasc Biol. 2024;44:1906-15.
68. Mulholland M, Kritikou E, Katra P, et al. LAG3 regulates T cell activation and plaque infiltration in atherosclerotic mice. JACC CardioOncol. 2022;4:635-45.
69. Pryshchep S, Goronzy JJ, Parashar S, Weyand CM. Insufficient deactivation of the protein tyrosine kinase lck amplifies T-cell responsiveness in acute coronary syndrome. Circ Res. 2010;106:769-78.
70. Singh P, Kashyap A, Silakari O. Exploration of the therapeutic aspects of Lck: a kinase target in inflammatory mediated pathological conditions. Biomed Pharmacother. 2018;108:1565-71.
71. Luo T, Hu J, Xi D, et al. Lck inhibits heat shock protein 65-mediated reverse cholesterol transport in T cells. J Immunol. 2016;197:3861-70.
72. Liu J, Guo Z, Zhang Y, et al. LCK inhibitor attenuates atherosclerosis in ApoE-/- mice via regulating T cell differentiation and reverse cholesterol transport. J Mol Cell Cardiol. 2020;139:87-97.
73. Karadimou G, Gisterå A, Gallina AL, et al. Treatment with a Toll-like Receptor 7 ligand evokes protective immunity against atherosclerosis in hypercholesterolaemic mice. J Intern Med. 2020;288:321-34.
74. Kyaw T, Cui P, Tay C, et al. BAFF receptor mAb treatment ameliorates development and progression of atherosclerosis in hyperlipidemic ApoE-/- mice. PLoS One. 2013;8:e60430.
75. Zhao TX, Aetesam-Ur-Rahman M, Sage AP, et al. Rituximab in patients with acute ST-elevation myocardial infarction: an experimental medicine safety study. Cardiovasc Res. 2022;118:872-82.
76. Tsioulos G, Vallianou NG, Skourtis A, et al. Vaccination as a promising approach in cardiovascular risk mitigation: are we ready to embrace a vaccine strategy? Biomolecules. 2024;14:1637.
77. Taylor JA, Hutchinson MA, Gearhart PJ, Maul RW. Antibodies in action: the role of humoral immunity in the fight against atherosclerosis. Immun Ageing. 2022;19:59.
78. Kovanen PT, Mänttäri M, Palosuo T, Manninen V, Aho K. Prediction of myocardial infarction in dyslipidemic men by elevated levels of immunoglobulin classes A, E, and G, but not M. Arch Intern Med. 1998;158:1434-9.
79. Khamis RY, Hughes AD, Caga-Anan M, et al. High serum immunoglobulin G and M levels predict freedom from adverse cardiovascular events in hypertension: a nested case-control substudy of the anglo-scandinavian cardiac outcomes trial. EBioMedicine. 2016;9:372-80.
80. Moffett HF, Harms CK, Fitzpatrick KS, Tooley MR, Boonyaratanakornkit J, Taylor JJ. B cells engineered to express pathogen-specific antibodies protect against infection. Sci Immunol. 2019;4:eaax0644.
81. Libby P. Interleukin-1 beta as a target for atherosclerosis therapy: biological basis of CANTOS and beyond. J Am Coll Cardiol. 2017;70:2278-89.
82. Mai W, Liao Y. Targeting IL-1β in the treatment of atherosclerosis. Front Immunol. 2020;11:589654.
83. Weber A, Wasiliew P, Kracht M. Interleukin-1beta (IL-1beta) processing pathway. Sci Signal. 2010;3:cm2.
84. Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519-50.
85. Abbate A, Van Tassell BW, Biondi-Zoccai GG. Blocking interleukin-1 as a novel therapeutic strategy for secondary prevention of cardiovascular events. BioDrugs. 2012;26:217-33.
86. Xue Y, Luo M, Hu X, et al. Macrophages regulate vascular smooth muscle cell function during atherosclerosis progression through IL-1β/STAT3 signaling. Commun Biol. 2022;5:1316.
87. Vromman A, Ruvkun V, Shvartz E, et al. Stage-dependent differential effects of interleukin-1 isoforms on experimental atherosclerosis. Eur Heart J. 2019;40:2482-91.
88. Cheng CY, Kuo CT, Lin CC, Hsieh HL, Yang CM. IL-1beta induces expression of matrix metalloproteinase-9 and cell migration via a c-Src-dependent, growth factor receptor transactivation in A549 cells. Br J Pharmacol. 2010;160:1595-610.
89. Furuyama A, Hosokawa T, Mochitate K. Interleukin-1beta and tumor necrosis factor-alpha have opposite effects on fibroblasts and epithelial cells during basement membrane formation. Matrix Biol. 2008;27:429-40.
90. Aday AW, Ridker PM. Antiinflammatory therapy in clinical care: the CANTOS trial and beyond. Front Cardiovasc Med. 2018;5:62.
91. Ku EJ, Kim BR, Lee JI, et al. The anti-atherosclerosis effect of anakinra, a recombinant human interleukin-1 receptor antagonist, in apolipoprotein E knockout mice. Int J Mol Sci. 2022;23:4906.
92. Harouki N, Nicol L, Remy-Jouet I, et al. The IL-1β antibody gevokizumab limits cardiac remodeling and coronary dysfunction in rats with heart failure. JACC Basic Transl Sci. 2017;2:418-30.
93. Roubille F, Busseuil D, Shi Y, et al. The interleukin-1β modulator gevokizumab reduces neointimal proliferation and improves reendothelialization in a rat carotid denudation model. Atherosclerosis. 2014;236:277-85.
