


Complex LDLR mutations in Taiwanese familial hypercholesterolemia
 0
0 Abstract
Aim: Familial hypercholesterolemia (FH) is a rare autosomal dominant lipid disorder, usually caused by LDLR (low-density lipoprotein receptor) mutations and strongly associated with premature coronary heart disease (CHD). Despite its clinical impact, FH remains underdiagnosed, and data on complex mutations in Asian populations are scarce. This study aimed to determine the prevalence and clinical significance of complex LDLR mutations in Taiwanese patients with heterozygous FH (HeFH), to evaluate the diagnostic utility of a two-step genetic strategy [FHChip plus MLPA (multiplex ligation-dependent probe amplification)], and to explore therapeutic implications for intensive lipid-lowering regimens and orphan drugs.
Methods: We recruited 100 index patients with phenotypic FH from National Taiwan University Hospital. Genetic testing included FHChip (Vita Genomics, Taiwan) for LDLR, apolipoprotein B (APOB), proprotein convertase subtilisin/kexin type 9 (PCSK9), and known InDels, followed by multiplex ligation-dependent probe amplification (MLPA) for mutation-negative cases. Complex mutations were defined as compound heterozygous, double mutations in one allele, or large LDLR rearrangements. Clinical and biochemical features were compared between patients with complex and single mutations.
Results: Pathogenic variants were detected in 76% of patients, with 12 (18.2%) harboring complex mutations. Compared with single mutations, complex mutations were associated with higher Low-density lipoprotein cholesterol (LDL-C) (319 vs. 210 mg/dL), more premature CHD (34.8% vs. 6.5%), and tendon xanthomas (69.6% vs. 1.1%). Intensive lipid-lowering therapy (statin-ezetimibe ± additional agents) achieved ~60% LDL-C reduction.
Conclusion: Complex mutations are not rare in Taiwanese HeFH and predict severe outcomes. Timely detection with FHChip-MLPA and aggressive therapy, including orphan drugs, is critical to lowering premature CHD risk in rare dyslipidemia.
Keywords
INTRODUCTION
According to data from the Taiwanese Survey on Hypertension, Hyperglycemia, and Hyperlipidemia (TwSHHH), approximately 10.9% of individuals aged 15 years or older in Taiwan had total cholesterol levels ≥ 240 mg/dL[1]. Globally, familial hypercholesterolemia (FH) is estimated to affect approximately 1 in 200-500 individuals. In certain founder populations, however, the frequency may be substantially higher. In Taiwan, a hospital-based genetic screening study reported a prevalence of 1.13% for genetically confirmed FH[2]. The high disease burden highlights the urgent need for public health strategies aimed at the prevention and management of atherosclerotic cardiovascular complications associated with severe hypercholesterolemia[2-4].
Given the substantial burden of FH in Taiwan, clarifying its molecular and clinical characteristics is of particular importance. The most severe forms of dyslipidemia arise from inherited abnormalities in low-density lipoprotein (LDL) metabolism, most commonly involving pathogenic variants in the LDLR (low density lipoprotein receptor) gene. Impaired LDL receptor function reduces hepatic clearance of circulating LDL cholesterol, resulting in markedly elevated Low-density lipoprotein cholesterol (LDL-C) levels and increased cardiovascular risk[5]. These mutations impair hepatic clearance of plasma LDL cholesterol[3-5], leading to markedly elevated LDL-C levels and a substantially increased risk of premature cardiovascular disease (CVD). Clinically, FH is one of the most readily identifiable forms of genetic dyslipidemia. Although often underdiagnosed, FH can be effectively managed, and when treatment is initiated early, most patients achieve a near-normal life expectancy. Cascade family screening is recommended to identify affected relatives for early intervention[3,4]. Phenotypic expression of FH varies by sex and between individuals. The clinical manifestations of FH show considerable heterogeneity. Female carriers generally experience coronary events at a later age than male carriers with the same LDLR variant, and substantial variation in age of onset is observed even among individuals with similar LDL-C levels at diagnosis. FH represents a significant global health burden, with an estimated 10 million individuals affected worldwide[6,7]. Approximately at least 20,000 premature deaths from myocardial infarction are attributed to FH each year[8]. Despite its clinical importance, around 80% of heterozygous FH (HeFH) cases remain undiagnosed, and 84% of affected individuals do not receive lipid-lowering therapy. The World Health Organization (WHO) Human Genome Program for FH (Paris, 1997) emphasized that early detection and treatment are cost-effective and that FH accounts for up to 9% of premature coronary heart disease (CHD)[8]. Sustained lipid-lowering treatment has been associated with substantial improvements in survival among patients with FH[8,9]. More than 2000 unique point mutations have been reported across different ethnic populations in HeFH. However, genetic testing confirms only 40%-70% of clinically diagnosed FH cases[10,11]. Patients with more severe hypercholesterolemia (e.g., LDL-C > 350 mg/dL) carry a particularly high risk of premature CHD[10,11]. Identifying causal genetic variants in such patients is essential for advancing the understanding of molecular mechanisms in FH and may provide novel insights for targeted therapies.
FH is a common yet under-recognized genetic disorder in Taiwan, with prevalence rates higher than those reported in many other regions worldwide. Given its significant contribution to premature CVD, FH should be prioritized as an urgent public health concern. National strategies integrating early detection, cascade family screening, genetic testing, and long-term lipid-lowering therapy are essential to reduce preventable cardiovascular morbidity and mortality. Establishing a nationwide FH registry and incorporating personalized treatment approaches into clinical practice may further improve outcomes and provide valuable insights for managing this rare but impactful disorder.
PATIENTS AND METHODS
Patients
We recruited patients with phenotypic FH from the lipid clinic of National Taiwan University Hospital between 2002 and 2011. Patients met the following criteria: (i) hypercholesterolemia with serum total cholesterol ≥ 290 mg/dL and LDL-C ≥ 190 mg/dL; and (ii) at least one first-degree relative with similarly elevated cholesterol levels, premature CHD, cutaneous xanthoma, or corneal arcus.
Exclusion criteria were active hypothyroidism, cholestatic jaundice, nephrotic syndrome, malignancy, or current treatment with chemotherapy or corticosteroids. A total of 100 families comprising 450 family members were enrolled. Cascade screening was performed for the index cases of phenotypic FH.
Genetic analysis was conducted using the FHChip assay to detect single-gene mutations in index cases. Positive findings were confirmed in first-degree relatives by direct sequencing. For index patients without identified single-gene mutations, multiplex ligation-dependent probe amplification (MLPA) was performed to detect large genomic rearrangements of the LDLR gene.
