


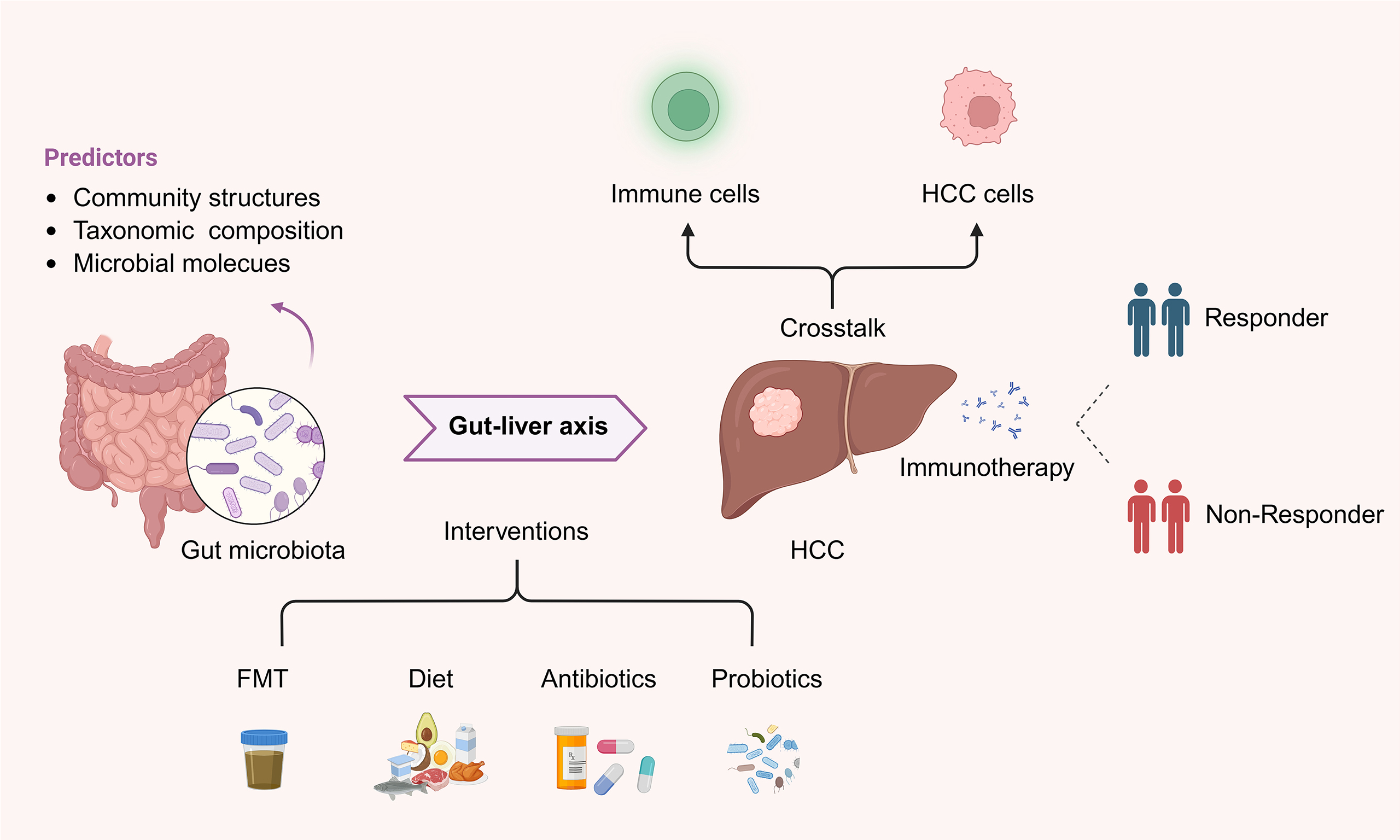
Gut microbiota as predictors of immunotherapy response in hepatocellular carcinoma
 0
0 
Abstract
Hepatocellular carcinoma (HCC) remains a major cause of global cancer-related mortality, with limited therapeutic options for advanced disease. The advent of immune checkpoint inhibitors (ICIs) has revolutionized HCC treatment; however, only a subset of patients achieve durable responses, underscoring the urgent need for reliable biomarkers to guide precision immunotherapy. Emerging evidence highlights the gut microbiota as a determinant of immunotherapy efficacy through the gut-liver axis. Community diversity, specific microbial taxa, and functional metabolites have been associated with responses to ICIs. Mechanistically, gut microbes influence antigen presentation, T cell activation, and immune tolerance within the hepatic microenvironment. Moreover, microbiota-targeted interventions hold promise for restoring responsiveness in non-responders. This review summarizes the regulatory roles of the gut microbiota in HCC immunotherapy and discusses the potential of microbiota-based strategies as predictive and therapeutic tools. Integrating multi-omics, strain-level analysis, and high-resolution microbial profiling is crucial for realizing microbiota-informed precision immunotherapy in HCC.
Keywords
INTRODUCTION
Hepatocellular carcinoma (HCC) accounts for 75%-85% of the approximately 0.9 million liver cancer cases diagnosed in 2022[1]. It ranks third among the global leading causes of cancer-related mortality due to its heterogeneity and coexistence with other underlying liver diseases[2], with an estimated 5-year survival rate of 20%[1,3,4]. Despite surgical resection, liver transplantation, and locoregional ablation therapies being effective for patients with early-stage HCC, most patients are diagnosed at an advanced stage and hence are ineligible for curative treatment[5]. Recently, immune checkpoint inhibitor (ICI)-based combinations have revolutionized the first-line treatment landscape for unresectable HCC. Pivotal Phase III trials, including IMbrave150 (atezolizumab plus bevacizumab)[6], HIMALAYA (durvalumab plus tremelimumab)[7], and the recent CheckMate-9DW (nivolumab plus ipilimumab)[8], have achieved objective response rates (ORRs) of approximately 20.1%-36%, compared with 5.1%-18.8% for multi-target kinase inhibitors, such as sorafenib and lenvatinib, in their respective pivotal trials. However, durable clinical benefit (DCB) is limited to a subset of patients, reflecting the lack of robust biomarkers for response prediction[9]. Current biomarkers - including programmed death-ligand 1 (PD-L1) expression, tumor mutational burden, and immune gene signatures - are of limited predictive value[4,10-14], underscoring the urgent need for more effective tools to identify responders and guide individualized treatment strategies.
The gut microbiota plays a crucial role in regulating host immunity and metabolism, and may also impact the efficacy of cancer immunotherapy. In non-small cell lung cancer (NSCLC) and renal cell carcinoma, patients with higher gut microbial diversity respond more favorably to anti-PD-1 therapy[15]. In melanoma, patients with prolonged progression-free survival (PFS ≥ 12 months) exhibit increased abundances of Bacteroides and Lactobacillus species in their gut microbiota[16]. Several clinical trials of fecal microbiota transplantation (FMT) from immune checkpoint blockade (ICB) responders or healthy donors to non-responders have demonstrated that this approach can restore sensitivity to anti-PD-1 treatment in melanoma[17,18].
Although the relationship between the gut microbiota and immunotherapy has been extensively studied in multiple cancer types, its role in HCC, a malignancy uniquely shaped by the gut-liver axis[19], remains underexplored and lacks a systematic synthesis, which hinders clinical translation. Considering that the liver maintains a close bidirectional interaction with the gut microbiota and its metabolites, it is crucial to understand this interplay. In this review, we summarize the current knowledge on the relationship between gut microbiota and immunotherapy response in HCC, discuss the underlying mechanisms, and highlight the potential of microbiota-based strategies as predictive biomarkers and therapeutic targets in precision oncology.
GUT-LIVER AXIS AND MICROBIAL DYSBIOSIS IN HCC
The gut and liver are anatomically and functionally linked through the portal venous system, which drains nutrient-rich blood, microbial products, and metabolites from the intestine directly to the liver. This portal circulation provides approximately 75% of the liver’s blood supply, positioning the liver as the first organ to encounter gut-derived signals under both physiological and pathological conditions[20,21]. The gut microbiota and its metabolites, such as short-chain fatty acids (SCFAs) and bile acids (BAs), profoundly influence hepatic immune homeostasis, bile acid metabolism, and energy homeostasis[22,23], whereas liver-derived BAs and cytokines reciprocally shape the intestinal microbial composition and mucosal integrity[24,25]. When this delicate equilibrium is disrupted, microbial translocation and altered metabolite profiles can trigger chronic inflammation and tumorigenic signaling within the liver[26]. For instance, impairment of the intestinal barrier can facilitate the translocation of Klebsiella pneumoniae from the gut to distal hepatic tumors, thereby promoting local tumor proliferation[27]. In parallel, microbiota-derived indole metabolites, including indole-3-acetic acid (IAA) and indole-3-propionate, suppress inflammation and attenuate hepatic steatosis in nonalcoholic fatty liver disease (NAFLD) or nonalcoholic steatohepatitis[28,29].