94. Coll RC, Robertson AA, Chae JJ, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21:248-55.
95. Zeng W, Wu D, Sun Y, et al. The selective NLRP3 inhibitor MCC950 hinders atherosclerosis development by attenuating inflammation and pyroptosis in macrophages. Sci Rep. 2021;11:19305.
96. Romero A, Dongil P, Valencia I, et al. Pharmacological blockade of NLRP3 inflammasome/IL-1β-positive loop mitigates endothelial cell senescence and dysfunction. Aging Dis. 2022;13:284-97.
97. Feng H, Li D, Zhu F, et al. Design, synthesis and biological evaluation of sulfonylurea derivatives as NLRP3 inflammasome inhibitors. Bioorg Med Chem Lett. 2024;114:129987.
98. Gatlik E, Mehes B, Voltz E, et al. First-in-human safety, tolerability, and pharmacokinetic results of DFV890, an oral low-molecular-weight NLRP3 inhibitor. Clin Transl Sci. 2024;17:e13789.
99. Novartis Pharmaceuticals. A study to investigate the efficacy, safety, and tolerability of DFV890 for inflammatory marker reduction in adult participants with coronary heart disease and elevated hsCRP; 2025. Available from: https://clinicaltrials.gov/study/NCT06031844 [Last accessed on 31 Jul 2025].
100. Ye B, Cai X, Liang X, et al. Emodin suppresses NLRP3/GSDMD-induced inflammation via the TLR4/MyD88/NF-κB signaling pathway in atherosclerosis. Cardiovasc Drugs Ther. 2024:Online ahead of print.
101. Huang Y, Jiang H, Chen Y, et al. Tranilast directly targets NLRP3 to treat inflammasome-driven diseases. EMBO Mol Med. 2018;10:e8689.
102. Chen S, Wang Y, Pan Y, et al. Novel role for tranilast in regulating NLRP3 ubiquitination, vascular inflammation, and atherosclerosis. J Am Heart Assoc. 2020;9:e015513.
103. Vrints C, Andreotti F, Koskinas KC, et al. 2024 ESC guidelines for the management of chronic coronary syndromes. Eur Heart J. 2024;45:3415-537.
104. Fiolet ATL, Opstal TSJ, Mosterd A, et al. Efficacy and safety of low-dose colchicine in patients with coronary disease: a systematic review and meta-analysis of randomized trials. Eur Heart J. 2021;42:2765-75.
105. Opstal TSJ, van Broekhoven A, Fiolet ATL, et al. Long-term efficacy of colchicine in patients with chronic coronary disease: insights from LoDoCo2. Circulation. 2022;145:626-8.
106. Nidorf SM, Fiolet ATL, Mosterd A, et al. Colchicine in patients with chronic coronary disease. N Engl J Med. 2020;383:1838-47.
107. Tardif JC, Kouz S, Waters DD, et al. Efficacy and safety of low-dose colchicine after myocardial infarction. N Engl J Med. 2019;381:2497-505.
108. Feng Y, Ye D, Wang Z, et al. The role of interleukin-6 family members in cardiovascular diseases. Front Cardiovasc Med. 2022;9:818890.
109. Reiss AB, Siegart NM, De Leon J. Interleukin-6 in atherosclerosis: atherogenic or atheroprotective? Clin Lipidol. 2017;12:14-23. Available from: https://www.tandfonline.com/doi/full/10.1080/17584299.2017.1319787#abstract [Last accessed on 1 Aug 2025].
110. Fraser A, May M, Lowe G, et al. Interleukin-6 and incident coronary heart disease: results from the British Women's Heart and Health Study. Atherosclerosis. 2009;202:567-72.
111. Amar J, Fauvel J, Drouet L, et al. Interleukin 6 is associated with subclinical atherosclerosis: a link with soluble intercellular adhesion molecule 1. J Hypertens. 2006;24:1083-8.
112. Cau R, Saba L. Interlinking pathways: a narrative review on the role of IL-6 in cancer and atherosclerosis. Cardiovasc Diagn Ther. 2024;14:1186-201.
113. Feng M, Cai XJ, Zhang W, et al. Interleukin-6 enhances matrix metalloproteinase-14 expression via the RAF-mitogen-activated protein kinase kinase-extracellular signal-regulated kinase 1/2-activator protein-1 pathway. Clin Exp Pharmacol Physiol. 2010;37:162-6.
114. Akita K, Isoda K, Sato-Okabayashi Y, et al. An interleukin-6 receptor antibody suppresses atherosclerosis in atherogenic mice. Front Cardiovasc Med. 2017;4:84.
115. Papastamos C, Antonopoulos AS, Simantiris S, et al. Interleukin-6 signaling in atherosclerosis: from molecular mechanisms to clinical outcomes. Curr Top Med Chem. 2023;23:2172-83.
116. Ou G, Cai H, Yao K, et al. Exploring the therapeutic potential of interleukin-6 receptor blockade in cardiovascular disease treatment through Mendelian randomization. Sci Rep. 2024;14:21452.
117. Kim SC, Solomon DH, Rogers JR, et al. Cardiovascular safety of tocilizumab versus tumor necrosis factor inhibitors in patients with rheumatoid arthritis: a multi-database cohort study. Arthritis Rheumatol. 2017;69:1154-64.
118. Ruiz-Limón P, Ortega R, Arias de la Rosa I, et al. Tocilizumab improves the proatherothrombotic profile of rheumatoid arthritis patients modulating endothelial dysfunction, NETosis, and inflammation. Transl Res. 2017;183:87-103.