FHChip resequencing microarray design
FHChip, a resequencing microarray designed for the diagnostic purpose of FH[12], was designed by Vita Genomics, Inc. and manufactured by Affymetrix (Santa Clara, California) using photolithography and solid-phase DNA synthesis. Each microarray contained 12.6 kb in duplication of coding exon and flanking intron sequence (both sense and antisense) of the three most relevant genes for FH including LDLR, APOB, and PCSK9 genes. In order to expand the insertion/deletion (InDel) mutation detection function of the chips, specific probes for 64 previously reported InDel mutations in LDLR gene were also designed and tiled on the FHChip. Each microarray contains 240,000 features that cover all of the specific sequences of genes to be tested, with each feature consisting of 106 copies of a 25 bp specific probe. For each position of the nucleotide sequences, four 25-mer probes are represented on the chip, each with a different nucleotide in the middle (A, G, C, or T) allowing for the detection of all possible nucleotide substitutions. The probe with the correct corresponding nucleotide in the middle for each position will give the highest signal intensity after hybridization and scanning. The sequence for each candidate gene was obtained from GenBank or the Human Genome database, then subjected to two programs to remove repetitive sequences: Repeat Masker (Institute for Systems Biology, Seattle, Wash) to identify repeat regions unsuitable for analysis (i.e., short interspersed nuclear elements (SINEs), long interspersed nuclear elements (LINEs), and Alu elements); and Micropeats (Bioinformatics Computational Core Laboratories, Virginia Commonwealth University, Richmond, Va) to mark the repeats (overlap) among the sequences for the same chip design. Before microarray production, polymerase chain reaction (PCR) conditions were optimized to ensure that successful amplification would not be a limiting factor in the experiments.
Experimental procedure of FHChip assay
Individual PCR was performed on 20 ng of DNA to amplify 30 fragments of 226 to 929 bp using 0.5 U AmpliTaq Gold (Applied Biosystems, Foster City, CA), 20 µmol/L deoxynucleotide triphosphates (dNTPs), magnesium concentrations of 2.5 mmol/L MgCl2, and 10 pmol of each primer. Primers were designed using Primer3 (Whitehead Institute, Cambridge, Mass). PCR condition was initiated at 94 °C for 10 min, 40 cycles of repetitive conditions (94 °C for 30 s, 59 °C for 30 s, and 72 °C for 45 s, followed by 72 °C for 10 min. Negative controls, containing all reagents except DNA, were included in each PCR run. PCR products were electrophoresed on a 2% agarose gel to document adequate amplification. An 840bp Affy Taq IQ-EX control sequence was amplified using the primers and template from the CustomseqTM control kit (Affymetrix). To ensure a uniform hybridization signal across the microarray, equimolar amounts of the amplified fragments were purified together using the QIAquick PCR Cleanup Kit (QIAGEN) and then quantified by spectrophotometry. A total of 600 ng of purified PCR products was fragmented using DNase I (0.2 U/μg DNA) at 37 °C, followed by heat inactivation at 95 °C. Aliquots randomly taken from the samples were electrophoresed on a gel to confirm fragment size (15-100 bp).
Microarray hybridization was accomplished according to conditions published by Cutler et al.[13]. The fragmented DNA samples were then labeled using biotinylated N6-dideoxyadenosine triphosphate (Biotin-N6-ddATP) and recombinant terminal deoxynucleotidyl transferase (rTdT) enzyme at 37 °C followed by
FHChip data analysis
Automated base calling was conducted using the Affymetrix GeneChip DNA Analysis Software (GDAS) version 2.0., which employs the ABACUS (Adaptive Background genotype Calling Scheme) algorithm[14]. This algorithm utilizes the hybridization intensities for base calling. It assigns a quality score to each potential base call (A, C, G, T), which is the difference between the log of the best-fitting model and the second-best model[15]. The base calls were directly deposited into a database, which has a user interface “VitaMINE” presenting all the nucleotide differences of the called sequence in comparison with the Reference Sequence obtained from GenBank. The nucleotide differences, as well as those bases that cannot be called by the algorithm (i.e., “no calls”, were evaluated again by users who directly look into the signal intensity plots (i.e., the manual curation process). All the nucleotide differences are then summarized in a report, with annotations and literature references linking to each nucleotide difference (i.e., the mutation) found.
MLPA
MLPA (SALSA MLPA Kit P062B LDLR, MRC Holland) is used to detect the presence of large genomic deletions and duplications of all 18 exons and the promoter of LDLR. MLPA requires the hybridization of two adjacent probes to each exon; these probes are then amplified by PCR. Each probe is attached to a set of universal primers, and one of the oligonucleotides contains a stuffer sequence of variable length that enables separation of the individual fragments according to their length by gel electrophoresis. The area under peak for each fragment was measured with the GeneScan Analysis Software Version 3.1.2 and Genotyper Software Version 2.5 (Applied Biosystems) and exported to Excel sheets for storing and for further processing. The peak area was normalized by dividing it by the combined area of all peaks in that lane. This normalized peak area was then divided by the average normalized peak area from five normal control subjects. With this method, the results are given as allele copy numbers as compared to normal controls, and a ratio of 1 is obtained if both alleles are present, a ratio of 0.5 if one allele is absent, and a ratio of 1.5 if one allele is duplicated.
Statistical analysis
Continuous variables were expressed as mean ± standard deviation and compared using Student’s t-test. Categorical variables were analyzed using the chi-square test. Multivariate logistic regression was performed to assess the association between complex mutations and clinical phenotypes, with age, sex, LDL-C level, premature CHD, and tendon xanthomas as covariates. All novel variants were classified according to the ClinGen Familial Hypercholesterolemia Variant Curation Expert Panel (FH VCEP) specifications of the American College of Medical Genetics and Genomics and Association for Molecular Pathology (ACMG/AMP) guidelines.
Odds ratios (ORs) with 95% confidence intervals (CIs) were calculated. A two-tailed P value < 0.05 was considered statistically significant. All analyses were performed using SAS 9.4 (SAS Institute, Cary, NC).
RESULTS
Mutation spectrum
Among the 66 patients with HeFH, single LDLR mutations were the predominant finding [Table 1]. Several recurrent variants were observed, most notably c.1747C>T (H583Y), c.986G>A (C329Y), and c.1432G>A (G478R), which together accounted for a substantial proportion of cases. In addition, multiple novel mutations were identified, including splicing defects (e.g., IVS10+5G>C, IVS12-1G>C) and frameshift mutations (e.g., c.1726delT, Y576Fs). Overall, pathogenic variants were detected in 76% of patients, comprising LDLR mutations in 68%, APOB in 2%, and PCSK9 in 6% [Table 2]. Within the HeFH cohort, 12 patients (18.2%) carried complex mutations, including seven who were compound heterozygotes or had double mutations on a single allele, and five who harbored large LDLR duplications or deletions. Two additional patients were diagnosed with homozygous FH [Table 3]. A founder effect involving c.1747C>T (p.H583Y) was identified in 15 of 66 LDLR-positive index cases [Figure 1], highlighting the contribution of ethnogenetic clustering to FH prevalence in Taiwan.
