


Plexiform neurofibromas in neurofibromatosis type 1: current and emergent therapeutic strategies
 0
0 Abstract
Neurofibromatosis type 1-associated plexiform neurofibromas cause significant morbidity and carry a risk of malignant transformation. Early targeted agents failed to demonstrate efficacy, safety, or durable responses until the discovery of mitogen-activated protein kinase kinase (MEK 1/2) inhibitors. Selumetinib and mirdametinib are approved medical therapies that can potentially reduce tumor volume and symptoms, improve patient-reported outcomes, and have manageable toxicities. An indirect comparison between selumetinib and mirdametinib suggests differences in efficacy and safety, but direct confirmatory head-to-head trials are needed. Active clinical trials in the pipeline are exploring other targets in the MEK pathway, along with combination therapies. Key priorities include the impact of malignant peripheral nerve sheath tumor risk, defining long-term safety, durability off therapy, predictors of response and resistance, and implementation of multidisciplinary care.
Keywords
INTRODUCTION
Neurofibromatosis type 1 (NF1) is a rare neurogenetic disorder that affects approximately 1 in 2,500 individuals worldwide[1]. This genetic defect causes a wide range of clinical manifestations, including, but not limited to, nervous system tumors, skeletal manifestations, vasculopathies, seizures, headaches, and learning disabilities. One hallmark manifestation of NF1 is the development of plexiform neurofibromas (PNs), which occurs in 30%-60% of individuals with NF1[1,2]. Although benign, PNs can cause substantial morbidity due to pain, motor or sensory impairment, disfigurement, and compression of vital anatomical structures including the spinal cord, airway, or major blood vessels. More importantly, 8%-13% of the PNs may transform into malignant peripheral nerve sheath tumors (MPNSTs), a type of aggressive sarcoma associated with high morbidity and mortality rates[1]. Historically, treatment options have been limited to surgical resection, which carries significant risk in many patients because of the location of the tumors. Recent targeted therapies have introduced a new era of pharmacologic management.
DRUGS THAT WERE PREVIOUSLY TRIALED
Developing effective treatments for NF1-associated PNs has remained an ongoing challenge[3]. Although several targeted agents showed preclinical or early-phase promise, most failed to demonstrate sufficient efficacy or acceptable safety to support regulatory approval, reflecting the complexity of PN biology and underscoring the limitations inherent to dysregulated Ras signaling[3].
For example, sirolimus, a mechanistic target of rapamycin (mTOR) inhibitor, was evaluated in a Phase II trial for patients with progressive PNs and showed no significant clinical benefit or tumor shrinkage
Summary of drugs previously trialed, FDA-approved, and currently in the pipeline. Partial response for all trials was defined as ≥ 20% reduction
| Drug name | Mechanism of action | Trial phase | Outcome | Refs. |
| Sirolimus | mTOR inhibitor | Phase II, completed | No significant tumor shrinkage or clinical benefit | [4] |
| Imatinib | Tyrosine kinase inhibitor | Phase II, completed | Partial response (PR) in 17% of intent-to-treat, and 26% in patients treated for ≥ 6 months; 30% subjective improvement in symptoms and function | [5] |
| Pirfenidone | Antifibrotic, anti-inflammatory, antioxidant | Phase II, completed | No objective responses (no tumor volume reduction or improvement in QoL) | [6] |
| Tipifarnib | Farnesyltransferase inhibitor | Phase II, completed | Improvement in QoL but no significant change in time to progression of tumor volume | [7] |
| Sunitinib | Multi-targeted tyrosine kinase inhibitor | Phase II, terminated | Trial terminated because of death of uncertain cause but possibly related to drug | [8] |
| Selumetinib | MEK 1/2 inhibitor | FDA approved (pediatrics and adults) | 70% of the patients achieved PR; 48% improvement in child-reported health-related QoL 20% objective response rate; meaningful pain intensity reduction | [9,12-14] |
| Mirdametinib | MEK 1/2 inhibitor | FDA approved (pediatrics and adults) | 52% PR in pediatrics, 41% PR in adults, defined as ≥ 20% reduction in tumor volume | [10,15] |
| Binimetinib | MEK 1/2 inhibitor | Phase II, completed | 74% of the patients achieved PR | [19,25] |
| Cabozantinib | Receptor tyrosine kinase inhibitor (VEGF, MET, TAM, RET) | Phase II, completed | 42% (8/19) in adolescents and adults; No significant change in global QoL scores; 3-point decrease in worst tumor-pain in the PR group | [21] |
| Cabozantinib + Selumetinib | Combination therapy with receptor tyrosine kinase inhibitor and MEK 1/2 inhibitor | Phase I, Ib, 2; Not yet recruiting | No data yet but assessing the synergistic effects of cabozantinib with selumetinib | [22] |
| Healx | Mitochondrial modulation, multi-pathway | Phase 2, recruiting | No data yet | [20] |
Other pathway-directed strategies also proved unsuccessful. Tipifarnib, a farnesyltransferase inhibitor, did not significantly prolong time to progression of PNs; however, it was well tolerated and was associated with some improvements in quality of life, suggesting a potential in supportive care[7]. Sunitinib, a multi-targeted tyrosine kinase inhibitor, was terminated early due to a patient death of uncertain cause with no meaningful response observed[8]. Given limited efficacy, safety concerns, and non-durable response, earlier agents were not adopted into practice until the recent success of mitogen-activated protein kinase kinase (MEK 1/2) inhibitors.
