


Incretin-based therapies: a new era in metabolic liver disease management
 0
0 
Abstract
Metabolic dysfunction-associated steatotic liver disease (MASLD) is characterized by hepatic steatosis and strongly associated with cardiometabolic risk factors, including obesity, type 2 diabetes (T2D), and hypertension. Its global prevalence continues to rise, and pharmacologic interventions are gaining attention, particularly the incretin-based agents, which offer benefits beyond weight loss and glycemic control, such as potential improvements in hepatic inflammation and fibrosis. This review collates current evidence on incretin-based therapies for MASLD and metabolic dysfunction-associated steatohepatitis (MASH), emphasizing their therapeutic potential and the need for robust data on long-term hepatic outcomes. A comprehensive literature search across multiple databases was conducted to evaluate the role of incretin-based agents in MASLD/MASH management. Incretin mimetics demonstrate broad metabolic benefits, with growing evidence supporting their ability to resolve steatohepatitis and mitigate liver fibrosis. While weight reduction and improved insulin sensitivity are primary mechanisms, additional pathways may contribute. Gastrointestinal adverse effects are common but generally manageable, allowing preservation of hepatic and cardiometabolic benefits. Early-phase trials reporting reductions in hepatic fat and favorable histologic changes show promising efficacy, however, most data are limited to small phase 2 studies. Large-scale phase 3 trials are essential to confirm effectiveness, establish long-term safety, and guide clinical integration.
Keywords
INTRODUCTION
Metabolic dysfunction-associated steatotic liver disease (MASLD) is defined by hepatic steatosis, characterized by triglyceride accumulation in more than 5% of hepatocytes, alongside any cardiometabolic risk factor establishes a diagnosis of MASLD[1]. If left untreated, MASLD can progress to MASH and hepatic fibrosis, with some patients ultimately developing cirrhosis and hepatocellular carcinoma (HCC)[2]. In 2023, major international societies endorsed a shift from the previous terminology, non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH), to MASLD and MASH[1]. This change was made to better reflect the central role of metabolic dysfunction in pathogenesis, reduce stigmatizing “non-alcoholic” language, and improve diagnostic clarity, while aligning disease definitions with clinical risk stratification. Incorporating this updated nomenclature is essential for accurately describing disease burden and guiding therapeutic development.
MASLD represents the hepatic manifestation of systemic insulin resistance and adipose tissue dysfunction, driven by excess hepatic lipid influx, de novo lipogenesis, oxidative stress, inflammation, and fibrogenesis[3,4]. Its development is strongly associated with metabolic risk factors such as obesity, type 2 diabetes (T2D), and hypertension, and its prevalence has risen markedly over recent decades. Current estimates suggest MASLD affects approximately 55.5% of individuals with T2D and up to 64.4% of those with obesity[5].
In its early stages, MASLD is typically asymptomatic and often detected incidentally through imaging or routine laboratory tests, leading to delayed diagnosis and silent progression that disrupts metabolic homeostasis, including glycemic and lipid regulation[6]. Globally, MASLD now affects nearly one-third of the population, with prevalence increasing by 50% over the past 30 years[7].
Cardiovascular disease remains the leading cause of death in individuals with MASLD or diabetes, particularly when both coexist. Histology-based cohort studies and meta-analyses consistently identify fibrosis stage as the strongest predictor of liver-related outcomes, including cirrhosis, HCC, and liver-related mortality[8-10]. In patients with advanced fibrosis (≥ F2), termed “at-risk MASH,” liver disease becomes a major contributor to mortality[11].
The renaming of NAFLD/NASH to MASLD/MASH in June 2023 underscores the central role of metabolic dysfunction and has been adopted by major liver societies, with updated guidelines emphasizing non-invasive risk stratification and integrated cardiometabolic care. Progression from simple steatosis to MASH and fibrosis involves complex interactions among host factors (e.g., T2D, obesity, sex, ethnicity, genetic and epigenetic variations, immune responses, gut microbiome) and environmental determinants (e.g., diet, alcohol intake, physical activity, socioeconomic status)[12]. Among these, T2D is a potent driver of fibrogenesis and connotes poorer outcomes[13-15].
Management of MASLD and MASH targets four evidence-based goals: (1) resolution of MASH by reducing hepatic inflammation and hepatocyte ballooning without worsening fibrosis, assessed via biopsy or non-invasive tests; (2) regression of MASLD by reducing hepatic fat, inflammation, or fibrosis;
This review summarizes current therapeutic strategies, with a focus on incretin-based agents that target both hepatic and cardiometabolic risk, highlighting their emerging role in MASLD/MASH management.
PATHOPHYSIOLOGY
Disordered hepatic energy metabolism
MASLD arises predominantly from disrupted hepatic energy handling. Under normal physiology, the liver coordinates the processing and storage of dietary carbohydrates and lipids. Excess energy intake, typical of Western-style diets (rich in sucrose, fructose, and saturated fats), creates an intrahepatic energy surplus. High carbohydrate consumption, especially monosaccharides and disaccharides, drives de novo lipogenesis (DNL), converting sugars into triglycerides and accelerating steatosis[22].
At the same time, increased dietary fat augments the influx of fatty acids into the liver, promoting esterification and further triglyceride accumulation. Elevated free fatty acids induce lipotoxicity, precipitating hepatocyte dysfunction and death. The buildup of lipids heightens susceptibility to oxidative and endoplasmic reticulum stress, fostering hepatocyte injury, apoptosis, and dysregulation of adipokines, thereby amplifying inflammation and fibrogenic signaling[23,24].
Insulin resistance as the central node
Insulin resistance (IR), marked by impaired suppression of hepatic glucose production, links MASLD with type 2 diabetes mellitus (T2DM) and is widely recognized as a defining and early event in disease development[25]. IR disrupts insulin signaling, upregulating lipogenic transcriptional programs and enhancing adipose tissue lipolysis, which substantially increases free fatty acid flux to the liver and fuels intrahepatic lipid accumulation[26-28].
Early murine studies proposed that insulin resistance impairs insulin-stimulated tyrosine phosphorylation, triggering activation of protein kinase C-epsilon (C-ε) and c-Jun N-terminal kinase (JNK), key mediators of insulin resistance[29,30]. Furthermore, hepatic accumulation of diacylglycerol, a triglyceride synthesis intermediate, exacerbates insulin resistance by activating protein kinase C-ε, which inhibits insulin receptor kinase activity[31,32]. Concurrently, c-Jun N-terminal kinase in hepatocytes can become hyperactivated by proinflammatory cytokines, endoplasmic reticulum stress, and reactive oxygen species originating in adipose tissue and skeletal muscle, compounding hepatic insulin resistance[33].
Given the pivotal role of IR in glucotoxicity and lipotoxicity, therapies that modulate incretin pathways, particularly glucagon-like peptide-1 (GLP-1) receptor agonists, may attenuate hepatic DNL and improve peripheral insulin sensitivity indirectly through weight loss, enhanced glycemic control, and reduced inflammatory tone[34]. The incretin effect describes meal-stimulated augmentation of insulin secretion and suppression of glucagon mediated by gut-derived GLP-1 and glucose-dependent insulinotropic polypeptide (GIP), primarily acting on pancreatic β-cells.
Innate immunity, inflammation, and fibrogenesis
Persistent hepatic lipid accumulation triggers chronic inflammation, a key driver of progression from steatosis to steatohepatitis and ultimately to fibrosis and malignancy[35]. In the early inflammatory phase, Kupffer cells adopt a proinflammatory M1 phenotype and release cytokines that recruit immune cells, creating an inflammatory microenvironment. In reparative contexts, macrophages transition toward an anti-inflammatory M2 phenotype to facilitate tissue remodeling. Loss of M1-M2 equilibrium in MASLD/MASH results in sustained inflammation, steatosis, and fibrogenesis through secretion of mediators such as interleukin-1 beta (IL-1β), interleukin-6 (IL-6), tumor necrosis factor-alpha (TNF-α), and C-X-C motif chemokine ligand 10 (CXCL10)[36-39]. Following injury, Kupffer cells undergo metabolic reprogramming that activates inflammasomes and amplifies reactive oxygen species generation[22]. Beyond macrophages, hepatic natural killer cells promote MASH progression by engaging Janus kinase (JAK)/signal transducer and activator of transcription (STAT) and nuclear factor kappa-light-enhancer of activated B cells (NF-κB) signaling, elevating reactive oxygen species and proinflammatory cytokines[40]. Neutrophil infiltration appears necessary for the initiation and propagation of hepatic inflammation, with neutrophils contributing via reactive oxygen species, cytokine release, and formation of neutrophil extracellular traps. Chronic inflammatory signaling activates hepatic stellate cells, leading to extracellular matrix deposition, architectural distortion, and progressive fibrosis[41,42].
Genetic susceptibility and disease modifiers
Genetic and heritable factors substantially influence susceptibility, onset, and progression of MASLD, as demonstrated in family and twin studies[43,44]. Recent progress in genome-wide association studies and next-generation sequencing has identified variants that perturb lipid droplet biology, very low-density lipoproteins (VLDL) assembly, DNL, glucose homeostasis, and innate immune responses[45,46]. Among genetic alterations, single-nucleotide polymorphisms (SNPs) are most implicated, with five reproducible risk loci most consistently associated with MASLD/MASH: patatin-like phospholipase domain-containing protein 3 (PNPLA3), hydroxysteroid 17-beta dehydrogenase 13 (HSD17B13), transmembrane 6 superfamily member 2 (TM6SF2), membrane-bound O-acyltransferase domain-containing 7 (MBOAT7), and glucokinase regulatory protein (GCKR)[47]. Among these genetic risk factors, PNPLA3 and HSD17B13 have emerged as primary therapeutic targets for MASH in clinical trials. PNPLA3 encodes a lipase that hydrolyzes lipid droplets; the Isoleucine 148 methionine (I148M) variant markedly reduces enzymatic activity, favoring steatosis[48]. In murine models, overexpression of human PNPLA3 I148M induces steatosis with insulin resistance and hepatocyte ballooning[49,50]. This impairs triglyceride mobilization by interfering with PNPLA3 ubiquitylation and proteasomal degradation[51], may suppress adipose triglyceride lipase activity to reduce hepatic triglyceride hydrolysis[52,53], and can activate proinflammatory pathways such as STAT1 and NF-κB[54,55].