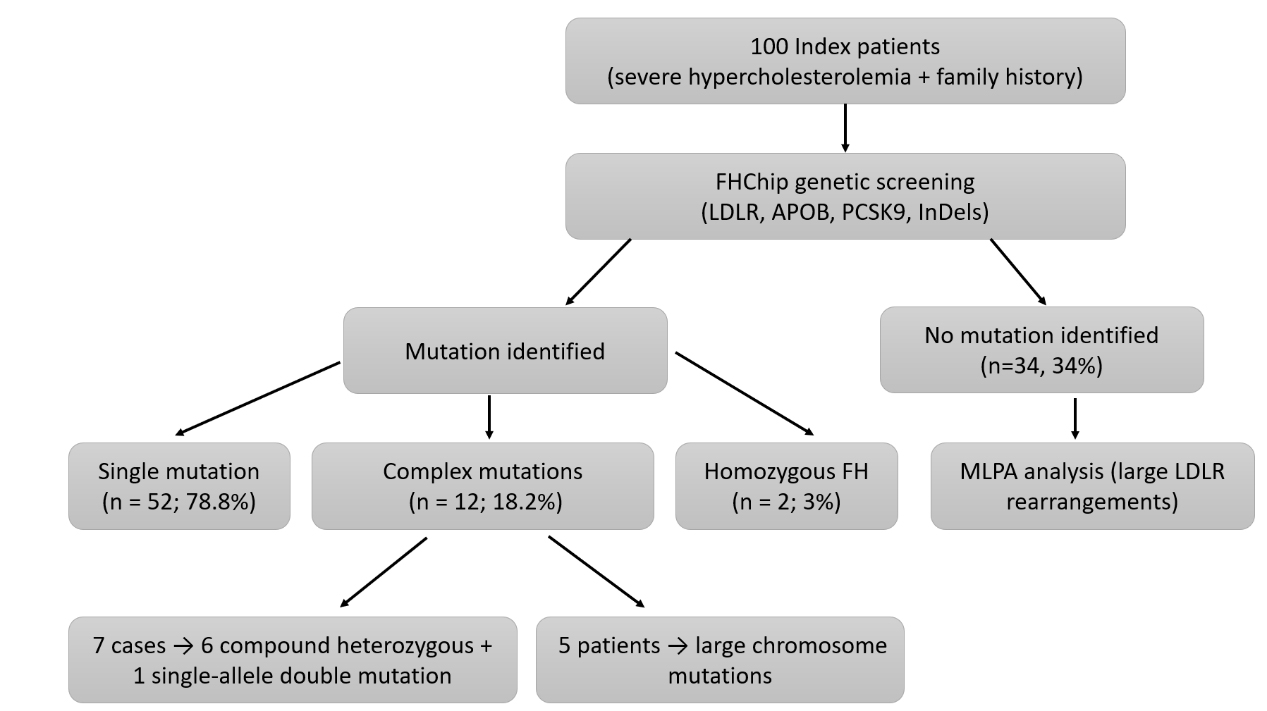
Figure 1. Flow diagram of patient recruitment and genetic testing. A total of 100 index patients with severe hypercholesterolemia were enrolled. All patients underwent initial screening using FHChip targeting LDLR, APOB, PCSK9, and InDel mutations. Cases with negative FHChip results were subsequently analyzed using multiplex ligation-dependent probe amplification (MLPA) to detect large LDLR rearrangements. Pathogenic or likely pathogenic LDLR variants were identified in 66 patients (66%), including 52 with single mutations, 12 with complex mutations, and 2 with homozygous FH. The remaining 34 patients (34%) had no mutation identified after FHChip and MLPA analyses. LDLR: Low-density lipoprotein receptor; APOB: apolipoprotein B; PCSK9: proprotein convertase subtilisin/kexin type 9; InDels: insertions and deletions; FHChip: Familial hypercholesterolemia resequencing microarray; FH: familial hypercholesterolemia. MLPA: multiplex ligation-dependent probe amplification.
Index patients with single mutation of heterozygous familial hypercholesterolemia
| Fm # | Age | Sex | CHO | TG | HDL | LDL | Exon No. | nt change | aa change, (860 a.a.) | Previously reported | ClinGen FH VCEP/ACMG classification/type |
| 6 | 46 | F | 7.71 | 1.06 | 1.89 | 5.33 | LDLR exon 12 | c.1747C>T | H583Y | Y | |
| 10 | 68 | F | 8.69 | 2.39 | 1.27 | 6.34 | LDLR exon10 | c.1432+5G>C | Splicing aberration | Novel | Likely pathogenic/PM2 + PP3/splice region |
| 11 | 61 | F | 8.9 | 1.91 | 1.58 | 5.87 | LDLR exon 13 | c.1953_1954delTA | D651Fs, PTC 667 | Y | |
| 12 | 58 | M | 10.42 | 1.7 | 1.37 | 7.21 | LDLR exon 12 | c.1726delT | Y576Fs, PTC 664 | Novel | Likely Pathogenic/PVS1 + PM2/ frameshift |
| 14 | 31 | F | 11.12 | 0.86 | 1.4 | 8.82 | LDLR exon 10 | c.1434_1448delGCTGGCTGTGGACTG insTCCAGTA | G478Fs, PTC 532 | Novel | Likely Pathogenic/PVS1 + PM2/frameshift |
| 15 | 51 | F | 9.1 | 0.64 | 1.11 | 9.08 | LDLR exon 13 | c.1953_1954delTA | D651Fs, PTC 667 | Y | |
| 19 | 39 | F | 10.78 | 1.47 | 1.19 | 8.92 | LDLR exon 12 | IVS12-1G>C | Splicing aberration | Novel | Pathogenic/PVS1 + PM2/splice |
| 20 | 61 | F | 8.2 | 1.59 | 2.3 | 5.17 | LDLR exon 12 | c.1747C>T | H583Y | Y | |
| 21 | 62 | M | 9 | 0.14 | 1.03 | 8.35 | LDLR exon 14 | c.2054C>T | P685L | Y | |
| 22 | 55 | F | 8.17 | 1.94 | 1.37 | 5.92 | LDLR exon 12 | c.1747C>T | H583Y | Y | |
| 24 | 62 | F | 8.35 | 0.91 | 1.94 | 6 | LDLR exon 09 | c.1246C>T | R416W | Y | |
| 27 | 73 | F | 8.69 | 1.32 | 1.99 | 6.1 | LDLR exon 12 | c.1747C>T | H583Y | Y | |
| 29 | 50 | F | 8.43 | 0.9 | 1.27 | 6.75 | LDLR exon 09 | c.1268T>C | I423T | Y | |
| 30 | 61 | F | 9.18 | 2 | 1.14 | 7.14 | LDLR exon 06 | c.828C>A | C276X | Y | |
| 31 | 22 | M | 9.28 | 0.97 | 1.32 | 7.53 | LDLR exon 07 | c.986G>A | C329Y | Y | |
| 35 | 24 | F | 9.1 | 0.89 | 1.45 | 5.84 | LDLR exon 11 | c.1618G>A | A540T | Y | |
| 39 | 41 | F | 10.4 | 1.33 | 1.47 | 8.04 | LDLR exon 14 | c.2054C>T | P685L | Y | |
| 40 | 51 | F | 10.29 | 1.45 | 1.32 | 8.15 | LDLR exon 07 | c.1016T>C | L339P | Y | |
| 41 | 49 | F | 8.66 | 0.67 | 1.68 | 6.8 | LDLR exon 05 | c.809G>A | C270Y | Y | |
| 42 | 53 | F | 8.61 | 1.24 | 1.29 | 6.75 | LDLR exon 04 | c.516C>G | D172E | Y | |
| 44 | 55 | F | 9.54 | 1.65 | 1.55 | 6.05 | LDLR exon 12 | c.1747C>T | H583Y | Y | |
| 46 | 50 | M | 9 | 0.86 | 1.47 | 6.05 | LDLR exon 12 | c.1747C>T | H583Y | Y | |
| 47 | 55 | F | 8.9 | 1.6 | 1.42 | 6.62 | LDLR exon 12 | c.1747C>T | H583Y | Y | |
| 48 | 57 | F | 9.13 | 2.13 | 1.42 | 6.13 | LDLR exon 12 | c.1747C>T | H583Y | Y | |
| 53 | 71 | M | 11.27 | 1.58 | 1.14 | 9.41 | LDLR exon 09 | c.1241T>G | L414R | Y | |