119. Bacchiega BC, Bacchiega AB, Usnayo MJ, Bedirian R, Singh G, Pinheiro GD. Interleukin 6 inhibition and coronary artery disease in a high-risk population: a prospective community-based clinical study. J Am Heart Assoc. 2017;6:e005038.
120. Ridker PM, Devalaraja M, Baeres FMM, et al. IL-6 inhibition with ziltivekimab in patients at high atherosclerotic risk (RESCUE): a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet. 2021;397:2060-9.
121. Wada Y, Jensen C, Meyer ASP, Zonoozi AAM, Honda H. Efficacy and safety of interleukin-6 inhibition with ziltivekimab in patients at high risk of atherosclerotic events in Japan (RESCUE-2): a randomized, double-blind, placebo-controlled, phase 2 trial. J Cardiol. 2023;82:279-85.
122. McKellar GE, McCarey DW, Sattar N, McInnes IB. Role for TNF in atherosclerosis? Lessons from autoimmune disease. Nat Rev Cardiol. 2009;6:410-7.
123. Prasongsukarn K, Chaisri U, Chartburus P, et al. Phenotypic alterations in human saphenous vein culture induced by tumor necrosis factor-alpha and lipoproteins: a preliminary development of an initial atherosclerotic plaque model. Lipids Health Dis. 2013;12:132.
124. Brånén L, Hovgaard L, Nitulescu M, Bengtsson E, Nilsson J, Jovinge S. Inhibition of tumor necrosis factor-alpha reduces atherosclerosis in apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol. 2004;24:2137-42.
125. Tsioufis P, Theofilis P, Tsioufis K, Tousoulis D. The impact of cytokines in coronary atherosclerotic plaque: current therapeutic approaches. Int J Mol Sci. 2022;23:15937.
126. Boesten LS, Zadelaar AS, van Nieuwkoop A, et al. Tumor necrosis factor-alpha promotes atherosclerotic lesion progression in APOE*3-Leiden transgenic mice. Cardiovasc Res. 2005;66:179-85.
127. Bian N, Du G, Ip MF, Ding J, Chang Q, Li Z. Pituitary adenylate cyclase-activating polypeptide attenuates tumor necrosis factor-α-induced apoptosis in endothelial colony-forming cells. Biomed Rep. 2017;7:11-6.
128. Missiou A, Köstlin N, Varo N, et al. Tumor necrosis factor receptor-associated factor 1 (TRAF1) deficiency attenuates atherosclerosis in mice by impairing monocyte recruitment to the vessel wall. Circulation. 2010;121:2033-44.
129. Lei L, Xiong Y, Chen J. TNF-alpha stimulates the ACAT1 expression in differentiating monocytes to promote the CE-laden cell formation. J Lipid Res. 2009;50:1057-67.
130. Chen C, Khismatullin DB. Oxidized low-density lipoprotein contributes to atherogenesis via co-activation of macrophages and mast cells. PLoS One. 2015;10:e0123088.
131. Minno MN, Iervolino S, Peluso R, Scarpa R, Di Minno G; CaRRDs study group. Carotid intima-media thickness in psoriatic arthritis: differences between tumor necrosis factor-α blockers and traditional disease-modifying antirheumatic drugs. Arterioscler Thromb Vasc Biol. 2011;31:705-12.
132. Ahlehoff O, Skov L, Gislason G, et al. Cardiovascular disease event rates in patients with severe psoriasis treated with systemic anti-inflammatory drugs: a Danish real-world cohort study. J Intern Med. 2013;273:197-204.
133. Jacobsson LTH, Turesson C, Gülfe A, et al. Treatment with tumor necrosis factor blockers is associated with a lower incidence of first cardiovascular events in patients with rheumatoid arthritis. J Rheumatol. 2005;32:1213-8.
134. Greenberg JD, Kremer JM, Curtis JR, et al. Tumour necrosis factor antagonist use and associated risk reduction of cardiovascular events among patients with rheumatoid arthritis. Ann Rheum Dis. 2011;70:576-82.
135. Medler J, Kucka K, Wajant H. Tumor necrosis factor receptor 2 (TNFR2): an emerging target in cancer therapy. Cancers. 2022;14:2603.
136. Lainez B, Fernandez-Real JM, Romero X, et al. Identification and characterization of a novel spliced variant that encodes human soluble tumor necrosis factor receptor 2. Int Immunol. 2004;16:169-77.
137. Kim HL, Lee JP, An JN, et al. Soluble tumor necrosis factor receptors and arterial stiffness in patients with coronary atherosclerosis. Am J Hypertens. 2017;30:313-8.
138. Iversen PL, Kipshidze N, Kipshidze N, et al. A novel therapeutic vaccine targeting the soluble TNFα receptor II to limit the progression of cardiovascular disease: AtheroVax™. Front Cardiovasc Med. 2023;10:1206541.
139. Mahmoudi M, Siassi F, Mahmoudi MJ, et al. Defective T-cell proliferation and IL-2 production in a subgroup of patients with coronary artery disease. Iran J Allergy Asthma Immunol. 2010;9:133-40.
140. Ross SH, Cantrell DA. Signaling and function of interleukin-2 in T lymphocytes. Annu Rev Immunol. 2018;36:411-33.
141. Elkind MS, Rundek T, Sciacca RR, et al. Interleukin-2 levels are associated with carotid artery intima-media thickness. Atherosclerosis. 2005;180:181-7.
142. Fisman EZ, Adler Y, Tenenbaum A. Biomarkers in cardiovascular diabetology: interleukins and matrixins. In: Fisman E, Tenenbaum A, editors. Cardiovascular Diabetology: Clinical, Metabolic and Inflammatory Facets. Basel: KARGER; 2008. pp. 44-64.
