


Rewiring neurotrophic signaling to escape targeted therapy in hepatocellular carcinoma
 0
0 
Acquired resistance remains the major barrier to achieving durable responses in hepatocellular carcinoma (HCC) treated with targeted therapy[1,2]. Although lenvatinib has become a standard first-line option for advanced disease, most patients ultimately experience progression[3,4]. A recent study by Xu et al. provides compelling evidence that HCC cells can evade lenvatinib treatment through activation of a neurotrophic signaling program centered on nerve growth factor (NGF), while also highlighting a potentially actionable combination strategy [Figure 1][5].
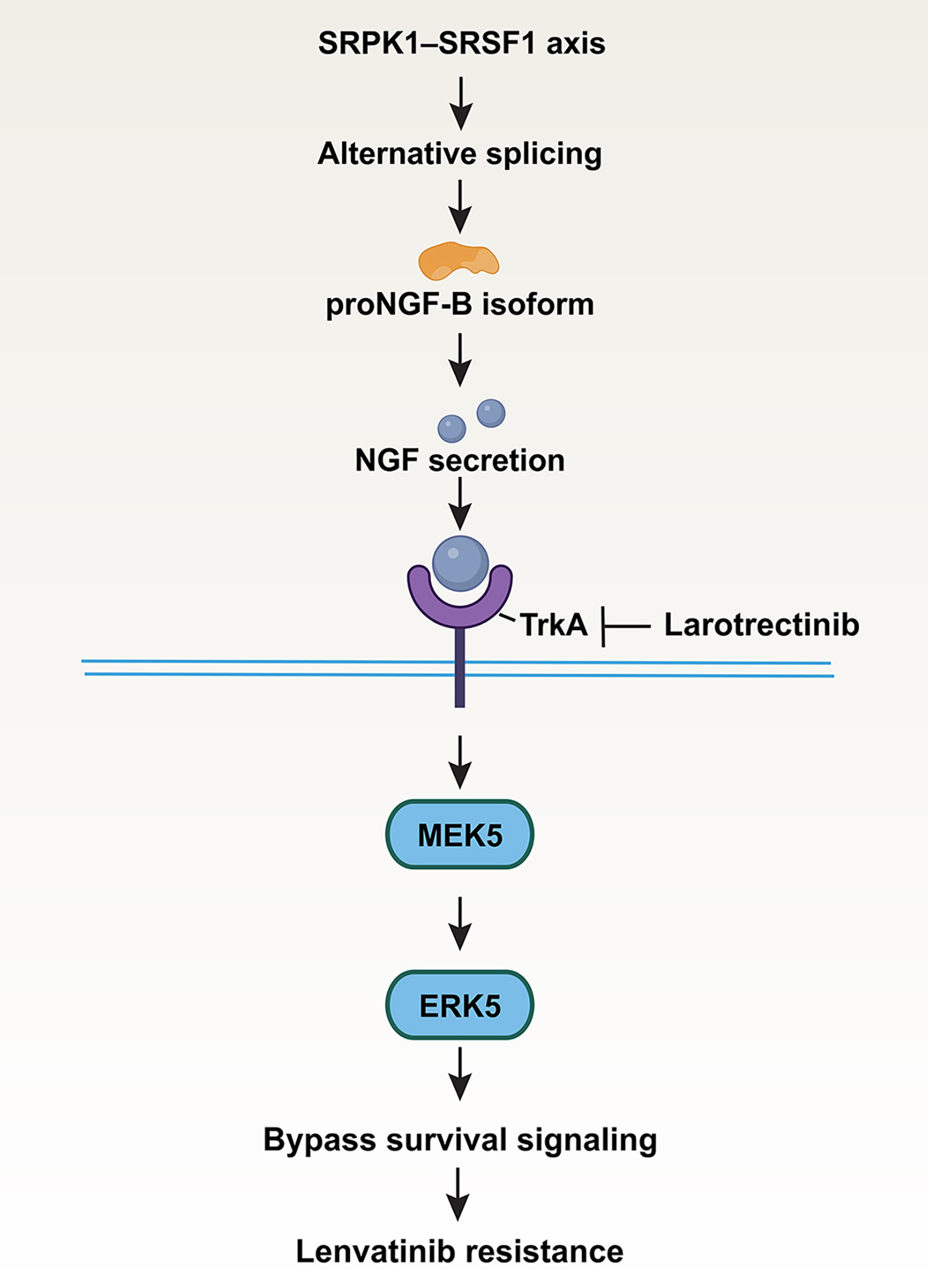
Figure 1. Schematic model of the NGF-driven adaptive signaling pathway underlying lenvatinib resistance in hepatocellular carcinoma. Chronic lenvatinib exposure activates the SRPK1-SRSF1 axis, promoting alternative splicing of NGF pre-mRNA toward the proNGF-B isoform, which exhibits enhanced translational efficiency and leads to increased NGF secretion. Secreted NGF activates TrkA on hepatocellular carcinoma cells, resulting in preferential engagement of the MEK5-ERK5 pathway. This signaling rewiring establishes a bypass survival program that enables tumor cells to evade continued lenvatinib treatment and develop acquired resistance. Pharmacologic inhibition of TrkA with larotrectinib interrupts this adaptive pathway and restores drug sensitivity. The figure was created using Adobe Illustrator. NGF: Nerve growth factor; SRPK1: serine/arginine-rich protein-specific kinase 1; SRSF1: serine/arginine-rich splicing factor 1; TrkA: tropomyosin receptor kinase A; MEK5: mitogen-activated protein kinase kinase 5; ERK5: extracellular signal-regulated kinase 5.