Beyond disease risk, emerging evidence indicates that common genetic variants can also modulate treatment response and thus represent an important avenue for future personalized therapeutic strategies. For example, carriers of the PNPLA3 I148M variant appear to have differential responses to both lifestyle-based and pharmacologic interventions. Human metabolic flux studies demonstrate that individuals homozygous for PNPLA3 I148M exhibit impaired hepatic mitochondrial function, reduced de novo lipogenesis, and a metabolic shift toward ketogenesis, features that may diminish the metabolic adaptability required for efficient intrahepatic triglyceride mobilization during weight loss interventions. In a detailed physiological investigation using stable isotope tracers, Luukkonen et al. reported that homozygous carriers display increased intrahepatic lipolysis and elevated fasting β-hydroxybutyrate, suggesting altered lipid flux patterns that may limit the steatosis-reducing effects typically achieved through caloric restriction and weight loss; these intrinsic differences in hepatocellular lipid handling help explain why PNPLA3 risk-allele carriers often require more substantial weight reduction to achieve comparable improvements in hepatic steatosis relative to non-carriers[56]. Evidence from interventional studies and systematic reviews further supports genotype-specific variability in therapeutic response: a recent systematic synthesis found moderate evidence that PNPLA3 I148M homozygotes benefit substantially less, or not at all, from omega-3 carboxylic acid supplementation, whereas individuals with the ancestral allele demonstrate measurable reductions in hepatic fat; low-certainty evidence in the same review suggests that lifestyle-based interventions (diet and exercise) remain beneficial in I148M carriers and may even be more effective than in wild-type individuals in some analyses[57]. These observations align with mechanistic models proposing that the I148M mutant protein on hepatocellular lipid droplets interferes with triglyceride hydrolysis, potentially attenuating the efficacy of interventions that rely on enhancing lipolysis or hepatic fat oxidation. Collectively, these data indicate that PNPLA3 I148M homozygotes exhibit a distinct metabolic phenotype, marked by impaired lipid droplet turnover, altered mitochondrial function, and modified hepatic lipid flux, that may blunt expected therapeutic responses to some steatosis-targeting strategies. Conversely, early data indicate that therapies targeting hepatic lipogenesis or fibrosis pathways may have enhanced therapeutic potential in this genetically defined subgroup.
HSD17B13 encodes a hepatic retinol dehydrogenase predominantly localized on lipid droplet surfaces. Silencing HSD17B13 has been reported to attenuate fibrosis, potentially via effects on pyrimidine catabolism, although complete deficiency does not uniformly protect against MASH in mice[58]. Emerging evidence suggests protein kinase A mediated phosphorylation of HSD17B13 enhances its interaction with adipose triglyceride lipase, promoting lipolysis and attenuating MASH progression[59]. The rs72613567 variant may increase hepatic phospholipid content, a core lipid droplet component, and is associated with reduced MASH risk[60]. Collectively, these findings highlight HSD17B13 as a promising therapeutic target for MASH. Other risk loci are implicated in complementary pathways: TM6SF2 affects lipoprotein secretion and VLDL assembly; MBOAT7 modulates phospholipid remodeling; GCKR influences glucose and lipid flux, each contributing to steatosis and progression through distinct metabolic and inflammatory mechanisms. As the field advances, integrating genetic and epigenetic profiling, including polygenic risk scores, transcriptomic signatures, and methylation markers, may enable more nuanced risk stratification and guide personalized treatment selection. Future clinical trials incorporating genotype-specific subgroup analyses will be essential for establishing precision approaches to MASLD management. Figure 1 outlines the various mechanisms involved in the pathophysiology of MASLD.
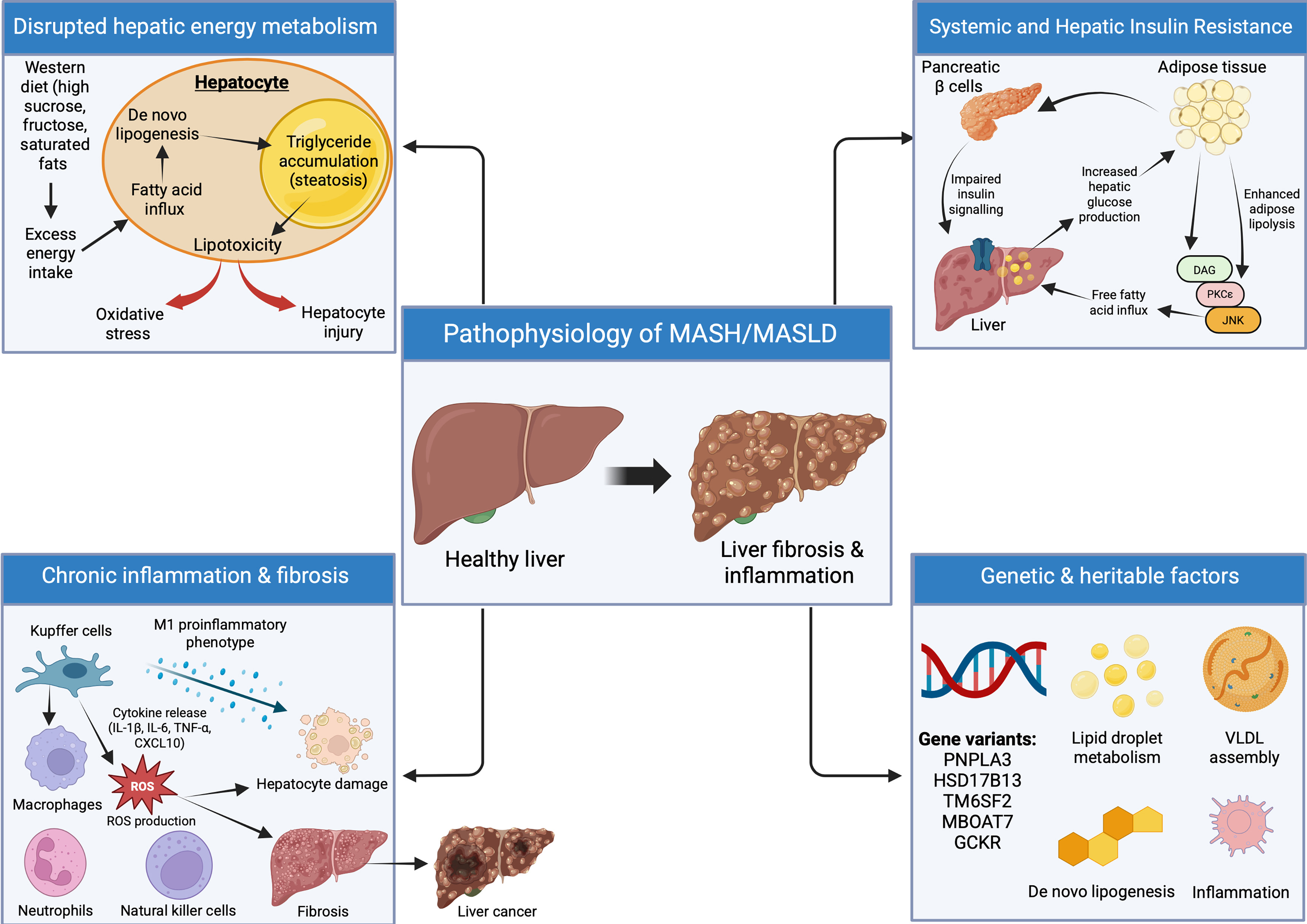
Figure 1. Pathophysiology of MASH/MASLD. The key pathophysiologic mechanisms involved in MASH/MASLD, including nutrient-driven hepatic lipid accumulation, insulin resistance mediated increases in fatty acid flux, and activation of inflammatory and fibrogenic pathways. Oxidative stress, cytokine signalling, and immune cell activation promote hepatocellular injury and fibrosis. Genetic variants (PNPLA3, TM6SF2, MBOAT7, HSD17B13, GCKR) modulate disease susceptibility through effects on lipid metabolism and inflammation. These combined mechanisms contribute to progression from steatosis to steatohepatitis and fibrosis, with GLP-1 receptor agonism highlighted as a potential therapeutic target. Created in BioRender. Elangovan, S. (2026) https://app.biorender.com/illustrations/697b672051a9c9e287df5dab. MASH: Metabolic dysfunction-associated steatohepatitis; MASLD: metabolic dysfunction-associated steatotic liver disease; GLP-1: glucagon-like peptide-1; VLDL: very-low-density lipoprotein; DAG: diacylglycerol; PKCε: protein kinase C epsilon; JNK: Jun N-terminal kinase; ROS: reactive oxygen species; PNPLA3: patatin-like phospholipase domain-containing protein 3; TM6SF2: transmembrane 6 superfamily member 2; MBOAT7: membrane-bound O-acyltransferase domain-containing 7; HSD17B13: hydroxysteroid 17-beta dehydrogenase 13; GCKR: glucokinase regulatory protein; IL-1β: interleukin-1 beta; IL-6: interleukin-6; TNF-α: tumor necrosis factor-alpha; CXCL10: C-X-C motif chemokine ligand 10.
Therapeutic implications
The multifactorial nature of MASLD/MASH, spanning energy surplus, insulin resistance, innate immune activation, and genetic predisposition, supports a multi-target therapeutic strategy. While numerous agents aim to reduce hepatic steatosis, inflammation, and fibrosis, only a small proportion have advanced to phase III trials, underscoring the translational challenges. Incretin-based therapies, by addressing weight, glycemia, and inflammatory milieu, hold promise as integrative treatments that intersect key pathogenic nodes.