143. Ding R, Gao W, Ostrodci DH, et al. Effect of interleukin-2 level and genetic variants on coronary artery disease. Inflammation. 2013;36:1225-31.
144. Dinh TN, Kyaw TS, Kanellakis P, et al. Cytokine therapy with interleukin-2/anti-interleukin-2 monoclonal antibody complexes expands CD4+CD25+Foxp3+ regulatory T cells and attenuates development and progression of atherosclerosis. Circulation. 2012;126:1256-66.
145. Mulholland M, Jakobsson G, Lei Y, et al. IL-2Rβγ signalling in lymphocytes promotes systemic inflammation and reduces plasma cholesterol in atherosclerotic mice. Atherosclerosis. 2021;326:1-10.
146. Zhao TX, Newland SA, Mallat Z. 2019 ATVB plenary lecture: interleukin-2 therapy in cardiovascular disease: the potential to regulate innate and adaptive immunity. Arterioscler Thromb Vasc Biol. 2020;40:853-64.
147. Sriranjan R, Zhao TX, Tarkin J, et al. Low-dose interleukin 2 for the reduction of vascular inflammation in acute coronary syndromes (IVORY): protocol and study rationale for a randomised, double-blind, placebo-controlled, phase II clinical trial. BMJ Open. 2022;12:e062602.
148. Zhou P. Emerging mechanisms and applications of low-dose IL-2 therapy in autoimmunity. Cytokine Growth Factor Rev. 2022;67:80-8.
149. Case A, O'brien J, Sriranjan R, et al. The effect of low-dose interleukin-2 on the T cell receptor landscape in patients with acute myocardial infarction. Atherosclerosis. 2024;395:118476.
150. Zhao TX, Kostapanos M, Griffiths C, et al. Low-dose interleukin-2 in patients with stable ischaemic heart disease and acute coronary syndromes (LILACS): protocol and study rationale for a randomised, double-blind, placebo-controlled, phase I/II clinical trial. BMJ Open. 2018;8:e022452.
151. Kobayashi T, Tahara Y, Matsumoto M, et al. Roles of thromboxane A2 and prostacyclin in the development of atherosclerosis in apoE-deficient mice. J Clin Invest. 2004;114:784-94.
152. van den Borne P, van der Laan SW, Bovens SM, et al. Leukotriene B4 levels in human atherosclerotic plaques and abdominal aortic aneurysms. PLoS One. 2014;9:e86522.
153. Jala VR, Haribabu B. Leukotrienes and atherosclerosis: new roles for old mediators. Trends Immunol. 2004;25:315-22.
154. Bäck M. Leukotriene signaling in atherosclerosis and ischemia. Cardiovasc Drugs Ther. 2009;23:41-8.
155. Sansbury BE, Spite M. Resolution of acute inflammation and the role of resolvins in immunity, thrombosis, and vascular biology. Circ Res. 2016;119:113-30.
156. Wang YF, Zhu XT, Hu ZP. Decreased plasma lipoxin A4, resolvin D1, protectin D1 are correlated with the complexity and prognosis of coronary heart disease: a retrospective cohort study. Prostaglandins Other Lipid Mediat. 2025;178:106990.
157. Chandrasekharan JA, Sharma-Walia N. Lipoxins: nature's way to resolve inflammation. J Inflamm Res. 2015;8:181-92.
158. Li Y, Li W, Guo S. Targeting proprotein convertase subtilisin/Kexin type 9 (PCSK9) for lipid-lowering. Chin Pharm J. 2023;58:2228-34.
159. Jeswani BM, Sharma S, Rathore SS, Nazir A, Bhatheja R, Kapoor K. PCSK9 inhibitors: the evolving future. Health Sci Rep. 2024;7:e70174.
160. Fitzgerald G, Kiernan T. PCSK9 inhibitors and LDL reduction: pharmacology, clinical implications, and future perspectives. Expert Rev Cardiovasc Ther. 2018;16:567-78.
161. Ding Z, Pothineni NVK, Goel A, Lüscher TF, Mehta JL. PCSK9 and inflammation: role of shear stress, pro-inflammatory cytokines, and LOX-1. Cardiovasc Res. 2020;116:908-15.
162. Mohyeldin M, Abuelgasim AS, Mustafa AM. Proprotein convertase subtilisin/kexin type 9 inhibitors in peripheral artery disease: a review of efficacy, safety, and outcomes. World J Cardiol. 2024;16:397-401.
163. Keech AC, Oyama K, Sever PS, et al. Efficacy and safety of long-term evolocumab use among asian subjects - a subgroup analysis of the further cardiovascular outcomes research with PCSK9 inhibition in subjects with elevated risk (FOURIER) trial. Circ J. 2021;85:2063-70.
164. Basutkar RS, Tsundue T, Siva H, Durai R, Ponnusankar S. Evolocumab in combination with statins for CVD risk reduction: an evidential review. J Cardiovasc Dis Res. 2018;9:45-53.
165. Liang D, Li C, Tu Y, Li Z, Zhang M. Additive effects of ezetimibe, evolocumab, and alirocumab on plaque burden and lipid content as assessed by intravascular ultrasound: a PRISMA-compliant meta-analysis. Medicine. 2022;101:e31199.
167. Kong Q, Liu M, Li Y, Zhu Q, Su G. Effect of evolocumab on the progression and stability of atherosclerotic plaques as evaluated by grayscale and iMAP-IVUS. Ann Palliat Med. 2020;9:3078-88.