HCC usually develops as the end stage of chronic liver diseases with various etiologies, including NAFLD, alcohol-associated liver disease (ALD), and liver cirrhosis[30-32]. In metabolic dysfunction-associated steatotic liver disease (MASLD), the gut microbiota is characterized by a decrease in the abundance of beneficial and SCFA-producing bacteria, such as Ruminococcaceae and Faecalibacterium[33]. In ALD, dysbiosis is mainly marked by the overgrowth of pathogenic bacteria (Enterococcus[34], Klebsiella[35]) and fungi (notably Candida albicans)[36]. Liver cirrhosis is associated with severe loss of microbial diversity, small intestinal bacterial overgrowth[37], and enrichment of potentially pathogenic taxa, including Escherichia coli and Klebsiella pneumoniae[38]. During progression to HCC, tumor-associated microbial signatures emerge, featuring the enrichment of Enterococcus and Klebsiella, along with the depletion of beneficial taxa such as Bifidobacterium and Akkermansia[39,40]. Although the patterns of gut microbiota dysbiosis vary across different etiologies and stages of liver disease, their overall functional consequences are largely convergent. These variations contribute to intestinal barrier disruption, increased gut permeability, and translocation of microbial-associated molecular patterns and toxins in portal circulation[41].
GUT MICROBIAL SIGNATURES PREDICTING HCC IMMUNOTHERAPY RESPONSE
Specific gut microbial features are linked to immunotherapy efficacy in HCC, allowing the stratification of patients into responders and non-responders[42-44]. Stool-derived microbiome profiling is noninvasive and readily accessible, providing baseline microbial information[42,43]. The potential predictive features of the gut microbiome can be broadly classified into the following three categories: (i) community structure, capturing global characteristics, such as microbial diversity; (ii) taxonomic composition, referring to individual microbial taxa which can be specifically targeted or modulated; and (iii) microbial molecules, encompassing proteins and metabolites, which may serve as mechanistically relevant biomarkers.
Community structure
A high-diversity baseline and community stability are recurrent hallmarks of clinical response in HCC immunotherapy. While alpha- and beta-diversity metrics serve as valuable population-level descriptors, their transition into individualized predictive biomarkers has been underwhelming. In a pilot study enrolling eight patients with HCC at Barcelona Clinic Liver Cancer (BCLC) Stage C receiving anti-PD-1 treatment after sorafenib failure, responders (complete or partial response, or stable disease lasting > 6 months; N = 3) showed higher taxa richness than that in non-responders (progressed disease or stable disease lasting < 6 months; N = 5)[43]. Consistently, in another cohort consisting of 35 patients with HCC, 16 were classified as responders and 19 as non-responders. During treatment, responders exhibited higher alpha diversity than that in non-responders, as indicated by both Shannon and inverse Simpson indices. Furthermore, beta diversity analyses demonstrated distinct clustering of gut microbial communities between the two groups[44]. Longitudinal analyses further indicated that the overall community structure of the gut microbiota remains largely stable during immunotherapy. For instance, in studies involving patients with NSCLC, repeated sampling throughout anti-PD-1 therapy revealed that alpha and beta diversity metrics did not fluctuate significantly over time, while responders and non-responders maintained distinct community profiles[45,46]. Although longitudinal data in HCC are limited, similar patterns in other cancers support the potential of the gut microbiome as a stable indicator for patient stratification[16]. Overall, these studies report statistical significance in diversity shifts but fail to provide robust performance metrics for patient stratification.
Taxonomic composition
At the taxonomic level, the definitive microbial signatures for ICI responders in HCC are characterized by significant inter-study heterogeneity. For example, a small-scale study involving eight patients with advanced HCC found that, at the genus level, responders more frequently exhibit a balanced Firmicutes/Bacteroidetes ratio (generally 0.5-1.5, 66.7% vs. 10%) and higher Prevotella/Bacteroides ratio (22.99 vs. 2.312) than those in non-responders[47]. In addition, responders exhibit the enrichment of Lactobacillus species, Lachnospiraceae bacterium, Alistipes sp., and Ruminococcaceae spp. Specifically, patients enriched for Lachnospiraceae bacterium achieve longer PFS [median PFS (mPFS): 7.9 months vs. 5.1 months] and overall survival [OS; median OS (mOS): not reached (NR) vs. 13.8 months]. Similar clinical benefits were observed in patients with a higher abundance of Alistipes sp. (mPFS: 9.0 months vs. 5.2 months; mOS: NR months vs. 13.8 months)[42,43]. However, one cohort study (N = 65) associated Veillonellaceae enrichment with worsening PFS (mPFS: 3.6 months vs. 10.8 months) and OS (mOS: 7.8 months vs. NR)[42], which is consistent with the reported role of Veillonellaceae in liver cirrhosis progression, whereas another study (N = 41) reports its enrichment in responders[48]. Such discrepancies may stem from variations in prior sorafenib exposure or underlying viral etiologies [hepatitis B virus (HBV) vs. hepatitis C virus (HCV)], which profoundly shape the baseline microbiome. Despite these localized inconsistencies, the following robust consensus is emerging from pan-cancer meta-analyses: biomarkers such as Akkermansia muciniphila and Faecalibacterium prausnitzii are consistently enriched in responders, indicating their universal role in anti-tumor immunity[43,47]. While ICI-associated biomarkers are still being refined, these key species represent the most reliable mechanistic anchors for current microbiome-based stratification.
Microbial metabolites
Beyond taxonomic inconsistencies, microbial functional molecules have emerged as more stable and biologically relevant predictors of ICI efficacy. While microbial compositions may vary across different HCC etiologies, their downstream metabolic outputs, particularly BAs and SCFAs, exhibit remarkable predictive consistency[42].
Primary BAs are produced from cholesterol in the liver, conjugated with glycine or taurine, and released into the intestine, where they are deconjugated and dehydroxylated by gut microbiota to secondary BAs[49]. Higher levels of ursodeoxycholic acid (UDCA), tauro-UDCA (TUDCA), ursocholic acid (UCA), and murideoxycholic acid (MDCA) have been observed in HCC immunotherapy responders (OR, N = 20) than those in non-responders [progressive disease (PD), N = 21], suggesting that BA profiles may serve as predictive metabolic signatures for treatment outcomes[48].
SCFAs contain fewer than six carbon atoms. In the proximal intestine, dietary fiber cannot be degraded by host digestive enzymes and thus passes into the cecum and colon, where it is fermented by the gut microbiota to generate SCFAs (primarily acetate, propionate, and butyrate)[50,51]. A prospective cohort study included 77 patients with virus-related HCC (V-HCC) and 25 patients with MASLD-associated HCC who initiated first-line combination immunotherapy. Pre-treatment fecal microbiota and metabolites, as well as serum cytokine and chemokines, were analyzed in relation to durable tumor response, OS, and PFS. Patients with MASLD-HCC showed lower baseline levels of SCFAs and UDCA than those in patients with V-HCC, whereas durable responders (DRs) exhibited higher levels of SCFAs (including acetate, propionate, butyrate, and isobutyrate) and UDCA. Similarly, DRs in the V-HCC cohort also displayed higher SCFA levels despite differing microbial backgrounds. In both MASLD-HCC and V-HCC cases, fecal acetate levels were a common predictor of DR, PFS, and OS. Patients with relatively higher acetate levels exhibited significantly longer OS (mOS: 25.2 months vs. 11.3 months) and PFS (mPFS: 15.3 months vs. 4.2 months)[52].
This functional convergence suggests that metabolic signatures transcend the inherent noise of taxonomic profiling, offering a more robust framework for patient stratification. Therefore, the metabolic profiling of fecal or serum samples represents a critical frontier in moving from associative observations to mechanistically grounded clinical predictions.
REGULATORY ROLES OF GUT MICROBIOTA IN HCC IMMUNOTHERAPY RESPONSE
Microbiota-associated immune microenvironment in HCC immunotherapy
Cancer immunoediting suggests that tumor immune responses undergo the following three phases: elimination, equilibrium, and escape[53]. In HCC, the final escape phase is characterized by the establishment of an immunosuppressive tumor microenvironment (TME). To evade immune recognition, tumor cells downregulate antigen presentation and secrete inhibitory factors [e.g., arginase, transforming growth factor-beta (TGF-β)], while upregulating checkpoint molecules such as PD-L1[54]. ICIs are designed to disrupt these evasive tactics by releasing the “molecular brakes” on T-cell function. Within the HCC TME, PD-1 is overexpressed on exhausted T cells, with its binding to PD-L1 on tumor cells or suppressive populations (e.g., myeloid-derived suppressor cells and tumor-associated macrophages) restricting cytokine production and cytotoxicity. Anti-PD-1/PD-L1 antibodies (e.g., nivolumab, pembrolizumab) reinvigorate these effector cells by blocking this inhibitory axis. Similarly, cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) competes with CD28 for B7 molecules on antigen-presenting cells, thereby limiting T-cell priming. CTLA-4 inhibitors (e.g., ipilimumab, tremelimumab) enhance T-cell priming and reduce Treg-mediated immunosuppression, thereby promoting a more immune-active TME[55,56].
Gut microbiota-immune cell crosstalk
Accumulating evidence highlights specific gut microbial signatures and their metabolites as pivotal orchestrators of the hepatic immune microenvironment, directly modulating both innate and adaptive immunity [Figure 1A-D].