GLP-1 receptor agonists
Over the past two decades, treatment options for type 2 diabetes have evolved significantly. Incretin-based therapies, particularly GLP-1 receptor agonists (GLP-1 RAs), have provided clinicians with effective strategies to manage hyperglycemia while addressing underlying metabolic disturbances such as obesity, insulin resistance, and NAFLD/MASLD.
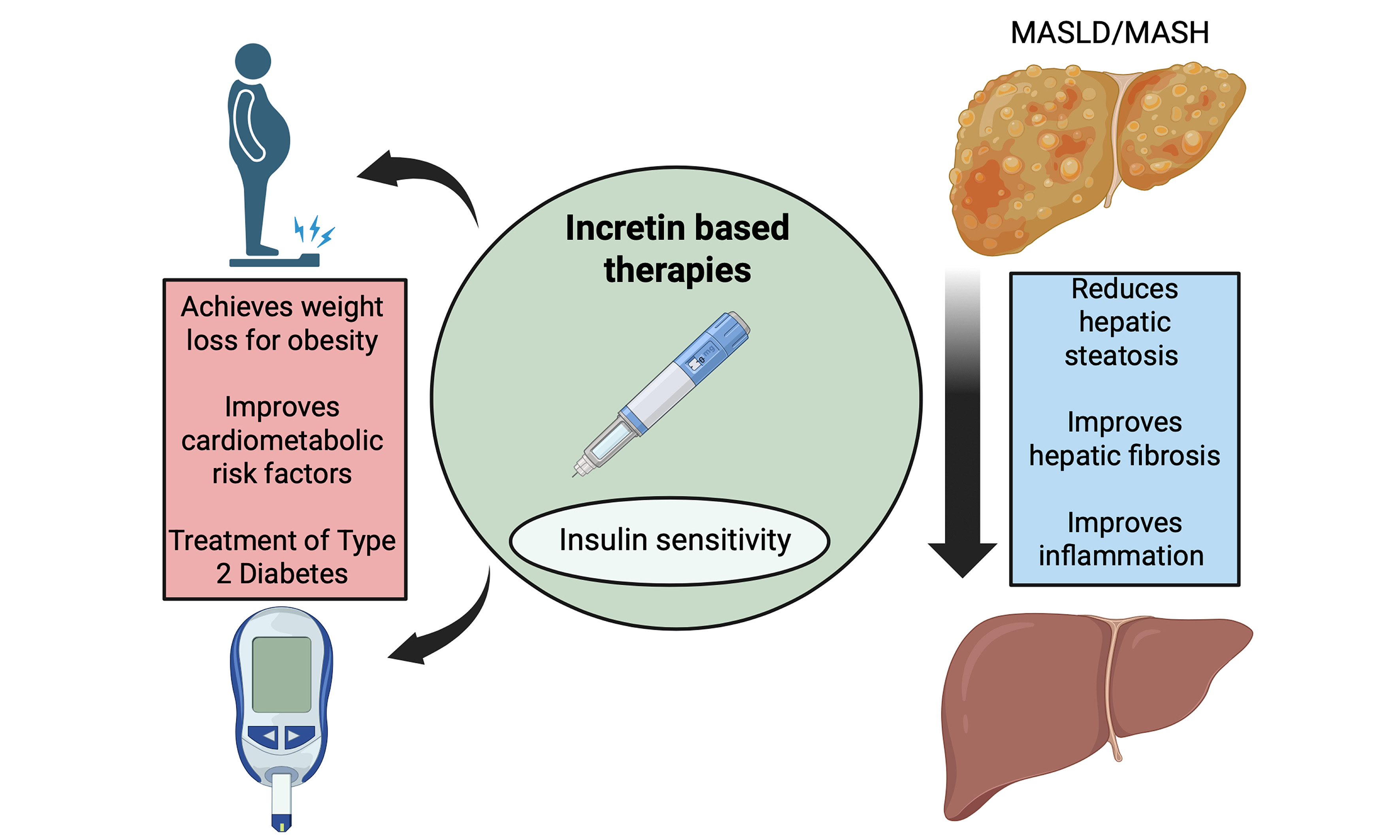
GLP-1 RAs mimic its actions by enhancing insulin secretion from pancreatic β-cells, suppressing glucagon release, delaying gastric emptying, and promoting satiety, leading to weight reduction and improved glycemic control. These effects improve hepatic glucose uptake, reduce hepatic glucose production, and decrease DNL. Additionally, GLP-1 signaling modulates hepatic lipid metabolism by downregulating genes involved in lipid processing, reducing triglyceride release and very low-density lipoproteins secretion. Collectively, these mechanisms help mitigate hepatic steatosis and inflammation, positioning GLP-1 RAs as promising therapeutic agents for MASLD[61,62]. Figure 2 summarizes the multisystem actions of incretin-based therapy relevant to MASLD.
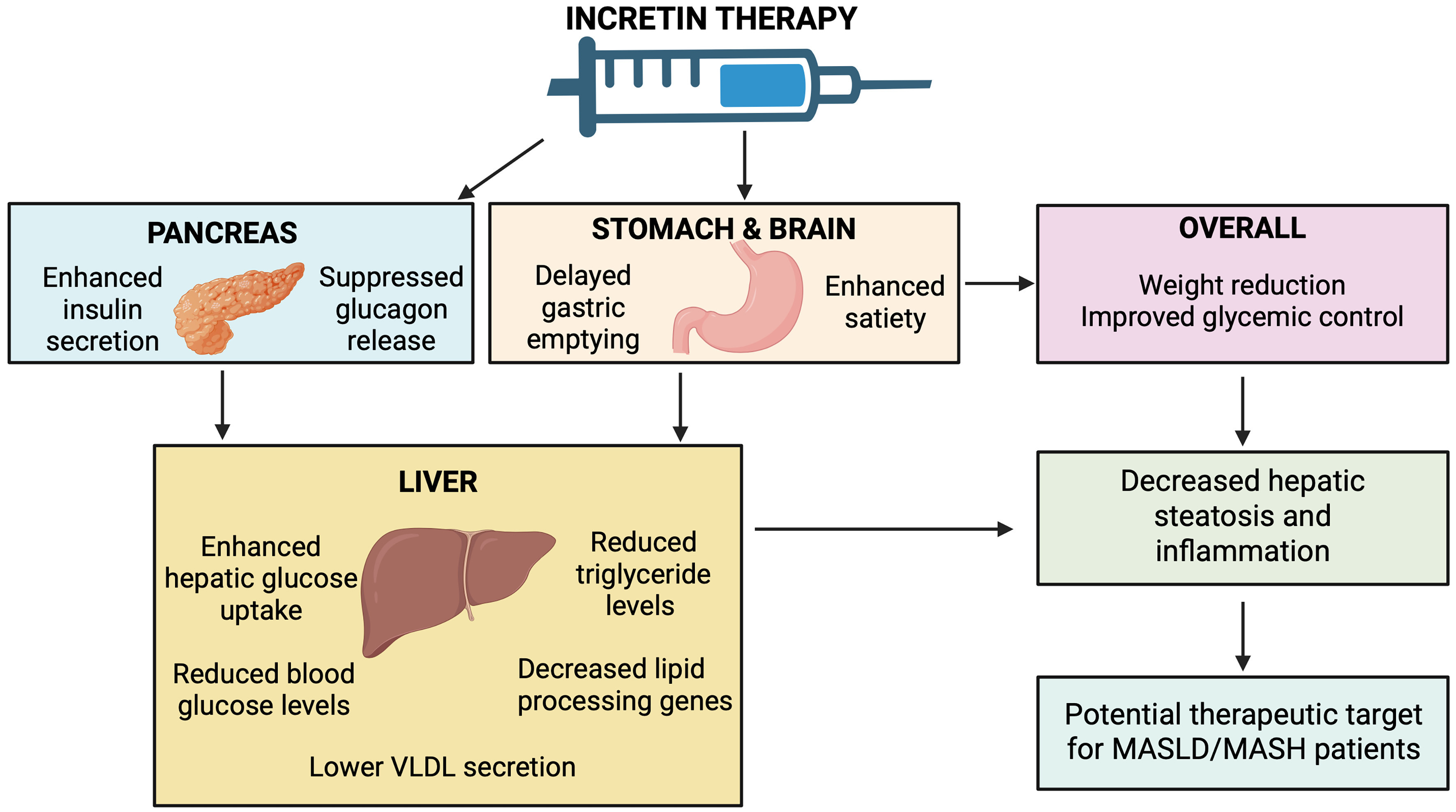
Figure 2. Metabolic and hepatic effects of incretin-based therapy. In the pancreas, incretin therapy enhances insulin secretion and suppresses glucagon release. In the stomach and brain, it delays gastric emptying and increases satiety, promoting weight reduction. These effects improve hepatic glucose uptake, reduce glucose and triglyceride release, and decrease lipid-processing pathways, lowering VLDL secretion. Collectively, these metabolic actions reduce hepatic steatosis and inflammation, supporting incretin therapy as a potential option for MASLD. Created in BioRender. Elangovan, S. (2026) https://app.biorender.com/illustrations/697b04b9ede664bc1684c975. MASLD: Metabolic dysfunction-associated steatotic liver disease; VLDL: very-low-density lipoprotein.
Clinical evidence
Table 1 provides a consolidated overview of the key clinical trials evaluating incretin-based therapies in MASLD/MASH, including their study populations, dosing regimens, and primary histologic or metabolic outcomes.
