


Incretin-based therapies in MASLD/MASH: from GLP-1 receptor agonists to dual and triple agonists - mechanisms, clinical evidence, and therapeutic perspectives
 0
0 Abstract
Metabolic dysfunction-associated steatotic liver disease (MASLD) and its progressive inflammatory form, metabolic dysfunction-associated steatohepatitis (MASH), constitute an escalating worldwide health challenge, with prevalence estimates reaching one-quarter of adults globally. The pathophysiology of MASLD/MASH encompasses complex interactions among genetic background, impaired insulin signaling, hepatic lipid accumulation, inflammatory cascades, and progressive fibrosis. Until recently, therapeutic strategies remained limited to lifestyle interventions, with few pharmacological options demonstrating meaningful clinical benefit. Glucagon-like peptide-1 receptor agonists (GLP-1 RAs) have garnered recognition as potentially revolutionary therapeutic modalities for MASLD/MASH management, offering multifaceted benefits extending beyond glycemic regulation. These medications exert (in)direct hepatic actions by reducing steatosis, attenuating inflammation, and potentially reversing fibrosis, while simultaneously addressing systemic metabolic abnormalities including excessive obesity, insulin resistance, and cardiovascular risk. Recent phase 2 and phase 3 clinical trials have established the therapeutic value of various GLP-1 RAs in improving liver histology and metabolic parameters in patients with MASH. Furthermore, dual-action compounds engaging GLP-1 combined with glucose-dependent insulinotropic polypeptide (GIP) or glucagon pathways, alongside triple agonists incorporating all three receptors activation, have exhibited enhanced therapeutic potential. This review examines the biological mechanisms underlying GLP-1 RA therapy in MASLD/MASH, analyzes the mechanistic distinctions between mono-, dual-, and triple agonists, and synthesizes current clinical evidence supporting their utilization in individuals with this condition.
Keywords
INTRODUCTION
Metabolic dysfunction-associated steatotic liver disease (MASLD), previously termed nonalcoholic fatty liver disease (NAFLD), has emerged as the predominant hepatic pathology worldwide, impacting roughly twenty-five percent of the adult population[1,2]. This disease spectrum ranges from isolated lipid deposition within hepatocytes through metabolic dysfunction-associated steatohepatitis (MASH), potentially advancing toward cirrhotic transformation, hepatic functional deterioration, and malignant hepatocellular transformation. The increasing prevalence of MASLD mirrors the global obesity and type 2 diabetes mellitus epidemic, reflecting common pathophysiological pathways rooted in metabolic dysfunction.
The clinical features of MASLD extend beyond hepatic manifestations. Individuals with this condition face substantially elevated risks of cardiovascular and chronic renal impairment, extrahepatic malignancies, and overall mortality[3-5]. The progressive form, MASH, exhibits hepatocyte damage, inflammatory cell infiltration, and variable degrees of tissue fibrosis, with advanced fibrosis representing the predominant prognostic determinant for hepatic morbidity and mortality, as well as an additional risk factor for extrahepatic complications. Despite the significant burden of disease, effective pharmacological options remain limited, leaving lifestyle interventions based on dietary modification and physical activity as the mainstay of clinical management[6].
The therapeutic landscape for MASLD is undergoing substantial transformation with emerging incretin-derived therapeutics. Glucagon-like peptide-1 receptor agonists (GLP-1 RAs), initially developed for type 2 diabetes management, have demonstrated remarkable efficacy in addressing the multifactorial pathogenesis of MASLD/MASH[7-10]. These agents operate through various mechanisms affecting both hepatic and systemic metabolism, offering a comprehensive approach to disease management. More recently, dual and triple agonists targeting additional incretin and metabolic hormone receptors have demonstrated enhanced therapeutic potential, revolutionizing the landscape of MASLD treatment[11,12].
This review provides an analysis of GLP-1 RAs and their multi-agonist derivatives as therapeutic agents for MASLD, examining their biological mechanisms and clinical efficacy based on current evidence.
EPIDEMIOLOGY AND DISEASE BURDEN OF MASLD/MASH
MASLD is estimated to impact around 32.4% of adults across the world and is currently considered the leading chronic liver disease globally, with estimates exceeding one billion individuals affected[13,14]. Prevalence is highest in South America and the Middle East (~44%), followed by Asia (27%), Europe (25%), North America (24%), and Africa (14%). The global burden has increased substantially over the last twenty years, mirroring the parallel epidemics of obesity and type 2 diabetes mellitus. Among individuals with obesity, MASLD prevalence exceeds 75%, whereas type 2 diabetes is associated with MASLD in 55%-70% of cases. Approximately 20%-30% of individuals with MASLD progress to MASH, characterized by hepatocyte ballooning, lobular inflammation, and variable fibrosis, and 10%-20% of MASH patients develop advanced fibrosis (stages F3-F4) or cirrhosis over their lifetime[6,13]. Hepatic fibrosis stage is the main predictor of liver-related morbidity and mortality: each unit increase in fibrosis stage confers a 2-fold increase in liver-related mortality and an approximately 1.5-fold increase in overall mortality[14].
Beyond liver-specific outcomes, MASLD exerts a substantial extrahepatic disease burden. Cardiovascular disease is the main cause of mortality in individuals with MASLD, who face a two- to threefold greater risk of both fatal and non-fatal cardiovascular events than the general population[14]. Metabolic comorbidities are highly prevalent and bidirectionally linked to MASLD: type 2 diabetes, hypertension, dyslipidemia, and chronic kidney disease (CKD) cluster together, collectively amplifying hepatic and systemic disease progression. CKD affects 20%-55% of MASLD patients and shares pathogenic mechanisms with MASLD through insulin resistance, oxidative stress, and systemic inflammation[15]. Extrahepatic malignancies - particularly colorectal, breast, and pancreatic cancer - are more common in MASLD patients independent of metabolic syndrome components[3-5]. Health-related quality of life is significantly impaired across all fibrosis stages, with impairments intensifying with disease severity. The economic burden is considerable: annual direct healthcare costs attributable to MASLD in the United States and Europe are estimated in the tens of billions of dollars, driven by hospitalizations, liver-related procedures, and management of comorbid conditions[6]. This context of high prevalence, progressive natural history, and multisystemic impact underscores the urgent need for effective, broadly applicable pharmacotherapies.
PATHOGENESIS OF MASLD/MASH
The development and progression of MASLD/MASH involve intricate interconnections among multiple metabolic, inflammatory, and fibrogenic mechanisms[2,6,16]. The pathogenesis originates from an imbalance in hepatic lipid homeostasis, where excessive triglyceride accumulation results from increased lipid influx, enhanced de novo lipogenesis, and impaired lipid export or oxidation. A key driver of this process is insulin resistance, which affects both hepatic and peripheral tissues, creating a metabolic environment that simultaneously promotes hyperglycemia and hepatic steatosis through selective pathway activation.
Progression from simple steatosis to MASH involves cellular stress pathways triggered by lipotoxicity, endoplasmic reticulum stress, mitochondrial dysfunction, and oxidative injury. These cellular stressors activate inflammatory cascades involving Kupffer cells, recruited macrophages, and other immune cells, perpetuating hepatic inflammation through secretion of inflammatory mediators and cellular damage signals[7]. The inflammatory milieu subsequently drives hepatic stellate cell activation and transdifferentiation into myofibroblasts, leading to excessive extracellular matrix deposition and progressive fibrosis, which represents the primary determinant of long-term liver-related outcomes[17].
Genetic analyses have demonstrated that MASLD is a heterogeneous disorder driven by multiple molecular mechanisms rather than a single pathogenic pathway. Genome-wide association analyses have detected critical polymorphisms in genes regulating hepatic lipid handling, including patatin-like phospholipase domain-containing 3 (PNPLA3) and transmembrane 6 superfamily member 2 (TM6SF2), which promote intrahepatic triglyceride accumulation by impairing lipid remodeling or very-low-density lipoprotein (VLDL) secretion[16,17]. Other loci, such as glucokinase regulator (GCKR) and membrane bound O-acyltransferase domain-containing 7 (MBOAT7), influence glucose metabolism, phospholipid remodeling, and insulin sensitivity, highlighting the tight link between metabolic dysfunction and liver injury. Importantly, not all variants confer increased risk: loss of function variants in HSD17B13, a lipid droplet-associated protein in hepatocytes, have been consistently associated with reduced hepatic inflammation and a lower risk of fibrosis and advanced liver disease[16]. These insights provide a framework for precision medicine, where genetic profiling may help refine risk stratification and guide targeted therapeutic strategies.
The intestinal-hepatic crosstalk constitutes a pivotal component of MASLD development, linking intestinal dysbiosis to hepatic inflammation and fibrosis[1]. Modifications in intestinal microbial structure compromise barrier integrity, facilitating bacterial product migration into the portal circulation. These microbial-derived signals activate hepatic immune pathways, including toll-like receptor-mediated responses, thereby promoting inflammation, hepatocellular injury, and fibrogenesis. In addition, changes in microbial metabolism influence bile acid signaling and the production of short-chain fatty acids, further modulating lipid metabolism, insulin resistance, and inflammatory pathways[1]. Together, these mechanisms highlight the bidirectional interaction between gut and liver and support therapeutic strategies targeting microbiota composition and intestinal barrier integrity in MASLD.
Beyond hepatic manifestations, MASLD is intrinsically linked to systemic metabolic disorder. Dysfunction of adipose tissue is a primary driver of MASLD development and progression. When subcutaneous adipose tissue loses its capacity to safely store excess energy, lipid spillover toward the liver increases, promoting hepatic steatosis. Concurrently, inflamed adipose tissue secretes elevated free fatty acids, pro-inflammatory cytokines, and adipokines that exacerbate hepatic insulin resistance, lipotoxicity, and fibrogenesis[2]. This framework highlights inter-organ crosstalk and positions adipose tissue health as a key determinant of liver outcomes, with important implications for risk stratification and therapeutic targeting. This systemic nature necessitates therapeutic approaches that address both hepatic and extra-hepatic metabolic abnormalities, positioning incretin-based therapies as particularly promising interventions[2].
MECHANISMS OF ACTION OF GLP-1 RAs AND MULTI-AGONIST THERAPIES
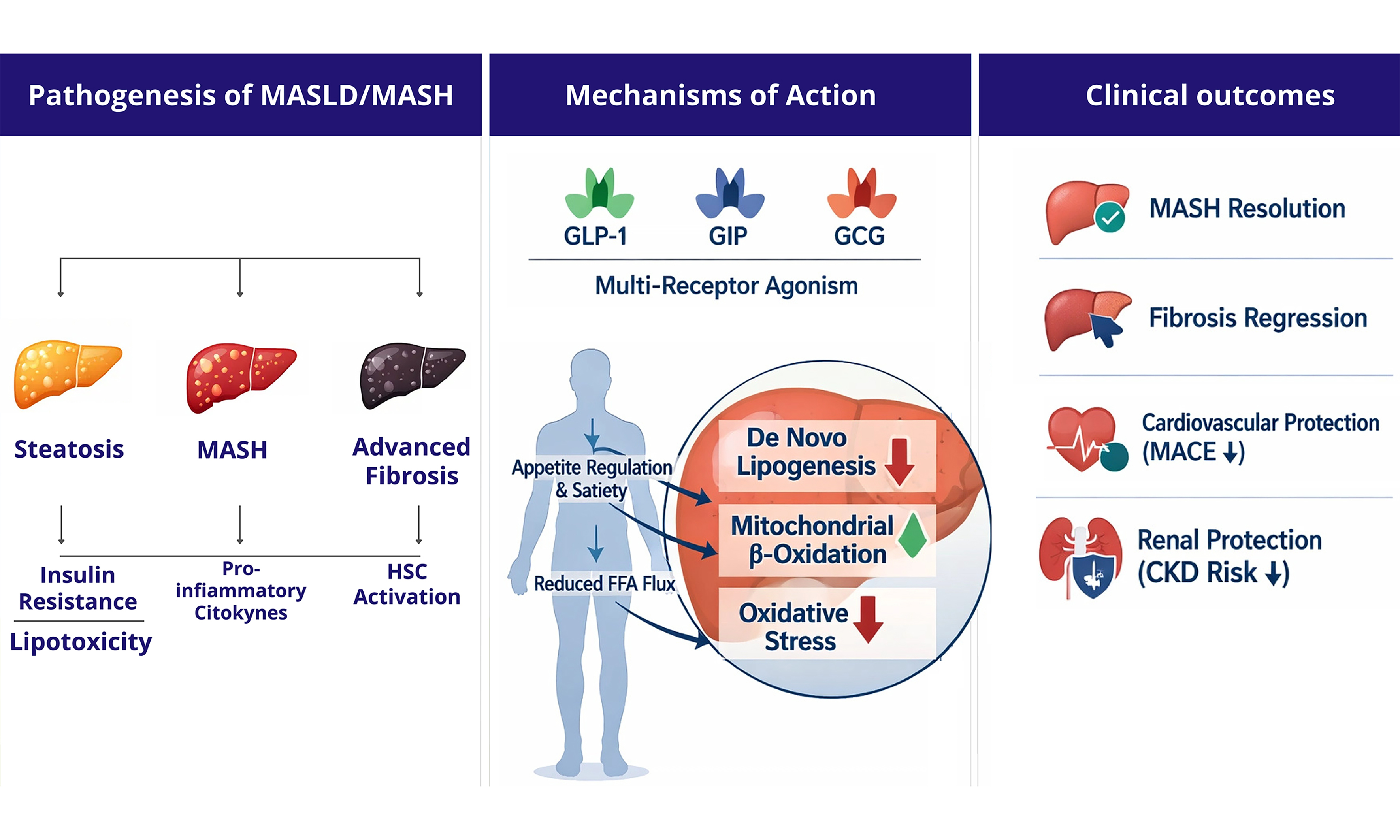
Incretin-based therapies leverage the physiological roles of gut-derived peptide hormones, principally glucagon-like peptide-1 (GLP-1), glucose-dependent insulinotropic polypeptide (GIP), and glucagon receptor (GCG) to address the multifaceted pathophysiology of MASLD. Understanding the distinct and complementary contributions of each receptor system is essential to appreciating how mono-, dual-, and triple agonist formulations achieve their therapeutic effects[7-9,18,19] [Figure 1 and Table 1].