One of the strengths of this study is the resistance model itself. Instead of relying solely on long-term in vitro drug selection, the authors established an in vivo-in vitro cross-circulated model, repeatedly cycling orthotopic tumors through lenvatinib treatment in mice followed by ex vivo expansion[5]. This design better captures the selective pressures imposed by the tumor microenvironment and allows resistance to evolve in a setting that more closely resembles the clinical scenario.
Using conditioned medium transfer and proteomic analysis, the authors identify NGF as a secreted factor that drives resistance. NGF is a well-characterized neurotrophic factor that primarily supports neuronal survival and differentiation, yet in the context of cancer it can be co-opted to promote tumor development[6,7]. Conditioned medium from resistant cells is sufficient to blunt lenvatinib sensitivity in parental HCC cells, and this effect is largely recapitulated by NGF[5]. Conversely, loss of NGF restores drug sensitivity both in vitro and in xenograft models. The observation that recurrent human HCC samples also show elevated NGF expression further strengthens the clinical relevance of this pathway.
The mechanistic work is particularly elegant. Rather than reactivating the canonical mitogen-activated protein kinase kinase 1/2 (MEK1/2)-extracellular signal-regulated kinase 1/2 (ERK1/2) pathway, resistant cells appear to shift their signaling dependence toward the mitogen-activated protein kinase kinase 5 (MEK5)-extracellular signal-regulated kinase 5 (ERK5) axis through activation of tropomyosin receptor kinase A (TrkA)[5]. This rewiring of downstream signaling provides an effective bypass route under continued kinase inhibitor pressure. Such pathway plasticity is increasingly recognized as a hallmark of acquired resistance, and the present study adds a new dimension by implicating neurotrophic signaling in this process.
Although multiple mechanisms of lenvatinib resistance have recently been described in HCC, including metabolic reprogramming driven by protein lactylation, epigenetic remodeling, and suppression of ferroptosis, these studies primarily focus on alterations in metabolism, chromatin regulation, or cell death pathways. In contrast, Xu et al. identify an adaptive mechanism initiated at the post-transcriptional level through serine/arginine-rich protein-specific kinase 1 (SRPK1)-serine/arginine-rich splicing factor 1 (SRSF1)-dependent alternative RNA splicing, resulting in enhanced production of the proNGF-B isoform and activation of an extracellular neurotrophic signaling circuit[5]. This work therefore extends the current landscape of resistance mechanisms by demonstrating that RNA splicing reprogramming can directly coordinate secreted growth factor signaling and bypass kinase inhibition. Importantly, because NGF activates the clinically druggable TrkA receptor, this pathway provides a direct mechanistic rationale for therapeutic intervention with the approved Trk inhibitor larotrectinib, thereby distinguishing it from several previously reported resistance mechanisms.
An additional conceptual advance comes from the upstream regulation of NGF itself. The authors trace NGF upregulation not to transcriptional activation, but to alternative splicing, with a shift toward a shorter proNGF-B isoform that is translated more efficiently. This switch is controlled by the SRPK1-SRSF1 pathway, linking post-transcriptional regulation directly to therapeutic adaptation[5]. This layer of regulation substantially deepens the mechanistic insight of the study and suggests that resistance may emerge through RNA processing programs as much as through classical signaling alterations.