| 55 | 55 | F | 6.9 | 1.11 | 1.55 | 7.21 | LDLR exon 12 | c.1747C>T | H583Y | Y | |
| 57 | 57 | F | 9.05 | 1.19 | 1.22 | 5.03 | LDLR exon 10 | c.1432G>A | G478R | Y | |
| 58 | 51 | M | 5.66 | 0.84 | 1.11 | 5.4 | LDLR exon 13 | c.1953_1954delTA | D651Fs, PTC 667 | Y | |
| 59 | 36 | M | 7.81 | 1.17 | 1.29 | 9.34 | LDLR exon 05 | c.769C>T | R257W | Y | |
| 62 | 54 | F | 14.3 | 1.83 | 1.84 | 6.39 | LDLR exon 14 | c.2054C>T | P685L | Y | |
| 63 | 58 | F | 10.86 | 0.62 | 2.35 | 4.75 | LDLR exon 04 | c.682G>A | E228K | Y | |
| 65 | 40 | F | 7.47 | 0.58 | 1.74 | 5.38 | LDLR exon 12 | c.1747C>T | H583Y | Y | |
| 68 | 61 | M | 6.98 | 0.47 | 1.32 | 5.28 | LDLR exon 07 | c.986G>A | C329Y | Y | |
| 70 | 56 | M | 8.02 | 2.83 | 0.96 | 5.35 | LDLR exon 12 | c.1747C>T | H583Y | Y | |
| 71 | 55 | F | 9.26 | 1.8 | 1.03 | 6.8 | LDLR exon 10 | c.1432G>A | G478R | Y | |
| 72 | 54 | M | 7.37 | 0.91 | 1.1 | 6.02 | LDLR exon 10 | c.1432G>A | G478R | Y | |
| 73 | 42 | M | 9.7 | 1.3 | 1.66 | 5.53 | LDLR exon 07 | c.986G>A | C329Y | Y | |
| 78 | 47 | M | 8.71 | 1.26 | 1.42 | 5.52 | LDLR exon 12 | c.1747C>T | H583Y | Y | |
| 81 | 51 | M | 9.26 | 1.96 | 1.09 | 7.91 | LDLR exon 12 | c.1747C>T | H583Y | Y | |
| 82 | 34 | M | 8.28 | 1.35 | 1.03 | 6.47 | LDLR exon 10 | c.1474G>A | D492N | Y | |
| 85 | 47 | M | 7.21 | 1.56 | 0.88 | 5.87 | LDLR exon 10 | c.1567G>A | V523M | Y | |
| 86 | 50 | M | 10.91 | 0.96 | 1.01 | 8.33 | LDLR exon 10 | c.1474G>A | D492N | Y | |
| 90 | 32 | M | 8.9 | 0.86 | 0.67 | 7.84 | LDLR exon 07 | c.986G>A | C329Y | Y | |
| 92 | 50 | F | 10.27 | 0.49 | 1.5 | 8.53 | LDLR exon 13 | 1953_1954delTA | D651Fs, PTC 667 | Y | |
| 93 | 61 | F | 10.09 | 0.85 | 1.29 | 8.02 | LDLR exon 04 | IVS4+2 T>C | Splicing aberration | Y | |
| 94 | 50 | F | 9.26 | 0.62 | 1.57 | 6.85 | LDLR exon 04 | c.523G>A | D175N | Y | |
| 95 | 59 | F | 11.9 | 1.03 | 2.11 | 8.95 | LDLR exon 07 | c.1016 T>C | L339P | Y | |
| 96 | 56 | F | 12.34 | 0.81 | 2.38 | 8.22 | LDLR exon 10 | c.1432 G>A | G478R | Y | |
| 97 | 28 | F | 10.16 | 0.79 | 2.49 | 6.13 | LDLR exon 10 | c.1432 G>A | G478R | Y | |
| 98 | 25 | F | 10.68 | 1.1 | 2.05 | 7.14 | LDLR exon 10 | c.1448 G>A | W483X | Y | |
| 99 | 51 | F | 12.21 | 3.27 | 2.1 | 8.66 | LDLR exon 07 | c.1027G>A | G343S | Y | |
| 100 | 52 | F | 9.41 | 2.83 | 1.11 | 7.42 | LDLR exon 07 | c.986G>A | C329Y | Y |
Twelve index patients with complex mutations of heterozygous familial hypercholesterolemia
| Age/Sex | CHO | TG | HDL | LDL | CAD | Xan | Exon No. | nt change | aa change | Previously reported | Medication | TxCHO | TxLDL |
| a21/M | 16.37 | 0.69 | 1.40 | 11.77 | No | Yes | LDLR exon 03 LDLR exon 07 | c.268G>A c.986G>A | D90N C329Y | Y Y | Lipitor 40 mg QD Ezetrol 10 mg QD | 5.46 | 3.85 |
| c51/M | 13.24 | 1.94 | 1.11 | 11.56 | No | Yes | LDLR exon 02-06 | Dup | Nov | Olbetam 250 mg QD Ezetrol 15 mg QD Crestor 10 mg BID | 5.90 | 4.24 | |
| a11/M | 16.91 | 0.89 | 2.25 | 13.06 | No | Yes | LDLR exon 03 LDLR exon 06 | c.268G>A c.880A>T | D90N K294X | Y N | Lipitor 40 mg QD Ezetrol 10 mg QD | 6.18 | 4.55 |
| c31/M | 13.03 | 1.08 | 1.42 | 10.71 | No | Yes | LDLR exon 02-06 | Dup | Nov | Lipitor 20 mg QD Ezetrol 10 mg QD | 6.10 | 4.42 | |
| b62/M | 8.87 | 1.60 | 2.02 | 6.13 | Yes | No | LDLR exon 08 LDLR exon10 | c.1084G>A c.1432G>A | D362N G478R | Y Y | Crestor 20 mg QD | 4.58 | 2.69 |
| a49/M | 10.68 | 0.95 | 0.98 | 9.26 | Yes | No | LDLR exon 05 LDLR exon 12 LDLR exon 13 | c.769C>T c.1765G>A c.1879G>A | R257W D589N A627T | Y Y Y | Crestor 10 mg QD Ezetrol 10 mg QD | 4.60 | 3.32 |