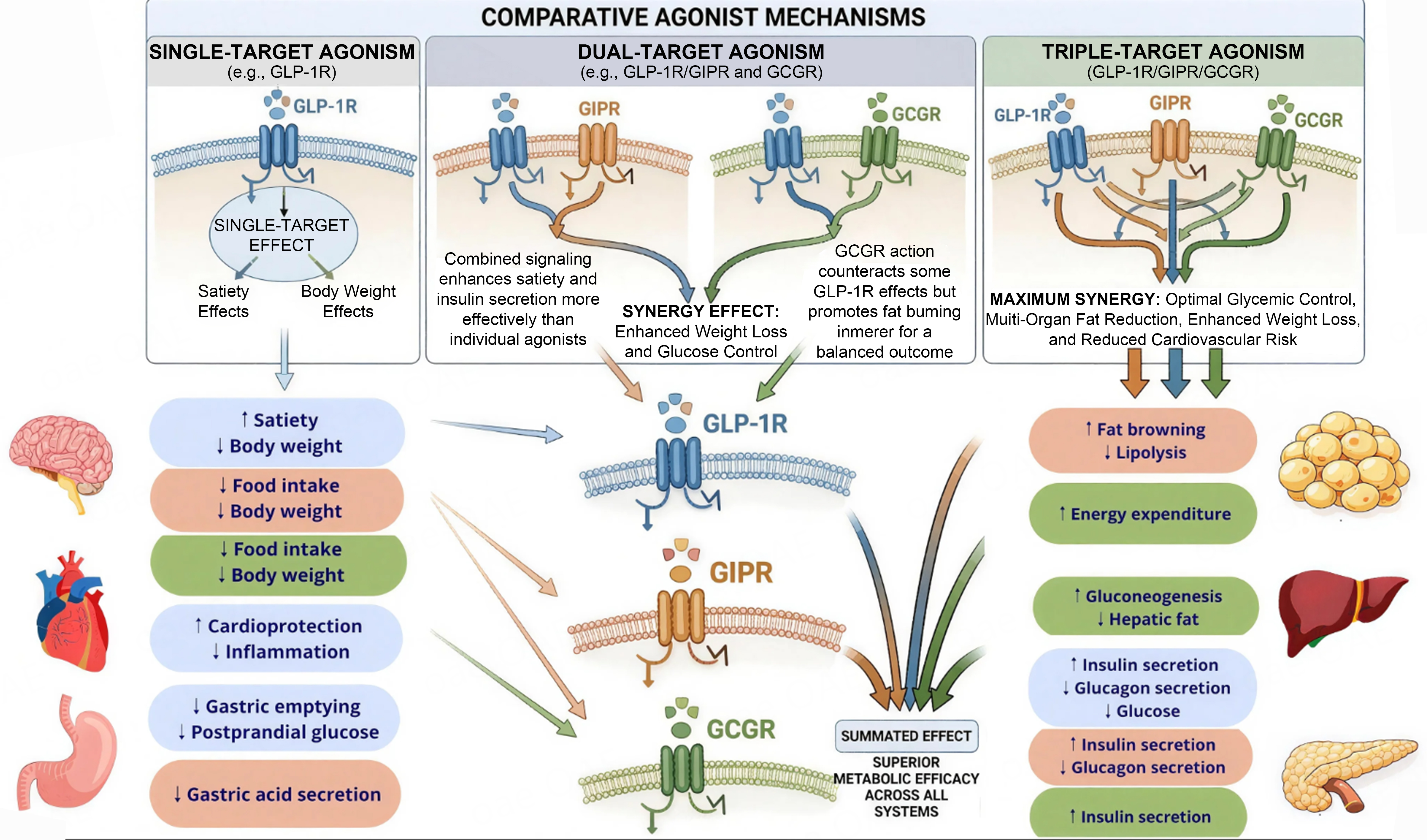
Figure 1. Mechanisms of action of GLP-1, GIP and GCG receptors in human target organs. This image was created by us with Canva. Figure illustrates the mechanisms of action of three incretin and metabolic hormone receptor systems - GLP-1R, GIPR, and GCGR - across key target organs involved in metabolic regulation. GLP-1R activation, depicted in blue, exerts a broad spectrum of effects spanning multiple organ systems. In the central nervous system, GLP-1R signaling within hypothalamic circuits increases satiety and reduces food intake, ultimately promoting body weight reduction. At the gastric level, it slows gastric emptying and reduces both postprandial glucose excursions and gastric acid secretion. In the cardiovascular system, GLP-1R engagement exerts cardioprotective effects and attenuates systemic inflammation. At the pancreas, it potentiates glucose-stimulated insulin secretion while suppressing glucagon release, thereby lowering circulating glucose concentrations. GIPR activation, shown in orange, primarily targets adipose tissue and the pancreas. In adipose tissue, GIPR signaling promotes fat browning, modulates lipolysis, and increases energy expenditure, thereby enhancing the metabolic flexibility of adipose depots and reducing ectopic lipid accumulation. At the pancreatic level, GIPR provides an additive insulinotropic stimulus complementing GLP-1R-mediated effects. GCGR activation, represented in green, is unique in that glucagon receptors are robustly expressed on hepatocytes, enabling direct liver-directed metabolic effects - a mechanistic feature absent from both GLP-1R and GIPR signaling. Hepatic GCGR activation stimulates gluconeogenesis while simultaneously reducing hepatic fat content through inhibition of de novo lipogenesis and promotion of mitochondrial fatty acid oxidation. Additionally, GCGR engagement at the pancreas further supports insulin secretion through paracrine signaling; at brain level, increases satiety and reduces food intake. Taken together, the figure highlights a key mechanistic principle underlying the therapeutic rationale for multi-agonist strategies: while GLP-1R monotherapy achieves meaningful metabolic and hepatic benefits exclusively through indirect, systemic pathways, the addition of GIPR and - most importantly - GCGR co-agonism introduces complementary and increasingly direct hepatic mechanisms, providing the pharmacological basis for the superior efficacy observed with dual and triple agonists in MASLD/MASH. GCG: Glucagon; GCGR: glucagon receptor; GIP: glucose-dependent insulinotropic polypeptide; GIPR: glucose-dependent insulinotropic polypeptide receptor; GLP-1: glucagon-like peptide-1; GLP-1R: glucagon-like peptide-1 receptor; MASLD: metabolic dysfunction-associated steatotic liver disease; MASH: metabolic dysfunction-associated steatohepatitis.
Comparative metabolic and hepatic effects of GLP-1-based mono-, dual-, and triple-agonist therapies
| Feature | GLP-1 RA (mono) | GLP-1/GIP (dual) | GLP-1/Glucagon (dual) | GLP-1/GIP/Glucagon (triple) |
| Insulin secretion | ↑↑ | ↑↑↑ | ↑↑ | ↑↑↑ |
| Appetite/weight loss | ↑↑ (5%-15%) | ↑↑↑ (15%-22%) | ↑↑↑ (10%-15%) | ↑↑↑↑ (~24%) |
| Hepatic fat reduction | Moderate (indirect via insulin sensitization, weight loss) | Pronounced (↑adipose lipid buffering + weight loss) | Pronounced (direct hepatic ↑β-oxidation, ↓lipogenesis) | Maximal (synergistic mechanisms; ~90% normalization at high dose)[20] |
| Direct hepatic action | Absent (no hepatocyte GLP-1R); systemic/indirect only[7,8] | Mainly indirect (adipose GIP-R); ↑lipid buffering[21,22] | Direct hepatic (glucagon-R expressed in hepatocytes): ↑β-oxidation, ↓lipogenesis, ↑follistatin | Direct + indirect (all three) |
| Anti-inflammatory effect | Systemic: ↓TNF-α, IL-1β, IL-6; modulation of hepatic macrophages (indirect) | Systemic (similar to GLP-1 RA) + possible adipose anti-inflammatory effects[11] | Systemic; hepatic inflammation reduction secondary to steatosis improvement[23] | Synergistic systemic + hepatic anti-inflammatory effects |
| Antifibrotic effect | Indirect (via steatosis/inflammation resolution); preclinical evidence of HSC inhibition | Indirect; additive benefit vs. GLP-1RA monotherapy | Indirect (secondary to steatosis); preclinical fibrosis improvement | Strongest preclinical and emerging clinical antifibrotic signal[24,25] |
| Cardiovascular benefit | Established (SELECT)[3,4] | Established | Data emerging; CV outcomes trial ongoing | Data emerging; CV outcomes trial planned |
| Main tolerability concern | GI (nausea, vomiting; ~5%-15% rate) | GI (similar or slightly attenuated vs. GLP-1 RA due to GIP co-agonism) | GI more pronounced (nausea/vomiting up to 66%); higher discontinuation rate | GI; dose-titration required; data still emerging |
GLP-1 receptor signaling
Intestinal L-cells serve as the primary source of GLP-1 synthesis, which operates via G-protein coupled receptors present in pancreatic beta cells, gastrointestinal and central neural systems, cardiac tissues, and kidneys[26]. GLP-1 originates from the cleavage of preproglucagon, a 160-amino acid precursor whose expression, driven by the GCG gene, occurs in pancreatic α-cells and intestinal cells. Its metabolic journey is a prime example of tissue-specific post-translational processing, where the same precursor is cleaved into different bioactive peptides depending on the enzymes present in the cell[27] [Figure 2].