168. Olivares-García JD, Román-Hernández R, Meráz-Martínez M. Efecto del tratamiento combinado de estatinas y evolocumab en pacientes con enfermedad arterial periférica. Rev Mex Angiol. 2023;51:85-92.
169. Pouwer MG, Pieterman EJ, Worms N, et al. Alirocumab, evinacumab, and atorvastatin triple therapy regresses plaque lesions and improves lesion composition in mice. J Lipid Res. 2020;61:365-75.
170. Marfella R, Prattichizzo F, Sardu C, et al. Evidence of an anti-inflammatory effect of PCSK9 inhibitors within the human atherosclerotic plaque. Atherosclerosis. 2023;378:117180.
171. Zhang Y, Chen H, Hong L, et al. Inclisiran: a new generation of lipid-lowering siRNA therapeutic. Front Pharmacol. 2023;14:1260921.
172. Tsouka AN, Tellis CC, Tselepis AD. Pharmacology of PCSK9 inhibitors: current status and future perspectives. Curr Pharm Des. 2018;24:3622-33.
173. Graham MJ, Lemonidis KM, Whipple CP, et al. Antisense inhibition of proprotein convertase subtilisin/kexin type 9 reduces serum LDL in hyperlipidemic mice. J Lipid Res. 2007;48:763-7.
174. Barter PJ, Brewer HB Jr, Chapman MJ, Hennekens CH, Rader DJ, Tall AR. Cholesteryl ester transfer protein: a novel target for raising HDL and inhibiting atherosclerosis. Arterioscler Thromb Vasc Biol. 2003;23:160-7.
175. Barter P. Lessons learned from the investigation of lipid level management to understand its impact in atherosclerotic events (ILLUMINATE) trial. Am J Cardiol. 2009;104:10E-5E.
176. Kotlyarov S. High-density lipoproteins: a role in inflammation in COPD. Int J Mol Sci. 2022;23:8128.
177. Johns DG, Duffy J, Fisher T, Hubbard BK, Forrest MJ. On- and off-target pharmacology of torcetrapib: current understanding and implications for the structure activity relationships (SAR), discovery and development of cholesteryl ester-transfer protein (CETP) inhibitors. Drugs. 2012;72:491-507.
178. Watts GF, Chan DC, Raal FJ. Inhibition of ANGPTL3 as a target for treating dyslipidemias. Elsevier; 2024. pp. 253-67.e1.
179. Wang X, Musunuru K. Angiopoietin-like 3: from discovery to therapeutic gene editing. JACC Basic Transl Sci. 2019;4:755-62.
180. Tikka A, Jauhiainen M. The role of ANGPTL3 in controlling lipoprotein metabolism. Endocrine. 2016;52:187-93.
181. Hatsuda S, Shoji T, Shinohara K, et al. Association between plasma angiopoietin-like protein 3 and arterial wall thickness in healthy subjects. J Vasc Res. 2007;44:61-6.
182. Liu YZ, Zhang C, Jiang JF, et al. Angiopoietin-like proteins in atherosclerosis. Clin Chim Acta. 2021;521:19-24.
183. Luo F, Wu P, Chen J, et al. ANGPTL3 possibly promotes cardiac angiogenesis through improving proangiogenic ability of endothelial progenitor cells after myocardial infarction. Lipids Health Dis. 2018;17:184.
184. Chan DC, Watts GF. Inhibition of the ANGPTL3/8 complex for the prevention and treatment of atherosclerotic cardiovascular disease. Curr Atheroscler Rep. 2024;27:6.
185. Lang W, Frishman WH. Angiopoietin-like 3 protein inhibition: a new frontier in lipid-lowering treatment. Cardiol Rev. 2019;27:211-7.
186. Xu S, Ogura S, Chen J, Little PJ, Moss J, Liu P. LOX-1 in atherosclerosis: biological functions and pharmacological modifiers. Cell Mol Life Sci. 2013;70:2859-72.
187. Kattoor AJ, Kanuri SH, Mehta JL. Role of Ox-LDL and LOX-1 in atherogenesis. Curr Med Chem. 2019;26:1693-700.
188. Duprat F, Robles C, Castillo MP, et al. LOX-1 activation by oxLDL induces AR and AR-V7 expression via NF-κB and STAT3 signaling pathways reducing enzalutamide cytotoxic effects. Int J Mol Sci. 2023;24:5082.
189. Vohra RS, Murphy JE, Walker JH, Ponnambalam S, Homer-Vanniasinkam S. Atherosclerosis and the lectin-like OXidized low-density lipoprotein scavenger receptor. Trends Cardiovasc Med. 2006;16:60-4.
190. Ran XL, Wu Q, Shi JS, Chen XP. Role of lectin-like oxidized low-density lipoprotein receptor-1 in atherosclerosis. Chin J Pharmacol Toxicol. 2013;27:865-71.
191. Cilingiroglu M, Ozer K. The lectin-like oxidized low-density lipoprotein receptor and its role in atherosclerosis. Curr Atheroscler Rep. 2005;7:103-7.
192. Navarra T, Del Turco S, Berti S, Basta G. The lectin-like oxidized low-density lipoprotein receptor-1 and its soluble form: cardiovascular implications. J Atheroscler Thromb. 2010;17:317-31.
193. Yurina V, Raras TYM, Rudijanto A, Lyrawati D, Handono K. Design and construction of DNA vaccine expressing lectin-like oxidize-LDL receptor-1 (LOX-1) as atherosclerosis vaccine candidate. J Biotech Res. 2017;8:103-12. Available from: https://www.btsjournals.com/assets/2017v8p103-11272098.pdf [Last accessed on 1 Aug 2025].
