


Kupffer cells and monocyte-derived macrophages in the pathogenesis and treatment of hepatocellular carcinoma
 0
0 Abstract
Kupffer cells, the resident liver macrophages, are characterized by self-renewal capacity and extensive plasticity. Kupffer cells are involved in the regulation of several liver functions such as the maintenance of liver tolerance against antigens arriving at the liver through the portal vein, the autophagy and apoptosis of hepatocytes and other liver sinusoidal cells.and various metabolic functions such as iron homeostasis and lipid metabolism. In acute liver injury, Kupffer cells are involved in both liver damage and resolution. During chronic liver injury, resident and bone marrow-derived macrophages drive either the progression or resolution of fibrosis and cirrhosis. Inevitably, they are implicated in the initiation, progression or defense against hepatocellular carcinoma (HCC) as members of the tumor microenvironment together with bone marrow-derived macrophages. The present review describes the heterogeneity of Kupffer cells and liver-infiltrating macrophages, their functions and their participation in liver cancer with particular emphasis on factors modulating their pro-tumoral and anti-tumoral differentiation. The review is further focused on their involvement in HCC as they participate in the population of tumor-associated macrophages. Finally, the potential use of Kupffer cells and macrophages for the treatment of HCC, including the potentiation of immune checkpoint inhibitors, is presented.
Keywords
INTRODUCTION
Lung, breast, and colorectal cancer (CRC) are the three leading causes of cancer-related death worldwide. However, liver cancer is the fourth leading cause of cancer-related mortality and the second in men, despite its relatively low prevalence compared with the other three. Its incidence is almost three times higher in men. Global Cancer Observatory (GLOBOCAN) estimated that approximately 684,659 new cases and 597,434 deaths were related to hepatocellular carcinoma (HCC) in 2022[1]. It should be stressed that the incidence and mortality of HCC are approximately the same, indicating a high fatality rate, despite some progress in treatment modalities in recent years. Sub-Saharan Africa and Asia have the highest incidence and mortality rates, with the highest incidence reported in Mongolia. In absolute numbers, approximately three-quarters of liver cancers occur in Asia, with the highest numbers reported from China, due to both a high incidence rate and the large population. Approximately 73% of HCC cases are hepatitis B- related in Eastern Asia, 44% were related to Hepatitis C virus (HCV) in Northern Africa and 25% were alcohol-related in Western Europe. Worldwide, 57% were due to Hepatitis B virus (HBV) and 19% were due to HCV[1]. Alcoholic liver disease and metabolic-associated fatty liver disease (MAFLD) are among the leading causes in the western world, where the etiological background is rapidly changing[2].
Improved mortality outcome of HCC can only be achieved when the mechanisms underlying the pathogenesis of HCC are clarified in depth. Kupffer cells (KCs) have a critical role in the pathophysiology of liver diseases including HCC. KCs are the resident macrophages of the liver and the largest number of tissue-resident macrophages (TRMs) in the body. They represent approximately 80%-90% of tissue macrophages[3].
The first description of KCs was done in 1876 by Carl Wilhem von Kupffer[4,5] although some investigators believe that his method actually identified stellate or Ito cells in the sinusoids. Von Kupffer used Gerlach’s gold chloride method, which allows for selective staining of vitamin A-storing stellate cells. Kupffer’s original stellate cells were not the sinusoidal endothelial cells nor phagocytic macrophages KCs; However, they were perisinusoidal cells that store vitamin A and are identical to Ito’s “fat-storing cells”, or hepatic stellate cells (HSCs) as they are known today[5]. Tadeusz Browicz designated them as macrophages[6], described for the first time by Ilya Metchnikoff[7].
Tissue macrophages, in general, are characterized by considerable plasticity that allows them to respond to alterations of the tissue environment, assuming different phenotypes[8]. Plasticity has been unequivocally demonstrated in vitro for bone marrow-derived macrophages (BMDMs); however, recent techniques such as single-cell RNA sequencing showed that the same is true for the resident macrophages in various tissues, including KCs[9-11].
In the liver, two distinct macrophage subpopulations of different origin can be identified: KCs, which have self-renewing capacity and are responsible for immune tolerance, and monocyte-derived macrophages (MoMFs), which are recruited to the liver when tolerance is breached, usually after infection. The latter eventually differentiate into pro-inflammatory macrophages[12].
KCs are constituents of innate immunity and express scavenger, complement, and pattern recognition receptors (PRRs) such as the toll-like receptors (TLRs) TLR4 and TLR9. As a consequence, they recognize a large number of molecules such as pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs)[13].
However, certain studies have indicated that TRMs[14,15] may not respond as extensively to foreign molecules as previously believed.
In the current review, the biology of KCs, including the heterogeneity and functions of KCs, is presented, and their role in the development and progression of HCC is analyzed.
HETEROGENEITY OF KCS AND MOMFS
KCs comprise approximately 80% of the immune cells in the healthy murine liver, while large numbers of MoMFs infiltrate the liver after acute and chronic liver damage[16].
Current evidence suggests that TRMs originate from erythromyeloid progenitors bearing the macrophage colony-stimulating factor 1 receptor (CSF1R) in the yolk sac or fetal liver[17].
KCs are mostly located in Zones 1 and 2 in normal livers. Phagocytic potential is higher in Zone 1 KCs compared to Zone 2 KCs and MoMFs. Apoptosis of KCs increases in acutely damaged livers when different heterogeneous MoMFs are found around necrotic areas and promotes dead cell clearance and necrosis resolution[18-20]. The resident KC pool is diminished during inflammation[21-23]. This has been attributed either to absolute number reduction[22,23], or to reduction of proportion rather than a decline in absolute numbers of KCs[21,24]. Irrespective of the reason for the reduction, this is associated with the recruitment of monocytes characterized by the markers Lymphocyte Antigen 6, Cluster of differentiation 11b and 64 and F4/80 (Ly6Chi, CD11b+, F4/80-, CD64lo), which eventually will differentiate into hepatic macrophages (Ly6C-, CD11b-/+, F4/80+, CD64+). They may further differentiate into monocyte-derived KCs (MoKCs) separated from resident KCs due to the lack of T-cell immunoglobulin and mucin domain containing 4 (TIM4) expression at least at the initial stages of their differentiation[21,22,24]. However, recent in vivo experimental evidence indicated that repopulation of the liver after depletion of KCs is derived directly from hematopoietic stem cells and not from pre-existing KCs or from MoMFs. This is a very interesting suggestion that may alter the current ideas of KC biology[25].
The identification of markers able to discriminate between resident KCs and MoMFs in humans and animal species is still far from complete. Data generated by novel methods, such as single-cell RNA sequencing, revealed a highly complex macrophage polarization profile, well beyond the old classification of inflammatory (M1) and anti-inflammatory (M2) macrophages[26].
C-type lectin domain family 4 member F (CLEC4F) was recognized as a specific marker of murine KCs, leading to the generation of murine models with either specific depletion or specific targeting of the KCs[27]. It has its limitations. It is expressed late in KC development, hindering thus the early identification of cells developing into KCs[10]. This is not a problem in the normal liver, where KCs are self-maintained throughout life[28]. In a diseased liver, though, KCs are replaced by moKCs that may exist for approximately a week before they can be separated from other liver macrophages as CLEC4F+ cells[22]. Moreover, CLEC4F is not present in human KCs[29].
Murine KCs are distinguished from other liver macrophages by the expression of additional surface molecules such as TIM4, V-set and immunoglobulin domain containing 4 (VSIG4), and C-type lectin domain family 1 member B (CLEC2). CLEC2 (encoded by Clec1b) is an early marker of KCs that remains throughout their life[21]. Moreover, recent reports have demonstrated that KCs express marker genes specific to the mature KCs. Marker genes such as inhibitor of DNA binding 3 (id3), cd5l, vsig4, macrophage receptor with collagenous structure (marco), cd163 and folate receptor beta/FRβ (forl2) are found in both humans and mice[29,30] suggesting the existence of KC subtypes[31-33] with immunomodulatory and metabolic functions[29,30]. A caution, therefore, should be exercised in previous studies where only one of these subpopulations was interpreted as KCs, and the observed KC heterogeneity may be due to the contamination of the genuine KCs with other populations[34].
Two diverse KCs have been described: the embryonic-derived KCs (EmKCs) and the MoKCs. EmKCs express TIM4 and MARCO, while MoKCs have a much lower expression of MARCO. EmKCs have increased phagocytic and pro-inflammatory capacity[35-37]. However, in similarity with the CLEC4F+ marker, recruited KCs can express TIM4 over time. Therefore, identification of these cells is difficult at least one month after recruitment[38]. The problem of differentiating between MoKCs and EmKCs has not been solved. It was reported that the expression of genes associated with KCs such as Id3, Marco and Timd4 was upregulated in MoKCs when co-cultured with HSCs, liver sinusoidal endothelial cells (LSECs) or incubated with Macrophage colony-stimulating factor and Hepatocyte growth factor (HGF). Signals from LSECs such as delta-like canonical notch ligand 4 (DLL4) and transforming growth factor beta (TGF-β), and from HSC through tumor necrosis factor α (TNF-α)-Receptor and interleukin receptor 1 (IL1b-R), led to expression of KC-specific markers such as liver X receptor alpha (LXRα), and inhibitor of DNA binding 3 (-ID3). However, the expression of these genes is always higher in resident KCs.
A complete similarity in the gene profile between MoKCs and EmKCs was not achieved even after three days of culture. It should also be stressed that MoKCs differ from BMDMs recruited after liver injury. MoKCs express KC markers such as CLEC4F, CLEC2, and VSIG4, while BMDMs mostly express C-C chemokine receptor type 2 (CCR2) without KC marker expression[27,39,40].
EmKCs themselves seem to be heterogeneous. In the irradiation-exhausted model, radio-resistant KCs (Cdkn1ahi) have been demonstrated, but their function remains to be established[41]. Furthermore, single-cell RNA studies have identified two distinct EmKC subsets in rodents. KC1s are characterized as CD206loESAM- cells and occupy approximately 80% of EmKCs, while KC2s are CD206hiESAM+ cells. Both are similarly distributed with similar phagocytic capacity[32]. KC1s mainly express genes implicated in immune functions. KC2s mainly express LSEC-associated genes indicating implication with lipid metabolism and lipid-associated oxidative stress through CD36[32]. However, the same CD206hiESAM+ KC2s seem to be necessary for an efficient T-CD8+ response in mice livers[33]. Additional studies suggested that the KC2 probably represents endothelial cell-KC doublets, something that requires further research[29,34,42].
Therefore, the new definition for murine KCs refers to liver macrophages that express CLEC4F regardless of origins[38]. Additionally, CLEC2[21], TIM4[43] and VSIG4[29] are also accepted as murine KC markers. It should be noted that VSIG4 has recently been reported to be conserved between mouse and human[29]. Several reports have demonstrated an initial secretion of cytokines, chemokines, complement factors and prostaglandins by KCs stimulated by inflammatory factors[44,45].
Human KC heterogeneity has been demonstrated as well. Human KCs are recognized as CD163+ MARCO+ CD5L+ TIMD4+ cells[46-48]. MARCO is a scavenger receptor that takes up inflammatory PAMPs and DAMPs, leading to suppression of immune responses. In analogy to rodents, two different subsets of human liver macrophages have been described[46,49]. They were designated as KCs because they were CD68+ and were described in the healthy liver. The transcriptome of CD68+ MARCO+ KCs seem to be the equivalent of the murine resident KCs. MARCO+ macrophages were also localized in the periportal areas being almost absent from pericentral areas[46]. MARCO+ KCs in the normal human liver are also divided into two subsets. Both expressed CD163 and MARCO and were designated as KC1 and KC2[47]. Lack of TIMD4 differentiates KC2 from KC1. TIMD4+ cell numbers were similar between normal and cirrhotic livers, but a selective reduction in MARCO + TIMD4- KC2 was observed in the fibrotic liver[48]. CD68+ MARCO- KCs were also recognized in the normal human liver. They expressed monocyte-associated genes such as Ficolin-1 (FCN1), S100 calcium-binding protein A8 (S100A8) and Lysozyme (LYZ) and produced increased amounts of TNF-α after stimulation with lipopolysaccharide (LPS) and interferon gamma (IFN-γ). MARCO+ KCs express reduced inflammatory mediators such as TNF-α but increased immunosuppressive genes such as Programmed Death-Ligand 1 (PD-L1) after LPS or IFN-γ stimulation[46]. Therefore, MARCO- cells were designated as inflammatory KCs. In a recent study, VSIG4 was proposed as the best marker of human KCs[29].
Gut microbiota affects the induction of MARCO+ macrophages. The bacterial family, Odoribacteraceae, was reported to induce this subpopulation and certainly this is not the only bacterial family that does the same when the integrity of the intestinal barrier is breached. Commensal bacteria induce MACRO+ immunosuppressive macrophages, which in turn reduce liver inflammation. Disruption of this system may participate in the pathogenesis of liver diseases such as Primary Sclerosing Cholangitis (PSC) or MAFLD[31,50].
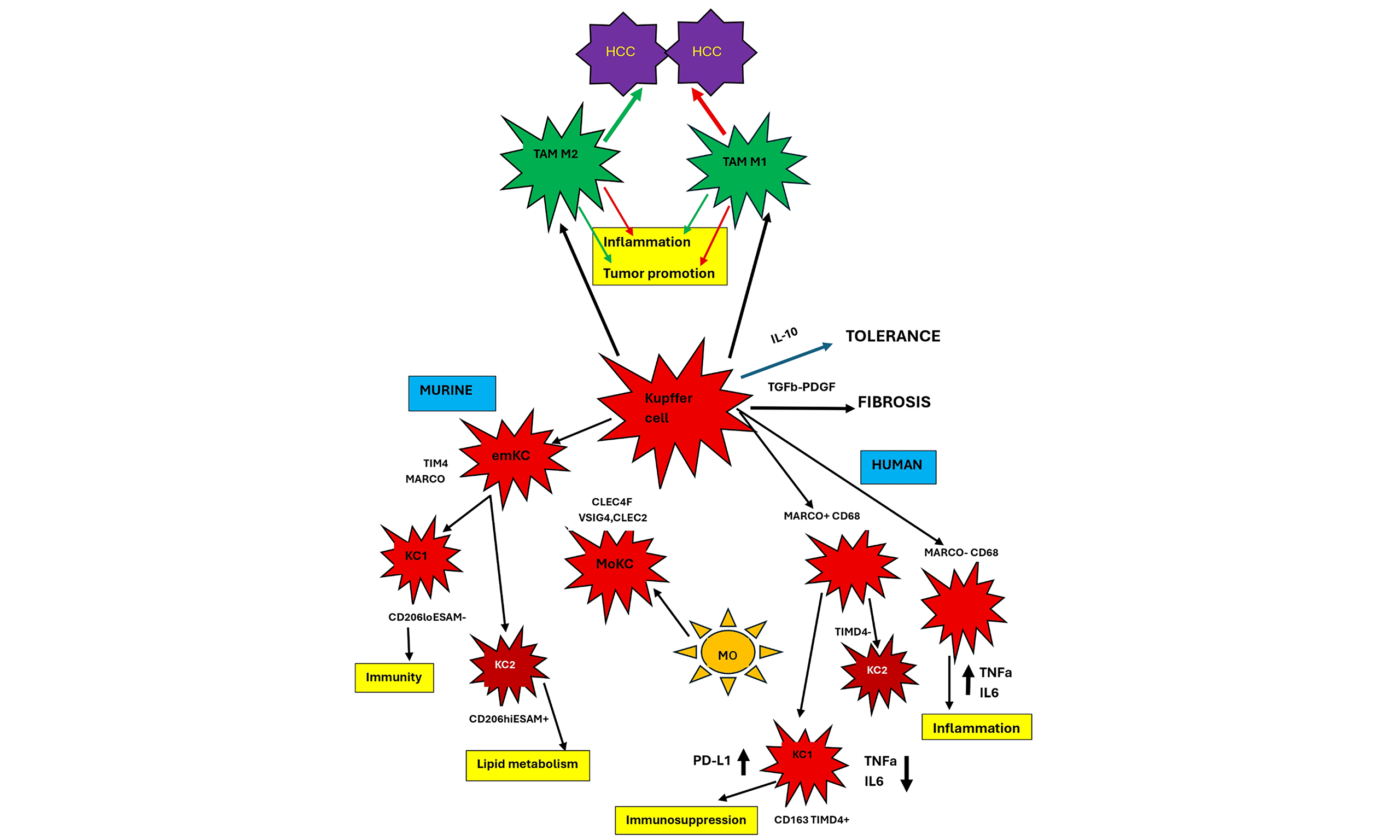
Other markers have been proposed in human KCs. Flow cytometry has identified CD32 as an additional marker capable of differentiating these two KC subsets. KCs that are characterized as CD32intCD68+ CD14+ regulate the immune response[51]. In addition, a subset of CD49a+, VSIG4+, and/or MARCO+ intrahepatic macrophages (ihMφ) not present in peripheral blood has also been identified in human liver. CD49a+ ihMφ were the most prominent macrophage population in livers with baseline TNF-α, IL-12, and IL-10 secretion before stimulation of TLRs. After stimulation, the proportion of TNF-α, IL-12, and IL-10 secreting cells was increased mostly within the CD49a- ihMφ cells[52] [Figure 1].