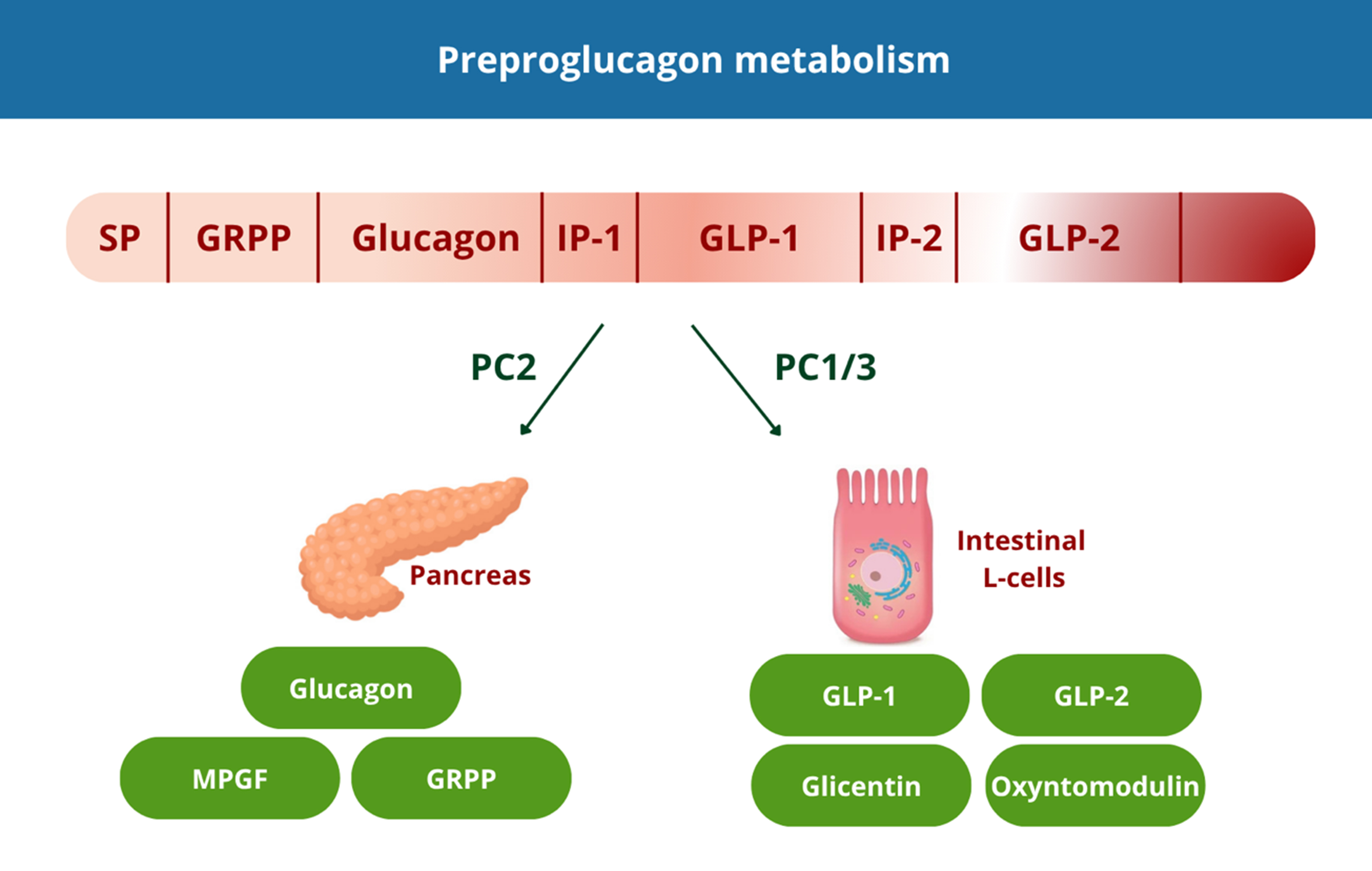
Figure 2. Tissue specific processing of preproglucagon. This image was created by us with Canva. SP: Signal peptide; GRPP: glicentin-related pancreatic polypeptide; IP-1: intervening peptide 1; GLP-1: glucagon-like peptide-1; IP-2: intervening peptide 2; GLP-2: glucagon-like peptide-2. MPGF: major proglucagon fragment.
Preproglucagon is a 160-amino acid precursor encoded by the GCG gene and expressed in pancreatic α-cells, intestinal cells. The same precursor is differentially cleaved by two prohormone convertases depending on the tissue context. In pancreatic α-cells, PC2 cleaves proglucagon to yield mature glucagon (29 aa), glicentin-related pancreatic polypeptide (GRPP) and the major proglucagon fragment (MPGF). Glucagon promotes hepatic glucose output and is secreted primarily during fasting. In intestinal L-cells - concentrated in the distal ileum and colon - PC1/3 processes proglucagon into GLP-1, GLP-2, glicentin, and oxyntomodulin. GLP-1 acts as a potent incretin, stimulating glucose-dependent insulin secretion postprandially.
While GLP-1 receptor expression is absent in hepatocytes, GLP-1 receptor agonism exerts profound hepatic benefits through multiple indirect mechanisms. The primary pathway involves amplification of glucose-triggered insulin production by pancreatic beta cells, which enhances adipose tissue nutrient uptake and reduces circulating free fatty acid flux to the liver[28]. This insulin-mediated improvement in systemic insulin sensitivity reduces hepatic lipid accumulation from peripheral sources.
Additionally, GLP-1 receptor activation indirectly suppresses hepatic de novo lipogenesis through downregulation of lipogenic transcription factors and enzymes, independent of weight loss[29]. GLP-1 also reduces hepatic VLDL release and decreases enterocyte chylomicron production, further limiting hepatic triglyceride delivery[30]. These coordinated actions on lipid metabolism contribute to steatosis resolution. Furthermore, GLP-1 RAs exhibit anti-inflammatory effects, lowering circulating levels of pro-inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β), and IL-6, while promoting anti-inflammatory mediator production[26]. Although precise hepatic anti-inflammatory mechanisms remain incompletely defined, proposed pathways include reduction of lipotoxicity-induced cellular stress, modulation of hepatic macrophage activity, and direct effects on inflammatory signaling cascades[26].
Regarding fibrosis, while direct anti-fibrotic mechanisms are less established, preclinical studies indicate that GLP-1 RAs could suppress fibrogenic cell activation and reduce expression of pro-fibrotic genes encoding extracellular matrix proteins, potentially facilitating fibrosis regression in conjunction with improvements in steatosis and inflammation[29-32]. In a recent study, Jara et al. evaluated the molecular mechanisms through which semaglutide mediated resolution of MASH[17]. They showed that semaglutide enhanced histological parameters of fibrosis and inflammation while decreasing hepatic expression of genes associated with fibrotic and inflammatory cascade: through proteomic profiling of serum from MASH patients treated with semaglutide, they detected 72 proteins linked to disease resolution and validated these findings in preclinical mouse models and an independent patient cohort[28].
Preclinical studies in accelerated mouse models of diet-induced steatohepatitis have demonstrated that retatrutide improves steatohepatitis through mechanisms involving enhanced hepatic fat oxidation, reduced lipogenesis, decreased inflammation, and attenuation of fibrosis markers, supporting the rationale for clinical development in MASH[33]. Combined activation of GLP-1, GIP, and GCG systems theoretically provides complementary mechanisms: GLP-1-mediated insulin sensitization and satiety promotion, GIP-mediated enhancement of insulin secretion and adipose lipid buffering, and glucagon-mediated direct hepatic lipid oxidation and energy expenditure augmentation.
GLP-1 receptor activation exerts profound systemic effects that indirectly benefit hepatic steatosis and MASH through multiple complementary mechanisms. At the systemic level, GLP-1 RAs improve insulin sensitivity in peripheral tissues (muscle, adipose), reduce circulating free fatty acid flux to the liver, suppress hypothalamic appetite circuits to promote weight loss, and lower systemic pro-inflammatory cytokines (TNF-α, IL-1β, IL-6), thereby reducing the hepatic inflammatory milieu indirectly[7,26,30]. Central nervous system-mediated appetite regulation represents a primary mechanism through which GLP-1 RAs induce weight reduction and subsequently improve hepatic steatosis. Key hypothalamic centers, including the arcuate and paraventricular nuclei, exhibit high GLP-1 receptors expression, which coordinate peripheral metabolic cues to modulate feeding behavior and energy balance[34]. Through stimulation of pro-opiomelanocortin (POMC) neurons while simultaneously inhibiting neuropeptide Y/agouti-related peptide neurons in the arcuate nucleus, GLP-1 receptor agonism promotes satiety, reduces meal size, and decreases overall caloric intake[35]. Importantly, GLP-1 appears to attenuate reward-driven food consumption by modulating dopaminergic signaling in the mesolimbic system, potentially reducing preference for high-fat, energy-dense foods[36]. These central effects translate into substantial weight loss - typically 5%-15% of body weight with current GLP-1 RAs - which directly ameliorates hepatic steatosis by reducing adipose tissue mass, reducing hepatic free fatty acid influx, and improving insulin sensitivity.
Beyond appetite suppression, GLP-1 RAs improve systemic glucose homeostasis through pancreatic and extra-pancreatic mechanisms. Potentiation of glucose-stimulated insulin release from pancreatic beta cells embodies the classic incretin action, reducing postprandial hyperglycemia and chronic hyperinsulinemia[35,36]. Concomitant suppression of inappropriate glucagon secretion from alpha cells further improves glycemic control by reducing hepatic glucose production during fed states[36]. These pancreatic effects are complemented by improved peripheral insulin responsiveness in muscle tissue and lipid storage sites, facilitating glucose disposal and reducing systemic insulin resistance - a key driver of hepatic lipid accumulation and MASH progression[28,31]. Moreover, GLP-1 RAs slow gastric emptying, thereby blunting postprandial glucose and lipid excursions, though diminished tolerance to this effect may develop during prolonged administration of extended-release preparations.
Cardiovascular therapeutic effects of GLP-1 RAs extend beyond traditional cardiometabolic risk factor improvement. Trial data have shown decreases in serious cardiovascular complications, including cardiovascular mortality, nonlethal heart attacks, and nonfatal cerebrovascular accidents, among individuals with type 2 diabetes and pre-existing cardiovascular pathology or high cardiovascular risk[3,5,13,14,37].
These benefits appear to reflect both improvements in traditional risk factors - weight, blood pressure, lipid profiles, glycemic control - and potentially direct cardioprotective effects mediated by GLP-1 receptors expressed in cardiomyocytes and vascular endothelium. Given the strong association between MASLD and cardiovascular pathology, that represents the primary cause of death in this population, the cardioprotective benefits of GLP-1 RAs provide important additional clinical value beyond hepatic benefits.
Renal protective effects have emerged as another important extra-hepatic benefit[15].
Collectively, these extra-hepatic effects create a favorable systemic metabolic environment that indirectly but substantially benefits hepatic steatosis and MASH.
GIP receptor signaling
GIP, produced by duodenal K-cells, shares insulinotropic properties with GLP-1 but exhibits distinct receptor expression patterns and physiological effects. GIP receptors are highly expressed in pancreatic beta cells and adipose tissue, but are present at minimal or negligible levels in the liver and skeletal muscle, suggesting tissue-specific mechanisms. The additive insulinotropic effect of GIP combined with GLP-1 results from direct beta-cell stimulation and enhancement of alpha-to-beta cell paracrine communication, with GIP contributing substantially to postprandial insulin secretion[30,31]. Importantly, GIP receptor sensitivity is impaired in prolonged hyperglycemic states due to receptor downregulation but can be restored upon achieving euglycemia through complementary GLP-1 action[38,39].
Beyond pancreatic effects, GIP receptor agonism enhances peripheral insulin sensitivity, particularly in adipose tissue, where it promotes healthy white adipose tissue (WAT) expansion, facilitates dietary triglyceride clearance through increased lipoprotein lipase activity and adipose blood flow, and improves lipid buffering capacity[40]. This enhanced adipose function reduces ectopic lipid deposition in liver and other tissues, indirectly ameliorating hepatic steatosis. Additionally, GIP receptor expression in hypothalamic regions and oligodendrocytes suggests central nervous system actions that may synergize with GLP-1 to reduce appetite and food intake while potentially mitigating GLP-1-associated nausea, thereby improving treatment tolerability[36,41,42].
GCG signaling
Unlike GLP-1 and GIP receptors, GCG show robust hepatic expression, enabling direct hepatic metabolic effects. Hepatic glucose homeostasis during fasting or hypoglycemia depends critically on GCG signaling, which activates both glycogen breakdown and gluconeogenic pathways to prevent dangerous decreases in circulating glucose levels. GCG activation suppresses endogenous hepatic lipid production by inhibiting lipogenic enzymatic pathways while simultaneously enhancing mitochondrial oxidative lipid catabolism, thereby promoting lipid catabolism and reducing hepatic triglyceride content[18].
Furthermore, glucagon stimulates the secretion of follistatin, a hepatokine that inhibits hepatic lipid influx and de novo lipogenesis, providing additional hypolipidemic effects[20]. While the inflammation-reducing and fibrosis-attenuating effects of glucagon agonism are less well-characterized, the reductions in hepatic inflammation parameters and fibrosis observed in preclinical models likely reflect secondary effects consequent to improved steatosis, rather than direct anti-inflammatory actions[21]. Glucagon also contributes to energy homeostasis by promoting satiety through vagal signaling to brainstem nuclei and by increasing resting energy expenditure through facultative thermogenesis in brown adipose tissue, skeletal muscle, and through promotion of WAT browning[22]. These effects on energy balance complement incretin-mediated appetite suppression, facilitating greater weight loss.
Boland et al. employed a diet-induced NASH mouse model to compare the efficacy of cotadutide (a dual GLP-1/GCG agonist) against liraglutide (selective GLP-1 receptor agonist) and pair-fed controls. Both cotadutide and liraglutide markedly diminished liver steatosis, inflammatory activity, and fibrosis compared to ad libitum-fed controls. However, cotadutide demonstrated superior performance in decreasing liver triglyceride levels and ameliorating mitochondrial functional markers in the liver compared to liraglutide, despite comparable reductions in body weight and food intake between the two treatments[43]. Mechanistic analyses revealed that cotadutide’s enhanced hepatic benefits were associated with increased mitochondrial β-oxidation capacity, improved mitochondrial respiratory chain function and more pronounced suppression of de novo lipogenesis enzymes. These findings suggest that the additional GCG agonism in cotadutide provides direct metabolic benefits to hepatocytes beyond those achievable through GLP-1 receptor activation alone, even when caloric restriction is matched[43].