APPROVED MEK INHIBITORS
The emergence of MEK1/2 inhibitors, selumetinib and mirdametinib, has led to Food and Drug Administration (FDA)-approved therapies for patients with symptomatic, inoperable PNs [Table 1][9,10]. MEK inhibitors are allosteric inhibitors of MEK1 and MEK2. MEK1/2 function within the mitogen-activated protein kinase (MAPK) signaling cascade, referred to as the Rat sarcoma (RAS)-rapidly accelerated fibrosarcoma (RAF)-mitogen-activated protein kinase kinase (MEK)-extracellular signal-regulated kinase (ERK) pathway, which regulates cellular growth, differentiation, proliferation, and survival [Figure 1][9,11]. In NF1, Ras signaling becomes dysregulated due to loss or reduced activity of neurofibromin, leading to prolonged and uncontrolled downstream MAPK activation and growth of peripheral nerve sheath tumors in some individuals. By inhibiting MEK1/2, selumetinib and mirdametinib prevent downstream activation of ERK, thereby suppressing cell proliferation and angiogenesis and producing tumor shrinkage and disease stabilization[9,11].
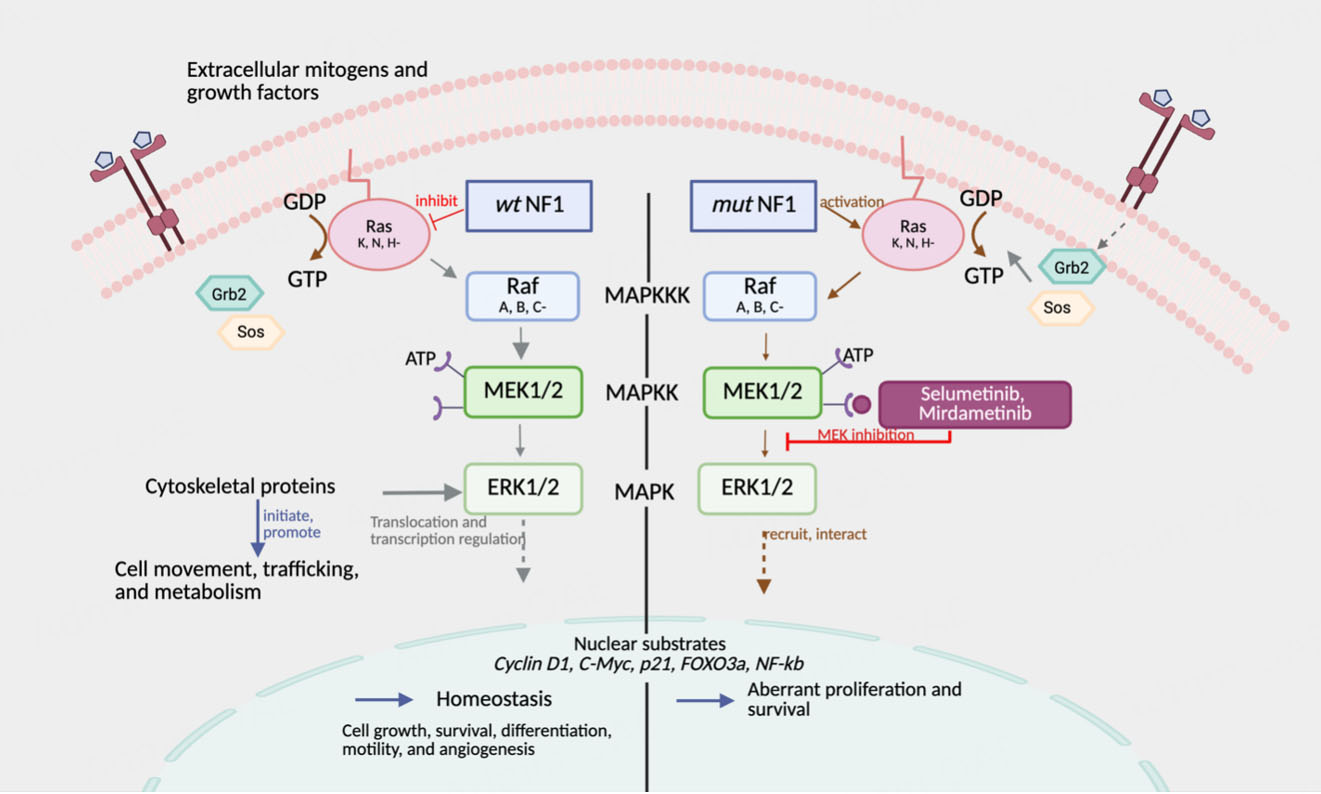
Figure 1. Mechanism of MEK inhibition by selumetinib and mirdametinib. Loss of NF1 function leads to overactive Ras signaling, promoting tumor growth. Created in BioRender. Kim, H. (2025) https://BioRender.com/796ondy. NF1: Neurofibromatosis type 1; MEK 1/2: mitogen-activated protein kinase kinase; ERK: extracellular signal-regulated kinase.
Selumetinib (Koselugo), approved on April 10, 2020, was the first FDA-approved therapy for PNs for pediatric patients ≥ 2 years with symptomatic, inoperable PNs [Table 1][9]. The approval was based on results from the Phase II SPRINT trial (NCT01362803), in which 70% of the patients achieved partial response, defined as a ≥ 20% reduction in tumor volume on volumetric magnetic resonance imaging (MRI), with the median tumor volume-reduction of 27.9%[9,12]. Notably, 56% of participants had a durable response lasting
Mirdametinib (Gomekli) was recently approved on February 11, 2025, for adults and pediatric patients