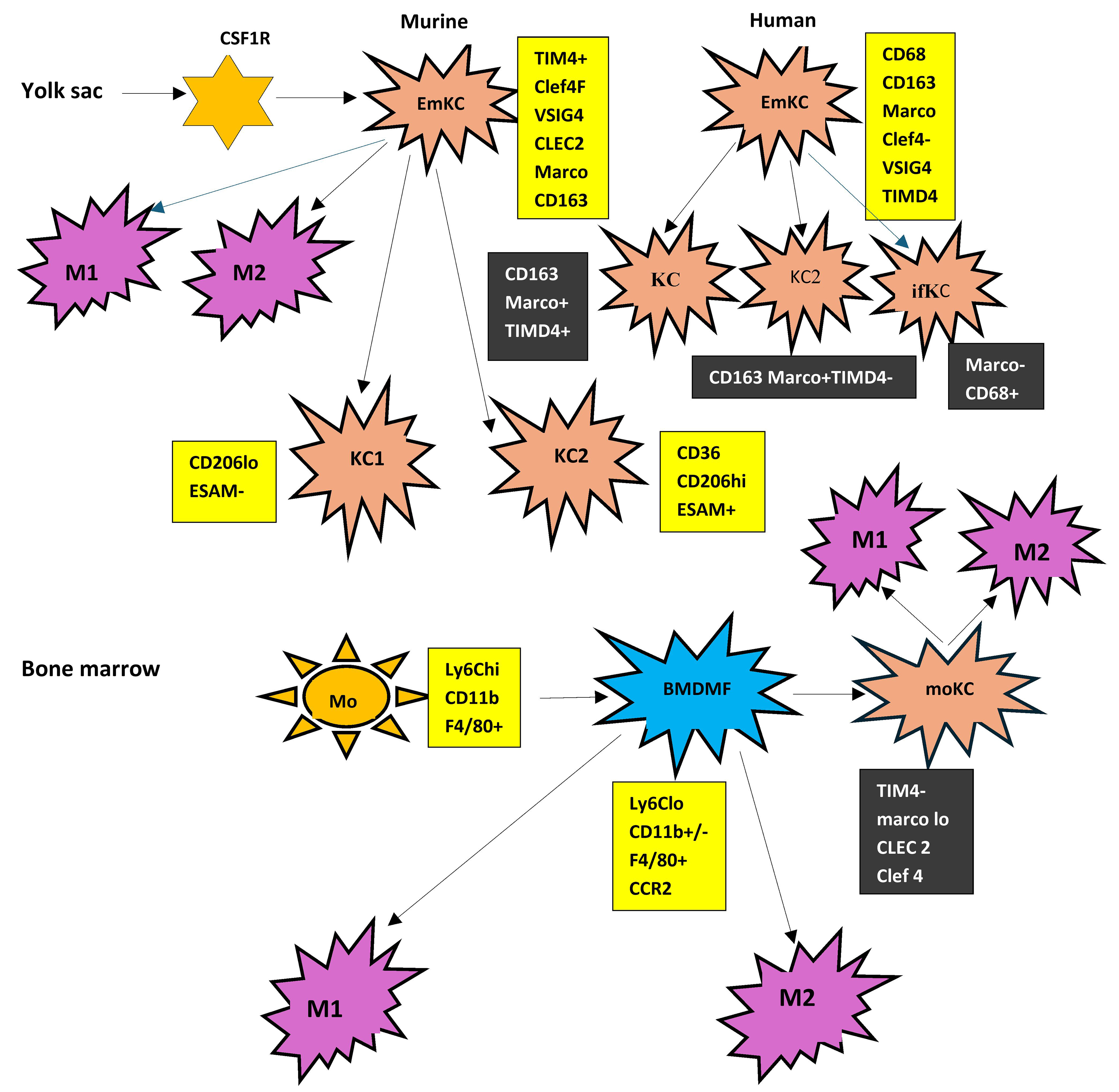
Figure 1. Heterogeneity of Kupffer cells and liver macrophages. Kupffer cells in both rodents and humans originate from yolk sac. Resident Kupffer cells (emKCs) are further classified into KC1 and KC2 in mice and into additional subsets in humans. A particular subset (ifKC) is strongly pro-inflammatory. In contrast, liver-infiltrating macrophages (BMDMs) originate from bone marrow monocytes and can differentiate into moKCs, which may be indistinguishable from emKCs. Irrespective of origin, liver macrophages are functionally classified as M1 and M2 macrophages. Boxes indicate the markers expressed in each subset. CSF1R: Colony-stimulating factor 1 receptor; EmKC/eMKC: embryonically derived Kupffer cell; KC: Kupffer cell; KC1: Kupffer cell subset 1; KC2: Kupffer cell subset 2; ifKC: inflammatory Kupffer cell; Mo: monocyte; BMDMF: bone marrow-derived macrophage; moKC: monocyte-derived Kupffer cell; M1: classically activated macrophage; M2: alternatively activated macrophage; TIM4: T-cell immunoglobulin and mucin domain-containing protein 4; VSIG4: V-set and immunoglobulin domain containing 4; CLEC2: C-type lectin domain family 2; MARCO: macrophage receptor with collagenous structure; CD68: cluster of differentiation 68; CD163: cluster of differentiation 163; CD36: cluster of differentiation 36; CD206: cluster of differentiation 206; ESAM: endothelial cell-selective adhesion molecule; Ly6Chi: lymphocyte antigen 6 complex, high expression; Ly6Clo: lymphocyte antigen 6 complex, low expression; CD11b: integrin alpha M; F4/80: epidermal growth factor-like module-containing mucin-like hormone receptor-like 1; CCR2: C-C motif chemokine receptor 2; EmKCs: embryonic-derived KCs.
The heterogeneity of macrophage phenotypes is further complicated by the identification of another subpopulation of macrophages in both mouse and human liver fibrotic tissue. In vitro, Granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-17A, and TGF-β1 were able to differentiate human monocytes into macrophages with scar-associated markers. These macrophages degraded collagen IV but not collagen I. This subpopulation was designated as scar-associated macrophages (SAMs) and was shown to activate HSCs to produce collagen I. Their relation with KCs remains to be clarified[53]. [Figure 1].
FUNCTIONS OF KUPFFER CELLS
KCs belong to the innate immunity response system and are the first line of defense against various pathogens originating in the gastrointestinal tract[31]. They also phagocytose damaged hepatocytes and injured red blood cells[30,54]. Unlike other macrophages, KCs are equipped with a complement receptor that binds C3b, leading to detention of bacteria to be later killed by other immune cells[55,56]. They also phagocytose and clear activated neutrophils. Furthermore, KCs can be activated by different receptors to secrete either pro- or anti-inflammatory cytokines in order to regulate inflammation[57].
Traditionally, macrophages including KCs are functionally divided as inflammatory or M1 and anti-inflammatory or M2 cells. M1 KCs are antigen presenters, while activation of M1 KCs by TNF-α and IFN-γ leads to overproduction of inflammatory factors such as IL-6, IL-1β and TNF-α. The inhibition of TNF-α/Tumor necrosis factor receptor1(TNFR1) alters the polarization of liver M1 KC, reducing the production of inflammatory mediators[58].
The identification of new phenotypes, as mentioned before, led many investigators to consider the M1 and M2 classification an oversimplification[59,60]. However, this binary consideration of macrophage function remains valuable because it represents the net result of all distinct phenotypes that may participate in each subset. Thus, it allows comparisons between diseases[61,62]. M1 macrophages suppress tumors, while M2 macrophages promote tumor expansion[63]. Nonetheless, the widely accepted current idea is that KCs actually represent a mixture of liver phagocytic cells, whose functional response may alter according to the type of injury and the activation of different receptors[26,47].
The principal function of KCs in the normal liver is the maintenance of immunological tolerance. This is accomplished by the low levels of major histocompatibility complex (MHC) class I and II and co-stimulatory molecules expressed in KCs that lead to defective T-cell activation[64,65]. In addition, KCs maintain an M2 macrophage phenotype when the levels of portal vein endotoxins are physiological. This is accompanied by increased secretion of anti-inflammatory molecules such as IL-10, TGF-β, nitric oxide (NO) and prostaglandins that also prevent the activation of T cells by other antigen-presenting cells (APCs)[66]. Moreover, increased levels of IL-10 and TGF-β upregulate the proliferation of regulatory T cells (Tregs)[67].
Chronic liver injury, regardless of etiology, may lead to fibrosis and cirrhosis. KCs play a central role in the development of cirrhosis. After the initial insult, damaged hepatocytes liberate molecules, such as DAMPs, reactive oxygen species (ROS) and pro-inflammatory extracellular vesicles (EVs)[68]. KCs are in turn activated accompanied by moKCs and BM-derived macrophages at the damage site[69-71]. Activated KCs coordinate the immune response through injury resolution and the production of cytokines and chemokines such as TNF-α, IL-6, and IL-12, IL-8, monocyte chemoattractant protein-1 (MCP-1) and macrophage inflammatory protein-1 alpha (MIP-1α)[26,72]. However, KCs secrete pro-fibrotic cytokines such as TGF-β and platelet-derived growth factor (PDGF) as well[72]. They also express the enzyme inducible nitric oxide synthase (iNOS), leading to increased NO production and the formation of reactive nitrogen species (RNS) after reaction with ROS. RNS, in turn, activate HSCs to transdifferentiate into collagen-producing myofibroblasts[54]. KCs themselves may transdifferentiate into fibrocytes, further promoting fibrosis[73]. Inflammatory signals can also activate inflammasomes in KCs, resulting in activation of caspase-1 and initiation of the maturation of pro-inflammatory cytokines IL-1β and IL-18. Both cytokines directly activate HSCs, to increase deposition of the extracellular matrix (ECM)[74-76]. Recruited macrophages comprise the majority of cells in the fibrotic septa that promote liver fibrosis by producing pro-inflammatory mediators[77,78].
In certain pathological situations, however, activation of KCs plays an anti-fibrotic role. In the carbon tetrachloride (CCL4)-induced liver fibrosis model, production of IL-22 promoted KCs polarization towards the M2 phenotype and attenuated inflammation and development of fibrosis[79]. In the cholesterol high-fat diet model, the same M2 polarization was reported after the inhibition of dipeptidyl peptidase 4 (DPP4), leading to improvement of insulin resistance, steatohepatitis, and fibrosis[80].
Apart from KCs, BM-derived macrophages are implicated in liver fibrosis. They express the transmembrane receptor CCR2 as mentioned above. Binding to its ligand Chemokine C-C motif ligand 2 (CCL2), CCR2 promoted the BM-derived macrophage infiltration into the liver[81,82]. It has been reported that inhibition of CCR2+ monocyte infiltration by inhibiting CCL2 could downregulate ECM deposition and liver fibrosis[83-85]. Progress of fibrosis leads to remodeling of the liver and loss of contact of KCs with neighboring hepatic cells with final loss of their distinct identity. KCs are in contact with LSECs, HSCs and hepatocytes, that and the whole constitutes the KC niche. Dying KCs release TNF-α and IL-1, which activate LSECs and HSCs to transiently express monocyte chemoattractants. KC niche is transiently open and depleted KCs are replaced by transient monocyte recruitment to the liver and their differentiation into moKCs, which proliferate to fill the niche[27,39,40].
Recruited monocytes inside intrahepatic vessels fuse into large cell aggregates that have a KC-like phenotype, and replenish resident KCs. These syncytia are identified irrespective of etiology of cirrhosis, and are characterized by increased bacterial capture capacity[86,87].
An additional mechanism that participates in liver fibrosis is autophagy. During the initial stages of fibrosis, autophagy in KCs is protective. This has been demonstrated in the experimental models of Carbon tetrachloride (CCl4) and N-diethylnitrosamine (DEN) induced cirrhosis. Autophagy of KCs reduces inflammation and fibrosis by inhibiting IL-1α and IL-1β production induced by ROS[88-90]. Moreover, deletion of Autophagy protein 5 (Atg5), a critical gene in the cascade of autophagy led to the aggravation of fibrosis[91]. On the other hand, IL-7 promotes inflammation and fibrosis in the Schistosoma japonicum mouse model by inhibiting the autophagy of KCs through the adenosine monophosphate-activated protein kinase (AMPK) pathway. Moreover, activation of autophagy by spermine can ameliorate liver injury in the same model, through inhibition of the pro-inflammatory response of KCs[92].
An additional implication of KCs in liver fibrosis is via the nicotinamide adenine dinucleotide phosphate hydrogen oxidase (NADPH oxidase or NOX). The isoforms of NOX are involved in liver fibrosis through the generation of ROS in both the HSCs and KCs[93]. Of the seven known NOX isoforms, KCs express only NOX2, while HSCs express NOX1, NOX2, and NOX4[94].
KCs are involved in several other functions in addition to tolerance and fibrosis. They are major factors in the regulation of iron turnover as they phagocytose aged red blood cells. Importantly, KCs secrete vasodilators, such as carbon monoxide, as a result of hemoglobin degradation[95,96]. They are also involved in the regulation of hemostasis as they clear aged platelets[97]. Additionally, KCs are implicated in lipid metabolism[98,99]. They do not function in isolation. KCs and sinusoidal cells interact both in normal liver homeostasis and during liver disease. KCs and LSECs are activated after liver injury and their crosstalk increase inflammation and tissue damage. KCs and other liver macrophages produce inflammatory cytokines and chemokines while LSECs increase expression of adhesion molecules such as Vascular cell adhesion protein (VCAM) and Intercellular Adhesion Molecule (ICAM) that attract immune cells in the liver. In addition, attracted macrophages initiate endothelial to mesenchymal transition (EndMT) of endothelial cells, an important step for fibrosis initiation and progression, and HSC activation that transdifferentiate into myofibroblasts as mentioned before[100,101].
Extensive reviews of KC heterogeneity and functions have recently been published[102-105].