In summary, the distinct receptor expression patterns and physiological roles of GLP-1, GIP, and glucagon create complementary mechanisms that, when engaged through mono-, dual-, or triple agonist formulations, address multiple pathogenic pathways in MASLD. GLP-1 receptor activation chiefly enhances systemic insulin sensitivity and decreases liver lipid accumulation through indirect mechanisms. GIP receptor agonism amplifies insulinotropic effects, enhances adipose tissue function to prevent ectopic lipid deposition, and may improve treatment tolerability. GCG agonism provides direct hepatic effects by inhibiting lipogenesis and promoting fat oxidation while augmenting energy expenditure. These integrated actions position incretin-based therapies as uniquely suited to address the complex, multifactorial pathophysiology of MASLD[19].
CLINICAL TRIAL EVIDENCE FOR GLP-1 RAs IN MASH
Table 2 provides a summary of phase 2 and phase 3 studies examining GLP-1 RAs and dual or triple agonists (aiming glucagon, GIP, and GLP-1 receptors) in individuals with MASLD/MASH, where hepatic biopsies were used to evaluate liver outcomes[3,5,10,11,15,22-25,44-52].
Overview of main clinical studies for MASLD/MASH with GLP-1 receptor agonists and dual or triple agonists targeting glucagon/GIP/GLP-1 receptors
| Drug | Phase | Population | Study arms | Patients, n | Duration (weeks) | Results | Main adverse events |
| Liraglutide | 2 | MASH | Placebo | 26 | 48 | MASH resolution without fibrosis worsening: 9% Fibrosis improvement: 14%[44] | Nausea: 46% Diarrhea: 38% Constipation: 27% Serious adverse events: 8%[44] |
| 1.8 mg daily | 26 | 48 | MASH resolution without fibrosis worsening: 39% Fibrosis improvement: 26%[44] | Nausea: 38% Diarrhea: 19% Constipation: 0% Serious adverse events: 8%[44] | |||
| Semaglutide | 3 | MASH with fibrosis F2-F3 | Placebo | 266 | 72 | MASH resolution without fibrosis worsening: 34.3% Fibrosis improvement ≥ 1 stage: 22.4%[10] | Nausea: 13.2% Diarrhea: 12.2% Constipation: 8.4% Gallbladder disorders: 1.5% Serious adverse events: 13.4%[10] |
| 2.4 mg weekly | 534 | 72 | MASH resolution without fibrosis worsening: 62.9% Fibrosis improvement ≥ 1 stage: 36.8%[10] | Nausea: 36.2% Diarrhea: 26.9% Constipation: 22.2% Gallbladder disorders: 2.5% Serious adverse events: 13.4%[10] | |||
| Tirzepatide | 2 | MASH with fibrosis F2-F3 | Placebo | 48 | 52 | MASH resolution without fibrosis worsening: 10% Fibrosis improvement ≥ 1 stage: 30%[11] | Nausea: 12% Diarrhea: 23% Constipation: 6% Serious adverse events: 6%[11] |
| 5 mg weekly | 47 | 52 | MASH resolution without fibrosis worsening: 44% Fibrosis improvement ≥ 1 stage: 55%[11] | Nausea: 36% Diarrhea: 32% Constipation: 23% Serious adverse events: 11%[11] | |||
| 10 mg weekly | 47 | 52 | MASH resolution without fibrosis worsening: 56% Fibrosis improvement ≥ 1 stage: 51%[11] | Nausea: 34% Diarrhea: 36% Constipation: 19% Serious adverse events: 9%[11] | |||
| 15 mg weekly | 48 | 52 | MASH resolution without fibrosis worsening: 62% Fibrosis improvement ≥ 1 stage: 51%[11] | Nausea: 44% Diarrhea: 27% Constipation: 15% Serious adverse events: 0%[11] | |||
| Survodutide | 2 | MASH with fibrosis F1-F3 | Placebo | 74 | 48 | MASH improvement without fibrosis worsening: 14% Fibrosis improvement ≥ 1 stage: 22%[23] | Nausea: 23% Diarrhea: 23% Constipation: 15% Serious adverse events: 7%[23] |
| 2.4 mg weekly | 73 | 48 | MASH improvement without fibrosis worsening: 47% Fibrosis improvement ≥ 1 stage: 34%[23] | Nausea: 63% Diarrhea: 41% Constipation: 21% Serious adverse events: 5%[23] | |||
| 4.8 mg weekly | 72 | 48 | MASH resolution without fibrosis worsening: 62% Fibrosis improvement ≥ 1 stage: 36%[23] | Nausea: 68% Diarrhea: 56% Constipation: 17% Serious adverse events: 10%[23] | |||
| 6 mg weekly | 72 | 48 | MASH resolution without fibrosis worsening: 43% Fibrosis improvement ≥ 1 stage: 34%[23] | Nausea: 66% Diarrhea: 50% Constipation: 26% Serious adverse events: 8%[23] | |||
| Pemvidutide | 2b | MASH with fibrosis F2-F3 | Placebo | 86 | 24 | MASH resolution without fibrosis worsening: 20% Fibrosis improvement without worsening of MASH: 28%[49-51] | Nausea: 14% Diarrhea: 8% Constipation: 9% Serious adverse events: 3%[49-51] |
| 1.2 mg weekly | 41 | 24 | MASH resolution without fibrosis worsening: 58% Fibrosis improvement without worsening of MASH: 33%[49-51] | Nausea: 22% Diarrhea: 10% Constipation: 12% Serious adverse events: 2%[49-51] | |||
| 1.8 mg weekly | 85 | 24 | MASH resolution without fibrosis worsening: 52% Fibrosis improvement without worsening of MASH: 36%[49-51] | Nausea: 41% Diarrhea: 21% Constipation: 13% Serious adverse events: 4%[49-51] | |||
| Efinopegdutide | 2a | NAFLD (LFC ≥ 10%) | Semaglutide 1 mg weekly | 73 | 24 | Liver fat content reduction: 42.3%[52] | Nausea: 31.5% Diarrhea: 17.8% Constipation: 5.5% Serious adverse events: 1.4%[52] |
| Efinopegdutide 10 mg weekly | 72 | 24 | Liver fat content reduction: 72.7%[52] | Nausea: 27.8% Diarrhea: 16.7% Constipation: 16.7% Serious adverse events: 1.4%[52] | |||
| Retatrutide | 2 | Obesity + MASLD (LFC ≥ 10%) | Placebo | 19 | 48 | Mean relative change: 0.3%[24,25] | Nausea: 5.3% Diarrhea: 5.3% Constipation: 5.3% Serious adverse events: 0%[24,25] |
| 1 mg weekly | 20 | 48 | Mean relative change: -42.9%[24,25] | Nausea: 20% Diarrhea: 20% Constipation: 5% Serious adverse events: 0%[24,25] | |||
| 4 mg weekly | 19 | 48 | Mean relative change: -57%[24,25] | Nausea: 21.1% Diarrhea: 0% Constipation: 5.3% Serious adverse events: 5.3%[24,25] | |||
| 8 mg weekly | 22 | 48 | Mean relative change: -81.4%[24,25] | Nausea: 54.5% Diarrhea: 27.3% Constipation: 18.2% Serious adverse events: 4.5%[24,25] | |||
| 12 mg weekly | 18 | 48 | Mean relative change: -82.4%[24,25] | Nausea: 38.9% Diarrhea: 16.7% Constipation: 27.8% Serious adverse events: 0%[24,25] |
Liraglutide
Liraglutide, a once-daily GLP-1 receptor agonist with authorized indications for type 2 diabetes and obesity, was evaluated in the LEAN trial (NCT01237119), a phase 2 randomized controlled study enrolling 52 adults with biopsy-proven MASH[22]. Participants received subcutaneous liraglutide 1.8 mg daily or placebo for a 48 week period. Histological resolution of MASH without fibrosis worsening - the study’s primary outcome - occurred in 39% of liraglutide recipients vs. only 9% of placebo-treated patients, achieving statistical significance (P = 0.019). While liraglutide demonstrated lower rates of fibrosis worsening compared to placebo (9% vs. 36%, P = 0.04), improvement in fibrosis stage did not differ significantly between groups (26% with liraglutide vs. 14% with placebo, P = 0.46)[44].
Secondary outcomes demonstrated significant improvements in metabolic parameters. Body weight reduction averaged 5.5% in the liraglutide arm compared to 0.7% with placebo (P = 0.003), while glycemic control improved as reflected by a mean HbA1c decrease of 5.7 mmol/mol (P = 0.03). Liraglutide therapy exhibited good safety tolerability, with gastrointestinal symptoms representing the predominant adverse events: nausea affected 46% of liraglutide users vs. 38% receiving placebo, diarrhea occurred in 38% vs. 19%, constipation in 27% vs. 0%, and vomiting in 19% vs. 12%. These gastrointestinal events were predominantly of minor to intermediate severity. Adverse event-related discontinuation rates were 8% among liraglutide-treated patients compared to 4% of placebo recipients[44].
Semaglutide
Semaglutide, a weekly-administered GLP-1 receptor agonist with enhanced potency and prolonged half-life, has been extensively evaluated in MASH populations. The phase 2 dose-ranging trial (NCT02970942) included 320 individuals with histologically proven MASH and hepatic fibrosis F1-F3, randomizing them 3:3:3:1:1:1 to daily subcutaneous administration of semaglutide 0.1, 0.2, or 0.4 mg, or placebo for 72 weeks[18]. The principal outcome - resolution of MASH in absence of fibrosis worsening - occurred in 40%, 36%, and 59% of individuals treated with semaglutide 0.1, 0.2, and 0.4 mg, respectively, vs. 17% in the placebo arm (P < 0.001 for 0.4 mg compared to placebo). However, the secondary outcome of hepatic fibrosis amelioration by at least one stage in the absence of MASH progression did not demonstrate statistical significance (49%, 32%, and 43% for semaglutide 0.1, 0.2, and 0.4 mg vs. 33% for placebo; P = 0.48 for 0.4 mg compared to placebo)[18].
Metabolic benefits were substantial, including mean weight decrease of 13% in the semaglutide 0.4 mg cohort relative to 1% with placebo. Dose-dependent reductions in HbA1c were observed regardless of diabetes status. Semaglutide 0.4 mg was associated with elevated rates of gastrointestinal side effects as opposed to placebo (nausea 42% vs. 11%; diarrhea 20% vs. 14%; constipation 22% vs. 12%; vomiting 15% vs. 2%), but were generally mild to moderate, leading to discontinuation in only 4% of semaglutide-treated participants. Biliary tract complications demonstrated higher incidence among semaglutide group (6%-7%) vs. placebo (2%), consistent with known GLP-1 RA class effects. No acute pancreatitis events were reported[18].
Building upon phase 2 findings, the ongoing phase 3 ESSENCE trial (NCT04822181) is assessing weekly subcutaneous semaglutide 2.4 mg compared to placebo in 1197 individuals with biopsy-proven MASH and fibrosis stages F2-F3 over 240 weeks, with primary endpoints of MASH resolution without fibrosis progression, fibrosis improvement in absence of MASH exacerbation, and cirrhosis-free survival[10]. A scheduled intermediate evaluation at week 72 in the earliest 800 enrollees (534 semaglutide, 266 placebo) demonstrated that among semaglutide-treated patients, 62.9% obtained MASH resolution absent fibrosis deterioration, compared with 34.3% in the placebo group (P < 0.001). Importantly, fibrosis reduction without MASH progression was documented in 36.8% of semaglutide recipients compared to 22.4% of placebo recipients (P < 0.001), representing the first demonstration of significant fibrosis benefit with a GLP-1 receptor agonist in a phase 3 MASH trial[10]. Subgroup analyses demonstrated that treatment responses with semaglutide were consistent across different patient populations. Specifically, the dual primary outcomes - steatohepatitis resolution without concurrent fibrosis worsening, and fibrosis improvement without concurrent steatohepatitis worsening - remained consistent regardless of age, sex, type 2 diabetes status, or baseline fibrosis severity (stage F2 vs. F3). This consistency suggests that semaglutide’s beneficial effects on liver histology are robust and applicable to a diverse patient population with MASH and intermediate-to-advanced fibrosis. However, semaglutide appeared less effective at reducing fibrosis without MASH worsening in patients with a body mass index (BMI) below 27 kg/m2, and showed attenuated efficacy for both histological endpoints in Hispanic patients; these findings merit further exploration and prospective validation. Additionally, an analysis of insulin resistance improvement [measured by homeostatic model assessment for insulin resistance (HOMA-IR)], performed after excluding patients using insulin, suggested that semaglutide reduced insulin resistance to a slightly greater extent in patients with type 2 diabetes than in those without, although both groups demonstrated improvement[10].