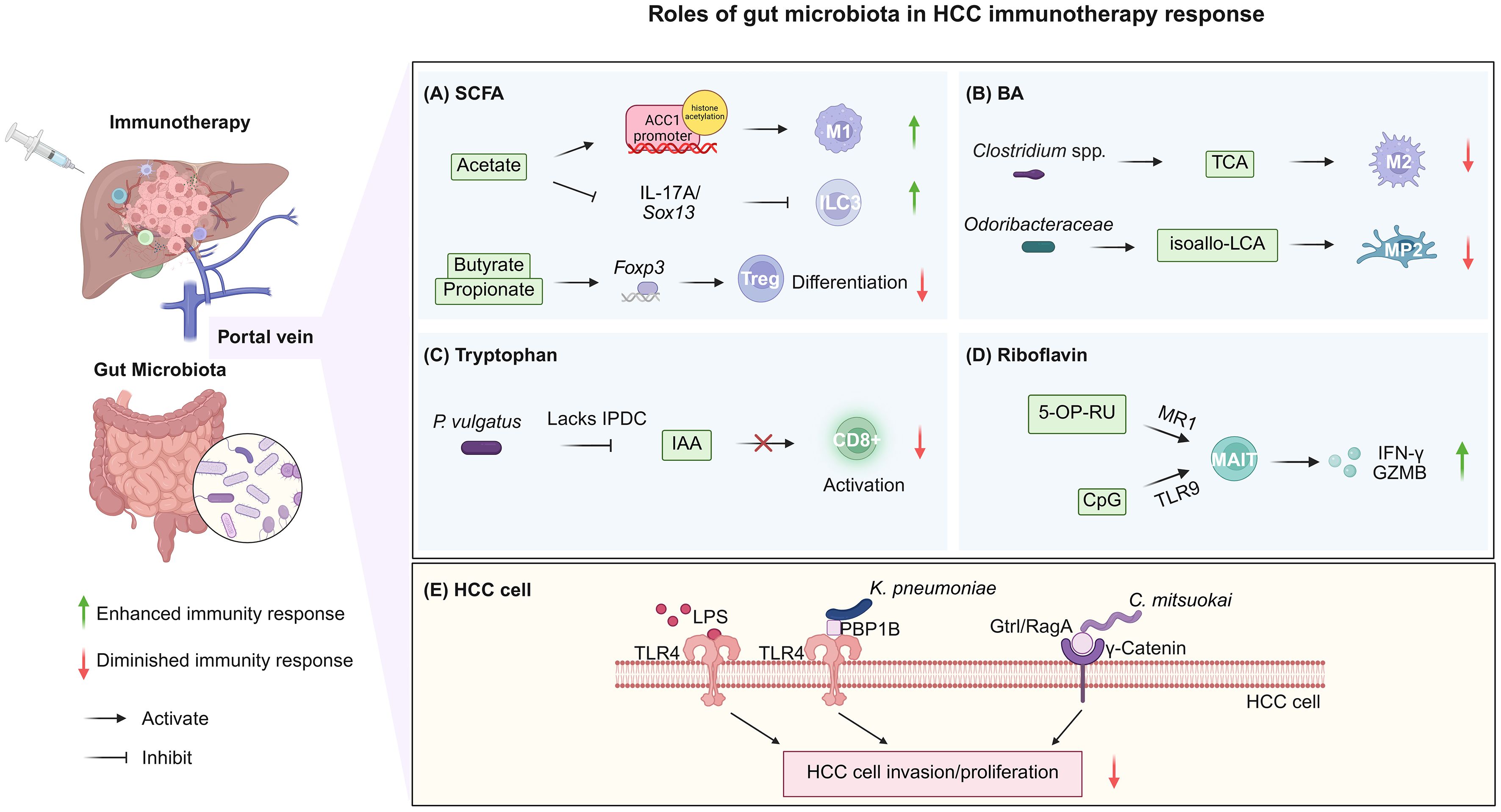
Figure 1. Roles of the gut microbiota in HCC immunotherapy response. (A) Microbiota-derived SCFAs exert context-dependent effects on hepatic immunity, regulating M1 macrophage polarization, ILC3 function, and Treg differentiation; (B) Microbiota-derived BAs induce immunosuppressive reprogramming of macrophages and Kupffer cells, suppressing antitumor immunity; (C) Microbial reprogramming of IAA production impairs CD8+ T-cell function, contributing to immunotherapy resistance; (D) Co-stimulation of microbiota-derived riboflavin metabolites via MR1 and CpG (TLR9 agonist) activates MAIT cells, thus enhancing antitumor immune responses; (E) Microbial components and intrahepatic bacteria promote HCC cell invasion and proliferation, thereby impairing antitumor immune responses. Created in BioRender. Hu, S. (2026) https://BioRender.com/rh59ehn. HCC: Hepatocellular carcinoma; SCFA: short-chain fatty acid; ILC3: group 3 innate lymphoid cell; Treg: regulatory T cell; BA: bile acid; TCA: taurocholic acid; isoallo-LCA: isoallolithocholic acid; MP2: Marco+ IL-10+ Kupffer cells; IAA: indole-3-acetic acid; 5-OP-RU: 5-(2-oxopropylideneamino)-6-D-ribitylaminouracil; MAIT: mucosal-associated invariant T cell; IFN-γ: interferon-γ; GZMB: granzyme B; LPS: microbial-derived lipopolysaccharide; TLR: Toll-like receptor.
SCFAs, particularly acetate and butyrate, potentially exert divergent, context-dependent effects on hepatic immunity. Acetate potentially enhances antitumor surveillance. For instance, Bacteroides thetaiotaomicron-derived acetate may promote M1 macrophage polarization via histone acetylation at the ACC1 promoter, subsequently augmenting CD8+ T-cell effector functions[57]. Correspondingly, acetate depletion, often associated with Lactobacillus reuteri reduction, increases interleukin (IL)-17A production by hepatic ILC3s via Sox13-mediated transcriptional reprogramming. Restoring acetate levels, particularly when combined with PD-1/PD-L1 blockade, alleviates this pro-tumorigenic inflammation and significantly enhances immunotherapy efficacy[58]. In contrast, butyrate enrichment in MASLD-HCC is associated with an immunosuppressive microenvironment. High butyrate concentrations correlate negatively with cytotoxic CD8+ T-cell infiltration[59]. Mechanistically, SCFAs such as propionate and butyrate may function as histone deacetylase inhibitors, thereby enhancing Foxp3 expression and facilitating Treg-mediated immune suppression[22,60-63].
BA derivatives shaped by specific gut taxa serve as signaling molecules that reprogram hepatic macrophages toward an immunosuppressive phenotype. Primary BAs such as taurocholic acid, whose metabolism is influenced by gut microbes, including Clostridium spp. and Akkermansia muciniphila, may favor M2-like macrophage polarization via FXR/TGR5 signaling[64-66]. M2-like macrophages secrete immunosuppressive cytokines (IL-10, TGF-β), which inhibit CD8+ T cell cytotoxicity, thereby counteracting T cell-mediated tumor cytotoxicity and facilitating immune evasion[67,68]. Similarly, isoallo-LCA, a secondary BA produced by Odoribacteraceae, induces a specialized subset of Marco+ IL-10+ Kupffer cells (MP2) through activation of the nuclear hormone receptor, NR4A1[69,70]. The NR4A1-mediated induction of MP2 cells may foster an immunosuppressive microenvironment characterized by increased IL-10 production, thereby potentially dampening antitumor immunity[71].
The reprogramming of tryptophan metabolism by gut commensals represents another pivotal mechanism underlying immunotherapy resistance. Higher Phocaeicola vulgatus abundance is associated with shorter PFS and OS[72]. Mechanistic validation in HCC mouse models suggests that P. vulgatus colonization markedly reduced the efficacy of PD-1 blockade. This resistance potentially correlates with a metabolic rerouting wherein, unlike commensals, which convert tryptophan to IAA via the indolepyruvate decarboxylase enzyme[73], P. vulgatus utilizes an indolepyruvate oxidoreductase-mediated pathway that diverts metabolic flux away from IAA production. Considering that IAA may enhance CD8+ T-cell effector function[74,75], its depletion by P. vulgatus may compromise the potency of ICB and serve as a mechanistic basis for immunotherapy resistance in HCC[72].
Microbiota-derived riboflavin (vitamin B2) metabolites represent a unique class of antigens which specifically modulate mucosal-associated invariant T (MAIT) cells, an innate-like T-cell population highly abundant in the human liver[76-78]. While tumor-infiltrating MAIT cells in HCC frequently exhibit an exhausted phenotype characterized by elevated PD-1/CTLA-4 expression and diminished effector function, leveraging microbial-derived signals offers a potential strategy to reverse this dysfunction[76,79]. Commensal bacteria from the Bacteroidetes, Proteobacteria, and Fusobacteria phyla provide the requisite metabolic precursor, 5-OP-RU. Upon MR1-mediated presentation and co-stimulation with the Toll-like receptor 9 (TLR9) agonist CpG, this microbial antigen robustly expands MAIT cells and restores their interferon-γ and granzyme B production. These findings suggest that the riboflavin biosynthetic capacity of gut microbiota may serve as a critical determinant of innate-like T-cell-mediated antitumor immunity in HCC[80].
Gut microbiota-HCC cell crosstalk
Gut microbes and their metabolites not only modulate immune cells but also directly affect HCC cells and the surrounding TME [Figure 1E].
Microbiota-derived lipopolysaccharide (LPS) is a potent facilitator of tumor cell reprogramming following gut barrier dysfunction[81,82]. In HCC cells characterized by high TLR4 expression, translocated LPS potentially activates the nuclear factor kappa B-Snail signaling axis, thereby inducing epithelial-mesenchymal transition and enhancing metastatic potential[83].