KUPFFER CELLS AND TUMOR BIOLOGY
Tumor progression depends on the creation of the tissue microenvironment (TME) that is critical for the expansion or containment of the tumor. TME contains several immune cells including macrophages. In the liver, KCs, MoKCs and BMDMs constitute the pool of tumor-associated macrophages (TAMs)[106-108]. KCs cannot be totally substituted by BMDMs[28,109]. TRMs can be pro-tumorigenic or anti-tumorigenic not only in the liver but in other tissues as well[110,111]. TAMs, including all macrophages, exhibit a binary function. M1-like TAMs are pro-inflammatory and have anti-tumor effects, while M2-like TAMs are anti-inflammatory and have tumor-promoting effects[112-114]. M1-like macrophages are able to present antigens and attract type 1 helper T cells (Th1) to kill and suppress tumors[8,115,116]. M2-like macrophages repress effector T-cell recruitment, activate T helper 2 (Th2)- immune responses, stimulate angiogenesis, and enhance tumor expansion[117,118]. Due to the difficulty in accurately identifying the role of the individual constituents of TAMs, the use of TAM includes KCs and MoKCs in many instances below [Figure 2].
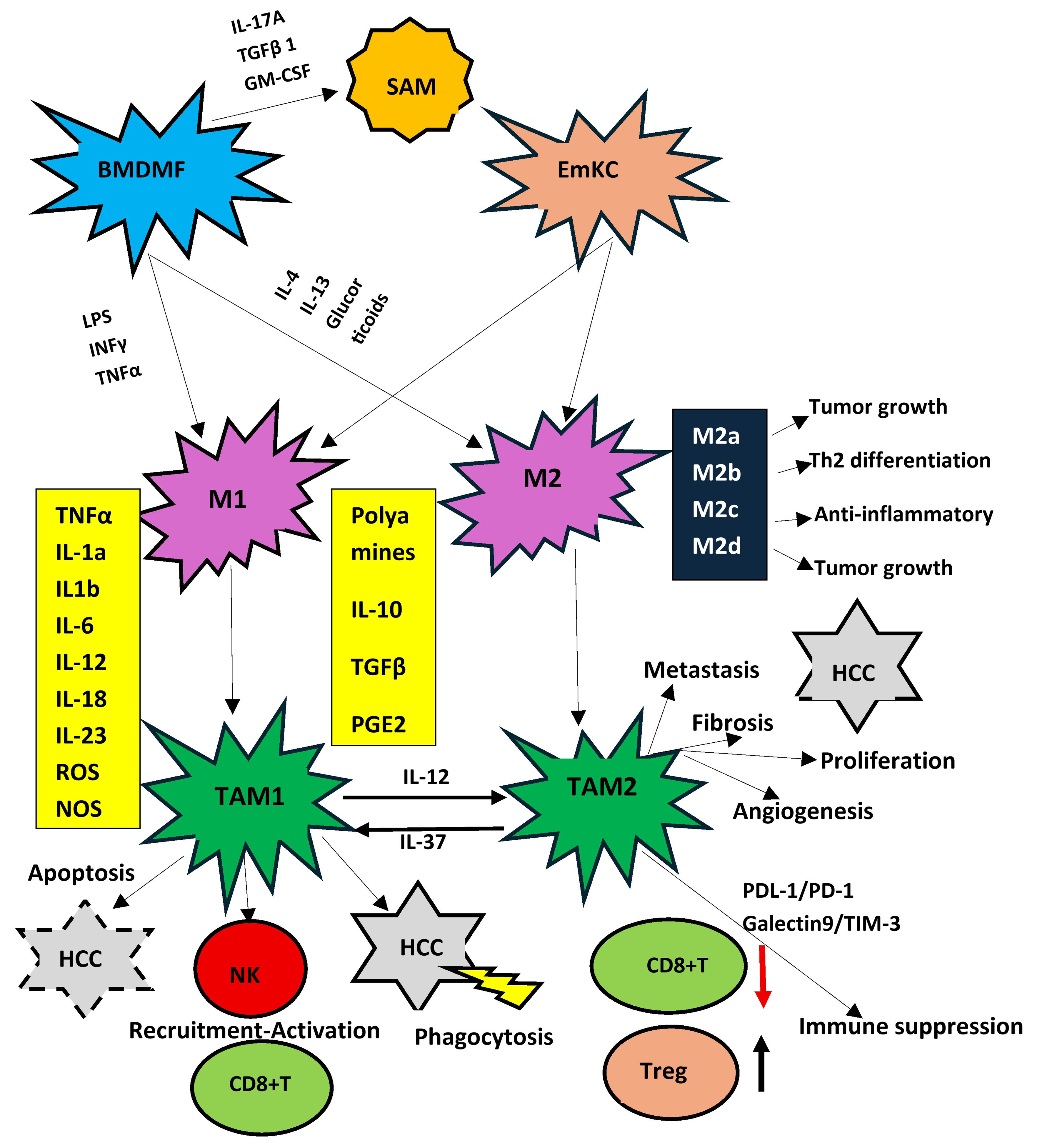
Figure 2. Function of M1 and M2 subsets of liver macrophages. Although this classification is considered obsolete, as several novel functional subsets have been described, it remains the best studied to date. M1 cells are the origin of TAM1, which are pro-inflammatory and anti-tumoral and promote apoptosis and phagocytosis of HCC cells. Moreover, they recruit and activate killer immune cells such as NK cells and effector CD8+Tcells. On the other hand, M2 cells are the origin of TAM2 cells that are pro-tumoral and favor HCC proliferation and metastasis. Moreover, they cause exhaustion of CD8+T cells and increase the proportion of T reg cells. All subsets of M2 cells (M2a-M2d) also favor tumor development. BMDMF: Bone marrow-derived macrophage; EmKC: embryonically derived Kupffer cell; SAM: scar-associated macrophage; M1: classically activated macrophage; M2: alternatively activated macrophage; M2a-M2d: M2 macrophage subsets; TAM: tumor-associated macrophage; TAM1: M1-like tumor-associated macrophage; TAM2: M2-like tumor-associated macrophage; HCC: hepatocellular carcinoma; NK: natural killer cell; CD8+ T: CD8-positive T lymphocyte; Treg: regulatory T cell; IL: interleukin; IL-1α: interleukin-1 alpha; IL-1β: interleukin-1 beta; IL-4: interleukin-4; IL-6: interleukin-6; IL-10: interleukin-10; IL-12: interleukin-12; IL-13: interleukin-13; IL-17A: interleukin-17A; IL-18: interleukin-18; IL-23: interleukin-23; IL-37: interleukin-37; TNFα: tumor necrosis factor alpha; IFNγ: interferon gamma; TGFβ: transforming growth factor beta; GM-CSF: granulocyte-macrophage colony-stimulating factor; PGE2: prostaglandin E2; ROS: reactive oxygen species; NOS: nitric oxide synthase; LPS: lipopolysaccharide; PD-1: programmed cell death protein 1; PD-L1: programmed death-ligand 1; TIM-3: T-cell immunoglobulin and mucin-domain containing-3.
Cancers of the gastrointestinal tract and other organs frequently metastasize in the liver. KCs play an important role in liver metastases. Early papers demonstrated that isolated KCs were cytotoxic against human colon adenocarcinoma cells particularly after stimulation with INF-γ and endotoxin[119,120] and induced their phagocytosis[121]. KCs induce Fas cell surface death receptor (Fas) and Fas-mediated apoptosis in colon cancer cells and malignant glioma cells[122,123]. Depletion of KCs by Gadolinium Chloride (GdCl3) increased the size and number of metastases, while activation of KCs by Zymosan had the opposite effect[124]. Production of NO by KCs after stimulation with endotoxin or TNF-α is also an anti-tumor mechanism[125,126]. This is usually the case at the initial stages of tumor induction. Later, tissue-resident KCs and BMDMs can differentiate to TAMs within the TME[127-129]. At later tumor stages, the behavior of TRMs is organ-specific. A reduction of their population is observed in some cancer models, but in others, they promote cancer cell expansion[118,130]. TAMs are polarized into an M2 phenotype by the metabolic products of tumor cells such as lactic acid that induces hypoxia-inducible factor 1α (HIF1α) signaling. HIF1α, in turn, upregulates the expression of vascular endothelial growth factor (VEGF) and macrophage galactose-type lectin-1 (Gal-1) in M2 TAMs, further promoting tumor development[131,132].
KC behavior may be either tumoricidal or tumor-promoting depending on the balance between activating and inhibitory receptors. Inhibition of receptors usually leads to promotion of metastatic cancer[133]. It was recently shown in the liver that the nuclear factor ID3 shifts this balance towards promotion of phagocytosis of tumor cells and recruitment and activation of natural killer (NK) and CD8 T effector cells to reduce the growth of tumors[134]. NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome activation in KCs primed by tumor cells leads to the release of IL-18, which mediates an upregulation of Fas ligand (FasL) death ligand by liver NK cells, leading to elimination of FasL-sensitive tumors[64,135]. It was also demonstrated that Galectin-9 and transmembrane receptor dectin-2found on the membrane of KCs form a complex with erythroid membrane-associated protein (ERMAP) expressed in several cancer cells. The complex is recognized by KCs leading to phagocytosis of tumor cells. Importantly, deficient expression of ERMAP on patient tumors was associated with increased liver metastases[136]. Additional factors that regulate the tumoricidal activity of KCs have been reported. EVs from different cells may contain DNA (EV-DNA) of the complete genome. In a CRC model without liver metastases, EV-DNA was taken up by KCs leading to the secretion of anti-tumor cytokines. This ultimately reduced the size and number of liver metastases. On the contrary, deletion of the apoptotic peptidase activating factor 1 (APAF1) decreased EV-DNA and increased liver metastases[137]. Other exosomes from CRC tumors had decreased levels of angiopoietin-like protein 1 (ANGPTL1) compared to exosomes from normal tissue. ANGPTL1 has been shown to repress liver metastases from various cancers. Exosomal ANGPTL1 was taken up by KCs, and decreased the metalloprotease (MMP)-9 secretion by inhibiting the Janus kinase 2 /Signal transducer and activator of transcription 3 signaling pathway (JAK2-STAT3) signaling pathway leading to reduction of the metastatic outgrowth[138].
On the other hand, cooperation of all sinusoidal cells increases the expansion of liver metastases. Production of pro-inflammatory mediators upregulates the expression of cell adhesion molecules such as vascular cell adhesion molecule-1 (VCAM-1) on LSECs, thus increasing the catchment of cancer cells and the extravasation from the sinusoids. Liver-infiltrating BMDMs, KCs and activated HSCs collaborate to secrete growth factors such as TGF-β and VEGF, MMPs such as MMP-2, -9, and -13 and ECM proteins such as fibronectin and periostin, thus promoting cancer cell expansion and neovascularization[64].
In addition to receptor activation, metabolic reprogramming of KCs and macrophages is a critical factor that regulates their function[139,140]. Upregulation of glycolytic metabolism, fatty acid synthesis and the pentose phosphate pathway leads to M1-polarization[141]. Furthermore, a disruption of the tricarboxylic (TCA) cycle is responsible for the increased concentrations of citrate, and succinate that maintain macrophage inflammatory responses by increasing the production of lipids, prostaglandins, NO, and ROS mediators that are cytotoxic against cancer cells[142,143]. However, a shift of macrophage metabolism towards oxidative phosphorylation and fatty acid oxidation leads to M2 polarization[144]. Glutamine metabolism contributes to this macrophage polarization[145]. Alpha-ketoglutarate derived from glutaminolysis destabilizes HIF1α, enhances fatty acid oxidation, and epigenetically reprograms TAMs[146]. In addition, the catabolism of arginine, via arginase 1 (ARG1), prevails over iNOS and NO production in pro-tumoral macrophages, leading to the production of tumor-promoting factors such as ornithine and polyamines[147,148].
LIVER MACROPHAGES AND HCC
HCC is tightly associated with TAMs, including the minority of resident KCs and the majority of MoKCs and BMDMs. TAMs of monocyte origin are usually suppressor cells[149-151]. During liver cancer progression, tissue-resident KCs that are initially tumoricidal are stimulated by factors promoting the progression of HCC. They undergo a phenotypic shift eventually becoming TAMs that enhance tumor progression. MoMFs undergo sequential evolution starting as erythro-myeloid progenitors, monocyte progenitors, mature monocytes, MoKCs, and TAMs[152]. The GdCl3 repression of KCs had no effect on the number of KCs, but clearly altered their function with reduction of TNF-α and IFN-γ levels and elevation of MMP-2 in experimental HCC. Tumor immunity was seriously repressed, and tumor growth was increased. Most importantly, survival of trial mice was significantly shortened[153].
An earlier study also demonstrated that KCs can support the initiation and progression of HCC by increasing IL-6 production in a manner dependent on the Toll-like receptor adaptor protein Myeloid Differentiation Primary Response 88 (MyD88). Differences in this pathway between sexes may explain the increased prevalence of HCC in males[154-156].
HCC cells produce several cytokines or chemokines such as CCL2, Chemokine C-X-C motif ligand 1(CXCL1), Chemokine (C-C motif) ligand 14 (CCL14), which attract monocytes into the TME through interactions with their receptors. Apoptosis of TME KCs leading to reduction of KCs also induces the infiltration by monocytes through the CCL2/CCR2 axis. Moreover, KCs produce IL-1 and TNF-α, which act on HCC cells that, in turn, secrete IL-8, thereby recruiting monocytes. HGF produced by M2 TAMs also contributes to monocyte attraction in HCC. TAMs simultaneously produce CCL20 to attain self-recruitment[157-159].
As mentioned above, HCC-associated TAMs are also divided into M1 and M2 subpopulations. M1-TAMs are mostly localized in the tissue next to the tumor, while M2-TAMs are mostly localized within the liver cancer tissue. HCC cells produce Wnt Family Member 3A Wnt3A ligands and EVs containing miR4458H, which can shift the polarization into M2-TAMs. Similarly, TAMs secrete IL-12 that indirectly switches the polarization toward M2-TAMs. Alternatively, when macrophages are stimulated with IFN-γ, or LPS, polarization turns into M1-TAMs. Moreover, IL-37 initiates reversion of M2-TAM polarization toward M1-TAMs within TME[157]. TAMs also directly affect HCC. Upregulation of PD-L1 expression in TAMs represses the activation of CD8+ T cells, and increases T regulatory cells recruitment, which is a crucial mechanism to inhibit the elimination of HCC cells by CD8+ T- lymphocytes[157]. The M2-TAMs are traditionally classified into four subsets (M2a, M2b, M2c, and M2d) according to the type of stimulus. M2a is induced by Th2 cytokines, M2b by immune complexes, M2c by anti-inflammatory cytokines or glucocorticoids, and M2d by IL-6. Their distinct roles and the biological roles of M2 subtypes in HCC development have not been clarified[160,161]. However, evidence indicates that all four subtypes promote tumor progression[162,163]. [Figure 2].
M2 macrophages have mannose receptors (MR) and the Arginase-1 gene (Arg-1), which promote anti-inflammatory Th2 reaction and mediate HCC development[164]. Both M1-TAMs and M2-TAMs interchange with each other upon stimulation with certain stimuli[113]. M1-TAMs appear to eliminate HCC cells in the primary stages of tumorigenesis, but as tumor progression advances, M1-TAMs are replaced by M2-TAMs. Several recent reports have revealed that TAMs comprise multiple subpopulations with variable phenotypes. Single-cell RNA sequencing technology identified TAM subtypes with independent functional characteristics based on differences in gene expression profiles. TAMs represent a heterogeneous population that is difficult to classify using the M1/M2 bipolarization theory[165,166].
Therefore, several investigations have tried to identify the significance of different subsets in the development of HCC. Intra-tumoral TAMs in HCC express low levels of CD169 and high levels of CD204. A reverse of this profile can improve prognosis of HCC patients[167]. Chemokine receptor CCR2+ monocytes induce the upregulation of the triggering receptor expressed on myeloid cells-1 (TREM1) in macrophages of the TME leading to MMP-9+ TAMs and the progression of HCC[168]. Siglec-10hi TAMs can inhibit the function of CD8+ T cells. Blocking them may improve anti-tumor immunity[169]. Macrophages in the TME produce more IFN-γ, which helps CD8+ T cells to kill tumors[170]. Moreover, forkhead box O1 (FOXO1) expressed in TAMs inhibits IL-6 and represses the progression of HCC[171].