Mean weight loss in the ESSENCE interim analysis was 10.5% with semaglutide vs. 2% with placebo (P < 0.001). Improvements in cardiometabolic parameters included reductions in HbA1c, high-sensitivity C-reactive protein, and lipid profiles. Non-invasive fibrosis markers improved significantly: liver stiffness by transient elastography decreased by 20%, improved hepatic fibrosis index decreased by 0.6 units, and Pro-C3 biomarker decreased by 17% (all P < 0.001). Gastrointestinal complications persisted as the predominant adverse effects (nausea 36.2% vs. 13.2%; diarrhea 26.9% vs. 12.2%; constipation 22.2% vs. 8.4%; vomiting 18.6% vs. 5.6%), although severe manifestations comprised only 13.4%. Treatment withdrawal secondary to adverse events was observed in 3% of both groups. Biliary tract events (2.5% vs. 1.5%) and acute pancreatitis inflammation (< 1%, 3 cases) were infrequent. Long-term outcomes are anticipated in 2028[10].
The landmark SELECT trial demonstrated that semaglutide decreases cardiovascular event rates in non-diabetic obesity who have prior cardiovascular disease, establishing cardiovascular benefits beyond glycemic effects. In this multicenter, double-masked, randomized, placebo-controlled, event-based superiority study, 17,604 participants were recruited; 8,803 were randomized to semaglutide and 8,801 to placebo. Among participants, 569 of 8,803 (6.5%) receiving semaglutide and 701 of 8,801 (8.0%) receiving placebo experienced a primary cardiovascular endpoint [hazard ratio (HR): 0.80; 95% confidence interval (CI): 0.72 to 0.90; P < 0.001][4]. Combined analysis across SELECT, FLOW, STEP-HFpEF (heart failure with preserved ejection fraction), and STEP-HFpEF DM studies confirmed these cardiovascular benefits across diverse patient populations. Across the four investigations, 3,743 participants (16.8%) among 22,282 total presented with established HFpEF (1,914 randomized to semaglutide, 1,829 to placebo). Among HFpEF subset, semaglutide reduced the composite endpoint risk of cardiovascular death or cardiac decompensation cases [103 (5.4%) of 1,914 in semaglutide arm vs. 138 (7.5%) of 1,829 in placebo arm; HR: 0.69 (95%CI: 0.53-0.89); P = 0.0045]. Furthermore, semaglutide decreased the risk of heart failure exacerbation events [54 (2.8%) vs. 86 (4.7%); HR: 0.59 (0.41-0.82); P = 0.0019]. Cardiovascular mortality as an isolated endpoint showed no meaningful effect [59 (3.1%) vs. 67 (3.7%); HR: 0.82 (0.57-1.16)][3-5].
The FLOW trial specifically demonstrated semaglutide’s impact on kidney function in subjects with type 2 diabetes, showing renal protective effects[15]. Proposed mechanisms include hemodynamic effects reducing intraglomerular pressure, anti-inflammatory actions, and metabolic improvements reducing oxidative stress and advanced glycation end-product formation. Since CKD frequently coexists with MASLD as part of the broader metabolic syndrome, renal protection represents a clinically relevant pleiotropic benefit[15].
A phase 2 trial in MASH-related cirrhosis (NCT03987451) assessed semaglutide 2.4 mg weekly vs. placebo in 71 adults with histologically confirmed cirrhosis for 48 weeks. Despite the primary endpoint of fibrosis improvement without worsening of MASH failing to reach statistical significance, the trial established safety in this advanced population without significant concerns[46]. However, the limited enrollment in this study precludes drawing definitive conclusions regarding semaglutide 2.4 mg weekly efficacy and safety in patients with MASH-related compensated cirrhosis.
CLINICAL TRIAL EVIDENCE FOR DUAL GLP-1/GIP RAs IN MASH
Tirzepatide
Tirzepatide, the first food and drug administration (FDA)-approved dual GLP-1/GIP receptor agonist for type 2 diabetes and obesity, was evaluated in the SYNERGY-NASH trial (NCT04166773), a phase 2, double-masked, dose-finding study in 190 participants with histologically confirmed MASH and fibrosis stages F2-F3[11]. Participants were randomized 1:1:1:1 to weekly subcutaneous tirzepatide 5, 10, or 15 mg, or placebo for 52 weeks. The primary endpoint - MASH resolution absent fibrosis progression - was documented in 44%, 56%, and 62% of individuals receiving tirzepatide 5, 10, and 15 mg, contrasting with 10% among placebo participants (P < 0.001 for all tirzepatide doses vs. placebo). Notably, the secondary endpoint of fibrosis amelioration by ≥ 1 stage without concurrent MASH deterioration was met in 55%, 51%, and 51% of the 5, 10, and 15 mg cohorts vs. 30% receiving placebo (P < 0.05 for all doses), representing substantial fibrosis benefit[11].
Non-invasive imaging demonstrated a marked reduction in liver fat fraction and intrahepatic lipid content measured by magnetic resonance imaging-proton density fat fraction (MRI-PDFF), with absolute reductions spanning 41.3% to 57% among tirzepatide recipients compared to 9.8% in placebo-treated individuals (P < 0.05 for all doses). Iron-corrected T1 (cT1), a biomarker of hepatic inflammation and fibrosis, decreased by 70.7-107.2 msec across tirzepatide groups (P < 0.05). Liver stiffness quantified by vibration-controlled transient elastography decreased by 3.1-3.5 kPa with tirzepatide vs. minimal change with placebo (P < 0.05). Biomarkers of fibrosis including enhanced liver fibrosis score (0.45-0.50 unit reduction) and Pro-C3 (40.1-45.7 μg/L reduction) improved significantly with all tirzepatide doses (P < 0.001)[11]. Subgroup analysis from the SYNERGY-NASH trial revealed consistent efficacy of tirzepatide across different patient populations. Weight reduction at 52 weeks showed a dose-dependent response in both participants with and without type 2 diabetes mellitus, with mean percentage changes ranging from -10.7% to -15.6% across the three tirzepatide doses compared to -0.8% with placebo. Regarding fibrosis stage, liver fibrosis improvement appeared more pronounced in participants with stage F3 fibrosis relative to stage F2 counterparts, potentially reflecting reduced placebo effect in the more advanced fibrosis group[47].
Mean weight loss was dose-dependent: 10.7%, 13.3%, and 15.6% in the 5, 10, and 15 mg groups, respectively, vs. 0.8% with placebo (P < 0.001 for all doses). Even participants with type 2 diabetes achieved substantial weight reduction reaching 13.7% among the maximum dose cohort. Gastrointestinal adverse events were more frequent with tirzepatide (nausea 44% vs. 12%; diarrhea 27% vs. 23%; constipation 15% vs. 6%; vomiting 15% vs. 2%) though generally mild or moderate (> 90%). Treatment withdrawal secondary to adverse events was observed in 4% of participants across both treatment arms. Cirrhotic progression was documented in 3% and 4% of tirzepatide and placebo cohorts, respectively. No events of hepatic decompensation, drug-induced liver injury, or acute pancreatitis were reported. Gallbladder-related events (3% vs. 2%) were infrequent[11]. Tirzepatide demonstrates significant extrahepatic benefits beyond its well-established hepatic effects, particularly in the cardiovascular and metabolic domains. The SUMMIT trial, involving 731 individuals with HFpEF and obesity followed for a median of 104 weeks, revealed a 38% decrease in the composite endpoint of cardiovascular mortality or heart-failure deterioration events (HR: 0.62, 95%CI: 0.41-0.95, P = 0.026), driven primarily by fewer worsening heart-failure events (8.0% vs. 14.2%, HR: 0.54). These cardiovascular benefits were accompanied by substantial metabolic improvements including 13.9% weight reduction, a 34.9 percentage point greater decrease in high-sensitivity C-reactive protein levels reflecting potent anti-inflammatory effects (P < 0.001), and a 4.7 mm Hg reduction in systolic blood pressure[48].
CLINICAL TRIAL EVIDENCE FOR DUAL GLP-1/GLUCAGON RAs AND TRIPLE AGONISTS IN MASH
Survodutide
Survodutide, a weekly dual GLP-1/GCG agonist, was assessed in a phase 2 dose-finding trial (NCT04771273) including 293 individuals with histologically confirmed MASH and fibrosis stages F1-F3[46]. Patients were randomized 1:1:1:1 to survodutide 2.4, 4.8, or 6 mg weekly, or placebo for 48 weeks. The primary endpoint - MASH improvement absent fibrosis progression - was observed in 47%, 62%, and 43% of the 2.4, 4.8, and
MRI-PDFF fat fraction assessment revealed marked hepatic fat decreases of 57%-67% among survodutide recipients vs. 14% in placebo-treated individuals. Absolute body weight reductions measured 10.2, 12.9, and 13.1 kg across the 2.4, 4.8, and 6 mg cohorts respectively vs. 0.6 kg with placebo. Of note, glycemic control improved with HbA1c reductions reaching 0.8% with survodutide therapy, indicating preserved glycemic benefits despite GCG agonism, likely reflecting the predominant GLP-1 receptor activity ratio. Gastrointestinal side effects demonstrated higher incidence compared to GLP-1 monotherapy (nausea 66% vs. 23%; diarrhea 49% vs. 23%; constipation 21% vs. 15%; vomiting 41% vs. 4%), leading to therapy interruption in 16% of survodutide recipients vs. 1.4% of placebo, highlighting tolerability challenges that may require optimization of dosing strategies or formulations[23].
Pemvidutide
Pemvidutide, another weekly GLP-1/glucagon dual agonist, is under evaluation in the ongoing phase 2 IMPACT trial (NCT05989711) in 190 individuals with histologically confirmed MASH and fibrosis stages F2-F3[47]. Patients are randomized to pemvidutide 1.2 or 1.8 mg weekly, or placebo for 24 weeks, with primary endpoints of MASH resolution without fibrosis progression and fibrosis amelioration without MASH deterioration. Preliminary data from a phase 1b extension study (NCT05292911) involving 64 individuals with MRI-PDFF ≥ 10% and BMI ≥ 28 kg/m2 demonstrated that 50%, 44%, and 60% of participants presenting baseline alanine aminotransferase (ALT) ≥ 30 IU/L reached combined MRI-PDFF reduction ≥ 30% and ALT reduction ≥ 17 IU/L at pemvidutide doses of 1.2, 1.8, and 2.4 mg, respectively, vs. 0% with placebo, suggesting promising early efficacy signals[49].
In the IMPACT trial, pemvidutide achieved mean weight losses of 4.8% and 5.8% with 1.2 and 1.8 mg doses respectively at 24 weeks, compared to 0.5% with placebo, with weight loss curves showing no evidence of plateauing, suggesting continued benefit with longer treatment[49-51].
Efinopegdutide
Emerging data on novel agents continue to expand the evidence base. Studies of efinopegdutide, another dual GLP-1/GCGR agonist, have shown preliminary efficacy. In a phase IIa, randomized, open-label study with an active comparator, efinopegdutide was compared with semaglutide over 24 weeks in individuals with elevated liver fat content (LFC). A total of 145 participants (33.1% with type 2 diabetes) were randomly assigned to once weekly efinopegdutide 10 mg or semaglutide 1 mg following dose titration. Efinopegdutide administered once weekly at 10 mg produced a greater hepatic fat decrease than semaglutide 1 mg in patients with NAFLD (LS mean -72.7% vs. -42.3%; P < 0.001). The safety profile showed modestly elevated rates of overall side effects and drug-attributable side effect in the efinopegdutide cohort relative to semaglutide, attributable predominantly to a higher frequency of gastrointestinal manifestations[52].
Retatrutide
Retatrutide represents the most advanced triple agonist aiming GLP-1, GIP, and GCG simultaneously. A phase 2 trial in 338 adults with obesity showed remarkable 24.2% mean weight loss (vs. -2.1% in the placebo group) at 48 weeks, the highest pharmacologic weight loss reported to date[24]. Enrollees underwent random allocation in a 2:1:1:1:1:2:2 distribution to receive weekly subcutaneous retatrutide at doses of 1, 4 mg (starting dose 2 mg), 4 mg (starting dose 4 mg), 8 mg (starting dose 2 mg), 8 mg (starting dose 4 mg), or
A MASLD substudy within the phase 2 obesity trial of retatrutide provided compelling evidence of hepatic benefits. Ninety-eight patients with obesity and MASLD (defined as ≥ 10% liver fat by MRI-PDFF) were contained in this substudy. After 24 weeks, retatrutide achieved mean relative reductions in liver fat of 42.9% (1 mg), 57.0% (4 mg), 81.4% (8 mg), and 82.4% (12 mg), compared to 0.3% increase with placebo (all P < 0.001 vs. placebo). At 48 weeks, the liver fat reductions were even more pronounced, with approximately 90% of individuals receiving maximum dose levels achieving normalization of liver fat[25].