Intrahepatic colonization by specific bacterial taxa provides a more direct mechanistic link between gut dysbiosis and hepatocarcinogenesis. For instance, Klebsiella pneumoniae and Catenibacterium mitsuokai can breach the compromised mucosal barrier to colonize the liver[27,84]. Once established within the liver, these microbes utilize distinct surface proteins to activate tumor-intrinsic signaling pathways. The K. pneumoniae protein, PBP1B, potentially activates TLR4, whereas C. mitsuokai employs Gtr1/RagA to interact with γ-catenin on HCC cells. Both mechanisms may promote tumor cell proliferation and survival[27,84]. Notably, C. mitsuokai, which is consistently enriched in both the feces and tumors of patients with HCC, further amplifies oncogenic signaling by secreting quinolinic acid, an oncometabolite which triggers the TIE2-PI3K/AKT axis[84].
In summary, the gut microbiota-HCC crosstalk operates through both tumor-intrinsic and immune-mediated mechanisms, shaping disease progression and immunotherapy responsiveness. This complex interplay highlights gut microbes as potential predictive markers and therapeutic targets for improving treatment response.
GUT MICROBIOTA-TARGETED INTERVENTIONS IN HCC IMMUNOTHERAPY
Targeting the gut microbiota has emerged as a promising strategy to enhance the efficacy of immunotherapy. This section reviews recent studies on the impact of targeted gut microbiota modulation on HCC immunotherapy outcomes, focusing on FMT, diet, antibiotics, and probiotics [Figure 2].
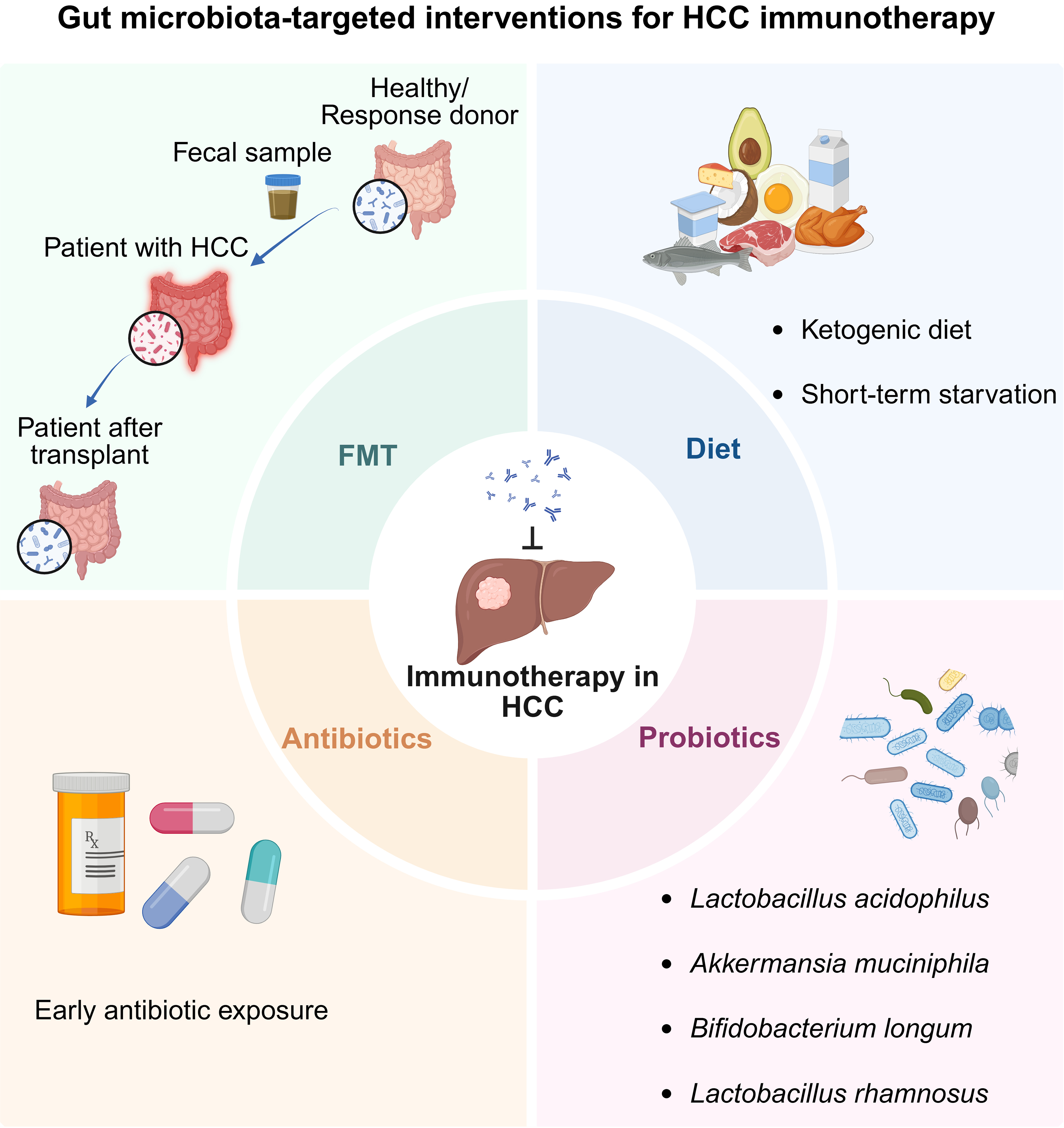
Figure 2. Gut microbiota-targeted interventions to potentiate HCC immunotherapy. Created in BioRender. Hu, S. (2026) https://BioRender.com/mqw7shd. HCC: Hepatocellular carcinoma; FMT: fecal microbiota transplantation.
FMT
Considering the distinct gut microbial signatures associated with responses to ICIs, FMT, which involves the transfer of stool from healthy individuals or ICI responders, typically via oral lyophilized/frozen capsules or endoscopic delivery, has emerged as a potential approach to enhance immunotherapy efficacy[85]. Accumulating clinical evidence from melanoma demonstrates that FMT can restore or potentiate responsiveness to anti-PD-1 therapy[17,86]. For instance, a phase I clinical trial (NCT03772899) evaluated the combination of FMT with anti-PD-1 treatment in advanced melanoma and reported an encouraging ORR of 65%, underscoring the therapeutic promise and clinical feasibility of microbiome modulation[18].
However, clinical data on FMT in HCC remain limited. A prospective clinical study evaluated the therapeutic impact of FMT combined with anti-PD-1 therapy in patients with unresectable or metastatic solid tumors. Six donors (four with HCC) were selected based on achieving DCB (complete response or partial response ≥ 6 months) from prior anti-PD-1 monotherapy. Thirteen recipients (four with HCC) had confirmed progressive disease despite ongoing PD-1-based treatment. All recipients received a 5-day course of amoxicillin-clavulanate before the first FMT to deplete the native microbiota, followed by colonoscopy-based FMT and continuation of standard-dose nivolumab until toxicity or further progression. Additional FMTs from the same or different donors were permitted when early clinical benefit was not observed. Among the 13 recipients, one patient achieved a partial response and five exhibited stable disease, resulting in an ORR of 7.7% and a disease control rate of 46.2%. Notably, the only partial responder was a patient with metastatic HCC who experienced substantial tumor regression after receiving FMT alongside continued nivolumab therapy[87]. Considering these preliminary findings, large-scale clinical trials are warranted to further evaluate the feasibility of FMT for enhancing ICI efficacy in patients with HCC and to elucidate the underlying mechanisms.
Diet
Dietary patterns can shape the gut microbiome and, in turn, affect hepatocarcinogenesis and therapeutic responses[88,89]. Fermentable fiber-enriched refined diets, such as inulin, induce cholestatic HCC resembling human icteric HCC in Toll-like receptor 5-deficient mice, which are prone to gut microbiota dysbiosis. This tumorigenesis is associated with gut dysbiosis and elevated secondary BA levels, while SCFAs, particularly butyrate, may further promote HCC under conditions of inflammation and cholestasis[90]. In contrast, the ketogenic diet, characterized by high fat and very low carbohydrate and protein intake, can alleviate tumor-induced immunosuppression[91]. Mice fed the ketogenic diet exhibit markedly reduced subcutaneous HCC tumor growth than that in mice fed a normal diet[92]. Moreover, a study in murine HCC models revealed that short-term starvation (STS) can potentiate responsiveness to ICIs, with mice undergoing two treatment cycles, each consisting of three repeated fasting periods. Under this regimen, the combination of STS with anti-PD-L1 produces greater tumor suppression than that with anti-PD-1 or other monotherapies, and survival analyses further support the superior efficacy of this combination[93].
Antibiotics
Antibiotics can selectively eliminate gut bacteria, thereby potentially influencing immunotherapy outcomes. In 2021, two studies reported seemingly conflicting findings regarding the impact of antibiotics on the efficacy of ICIs in HCC. One retrospective cohort study from Hong Kong, including 395 patients with advanced HCC, found that the concurrent use of antibiotics during ICI therapy is associated with higher cancer-related and all-cause mortality[94]. In contrast, an international cohort study including 450 patients with HCC from Europe, North America, and Asia evaluated patients receiving either monotherapy or combination ICI. Early antibiotic exposure was defined as antibiotic use within 30 day before or after ICI initiation. This study found that early antibiotic exposure did not compromise OS or treatment efficacy and was even associated with prolonged PFS[95]. However, more recent studies published in 2023, including two large retrospective cohorts comprising 4,098 patients with unresectable HCC receiving ICI therapy[96] and 105 patients treated with atezolizumab plus bevacizumab[97], consistently demonstrated that early antibiotic exposure is associated with reduced treatment efficacy, including shorter PFS and OS. The causal relationship between antibiotic use and outcomes in HCC patients is yet to be definitively established.