| a76/F | 13.45 | 0.45 | 2.20 | 11.04 | Yes | Yes | LDLR exon 05 LDLR exon 12 | c.769C>T c.1765G>A | R257W D589N | Y | Ezetrol 10 mg QD Crestor 20 mg QD | 4.22 | 1.34 |
| c65/F | 10.68 | 0.98 | 1.42 | 7.09 | No | Yes | LDLR exon 13.14 | Dup | Nov | Lipitor 20 mg QD | 4.84 | 2.79 | |
| a54/M | 12.05 | 8.74 | 1.27 | 6.96 | Yes | Yes | LDLR exon 05 LDLR exon 12 | c.769C>T c.1765G>A | R257W D589N | Y Y | Ezetrol 10 mg QD Lipanthyl 160 mg QD Crestor 10 mg QD | 4.09 | 1.81 |
| c51/F | 9.36 | 0.79 | 1.63 | 7.29 | No | Yes | LDLR exon 02-06 | Dup | Nov | Lipitor 40 mg QD Ezetrol 20 mg QD | 6.78 | 5.09 | |
| c57/M | 9.83 | 1.23 | 0.98 | 8.15 | Yes | No | LDLR exon 06.07.08 | Dup | Nov | Crestor 20 mg HS | 6.75 | 5.25 | |
| a19/M | 11.25 | 0.49 | 1.34 | 8.97 | No | No | LDLR exon 07 LDLR exon 12 | c.1048C>T c.1747C>T | R350X H583Y | Y | Ezetrol 10 mg QD Crestor 10 mg Hs | 6.05 | 3.93 |
Patients with true homozygous familial hypercholesterolemia
| No. | TCHO | TG | HDL | LDL | Exon No. | nt change | aa change | Previously reported |
| 36 | 13.03 | 2.68 | 0.85 | 8.69 | LDLR exon 09 | c.1246C>T | R416W | Y |
| 49 | 21.15 | 0.77 | 1.34 | 19.27 | LDLR exon 15 | c.2205_2206insTT | V736Fs, PTC737 | Novel |
Clinical characteristics of complex vs. single mutations
Patients with complex mutations (n = 23) displayed a significantly more severe lipid and clinical phenotype compared with those carrying single mutations (n = 185). Mean LDL-C was markedly higher in the complex mutation group (319 ± 100 vs. 210 ± 71 mg/dL, P < 0.001), as was total cholesterol (415 ± 115 vs.
Comparisons of lipid profiles and cardiovascular characteristics between single mutation and complex mutations of heterozygous familial hypercholesterolemia
| Complex mutations | Single mutations | Effect size | P-value | |
| Characteristics | n = 23 | n = 185 | (95%CI) | |
| Cholesterol, mg/dL | 414.74 ± 114.55 | 303.72 ± 74.21 | 111.02 (60.49-161.55) | 0.0001 |
| Triglycerides, mg/dL | 135.87 ± 152.54 | 115.21 ± 66.76 | 20.66 (-45.28-86.60) | 0.5272 |
| HDL-C, mg/dL | 54.82 ± 14.99 | 57.57 ± 21.05 | -2.75 (-10.75-5.25) | 0.5526 |
| LDL-C, mg/dL | 319.18 ± 99.54 | 210.00 ± 71.44 | 109.18 (65.08-153.28) | < 0.0001 |
| Age, years | 43.35 ± 18.37 | 42.48 ± 16.62 | 0.87 (-6.72-8.46) | 0.8160 |
| Male, % | 69.57 | 41.08 | 3.28 (1.29-8.34) | 0.0095 |
| Body mass index, kg/m2 | 21.73 ± 2.60 | 22.86 ± 4.14 | -1.13 (-2.23- -0.03) | 0.0792 |
| Waist circumference, cm | 74.91 ± 10.29 | 75.43 ± 11.93 | -0.52 (-5.13-4.09) | 0.8443 |
| Hypertension, % | 26.09 | 16.76 | 1.75 (0.64-4.80) | 0.2585 |
| Systolic blood pressure, mmHg | 110.80 ± 15.91 | 114.63 ± 17.86 | -3.83 (-11.36-3.70) | 0.3602 |
| Diastolic blood pressure, mmHg | 67.57 ± 12.47 | 71.25 ± 10.80 | -3.68 (-9.17-1.81) | 0.1570 |
| Diabetes mellitus, % | 8.70 | 4.32 | 2.11 (0.42-10.60) | 0.3049 |
| Fasting glucose, mg/dL | 97.91 ± 28.78 | 92.92 ± 20.87 | 4.99 (-7.68-17.66) | 0.4380 |
| Current smoking habit, % | 26.09 | 11.35 | 2.75 (0.98-7.73) | 0.0905 |
| Current alcohol habit, % | 8.70 | 8.70 | 1.01 (0.22-4.66) | 1.0000 |
| Coronary heart diseases, % | 34.78 | 6.52 | 7.69 (2.72-21.71) | 0.0004 |
| Xanthoma, % | 69.56 | 1.08 | 209.14 (40.1-1093.0) | <0.0001 |
Predictors of complex mutations
Multivariate logistic regression identified total cholesterol ≥ 361 mg/dL (OR 9.87, 95%CI 3.23-30.14), LDL-C ≥ 237 mg/dL (OR 10.82, 95%CI 2.70-43.40), male sex (OR 3.28, 95%CI 1.29-8.35), premature CHD (OR 5.12, 95%CI 1.34-19.55), and tendon xanthomas (OR 16.15, 95%CI 2.84-91.78) as independent predictors of complex mutations [Table 5 and Figure 2]. Tendon xanthomas showed the strongest association.