REAL-WORLD EVIDENCE ON GLP-1 RAs AND LIVER OUTCOMES
Beyond randomized controlled trials, emerging real-world data from large observational cohorts and healthcare databases provide important insights into the long-term effectiveness and safety of GLP-1 RAs in patients with MASLD and type 2 diabetes. These studies, while bearing inherent observational study constraints including potential confounding and enrollment bias, offer valuable complementary evidence regarding major adverse liver outcomes (MALOs) in real-world clinical practice.
Comparative effectiveness studies have provided insights into the relative benefits of GLP-1 RAs vs. other therapies. An analysis examining liver-related event risks among MASLD patients receiving sodium-glucose cotransporter-2 inhibitors vs. GLP-1 RAs and thiazolidinediones showed favorable outcomes with GLP-1 RAs[53]. The potential synergistic benefits of combination therapy have been explored. Dual metformin and GLP-1 receptor agonist therapy was found to reduce death rates and hepatic adverse outcomes in diabetic patients with cirrhosis[54].
A large-scale retrospective cohort analysis by Kanwal et al. employed the Veterans Affairs healthcare database, including patients with metabolic dysfunction-associated steatotic liver disease[55]. Among 169,649 patients with MASLD, GLP-1 receptor agonist initiation was linked to significantly lower probability of incident cirrhosis [adjusted hazard ratio (aHR): 0.86, 95%CI: 0.80-0.93], hepatic decompensation (aHR: 0.77, 95%CI: 0.65-0.90), and hepatocellular carcinoma (HCC) (aHR: 0.79, 95%CI: 0.68-0.92) over a median follow-up of 3.5 years compared to non-users. These benefits persisted after adjusting for multiple potential confounders including baseline liver disease severity, comorbidities, and use of other medications[55].
In a separate investigation of the TriNetX database involving 15,176 propensity-score matched patients pairs with type 2 diabetes and MASLD, Kuo et al. found that GLP-1 receptor agonist use, compared with initiation of sodium-glucose cotransporter 2 (SGLT2) inhibitors, was associated with a lower risk of composite MALOs (aHR: 0.84, 95%CI: 0.74-0.94), including specifically lower occurrence of liver decompensation (aHR: 0.83) and all-cause mortality (aHR: 0.84)[56]. This head-to-head comparison suggests potential superiority of GLP-1 RAs over SGLT2 inhibitors for liver-related outcomes, although both medication classes demonstrate metabolic benefits.
Yang et al. performed a population-based cohort study in Taiwan employing a target trial emulation design to reduce confounding in 97,628 patients with type 2 diabetes[57]. This study showed that GLP-1 receptor agonist use was associated with markedly diminished risks of cirrhosis (aHR: 0.72, 95%CI: 0.66-0.79) and HCC (aHR: 0.65, 95%CI: 0.56-0.76) compared to non-use throughout a mean observation period of 4.8 years, with benefits observed across different baseline liver disease stages[57].
An analysis by Elsaid et al. specifically examined patients with MASLD cirrhosis and type 2 diabetes, finding that GLP-1 receptor agonist initiation was linked to reduced risk of unfavorable liver outcomes including hepatic decompensation events and mortality from any cause (aHR: 0.78, 95%CI: 0.68-0.90) relative to non-users, suggesting potential benefits even in advanced liver disease populations[58]. Similarly, Chen et al. reported in a propensity score-matched population-based cohort analysis that GLP-1 receptor agonist use in patients with MASLD and type 2 diabetes was associated with a lower risk of HCC (aHR: 0.55, 95%CI: 0.44-0.69) and all-cause mortality (aHR: 0.62, 95%CI: 0.55-0.71)[59].
A Scandinavian cohort study by Engström et al. assessed the relationship between GLP-1 RAs and severe hepatic complications through 88,371 individuals with type 2 diabetes. In this study, GLP-1 receptor agonist therapy correlated with a lower incidence of severe liver events compared with dipeptidyl peptidase-4 (DPP-4) inhibitors (16.9 vs. 19.2 events per 10,000 person-years), corresponding to an aHR of 0.85 (95%CI: 0.75-0.97). This association was largely explained by a reduced risk of compensated and decompensated cirrhosis (aHR: 0.85), whereas no significant effect was observed for HCC (aHR: 1.05)[60].
Real-world evidence has also emerged regarding dual GLP-1/GIP agonists. Henney et al. conducted target trial emulations comparing dual GLP-1/GIP agonists to GLP-1 monotherapy in patients with type 2 diabetes, finding that dual agonists were linked to numerically lower risks of MALOs, although confidence intervals overlapped, suggesting that extended observation period and larger patient cohorts may be needed to definitively establish comparative effectiveness[61].
Regarding safety in real-world practice, these large observational studies have not identified unexpected safety signals beyond those known from clinical trials. Gastrointestinal adverse events represent the leading cause of treatment withdrawal, but serious adverse events including pancreatitis remain rare. Importantly, no signals of drug-induced liver injury have emerged despite extensive real-world use, supporting the favorable hepatic safety profile of GLP-1 RAs[62].
Meta-analytic synthesis of randomized controlled trials has also complemented observational evidence. Wang et al. performed a systematic review and meta-analysis of GLP-1-based therapies on MASLD/MASH outcomes, incorporating both clinical trials and real-world studies[63]. This comprehensive analysis confirmed substantial reductions in hepatic fat content measured by MRI-PDFF (weighted mean difference -5.8%, 95%CI: -7.1% to -4.5%), liver enzymes, and histological parameters including steatosis and inflammation scores. The meta-analysis demonstrated consistent benefits across different GLP-1 receptor agonist preparations and dosing protocols[63].
A comprehensive meta-analysis of observational cohort studies by Celsa et al. examined GLP-1 receptor agonist use and risk of MALOs[64]. This work demonstrated that GLP-1RA treatment was associated with a significant 29% reduction in MALOs compared to non-use (IRR: 0.71, 95%CI: 0.57-0.88, P = 0.003) over a median follow-up of approximately 70,045 patient-years. This protective effect was primarily driven by a 30% reduction in liver decompensation events, comprising ascites, hepatic encephalopathy, variceal bleeding, and hepatorenal syndrome (IRR: 0.70, 95%CI: 0.52-0.94, P = 0.020), based on six studies comprising 877,488 participants. Although a trend toward reduced HCC risk was observed, this failed to achieve statistical significance in the overall analysis (IRR: 0.82, 95%CI: 0.61-1.11, P = 0.189), possibly reflecting insufficient follow-up duration to capture the full impact on hepatocarcinogenesis. Subgroup analyses revealed important differences based on comparator drugs: GLP-1RAs demonstrated greater effectiveness than SGLT2 inhibitors in preventing MALOs (IRR: 0.93, 95%CI: 0.87-0.99, P = 0.043), than DPP-4 inhibitors in preventing hepatic decompensation events (IRR: 0.74, 95%CI: 0.66-0.83, P = 0.003), and than insulin therapy in averting HCC development (IRR: 0.32, 95%CI: 0.13-0.80, P = 0.033)[64].
Collectively, real-world evidence strongly validates the effectiveness of GLP-1 RAs in decreasing MALOs in patients with MASLD and type 2 diabetes. Reviews focusing on semaglutide specifically[65] and broader examinations of GLP-1 RA mechanisms and clinical data have consistently concluded that these agents represent important therapeutic advances. While these observational data cannot definitively establish causality and may be subject to residual confounding despite sophisticated analytical approaches, the consistency of findings across multiple large, geographically diverse cohorts using different methodologies substantially strengthens confidence in the hepatoprotective benefits of GLP-1-based therapies. Ongoing long-term randomized trials will be essential to confirm these observational data and establish the durability of liver-related benefits.
SAFETY, TOLERABILITY, AND SPECIAL POPULATIONS
The overall safety profile of GLP-1 RAs and multi-agonists is well-characterized from large clinical trials and real-world data. Gastrointestinal adverse events - predominantly nausea, vomiting, diarrhea, and constipation - are the most common class effects, occurring in 20%-60% of patients depending on the agent and dose, and are typically of mild-to-moderate severity, dose-dependent, and most prominent during titration. Slow dose escalation mitigates these effects substantially, and discontinuation due to gastrointestinal intolerance ranges from 3% to 16% across agents, being highest for GLP-1/glucagon dual agonists (e.g., survodutide). Biliary complications (cholelithiasis, cholecystitis) represent a class effect attributable to reduced gallbladder motility, with incidence approximately 1.5-2.5-fold higher than placebo in long-term trials; prophylactic measures in high-risk individuals may be considered[8,9]. Serious events including acute pancreatitis are rare (< 1%) and are not causally established, though GLP-1 RAs should not be used in individuals with a personal or family history of medullary thyroid carcinoma or multiple endocrine neoplasia type 2.
Compensated cirrhosis
Patients with MASH-related compensated cirrhosis (Child-Pugh A) represent a clinically important subgroup given the risk of hepatic decompensation and HCC. A phase 2 trial of semaglutide 2.4 mg in 71 cirrhotic patients confirmed an acceptable safety profile without significant decompensation events, although the primary histological endpoint was not met, possibly due to the limited ability of antifibrotic remodeling to achieve biopsy-detectable fibrosis stage improvement in established cirrhosis over 48 weeks[46]. Real-world evidence from observational studies consistently shows that GLP-1 RA use is associated with reduced risk of hepatic decompensation even in patients with established cirrhosis, supporting cautious use in this population. GLP-1 RAs are not renally cleared and do not require dose adjustment for hepatic impairment per se; however, close monitoring for gastrointestinal adverse events is warranted in cirrhotic patients, who may have reduced tolerance. Dual and triple agonists have not been formally evaluated in compensated cirrhosis in dedicated trials, and their use in this setting should await further evidence.
Renal impairment
GLP-1 RAs are generally safe in patients with CKD and have demonstrated nephroprotective effects in the FLOW trial, where semaglutide significantly reduced the composite renal endpoint (hospitalization for acute kidney injury, sustained ≥ 50% decline in estimated glomerular filtration rate (eGFR), end-stage kidney disease, or renal death) in patients with type 2 diabetes and CKD[15]. Semaglutide and liraglutide are not renally excreted and can be used across eGFR categories without dose adjustment, though caution is advised in severe CKD (eGFR < 15 mL/min/1.73m2) due to limited data. Gastrointestinal side effects (nausea, vomiting) may exacerbate dehydration and prerenal acute kidney injury in susceptible patients, requiring hydration counseling. Tirzepatide’s renal outcomes trial is ongoing. Given the high prevalence of CKD in MASLD patients and the shared pathophysiology, GLP-1-based therapies are particularly attractive in this comorbid population[15].
Cardiovascular disease
Patients with established cardiovascular disease represent a population that derives both hepatic and cardiovascular benefit from GLP-1 RAs. The SELECT trial demonstrated a 20% reduction in major adverse cardiovascular events (MACE) with semaglutide in non-diabetic patients with obesity and prior cardiovascular events[4,5]. In patients with HFpEF and obesity, both semaglutide (STEP-HFpEF) and tirzepatide (SUMMIT) significantly reduced cardiovascular death and worsening heart failure events, with concomitant improvements in quality of life and functional capacity[3,47]. Importantly, hemodynamic effects - modest reductions in heart rate (+2-4 bpm) and blood pressure decreases - are generally well tolerated and beneficial in this population. GLP-1 RAs are not contraindicated in patients with prior atrial fibrillation, although the modest heart rate increase warrants monitoring. In patients with MASLD and established CVD, GLP-1-based therapies are strongly supported as a cornerstone treatment addressing both hepatic and cardiometabolic disease burden simultaneously.
Elderly patients and other special populations
Older adults (≥ 65 years) with MASLD may experience greater gastrointestinal intolerance and dehydration risk; slower dose titration and increased monitoring are advisable. Muscle mass preservation is an important consideration given the risk of sarcopenic obesity: weight loss with GLP-1 RAs includes a lean mass component (approximately 25%-40% of total weight loss), though the clinical significance in MASLD patients without sarcopenia is uncertain. Patients with very low body mass index (< 27 kg/m2) showed attenuated histological responses in the ESSENCE trial subgroup analysis, and the benefit-risk profile in non-obese MASLD warrants further evaluation[10]. In patients with type 2 diabetes receiving insulin, GLP-1 RA initiation may require insulin dose reduction to minimize hypoglycemia risk. Drug-drug interactions are minimal given predominant non-renal, non-hepatic elimination pathways, though delayed gastric emptying may affect absorption of oral medications with narrow therapeutic windows. Overall, the benefit-risk profile supports broad use of GLP-1-based therapies in MASLD across most patient subgroups, with individualized monitoring plans for high-risk populations.