Probiotics
Probiotics are defined as “live microorganisms which, when administered in adequate amounts, confer a health benefit on the host”[98]. Unlike FMT, transplantation of specific gut bacteria, alone or in combination, offers a more targeted and safer approach to modulate the gut microbiota in patients with HCC. In mouse models of MASLD-HCC, the administration of Lactobacillus acidophilus[99] and Akkermansia muciniphila[100] effectively suppresses hepatocarcinogenesis and substantially reduces tumor burden, respectively. Notably, the combination of Akkermansia muciniphila with anti-PD-1 therapy elicits a more robust antitumor response across multiple MASLD-HCC models[100]. In addition to antitumor efficacy, probiotics can potentially improve postoperative outcomes. In a clinical trial involving patients with HCC, the oral administration of a probiotic bacteria cocktail containing Bifidobacterium longum reduced the rates of delayed recovery, shortened hospital stays, and improved overall 1-year survival[101]. Furthermore, although PD-1 inhibitors exhibit antitumor activity, their use is often limited by immune-related adverse events (irAEs) such as colitis and hepatitis[102]. Lactobacillus rhamnosus modulates inflammatory signaling in both the gut and liver in HCC murine models[103], suggesting its potential as a complementary approach to mitigate irAEs while enhancing immunotherapeutic efficacy.
CHALLENGES AND FUTURE PERSPECTIVES
Limitations of current microbial predictors of ICI response in HCC
As summarized in Table 1, current microbial studies in HCC are skewed toward East Asian populations and primarily focus on taxonomic abundance and simple diversity metrics, providing a foundational but fragmented and geographically biased understanding regarding the microbial predictors of ICI response.
Summary of predictive gut microbial signatures for ICI efficacy in HCC cohorts
| Patient characteristics | Location | Cohort size | Evaluation criteria | Sequencing | Predictive signatures | Remarks |
| Unresectable HCC or advanced BTC (after failure of gemcitabine + cisplatin)[42] | Beijing, China | 65 | CBR: CR, PR, or SD ≥ 6 months (N = 17 HCC, N = 15 BTC); NCB: SD < 6 months or PD (N = 13 HCC, N = 20 BTC) | Metagenomics | Lachnospiraceae bacterium-GAM79, Alistipes sp Marseille-P5997, Ruminococcus calidus and Erysipelotichaceae bacterium-GAM147 enriched in CBR; Veillonellaceae family enriched in NCB | Higher diversity and Firmicutes abundance identified as potential protective factors against irAEs (e.g., diarrhea) |
| HCC (BCLC Stage C; Sorafenib failure)[43] | Zhejiang, China | 8 | R: CR, PR, or SD > 6 months (N = 3); NR: PD or SD < 6 months (N = 5) | Metagenomics | Higher taxa richness in R | Small sample size |
| Lactobacillus spp., Akkermansia muciniphila and Lachnospiraceae enriched in R; Escherichia coli enriched in NR | ||||||
| HCC[44] | Guangdong, China | 35 | R: CR, PR, or SD ≥ 6 months (N = 16); NR: PD or SD < 6 months (N = 19) | Fecal 16S rRNA; Serum metabolomics | Higher alpha diversity in R; distinct beta diversity clustering between R and NR | AUC of 0.827-0.837 for predicting the R status |
| Faecalibacterium, Blautia, Lachnospiraceae incertae sedis, Megamonas and Ruminococcus enriched in R | ||||||
| HCC (Sorafenib failure)[47] | South Korea | 8 | R: CR, PR, or SD > 6 months (N = 5); NR: PD or SD < 6 months (N = 3) | Fecal 16S rRNA | Higher taxa richness in Rs | |
| Proper Firmicutes/Bacteroidetes ratio (0.5-1.5) and higher Prevotella/Bacteroides ratio in R. Akkermansia species enriched in R | ||||||
| HCC[48] | Taiwan, China | 41 | OR: CR or PR (N = 20); PD: N = 21 | Fecal 16S rRNA; Fecal BAs and SCFAs | Veillonella, Lachnospiraceae, and Lachnoclostridium enriched in OR; Prevotella 9 in PD | Co-existence of Lachnoclostridium enrichment and Prevotella 9 depletion predicted better OS |
| UDCA, TUDCA, UCA and MDCA enriched in OR | ||||||
| V-HCC,MASLD-HCC[52] | Taiwan, China | 102 | DR: PR or SD ≥ 8 months (N = 32, V-HCC; N = 2, MASLD-HCC) | Fecal 16S rRNA; Fecal BAs and SCFAs; Serum cytokines | Mediterraneibacter gnavus ATCC 29149 enriched in DR (MASLD-HCC); Bifidobacterium enriched in DR (v-HCC) | Baseline metabolic levels significantly differ between V-HCC and MASLD-HCC etiologies |
| Acetate enriched in DR |
First, the restricted scale and observational nature of existing research remain primary hurdles[43,47]. With only one study incorporating an external validation cohort[48], these inherent limitations increase the risk of overfitting and restrict the statistical power required to define robust biomarkers. Furthermore, the majority of the identified microbial-metabolite correlations lack mechanistic confirmation; therefore, further in vivo and in vitro validation experiments are needed.
Second, the clinical landscape of HCC introduces etiological and therapeutic heterogeneity. For instance, the cohort of 35 patients involves mixed treatment backgrounds (e.g., combinations of ICI, TACE, and targeted therapies)[44]. Such therapeutic confounding, alongside the baseline divergence between V-HCC and MASLD-HCC etiologies[52], suggests that many “responder” signatures are highly context-dependent. This is further exacerbated by the ability of concurrent agents, such as anti-CTLA-4, to modify gut barrier integrity and microbial compositions[104].
Third, technical discrepancies in protocols and sequencing methodologies hinder cross-cohort consistency. The reliance on 16S rRNA sequencing in several cohorts[44,47,48,52] introduces a “resolution gap” that fails to distinguish between closely related species or functional strains. The genus Veillonella epitomizes this fragility, serving as a positive predictor in some reports[47], whereas other reports suggesting its enrichment in non-responders[42,43]. As highlighted by large-scale meta-analyses in melanoma, inherent population and microbiome heterogeneity make the identification of universal biomarkers exceptionally difficult, even with unified analytical pipelines[105].
Finally, robust predictive modeling for microbe-based ICI efficacy remains scarce. Here, only one study attempted to construct a predictive framework using a Random Forest model[44]. The development of such models faces significant technical hurdles, particularly the risk of overfitting due to the relatively small sample sizes in current HCC cohorts. Furthermore, the lack of cross-cohort generalizability, mainly driven by geographical and etiological heterogeneity, remains a critical barrier to clinical implementation. Transitioning toward multi-dimensional frameworks to capture the functional output of the tumor-immune-microbe axis remains a challenge.
Emerging microbial predictive signatures
Accumulating research in other malignancy contexts has identified more sophisticated microbial signatures that hold predictive value for HCC immunotherapy.
Microbial structural variations (SVs), comprising intra-species genomic deletions or insertions, may serve as a novel predictive dimension for immunotherapy efficacy. Using data from the gut microbiome of 996 patients from seven datasets, researchers have demonstrated that specific SVs are significantly associated with OR, PFS, and OS[106]. Crucially, these microbial SV associations are independent of taxonomic abundance. This shift toward genomic granularity is further supported by evidence that strain-resolved microbial abundances significantly improve machine learning-based predictions of ICB response and landmark PFS compared to species-rank quantifications or comprehensive clinical factors. Furthermore, meta-analyses across diverse cohorts have confirmed the cross-cancer and cross-country validity of these strain-response signatures, provided that the training and test cohorts utilize concordant ICB regimens[107].
Beyond microbial genomic markers, the characterization of immunotherapy sensitivity is increasingly focused on the ecological network. Enterotype classification provides a framework to capture individual microbiome heterogeneity, linking compositional patterns to immune phenotypes and therapeutic outcomes. The identification of favorable enterotypes with heightened microbial diversity signifies a potential biomarker for predicting treatment response in pan-cancer cohorts, since FMT from favorable-type donors enhances anti-PD-1 sensitivity and immune activation[108,109]. Consequently, emerging ecosystem-based strategies, such as TOPOSCORE, evaluate the stability and connectivity of microbial co-abundance networks. By identifying “guilds” or functional groups of cooperative bacteria, this approach reveals a balance between antagonistic sub-communities, defining a state of detrimental dysbiosis associated with immunotherapy resistance[110]. This topology-based scoring system has demonstrated predictive power for patient survival across multiple malignancies, moving the field from identifying “who is there” to characterizing “how they interact” within the tumor-immune-microbe axis.