Summary of major clinical trials of incretin-based therapies in MASLD/MASH
| Incretin therapy | Trial (Phase) | Dose(s) studied | Study population | Primary/Key endpoints | Key findings |
| Liraglutide (GLP-1 RA) | LEAN Trial (Phase 2)[63] | Daily 1.8 mg | 52 patients with biopsy-proven MASH | 1. NASH resolution without worsening fibrosis 2. Fibrosis progression | ·NASH resolution: 39% vs. 9% placebo ·Less fibrosis progression: 9% vs. 36% |
| Semaglutide (GLP-1 RA) | NCT02970942 (Phase 2)[65] | Daily 0.1, 0.2, 0.4 mg | 320 patients with biopsy-confirmed MASH, F2-F3 | MASH resolution without fibrosis worsening | ·Dose-dependent MASH resolution: 40%, 36%, 59% vs. 17% placebo ·Big improvements in ALT/AST, weight loss (~12 kg) |
| NCT03987451 (Phase 2)[66] | Weekly 2.4 mg | 71 patients with MASH cirrhosis | ·Fibrosis improvement ·MASH resolution | No significant fibrosis or NASH improvement despite metabolic benefits | |
| ESSENCE Trial (Phase 3)[69] | Weekly 2.4 mg | ~1,200 pts with MASH, F2-F3 | ·Fibrosis improvement ·MASH resolution | ·MASH resolution: 62.9% vs. 34.1% ·Fibrosis improvement: 37% vs. 22.5% ·Met both endpoints: 32.8% vs. 16.2% | |
| Tirzepatide (GLP-1/GIP-RA) | SYNERGY-NASH (Phase 2)[76] | Weekly 5, 10, 15 mg | 157 pts with biopsy-confirmed MASH, F2-F3 | MASH resolution without fibrosis progression | ·Resolution: 44%, 56%, 62% vs. 10% placebo ·≥ 1-stage fibrosis improvement: 55%, 51%, 51% vs. 30% |
| SURPASS-3 MRI substudy (Phase 3)[77] | Weekly 5-15 mg | 296 T2D patients on metformin ± SGLT2i | Percentage liver fat reduction (MRI-PDFF) | ·~47% reduction in liver fat ·Reduced VAT, SAT, improved lipids | |
| Efinopegdutide (GLP-1/Glucagon Co-agonist) | NCT04944992 (Phase 2a)[81] | Weekly 10 mg | 145 patients with MAFLD defined by MRI-PDFF confirmed liver fat ≥ 10%, in individuals with metabolic risk factors | Liver fat reduction (MRI-PDFF) | ·72.7% liver fat reduction vs. 42.3% semaglutide (low-dose comparator) ·~8.5 kg weight loss |
| Survodutide (GLP-1/Glucagon RA) | Obesity (Phase 2)[82] | Weekly 2.4-4.8 mg | 387 patients with obesity | Percentage weight reduction | 12.5%-14.9% weight loss |
| MASH Trial (Phase 2)[83] | 2.4, 4.8, 6 mg | 293 patients with biopsy-confirmed F1-F3 MASH | ·Fibrosis improvement ·MASH resolution | ·Resolution: 47%, 62%, 43% vs. 14% placebo ·Fibrosis improvement: 34%-36% vs. 22% placebo | |
| Pemividutide (GLP-1/Glucagon RA) | IMPACT (Phase 2b)[84] | Weekly 1.2, 1.8 mg | 212 patients with biopsy confirmed MASH | MASH resolution without fibrosis progression | ·Resolution: 58% & 52% vs. 20% placebo ·Fibrosis improvement similar to placebo |
| Retatrutide (Triple GIP/GLP-1/Glucagon RA) | Obesity trial with MASLD substudy (Phase 2)[85] | Weekly 4, 8, 12 mg | 338 patients with obesity (98 patients in the MASLD substudy) | ·Percentage liver fat reduction ·Steatosis resolution | ·Liver fat normalization in > 85% at 8-12 mg ·Up to 24% weight loss |
The Liraglutide Efficacy and Action in Non-alcoholic Steatohepatitis (LEAN) trial, a phase 2 randomized clinical trial (RCT), was the first to assess histologic outcomes of a GLP-1 RA in NASH. Among 52 participants, NASH resolution occurred in 39% of those receiving liraglutide vs. 9% on placebo (RR 4.3; 95%CI: 1.0-17.7; P = 0.019). Fibrosis progression was also less frequent with liraglutide (9% vs. 36%; RR 0.2; 95%CI: 0.1-1.0; P = 0.04)[63].
Semaglutide, a long-acting GLP-1 RA, is available as a once-weekly injection and a daily oral formulation. Both are approved for type 2 diabetes, while the higher-dose injectable form is indicated for obesity. Among GLP-1 mono-agonists, semaglutide demonstrates the greatest efficacy in glycemic control and weight reduction[64] and has shown promise in MASH resolution. In the first phase 2 trial (NCT02970942) involving 320 patients with biopsy-confirmed NASH and F2-F3 fibrosis, semaglutide significantly increased NASH resolution without fibrosis worsening at 72 weeks: 40%, 36%, and 59% for daily doses of 0.1, 0.2, and 0.4 mg, respectively, vs. 17% with placebo (P < 0.001). Improvements were also noted in steatosis scores, liver enzymes [60% reduction in alanine aminotransferase (ALT), 50% reduction in aspartate transaminase (AST) and 52% reduction in gamma-glutamyl transferase (GGT)], glycated hemoglobin A1c (HbA1c) (1.2% reduction), and body weight (12.3 kg reduction). However, fibrosis improvement was similar across groups, likely due to short treatment duration and high placebo response[65].
In the second phase 2 trial (NCT03987451) involving 71 patients with NASH cirrhosis, once-weekly semaglutide 2.4 mg did not significantly improve fibrosis at 48 weeks (11% vs. 29% placebo), nor NASH resolution, despite meaningful weight loss and metabolic benefits[66]. Advanced liver disease stage and limited duration of therapy, particularly for an agent without direct antifibrotic activity, likely explain these findings[67]. Semaglutide has since advanced to phase 3 evaluation.
A recent meta-analysis of 11 RCTs confirmed a strong correlation between GLP-1 RA induced reductions in liver fat based on magnetic resonance imaging (MRI) and body mass index (BMI) (r² = 0.791)[68]. The ESSENCE trial (NCT04822181) is a pivotal, phase 3, randomized, double-blind, placebo-controlled study evaluating once-weekly subcutaneous semaglutide
GLP-1/GIP receptor coagonism
GIP is a short-acting incretin hormone secreted by intestinal K cells in response to carbohydrate- and fat-rich meals, with a plasma half-life of approximately seven minutes. While GIP exerts insulinotropic effects on pancreatic β-cells, its potency is lower than that of GLP-1. GIP acts through the GIP receptor, a class B G protein-coupled receptor that is minimally expressed in hepatic tissue[71].
Recent advances in peptide engineering have enabled the development of single molecules capable of activating multiple incretin receptors simultaneously. Dual agonism of GLP-1 and GIP receptors enhances the metabolic benefits of GLP-1 receptor agonists, improving glycemic control and promoting greater weight reduction. Emerging evidence also suggests that this approach may confer additional benefits in managing MASH, partly through direct effects on white adipose tissue and systemic lipid metabolism[72].
Tirzepatide, the first GLP-1/GIP receptor coagonist, is administered as a once-weekly subcutaneous injection and has received FDA approval for type 2 diabetes and obesity management. Clinical trials demonstrate that tirzepatide achieves superior glycemic control and weight loss compared with GLP-1 receptor agonists alone[73]. In obese individuals, weekly doses of 5, 10, or 15 mg over 72 weeks produce substantial and sustained weight reduction[74]. Enhanced incretin signaling not only supports prolonged weight loss but also improves glycemic parameters, underscoring its therapeutic potential for MASH. Notably, tirzepatide has been associated with dose-dependent increases in adiponectin and reductions in MASH-related biomarkers, including ALT, cytokeratin-18, and procollagen III, in patients with type 2 diabetes[75].
Clinical evidence
The SYNERGY-NASH trial (NCT04166773), a 52-week phase 2 randomized controlled study, evaluated tirzepatide in 157 patients with biopsy-confirmed MASH and stage F2-F3 fibrosis. MASH resolution without fibrosis progression occurred in 44%, 56%, and 62% of patients receiving tirzepatide at 5, 10, and 15 mg, respectively, compared with 10% in the placebo group. Tirzepatide also improved histologic features of steatosis, lobular inflammation, hepatocellular ballooning, and fibrosis. An improvement of at least one fibrosis stage was observed in 55%, 51%, and 51% of patients across the three dose groups vs. 30% with placebo. These findings were accompanied by reductions in body weight, liver enzymes, and biomarkers of hepatic fat content[76].
Further evidence comes from a sub study of SURPASS-3, which included 296 participants with type 2 diabetes receiving metformin with or without an SGLT2 inhibitor. Using MRI-derived proton density fat fraction, tirzepatide was shown to reduce liver fat by approximately 47% relative to baseline, alongside significant reductions in subcutaneous and visceral adipose tissue and improvements in plasma lipid profiles[77]. Collectively, tirzepatide demonstrates superior efficacy in achieving MASH resolution and promoting fibrosis regression, highlighting the promise of dual incretin-based therapies in MASLD treatment.
GLP-1 and glucagon receptor agonism
Glucagon acts through its receptor, a class B G protein-coupled receptor highly expressed in hepatic tissue. Activation of this receptor exerts diverse metabolic effects, including stimulation of lipolysis, enhancement of basal energy expenditure, and modulation of hepatic lipid processing[78-80]. In the context of MASLD, glucagon receptor activation may complement GLP-1 RA by promoting hepatic fatty acid oxidation and suppressing lipogenesis, thereby reducing intrahepatic fat accumulation. Additionally, glucagon signaling may attenuate hepatic inflammation and fibrosis, highlighting its potential as a therapeutic strategy for MASLD.
Several dual GLP-1/glucagon receptor agonists are currently under clinical investigation. Efinopegdutide and survodutide have advanced into phase 2 and phase 3 trials targeting MASH-related fibrosis. In a phase 2a study (NCT04944992) comparing efinopegdutide with semaglutide in patients with NAFLD, weekly administration of efinopegdutide (10 mg for 24 weeks) achieved a 72.7% reduction in liver fat content vs. 42.3% with semaglutide[81]. The caveat was that lower doses of semaglutide were used as comparator, rather than the effective 2.4 mg dose. Beyond steatosis reduction and an average weight loss of 8.5 kg, efinopegdutide significantly improved lipid parameters, including total cholesterol (-15.2%), LDL (-13.0%), HDL (-8.1%), triglycerides (-30.9%), and apolipoprotein B (-14.7%). These findings suggest that glucagon receptor agonism augments hepatic lipid metabolism beyond the effects of GLP-1 receptor agonism alone. While semaglutide and tirzepatide have demonstrated benefits in MASLD, direct comparative data on liver-specific outcomes remain limited.