194. Adianingsih OR, Tamara F, Putri AP, et al. Lectin-like oxidized LDL receptor-1 (LOX-1) protein vaccination reduces inflammation and attenuates atherosclerosis progression in atherogenic-diet wistar rats. Int Cardiovasc Res J. 2016;10:e9803. Available from: https://brieflands.com/articles/ircrj-9803 [Last accessed on 1 Aug 2025].
195. Babakr AT. Oxidized low-density lipoproteins and their contribution to atherosclerosis. Rev Clin Pharm Drug Ther. 2024;22:351-60.
196. Babakr AT. Scavenger receptors: different classes and their role in the uptake of oxidized low-density lipoproteins. Biomed Pharmacol J. 2024;17:699-712.
197. Yang X, Hu Y. Role of oxidized low density lipoprotein in the diagnosis of atherosclerotic cardiovascular disease. Chin J Lab Med. ;44:563-8.
198. Maiolino G, Rossitto G, Caielli P, Bisogni V, Rossi GP, Calò LA. The role of oxidized low-density lipoproteins in atherosclerosis: the myths and the facts. Mediators Inflamm. 2013;2013:714653.
199. Jiang H, Zhou Y, Nabavi SM, et al. Mechanisms of oxidized LDL-mediated endothelial dysfunction and its consequences for the development of atherosclerosis. Front Cardiovasc Med. 2022;9:925923.
200. Nilsson J, Nordin Fredrikson G, Schiopu A, Shah PK, Jansson B, Carlsson R. Oxidized LDL antibodies in treatment and risk assessment of atherosclerosis and associated cardiovascular disease. Curr Pharm Des. 2007;13:1021-30.
201. Nilsson J, Fredrikson GN, Björkbacka H, Chyu KY, Shah PK. Vaccines modulating lipoprotein autoimmunity as a possible future therapy for cardiovascular disease. J Intern Med. 2009;266:221-31.
202. Habets KL, van Puijvelde GH, van Duivenvoorde LM, et al. Vaccination using oxidized low-density lipoprotein-pulsed dendritic cells reduces atherosclerosis in LDL receptor-deficient mice. Cardiovasc Res. 2010;85:622-30.
203. Hulthe J. Antibodies to oxidized LDL in atherosclerosis development-clinical and animal studies. Clin Chim Acta. 2004;348:1-8.
204. Carvalho JF, Sherer Y, Shoenfeld Y. The fine-tuning of anti-oxidized low-density lipoprotein antibodies in cardiovascular disease and thrombosis. Thromb Haemost. 2007;98:1157-9.
205. Kimura T, Tse K, Sette A, Ley K. Vaccination to modulate atherosclerosis. Autoimmunity. 2015;48:152-60.
206. Zhong Y, Wang X, Ji Q, et al. CD4+LAP + and CD4 +CD25 +Foxp3 + regulatory T cells induced by nasal oxidized low-density lipoprotein suppress effector T cells response and attenuate atherosclerosis in ApoE-/- mice. J Clin Immunol. 2012;32:1104-17.
207. van Puijvelde GH, Hauer AD, de Vos P, et al. Induction of oral tolerance to oxidized low-density lipoprotein ameliorates atherosclerosis. Circulation. 2006;114:1968-76.
208. Kotlyarov S, Kotlyarova A. Clinical significance of polyunsaturated fatty acids in the prevention of cardiovascular diseases. Front Nutr. 2022;9:998291.
209. Aubeux D, Tessier S, Pérez F, Geoffroy V, Gaudin A. In vitro phenotypic effects of Lipoxin A4 on M1 and M2 polarized macrophages derived from THP-1. Mol Biol Rep. 2023;50:339-48.
210. Yuan J, Lin F, Chen L, et al. Lipoxin A4 regulates M1/M2 macrophage polarization via FPR2-IRF pathway. Inflammopharmacology. 2022;30:487-98.
211. Filep JG, Zouki C, Petasis NA, Hachicha M, Serhan CN. Lipoxin A4 and aspirin-triggered 15-Epi-lipoxin A4 modulate adhesion molecule expression on human leukocytes in whole blood and inhibit neutrophil-endothelial cell adhesion. In: Honn KV, Marnett LJ, Nigam S, Dennis E, Serhan C, editors. Eicosanoids and Other Bioactive Lipids in Cancer, Inflammation, and Radiation Injury, 5. Boston: Springer US; 2002. pp. 223-8.
212. Baylor College of Medicine. The effect of ticagrelor on 15-Epi-lipoxin A4 and inflammation. 2019. Available from: https://clinicaltrials.gov/study/NCT02626169 [Last accessed on 31 Jul 2025].
213. University of California, San Francisco. The effects of omega-3 fatty acids supplementation on endothelial function and inflammation (OMEGA-PAD). 2021. Available from: https://clinicaltrials.gov/study/NCT01310270 [Last accessed on 31 Jul 2025].
214. Grenon SM, Owens CD, Nosova EV, et al. Short-term, high-dose fish oil supplementation increases the production of omega-3 fatty acid-derived mediators in patients with peripheral artery disease (the OMEGA-PAD I Trial). J Am Heart Assoc. 2015;4:e002034.
215. Millar B, de Gaetano M. Posing the rationale for synthetic lipoxin mimetics as an adjuvant treatment to gold standard atherosclerosis therapies. Front Pharmacol. 2023;14:1125858.
216. Gerlach BD, Marinello M, Heinz J, et al. Resolvin D1 promotes the targeting and clearance of necroptotic cells. Cell Death Differ. 2020;27:525-39.