KCs are a subset of TAMs that can also facilitate the progression of HCC. KCs accumulate in TME, and repress the anti-tumor effect by inducing T-cell dysfunction through PD-L1/Programmed cell death protein 1 (PD-1) and galectin-9/T-cell immunoglobulin and mucin-domain containing-3 (TIM-3) signaling. Moreover, HCC cell stimulation directly increased the expression of TREM1 in KCs, which, in turn, increased KCs activation and HCC progression. On the contrary, TREM1 deletion reduced IL-1β, IL-6, TNF-α, CCL2, and C-X-C motif chemokine ligand 10 (CXCL10) production by KCs and repressed HCC progression[172,173]. Signals from TME activate KCs and induce their functional heterogeneity. The secretion of CCL2 by HCC cells leads to the reduction of EmKCs, increasing the infiltration by MoKCs and immature monocytes[174,175]. The loss of Src-homology 2 domain-containing phosphatase 2 (Shp2) expression in KCs leads to apoptosis of KCs and increases the differentiation of CCR2+ monocytes into TAMs, altering the immunosuppressive TME[176]. Overall, six subsets of MoMFs (Macro1-Macro6) and four subsets of KCs (KC1-KC4) were recently described in HCC. Their relation with the M1/M2 classification and their functional role in HCC progression have not been studied. Recent evidence indicated that at least one of the six MoMFs expressing CXCL10 and one of the four KC subsets expressing MT1G were localized within tumor tissues and were related to HCC development[177]. However, at present, the functional M1/M2 classification provides a better framework for understanding HCC development.
Additional mechanisms favor M2 polarization of KCs and macrophages in HCC. Tumor-derived alpha fetoprotein (AFP) drove immature macrophage polarization into M2 macrophages and suppressed phagocytosis of HCC cells by M1 macrophages. AFP also inhibited the apoptosis of HCC cells when co-cultured with M1-like macrophages[178]. M2 polarization was also induced by exosomes produced by HCC cells that contain miRNA-21-5p[179]. HCC cells are competitors with macrophages for iron to increase the expression of M2 genes. Transferrin receptor (TFRC) is significantly upregulated in HCC, and high TFRC levels are associated with reduced overall survival. TFRC is positively correlated to HCC-infiltrating M2 macrophages[180]. Oncoprotein-induced transcript 3 (OIT3) is a recently described marker of M2 macrophages. Overexpression of OIT3 reprogrammed the metabolism of M2 macrophages and increased the migration and invasion of co-cultured HCC cells. In vivo, overexpression of OIT3 in macrophages enhanced initiation and progression of HCC[181].
Single cell RNA analysis on blood-derived and liver-derived macrophages in human HCC revealed that liver macrophages had an anti-inflammatory profile in comparison to blood-derived macrophages[182]. Two additional studies demonstrated an overlap in M1 and M2 polarizations in patients with HCC a further indication that macrophage biology goes well beyond the binary view of M1 and M2 macrophages[183-185]. Liver macrophages also produce pro-angiogenic factors such as VEGF, and PDGF, which promote angiogenesis and HCC expansion [Figure 2][12,186,187].
In the DEN-CCl4 murine model of chronic liver inflammation and HCC development, a dynamic alteration of the immune landscape was observed similar to that found in human HCC. Characteristically, levels of Tregs were increased up to 125-fold in the invasive margin (IM) of the tumor accompanied by a 5-fold increase of macrophages while the number of CD8+T cells was unchanged. Interactions between the T lymphocytes and macrophages were identified. In the IM area, KCs and macrophages were positively associated with CD8+ T cells, indicating an anti-tumor cooperation. In contrast, in the tumor region, Tregs were negatively associated with KCs, repressing this response[188]. Interestingly, a recent report suggested that temporal ablation of resident KCs has no effect on HCC pathogenesis, neither at the initiation stage nor at the later stages of progression because moKCs quickly replenish the reduced resident KCs adopting their phenotype[175]. Another activating pathway for KCs that is particularly important in Metabolic dysfunction-associated steatohepatitis (MASH)-associated HCC initiation and progression is the hyaluronan-CD44-dependent binding of platelets. Therapeutic inhibition of platelet adhesion activation decreased KC activation, and reduced carcinogenesis in murine models of MASH-associated HCC[189,190]. Furthermore, the increased presence of anti-inflammatory macrophages is associated with poor prognosis in HCC patients[191].
Despite the general agreement that KCs in TME are promoters of HCC development, there is limited evidence that, in some cases, KCs may be protective. The better documented evidence is a study of 302 HCC patients, which showed that high numbers of CD68+ macrophages are associated with better survival[192]. This is supported by another study indicating that CD68+CD169+ macrophage infiltration of the liver was also associated with improved overall survival[193]. The opposite conclusion has also been reported as high infiltration of the liver by CD68+ macrophages was associated with poor prognosis of HCC patients[194]. Therefore, three clinical studies with similar patient characteristics led to contradictory results, probably indicating the difficulty in assessing the heterogeneous KC subpopulations. A protective function of liver macrophages has also been demonstrated in a murine model. Pre-malignant senescent hepatocytes secrete chemokines and cytokines and are cleaned by CD4(+) T-cell-mediated adaptive immune response. CD4+ T cells require the assistance of hepatic macrophages to clean the pre-malignant senescent hepatocytes that favor HCC initiation[195]. Increased microRNA-206 expression increases the recruitment of CD8+ T lymphocytes. Overexpression of microRNA-206 ameliorates the tumor-promoting effect of KCs by driving them into M1 polarization[196].
Further support for a protective role of KCs was produced in the partial hepatectomy (PH) experimental model. Mice without hepatectomy rejected implanted Hepa1-6 hepatoma cells. In contrast, hepatectomized animals increased tumor proliferation associated with a reduction in the number of KCs. KC reduction after PH was associated with TNF-α leading to KC apoptosis through caspase-8 activation, whereas interleukin IL-6 promoted KC survival[197]. Moreover, upregulated expression of miR-26a in either hepatocytes or KCs led to resistance in HCC initiation in a murine model of carcinogenesis. This was attributed to repression of nuclear factor kappa B (NF-κB) or STAT3 pathways by miR-26a[198].
In that respect, the role of long non-coding RNAs (lncRNAs) should be examined as they can affect the behavior of KCs and TAMs in HCC. lncRNAs act in the nucleus or in the cytoplasm, regulating gene expression through interference with DNA, RNA, or proteins. They may function as sponges to absorb microRNAs (miRNAs) or take part in transcriptional modifications. In HCC, lncRNAs may act as tumor suppressors or as tumor promoters when they interact with liver macrophages. They usually interfere with polarization of TAMs[199].
Several lncRNAs act as tumor suppressors and improve survival in HCC. Thus, the upregulation of lncRNA Five Prime to Xist (FTX) repressed the development of HCC during the evolution of MAFLD by promoting M1 polarization of KCs[200]. The lncRNA Maternally Expressed 3 (MEG3) expression increased during LPS/IFN-γ-induced M1 polarization. Overexpression of MEG3 repressed M2 polarization and inhibited HCC metastasis and angiogenesis repressing tumor expansion in vivo[201]. Downregulation of the lncRNA Metastasis Associated Lung Adenocarcinoma Transcript 1 (MALAT1) suppressed VEGF-induced angiogenesis in a LSEC cell line and switched the polarization of macrophages toward the M1 subgroup. This was accomplished through the interaction of MALAT1 with miR-140. On the other hand, inhibition of miR-140 promoted angiogenesis in the LSEC cell line, and polarized macrophages toward the M2 subset[202]. The lncRNA LINC00261 acts as a tumor suppressor in several cancers. In HCC, LINC00261 expression was reduced in tumors and significantly correlated with improved prognosis. LINC00662 activated Wnt/β-catenin signaling pathway in macrophages leading to M2 polarization[203]. lncRNA Cyclooxygenase-2 (cox-2) drives M1 polarization, and reduces M2 macrophage presence in a murine model. High expression of the same cox-2 was associated with a better prognosis in HCC patients[204]. Likewise, overexpression of lncRNA Growth Arrest Specific 5 (GAS5) supported the M1- polarization of TAMs and repressed M2 polarization indicating its antitumor activity in human liver cancer cell lines[205].
By contrast, several other lncRNAs support HCC progression either by promoting M2 polarization of TAMs or by reprogramming the metabolism of macrophages and HCC cells. Thus, TAMs produce exosomes that contain the M2 macrophage polarization associated lncRNA (lncMMPA), which upregulates aerobic glycolysis and growth of HCC cells by acting as a sponge for miR-548. Moreover, lncMMPA promotes M2 macrophage polarization. Clinical studies indicated that lncMMPA expression was strongly associated with TAM glycolysis and reduced survival rates in HCC patients[206]. Similarly, HCC cell-derived exosomes with the lncRNA TUC339 were absorbed by macrophages and suppressed the secretion of pro-inflammatory factors, the expression of co-stimulatory molecules, and the phagocytic capability of macrophages. TUC339 is overexpressed in M2 macrophages and is implicated in M1/M2 polarization[207]. Also, exosomes from the serum of HCC patients were rich in the focally amplified lncRNA on chromosome 1 (lncRNA FAL1) and promoted tumor growth by activating the Wnt/β-catenin signaling pathway. They initiated M2 polarization in macrophages, increasing the proliferation of HCC cells. They also inhibited apoptosis of HCC cells[208]. A recent report demonstrated the implication of the hepatocellular carcinoma up-regulated EZH2-associated long non-coding RNA (lncRNA HEIH) in HCC. It is overexpressed in HCC tumor tissues and hepatoma cell lines and is transported to TAMs by HCC-derived exosomes, initiating M2 polarization[209]. Hyaluronan-mediated motility receptor -AS1(HMMR-AS1) is another lncRNA that promoted M2 polarization in macrophages by binding to miR-147a[210]. The lncRNA Prostate Androgen-Regulated Transcript 1(PART1) was also absorbed by macrophages via cancer cell-derived EVs in HCC. PART1 inhibited the miR-372-3p, which is a negative regulator of Toll-like receptor 4 (TLR4) in macrophages. EVs containing PART1, supported macrophage polarization towards M2[211]. The lncRNA miR4458HG promoted tumor growth in HCC, through glucose metabolism. Overexpression of miR4458HG activated glycolytic pathways, and induced M2 polarization of TAMs[212].
Extensive reports on lncRNAs in HCC have been recently published[213,214].
Another implication of KCs in the progression of HCC is through the promotion of fibrosis and cirrhosis[215]. Autophagy is involved in liver fibrosis as mentioned above. Autophagy-defective KCs increase liver fibrosis, and HCC development via the ROS/NF-κB/IL-1α/β signaling axis[88,216]. Toll-like receptor 2 (TLR2) ligands led to NF-κB degradation by autophagy, increasing M2 polarization in macrophages and progression of HCC[217]. By contrast, apoptosis has a protective role in the association of KCs with HCC. The inhibitor of macrophage Apoptosis Inhibitor of Macrophage (AIM) (encoded by cd5l) that is produced by KCs is a circulating protein promoting the survival of macrophages against apoptotic stimuli. AIM produced by liver KCs directly eliminated HCC cells as shown in a murine model[218].
An additional mediator of KC function in HCC is the solute carrier family 16 member 3 (SLC16A3), which is a lactic acid transporter. It can also transport other short-chain sugars such as pyruvate, maintaining intracellular pH and glycolysis. SLC16A3 is an important component in the glycolytic metabolism of tumor cells. Multi-omics data demonstrated that SLC16A3 expression is associated with the modulation of TME-related KCs and the creation of an inhibitory immune microenvironment. SLC16A3 can directly intervene in the proliferation of HBV-positive liver cancer cell lines in vitro[219]. Clinical studies demonstrated an upregulated expression of SLC16A3 in HCC tissues compared to adjacent normal liver. Most importantly, SLC16A3 was significantly associated with the size of the tumor and patient prognosis[220,221].
An important aspect of KC function in HCC is related to the effect of cholesterol. This is critical for the development of HCC in MAFLD. Entrance of cholesterol in KCs is achieved through endocytosis of low-density lipoproteins (LDL) via the LDL receptor (LDLR). The cholesterol-containing endosome is fused with lysosomes, liberating free cholesterol that is handled in the cytosol by Niemann-Pick type C proteins[222]. KCs are activated by cholesterol overloading, and enhance the chronic inflammation in HCC tumors by recruiting immune cells that increase HCC progression. HCC cells obtain cholesterol from the diet, by de novo synthesis, or through delivery from cells of TME[223]. Cholesterol initiates macrophage attraction and differentiation to M2 polarity in HCC. High cholesterol diet (HCD) induced M1 macrophages during HCC initiation, but a switch towards M2 polarity occurred in later stages. Cholesterol loading is correlated with an aggressive behavior of HCC[224,225].
TREATMENT OF HCC BASED ON KC AND TAM MANIPULATION
The use of macrophages as a therapeutic target in cancer has emerged as a promising approach to combat tumor progression; however, the overall results from the clinical trials may not be as optimistic as expected. Targeting macrophages for treatment of HCC is not so extensive as for treatment of other tumors, probably because of the complexity of the liver tumor microenvironment[162,226].
The four most promising strategies to combat tumor progression by targeting TAMs are categorized into inhibition of macrophage recruitment, repolarization of TAMs, depletion of TAMs, or promotion of phagocytosis[162]. [Figure 3].