Survodutide has shown promising results in obesity and MASH. In a large phase 2 trial involving 387 participants across 12 countries, weekly survodutide administration for 46 weeks produced dose-dependent weight reductions of 12.5% to 14.9% at doses of 2.4-4.8 mg[82]. Gastrointestinal adverse events were common, occurring in 75% of treated patients compared to 42% in controls. In the NCT04771273 trial, a multicenter phase 2 study in patients with biopsy-confirmed MASH and fibrosis stages F1-F3, survodutide achieved MASH resolution without fibrosis worsening in 47%, 62%, and 43% of patients receiving 2.4, 4.8, and 6 mg, respectively, vs. 14% with placebo after 48 weeks[83]. Improvement of at least one fibrosis stage without MASH progression was observed in 34%, 36%, and 34% of patients across dose groups compared to 22% in the placebo arm.
Pemividutide, another GLP-1/glucagon receptor coagonist, was evaluated in the phase 2b IMPACT trial[84]. At 24 weeks, MASH resolution without fibrosis worsening occurred in 58% and 52% of patients receiving 1.2 and 1.8 mg, respectively, compared to 20% with placebo. However, fibrosis improvement without MASH progression did not differ significantly between treatment and placebo groups.
GLP-1, GIP and glucagon receptor agonism
Retatrutide (RETA) is a once-weekly injectable triple agonist targeting GIP, GLP-1, and glucagon receptors, and it has demonstrated unprecedented efficacy in obesity management. In a phase 2 trial, RETA achieved weight reductions of up to 24% after 48 weeks, approaching outcomes typically associated with bariatric surgery. A sub-study in participants with NAFLD/MASLD revealed that all RETA doses significantly reduced liver fat compared with placebo, with the 8 and 12 mg doses resolving hepatic steatosis (liver fat content < 5%) in more than 85% of patients by week 48[85]. These findings underscore the transformative potential of multi-agonist therapies for treating MASH in overweight or obese individuals.
The strong metabolic benefits of incretin-based therapies, coupled with emerging histological improvements, highlight this drug class as a promising candidate for future MASLD treatment. In the absence of direct head-to-head trials, it remains impossible to conclusively favor one agent over the other based solely on liver-related outcomes. The development of GLP-1 receptor agonists with additional receptor activity presents theoretical advantages compared to GLP-1 RAs alone, making this an area of ongoing significant interest. However, it is important to recognize that pivotal trials in this therapeutic class, such as ESSENCE and SYNERGY-NASH, deliberately target patients at highest near-term risk of progression (fibrosis stage F2-F3), thereby enriching the study populations for severe obesity and type 2 diabetes. While this design strategy enhances histologic signal detection, it may under-represent non-obese MASLD or earlier-stage fibrosis phenotypes typically encountered in community practice, ultimately limiting the generalizability of these findings to the broader MASLD spectrum.
Although much of the therapeutic benefit of incretin-based therapies has been attributed to weight-loss-mediated metabolic improvement, there is evidence to suggest that these agents also exert direct hepatic effects that may be clinically meaningful in lean MASLD, a phenotype highly prevalent in Asian populations. Although hepatocytes express low levels of GLP-1 receptors, indirect mechanisms via weight-independent improvements in insulin resistance, altered gut-liver axis signaling, and changes in adipokine and inflammatory profiles appear to contribute meaningfully to reductions in steatosis and necroinflammation[86,87]. Clinical trials of agents such as semaglutide and dual incretin agonists demonstrate histological improvement in MASH that correlates not only with weight loss but also with glycemic and metabolic improvements, suggesting partially weight-independent effects. Nonetheless, the magnitude of benefit in truly lean individuals (BMI < 23 kg/m² in Asian populations) remains insufficiently characterized, as most pivotal trials enrolled overweight or obese participants. Therefore, while mechanistic and early clinical data support the plausibility of direct and weight-independent hepatic benefits, dedicated studies in lean MASLD are required to determine whether incretin-based therapies can achieve clinically meaningful MASH resolution when BMI reduction is not the primary therapeutic driver.
Rapid weight loss induced by potent incretin-based therapies has raised concerns regarding unintended reductions in lean body mass, particularly in vulnerable groups[88]. Given that both elderly individuals and patients with cirrhosis are at heightened risk for sarcopenia, it is important to incorporate routine monitoring of muscle mass during treatment. Dose attenuation is required to allow for milder and more gradual weight loss to mitigate the effects of weight-loss-related sarcopenia. Simple, clinically feasible approaches, such as handgrip strength assessment or bioelectrical impedance analysis (BIA), can help identify early declines in muscle function or composition. Incorporating these measures into therapeutic monitoring ensures that metabolic and hepatic benefits are balanced against the need to preserve functional status and avoid exacerbating sarcopenia.
It is essential to acknowledge the substantial cost and accessibility barriers associated with agents such as tirzepatide and the emerging triple agonist retatrutide. These medications are priced at levels that may limit availability in many healthcare settings, particularly in low-resource environments and in publicly funded systems where prioritization decisions must balance efficacy with budget constraints. Issues of equitable distribution are therefore critical, as disproportionate access may widen existing health disparities, especially among populations already experiencing a high burden of MASLD. In this context, prioritizing treatment for high-risk groups such as individuals with advanced fibrosis, multiple cardiometabolic comorbidities, or rapid disease progression may offer the greatest clinical benefit. However, implementing such prioritization poses challenges for health systems, which must integrate risk-stratification pathways, manage escalating medication costs, and ensure long-term treatment adherence. Addressing these structural and economic barriers is essential to maximizing the real-world impact of incretin-based therapies and ensuring that therapeutic advances translate into improved outcomes across diverse patient populations.
Safety profile
GLP-1 RAs are generally well tolerated and rarely cause hypoglycemia. The most common adverse effects are gastrointestinal (nausea, vomiting, altered bowel habits), affecting 40%-65% of patients, primarily due to delayed gastric emptying. A recent systematic review and meta-analysis of 55 trials by Chiang et al. evaluated the gastrointestinal and biliary safety of GLP-1RAs in people with type 2 diabetes, overweight/obesity, and MASH/MASLD[89]. The analysis found that GLP-1RAs increased the risk of cholelithiasis and likely increased the risk of GERD, though the absolute risk increases were small (2 extra cholelithiasis cases and 4 extra GERD cases per 1,000 treated patients). The heightened GERD risk was mainly seen in studies involving overweight/obese patients, those with MASH/MASLD, and those using higher dose or weight-loss focused GLP-1RAs. No increased risk was observed for other outcomes such as cholecystitis, pancreatitis, intestinal obstruction, or other serious gastrointestinal or biliary events[89]. Rare but serious risks include aspiration during anaesthesia, pancreatitis (especially in those with prior history), and potential sarcopenia, underscoring the need for resistance training and adequate protein intake (1.2-1.6 g/kg/day). GLP-1 RAs carry an FDA black box warning for medullary thyroid carcinoma based on preclinical data, though clinical incidence is extremely low; large cohort studies show no significant increase in thyroid cancer risk[90]. Table 2 provides a comparative summary of the safety profiles and discontinuation rates observed across major incretin-based therapy trials, complementing the safety considerations outlined in this section.
Comparative safety profile and discontinuation rates of incretin-based therapies in MASLD/MASH clinical trials
| Agent | Most common adverse events | Serious adverse events | Discontinuation rate |
| Liraglutide[63] | GI disorders in 81% vs. 65% placebo; diarrhea 38% vs. 19%; constipation 27% vs. 0%; decreased appetite 31% vs. 8% | Not elevated | ~13% overall |
| Semaglutide (NEJM 2021)[65] | Nausea 42% vs. 11%; constipation 22% vs. 12%; vomiting 15% vs. 2% (0.4mg vs. pooled placebo) | Neoplasms: 15% vs. 8% in placebo group | ~11% overall |
| Semaglutide (Lancet Gastro Hep 2023)[66] | Any adverse event 89% vs. 79% in placebo group | 13% vs. 8% in placebo group | Not reported |
| Tirzepatide[76] | GI events were most common, mostly mild-moderate | Not highlighted as excess vs. placebo. Safety profile consistent with class | ~13% overall Dose reductions in 20% (10 mg) and 7% (15 mg) |
| Survodutide[82,83] | Nausea 66% vs. 23%, diarrhea 49% vs. 23%, vomiting 41% vs. 4% any dose vs. placebo | 8% vs. 7% (placebo) | ~4% overall |
| Pemvidutide[84] | GI adverse events (class-typical) with details not quantified | Nil | 0% (1.2 mg), 1% (1.8 mg) vs. 2% (placebo) at 24 weeks |
| Efinopegdutide[81] | Not reported | Not reported | Not reported |
| Retatrutide[85] | Not reported | Not reported | Not reported |
In certain regions, nearly one-third of patients discontinue GLP-1 RA therapy within the first year of initiation[72]. Although high treatment cost remains a major barrier, adherence patterns require further investigation to inform strategies that improve long-term persistence. Expanding indications for incretin-based therapies may amplify these concerns, as premature discontinuation often results in significant weight regain, likely reflecting the composition lost during clinical trials. The physiological consequences of this rebound effect on organ health remain poorly understood but could have important implications for payer coverage decisions and patient selection criteria.