217. Kohli P, Levy BD. Resolvins and protectins: mediating solutions to inflammation. Br J Pharmacol. 2009;158:960-71.
218. Spite M, Clària J, Serhan CN. Resolvins, specialized proresolving lipid mediators, and their potential roles in metabolic diseases. Cell Metab. 2014;19:21-36.
219. Serhan CN, Hong S, Gronert K, et al. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J Exp Med. 2002;196:1025-37.
220. Bazan HA, Lu Y, Jun B, Fang Z, Woods TC, Hong S. Circulating inflammation-resolving lipid mediators RvD1 and DHA are decreased in patients with acutely symptomatic carotid disease. Prostaglandins Leukot Essent Fatty Acids. 2017;125:43-7.
221. Hosseini Z, Marinello M, Decker C, et al. Resolvin D1 enhances necroptotic cell clearance through promoting macrophage fatty acid oxidation and oxidative phosphorylation. Arterioscler Thromb Vasc Biol. 2021;41:1062-75.
222. Yang Y, Zhu Y, Xiao J, et al. Maresin conjugates in tissue regeneration 1 prevents lipopolysaccharide-induced cardiac dysfunction through improvement of mitochondrial biogenesis and function. Biochem Pharmacol. 2020;177:114005.
223. Saito-Sasaki N, Sawada Y, Nakamura M. Maresin-1 and inflammatory disease. Int J Mol Sci. 2022;23:1367.
224. Liu WC, Yang YH, Wang YC, Chang WM, Wang CW. Maresin: macrophage mediator for resolving inflammation and bridging tissue regeneration-a system-based preclinical systematic review. Int J Mol Sci. 2023;24:11012.
225. Marcon R, Bento AF, Dutra RC, Bicca MA, Leite DF, Calixto JB. Maresin 1, a proresolving lipid mediator derived from omega-3 polyunsaturated fatty acids, exerts protective actions in murine models of colitis. J Immunol. 2013;191:4288-98.
226. Liu M, Boussetta T, Makni-Maalej K, et al. Protectin DX, a double lipoxygenase product of DHA, inhibits both ROS production in human neutrophils and cyclooxygenase activities. Lipids. 2014;49:49-57.
227. Serhan CN, Dalli J, Colas RA, Winkler JW, Chiang N. Protectins and maresins: new pro-resolving families of mediators in acute inflammation and resolution bioactive metabolome. Biochim Biophys Acta. 2015;1851:397-413.
228. Hansen T, Serhan CN. Protectins: Their biosynthesis, metabolism and structure-functions. Biochem Pharmacol. 2022;206:115330.
229. Zeitlinger M, Bauer M, Reindl-Schwaighofer R, et al. A phase I study assessing the safety, tolerability, immunogenicity, and low-density lipoprotein cholesterol-lowering activity of immunotherapeutics targeting PCSK9. Eur J Clin Pharmacol. 2021;77:1473-84.
230. Li Z, Yang Y, Zong J, et al. Dendritic cells immunotargeted therapy for atherosclerosis. Acta Pharm Sin B. 2025;15:792-808.
231. Zhu R, Zhang F, Pan C, Yu K, Zhong Y, Zeng Q. Role of IL-37- and IL-37-treated dendritic cells in acute coronary syndrome. Oxid Med Cell Longev. 2021;2021:6454177.
232. Schiopu A, Bengtsson J, Söderberg I, et al. Recombinant human antibodies against aldehyde-modified apolipoprotein B-100 peptide sequences inhibit atherosclerosis. Circulation. 2004;110:2047-52.
233. Schiopu A, Frendéus B, Jansson B, et al. Recombinant antibodies to an oxidized low-density lipoprotein epitope induce rapid regression of atherosclerosis in apobec-1-/-/low-density lipoprotein receptor-/- mice. J Am Coll Cardiol. 2007;50:2313-8.
234. Lehrer-Graiwer J, Singh P, Abdelbaky A, et al. FDG-PET imaging for oxidized LDL in stable atherosclerotic disease: a phase II study of safety, tolerability, and anti-inflammatory activity. JACC Cardiovasc Imaging. 2015;8:493-4.
235. van Leeuwen M, Kemna MJ, de Winther MP, et al. Passive immunization with hypochlorite-oxLDL specific antibodies reduces plaque volume in LDL receptor-deficient mice. PLoS One. 2013;8:e68039.
236. Gounopoulos P, Merki E, Hansen LF, Choi SH, Tsimikas S. Antibodies to oxidized low density lipoprotein: epidemiological studies and potential clinical applications in cardiovascular disease. Minerva Cardioangiol. 2007;55:821-37.
237. Baganha F, Sluiter TJ, de Jong RCM, et al. Phosphorylcholine monoclonal antibody therapy decreases intraplaque angiogenesis and intraplaque hemorrhage in murine vein grafts. Int J Mol Sci. 2022;23:13662.
238. de Vries MR, Ewing MM, de Jong RCM, et al. Identification of IgG1 isotype phosphorylcholine antibodies for the treatment of inflammatory cardiovascular diseases. J Intern Med. 2021;290:141-56.
239. Fredrikson GN, Björkbacka H, Söderberg I, Ljungcrantz I, Nilsson J. Treatment with apo B peptide vaccines inhibits atherosclerosis in human apo B-100 transgenic mice without inducing an increase in peptide-specific antibodies. J Intern Med. 2008;264:563-70.
240. Klingenberg R, Lebens M, Hermansson A, et al. Intranasal immunization with an apolipoprotein B-100 fusion protein induces antigen-specific regulatory T cells and reduces atherosclerosis. Arterioscler Thromb Vasc Biol. 2010;30:946-52.