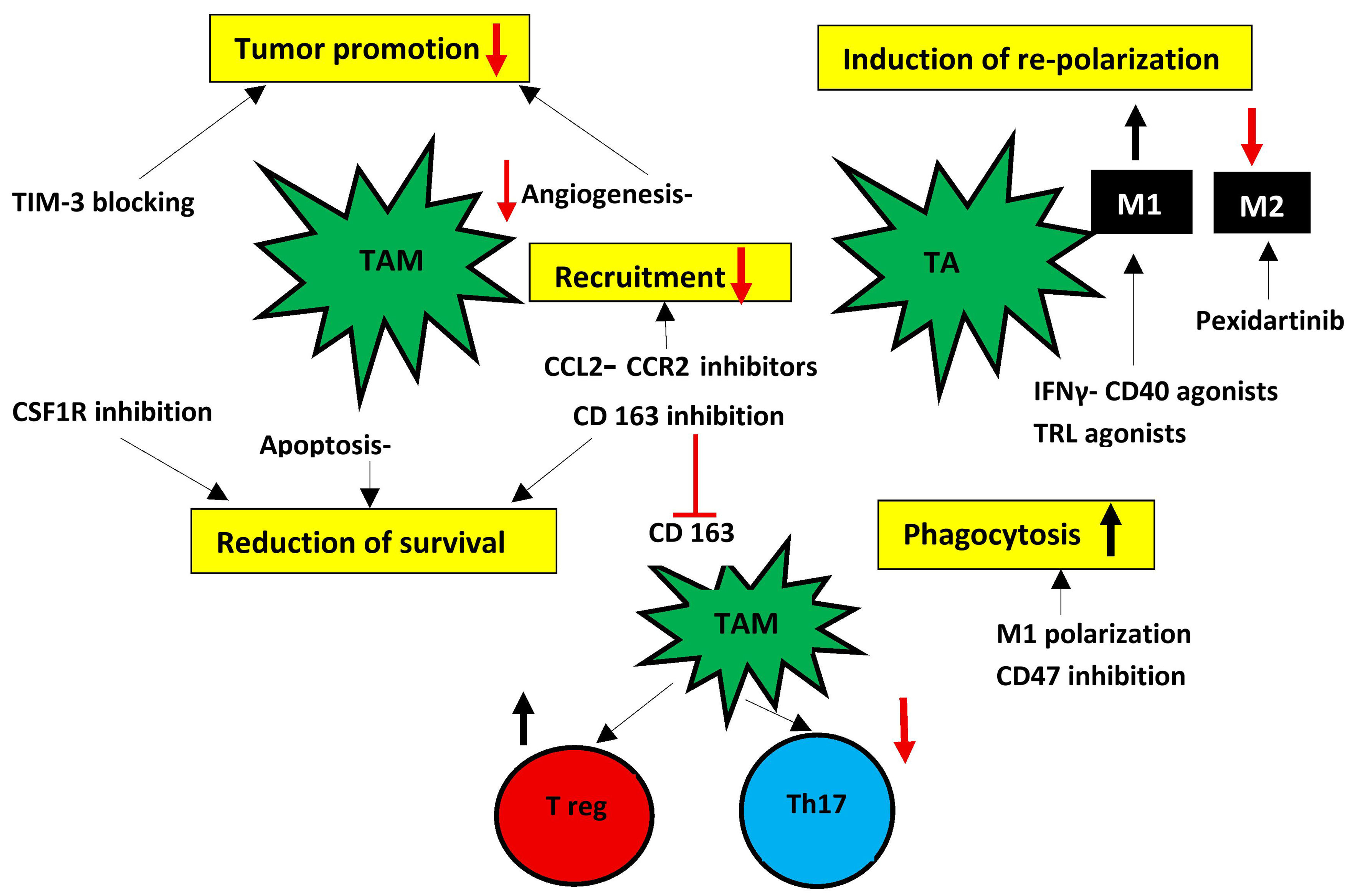
Figure 3. Targeting TAMs may be a promising therapeutic modality in the treatment of HCC. There are five main strategies to combat TAMs presented within the yellow boxes. However, only pexidartinib, which reduces M2 cells, has been approved by the FDA for use in human tenosynovial giant cell tumors. CD163 inhibitors appear promising, as their inhibition may reverse the immunocompromised response in HCC. M1: Classically activated macrophage; M2: alternatively activated macrophage; TAMs: tumor-associated macrophages; TA: tumor-associated; HCC: hepatocellular carcinoma; TIM-3: T-cell immunoglobulin and mucin-domain containing-3; CCL2: C-C motif chemokine ligand 2; CCR2: C-C motif chemokine receptor 2; IFNγ: interferon gamma; CD40: cluster of differentiation 40; TLR: Toll-like receptor; CSF1R: colony-stimulating factor 1 receptor; CD163: cluster of differentiation 163; CD47: cluster of differentiation 47; Treg: regulatory T cell; Th17: T helper 17 cell; FDA: Food and Drug Administration.
It has been suggested that KC targeting is a promising immunotherapy treatment of HCC, given the crucial role of KCs in the immune landscape of TME. It is expected that specific targeting of KCs will reduce tumor growth and sensitize tumors to anti-PD-1 treatment[227]. In that respect, it was shown in murine experiments that engineered bacteria can restore KC dysfunction and obtain substantial therapeutic results against several types of metastatic liver cancer[228].
Checkpoint inhibitors have been used to treat advanced HCC. However, KCs and high density of TAMs impair T cell responses and reduce immunotherapeutic sensitivity[227]. Moreover, KCs are implicated in the liver toxicity induced by neutrophils during immunotherapy[229]. Furthermore, TAMs produce HGF and HIF1α that confer tumor resistance to therapeutic agents such as sorafenib[230].
The mechanisms underlying this resistance show that KCs and TAMs are critical factors[231]. M2-immunosuppressive TAMs produced anti-inflammatory cytokines, which reduced the anti-tumoral activity of effector T cells, thus diminishing the efficacy of immune checkpoint inhibitors (ICIs)[232,233]. TAMs also enhance angiogenesis and epithelial-mesenchymal transition that favor tumor invasiveness and additionally diminish the efficacy of ICIs[216,234,235]. Targeting TAMs is an interesting strategy to overcome ICI resistance in HCC. There are different ways to target TAMs. Attempts have been made either to eliminate TAMs by inhibiting the Wnt/β-catenin pathway or to arrest recruitment of TAMs by inhibiting the CCR2 signaling. Another approach is the modulation of production of pro-tumoral molecules by TAMs through triggering TNFR1 signaling and inhibiting the NF-κB pathway. Restoration of the phagocytic capacity of TAMs or reprogramming of their polarization has also been tested[161,187,230]. It was reported that CD11b agonists can lead to a phenotypic switch in TAMs by inhibiting NF-κB signaling. Combination of immunotherapy and targeting TAMs is also tested in clinical trials. In these trials, Pexidartinib (PLX3397) and other drugs are being used in combination with PD-1/PD-L1 inhibitors, showing promising results[236,237]. Moreover, macrophage eliminators such as Trabectedin are combined with the PD-1 inhibitor Nivolumab in early clinical trials of sarcoma. The macrophage recruitment inhibitor Cabiralizumab has also been combined with Nivolumab for the treatment of HCC. The CD40 agonist antibody Selicrelumab, which reprograms macrophages into M1 phenotype, has also been used in combination with the PD-L1 inhibitor Atezolizumab in solid tumors[226]. Trials on treatment of HCC are expected in the near future, as macrophages are important in the pathogenesis of HCC.
Several attempts have been made to reprogram TAMs. TREM1 is involved in the transformation of M2 to M1 macrophages as previously mentioned. Reduction of TREM1 in macrophages switched M2 polarization into an M1 phenotype. TREM1 depletion in macrophages inhibited Phosphoinositide 3-kinase/AKT Serine/Threonine Kinase/Mammalian or Mechanistic Target of Rapamycin (PI3K/AKT/mTOR) activation. Downregulation of this receptor also led to inhibition of migration and invasion of HCC cell lines[238-240]. A polysaccharide from the spores of Ganoderma lucidum enhanced the polarization of macrophages to M1 type by also interfering with the PI3K/AKT pathway to induce tumor cell apoptosis[241]. Other exogenous molecules also interfere with macrophage polarization and may be useful in HCC treatment. Cantharidin increased M1 polarization by upregulating microRNA-214 expression[242]. Astragaloside IV, a constituent of Astragalus membranaceus, inhibited macrophage M2 polarization through the TLR4/NF-κB/STAT3 signaling pathway leading to the reduction of invasion and expansion of HCC[243]. A different approach is to use macrophages instead of macrophage elimination. This has been tested by engineering of autologous macrophages to create tumor-targeted Chimeric antigen receptor macrophages (CAR-macrophages). Macrophages can also be loaded with therapeutic agents and sent to upload the drugs into the tumor tissue[230,244].
Recently, a finding in the CTNNB1-mutated HCC patients has been used to restore the CD8+ anti-tumor activity and enhance the efficacy of immunotherapy. CTNNB1 mutations are identified in approximately a third of patients with HCC[245]. The gain-of-function CTNNB1 mutations (CTNNB1 GOF) in HCC favor the immune escape and resistance to anti-PD-1 treatment through overproduction of MMP-9 from M1 KCs and TAMs. MMP-9 reduced infiltration and anti-tumoral activity of CD8+ T cells. Targeting MMP-9 restored TME and enhanced anti-PD-1 efficacy in patients with CTNNB1 GOF HCC[246].
Other potential targets in HCC treatment have emerged from recent investigations. A novel mechanism of TAM infiltration in HCC has been recently reported. Mitochondrial Transcription Factor A (TFAM) is implicated in mitochondrial DNA (mtDNA) replication and transcription. TFAM expression was significantly reduced and was closely associated with CD163 expression in HCC tissues. Reduction of TFAM led to TAM infiltration by inducing cytosolic mtDNA stress and M2 polarization of the recruited TAMs. Moreover, inhibition of the mtDNA-NLRP3 pathway significantly attenuated TAM infiltration and HCC advancement. Interference with this mechanism, therefore, has the potential of a new approach to treatment of HCC[247]. Another potential approach of HCC treatment has been recently proposed. The liver is the principal organ of ketone production. Ketones are metabolized by the enzyme 3-oxoacid CoA-transferase 1 (OXCT1). Ketone metabolism is increased in HCC through the overexpression of OXCT1 in TAMs leading to a pro-tumoral polarization of TAMs. Drug targeting or genetic depletion of OXCT1 in TAMs increased anti-tumor immunity and repressed tumor growth[248]. Targeting lncRNAs may also be a treatment modality for the future. At the moment, only experimental data are available[213].
Beyond the traditional M1/M2 classification, single-cell studies have revealed distinct TAM subpopulations such as Secreted Phosphoprotein 1(SPP1+), Triggering Receptor Expressed on Myeloid cells 2 (TREM2+), and MARCO+ macrophages that attenuate cytotoxic immune activity, and are associated with poor prognosis and immunotherapy resistance. Their inhibition is an interesting prospect. Clinically, macrophage-based therapeutic strategies include blockade of macrophage recruitment. This was tried by CCL2/CCR2 inhibitors such as Carlumab, which inhibits macrophage infiltration or Emapticap pegol, which inhibits TAM recruitment (162). In addition, Colony-stimulating factor 1(CSF1)/CSF1R inhibitors such as Tinengotinib or Plerixafor may lead to depletion of immunosuppressive TAMs. Both are clinically tested for solid tumors other than HCC (162). Emactuzumab, a CSF1R inhibitor, and BMS-813160, which antagonizes CCL2/CCR5, are also being tested for TAM targeting in HCC[249].
Reprogramming of macrophages toward M1-like phenotypes also offers promising prospects to enhance the efficacy of immune checkpoint inhibitors, anti-angiogenic agents, and locoregional treatments[244,250,251]. However, translation into clinical practice requires multicenter validation, more integration of multi-omics data, and a better understanding of spatially and temporally dynamic macrophage states within the tumor microenvironment[249].
Most efforts are being tested in animals or are still at the preclinical stage[252]. Food and Drug Administration (FDA)-approved pexidartinib, a CSF1R inhibitor and M2 macrophage inhibitor, is under clinical investigation in liver, breast, and pancreatic cancers[253]. CD47 inhibitors that promote phagocytosis of TAMs are also in early trials on solid tumors[162]. Other drugs targeting macrophages are under clinical development. Zoledronic acid, which depletes macrophages, is being evaluated in combination with sorafenib in safety studies. Codrituzumab (GC33), which binds to human glypican-3 and inhibits M2 polarization, is also under clinical evaluation. Baicalin, an autophagy-induced p52 activator, promotes TAM repolarization into the M1 phenotype. Exosome modulators are also being tested[251].
Current treatment modalities targeting TAMs should overcome major issues such as TAM heterogeneity, plasticity, and therapeutic resistance, in addition to the excessive production costs associated with macrophage therapies. These obstacles remain critical if optimal clinical efficacy is to be achieved[254-256]. Moreover, while induction of TAM clearance appears to be a logical approach to eliminate tumor promotion by TAMs, a significant obstacle is the concomitant nonspecific depletion of monocytes. Selective targeting of TAMs with sparing of TRMs is critical to minimize adverse effects and ensure treatment safety and efficacy, but this has not been achieved so far[257].
CONCLUSIONS
KCs and BMDMs are fundamental factors in both innate and adaptive liver immunity as they are involved in several critical functions of liver biology. They are implicated in the presentation of antigens and the regulation of apoptosis of other liver cells. Deranged autophagy of KCs is involved in the progression of liver fibrosis in collaboration with LSECs and HSCs. Moreover, they are involved in the hepatic recruitment and function of lymphocytes. As a result of these complex functions, it is not surprising that macrophages are heavily involved in the initiation and progression of HCC. They are important cells in the creation of the tumor microenvironment, participating in the TAM cluster as either resident KCs or MoKCs. It is difficult to assess the exact role of KCs in the landscape of HCC microenvironment as the discrimination markers from the bone marrow-derived TAMs are not very specific so far. The function and fate of TAM1 and TAM2 are influenced by their niche in the tumor microenvironment, where distinct conditions shape macrophage behavior. This may be the explanation why TAMs may be either pro-tumoral or anti-tumoral[258].
The recent identification of several subpopulations of KCs whose precise function has not been clarified requires further research. At the moment, it is accepted that M1 and M2 polarization of KCs and other TAMs regulate the initiation and progression of HCC. There is an urgent need to identify more precise surface markers for identification of pro-tumoral KCs/TAM subsets. Moreover, a better understanding of the metabolic and epigenetic mechanisms that regulate their plasticity and variability of phenotypes in HCC is also urgently required. Since KCs are also implicated in the reduced potency of immune checkpoint inhibitors used in therapeutic regimes of HCC, several investigators have reported that reprogramming of KC and TAM functions could be a promising treatment of HCC, either alone or in combination with immunotherapy to reduce ICI resistance. However, these propositions are mostly based on animal studies and early phase I and phase II clinical studies. Therefore, more extensive trials in patients are required.
DECLARATIONS
Authors’ contributions
Made substantial contributions to conception and design of the study: Voumvouraki A
Performed data acquisition and wrote the first draft of the manuscript: Kouroumalis E
Performed data acquisition and finalized the paper: Tsomidis I
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Li P, Ding Z, Feng Y, et al. Global, regional, and national burden of hepatocellular carcinoma and contribution of nine modifiable risk factors across 185 countries/territories in 2022. Sci Bull. 2026;71:838-49.
2. Karageorgos SA, Stratakou S, Koulentaki M, et al. Long-term change in incidence and risk factors of cirrhosis and hepatocellular carcinoma in Crete, Greece: a 25-year study. Ann Gastroenterol. 2017;30:357-63.
3. Bilzer M, Roggel F, Gerbes AL. Role of Kupffer cells in host defense and liver disease. Liver Int. 2006;26:1175-86.
4. Kupffer C. Ueber sternzellen der leber: briefliche mittheilung an Prof. Waldeyer. Archiv f mikrosk Anat. 1876;12:353-8.
5. Wake K. Karl Wilhelm Kupffer and his contributions to modern hepatology. Comp Hepatol. 2004;3:S2.
6. Ueber intravasculäre Zellen in den Blutcapillaren der Leberacini. Arch f mikrosk Anat. 1899;55:420-6.
7. Underhill DM, Gordon S, Imhof BA, Núñez G, Bousso P. Élie Metchnikoff (1845-1916): celebrating 100 years of cellular immunology and beyond. Nat Rev Immunol. 2016;16:651-6.
8. Locati M, Curtale G, Mantovani A. Diversity, mechanisms, and significance of macrophage plasticity. Annu Rev Pathol. 2020;15:123-47.
9. Aegerter H, Kulikauskaite J, Crotta S, et al. Influenza-induced monocyte-derived alveolar macrophages confer prolonged antibacterial protection. Nat Immunol. 2020;21:145-57.
10. Remmerie A, Martens L, Thoné T, et al. Osteopontin expression identifies a subset of recruited macrophages distinct from kupffer cells in the fatty liver. Immunity. 2020;53:641-57.e14.
11. Remmerie A, Martens L, Scott CL. Macrophage subsets in obesity, aligning the liver and adipose tissue. Front Endocrinol. 2020;11:259.
12. Ju C, Tacke F. Hepatic macrophages in homeostasis and liver diseases: from pathogenesis to novel therapeutic strategies. Cell Mol Immunol. 2016;13:316-27.
14. Ydens E, Amann L, Asselbergh B, et al. Profiling peripheral nerve macrophages reveals two macrophage subsets with distinct localization, transcriptome and response to injury. Nat Neurosci. 2020;23:676-89.
15. Morgantini C, Jager J, Li X, et al. Liver macrophages regulate systemic metabolism through non-inflammatory factors. Nat Metab. 2019;1:445-59.