Tolerability and safety considerations
Both drugs were generally well tolerated, but adverse events (AEs) are common[9,12,15,16]. Common AEs include gastrointestinal upset, xerosis, fatigue, muscle pain, acne, stomatitis, paronychia, and headache. Most were Grade 1-2 with the use of NCI common terminology criteria for adverse events (CTCAE) v 4/5
Most common side effects of mirdametinib and selumetinib[26]
| Side effect | Selumetinib | Mirdametinib |
| Cardiac (decreased ejection fraction/shortening fraction) | 38% (Grade 1-2); 2% (Grade 3) | Not reported to date |
| Diarrhea | 54% (Grade 1-2); 4% (Grade 3) | Not reported to date |
| Fatigue | 56% (Grade 1-2) | 26% (Grade ≥ 2) |
| Nausea/vomiting | 44% (Grade 1-2) | 21% (Grade ≥ 2) |
| Ophthalmologic | No dose limiting toxicity | No dose limiting toxicity |
| Paronychia | 38% (Grade 1-2); 6% (Grade 3) | Not reported to date |
| Rash/skin toxicity | 52%-58% (Grade 1-2); 4%-10% (Grade 3) | 53% (Grade ≥ 2) |
Comparative effectiveness and implications
In an indirect treatment comparison, using matching-adjusted indirect comparison (MAIC) and simulated treatment comparison (STC), mirdametinib demonstrated a significantly greater mean best reduction in tumor volume compared to selumetinib (MAIC: -36.0% vs. -22.8%; STC: -36.2% vs. -22.8%)[17].
Dose reductions due to treatment-related adverse events (TRAEs) were significantly lower in mirdametinib (MAIC: 12% vs. 28%, odds ratio [OR] = 0.355, P = 0.048; STC: 11% vs. 28%, OR = 0.309, P = 0.028). Several common TRAEs, including acneiform rash, diarrhea, nausea, vomiting, fatigue, stomatitis, and elevated creatine phosphokinase, occurred less frequently. No significant differences in ORR or selected safety outcomes (paronychia, alopecia, neutropenia, asymptomatic left ventricular ejection fraction (LVEF) decline) were observed[17]. Since the drugs were not tested in the same study and patient differences may bias results, the findings should be validated in head-to-head trials and could be strengthened through biomarker-informed studies.