Moreover, the transition from associative descriptions to predictive systems is accelerated by integrative multi-omics modeling. For instance, in HBV-related HCC, integrating gut microbial signatures with associated BA profiles and host tumor transcripts provides mechanistic insights into how the gut microbiota contributes to tumor burden[111]. Such multi-dimensional predictive models, which incorporate strain-level variability, ecological topology, and host-microbe metabolic crosstalk, remain the frontier for establishing robust, cross-cohort microbial predictors in HCC.
Technical and methodological innovation
While numerous studies have established correlations between certain taxa or microbial patterns and immunotherapy responsiveness, it remains unclear whether these signatures are causal drivers of anti-tumor immunity or simply predictive markers of host physiology. Emerging technologies are now bridging this mechanistic gap.
Click chemistry enables selective labeling and enrichment of low-abundance microbial metabolites, allowing discrimination between microbiota-derived molecules and host-derived analogs which are otherwise indistinguishable by conventional metabolomics. This approach has facilitated the discovery of novel microbial BA derivatives, such as 3-succinylated cholic acid, and enabled functional characterization linking microbial metabolism to host immune regulation[66].
Serum antibodies can recognize both pathogens and commensal gut microbiota, yet the antigenic specificities of these responses remain incompletely characterized[112]. Phage Immunoprecipitation Sequencing is a high-throughput platform for profiling host antibody responses against microbial antigens at the epitope level[113]. These “immunological fingerprints” are longitudinally more stable than taxonomic compositions and enable the identification of systemic biomarkers and immunologically relevant microbial targets[114].
Moreover, the single-cell Bacteria Polygenic Score framework integrates microbial genome-wide association study with multi-organ human scRNA-seq data to map microbe-relevant genetic signals in the cellular context, yielding previously unperceived biological insights regarding involved cell types and biological pathways[115]. Complementing these novel technical and methodological improvements, large-scale multi-cohort integration is essential for validating microbial signatures across broader populations. For instance, performing de novo assembly across approximately 4,000 metagenomic samples from 38 studies spanning 15 diseases enables the recovery of high-quality metagenome-assembled genomes to define a stable “core microbiome signature”[116]. This interaction-focused analytical approach remains resilient to confounding factors without the need for conventional mathematical adjustments, providing a potent predictive backbone for immunotherapy outcomes across diverse global cohorts.
Clinical translation
Despite growing evidence linking gut microbiota to immunotherapy outcomes, the clinical translation of microbiota research in HCC is still in its early stages, and HCC cohorts remain small and largely retrospective. FMT has been proposed as a potential strategy to overcome resistance to ICIs. However, adverse events related to FMT have been reported, with an incidence of 19% and serious adverse events accounting for approximately 1.4% of all cases[117]. Moreover, portal hypertension and gastrointestinal bleeding in advanced HCC also influence the gut-liver axis. A recent systematic review revealed that all FMT-related serious adverse events occur in patients with damage to the mucosal barrier[117], a common feature in cirrhotic patients with HCC, which increases the risk of bacterial translocation and systemic infection. Furthermore, FMT acts as a “double-edged sword”; it can eradicate unfavorable microbial signatures but may inadvertently transmit procarcinogenic bacteria or harmful genetic elements that elude current donor screening protocols[118]. These risks necessitate more rigorous donor screening and longitudinal monitoring of intestinal barrier integrity.
The success of FMT is influenced by a combination of donor- and recipient-related variables, including microbial diversity, immunomodulatory taxa, baseline dysbiosis, host immune tone, and genetic background. Procedural parameters, such as dosing, administration schedule, delivery route, and adjunctive antibiotic use, also play important roles[85]. As evidenced by the TACITO and PERFORM trials, recruitment and logistical challenges often lead to unintentional treatment delays. While relying on a single “super-donor” ensures consistency, it exacerbates supply shortages[119,120]. Moreover, recent Phase II trials such as FMT-LUMINate highlight the challenge of microbial engraftment, where the potential loss of transplanted bacteria from the intestinal tract remains a critical concern for functional stability[121]. These constraints underscore the necessity of transitioning toward standardized Live Biotherapeutic Products. Such next-generation interventions should be designed to offer a more predictable and scalable alternative to conventional FMT.
The clinical impact of antibiotic exposure remains a contentious issue, which further complicates the translation of FMT. As discussed, the role of antibiotics as a “preconditioning” tool for FMT introduces a paradox. Systemic antibiotics (e.g., vancomycin and neomycin) can facilitate donor strain engraftment by reducing recipient microbial diversity to create a “niche,” whereas agents with minimal impact on diversity, such as rifaximin, often fail[122,123]. In HCC, identifying the optimal antibiotic class, dosage, and “window of vulnerability” is critical to balance the detrimental effects of dysbiosis against the necessity of microbial clearance for therapeutic colonization.
CONCLUSION
The gut microbiota shapes the immune landscape of HCC and influences responses to ICIs. Community structures, specific taxa, and microbial molecules not only correlate with treatment outcomes but also offer potential targets for therapeutic modulation through FMT, diet, antibiotics, and probiotics. While challenges remain, advances in large-scale, multi-omics profiling, strain-level analysis, and innovative techniques hold promise for precision microbiota-informed immunotherapy. Harnessing the gut microbiota as both a biomarker and therapeutic adjunct may ultimately optimize the efficacy of immunotherapy, mitigate adverse events, and advance personalized treatment strategies in HCC.
DECLARATIONS
Acknowledgments
The Graphical Abstract was created in BioRender. Hu, S. (2026) https://BioRender.com/423wmn3.
Authors’ contributions
Conceptualized the idea and planned the manuscript: Xia S, Yang Q, Liu Y, Yan A, Li S, Xu L, Guan H
Wrote the initial draft: Xia S
Revised, edited, and approved the final version: Xia S, Hu S, Xie Y, Liu Y
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
During the preparation of this manuscript, the AI tool ChatGPT (GPT-5.5) was used solely for language editing. The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship
This study was supported by the following grants: Guangdong Basic and Applied Basic Research Foundation (No. 2025B1515020059) and National Natural Science Foundation of China (No. 82570639; No. 82300623).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Vogel A, Meyer T, Sapisochin G, Salem R, Saborowski A. Hepatocellular carcinoma. Lancet. 2022;400:1345-62.
2. Llovet JM, Kelley RK, Villanueva A, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7:6.
3. El-Khoueiry AB, Sangro B, Yau T, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389:2492-502.
4. Greten TF, Villanueva A, Korangy F, et al. Biomarkers for immunotherapy of hepatocellular carcinoma. Nat Rev Clin Oncol. 2023;20:780-98.
5. Llovet JM, Castet F, Heikenwalder M, et al. Immunotherapies for hepatocellular carcinoma. Nat Rev Clin Oncol. 2022;19:151-72.
6. Finn RS, Qin S, Ikeda M, et al.; IMbrave150 Investigators. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med. 2020;382:1894-905.
7. Abou-Alfa GK, Lau G, Kudo M, et al. Tremelimumab plus durvalumab in unresectable hepatocellular carcinoma. NEJM Evid. 2022;1:EVIDoa2100070.
8. Yau T, Galle PR, Decaens T, et al.; CheckMate 9DW investigators. Nivolumab plus ipilimumab versus lenvatinib or sorafenib as first-line treatment for unresectable hepatocellular carcinoma (CheckMate 9DW): an open-label, randomised, phase 3 trial. Lancet. 2025;405:1851-64.
9. Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020;20:651-68.
10. Taube JM, Klein A, Brahmer JR, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res. 2014;20:5064-74.
11. Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124-8.
12. Borghaei H, Paz-Ares L, Horn L, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373:1627-39.
13. Zaretsky JM, Garcia-Diaz A, Shin DS, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. 2016;375:819-29.
14. Pinter M, Jain RK, Duda DG. The current landscape of immune checkpoint blockade in hepatocellular carcinoma: a review. JAMA Oncol. 2021;7:113-23.
15. Routy B, Le Chatelier E, Derosa L, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359:91-7.
16. Björk JR, Bolte LA, Maltez Thomas A, et al. Longitudinal gut microbiome changes in immune checkpoint blockade-treated advanced melanoma. Nat Med. 2024;30:785-96.
17. Baruch EN, Youngster I, Ben-Betzalel G, et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science. 2021;371:602-9.
18. Routy B, Lenehan JG, Miller WH Jr, et al. Fecal microbiota transplantation plus anti-PD-1 immunotherapy in advanced melanoma: a phase I trial. Nat Med. 2023;29:2121-32.
19. Hsu CL, Schnabl B. The gut-liver axis and gut microbiota in health and liver disease. Nat Rev Microbiol. 2023;21:719-33.
20. Hooper LV, Midtvedt T, Gordon JI. How host-microbial interactions shape the nutrient environment of the mammalian intestine. Annu Rev Nutr. 2002;22:283-307.
21. Fan Y, Pedersen O. Gut microbiota in human metabolic health and disease. Nat Rev Microbiol. 2021;19:55-71.
22. Arpaia N, Campbell C, Fan X, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. 2013;504:451-5.
23. Schulthess J, Pandey S, Capitani M, et al. The short chain fatty acid butyrate imprints an antimicrobial program in macrophages. Immunity. 2019;50:432-45.e7.
24. Sorrentino G, Perino A, Yildiz E, et al. Bile acids signal via TGR5 to activate intestinal stem cells and epithelial regeneration. Gastroenterology. 2020;159:956-68.e8.
25. Paik D, Yao L, Zhang Y, et al. Human gut bacteria produce ΤΗ17-modulating bile acid metabolites. Nature. 2022;603:907-12.