16. Perdiguero EG, Geissmann F. The development and maintenance of resident macrophages. Nat Immunol. 2016;17:2-8.
18. Gola A, Dorrington MG, Speranza E, et al. Commensal-driven immune zonation of the liver promotes host defence. Nature. 2021;589:131-6.
19. Ben-Moshe S, Veg T, Manco R, et al. The spatiotemporal program of zonal liver regeneration following acute injury. Cell Stem Cell. 2022;29:973-89.e10.
20. Feng D, Guan Y, Wang Y, Maccioni L, Mackowiak B, Gao B. Characterisation of macrophages in healthy and diseased livers in mice: identification of necrotic lesion-associated macrophages. eGastroenterology. 2025;3:e100189.
21. Tran S, Baba I, Poupel L, et al. Impaired kupffer cell self-renewal alters the liver response to lipid overload during non-alcoholic steatohepatitis. Immunity. 2020;53:627-40.e5.
22. Daemen S, Gainullina A, Kalugotla G, et al. Dynamic shifts in the composition of resident and recruited macrophages influence tissue remodeling in NASH. Cell Rep. 2021;34:108626.
23. Rolot M, M Dougall A, Javaux J, et al. Recruitment of hepatic macrophages from monocytes is independent of IL-4Rα but is associated with ablation of resident macrophages in schistosomiasis. Eur J Immunol. 2019;49:1067-81.
24. Seidman JS, Troutman TD, Sakai M, et al. Niche-specific reprogramming of epigenetic landscapes drives myeloid cell diversity in nonalcoholic steatohepatitis. Immunity. 2020;52:1057-74.e7.
25. Fan X, Lu P, Cui XH, et al. Repopulating Kupffer cells originate directly from hematopoietic stem cells. Stem Cell Res Ther. 2023;14:351.
26. Guillot A, Tacke F. Liver macrophages: old dogmas and new insights. Hepatol Commun. 2019;3:730-43.
27. Sakai M, Troutman TD, Seidman JS, et al. Liver-derived signals sequentially reprogram myeloid enhancers to initiate and maintain Kupffer cell identity. Immunity. 2019;51:655-70.e8.
28. Liu Z, Gu Y, Chakarov S, et al. Fate mapping via Ms4a3-expression history traces monocyte-derived cells. Cell. 2019;178:1509-25.e19.
29. Guilliams M, Bonnardel J, Haest B, et al. Spatial proteogenomics reveals distinct and evolutionarily conserved hepatic macrophage niches. Cell. 2022;185:379-96.e38.
31. Miyamoto Y, Kikuta J, Matsui T, et al. Periportal macrophages protect against commensal-driven liver inflammation. Nature. 2024;629:901-9.
32. Blériot C, Barreby E, Dunsmore G, et al. A subset of Kupffer cells regulates metabolism through the expression of CD36. Immunity. 2021;54:2101-16.e6.
33. De Simone G, Andreata F, Bleriot C, et al. Identification of a Kupffer cell subset capable of reverting the T cell dysfunction induced by hepatocellular priming. Immunity. 2021;54:2089-100.e8.
34. Hume DA, Offermanns S, Bonnavion R. Contamination of isolated mouse Kupffer cells with liver sinusoidal endothelial cells. Immunity. 2022;55:1139-40.
35. Beattie L, Sawtell A, Mann J, et al. Bone marrow-derived and resident liver macrophages display unique transcriptomic signatures but similar biological functions. J Hepatol. 2016;65:758-68.
36. Lee KJ, Kim MY, Han YH. Roles of heterogenous hepatic macrophages in the progression of liver diseases. BMB Rep. 2022;55:166-74.
37. Guo W, Li Z, Anagnostopoulos G, et al. Notch signaling regulates macrophage-mediated inflammation in metabolic dysfunction-associated steatotic liver disease. Immunity. 2024;57:2310-27.e6.
38. Scott CL, Zheng F, De Baetselier P, et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat Commun. 2016;7:10321.
39. Bonnardel J, T’Jonck W, Gaublomme D, et al. Stellate cells, hepatocytes, and endothelial cells imprint the Kupffer cell identity on monocytes colonizing the liver macrophage niche. Immunity. 2019;51:638-54.e9.
40. Elchaninov A, Vishnyakova P, Kuznetsova M, et al. Mimicking the cellular environment does not cause monocyte-derived macrophages to become phenotypically similar to Kupffer cells. Immunol Cell Biol. 2024;102:381-95.
41. Soysa R, Lampert S, Yuen S, et al. Fetal origin confers radioresistance on liver macrophages via p21(cip1/WAF1). J Hepatol. 2019;71:553-62.
42. Iannacone M, Blériot C, Andreata F, et al. Response to contamination of isolated mouse Kupffer cells with liver sinusoidal endothelial cells. Immunity. 2022;55:1141-2.
43. Ni M, Zhang J, Sosa R, et al. T-cell immunoglobulin and mucin domain-containing protein-4 is critical for Kupffer cell homeostatic function in the activation and resolution of liver ischemia reperfusion injury. Hepatology. 2021;74:2118-32.
44. Woolbright BL, Jaeschke H. The impact of sterile inflammation in acute liver injury. J Clin Transl Res. 2017;3:170-88.
45. Zwicker C, Bujko A, Scott CL. Hepatic macrophage responses in inflammation, a function of plasticity, heterogeneity or both? Front Immunol. 2021;12:690813.
46. MacParland SA, Liu JC, Ma XZ, et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat Commun. 2018;9:4383.
47. Aizarani N, Saviano A, Sagar, et al. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature. 2019;572:199-204.
48. Ramachandran P, Dobie R, Wilson-Kanamori JR, et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature. 2019;575:512-8.
49. Andrews TS, Atif J, Liu JC, et al. Single-cell, single-nucleus, and spatial RNA sequencing of the human liver identifies cholangiocyte and mesenchymal heterogeneity. Hepatol Commun. 2022;6:821-40.
50. Ramachandran P. Zonal control of liver inflammation is orchestrated by periportal Kupffer cells. J Hepatol. 2025;82:546-7.
51. Wu X, Hollingshead N, Roberto J, et al. Human liver macrophage subsets defined by CD32. Front Immunol. 2020;11:2108.
52. Martrus G, Goebels H, Langeneckert AE, et al. CD49a expression identifies a subset of intrahepatic macrophages in humans. Front Immunol. 2019;10:1247.
53. Fabre T, Barron AMS, Christensen SM, et al. Identification of a broadly fibrogenic macrophage subset induced by type 3 inflammation. Sci Immunol. 2023;8:eadd8945.
54. Wen Y, Lambrecht J, Ju C, Tacke F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell Mol Immunol. 2021;18:45-56.
55. Lee WY, Moriarty TJ, Wong CH, et al. An intravascular immune response to Borrelia burgdorferi involves Kupffer cells and iNKT cells. Nat Immunol. 2010;11:295-302.
56. Gorgani NN, He JQ, Katschke KJ Jr, et al. Complement receptor of the Ig superfamily enhances complement-mediated phagocytosis in a subpopulation of tissue resident macrophages. J Immunol. 2008;181:7902-8.
57. Chaudhry S, Emond J, Griesemer A. Immune cell trafficking to the liver. Transplantation. 2019;103:1323-37.
58. Zhang JX, Yang Y, Huang H, et al. TNF-α/TNFR1 regulates the polarization of Kupffer cells to mediate trichloroethylene-induced liver injury. Ecotoxicol Environ Saf. 2022;230:113141.
59. Strizova Z, Benesova I, Bartolini R, et al. M1/M2 macrophages and their overlaps - myth or reality? Clin Sci. 2023;137:1067-93.
60. Sanin DE, Ge Y, Marinkovic E, et al. A common framework of monocyte-derived macrophage activation. Sci Immunol. 2022;7:eabl7482.
61. Oshi M, Tokumaru Y, Asaoka M, et al. M1 macrophage and M1/M2 ratio defined by transcriptomic signatures resemble only part of their conventional clinical characteristics in breast cancer. Sci Rep. 2020;10:16554.
62. Cao F, Liu Y, Cheng Y, Wang Y, He Y, Xu Y. Multi-omics characteristics of tumor-associated macrophages in the tumor microenvironment of gastric cancer and their exploration of immunotherapy potential. Sci Rep. 2023;13:18265.
63. Cheng K, Cai N, Zhu J, Yang X, Liang H, Zhang W. Tumor-associated macrophages in liver cancer: from mechanisms to therapy. Cancer Commun. 2022;42:1112-40.
64. Keirsse J, Van Damme H, Geeraerts X, Beschin A, Raes G, Van Ginderachter JA. The role of hepatic macrophages in liver metastasis. Cell Immunol. 2018;330:202-15.
65. Heymann F, Peusquens J, Ludwig-Portugall I, et al. Liver inflammation abrogates immunological tolerance induced by Kupffer cells. Hepatology. 2015;62:279-91.
66. Doherty DG. Immunity, tolerance and autoimmunity in the liver: a comprehensive review. J Autoimmun. 2016;66:60-75.
67. Breous E, Somanathan S, Vandenberghe LH, Wilson JM. Hepatic regulatory T cells and Kupffer cells are crucial mediators of systemic T cell tolerance to antigens targeting murine liver. Hepatology. 2009;50:612-21.
68. Gong J, Tu W, Liu J, Tian D. Hepatocytes: a key role in liver inflammation. Front Immunol. 2022;13:1083780.
69. Campana L, Esser H, Huch M, Forbes S. Liver regeneration and inflammation: from fundamental science to clinical applications. Nat Rev Mol Cell Biol. 2021;22:608-24.
72. Dixon LJ, Barnes M, Tang H, Pritchard MT, Nagy LE. Kupffer cells in the liver. Compr Physiol. 2013;3:785-97.
73. Li X, Hollingshead N, Lampert S, et al. A conserved pathway of transdifferentiation in murine Kupffer cells. Eur J Immunol. 2021;51:2452-63.
74. Gaul S, Leszczynska A, Alegre F, et al. Hepatocyte pyroptosis and release of inflammasome particles induce stellate cell activation and liver fibrosis. J Hepatol. 2021;74:156-67.
75. Knorr J, Kaufmann B, Inzaugarat ME, et al. Interleukin-18 signaling promotes activation of hepatic stellate cells in mouse liver fibrosis. Hepatology. 2023;77:1968-82.
76. Gan C, Cai Q, Tang C, Gao J. Inflammasomes and pyroptosis of liver cells in liver fibrosis. Front Immunol. 2022;13:896473.
77. Vonderlin J, Chavakis T, Sieweke M, Tacke F. The multifaceted roles of macrophages in NAFLD pathogenesis. Cell Mol Gastroenterol Hepatol. 2023;15:1311-24.
78. Guillot A, Winkler M, Silva Afonso M, et al. Mapping the hepatic immune landscape identifies monocytic macrophages as key drivers of steatohepatitis and cholangiopathy progression. Hepatology. 2023;78:150-66.
79. Su SB, Qin SY, Xian XL, et al. Interleukin-22 regulating Kupffer cell polarization through STAT3/Erk/Akt crosstalk pathways to extenuate liver fibrosis. Life Sci. 2021;264:118677.
80. Sakai Y, Chen G, Ni Y, et al. DPP-4 inhibition with anagliptin reduces lipotoxicity-induced insulin resistance and steatohepatitis in male mice. Endocrinology. 2020;161:bqaa139.
81. Cao S, Liu M, Sehrawat TS, Shah VH. Regulation and functional roles of chemokines in liver diseases. Nat Rev Gastroenterol Hepatol. 2021;18:630-47.
82. She S, Ren L, Chen P, et al. Functional roles of chemokine receptor CCR2 and its ligands in liver disease. Front Immunol. 2022;13:812431.
83. Gao J, Wei B, Liu M, et al. Endothelial p300 promotes portal hypertension and hepatic fibrosis through C-C motif chemokine ligand 2-mediated angiocrine signaling. Hepatology. 2021;73:2468-83.
84. Guo Y, Zhao C, Dai W, et al. C-C motif chemokine receptor 2 inhibition reduces liver fibrosis by restoring the immune cell landscape. Int J Biol Sci. 2023;19:2572-87.
85. Lefere S, Devisscher L, Tacke F. Targeting CCR2/5 in the treatment of nonalcoholic steatohepatitis (NASH) and fibrosis: opportunities and challenges. Expert Opin Investig Drugs. 2020;29:89-92.
86. Peiseler M, Araujo David B, Zindel J, et al. Kupffer cell-like syncytia replenish resident macrophage function in the fibrotic liver. Science. 2023;381:eabq5202.
87. Arteel GE. When is a Kupffer cell not a Kupffer cell? Novel insight into macrophage fate and function in hepatic fibrosis. J Leukoc Biol. 2024;115:415-6.
88. Sun K, Xu L, Jing Y, et al. Autophagy-deficient Kupffer cells promote tumorigenesis by enhancing mtROS-NF-κB-IL1α/β-dependent inflammation and fibrosis during the preneoplastic stage of hepatocarcinogenesis. Cancer Lett. 2017;388:198-207.
89. Ilyas G, Zhao E, Liu K, et al. Macrophage autophagy limits acute toxic liver injury in mice through down regulation of interleukin-1β. J Hepatol. 2016;64:118-27.
90. Chen L, Kong D, Xia S, et al. Crosstalk between autophagy and innate immunity: a pivotal role in hepatic fibrosis. Front Pharmacol. 2022;13:891069.
91. Lodder J, Denaës T, Chobert MN, et al. Macrophage autophagy protects against liver fibrosis in mice. Autophagy. 2015;11:1280-92.
92. Zhu J, Zhang W, Zhang L, et al. IL-7 suppresses macrophage autophagy and promotes liver pathology in Schistosoma japonicum-infected mice. J Cell Mol Med. 2018;22:3353-63.
93. Matuz-Mares D, Vázquez-Meza H, Vilchis-Landeros MM. NOX as a therapeutic target in liver disease. Antioxidants. 2022;11:2038.
94. Herranz-Itúrbide M, Peñuelas-Haro I, Espinosa-Sotelo R, Bertran E, Fabregat I. The TGF-β/NADPH oxidases axis in the regulation of liver cell biology in health and disease. Cells. 2021;10:2312.
95. Nairz M, Theurl I, Swirski FK, Weiss G. “Pumping iron”-how macrophages handle iron at the systemic, microenvironmental, and cellular levels. Pflugers Arch. 2017;469:397-418.
96. Scott CL, Guilliams M. The role of Kupffer cells in hepatic iron and lipid metabolism. J Hepatol. 2018;69:1197-9.
97. Deppermann C, Kratofil RM, Peiseler M, et al. Macrophage galactose lectin is critical for Kupffer cells to clear aged platelets. J Exp Med. 2020;217:e20190723.
99. Gao H, Jin Z, Bandyopadhyay G, et al. MiR-690 treatment causes decreased fibrosis and steatosis and restores specific Kupffer cell functions in NASH. Cell Metab. 2022;34:978- 90.e4.
100. Coelho I, Duarte N, Macedo MP, Penha-Gonçalves C. Insights into macrophage/monocyte-endothelial cell crosstalk in the liver: a role for trem-2. J Clin Med. 2021;10:1248.
101. Gibert-Ramos A, Andrés-Rozas M, Pastó R, Alfaro-Retamero P, Guixé-Muntet S, Gracia-Sancho J. Sinusoidal communication in chronic liver disease. Clin Mol Hepatol. 2025;31:32-55.
102. Li W, Chang N, Li L. Heterogeneity and function of kupffer cells in liver injury. Front Immunol. 2022;13:940867.
103. Elchaninov A, Vishnyakova P, Menyailo E, Sukhikh G, Fatkhudinov T. An eye on Kupffer cells: development, phenotype and the macrophage niche. Int J Mol Sci. 2022;23:9868.