The translational implications are immediate. Pharmacologic inhibition of TrkA with larotrectinib restores lenvatinib sensitivity across multiple preclinical systems, including xenografts, patient-derived organoids, and patient-derived xenograft models. Given that larotrectinib is already clinically available[8,9], this combination strategy may be particularly attractive for rapid translation into biomarker-driven clinical studies.
At the same time, the study opens several important lines of inquiry. What initiates activation of the SRPK1-SRSF1 splicing program during chronic lenvatinib exposure remains unclear. It will also be important to determine whether the NGF-TrkA-ERK5 axis is specific to lenvatinib resistance or represents a broader adaptive pathway engaged by other targeted or immunotherapy-based regimens in HCC. In addition, the possibility that stromal or neural components of the tumor microenvironment contribute to NGF signaling deserves closer attention[10].
Despite these encouraging findings, several challenges should be considered before clinical translation. First, the prevalence of NGF-TrkA activation among patients receiving lenvatinib remains unknown, emphasizing the need for predictive biomarkers to identify those most likely to benefit from combination therapy. Second, although larotrectinib is clinically available, its efficacy and safety in combination with lenvatinib require prospective clinical evaluation. Finally, adaptive resistance to dual-target inhibition may eventually emerge, underscoring the need to identify additional escape mechanisms and optimize rational combination strategies.
More broadly, this work raises the intriguing possibility that tumors under therapeutic stress can co-opt neurotrophic pathways as an adaptive survival mechanism. Whether similar rewiring occurs in other solid tumors may represent an important direction for future investigation.
Taken together, this study expands the current framework of lenvatinib resistance by identifying neurotrophic signaling as an adaptive vulnerability governed by RNA splicing. Beyond revealing a previously unrecognized mechanism of therapeutic escape, it provides a compelling rationale for biomarker-guided combination strategies targeting the NGF-TrkA axis in HCC.
DECLARATIONS
Authors’ contributions
Drafted the manuscript: Tang D
Revised the manuscript: Kang R
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This work was supported by the US National Institutes of Health (Grant No. P50CA295495, Tang D).
Conflicts of interest
Both authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Chen Y, Chen D, Liang Z, et al. Immune checkpoint inhibitors in hepatocellular carcinoma therapy: resistance mechanisms, liver transplantation challenges and management strategies. Cancer Drug Resist. 2025;8:48.
2. Qi D, Qin Y, Zhu H, Li Y, Han S. Resistance of first-line targeted drugs in hepatocellular carcinoma: the epigenetic regulation mechanisms. Cell Death Dis. 2025;16:875.
3. Sun Y, Luo C, Yang H, et al. Lactylation converts ABHD6 into a mitochondrial regulator that drives lenvatinib resistance in hepatocellular carcinoma. Cancer Res. 2026;86:2970-89.
4. Zhang Y, Lin Y, Cai H, Zhou T. EZH2 confers lenvatinib resistance in hepatocellular carcinoma by suppressing ACSL1-Mediated ferroptosis. BMC Cancer. 2025;25:1638.
5. Xu, M. , Zheng, Y., Zhao, L., et al. Activation of nerve growth factor signaling limits the response to lenvatinib in hepatocellular carcinoma. Signal Transduct Target Ther. 2026;11:10.
6. Ferraguti G, Terracina S, Tarani L, et al. Nerve growth factor and the role of inflammation in tumor development. Curr Issues Mol Biol. 2024;46:965-89.
7. Yin T, Wang G, Wang L, et al. Breaking NGF-TrkA immunosuppression in melanoma sensitizes immunotherapy for durable memory T cell protection. Nat Immunol. 2024;25:268-81.
8. Balasubramanian K, Fung KM, McNall-Knapp RY, Balsara K. Sustained response to larotrectinib in a pediatric patient with recurrent STRN3::NTRK2 fusion-positive pilocytic astrocytoma. CNS Oncol. 2025;14:2558455.
9. Yamazaki H, Sugimori M, Saito A. Larotrectinib efficacy for liver metastases in papillary thyroid carcinoma patient harboring SQSTM1-NTRK1 fusion. Surg Case Rep. 2024;10:171.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].