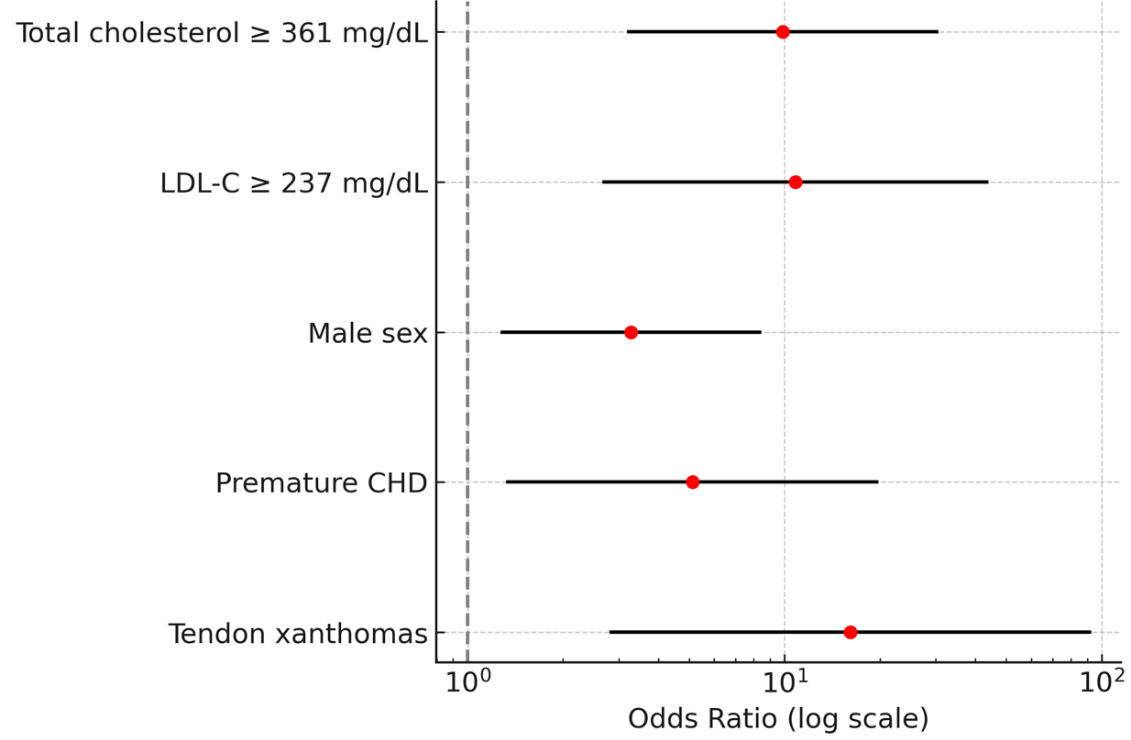
Figure 2. Forest plot of predictors of complex mutations in HeFH. Odds ratios (ORs) with 95% confidence intervals (CIs) from multivariate logistic regression are shown on a logarithmic scale. Tendon xanthomas showed the strongest association with complex mutations (OR 16.15, 95%CI 2.84-91.78). HeFH: Heterozygous familial hypercholesterolemia; LDL-C: low-density lipoprotein cholesterol; CHD: coronary heart disease.
Multivariate logistic regression analysis for the odds ratios of complex mutations in heterozygous familial hypercholesterolemia
| Univariate | Multivariate (Model 1) | Multivariate (Model 2) | Multivariate (Stepwise) | |
| Cholesterol 361 mg/dL | 11.36 (4.31-29.91)‡ | 12.92 (3.76-44.40)‡ | - | 9.87 (3.23-30.14)‡ |
| LDL-C 237 mg/dL | 14.66 (4.16-51.61)‡ | - | 10.82 (2.70-43.40)‡ | - |
| Coronary heart disease | 7.64 (2.71-21.60)‡ | 3.74 (0.81-17.19) | 3.18 (0.68-14.90) | 5.12 (1.34-19.55)* |
| Smoking habit | 2.76 (0.98-7.77) | 2.07 (0.43-10.05) | 1.16 (0.23-5.94) | - |
| Male | 3.28 (1.29-8.35)* | 2.44 (0.61-9.80) | 2.10 (0.54-8.15) | - |
| Hypertension | 1.75 (0.64-4.80) | 4.42 (1.02-19.08)* | 4.85 (1.12-20.96)* | - |
| Diabetes mellitus | 2.11 (0.42-10.59) | 3.35 (0.47-23.68) | 1.89 (0.25-14.16) | - |
| Age, years | 1.00 (0.98-1.03) | 0.99 (0.95-1.03) | 0.99 (0.95-1.03) | - |
| Cutaneous xanthoma | 48.08 (9.50-250.68)‡ | 18.21 (2.68-123.69)‡ | 17.61 (2.68-115.50)‡ | 16.15 (2.84-91.78)‡ |
Treatment response
Among index patients with complex mutations (n = 12), intensive lipid-lowering therapy (statin plus ezetimibe additional agents) produced a 53.2% reduction in total cholesterol (from 12.14 mmol/L to
DISCUSSION
This study underscores the importance of early diagnosis and treatment of FH, with particular emphasis on the clinical and genetic features of complex mutations in HeFH in Taiwan. We demonstrated that patients with severe hypercholesterolemia (total cholesterol ≥ 361 mg/dL or LDL-C ≥ 237 mg/dL), especially those presenting with tendon or skin xanthomas and premature CHD, should be strongly suspected of harboring complex LDLR mutations. These include compound heterozygous, single-allele double mutations, homozygous mutations, and large fragment rearrangements. Notably, large duplications such as LDLR exons 2-6 were confirmed in three index cases using MLPA, highlighting its diagnostic value when conventional sequencing fails.