Contextualizing incretin therapies within the expanding MASLD/MASH treatment pipeline
Beyond incretin-based therapies, the expanding therapeutic landscape for MASLD/MASH now includes several mechanistically distinct drug classes that warrant discussion to contextualize the relative efficacy and safety of incretins. Resmetirom, a selective thyroid hormone receptor-β (THR-β) agonist, was approved in the United States in 2024 following pivotal trials demonstrating MASH resolution in approximately 26%-27% of treated patients compared with 9%-13% with placebo, along with favorable effects on atherogenic lipids and acceptable tolerability[20]. These results provide an important benchmark when comparing the magnitude of benefit seen with GLP-1 RAs and dual/triple incretin agonists, which in phase 2 and 3 studies have shown higher rates of MASH resolution but with less consistent antifibrotic effects.
Fibroblast growth factor 21 (FGF21) analogues remain a major non-incretin class of emerging therapeutics. Agents such as aldafermin and pegozafermin have demonstrated dose-dependent reductions in liver fat, improvements in dyslipidemia, and signals toward fibrosis improvement[91]. Efruxifermin (EFX), another bivalent FGF21 analogue, has shown encouraging results across multiple fibrosis stages. Phase 2b trials in both pre-cirrhotic and compensated cirrhotic MASH demonstrated improvements in fibrosis in subsets of patients, with long-term data suggesting a potential benefit despite some studies not meeting the primary endpoint at 36 weeks[92]. Efruxifermin also continues to advance in large global Phase 3 programs (SYNCHRONY), evaluating its antifibrotic and metabolic efficacy across the spectrum of MASH/MASLD[93].
Additional metabolic-targeted approaches include diacylglycerol O-acyltransferase 2 (DGAT2) inhibitors, such as elafibranor, which act via direct inhibition of hepatic lipogenesis and have demonstrated early signals of therapeutic benefit[94].
A distinct mechanistic class is represented by lanifibranor, a pan-peroxisome proliferator-activated receptor (pan-PPAR) agonist that activates PPAR-α, PPAR-δ, and PPAR-γ to modulate inflammation, fibrosis, and metabolic pathways. Phase 2 studies showed significant reductions in intrahepatic triglyceride content, improvements in insulin resistance, and histologic markers of disease activity[95]. Lanifibranor is currently being evaluated in the large global Phase 3 NATiV3 trial, which has completed enrollment and is expected to publish results in the second half of 2026, potentially positioning lanifibranor as the next oral therapy for MASH.
Integrating these agents into the broader therapeutic discussion highlights the heterogeneity of mechanisms under development, ranging from enhancement of hepatic mitochondrial function (THR-β agonists), to modulation of adipose-liver crosstalk (FGF21 analogues), to direct suppression of lipogenesis (DGAT2 inhibitors), to systemic metabolic and antifibrotic modulation (pan-PPAR agonists such as lanifibranor). This comparative framework helps contextualize where incretin-based therapies fit within the evolving treatment paradigm, especially given their characteristic gastrointestinal tolerability profile, weight-centric mechanisms, and variable antifibrotic performance relative to other emerging modalities.
Incretin-based chemoprevention and evolving risk stratification in MASLD
Emerging evidence suggests that incretin-based therapies may exert chemopreventive effects against HCC in MASLD. GLP-1 RAs beneficially modulate several key pathways implicated in hepatocarcinogenesis, including metabolic dysfunction, insulin resistance, chronic inflammation, and fibrosis progression, thereby potentially reducing long-term oncogenic risk. In a large retrospective cohort study of nearly 1.9 million individuals with type 2 diabetes, Wang et al. demonstrated that GLP-1 RA use was associated with a significantly lower incidence of HCC compared with insulin, sulfonylureas, and metformin, alongside a reduced risk of hepatic decompensation across subgroups with and without underlying metabolic liver disease[96]. Similarly, a national cohort of more than 16,000 patients with MASLD and diabetes showed that GLP-1 RA therapy was linked to lower rates of cirrhosis progression, hepatic decompensation, and HCC, but only when initiated prior to the development of cirrhosis. Patients who began therapy after cirrhosis onset did not experience these protective effects, highlighting the importance of early initiation of GLP-1 RAs for potential chemoprevention[97]. Collectively, mechanistic data and early clinical evidence suggest that incretin-based therapies may influence long-term HCC risk in MASLD, complementing their established metabolic and histologic benefits.
Parallel advances in MASLD risk stratification and hepatocarcinogenesis underscore the need to integrate structured assessment pathways with emerging therapeutics. Prospective validation of the EASL-EASD algorithm indicates that systematic fibrosis-4 index (FIB-4)-based screening followed by elastography identifies advanced MASLD in approximately 21% of patients with elevated FIB-4, reinforcing the value of stepwise non-invasive evaluation in routine practice[98]. Real-world implementation studies further highlight both the feasibility and the challenges of applying this algorithm at scale: in a large retrospective cohort of 14,814 adults undergoing abdominal ultrasound, 3,052 (20.6%) met MASLD criteria, and age-adjusted FIB-4 stratification classified 15.2% as high-risk and 18.0% as indeterminate, yet more than half of patients in these elevated-risk categories did not receive appropriate hepatology referral or secondary testing, revealing persistent gaps in real-world care pathways[99]. Complementing these findings, the multinational vibration-controlled transient elastography (VCTE)-Prognosis study (n = 12,950) demonstrated that the two-step FIB-4 followed by elastography approach effectively delineates low-, intermediate-, and high-risk groups, with the high-risk cohort exhibiting a markedly increased 5-year incidence of liver-related events (10.8%), thereby validating the prognostic utility of the EASL-EASD algorithm in routine practice[100]. At the same time, recent multicenter evidence confirms that MASLD is now a major cause of non-cirrhotic HCC, emphasizing that carcinogenesis may occur even in early-stage disease. The multicenter study by Romero-Gutiérrez et al. demonstrated that a substantial proportion of MASLD-related HCC arises in non-cirrhotic livers, strengthening the rationale for expanding surveillance to high-risk individuals, particularly those with metabolic dysfunction, elevated fibrosis markers, or increasing liver stiffness, regardless of cirrhosis status[101]. Together, these findings highlight that MASLD confers a meaningful oncogenic risk across the fibrosis spectrum and that early identification and targeted surveillance, in combination with therapies that may mitigate carcinogenic drivers, are essential to reducing progression to HCC.
Linking early diagnostic strategies with emerging incretin-based treatments
To further strengthen the clinical relevance of incretin-based therapies in MASLD, it is important to recognize that their potential benefits extend beyond pharmacologic treatment and intersect meaningfully with broader disease-management strategies. Given that MASLD is highly prevalent among individuals with T2D, incorporating early detection strategies into routine diabetes care may substantially improve clinical outcomes. Early identification of hepatic steatosis, fibrosis risk, and related metabolic complications enables timely intervention, whether through lifestyle modification, optimization of glycemic control, or initiation of agents such as GLP-1 RAs, dual or triple incretin agonists, which have demonstrated favorable effects on steatosis, inflammation, and weight regulation across phase 2 and phase 3 trials included in this review. Integrating structured screening approaches (e.g., non-invasive fibrosis assessments, imaging-based liver fat quantification, and risk-stratification tools) into T2D management pathways therefore offers an opportunity to enhance patient selection, prevent disease progression, and leverage the therapeutic impact of incretin-based treatments more effectively. This broader, proactive approach complements the emerging evidence base and underscores the importance of early risk mitigation in the evolving landscape of MASLD care.
CONCLUSION
Effective management of MASLD requires a comprehensive approach that addresses its strong association with IR, metabolic syndrome, and T2D. While lifestyle modification remains the cornerstone of therapy, the integration of targeted pharmacologic interventions offers additional benefits, particularly in early disease stages when reversal and prevention of progression are most achievable.
Whilst the therapeutic landscape for MASH is rapidly evolving, with numerous agents demonstrating encouraging results in clinical trials, meaningful comparisons between therapies remain challenging due to heterogeneity in trial design and variability in placebo response rates.
Recent RCTs underscore the complexity of molecular pathways driving MASLD and the growing array of investigational agents targeting these mechanisms. Among these, incretin-based therapies have emerged as some of the most promising candidates. GLP-1 RAs and dual or triple agonists incorporating GIP and/or glucagon receptor activity exhibit broad metabolic benefits, including improvements in steatosis, insulin sensitivity, inflammation, and body weight. Early-phase studies report favorable outcomes in hepatic fat reduction, metabolic parameters, and even histologic endpoints; however, most evidence is derived from small phase 2 trials. Consequently, MASLD drug development remains at an exciting yet preliminary stage, and large-scale phase 3 studies are essential to confirm efficacy, establish long-term safety, and define evidence-based pharmacologic options for clinical practice.
Importantly, despite their substantial metabolic and hepatic benefits, incretin therapies have not yet been shown to reduce all-cause mortality or prevent HCC. As such, their role in altering long-term liver-related outcomes remains to be established, highlighting the need for continued investigation.
Given the multisystemic nature of MASLD, multidisciplinary collaboration bringing together hepatologists, endocrinologists, cardiologists, and primary care physicians, is essential to optimize patient care. Furthermore, broad implementation of structured screening pathways, including those informed by algorithms endorsed by societies such as EASL-EASD, will be critical to improving early detection, risk stratification, and timely initiation of therapy in individuals with T2D and other high-risk groups.
Continued scientific progress and translation of research into practical treatments bring the prospect of addressing MASH and improving patient outcomes closer to reality. The next decade will likely witness a paradigm shift in MASLD management, driven by incretin-based therapies and other innovative therapies that target the disease at its metabolic core.
DECLARATIONS
Acknowledgments
The graphical abstract was created with BioRender.com (Created in BioRender. Elangovan, S. (2026) https://app.biorender.com/illustrations/697e11b83d4f64b65cee985d.
Authors’ contributions
Contributed substantially to the conception, design, literature review, analysis, and writing of this manuscript: Elangovan S, Cheah CCM, Goh GBB
Reviewed and approved the final version of the manuscript prior to submission: Elangovan S, Cheah CCM, Goh GBB
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
None.
Conflict of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Rinella ME, Lazarus JV, Ratziu V, et al. ; NAFLD Nomenclature consensus group. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. Hepatology. 2023;78:1966-86.