104. Musrati MA, De Baetselier P, Movahedi K, Van Ginderachter JA. Ontogeny, functions and reprogramming of Kupffer cells upon infectious disease. Front Immunol. 2023;14:1238452.
105. Nusse Y, Kubes P. Liver macrophages: development, dynamics, and functions. Cell Mol Immunol. 2025;22:1178-89.
106. Cao M, Wang Z, Lan W, et al. The roles of tissue resident macrophages in health and cancer. Exp Hematol Oncol. 2024;13:3.
107. Vogel A, Weichhart T. Tissue-resident macrophages - early passengers or drivers in the tumor niche? Curr Opin Biotechnol. 2023;83:102984.
108. Mulder K, Patel AA, Kong WT, et al. Cross-tissue single-cell landscape of human monocytes and macrophages in health and disease. Immunity. 2021;54:1883-900.e5.
109. Lazarov T, Juarez-Carreño S, Cox N, Geissmann F. Physiology and diseases of tissue-resident macrophages. Nature. 2023;618:698-707.
110. Elfstrum AK, Bapat AS, Schwertfeger KL. Defining and targeting macrophage heterogeneity in the mammary gland and breast cancer. Cancer Med. 2024;13:e7053.
111. Hirano R, Okamoto K, Shinke M, et al. Tissue-resident macrophages are major tumor-associated macrophage resources, contributing to early TNBC development, recurrence, and metastases. Commun Biol. 2023;6:144.
112. Wu K, Lin K, Li X, et al. Redefining tumor-associated macrophage subpopulations and functions in the tumor microenvironment. Front Immunol. 2020;11:1731.
113. Xiang X, Wang J, Lu D, Xu X. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal Transduct Target Ther. 2021;6:75.
114. Boutilier AJ, Elsawa SF. Macrophage polarization states in the tumor microenvironment. Int J Mol Sci. 2021;22:6995.
115. Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32:593-604.
116. Galli SJ, Borregaard N, Wynn TA. Phenotypic and functional plasticity of cells of innate immunity: macrophages, mast cells and neutrophils. Nat Immunol. 2011;12:1035-44.
117. Griess B, Mir S, Datta K, Teoh-Fitzgerald M. Scavenging reactive oxygen species selectively inhibits M2 macrophage polarization and their pro-tumorigenic function in part, via Stat3 suppression. Free Radic Biol Med. 2020;147:48-60.
118. Chi J, Gao Q, Liu D. Tissue-resident macrophages in cancer: friend or foe? Cancer Med. 2024;13:e70387.
119. Roh MS, Wang L, Oyedeji C, et al. Human Kupffer cells are cytotoxic against human colon adenocarcinoma. Surgery. 1990;108:400-4.
120. Heuff G, van de Loosdrecht AA, Betjes MG, Beelen RH, Meijer S. Isolation and purification of large quantities of fresh human Kupffer cells, which are cytotoxic against colon carcinoma. Hepatology. 1995;21:740-5.
121. Kan Z, Ivancev K, Lunderquist A, McCuskey PA, McCuskey RS, Wallace S. In vivo microscopy of hepatic metastases: dynamic observation of tumor cell invasion and interaction with Kupffer cells. Hepatology. 1995;21:487-94.
122. Song E, Chen J, Ouyang N, Wang M, Exton MS, Heemann U. Kupffer cells of cirrhotic rat livers sensitize colon cancer cells to Fas-mediated apoptosis. Br J Cancer. 2001;84:1265-71.
123. Lau WY, Chen GG, Lai PB, et al. Induction of Fas and Fas ligand expression on malignant glioma cells by Kupffer cells, a potential pathway of antiliver metastases. J Surg Res. 2001;101:44-51.
124. Pearson HJ, Anderson J, Chamberlain J, Bell PR. The effect of Kupffer cell stimulation or depression on the development of liver metastases in the rat. Cancer Immunol Immunother. 1986;23:214-6.
125. Valatas V, Kolios G, Manousou P, et al. Secretion of inflammatory mediators by isolated rat Kupffer cells: the effect of octreotide. Regul Pept. 2004;120:215-25.
126. Aono K, Isobe K, Nakashima I, Kondo S, Miyachi M, Nimura Y. Kupffer cells cytotoxicity against hepatoma cells is related to nitric oxide. Biochem Biophys Res Commun. 1994;201:1175-81.
127. Hourani T, Holden JA, Li W, Lenzo JC, Hadjigol S, O’Brien-Simpson NM. Tumor associated macrophages: origin, recruitment, phenotypic diversity, and targeting. Front Oncol. 2021;11:788365.
128. Christofides A, Strauss L, Yeo A, Cao C, Charest A, Boussiotis VA. The complex role of tumor-infiltrating macrophages. Nat Immunol. 2022;23:1148-56.
129. Tian Z, Hou X, Liu W, Han Z, Wei L. Macrophages and hepatocellular carcinoma. Cell Biosci. 2019;9:79.
130. Cotechini T, Atallah A, Grossman A. Tissue-resident and recruited macrophages in primary tumor and metastatic microenvironments: potential targets in cancer therapy. Cells. 2021;10:960.
131. Wu JY, Huang TW, Hsieh YT, et al. Cancer-derived succinate promotes macrophage polarization and cancer metastasis via succinate receptor. Mol Cell. 2020;77:213-27.e5.
132. Casari M, Siegl D, Deppermann C, Schuppan D. Macrophages and platelets in liver fibrosis and hepatocellular carcinoma. Front Immunol. 2023;14:1277808.
133. Kuo TC, Chen A, Harrabi O, et al. Targeting the myeloid checkpoint receptor SIRPα potentiates innate and adaptive immune responses to promote anti-tumor activity. J Hematol Oncol. 2020;13:160.
134. Deng Z, Loyher PL, Lazarov T, et al. The nuclear factor ID3 endows macrophages with a potent anti-tumour activity. Nature. 2024;626:864-73.
135. Brodt P. Role of the microenvironment in liver metastasis: from pre- to prometastatic niches. Clin Cancer Res. 2016;22:5971-82.
136. Li J, Liu XG, Ge RL, et al. The ligation between ERMAP, galectin-9 and dectin-2 promotes Kupffer cell phagocytosis and antitumor immunity. Nat Immunol. 2023;24:1813-24.
137. Wortzel I, Seo Y, Akano I, et al. Unique structural configuration of EV-DNA primes Kupffer cell-mediated antitumor immunity to prevent metastatic progression. Nat Cancer. 2024;5:1815-33.
138. Jiang K, Chen H, Fang Y, et al. Exosomal ANGPTL1 attenuates colorectal cancer liver metastasis by regulating Kupffer cell secretion pattern and impeding MMP9 induced vascular leakiness. J Exp Clin Cancer Res. 2021;40:21.
139. Jin HR, Wang J, Wang ZJ, et al. Lipid metabolic reprogramming in tumor microenvironment: from mechanisms to therapeutics. J Hematol Oncol. 2023;16:103.
140. Ammarah U, Pereira-Nunes A, Delfini M, Mazzone M. From monocyte-derived macrophages to resident macrophages-how metabolism leads their way in cancer. Mol Oncol. 2024;18:1739-58.
141. Nagy C, Haschemi A. Time and demand are two critical dimensions of immunometabolism: the process of macrophage activation and the pentose phosphate pathway. Front Immunol. 2015;6:164.
142. Jha AK, Huang SC, Sergushichev A, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. 2015;42:419-30.
144. Zaidi NE, Shazali NAH, Leow TC, Osman MA, Ibrahim K, Rahman NMANA. Crosstalk between fatty acid metabolism and tumour-associated macrophages in cancer progression. Biomedicine. 2022;12:9-19.
145. Liu Y, Xu R, Gu H, et al. Metabolic reprogramming in macrophage responses. Biomark Res. 2021;9:1.
146. Liu PS, Wang H, Li X, et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol. 2017;18:985-94.
147. Van den Bossche J, Lamers WH, Koehler ES, et al. Pivotal Advance: Arginase-1-independent polyamine production stimulates the expression of IL-4-induced alternatively activated macrophage markers while inhibiting LPS-induced expression of inflammatory genes. J Leukoc Biol. 2012;91:685-99.
148. Van den Bossche J, O’Neill LA, Menon D. Macrophage Immunometabolism: where are we (going)? Trends Immunol. 2017;38:395-406.
149. Sharma A, Seow JJW, Dutertre CA, et al. Onco-fetal reprogramming of endothelial cells drives immunosuppressive macrophages in hepatocellular carcinoma. Cell. 2020;183:377-94.e21.
150. Casanova-Acebes M, Dalla E, Leader AM, et al. Tissue-resident macrophages provide a pro-tumorigenic niche to early NSCLC cells. Nature. 2021;595:578-84.
151. Shen KY, Zhu Y, Xie SZ, Qin LX. Immunosuppressive tumor microenvironment and immunotherapy of hepatocellular carcinoma: current status and prospectives. J Hematol Oncol. 2024;17:25.
152. Mass E, Ballesteros I, Farlik M, et al. Specification of tissue-resident macrophages during organogenesis. Science. 2016;353:aaf4238.
153. Li XY, Wang MY, Zhang JY, Li JZ, Gong JP, Zhang W. Kupffer cells suppress hepatocarcinogenesis and metastasis in tumor orthotopic implanted Kunming mice. Asian Pac J Cancer Prev. 2013;14:6393-8.
154. Hong GQ, Cai D, Gong JP, Lai X. Innate immune cells and their interaction with T cells in hepatocellular carcinoma. Oncol Lett. 2021;21:57.
155. Prieto J. Inflammation, HCC and sex: IL-6 in the centre of the triangle. J Hepatol. 2008;48:380-1.
156. Naugler WE, Sakurai T, Kim S, et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317:121-4.
157. Yuan Y, Wu D, Li J, et al. Mechanisms of tumor-associated macrophages affecting the progression of hepatocellular carcinoma. Front Pharmacol. 2023;14:1217400.
158. Lefere S, Degroote H, Van Vlierberghe H, Devisscher L. Unveiling the depletion of Kupffer cells in experimental hepatocarcinogenesis through liver macrophage subtype-specific markers. J Hepatol. 2019;71:631-3.
159. Degroote H, Lefere S, Vandierendonck A, et al. Characterization of the inflammatory microenvironment and hepatic macrophage subsets in experimental hepatocellular carcinoma models. Oncotarget. 2021;12:562-77.
160. Bosco MC. Macrophage polarization: Reaching across the aisle? J Allergy Clin Immunol. 2019;143:1348-50.
161. Zhang Y, Han G, Gu J, Chen Z, Wu J. Role of tumor-associated macrophages in hepatocellular carcinoma: impact, mechanism, and therapy. Front Immunol. 2024;15:1429812.
162. Toledo B, Zhu Chen L, Paniagua-Sancho M, Marchal JA, Perán M, Giovannetti E. Deciphering the performance of macrophages in tumour microenvironment: a call for precision immunotherapy. J Hematol Oncol. 2024;17:44.
163. Wang Q, Ma W. Revisiting TAM polarization: beyond M1- and M2-type TAM toward clinical precision in macrophage-targeted therapy. Exp Mol Pathol. 2025;143:104982.
164. Yang Y, Ye YC, Chen Y, et al. Crosstalk between hepatic tumor cells and macrophages via Wnt/β-catenin signaling promotes M2-like macrophage polarization and reinforces tumor malignant behaviors. Cell Death Dis. 2018;9:793.
165. Ma RY, Black A, Qian BZ. Macrophage diversity in cancer revisited in the era of single-cell omics. Trends Immunol. 2022;43:546-63.
166. Guo S, Chen X, Guo C, Wang W. Tumour-associated macrophages heterogeneity drives resistance to clinical therapy. Expert Rev Mol Med. 2022;24:e17.
167. Li JQ, Yu XJ, Wang YC, et al. Distinct patterns and prognostic values of tumor-infiltrating macrophages in hepatocellular carcinoma and gastric cancer. J Transl Med. 2017;15:37.
168. Lu Y, Yang A, Quan C, et al. A single-cell atlas of the multicellular ecosystem of primary and metastatic hepatocellular carcinoma. Nat Commun. 2022;13:4594.
169. Xiao N, Zhu X, Li K, et al. Blocking siglec-10hi tumor-associated macrophages improves anti-tumor immunity and enhances immunotherapy for hepatocellular carcinoma. Exp Hematol Oncol. 2021;10:36.
170. Ng HHM, Lee RY, Goh S, et al. Immunohistochemical scoring of CD38 in the tumor microenvironment predicts responsiveness to anti-PD-1/PD-L1 immunotherapy in hepatocellular carcinoma. J Immunother Cancer. 2020;8:e000987.
171. Cui X, Zhao H, Wei S, et al. Hepatocellular carcinoma-derived FOXO1 inhibits tumor progression by suppressing IL-6 secretion from macrophages. Neoplasia. 2023;40:100900.
172. Wu J, Li J, Salcedo R, Mivechi NF, Trinchieri G, Horuzsko A. The proinflammatory myeloid cell receptor TREM-1 controls Kupffer cell activation and development of hepatocellular carcinoma. Cancer Res. 2012;72:3977-86.
173. Dou L, Shi X, He X, Gao Y. Macrophage phenotype and function in liver disorder. Front Immunol. 2019;10:3112.
174. Thomann S, Weiler SME, Wei T, et al. YAP-induced Ccl2 expression is associated with a switch in hepatic macrophage identity and vascular remodelling in liver cancer. Liver Int. 2021;41:3011-23.
175. Vanderborght B, De Muynck K, Gijbels E, et al. Transient Kupffer cell depletion and subsequent replacement by infiltrating monocyte-derived cells does not alter the induction or progression of hepatocellular carcinoma. Int J Cancer. 2023;152:2615-28.
176. Du L, Ji Y, Xin B, et al. Shp2 deficiency in Kupffer cells and hepatocytes aggravates hepatocarcinogenesis by recruiting non-Kupffer macrophages. Cell Mol Gastroenterol Hepatol. 2023;15:1351-69.
177. Li X, Li R, Miao X, et al. Integrated single cell analysis reveals an atlas of tumor associated macrophages in hepatocellular carcinoma. Inflammation. 2024;47:2077-93.
178. Zhang M, Liu K, Zhang Q, et al. Alpha fetoprotein promotes polarization of macrophages towards M2-like phenotype and inhibits macrophages to phagocytize hepatoma cells. Front Immunol. 2023;14:1081572.
179. Yu H, Pan J, Zheng S, et al. Hepatocellular carcinoma cell-derived exosomal miR-21-5p induces macrophage M2 polarization by targeting RhoB. Int J Mol Sci. 2023;24:4593.
180. Sun JL, Zhang NP, Xu RC, et al. Tumor cell-imposed iron restriction drives immunosuppressive polarization of tumor-associated macrophages. J Transl Med. 2021;19:347.
181. Yang S, Zhang J, Xu Y, et al. OIT3 mediates macrophage polarization and facilitates hepatocellular carcinoma progression. Cancer Immunol Immunother. 2022;71:2677-89.
182. Zhao J, Zhang S, Liu Y, et al. Single-cell RNA sequencing reveals the heterogeneity of liver-resident immune cells in human. Cell Discov. 2020;6:22.
183. Sun Y, Wu L, Zhong Y, et al. Single-cell landscape of the ecosystem in early-relapse hepatocellular carcinoma. Cell. 2021;184:404-21.e16.
184. Zhang Q, He Y, Luo N, et al. Landscape and dynamics of single immune cells in hepatocellular carcinoma. Cell. 2019;179:829-45.e20.
185. Sas Z, Cendrowicz E, Weinhäuser I, Rygiel TP. Tumor microenvironment of hepatocellular carcinoma: challenges and opportunities for new treatment options. Int J Mol Sci. 2022;23:3778.