26. Zhao R, Fajardo J, Shen GX. Influence of northern wild rice on gut dysbiosis and short chain fatty acids: correlation with metabolic and inflammatory markers in mice on high fat diet. Nutrients. 2024;16:2834.
27. Wang X, Fang Y, Liang W, et al. Gut-liver translocation of pathogen Klebsiella pneumoniae promotes hepatocellular carcinoma in mice. Nat Microbiol. 2025;10:169-84.
28. Ma L, Li H, Hu J, et al. Indole alleviates diet-induced hepatic steatosis and inflammation in a manner involving myeloid cell 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3. Hepatology. 2020;72:1191-203.
29. Beaumont M, Neyrinck AM, Olivares M, et al. The gut microbiota metabolite indole alleviates liver inflammation in mice. FASEB J. 2018;32:fj201800544.
30. Roderburg C, Luedde T. The role of the gut microbiome in the development and progression of liver cirrhosis and hepatocellular carcinoma. Gut Microbes. 2014;5:441-5.
31. Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, Roberts LR. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol. 2019;16:589-604.
32. Refolo MG, Messa C, Guerra V, Carr BI, D’Alessandro R. Inflammatory mechanisms of HCC development. Cancers. 2020;12:641.
33. Aron-Wisnewsky J, Vigliotti C, Witjes J, et al. Gut microbiota and human NAFLD: disentangling microbial signatures from metabolic disorders. Nat Rev Gastroenterol Hepatol. 2020;17:279-97.
34. Duan Y, Llorente C, Lang S, et al. Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature. 2019;575:505-11.
35. Grander C, Adolph TE, Wieser V, et al. Recovery of ethanol-induced Akkermansia muciniphila depletion ameliorates alcoholic liver disease. Gut. 2018;67:891-901.
36. Ohtani N, Hara E. Gut-liver axis-mediated mechanism of liver cancer: a special focus on the role of gut microbiota. Cancer Sci. 2021;112:4433-43.
37. Bauer TM, Schwacha H, Steinbrückner B, et al. Small intestinal bacterial overgrowth in human cirrhosis is associated with systemic endotoxemia. Am J Gastroenterol. 2002;97:2364-70.
38. Song X, Sun X, Oh SF, et al. Microbial bile acid metabolites modulate gut RORγ+ regulatory T cell homeostasis. Nature. 2020;577:410-5.
39. Ren Z, Li A, Jiang J, et al. Gut microbiome analysis as a tool towards targeted non-invasive biomarkers for early hepatocellular carcinoma. Gut. 2019;68:1014-23.
40. Zheng R, Wang G, Pang Z, et al. Liver cirrhosis contributes to the disorder of gut microbiota in patients with hepatocellular carcinoma. Cancer Med. 2020;9:4232-50.
41. Tilg H, Adolph TE, Trauner M. Gut-liver axis: pathophysiological concepts and clinical implications. Cell Metab. 2022;34:1700-18.
42. Mao J, Wang D, Long J, et al. Gut microbiome is associated with the clinical response to anti-PD-1 based immunotherapy in hepatobiliary cancers. J Immunother Cancer. 2021;9:e003334.
43. Zheng Y, Wang T, Tu X, et al. Gut microbiome affects the response to anti-PD-1 immunotherapy in patients with hepatocellular carcinoma. J Immunother Cancer. 2019;7:193.
44. Wu H, Zheng X, Pan T, et al. Dynamic microbiome and metabolome analyses reveal the interaction between gut microbiota and anti-PD-1 based immunotherapy in hepatocellular carcinoma. Int J Cancer. 2022;151:1321-34.
45. Jin Y, Dong H, Xia L, et al. The diversity of gut microbiome is associated with favorable responses to anti-programmed death 1 immunotherapy in chinese patients with NSCLC. J Thorac Oncol. 2019;14:1378-89.
46. Zhang C, Wang J, Sun Z, Cao Y, Mu Z, Ji X. Commensal microbiota contributes to predicting the response to immune checkpoint inhibitors in non-small-cell lung cancer patients. Cancer Sci. 2021;112:3005-17.
47. Chung MW, Kim MJ, Won EJ, et al. Gut microbiome composition can predict the response to nivolumab in advanced hepatocellular carcinoma patients. World J Gastroenterol. 2021;27:7340-9.
48. Lee PC, Wu CJ, Hung YW, et al. Gut microbiota and metabolites associate with outcomes of immune checkpoint inhibitor-treated unresectable hepatocellular carcinoma. J Immunother Cancer. 2022;10:e004779.
49. Thomas C, Pellicciari R, Pruzanski M, Auwerx J, Schoonjans K. Targeting bile-acid signalling for metabolic diseases. Nat Rev Drug Discov. 2008;7:678-93.
50. Morrison DJ, Preston T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes. 2016;7:189-200.
51. den Besten G, van Eunen K, Groen AK, Venema K, Reijngoud DJ, Bakker BM. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J Lipid Res. 2013;54:2325-40.
52. Lee PC, Wu CJ, Hung YW, et al. Distinct gut microbiota but common metabolomic signatures between viral and MASLD HCC contribute to outcomes of combination immunotherapy. Hepatology. 2025;Epub ahead of print.
53. Choudhry H. The microbiome and its implications in cancer immunotherapy. Molecules. 2021;26:206.
55. Hargadon KM, Johnson CE, Williams CJ. Immune checkpoint blockade therapy for cancer: an overview of FDA-approved immune checkpoint inhibitors. Int Immunopharmacol. 2018;62:29-39.
56. Dai S, Chen Y, Cai W, et al. Combination immunotherapy in hepatocellular carcinoma: synergies among immune checkpoints, TKIs, and chemotherapy. J Hematol Oncol. 2025;18:85.
57. Ma H, Yang L, Liang Y, et al. B. thetaiotaomicron-derived acetic acid modulate immune microenvironment and tumor growth in hepatocellular carcinoma. Gut Microbes. 2024;16:2297846.
58. Hu C, Xu B, Wang X, et al. Gut microbiota-derived short-chain fatty acids regulate group 3 innate lymphoid cells in HCC. Hepatology. 2023;77:48-64.
59. Behary J, Amorim N, Jiang XT, et al. Gut microbiota impact on the peripheral immune response in non-alcoholic fatty liver disease related hepatocellular carcinoma. Nat Commun. 2021;12:187.
60. Waldecker M, Kautenburger T, Daumann H, Busch C, Schrenk D. Inhibition of histone-deacetylase activity by short-chain fatty acids and some polyphenol metabolites formed in the colon. J Nutr Biochem. 2008;19:587-93.
61. Park J, Kim M, Kang SG, et al. Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR-S6K pathway. Mucosal Immunol. 2015;8:80-93.
62. Smith PM, Howitt MR, Panikov N, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341:569-73.
63. Furusawa Y, Obata Y, Fukuda S, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504:446-50.
64. Xia JK, Tang N, Wu XY, Ren HZ. Deregulated bile acids may drive hepatocellular carcinoma metastasis by inducing an immunosuppressive microenvironment. Front Oncol. 2022;12:1033145.
65. Sun R, Zhang Z, Bao R, et al. Loss of SIRT5 promotes bile acid-induced immunosuppressive microenvironment and hepatocarcinogenesis. J Hepatol. 2022;77:453-66.
66. Nie Q, Luo X, Wang K, et al. Gut symbionts alleviate MASH through a secondary bile acid biosynthetic pathway. Cell. 2024;187:2717-34.e33.
67. Petty AJ, Owen DH, Yang Y, Huang X. Targeting tumor-associated macrophages in cancer immunotherapy. Cancers. 2021;13:5318.
68. Retracted: M2 macrophage-derived exosomes facilitate HCC metastasis by transferring αMβ2 integrin to tumor cells. Hepatology. 2021;74:3564.
69. Miyamoto Y, Kikuta J, Matsui T, et al. Periportal macrophages protect against commensal-driven liver inflammation. Nature. 2024;629:901-9.
70. Li W, Hang S, Fang Y, et al. A bacterial bile acid metabolite modulates T(reg) activity through the nuclear hormone receptor NR4A1. Cell Host Microbe. 2021;29:1366-77.e9.
71. Koenis DS, Medzikovic L, van Loenen PB, et al. Nuclear receptor Nur77 limits the macrophage inflammatory response through transcriptional reprogramming of mitochondrial metabolism. Cell Rep. 2018;24:2127-40.e7.
72. Zhao CN, Li SS, Yau T, et al. Phocaeicola vulgatus induces immunotherapy resistance in hepatocellular carcinoma via reducing indoleacetic acid production. Cell Rep Med. 2025;6:102370.
73. Roager HM, Licht TR. Microbial tryptophan catabolites in health and disease. Nat Commun. 2018;9:3294.
74. Renga G, Nunzi E, Pariano M, et al. Optimizing therapeutic outcomes of immune checkpoint blockade by a microbial tryptophan metabolite. J Immunother Cancer. 2022;10:e003725.
75. Zelante T, Iannitti RG, Cunha C, et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity. 2013;39:372-85.
76. Kjer-Nielsen L, Patel O, Corbett AJ, et al. MR1 presents microbial vitamin B metabolites to MAIT cells. Nature. 2012;491:717-23.