Clinically, patients with complex mutations exhibited significantly higher LDL-C levels and a greater prevalence of premature CHD and tendon xanthomas compared with those carrying single point mutations, confirming a strong genotype-phenotype correlation. This observation aligns with findings from the Dutch FH Screening Program, the UK Simon Broome Register[16,17], as well as studies from Japan[18] and Spain[18,19], all of which demonstrate that mutation type influences phenotypic severity and cardiovascular risk[20,21].
From a diagnostic standpoint, our results emphasize the utility of a two-step genetic testing strategy combining FHChip and MLPA, which substantially improved the detection rate of pathogenic variants. Recent advances in next-generation sequencing (NGS) have further refined the genetic diagnosis of FH, enabling detection of rare, intronic, and regulatory variants not captured by targeted platforms. The ClinGen Familial Hypercholesterolemia Variant Curation Expert Panel has also provided standardized criteria for variant classification, improving consistency and clinical interpretation of LDLR variants[22]. Emerging evidence suggests that a tiered diagnostic strategy - initial targeted screening followed by NGS in mutation-negative or clinically discordant cases - may optimize both cost-effectiveness and diagnostic sensitivity in FH populations[23]. This approach is consistent with international guidelines European Atherosclerosis Society (EAS) and National Lipid Association (NLA) that recommend MLPA for mutation-negative FH patients following sequencing. Along with our previous identification of a novel splice-site mutation (c.1186+2T>G) with founder effect in LDLR, these findings highlight the influence of family migration and ancestral origins on FH genetics in Taiwan[11]. In Taiwan - a region with high FH prevalence and potential founder effects - such strategies can significantly enhance early diagnosis, facilitate cascade screening, and enable timely initiation of aggressive lipid-lowering therapy[24,25]. Cascade screening also proved highly cost-effective in our cohort, echoing international evidence that early identification and treatment of affected relatives reduces cardiovascular morbidity and mortality.
Therapeutically, even in patients with complex mutations and markedly elevated baseline LDL-C, aggressive lipid-lowering therapy - particularly statin-ezetimibe combination - achieved substantial reductions in LDL-C levels. Given that over 98% of severe hypercholesterolemia patients in this study were HeFH[26], these findings highlight the effectiveness of intensive therapy and the opportunity for significant population-level CHD risk reduction[27,28]. The availability of orphan drugs such as PCSK9 inhibitors, lomitapide, and evinacumab further expands therapeutic options for high-risk patients with complex mutations. In particular, angiopoietin-like protein 3 (ANGPTL3) inhibition has shown significant LDL-C reduction in severe hypercholesterolemia[29].
From an ethnogenetic perspective, we identified a founder effect in the LDLR c.1747C>T (H583Y) mutation, which was clustered in 15 Minnan families in Taipei, despite different surnames. Along with our previous discovery of a novel splice-site variant (c.1186+2T>G) with founder effect[30], this finding reflects the impact of ancestral migration on FH genetics in Taiwan[31]. Similar founder mutations have been reported in other populations, such as the French Canadians[32] and South African Afrikaners, further supporting the role of population-specific mutations in FH epidemiology. No significant differences in LDL-C levels or CHD prevalence were observed between carriers of c.1747C>T (p.H583Y) and other LDLR mutation carriers, suggesting that the founder mutation confers a phenotype comparable in severity to other pathogenic LDLR variants. These data reinforce the need to consider ethnogenetic factors when designing genetic screening programs. In addition, growing evidence indicates that a subset of patients with a clinical FH phenotype but negative monogenic testing may harbor polygenic risk variants that contribute to elevated LDL-C levels. Incorporation of polygenic risk scores alongside monogenic analysis may further refine risk stratification and clinical management in such cases[33].
Limitations
This study has several limitations. First, the relatively modest sample size and single-center design may limit the generalizability of our findings. In particular, the relatively small number of patients with complex mutations may result in less stable effect estimates and wider confidence intervals, and these findings should therefore be interpreted with caution. Second, although the two-step genetic testing strategy combining FHChip and MLPA improved diagnostic yield and remains widely used in clinical FH screening, the probe-based design of FHChip restricts variant detection to predefined genomic regions. Compared with NGS, it offers lower genomic coverage and reduced sensitivity for detecting novel, intronic, regulatory, or rare structural variants. Consequently, certain pathogenic variants may have been missed. Third, the absence of parallel NGS validation limits comprehensive assessment of the full mutational spectrum and potential polygenic contributions to LDL-C levels. Finally, the cross-sectional nature of the study precludes evaluation of long-term cardiovascular outcomes and treatment durability.
As genetic diagnostics continue to advance, future studies incorporating NGS-based platforms - particularly whole-exome or whole-genome sequencing in mutation-negative cases - together with functional validation and polygenic risk assessment, will be essential to further elucidate the genetic architecture of FH and refine precision therapeutic strategies.
Conclusion
In conclusion, this study provides the first comprehensive molecular characterization of complex LDLR mutations in Taiwanese patients with HeFH. We demonstrated a strong genotype-phenotype correlation, with complex mutations associated with markedly elevated LDL-C levels, premature CHD, and tendon xanthomas. A two-step genetic testing strategy (FHChip plus MLPA) significantly improved the diagnostic yield, underscoring its value particularly in mutation-negative cases by conventional sequencing. These findings highlight the importance of population-specific genetic strategies and early therapeutic intervention - including the potential use of orphan drugs - to reduce the burden of premature CHD in this rare dyslipidemia.
DECLARATIONS
Authors’ contributions
Performed data analysis, contributed to the conception and design of the study, interpreted the data, and wrote the manuscript: Su TC
Contributed to the supervision of the study and made revisions: Liau CS
Critically revised the manuscript and approved the final version: Chien KL, Lin PC, Lee YT
Availability of data and materials
The datasets generated and analyzed during the current study are not publicly available due to patient privacy restrictions but are available from the corresponding author upon reasonable request.
AI and AI-assisted tools statement
During the preparation of this manuscript, the AI tool ChatGPT (version OpenAI, GPT-5.3, released 2026-03-03) was used solely for generating the Graphical Abstract. The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship
This study was supported by the National Science and Technology Council (NSTC 108-2314-B-002-204-MY2).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
The study was approved by the Research Ethics Committee of NTUH (IRB No. 201903123RINC). Written informed consent was obtained from all participants.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Tu YK, Tseng CD, Hsiao CK, et al. Evidence for improved control of hypertension in Taiwan: 1993-2002. J Hypertens. 2008;26:600-6.