CONCLUSIONS
GLP-1 RAs and multi-agonist therapies combining GLP-1 with GIP and/or GCG agonism embody transformative therapeutic approaches for MASLD/MASH, addressing multiple pathogenic pathways through complementary mechanisms of action. The distinct contributions of each receptor system - GLP-1-mediated improvements in insulin sensitivity and appetite regulation, GIP-mediated enhancement of insulin secretion and adipose tissue function, and glucagon-mediated direct hepatic metabolic modulation and increased energy expenditure - create synergistic benefits when engaged through dual or triple agonist formulations.
Phase 2 and interim phase 3 clinical trial data demonstrate robust effectiveness of GLP-1 RAs in achieving MASH resolution, with semaglutide showing additional promise for fibrosis improvement in the ongoing ESSENCE trial. Dual GLP-1/GIP agonist tirzepatide has exhibited impressive histological benefits including both MASH resolution and fibrosis improvement rates exceeding 50% in phase 2 studies. Dual GLP-1/glucagon agonists such as survodutide have demonstrated substantial hepatic fat reduction and MASH improvement, though tolerability optimization may be needed. Triple agonists including retatrutide show exceptional promise based on remarkable weight loss and liver fat normalization in early trials, with dedicated MASH histology studies ongoing.
Complementing randomized trial evidence, extensive real-world observational data from large healthcare databases consistently demonstrate associations between GLP-1 receptor agonist utilization and decreased risks of MALOs involving cirrhosis, hepatic decompensation, HCC, and death from hepatic causes in patients with MASLD and type 2 diabetes. These data, while subject to inherent limitations of observational research, provide important external validation of clinical trial results and suggest potential long-term hepatoprotective benefits[55-64].
Extended-duration effects of GLP-1 RAs and multi-agonists on liver-associated outcomes involving cirrhosis development, HCC incidence, and death from hepatic causes are being elucidated through ongoing observational studies and long-term trial extensions[65-70].
Critical uncertainties persist concerning ideal candidate identification, therapeutic sequence, combination strategies with other emerging MASH therapies, long-term durability of benefits, and cost-effectiveness. Future research directions include direct comparative trials between various GLP-1 RAs and multi-agonists in MASLD populations to identify optimal therapeutic choices for specific patient subgroups. Investigation of combination therapies pairing incretin-based agents with other mechanistically distinct therapies may yield additive or synergistic benefits[71].
The exploration of combination with resmetirom, the first FDA-approved agent specifically for MASH, embodies a particularly interesting avenue given the complementary mechanisms of action[71]. Resmetirom (RezdiffraTM), a liver-directed thyroid hormone receptor-β (THR-β) agonist, received accelerated FDA approval in March 2024 as the first agent specifically indicated for non-cirrhotic MASH with moderate-to-advanced fibrosis (F2-F3), based on the MAESTRO-NASH trial demonstrating MASH resolution and fibrosis improvement. GLP-1-based therapies and resmetirom operate through distinct and potentially complementary mechanisms: while resmetirom exerts direct hepatic effects by activating THR-β to enhance mitochondrial function, reduce hepatic lipogenesis, and promote lipid oxidation specifically in hepatocytes, GLP-1 RAs and multi-agonists address the upstream metabolic drivers of MASLD including insulin resistance, obesity, and systemic inflammation. Clinically, GLP-1-based therapies offer the additional advantage of established cardiovascular protection and renal benefits, making them particularly attractive for the large proportion of MASLD patients with comorbid cardiovascular disease or type 2 diabetes. Conversely, resmetirom may be preferable in patients with normal body weight or in those who have not responded adequately to GLP-1-based strategies. Secondary analyses of the MAESTRO-NASH trial suggest that resmetirom retains efficacy in patients concomitantly using GLP-1 RAs, supporting the feasibility of combination approaches[71]. Head-to-head comparative trials and studies of combination regimens are needed to establish optimal treatment sequencing and identify which patients benefit most from each approach or from their combination.
Durability of histological response and rebound effects
A critical unresolved question concerns the durability of histological improvement following treatment interruption or dose reduction. Available evidence from diabetes and obesity trials demonstrates substantial weight regain after discontinuation of GLP-1 RAs, reaching pre-treatment levels in some cohorts within 12-18 months of cessation, with accompanying deterioration in cardiometabolic parameters[72]. In the context of MASLD/MASH, analogous rebound phenomena would be expected: regression of achieved steatosis resolution and potential re-emergence of inflammatory activity and fibrogenic stimuli upon drug withdrawal. Dedicated histological reassessment studies after GLP-1 RA discontinuation in MASH patients are currently lacking, representing a major evidence gap. The ESSENCE trial will provide valuable data on the maintenance of histological benefits over 240 weeks of continuous treatment, but long-term extension studies and off-treatment follow-up protocols are needed to characterize rebound kinetics. Until durability data are available, chronic maintenance therapy analogous to treatment paradigms for other chronic liver conditions is likely required to sustain hepatic benefits. This has important implications for patient counseling, treatment planning, and cost-effectiveness analyses.
Future perspectives and outstanding questions
Despite substantial progress, several unmet needs and key questions require investigation to fully realize the therapeutic potential of GLP-1-based therapies in MASLD/MASH. First, optimal patient selection criteria remain poorly defined. Although current evidence supports broad applicability across BMI, diabetes status, and fibrosis stages F2-F3, subgroup analyses suggest attenuated responses in non-obese patients and in certain ethnic groups (Hispanic patients in ESSENCE), emphasizing the need for precision medicine approaches incorporating anthropometric, genetic (e.g., PNPLA3, HSD17B13), and metabolic profiling. Second, head-to-head comparative trials between GLP-1 RA monotherapy, dual agonists, and triple agonists in MASH populations with histological endpoints are entirely lacking: current selection among agents relies on indirect comparisons across trials with different inclusion criteria and follow-up periods. Such trials are essential to guide evidence-based prescribing. Third, the optimal positioning and sequencing of GLP-1-based therapies relative to resmetirom and other emerging MASH agents (e.g., lanifibranor, pegozafermin, semaglutide combination regimens) requires dedicated combination trials, of which preliminary data from the MAESTRO-NASH secondary analysis are encouraging. Fourth, long-term clinical outcomes data - cirrhosis incidence, HCC rates, liver transplantation, and liver-related mortality assessed as primary endpoints in randomized trials - are not yet available and are urgently needed to move beyond surrogate histological endpoints. The ESSENCE trial 240-week readout in 2028 will be a landmark, but longer follow-up and adequately powered studies in higher-risk populations (F3-F4 fibrosis) remain priorities. Fifth, cost-effectiveness analyses are critical given the high price of current GLP-1 RAs and the large potential treatment population: preliminary health-economic models suggest acceptable cost-effectiveness ratios in high-fibrosis patients with comorbid type 2 diabetes and cardiovascular disease, but robust data in broader MASLD populations are lacking. Finally, the biology of non-response and therapeutic resistance - including the role of gut microbiome composition, adipose tissue dysfunction phenotypes, and genetic determinants of drug metabolism - represents a frontier for translational investigation to improve patient outcomes.
Longer-duration randomized controlled trials with hard clinical endpoints including liver-related morbidity and mortality are essential to definitively establish the clinical value of incretin-based therapies in MASH. Nevertheless, current evidence supports GLP-1 RAs and multi-agonist formulations as highly promising pharmacotherapies that address the complex, multifactorial pathophysiology of MASLD while simultaneously improving cardiometabolic comorbidities, positioning them as cornerstone treatments in the evolving therapeutic landscape for this prevalent and burdensome disease.
DECLARATIONS
Authors’ contributions
Controlled the study design and preparation of the article: Ciancimino G, Pecorella F, Viscardi SM, La Mantia C, Caserta A, Pennisi G, Petta S
Participated in planning and drafting the article: Ciancimino G, Pecorella F, Viscardi SM, La Mantia C, Caserta A, Pennisi G, Petta S
Approved the final draft of the article: Ciancimino G, Pecorella F, Viscardi SM, La Mantia C, Caserta A, Pennisi G, Petta S
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
During the preparation of this manuscript, the AI tool OpenAI GPT (version 5.5, released 2026-04-23) was used solely for language editing. The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship
The research leading to these results has received funding from MIUR under PNRR M4C2I1.3 Heal Italia project (PE00000019, CUP B73C22001250006) to Petta S. The project is supported by the Italian PNRR-MAD project (PNRR-MAD-2022-12375656). The project is supported by PRIN 2022 (2022L273C9). The project is supported by RF-2021 (RF-2021-12372399).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Vallianou NG, Kounatidis D, Psallida S, et al. NAFLD/MASLD and the gut-liver axis: from pathogenesis to treatment options. Metabolites. 2024;14:366.
2. Lee E, Korf H, Vidal-Puig A. An adipocentric perspective on the development and progression of non-alcoholic fatty liver disease. J Hepatol. 2023;78:1048-62.
3. Kosiborod MN, Deanfield J, Pratley R, et al.; SELECT, FLOW, STEP-HFpEF, and STEP-HFpEF DM Trial Committees and Investigators. Semaglutide versus placebo in patients with heart failure and mildly reduced or preserved ejection fraction: a pooled analysis of the SELECT, FLOW, STEP-HFpEF, and STEP-HFpEF DM randomised trials. Lancet. 2024;404:949-61.
4. Lingvay I, Brown-Frandsen K, Colhoun HM, et al.; SELECT Study Group. Semaglutide for cardiovascular event reduction in people with overweight or obesity: SELECT study baseline characteristics. Obesity. 2023;31:111-22.
5. Lincoff AM, Brown-Frandsen K, Colhoun HM, et al.; SELECT Trial Investigators. Semaglutide and cardiovascular outcomes in obesity without diabetes. N Engl J Med. 2023;389:2221-32.
6. European Association for the Study of the Liver (EASL), European Association for the Study of Diabetes (EASD), European Association for the Study of Obesity (EASO). EASL-EASD-EASO clinical practice guidelines on the management of metabolic dysfunction-associated steatotic liver disease (MASLD). Obes Facts. 2024;17:374-444.
7. McLean BA, Wong CK, Campbell JE, Hodson DJ, Trapp S, Drucker DJ. Revisiting the complexity of GLP-1 action from sites of synthesis to receptor activation. Endocr Rev. 2021;42:101-32.
8. Zheng Z, Zong Y, Ma Y, et al. Glucagon-like peptide-1 receptor: mechanisms and advances in therapy. Signal Transduct Target Ther. 2024;9:234.
9. Singh A, Sohal A, Batta A. GLP-1, GIP/GLP-1, and GCGR/GLP-1 receptor agonists: novel therapeutic agents for metabolic dysfunction-associated steatohepatitis. World J Gastroenterol. 2024;30:5205-11.
10. Sanyal AJ, Newsome PN, Kliers I, et al.; ESSENCE Study Group. Phase 3 trial of semaglutide in metabolic dysfunction-associated steatohepatitis. N Engl J Med. 2025;392:2089-99.
11. Loomba R, Hartman ML, Lawitz EJ, et al. Tirzepatide for metabolic dysfunction-associated steatohepatitis with liver fibrosis. N Engl J Med. 2024;391:299-310.
12. Urias E, Tedesco NR, Oliveri A, Raut C, Speliotes EK, Chen VL; Michigan Genomics Initiative. PNPLA3 risk allele association with ALT response to semaglutide treatment. Gastroenterology. 2024;166:515-7.e2.
13. Akbari C, Dodd M, Stål P, et al. Long-term major adverse liver outcomes in 1,260 patients with non-cirrhotic NAFLD. JHEP Rep. 2024;6:100915.
14. Simon TG, Roelstraete B, Hagström H, Sundström J, Ludvigsson JF. Non-alcoholic fatty liver disease and incident major adverse cardiovascular events: results from a nationwide histology cohort. Gut. 2022;71:1867-75.
15. Perkovic V, Tuttle KR, Rossing P, et al.; FLOW Trial Committees and Investigators. Effects of semaglutide on chronic kidney disease in patients with type 2 diabetes. N Engl J Med. 2024;391:109-21.
16. Chen VL, Kuppa A, Oliveri A, et al. Human genetics of metabolic dysfunction-associated steatotic liver disease: from variants to cause to precision treatment. J Clin Invest. 2025;135:e186424.
17. Jara M, Norlin J, Kjær MS, et al. Modulation of metabolic, inflammatory and fibrotic pathways by semaglutide in metabolic dysfunction-associated steatohepatitis. Nat Med. 2025;31:3128-40.
18. Newsome PN, Ambery P. Incretins (GLP-1 receptor agonists and dual/triple agonists) and the liver. J Hepatol. 2023;79:1557-65.
19. Zafer M, Tavaglione F, Romero-Gómez M, Loomba R. Review article: GLP-1 receptor agonists and glucagon/GIP/GLP-1 receptor dual or triple agonists-mechanism of action and emerging therapeutic landscape in MASLD. Aliment Pharmacol Ther. 2025;61:1872-88.