3. Kanwal F, Neuschwander-Tetri BA, Loomba R, Rinella ME. Metabolic dysfunction-associated steatotic liver disease: update and impact of new nomenclature on the American Association for the Study of Liver Diseases practice guidance on nonalcoholic fatty liver disease. Hepatology. 2024;79:1212-9.
4. Powell EE. A new treatment and updated clinical practice guidelines for MASLD. Nat Rev Gastroenterol Hepatol. 2025;22:88-9.
5. Younossi ZM, Golabi P, de Avila L, et al. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: a systematic review and meta-analysis. J Hepatol. 2019;71:793-801.
6. Grander C, Grabherr F, Tilg H. Non-alcoholic fatty liver disease: pathophysiological concepts and treatment options. Cardiovasc Res. 2023;119:1787-98.
7. Chan WK, Chuah KH, Rajaram RB, Lim LL, Ratnasingam J, Vethakkan SR. Metabolic dysfunction-associated steatotic liver disease (MASLD): a state-of-the-art review. J Obes Metab Syndr. 2023;32:197-213.
8. Ekstedt M, Hagström H, Nasr P, et al. Fibrosis stage is the strongest predictor for disease‐specific mortality in NAFLD after up to 33 years of follow‐up. Hepatology. 2015;61:1547-54.
9. Taylor RS, Taylor RJ, Bayliss S, et al. Association between fibrosis stage and outcomes of patients with nonalcoholic fatty liver disease: a systematic review and meta-analysis. Gastroenterology. 2020;158:1611-25.e12.
10. Dulai PS, Singh S, Patel J, et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: Systematic review and meta‐analysis. Hepatology. 2017;65:1557-65.
11. Cusi K, Isaacs S, Barb D, et al. American Association of Clinical Endocrinology clinical practice guideline for the diagnosis and management of nonalcoholic fatty liver disease in primary care and endocrinology clinical settings: co-sponsored by the American Association for the Study of Liver Diseases (AASLD). Endocr Pract. 2022;28:528-62.
12. Hagström H, Shang Y, Hegmar H, Nasr P. Natural history and progression of metabolic dysfunction-associated steatotic liver disease. Lancet Gastroenterol Hepatol. 2024;9:944-56.
13. Younossi ZM, Golabi P, Paik JM, Henry A, Van Dongen C, Henry L. The global epidemiology of nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH): a systematic review. Hepatology. 2023;77:1335-47.
14. Stefan N, Cusi K. A global view of the interplay between non-alcoholic fatty liver disease and diabetes. Lancet Diabetes Endocrinol. 2022;10:284-96.
15. Goh GB, Pan A, Chow WC, Yuan JM, Koh WP. Association between diabetes mellitus and cirrhosis mortality: the Singapore Chinese Health Study. Liver Int. 2017;37:251-8.
16. Sanyal AJ, Van Natta ML, Clark J, et al. Prospective study of outcomes in adults with nonalcoholic fatty liver disease. N Engl J Med. 2021;385:1559-69.
17. Vilar-Gomez E, Martinez-Perez Y, Calzadilla-Bertot L, et al. Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology. 2015;149:367-78.e5.
18. Friedman SL, Neuschwander-tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. 2018;24:908-22.
19. Kanwal F, Shubrook JH, Adams LA, et al. Clinical Care Pathway for the Risk Stratification and Management of Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology. 2021;161:1657-69.
20. Chen VL, Morgan TR, Rotman Y, et al. Resmetirom therapy for metabolic dysfunction-associated steatotic liver disease: October 2024 updates to AASLD Practice Guidance. Hepatology. 2025;81:312-20.
21. Bansal MB, Patton H, Morgan TR, Carr RM, Dranoff JA, Allen AM. Semaglutide therapy for metabolic dysfunction-associated steatohepatitis: November 2025 updates to AASLD Practice Guidance. Hepatology. 2026;83:1326-40.
22. Loomba R, Friedman SL, Shulman GI. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell. 2021;184:2537-64.
23. Fabbrini E, Mohammed BS, Magkos F, Korenblat KM, Patterson BW, Klein S. Alterations in adipose tissue and hepatic lipid kinetics in obese men and women with nonalcoholic fatty liver disease. Gastroenterology. 2008;134:424-31.
24. Buzzetti E, Pinzani M, Tsochatzis EA. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism. 2016;65:1038-48.
26. Sanyal AJ, Campbell-Sargent C, Mirshahi F, et al. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001;120:1183-92.
27. Bugianesi E, McCullough AJ, Marchesini G. Insulin resistance: a metabolic pathway to chronic liver disease. Hepatology. 2005;42:987-1000.
28. Maher JJ, Leon P, Ryan JC. Beyond insulin resistance: Innate immunity in nonalcoholic steatohepatitis. Hepatology. 2008;48:670-8.
29. Samuel VT, Liu ZX, Qu X, et al. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem. 2004;279:32345-53.
30. Hirosumi J, Tuncman G, Chang L, et al. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333-6.
31. Petersen MC, Madiraju AK, Gassaway BM, et al. Insulin receptor Thr1160 phosphorylation mediates lipid-induced hepatic insulin resistance. J Clin Invest. 2016;126:4361-71.
32. Petersen MC, Shulman GI. Roles of diacylglycerols and ceramides in hepatic insulin resistance. Trends Pharmacol Sci. 2017;38:649-65.
33. Czaja MJ. JNK regulation of hepatic manifestations of the metabolic syndrome. Trends in Endocrinol Metab. 2010;21:707-13.
34. Bednarz K, Kowalczyk K, Cwynar M, et al. The role of Glp-1 receptor agonists in insulin resistance with concomitant obesity treatment in polycystic ovary syndrome. Int J Mol Sci. 2022;23:4334.
35. Heymann F, Tacke F. Immunology in the liver - from homeostasis to disease. Nat Rev Gastroenterol Hepatol. 2016;13:88-110.
36. Zhang X, Fan L, Wu J, et al. Macrophage p38α promotes nutritional steatohepatitis through M1 polarization. J Hepatol. 2019;71:163-74.
37. Zhang X, Han J, Man K, et al. CXC chemokine receptor 3 promotes steatohepatitis in mice through mediating inflammatory cytokines, macrophages and autophagy. J Hepatol. 2016;64:160-70.
38. Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol. 2017;17:306-21.
39. Grunhut J, Wang W, Aykut B, et al. Macrophages in nonalcoholic steatohepatitis: friend or foe? Eur Med J Hepatol. 2018;6:100-9.
40. Wang F, Zhang X, Liu W, et al. Activated natural killer cell promotes nonalcoholic steatohepatitis through mediating JAK/STAT pathway. Cell Mol Gastroenterol Hepatol. 2022;13:257-74.
41. Jorch SK, Kubes P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med. 2017;23:279-87.
42. Huby T, Gautier EL. Immune cell-mediated features of non-alcoholic steatohepatitis. Nat Rev Immunol. 2022;22:429-43.
43. Kozlitina J, Smagris E, Stender S, et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2014;46:352-6.
44. Eslam M, George J. Genetic contributions to NAFLD: leveraging shared genetics to uncover systems biology. Nat Rev Gastroenterol Hepatol. 2019;17:40-52.
45. Luukkonen PK, Qadri S, Ahlholm N, et al. Distinct contributions of metabolic dysfunction and genetic risk factors in the pathogenesis of non-alcoholic fatty liver disease. J Hepatol. 2022;76:526-35.
47. Pei Y, Goh GB. Genetic risk factors for metabolic dysfunction-associated steatotic liver disease. Gut Liver. 2025;19:8-18.
48. BasuRay S, Wang Y, Smagris E, Cohen JC, Hobbs HH. Accumulation of PNPLA3 on lipid droplets is the basis of associated hepatic steatosis. Proc Natl Acad Sci U S A. 2019;116:9521-6.
49. Li JZ, Huang Y, Karaman R, et al. Chronic overexpression of PNPLA3I148M in mouse liver causes hepatic steatosis. J Clin Invest. 2012;122:4130-44.
50. Kabbani M, Michailidis E, Steensels S, et al. Human hepatocyte PNPLA3-148M exacerbates rapid non-alcoholic fatty liver disease development in chimeric mice. Cell Rep. 2022;40:111321.
51. Basuray S, Smagris E, Cohen JC, Hobbs HH. The PNPLA3 variant associated with fatty liver disease (I148M) accumulates on lipid droplets by evading ubiquitylation. Hepatology. 2017;66:1111-24.
52. Wang Y, Kory N, Basuray S, Cohen JC, Hobbs HH. PNPLA3, CGI‐58, and Inhibition of Hepatic Triglyceride Hydrolysis in Mice. Hepatology. 2019;69:2427-41.
53. Cherubini A, Ostadreza M, Jamialahmadi O, et al. ; EPIDEMIC Study Investigators. Interaction between estrogen receptor-α and PNPLA3 p.I148M variant drives fatty liver disease susceptibility in women. Nat Med. 2023;29:2643-55.
54. Yuan S, Liu H, Yuan D, et al. PNPLA3 I148M mediates the regulatory effect of NF‐kB on inflammation in PA‐treated HepG2 cells. J Cell Mol Med. 2019;24:1541-52.
55. Banini BA, Kumar DP, Cazanave S, et al. Identification of a metabolic, transcriptomic, and molecular signature of Patatin-like phospholipase domain containing 3-mediated acceleration of steatohepatitis. Hepatology. 2021;73:1290-306.
56. Luukkonen PK, Porthan K, Ahlholm N, et al. The PNPLA3 I148M variant increases ketogenesis and decreases hepatic de novo lipogenesis and mitochondrial function in humans. Cell Metab. 2023;35:1887-96.e5.
57. Boeckmans J, Gatzios A, Schattenberg JM, Koek GH, Rodrigues RM, Vanhaecke T. PNPLA3 I148M and response to treatment for hepatic steatosis: a systematic review. Liver Int. 2023;43:975-88.
58. Luukkonen PK, Sakuma I, Gaspar RC, et al. Inhibition of HSD17B13 protects against liver fibrosis by inhibition of pyrimidine catabolism in nonalcoholic steatohepatitis. Proc Natl Acad Sci U S A. 2023;120:e2217543120.