2. Chen YJ, Chen IC, Chen YM, et al. Prevalence of genetically defined familial hypercholesterolemia and the impact on acute myocardial infarction in Taiwanese population: a hospital-based study. Front Cardiovasc Med. 2022;9:994662.
3. Nordestgaard BG, Chapman MJ, Humphries SE, et al.; European Atherosclerosis Society Consensus Panel. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013;34:3478-90a.
4. Sniderman AD, Tsimikas S, Fazio S. The severe hypercholesterolemia phenotype: clinical diagnosis, management, and emerging therapies. J Am Coll Cardiol. 2014;63:1935-47.
5. Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232:34-47.
6. Brautbar A, Ballantyne CM. Pharmacological strategies for lowering LDL cholesterol: statins and beyond. Nat Rev Cardiol. 2011;8:253-65.
7. Ito MK, McGowan MP, Moriarty PM. Management of familial hypercholesterolemias in adult patients: recommendations from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011;5:S38-45.
8. Koivisto UM, Hämäläinen L, Taskinen MR, Kettunen K, Kontula K. Prevalence of familial hypercholesterolemia among young north Karelian patients with coronary heart disease: a study based on diagnosis by polymerase chain reaction. J Lipid Res. 1993;34:269-77. Available from: https://www.sciencedirect.com/science/article/pii/S0022227520407540?via%3Dihub [Last accessed on 27 May 2026].
9. Goldberg AC, Hopkins PN, Toth PP, et al. Familial hypercholesterolemia: screening, diagnosis and management of pediatric and adult patients: clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011;5:S1-8.
10. Hinchcliffe M, Le H, Fimmel A, et al. Diagnostic validation of a familial hypercholesterolaemia cohort provides a model for using targeted next generation DNA sequencing in the clinical setting. Pathology. 2014;46:60-8.
11. Hsiung YC, Lin PC, Chen CS, et al. Identification of a novel LDLR disease-causing variant using capture-based next-generation sequencing screening of familial hypercholesterolemia patients in Taiwan. Atherosclerosis. 2018;277:440-7.
12. Chiou KR, Charng MJ, Chang HM. Array-based resequencing for mutations causing familial hypercholesterolemia. Atherosclerosis. 2011;216:383-9.
13. Cutler DJ, Zwick ME, Carrasquillo MM, et al. High-throughput variation detection and genotyping using microarrays. Genome Res. 2001;11:1913-25.
14. Yang KC, Su YN, Shew JY, et al. LDLR and ApoB are major genetic causes of autosomal dominant hypercholesterolemia in a Taiwanese population. J Formos Med Assoc. 2007;106:799-807.
15. Ademi Z, Watts GF, Pang J, et al. Cascade screening based on genetic testing is cost-effective: evidence for the implementation of models of care for familial hypercholesterolemia. J Clin Lipidol. 2014;8:390-400.
16. Slack J. Risks of ischaemic heart-disease in familial hyperlipoproteinaemic states. Lancet. 1969;2:1380-2.
17. Yoshida A, Morisaki H, Nakaji M, et al. Genetic mutation analysis in Japanese patients with non-syndromic congenital heart disease. J Hum Genet. 2016;61:157-62.
18. Sacramento-Pacheco J, Sánchez-Gómez MB, Gómez-Salgado J, Novo-Muñoz MM, Duarte-Clíments G. Prevalence of cardiovascular risk factors in Spain: a systematic review. J Clin Med. 2023;12:6944.
19. Guillén M, Corella D, Portolés O, González JI, Mulet F, Sáiz C. Prevalence of the methylenetetrahydrofolate reductase 677C > T mutation in the Mediterranean Spanish population. Association with cardiovascular risk factors. Eur J Epidemiol. 2001;17:255-61.
20. Kerr M, Pears R, Miedzybrodzka Z, et al. Cost effectiveness of cascade testing for familial hypercholesterolaemia, based on data from familial hypercholesterolaemia services in the UK. Eur Heart J. 2017;38:1832-9.
21. Chora JR, Iacocca MA, Tichý L, et al. The clinical genome resource (ClinGen) familial hypercholesterolemia variant curation expert panel consensus guidelines for LDLR variant classification. Genet Med. 2022;24:293-306.
22. Reeskamp LF, Tromp TR, Defesche JC, et al. Next-generation sequencing to confirm clinical familial hypercholesterolemia. Eur J Prev Cardiol. 2021;28:875-83.
23. Alonso R, Perez de Isla L, Muñiz-Grijalvo O, Diaz-Diaz JL, Mata P. Familial hypercholesterolaemia diagnosis and management. Eur Cardiol. 2018;13:14-20.
24. Ned RM, Sijbrands EJ. Cascade screening for familial hypercholesterolemia (FH). PLoS Curr. 2011;3:RRN1238.
25. Pisciotta L, Fasano T, Bellocchio A, et al. Effect of ezetimibe coadministered with statins in genotype-confirmed heterozygous FH patients. Atherosclerosis. 2007;194:e116-22.
26. Vavlukis M, Vavlukis A. Adding ezetimibe to statin therapy: latest evidence and clinical implications. Drugs Context. 2018;7:212534.
27. Zhan S, Tang M, Liu F, Xia P, Shu M, Wu X. Ezetimibe for the prevention of cardiovascular disease and all-cause mortality events. Cochrane Database Syst Rev. 2018;11:CD012502.
28. Raal FJ, Rosenson RS, Reeskamp LF, et al. Evinacumab for homozygous familial hypercholesterolemia. N Engl J Med. 2020;383:711-20.
29. Chiou KR, Charng MJ. Detection of mutations and large rearrangements of the low-density lipoprotein receptor gene in Taiwanese patients with familial hypercholesterolemia. Am J Cardiol. 2010;105:1752-8.
30. Huang CC, Niu DM, Charng MJ. Genetic analysis in a Taiwanese cohort of 750 index patients with clinically diagnosed familial hypercholesterolemia. J Atheroscler Thromb. 2022;29:639-53.
31. Davignon J, Roy M. Familial hypercholesterolemia in French-Canadians: taking advantage of the presence of a “founder effect”. Am J Cardiol. 1993;72:6D-10D.
32. D'Erasmo L, Minicocci I, Di Costanzo A, et al. Clinical Implications of monogenic versus polygenic hypercholesterolemia: long-term response to treatment, coronary atherosclerosis burden, and cardiovascular events. J Am Heart Assoc. 2021;10:e018932.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].