20. Koh B, Xiao J, Ng CH, et al. Comparative efficacy of pharmacologic therapies for MASH in reducing liver fat content: systematic review and network meta-analysis. Hepatology. 2026;83:117-26.
21. Yen FS, Hou MC, Cheng-Chung Wei J, et al. Glucagon-like peptide-1 receptor agonist use in patients with liver cirrhosis and type 2 diabetes. Clin Gastroenterol Hepatol. 2024;22:1255-64.e18.
22. Conceição-Furber E, Coskun T, Sloop KW, Samms RJ. Is glucagon receptor activation the thermogenic solution for treating obesity? Front Endocrinol. 2022;13:868037.
23. Sanyal AJ, Bedossa P, Fraessdorf M, et al.; 1404-0043 Trial Investigators. A phase 2 randomized trial of survodutide in MASH and fibrosis. N Engl J Med. 2024;391:311-9.
24. Jastreboff AM, Kaplan LM, Frías JP, et al.; Retatrutide Phase 2 Obesity Trial Investigators. Triple-hormone-receptor agonist retatrutide for obesity - a phase 2 trial. N Engl J Med. 2023;389:514-26.
25. Sanyal AJ, Kaplan LM, Frias JP, et al. Triple hormone receptor agonist retatrutide for metabolic dysfunction-associated steatotic liver disease: a randomized phase 2a trial. Nat Med. 2024;30:2037-48.
26. Yabut JM, Drucker DJ. Glucagon-like peptide-1 receptor-based therapeutics for metabolic liver disease. Endocr Rev. 2023;44:14-32.
27. Bethea M, Bozadjieva-Kramer N, Sandoval DA. Preproglucagon products and their respective roles regulating insulin secretion. Endocrinology. 2021;162:bqab150.
28. Xie C, Alkhouri N, Elfeki MA. Role of incretins and glucagon receptor agonists in metabolic dysfunction-associated steatotic liver disease: opportunities and challenges. World J Hepatol. 2024;16:731-50.
29. Tanday N, Flatt PR, Irwin N. Metabolic responses and benefits of glucagon-like peptide-1 (GLP-1) receptor ligands. Br J Pharmacol. 2022;179:526-41.
30. Targher G, Mantovani A, Byrne CD. Mechanisms and possible hepatoprotective effects of glucagon-like peptide-1 receptor agonists and other incretin receptor agonists in non-alcoholic fatty liver disease. Lancet Gastroenterol Hepatol. 2023;8:179-91.
31. Nevola R, Epifani R, Imbriani S, et al. GLP-1 receptor agonists in non-alcoholic fatty liver disease: current evidence and future perspectives. Int J Mol Sci. 2023;24:1703.
32. Ortiz C, Schierwagen R, Schaefer L, Klein S, Trepat X, Trebicka J. Extracellular matrix remodeling in chronic liver disease. Curr Tissue Microenviron Rep. 2021;2:41-52.
33. Viebahn GK, Khurana A, Freund L, et al. Retatrutide improves steatohepatitis in an accelerated mouse model of diet-induced steatohepatitis with a fructose binge. Am J Physiol Gastrointest Liver Physiol. 2025;329:G680-95.
34. Kanoski SE, Hayes MR, Skibicka KP. GLP-1 and weight loss: unraveling the diverse neural circuitry. Am J Physiol Regul Integr Comp Physiol. 2016;310:R885-95.
35. Péterfi Z, Szilvásy-Szabó A, Farkas E, et al. Glucagon-like peptide-1 regulates the proopiomelanocortin neurons of the arcuate nucleus both directly and indirectly via presynaptic action. Neuroendocrinology. 2021;111:986-97.
36. Zhang T, Perkins MH, Chang H, Han W, de Araujo IE. An inter-organ neural circuit for appetite suppression. Cell. 2022;185:2478-94.e28.
37. Huang DQ, Wong VWS, Rinella ME, et al. Metabolic dysfunction-associated steatotic liver disease in adults. Nat Rev Dis Primers. 2025;11:14.
38. Campbell JE, Müller TD, Finan B, DiMarchi RD, Tschöp MH, D’Alessio DA. GIPR/GLP-1R dual agonist therapies for diabetes and weight loss-chemistry, physiology, and clinical applications. Cell Metab. 2023;35:1519-29.
39. Samms RJ, Christe ME, Collins KA, et al. GIPR agonism mediates weight-independent insulin sensitization by tirzepatide in obese mice. J Clin Invest. 2021;131:e146353.
40. Nauck MA, Quast DR, Wefers J, Pfeiffer AFH. The evolving story of incretins (GIP and GLP-1) in metabolic and cardiovascular disease: a pathophysiological update. Diabetes Obes Metab. 2021;23:5-29.
41. Zhang Q, Delessa CT, Augustin R, et al. The glucose-dependent insulinotropic polypeptide (GIP) regulates body weight and food intake via CNS-GIPR signaling. Cell Metab. 2021;33:833-44.e5.
42. Borner T, Geisler CE, Fortin SM, et al. GIP receptor agonism attenuates GLP-1 receptor agonist-induced nausea and emesis in preclinical models. Diabetes. 2021;70:2545-53.
43. Boland ML, Laker RC, Mather K, et al. Resolution of NASH and hepatic fibrosis by the GLP-1R/GcgR dual-agonist Cotadutide via modulating mitochondrial function and lipogenesis. Nat Metab. 2020;2:413-31.
44. Armstrong MJ, Gaunt P, Aithal GP, et al.; LEAN trial team. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet. 2016;387:679-90.
45. Newsome PN, Buchholtz K, Cusi K, et al.; NN9931-4296 Investigators. A placebo-controlled trial of subcutaneous semaglutide in nonalcoholic steatohepatitis. N Engl J Med. 2021;384:1113-24.
46. Loomba R, Abdelmalek MF, Armstrong MJ, et al.; NN9931-4492 investigators. Semaglutide 2·4 mg once weekly in patients with non-alcoholic steatohepatitis-related cirrhosis: a randomised, placebo-controlled phase 2 trial. Lancet Gastroenterol Hepatol. 2023;8:511-22.
47. Hartman ML, Loomba R, Lawitz EJ, et al. Consistent improvements in liver histology across subgroups in a post hoc analysis of the SYNERGY-NASH trial with tirzepatide. JHEP Rep. 2025;7:101472.
48. Packer M, Zile MR, Kramer CM, et al.; SUMMIT Trial Study Group. Tirzepatide for heart failure with preserved ejection fraction and obesity. N Engl J Med. 2025;392:427-37.
49. Noureddin M, Harrison SA, Loomba R, et al. Safety and efficacy of weekly pemvidutide versus placebo for metabolic dysfunction-associated steatohepatitis (IMPACT): 24-week results from a multicentre, randomised, double-blind, phase 2b study. Lancet. 2025;406:2644-55.
50. Browne SK, Suschak JJ, Tomah S, et al. Safety and efficacy of 24 weeks of pemvidutide in metabolic dysfunction-associated steatotic liver disease: a randomized, controlled clinical trial. JHEP Rep. 2025;7:101483.
51. Harrison SA, Browne SK, Suschak JJ, et al. Effect of pemvidutide, a GLP-1/glucagon dual receptor agonist, on MASLD: a randomized, double-blind, placebo-controlled study. J Hepatol. 2025;82:7-17.
52. Romero-Gómez M, Lawitz E, Shankar RR, et al.; MK-6024 P001 Study Group. A phase IIa active-comparator-controlled study to evaluate the efficacy and safety of efinopegdutide in patients with non-alcoholic fatty liver disease. J Hepatol. 2023;79:888-97.
53. Bea S, Ko HY, Bae JH, et al. Risk of hepatic events associated with use of sodium-glucose cotransporter-2 inhibitors versus glucagon-like peptide-1 receptor agonists, and thiazolidinediones among patients with metabolic dysfunction-associated steatotic liver disease. Gut. 2025;74:284-94.
54. Huynh DJ, Renelus BD, Jamorabo DS. Dual metformin and glucagon-like peptide-1 receptor agonist therapy reduces mortality and hepatic complications in cirrhotic patients with diabetes mellitus. Ann Gastroenterol. 2023;36:555-63.
55. Kanwal F, Kramer JR, Li L, et al. GLP-1 receptor agonists and risk for cirrhosis and related complications in patients with metabolic dysfunction-associated steatotic liver disease. JAMA Intern Med. 2024;184:1314-23.
56. Kuo CC, Chuang MH, Li CH, et al. Glucagon-like peptide-1 receptor agonists and liver outcomes in patients with MASLD and type 2 diabetes. Aliment Pharmacol Ther. 2025;61:1163-74.
57. Yang CT, Yao WY, Yang CY, Peng ZY, Ou HT, Kuo S. Lower risks of cirrhosis and hepatocellular carcinoma with GLP-1RAs in type 2 diabetes: a nationwide cohort study using target trial emulation framework. J Intern Med. 2024;295:357-68.
58. Elsaid MI, Li N, Firkins SA, et al. Impacts of glucagon-like peptide-1 receptor agonists on the risk of adverse liver outcomes in patients with metabolic dysfunction-associated steatotic liver disease cirrhosis and type 2 diabetes. Aliment Pharmacol Ther. 2024;59:1096-110.
59. Chen WM, Ng HJ, Jao AT, Wu SY, Soong RS. GLP-1 receptor agonists and risk of hepatocellular carcinoma and all-cause mortality in patients with MASLD and type 2 diabetes: a propensity score-matched population-based cohort study. Diabetes Res Clin Pract. 2025;227:112407.
60. Engström A, Wintzell V, Melbye M, et al. Association of glucagon-like peptide-1 receptor agonists with serious liver events among patients with type 2 diabetes: a Scandinavian cohort study. Hepatology. 2024;79:1401-11.
61. Henney AE, Riley DR, Anson M, Azmi S, Alam U, Cuthbertson DJ. Target trial emulations of GLP-1 and dual GLP-1/GIP agonists to reduce major adverse liver outcomes in type 2 diabetes. Liver Int. 2025;45:e70367.
62. Mantovani A, Morandin R, Fiorio V, et al. Glucagon-like peptide-1 receptor agonists improve MASH and liver fibrosis: a meta-analysis of randomised controlled trials. Liver Int. 2025;45:e70256.
63. Wang Y, Zhou Y, Wang Z, Ni Y, Prud’homme GJ, Wang Q. Efficacy of GLP-1-based therapies on metabolic dysfunction-associated steatotic liver disease and metabolic dysfunction-associated steatohepatitis: a systematic review and meta-analysis. J Clin Endocrinol Metab. 2025;110:2964-79.
64. Celsa C, Pennisi G, Tulone A, et al. Glucagon-like peptide-1 receptor agonist use is associated with a lower risk of major adverse liver-related outcomes: a meta-analysis of observational cohort studies. Gut. 2025;74:815-24.
65. Petta S, Kim K, Targher G, et al. Focus on semaglutide 2.4 mg/week for the treatment of metabolic dysfunction-associated steatohepatitis. Liver Int. 2025;45:e70407.
66. Wang L, Berger NA, Kaelber DC, Xu R. Association of GLP-1 receptor agonists and hepatocellular carcinoma incidence and hepatic decompensation in patients with type 2 diabetes. Gastroenterology. 2024;167:689-703.
67. Wester A, Shang Y, Toresson Grip E, Matthews AA, Hagström H. Glucagon-like peptide-1 receptor agonists and risk of major adverse liver outcomes in patients with chronic liver disease and type 2 diabetes. Gut. 2024;73:835-43.
68. Wang L, Xu R, Kaelber DC, Berger NA. Glucagon-like peptide 1 receptor agonists and 13 obesity-associated cancers in patients with type 2 diabetes. JAMA Netw Open. 2024;7:e2421305.
69. Simon TG, Patorno E, Schneeweiss S. Glucagon-like peptide-1 receptor agonists and hepatic decompensation events in patients with cirrhosis and diabetes. Clin Gastroenterol Hepatol. 2022;20:1382-93.e19.
70. Al Ashi S, Shah R, Iftikhar N, Lingvay I, Kinaan M, Mansi IA. Association of GLP1-receptor agonist use with liver disease progression, major cardiovascular events, and mortality in people with hepatic steatosis and diabetes. Diabetes Obes Metab. 2025;27:5971-84.
71. Noureddin M, Rinella M, Taub R, et al. Effects of resmetirom on metabolic-dysfunction associated steatohepatitis in patients with weight loss and/or diabetes taking glucagon-like peptide-1 receptor agonists and other diabetes therapies: a secondary analysis of the MAESTRO-NASH trial. Aliment Pharmacol Ther. 2025;62:1089-99.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].