186. Capece D, Fischietti M, Verzella D, et al. The inflammatory microenvironment in hepatocellular carcinoma: a pivotal role for tumor-associated macrophages. Biomed Res Int. 2013;2013:187204.
187. Yu M, Yu H, Wang H, et al. Tumor‑associated macrophages activated in the tumor environment of hepatocellular carcinoma: characterization and treatment (Review). Int J Oncol. 2024;65:100.
188. Ajith A, Evraerts J, Bouzin C, et al. Progression to fibrosis and hepatocellular carcinoma in DEN CCl4 liver mice, is associated with macrophage and striking regulatory T cells infiltration. Front Immunol 2025;16:1601215.[PMID:40698077 DOI:10.3389/fimmu.2025.1601215 PMCID:12279789] Caution!.
189. Malehmir M, Pfister D, Gallage S, et al. Platelet GPIbα is a mediator and potential interventional target for NASH and subsequent liver cancer. Nat Med. 2019;25:641-55.
190. Roderburg C, Wree A, Demir M, Schmelzle M, Tacke F. The role of the innate immune system in the development and treatment of hepatocellular carcinoma. Hepat Oncol. 2020;7:HEP17.
191. Chen J, Zheng DX, Yu XJ, et al. Macrophages induce CD47 upregulation via IL-6 and correlate with poor survival in hepatocellular carcinoma patients. Oncoimmunology. 2019;8:e1652540.
192. Li YW, Qiu SJ, Fan J, et al. Tumor-infiltrating macrophages can predict favorable prognosis in hepatocellular carcinoma after resection. J Cancer Res Clin Oncol. 2009;135:439-49.
193. Zhang Y, Li JQ, Jiang ZZ, Li L, Wu Y, Zheng L. CD169 identifies an anti-tumour macrophage subpopulation in human hepatocellular carcinoma. J Pathol. 2016;239:231-41.
194. Ding T, Xu J, Wang F, et al. High tumor-infiltrating macrophage density predicts poor prognosis in patients with primary hepatocellular carcinoma after resection. Hum Pathol. 2009;40:381-9.
195. Kang TW, Yevsa T, Woller N, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011;479:547-51.
196. Liu N, Wang X, Steer CJ, Song G. MicroRNA-206 promotes the recruitment of CD8+ T cells by driving M1 polarisation of Kupffer cells. Gut. 2022;71:1642-55.
197. Hastir JF, Delbauve S, Larbanoix L, et al. Hepatocarcinoma induces a tumor necrosis factor-dependent Kupffer cell death pathway that favors its proliferation upon partial hepatectomy. Front Oncol. 2020;10:547013.
198. Tian Y, Zhang M, Fan M, et al. A miRNA-mediated attenuation of hepatocarcinogenesis in both hepatocytes and Kupffer cells. Mol Ther Nucleic Acids. 2022;30:1-12.
199. Herman AB, Tsitsipatis D, Gorospe M. Integrated lncRNA function upon genomic and epigenomic regulation. Mol Cell. 2022;82:2252-66.
200. Wu H, Zhong Z, Wang A, et al. LncRNA FTX represses the progression of non-alcoholic fatty liver disease to hepatocellular carcinoma via regulating the M1/M2 polarization of Kupffer cells. Cancer Cell Int. 2020;20:266.
201. Wei Q, Liu G, Huang Z, et al. LncRNA MEG3 inhibits tumor progression by modulating macrophage phenotypic polarization via miR-145-5p/DAB2 axis in hepatocellular carcinoma. J Hepatocell Carcinoma. 2023;10:1019-35.
202. Hou ZH, Xu XW, Fu XY, Zhou LD, Liu SP, Tan DM. Long non-coding RNA MALAT1 promotes angiogenesis and immunosuppressive properties of HCC cells by sponging miR-140. Am J Physiol Cell Physiol. 2020;318:C649-63.
203. Tian X, Wu Y, Yang Y, et al. Long noncoding RNA LINC00662 promotes M2 macrophage polarization and hepatocellular carcinoma progression via activating Wnt/β-catenin signaling. Mol Oncol. 2020;14:462-83.
204. Ye Y, Xu Y, Lai Y, et al. Long non-coding RNA cox-2 prevents immune evasion and metastasis of hepatocellular carcinoma by altering M1/M2 macrophage polarization. J Cell Biochem. 2018;119:2951-63.
205. Wang X, Li FY, Zhao W, et al. Long non-coding RNA GAS5 overexpression inhibits M2-like polarization of tumour-associated macrophages in SMCC-7721 cells by promoting PTEN expression. Int J Exp Pathol. 2020;101:215-22.
206. Xu M, Zhou C, Weng J, et al. Tumor associated macrophages-derived exosomes facilitate hepatocellular carcinoma malignance by transferring lncMMPA to tumor cells and activating glycolysis pathway. J Exp Clin Cancer Res. 2022;41:253.
207. Li X, Lei Y, Wu M, Li N. Regulation of macrophage activation and polarization by HCC-derived exosomal lncRNA TUC339. Int J Mol Sci. 2018;19:2958.
208. Lv S, Wang J, Li L. Extracellular vesicular lncRNA FAL1 promotes hepatocellular carcinoma cell proliferation and invasion by inducing macrophage M2 polarization. J Physiol Biochem. 2023;79:669-82.
209. Ai JH, Wen YZ, Dai SJ, Zhang LD, Huang ZJ, Shi J. Exosomal lncRNA HEIH, an essential communicator for hepatocellular carcinoma cells and macrophage M2 polarization through the miR-98-5p/STAT3 axis. J Biochem Mol Toxicol. 2024;38:e23686.
210. Wang X, Zhou Y, Dong K, Zhang H, Gong J, Wang S. Exosomal lncRNA HMMR-AS1 mediates macrophage polarization through miR-147a/ARID3A axis under hypoxia and affects the progression of hepatocellular carcinoma. Environ Toxicol. 2022;37:1357-72.
211. Zhou J, Che J, Xu L, Yang W, Zhou W, Zhou C. Tumor-derived extracellular vesicles containing long noncoding RNA PART1 exert oncogenic effect in hepatocellular carcinoma by polarizing macrophages into M2. Dig Liver Dis. 2022;54:543-53.
212. Ye Y, Wang M, Wang G, et al. lncRNA miR4458HG modulates hepatocellular carcinoma progression by activating m6A-dependent glycolysis and promoting the polarization of tumor-associated macrophages. Cell Mol Life Sci. 2023;80:99.
213. Zhao S, Chen F, Hu L, et al. Long non-coding rnas as key modulators of the immune microenvironment in hepatocellular carcinoma: implications for Immunotherapy. Front Immunol. 2025;16:1523190.
214. Ghorbani Vanan A, Nami MT, Ghorbaninezhad F, et al. Macrophage polarization in hepatocellular carcinoma: a lncRNA-centric perspective on tumor progression and metastasis. Clin Exp Med. 2025;25:173.
215. Delire B, Henriet P, Lemoine P, Leclercq IA, Stärkel P. Chronic liver injury promotes hepatocarcinoma cell seeding and growth, associated with infiltration by macrophages. Cancer Sci. 2018;109:2141-52.
216. Huang Y, Ge W, Zhou J, Gao B, Qian X, Wang W. The role of tumor associated macrophages in hepatocellular carcinoma. J Cancer. 2021;12:1284-94.
217. Chang CP, Su YC, Lee PH, Lei HY. Targeting NFKB by autophagy to polarize hepatoma-associated macrophage differentiation. Autophagy. 2013;9:619-21.
218. Sugisawa R, Komatsu G, Hiramoto E, et al. Independent modes of disease repair by AIM protein distinguished in AIM-felinized mice. Sci Rep. 2018;8:13157.
219. Zhang J, Pan Y, Zhao Z, et al. SLC16A3 as an immunosuppressive Kupffer cell marker predicts poor prognosis in HBV-positive hepatocellular carcinoma. J Transl Med. 2025;23:988.
220. Ohno A, Yorita K, Haruyama Y, et al. Aberrant expression of monocarboxylate transporter 4 in tumour cells predicts an unfavourable outcome in patients with hepatocellular carcinoma. Liver Int. 2014;34:942-52.
221. Alves VA, Pinheiro C, Morais-Santos F, Felipe-Silva A, Longatto-Filho A, Baltazar F. Characterization of monocarboxylate transporter activity in hepatocellular carcinoma. World J Gastroenterol. 2014;20:11780-7.
222. Ioannou GN. The role of cholesterol in the pathogenesis of NASH. Trends Endocrinol Metab. 2016;27:84-95.
223. Escobedo-calvario A, Chávez-rodríguez L, Simoni-nieves A, et al. Tumor immune microenvironment modulation by cholesterol in hepatocellular carcinoma. Explor Dig Dis. 2022;1:21-39.
224. Simoni-Nieves A, Salas-Silva S, Chávez-Rodríguez L, et al. The consumption of cholesterol-enriched diets conditions the development of a subtype of HCC with high aggressiveness and poor prognosis. Cancers. 2021;13:1721.
225. Goossens P, Rodriguez-Vita J, Etzerodt A, et al. Membrane cholesterol efflux drives tumor-associated macrophage reprogramming and tumor progression. Cell Metab. 2019;29:1376-89.e4.
226. Yang Y, Li S, To KKW, Zhu S, Wang F, Fu L. Tumor-associated macrophages remodel the suppressive tumor immune microenvironment and targeted therapy for immunotherapy. J Exp Clin Cancer Res. 2025;44:145.
227. Sheng J, Zhang J, Wang L, et al. Topological analysis of hepatocellular carcinoma tumour microenvironment based on imaging mass cytometry reveals cellular neighbourhood regulated reversely by macrophages with different ontogeny. Gut. 2022;71:1176-91.
228. Liu W, Zhou X, Yao Q, et al. In situ expansion and reprogramming of Kupffer cells elicit potent tumoricidal immunity against liver metastasis. J Clin Invest. 2023;133:e157937.
229. Siwicki M, Gort-Freitas NA, Messemaker M, et al. Resident Kupffer cells and neutrophils drive liver toxicity in cancer immunotherapy. Sci Immunol. 2021;6:eabi7083.
230. Xu W, Cheng Y, Guo Y, Yao W, Qian H. Targeting tumor associated macrophages in hepatocellular carcinoma. Biochem Pharmacol. 2022;199:114990.
231. Giannone G, Ghisoni E, Genta S, et al. Immuno-metabolism and microenvironment in cancer: key players for immunotherapy. Int J Mol Sci. 2020;21:4414.
232. Lemaitre L, Adeniji N, Suresh A, et al. Spatial analysis reveals targetable macrophage-mediated mechanisms of immune evasion in hepatocellular carcinoma minimal residual disease. Nat Cancer. 2024;5:1534-56.
233. Weng J, Wang Z, Hu Z, et al. Repolarization of immunosuppressive macrophages by targeting SLAMF7-regulated CCL2 signaling sensitizes hepatocellular carcinoma to immunotherapy. Cancer Res. 2024;84:1817-33.
234. Zhang Y, Rao Y, Lu J, et al. The influence of biophysical niche on tumor-associated macrophages in liver cancer. Hepatol Commun. 2024;8:e0569.
235. Li M, He L, Zhu J, Zhang P, Liang S. Targeting tumor-associated macrophages for cancer treatment. Cell Biosci. 2022;12:85.
236. Liu X, Hogg GD, Zuo C, et al. Context-dependent activation of STING-interferon signaling by CD11b agonists enhances anti-tumor immunity. Cancer Cell. 2023;41:1073-90.e12.
237. Liu F, Li X, Zhang Y, et al. Targeting tumor-associated macrophages to overcome immune checkpoint inhibitor resistance in hepatocellular carcinoma. J Exp Clin Cancer Res. 2025;44:227.
238. Chen M, Lai R, Lin X, Chen W, Wu H, Zheng Q. Downregulation of triggering receptor expressed on myeloid cells 1 inhibits invasion and migration of liver cancer cells by mediating macrophage polarization. Oncol Rep. 2021;45:37.
239. Yang Y, Jia X, Qu M, et al. Exploring the potential of treating chronic liver disease targeting the PI3K/Akt pathway and polarization mechanism of macrophages. Heliyon. 2023;9:e17116.
240. Ren L, Qiao GL, Zhang SX, Zhang ZM, Lv SX. Pharmacological inhibition or silencing of TREM1 restrains HCC cell metastasis by inactivating TLR/PI3K/AKT signaling. Cell Biochem Biophys. 2024;82:2673-85.
241. Song M, Li ZH, Gu HS, et al. Ganoderma lucidum spore polysaccharide inhibits the growth of hepatocellular carcinoma cells by altering macrophage polarity and induction of apoptosis. J Immunol Res. 2021;2021:6696606.
242. Lu S, Gao Y, Huang X, Wang X. Cantharidin exerts anti-hepatocellular carcinoma by miR-214 modulating macrophage polarization. Int J Biol Sci. 2014;10:415-25.
243. Min L, Wang H, Qi H. Astragaloside IV inhibits the progression of liver cancer by modulating macrophage polarization through the TLR4/NF-κB/STAT3 signaling pathway. Am J Transl Res. 2022;14:1551-66.
244. Bannister ME, Chatterjee DA, Shetty S, Patten DA. The role of macrophages in hepatocellular carcinoma and their therapeutic potential. Int J Mol Sci. 2024;25:13167.
245. Cancer Genome Atlas Research Network. Electronic address: [email protected]; Cancer Genome Atlas Research Network. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell. 2017;169:1327-41.e23.
246. Cai N, Cheng K, Ma Y, et al. Targeting MMP9 in CTNNB1 mutant hepatocellular carcinoma restores CD8+ T cell-mediated antitumour immunity and improves anti-PD-1 efficacy. Gut. 2024;73:985-99.
247. Zhao J, Cui M, Yao X, et al. Targeting TFAM downregulation mediated mtDNA-NLRP3 pathway suppresses TAM infiltration and HCC progression. Oncogene. 2025;44:2956-69.
248. Zhu CX, Yan K, Chen L, et al. Targeting OXCT1-mediated ketone metabolism reprograms macrophages to promote antitumor immunity via CD8+ T cells in hepatocellular carcinoma. J Hepatol. 2024;81:690-703.
249. Fu X, Pang M, Wang Z, Wang H. Macrophage polarization in the tumor microenvironment of hepatocellular carcinoma: from mechanistic insights to translational therapies. Cancer Control. 2025;32:10732748251406674.
250. Tan J, Fan W, Liu T, et al. TREM2+ macrophages suppress CD8+ T-cell infiltration after transarterial chemoembolisation in hepatocellular carcinoma. J Hepatol. 2023;79:126-40.
251. Yang H, Zhang Z, Zhao K, et al. Targeting the adenosine signaling pathway in macrophages for cancer immunotherapy. Hum Immunol. 2024;85:110774.
252. Karimova AF, Khalitova AR, Suezov R, et al. Immunometabolism of tumor-associated macrophages: a therapeutic perspective. Eur J Cancer. 2025;220:115332.
253. Li D, Rudloff U. Emerging therapeutics targeting tumor-associated macrophages for the treatment of solid organ cancers. Expert Opin Emerg Drugs. 2025;30:109-47.
254. Duan Z, Luo Y. Targeting macrophages in cancer immunotherapy. Signal Transduct Target Ther. 2021;6:127.
255. Yang S, Wang Y, Jia J, et al. Advances in engineered macrophages: a new frontier in cancer immunotherapy. Cell Death Dis. 2024;15:238.
256. Na YR, Kim SW, Seok SH. A new era of macrophage-based cell therapy. Exp Mol Med. 2023;55:1945-54.
257. Etzerodt A, Tsalkitzi K, Maniecki M, et al. Specific targeting of CD163+ TAMs mobilizes inflammatory monocytes and promotes T cell-mediated tumor regression. J Exp Med. 2019;216:2394-411.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0











Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].