59. Luukkonen PK, Tukiainen T, Juuti A, et al. Hydroxysteroid 17-β dehydrogenase 13 variant increases phospholipids and protects against fibrosis in nonalcoholic fatty liver disease. JCI Insight. 2020;5:e132158.
60. Verweij N, Haas ME, Nielsen JB, et al. Germline Mutations in CIDEB and Protection against Liver Disease. N Engl J Med. 2022;387:332-44.
61. Lee HA, Kim HY. Therapeutic mechanisms and clinical effects of glucagon-like peptide 1 receptor agonists in nonalcoholic fatty liver disease. Int J Mol Sci. 2023;24:9324.
62. Yabut JM, Drucker DJ. Glucagon-like peptide-1 receptor-based therapeutics for metabolic liver disease. Endocr Rev. 2023;44:14-32.
63. Armstrong MJ, Gaunt P, Aithal GP, et al.; LEAN trial team. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet. 2016;387:679-90.
64. Pratley RE, Aroda VR, Lingvay I, et al. Semaglutide versus dulaglutide once weekly in patients with type 2 diabetes (SUSTAIN 7): a randomised, open-label, phase 3b trial. Lancet Diabetes Endocrinol. 2018;6:275-86.
65. Newsome PN, Buchholtz K, Cusi K, et al. A placebo-controlled trial of subcutaneous semaglutide in nonalcoholic steatohepatitis. N Engl J Med. 2021;384:1113-24.
66. Loomba R, Abdelmalek MF, Armstrong MJ, et al. ; NN9931-4492 investigators. Semaglutide 2·4 mg once weekly in patients with non-alcoholic steatohepatitis-related cirrhosis: a randomised, placebo-controlled phase 2 trial. Lancet Gastroenterol Hepatol. 2023;8:511-22.
67. Armstrong MJ, Okanoue T, Sundby Palle M, Sejling AS, Tawfik M, Roden M. Similar weight loss with semaglutide regardless of diabetes and cardiometabolic risk parameters in individuals with metabolic dysfunction‐associated steatotic liver disease: post hoc analysis of three randomised controlled trials. Diabetes Obes Metab. 2025;27:710-8.
68. Mantovani A, Petracca G, Beatrice G, Csermely A, Lonardo A, Targher G. Glucagon-like peptide-1 receptor agonists for treatment of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis: an updated meta-analysis of randomized controlled trials. Metabolites. 2021;11:73.
69. Sanyal AJ, Newsome PN, Kliers I, et al. ; ESSENCE Study Group. Phase 3 trial of semaglutide in metabolic dysfunction-associated steatohepatitis. N Engl J Med. 2025;392:2089-99.
71. Deacon CF, Nauck MA, Meier J, Hücking K, Holst JJ. Degradation of endogenous and exogenous gastric inhibitory polypeptide in healthy and in type 2 diabetic subjects as revealed using a new assay for the intact peptide. J Clin Endocrinol Metab. 2000;85:3575-81.
72. Genua I, Cusi K. Pharmacological approaches to nonalcoholic fatty liver disease: current and future therapies. Diabetes Spectr. 2024;37:48-58.
73. Frías JP, Davies MJ, Rosenstock J, et al. ; SURPASS-2 Investigators. Tirzepatide versus semaglutide once weekly in patients with type 2 diabetes. N Engl J Med. 2021;385:503-15.
74. Jastreboff AM, Aronne LJ, Ahmad NN, et al. Tirzepatide once weekly for the treatment of obesity. N Engl J Med. 2022;387:205-16.
75. Hartman ML, Sanyal AJ, Loomba R, et al. Effects of novel dual GIP and GLP-1 receptor agonist tirzepatide on biomarkers of nonalcoholic steatohepatitis in patients with type 2 diabetes. Diabetes Care. 2020;43:1352-5.
76. Loomba R, Hartman ML, Lawitz EJ, et al. ; SYNERGY-NASH Investigators. Tirzepatide for metabolic dysfunction-associated steatohepatitis with liver fibrosis. N Engl J Med. 2024;391:299-310.
77. Gastaldelli A, Cusi K, Fernández Landó L, Bray R, Brouwers B, Rodríguez Á. Effect of tirzepatide versus insulin degludec on liver fat content and abdominal adipose tissue in people with type 2 diabetes (SURPASS-3 MRI): a substudy of the randomised, open-label, parallel-group, phase 3 SURPASS-3 trial. Lancet Diabetes Endocrinol. 2022;10:393-406.
78. Perry RJ, Zhang D, Guerra MT, et al. Glucagon stimulates gluconeogenesis by INSP3R1-mediated hepatic lipolysis. Nature. 2020;579:279-83.
79. Kim T, Nason S, Holleman C, et al. Glucagon receptor signaling regulates energy metabolism via hepatic farnesoid X receptor and fibroblast growth factor 21. Diabetes. 2018;67:1773-82.
80. Lv S, Qiu X, Li J, et al. Glucagon-induced extracellular cAMP regulates hepatic lipid metabolism. J Endocrinol. 2017;234:73-87.
81. Romero-Gómez M, Lawitz E, Shankar RR, et al. ; MK-6024 P001 Study Group. A phase IIa active-comparator-controlled study to evaluate the efficacy and safety of efinopegdutide in patients with non-alcoholic fatty liver disease. J Hepatol. 2023;79:888-97.
82. le Roux CW, Steen O, Lucas KJ, Startseva E, Unseld A, Hennige AM. Glucagon and GLP-1 receptor dual agonist survodutide for obesity: a randomised, double-blind, placebo-controlled, dose-finding phase 2 trial. Lancet Diabetes Endocrinol. 2024;12:162-73.
83. Sanyal AJ, Bedossa P, Fraessdorf M, et al. ; 1404-0043 Trial Investigators. A phase 2 randomized trial of survodutide in MASH and fibrosis. N Engl J Med. 2024;391:311-9.
84. Noureddin M, Harrison SA, Loomba R, et al. Safety and efficacy of weekly pemvidutide versus placebo for metabolic dysfunction-associated steatohepatitis (IMPACT): 24-week results from a multicentre, randomised, double-blind, phase 2b study. Lancet. 2025;406:2644-55.
86. Armstrong MJ, Hull D, Guo K, et al. Glucagon-like peptide 1 decreases lipotoxicity in non-alcoholic steatohepatitis. J Hepatol. 2016;64:399-408.
87. Petrovic A, Igrec D, Rozac K, et al. The role of GLP1-RAs in direct modulation of lipid metabolism in hepatic tissue as determined using in vitro models of NAFLD. Curr Issues Mol Biol. 2023;45:4544-56.
88. Memel Z, Gold SL, Pearlman M, Muratore A, Martindale R. Impact of GLP-1 receptor agonist therapy in patients high risk for sarcopenia. Curr Nutr Rep. 2025;14:63.
89. Chiang CH, Jaroenlapnopparat A, Colak SC, et al. Glucagon-like peptide-1 receptor agonists and gastrointestinal adverse events: a systematic review and meta-analysis. Gastroenterology. 2025;169:1268-81.
90. Pasternak B, Wintzell V, Hviid A, et al. Glucagon-like peptide 1 receptor agonist use and risk of thyroid cancer: Scandinavian cohort study. BMJ. 2024;385:e078225.
91. Jeong C, Han N, Jeon N, et al. Efficacy and safety of fibroblast growth factor‐21 analogs for the treatment of metabolic dysfunction‐associated steatohepatitis: a systematic review and meta‐analysis. Clin Pharmacol Ther. 2024;116:72-81.
92. Alkhouri N, Loomba R, Frias JP, et al. Pegozafermin led to significant metabolic benefits, in addition to robust beneficial effects on the liver, in an open-label cohort of a phase 1b/2a study in subjects with non-alcoholic steatohepatitis (NASH). Metabolism. 2023;142:155455. Available from: https://www.metabolismjournal.com/article/S0026-0495(23)00058-6/abstract. [Last accessed on 25 May 2026].
93. Kamrul-Hasan ABM, Borozan S, Jena S, et al. Safety and efficacy of efruxifermin in metabolic dysfunction-associated steatohepatitis: a systematic review. World J Gastrointest Pharmacol Ther. 2025;16:110709.
94. Longo M, Paolini E, Di Benedetto P, Tomassini E, Meroni M, Dongiovanni P. DGAT1 and DGAT2 inhibitors for metabolic dysfunction-associated steatotic liver disease (MASLD) management: benefits for their single or combined application. Int J Mol Sci. 2024;25:9074.
95. Barb D, Kalavalapalli S, Godinez Leiva E, et al. Pan-PPAR agonist lanifibranor improves insulin resistance and hepatic steatosis in patients with T2D and MASLD. J Hepatol. 2025;82:979-91.
96. Wang L, Berger NA, Kaelber DC, Xu R. Association of GLP-1 receptor agonists and hepatocellular carcinoma incidence and hepatic decompensation in patients with Type 2 diabetes. Gastroenterology. 2024;167:689-703.
97. Kanwal F, Kramer JR, Li L, et al. GLP-1 receptor agonists and risk for cirrhosis and related complications in patients with metabolic dysfunction-associated steatotic liver disease. JAMA Intern Med. 2024;184:1314-23.
98. Woodard JS, Abrams GA. Increased prevalence of advanced metabolic dysfunction-associated steatotic liver disease fibrosis in type 2 diabetics despite low-risk fibrosis-4 index scores. J Endocrinol Metab. 2024;14:40-7.
99. Ebiai R, Mcnair J, Shuaibi S, et al. Unrecognized fibrosis risk in MASLD: a real-world analysis and the case for AI-Augmented stratification. Gastro Hep Adv. 2025;5:100857.
100. Yip TC, Lee HW, Lin H, et al. Prognostic performance of the two-step clinical care pathway in metabolic dysfunction-associated steatotic liver disease. J Hepatol. 2025;83:304-14.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].