77. Legoux F, Salou M, Lantz O. MAIT cell development and functions: the microbial connection. Immunity. 2020;53:710-23.
78. Toubal A, Nel I, Lotersztajn S, Lehuen A. Mucosal-associated invariant T cells and disease. Nat Rev Immunol. 2019;19:643-57.
79. Duan M, Goswami S, Shi JY, et al. Activated and exhausted MAIT cells foster disease progression and indicate poor outcome in hepatocellular carcinoma. Clin Cancer Res. 2019;25:3304-16.
80. Ruf B, Catania VV, Wabitsch S, et al. Activating mucosal-associated invariant T cells induces a broad antitumor response. Cancer Immunol Res. 2021;9:1024-34.
81. Gäbele E, Dostert K, Hofmann C, et al. DSS induced colitis increases portal LPS levels and enhances hepatic inflammation and fibrogenesis in experimental NASH. J Hepatol. 2011;55:1391-9.
82. Szabo G, Petrasek J. Inflammasome activation and function in liver disease. Nat Rev Gastroenterol Hepatol. 2015;12:387-400.
83. Jing YY, Han ZP, Sun K, et al. Toll-like receptor 4 signaling promotes epithelial-mesenchymal transition in human hepatocellular carcinoma induced by lipopolysaccharide. BMC Med. 2012;10:98.
84. Zhang Y, Liu W, Wong CC, et al. Catenibacteriummitsuokai promotes hepatocellular carcinogenesis by binding to hepatocytes and generating quinolinic acid. Cell Metab. 2025;37:1998-2013.e7.
85. Porcari S, Benech N, Valles-Colomer M, et al. Key determinants of success in fecal microbiota transplantation: from microbiome to clinic. Cell Host Microbe. 2023;31:712-33.
86. Davar D, Dzutsev AK, McCulloch JA, et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science. 2021;371:595-602.
87. Kim Y, Kim G, Kim S, et al. Fecal microbiota transplantation improves anti-PD-1 inhibitor efficacy in unresectable or metastatic solid cancers refractory to anti-PD-1 inhibitor. Cell Host Microbe. 2024;32:1380-93.e9.
88. David LA, Maurice CF, Carmody RN, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505:559-63.
89. Falony G, Joossens M, Vieira-Silva S, et al. Population-level analysis of gut microbiome variation. Science. 2016;352:560-4.
90. Singh V, Yeoh BS, Chassaing B, et al. Dysregulated microbial fermentation of soluble fiber induces cholestatic liver cancer. Cell. 2018;175:679-94.e22.
91. Woolf EC, Syed N, Scheck AC. Tumor metabolism, the ketogenic diet and β-hydroxybutyrate: novel approaches to adjuvant brain tumor therapy. Front Mol Neurosci. 2016;9:122.
92. Wang YH, Liu CL, Chiu WC, Twu YC, Liao YJ. HMGCS2 mediates ketone production and regulates the proliferation and metastasis of hepatocellular carcinoma. Cancers. 2019;11:1876.
93. Cheng K, Cai N, Yang X, et al. Short-term starvation boosts anti-PD-L1 therapy by reshaping tumor-associated macrophages in hepatocellular carcinoma. Hepatology. 2025;82:1414-31.
94. Cheung KS, Lam LK, Seto WK, Leung WK. Use of antibiotics during immune checkpoint inhibitor treatment is associated with lower survival in hepatocellular carcinoma. Liver Cancer. 2021;10:606-14.
95. Fessas P, Naeem M, Pinter M, et al. Early antibiotic exposure is not detrimental to therapeutic effect from immunotherapy in hepatocellular carcinoma. Liver Cancer. 2021;10:583-92.
96. Pinato DJ, Li X, Mishra-Kalyani P, et al. Association between antibiotics and adverse oncological outcomes in patients receiving targeted or immune-based therapy for hepatocellular carcinoma. JHEP Rep. 2023;5:100747.
97. Maesaka K, Sakamori R, Yamada R, et al. Pretreatment with antibiotics is associated with reduced therapeutic response to atezolizumab plus bevacizumab in patients with hepatocellular carcinoma. PLoS One. 2023;18:e0281459.
98. Kaźmierczak-Siedlecka K, Skonieczna-Żydecka K, Hupp T, Duchnowska R, Marek-Trzonkowska N, Połom K. Next-generation probiotics - do they open new therapeutic strategies for cancer patients? Gut Microbes. 2022;14:2035659.
99. Lau HC, Zhang X, Ji F, et al. Lactobacillus acidophilus suppresses non-alcoholic fatty liver disease-associated hepatocellular carcinoma through producing valeric acid. EBioMedicine. 2024;100:104952.
100. Wu XQ, Ying F, Chung KPS, et al. Intestinal Akkermansia muciniphila complements the efficacy of PD1 therapy in MAFLD-related hepatocellular carcinoma. Cell Rep Med. 2025;6:101900.
101. Yu J, Zhu P, Shi L, et al. Bifidobacterium longum promotes postoperative liver function recovery in patients with hepatocellular carcinoma. Cell Host Microbe. 2024;32:131-44.e6.
102. Tang L, Wang J, Lin N, et al. Immune checkpoint inhibitor-associated colitis: from mechanism to management. Front Immunol. 2021;12:800879.
103. Nenu I, Baldea I, Coadă CA, et al. Lactobacillus rhamnosus probiotic treatment modulates gut and liver inflammatory pathways in a hepatocellular carcinoma murine model. A preliminary study. Food Chem Toxicol. 2024;183:114314.
104. Mager LF, Burkhard R, Pett N, et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science. 2020;369:1481-9.
105. Lee KA, Thomas AM, Bolte LA, et al. Cross-cohort gut microbiome associations with immune checkpoint inhibitor response in advanced melanoma. Nat Med. 2022;28:535-44.
106. Liu R, Zou Y, Wang WQ, et al. Gut microbial structural variation associates with immune checkpoint inhibitor response. Nat Commun. 2023;14:7421.
107. Piccinno G, Thompson KN, Manghi P, et al. Pooled analysis of 3,741 stool metagenomes from 18 cohorts for cross-stage and strain-level reproducible microbial biomarkers of colorectal cancer. Nat Med. 2025;31:2416-29.
108. Zhu X, Huang X, Hu M, et al. A specific enterotype derived from gut microbiome of older individuals enables favorable responses to immune checkpoint blockade therapy. Cell Host Microbe. 2024;32:489-505.e5.
109. Hu M, Zhu X, Huang X, et al. Optimizing anti-PD-1/PD-L1 therapy efficacy and fecal microbiota transplantation donor selection through gut mycobiome-based enterotype. Cell Rep. 2025;44:115589.
110. Derosa L, Iebba V, Silva CAC, et al. Custom scoring based on ecological topology of gut microbiota associated with cancer immunotherapy outcome. Cell. 2024;187:3373-89.e16.
111. Huang H, Ren Z, Gao X, et al. Integrated analysis of microbiome and host transcriptome reveals correlations between gut microbiota and clinical outcomes in HBV-related hepatocellular carcinoma. Genome Med. 2020;12:102.
112. Sterlin D, Fadlallah J, Slack E, Gorochov G. The antibody/microbiota interface in health and disease. Mucosal Immunol. 2020;13:3-11.
113. Larman HB, Zhao Z, Laserson U, et al. Autoantigen discovery with a synthetic human peptidome. Nat Biotechnol. 2011;29:535-41.
114. Vogl T, Klompus S, Leviatan S, et al. Population-wide diversity and stability of serum antibody epitope repertoires against human microbiota. Nat Med. 2021;27:1442-50.
115. Li J, Ma Y, Cao Y, et al. Integrating microbial GWAS and single-cell transcriptomics reveals associations between host cell populations and the gut microbiome. Nat Microbiol. 2025;10:1210-26.
116. Wu G, Xu T, Zhao N, et al. A core microbiome signature as an indicator of health. Cell. 2024;187:6550-65.e11.
117. Marcella C, Cui B, Kelly CR, Ianiro G, Cammarota G, Zhang F. Systematic review: the global incidence of faecal microbiota transplantation-related adverse events from 2000 to 2020. Aliment Pharmacol Ther. 2021;53:33-42.
118. Drewes JL, Corona A, Sanchez U, et al. Transmission and clearance of potential procarcinogenic bacteria during fecal microbiota transplantation for recurrent Clostridioides difficile. JCI Insight. 2019;4:130848.
119. Porcari S, Ciccarese C, Heidrich V, et al. Fecal microbiota transplantation plus pembrolizumab and axitinib in metastatic renal cell carcinoma: the randomized phase 2 TACITO trial. Nat Med. 2026;32:1316-24.
120. Fernandes R, Jabbarizadeh B, Rajeh A, et al. Fecal microbiota transplantation plus immunotherapy in metastatic renal cell carcinoma: the phase 1 PERFORM trial. Nat Med. 2026;32:1325-36.
121. Duttagupta S, Messaoudene M, Hunter S, et al. Fecal microbiota transplantation plus immunotherapy in non-small cell lung cancer and melanoma: the phase 2 FMT-LUMINate trial. Nat Med. 2026;32:1337-50.
122. Haifer C, Paramsothy S, Kaakoush NO, et al. Lyophilised oral faecal microbiota transplantation for ulcerative colitis (LOTUS): a randomised, double-blind, placebo-controlled trial. Lancet Gastroenterol Hepatol. 2022;7:141-51.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].