241. Pierides C, Bermudez-Fajardo A, Fredrikson GN, Nilsson J, Oviedo-Orta E. Immune responses elicited by apoB-100-derived peptides in mice. Immunol Res. 2013;56:96-108.
242. Wigren M, Kolbus D, Dunér P, et al. Evidence for a role of regulatory T cells in mediating the atheroprotective effect of apolipoprotein B peptide vaccine. J Intern Med. 2011;269:546-56.
243. Govea-Alonso DO, Beltrán-López J, Salazar-González JA, Vargas-Morales J, Rosales-Mendoza S. Progress and future opportunities in the development of vaccines against atherosclerosis. Expert Rev Vaccines. 2017;16:337-50.
244. Yang M, Tian S, Lin Z, Fu Z, Li C. Costimulatory and coinhibitory molecules of B7-CD28 family in cardiovascular atherosclerosis: a review. Medicine. 2022;101:e31667.
245. Foks AC, Bot I, Frodermann V, et al. Interference of the CD30-CD30L pathway reduces atherosclerosis development. Arterioscler Thromb Vasc Biol. 2012;32:2862-8.
246. Jung IH, Oh GT. The roles of CD137 signaling in atherosclerosis. Korean Circ J. 2016;46:753-61.
247. Olofsson PS, Söderström LA, Wågsäter D, et al. CD137 is expressed in human atherosclerosis and promotes development of plaque inflammation in hypercholesterolemic mice. Circulation. 2008;117:1292-301.
248. Bosmans LA, Bosch L, Kusters PJH, Lutgens E, Seijkens TTP. The CD40-CD40L dyad as Immunotherapeutic target in cardiovascular disease. J Cardiovasc Transl Res. 2021;14:13-22.
249. Seijkens TTP, van Tiel CM, Kusters PJH, et al. Targeting CD40-induced TRAF6 signaling in macrophages reduces atherosclerosis. J Am Coll Cardiol. 2018;71:527-42.
250. Matsumoto T, Sasaki N, Yamashita T, et al. Overexpression of cytotoxic T-lymphocyte-associated antigen-4 prevents atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2016;36:1141-51.
251. Doesch AO, Zhao L, Gleissner CA, et al. Inhibition of B7-1 (CD80) by RhuDex® reduces lipopolysaccharide-mediated inflammation in human atherosclerotic lesions. Drug Des Devel Ther. 2014;8:447-57.
252. Wu X, Pan X, Zhou Y, et al. Identification of key genes for atherosclerosis in different arterial beds. Sci Rep. 2024;14:6543.
253. Gummesson A, Lundmark P, Chen QS, et al. A genome-wide association study of imaging-defined atherosclerosis. Nat Commun. 2025;16:2266.
254. Chen H, Li X, Liu S, Gu L, Zhou X. MircroRNA-19a promotes vascular inflammation and foam cell formation by targeting HBP-1 in atherogenesis. Sci Rep. 2017;7:12089.
256. Churov A, Summerhill V, Grechko A, Orekhova V, Orekhov A. MicroRNAs as potential biomarkers in atherosclerosis. Int J Mol Sci. 2019;20:5547.
257. Tang H, Liu Y, Yan C, Petasis NA, Serhan CN, Gao H. Protective actions of aspirin-triggered (17R) resolvin D1 and its analogue, 17R-hydroxy-19-para-fluorophenoxy-resolvin D1 methyl ester, in C5a-dependent IgG immune complex-induced inflammation and lung injury. J Immunol. 2014;193:3769-78.
258. Serhan CN, Fredman G, Yang R, et al. Novel proresolving aspirin-triggered DHA pathway. Chem Biol. 2011;18:976-87.
259. Sun YP, Oh SF, Uddin J, et al. Resolvin D1 and its aspirin-triggered 17R epimer. Stereochemical assignments, anti-inflammatory properties, and enzymatic inactivation. J Biol Chem. 2007;282:9323-34.
260. Crane JM, Tamm LK. Role of cholesterol in the formation and nature of lipid rafts in planar and spherical model membranes. Biophys J. 2004;86:2965-79.
261. Ruysschaert JM, Lonez C. Role of lipid microdomains in TLR-mediated signalling. Biochim Biophys Acta. 2015;1848:1860-7.
262. Wang S, Xie X, Lei T, et al. Statins attenuate activation of the NLRP3 inflammasome by oxidized LDL or TNFα in vascular endothelial cells through a PXR-dependent mechanism. Mol Pharmacol. 2017;92:256-64.
263. Luo B, Li B, Wang W, et al. Rosuvastatin alleviates diabetic cardiomyopathy by inhibiting NLRP3 inflammasome and MAPK pathways in a type 2 diabetes rat model. Cardiovasc Drugs Ther. 2014;28:33-43.
264. Kong F, Ye B, Lin L, Cai X, Huang W, Huang Z. Atorvastatin suppresses NLRP3 inflammasome activation via TLR4/MyD88/NF-κB signaling in PMA-stimulated THP-1 monocytes. Biomed Pharmacother. 2016;82:167-72.
265. González-Herrera F, Cramer A, Pimentel P, et al. Simvastatin attenuates endothelial activation through 15-Epi-lipoxin A4 production in murine chronic chagas cardiomyopathy. Antimicrob Agents Chemother. 2017;61:e02137-16.
266. Zhang J, Hao N, Li W, et al. Simvastatin upregulates lipoxin A4 and accelerates neuroinflammation resolution after intracerebral hemorrhage. Curr Neurovasc Res. 2022;19:321-32.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0










Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].