Long-term considerations
Longer-term tolerability, resistance, and toxicity of MEK inhibitors have not been well studied. Emerging follow-up data on selumetinib demonstrate that only four participants (5.4%) experienced > 20% of tumor growth over seven years, with sustained improvements in PN-related pain intensity and reduced pain interference[18]. No new safety signals were identified during long-term follow-up; however, some known toxicities of selumetinib were observed for the first time after years of therapy, reinforcing the need for continued surveillance[9,19]. Further, little is known about tumor regrowth after treatment discontinuation and the mechanisms of primary or acquired MEK inhibitor resistance. While MEK1/2 inhibitors have marked significant advances in treatment of PNs, non-responders underscore the need for alternate RAS-pathway agents and other novel mechanisms, which are currently under investigation in clinical trials.
DRUGS IN THE PIPELINE
Several therapies are currently in clinical trials for NF1-associated PNs with efforts focused both on expanding the utility of MEK inhibitors and exploring alternative therapeutic targets. Within MEK inhibitors, binimetinib (MEK162) has shown partial responses (≥ 20% volumetric reduction) in Phase II cohorts
CONCLUSIONS AND FUTURE DIRECTIONS
The therapeutic landscape for PNs has rapidly evolved since 2020, largely driven by the success of MEK1/2 inhibitors such as selumetinib and mirdametinib. These agents have redefined the standard of care for NF1-associated inoperable PNs by demonstrating clinically meaningful tumor shrinkage, disease stabilization, and improved quality of life. However, no studies to date evaluate whether MEK inhibition can prevent or delay malignant transformation.
MPNSTs are aggressive sarcomas with high morbidity and mortality, which arise from pre-existing PNs[1,2]. Although all individuals with NF1 carry a lifetime risk of approximately 8%-13%, patients with internal PNs have a 20-fold greater risk of transformation compared with other patients without internal PNs and compared to the general public without NF1[1,2]. Complete surgical resection remains the only potentially curative option, yet 5-year overall survival remains poor even after excision, and no FDA-approved systemic therapies are available for MPNSTs[1,2]. Further research is needed to understand the mechanisms driving resistance to MEK inhibitors and biomarkers that can inform individualized strategies. Emerging liquid-biopsy approaches using cell-free DNA (cfDNA) suggest that copy-number alteration-based assays can only identify MPNST, whereas fragmentomic signatures may distinguish premalignant stages from benign PNs (e.g., atypical neurofibroma) noninvasively, enabling early intervention[23]. Prospective, multicenter validation is needed to establish cfDNA as a routine surveillance tool and enable risk-adapted management of PNs.
As the pipeline for novel and targeted therapies continues to grow, development should not only be evaluated for volumetric response and symptomatic improvement, but also for the reduction in malignant transformation risk and early detection of resistance using biomarkers, such as cfDNA or fragmentomic signatures. Finally, there is a critical need to develop therapeutic strategies beyond pharmaceutical interventions including early pain interventions, physical and occupational rehabilitation, psychosocial support groups, and integrative care models to fully address the needs of individuals with NF1-associated PNs across their life span.
Preclinical work suggests that restoring the missing neurofibromin gene could reduce PN burden and alleviate symptoms[24]. In an NF1 xenograft model, an engineered adeno-associated virus for NF (AAV-NF) (K55) vector showed reduced hepatic uptake and enhanced tumor targeting, improving intratumoral delivery bringing gene therapy a step closer to first-in-human clinical trials[24]. Sustained collaboration among patients, researchers, and philanthropic partners, such as the Children’s Tumor Foundation and the Gilbert Family Foundation, will accelerate the development of these promising treatment strategies and may ultimately lead to a cure for NF1.
DECLARATIONS
Authors’ contribution
Original draft preparation, reviewing, editing: Kim H
Assisted with draft preparation, reviewing and editing: Polen J
Assisted with draft preparation, supervision, reviewing, and editing: Kaur G
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
During the preparation of the manuscript, the authors used ChatGPT (OpenAI) solely for language clarity and readability. After using this tool, the authors reviewed and edited the content and take full responsibility for the content of the published article.
Financial support and sponsorship
None.
Conflicts of interest
Gurcharanjeet Kaur is a Guest Editor Assistant of the Special Issue "Topic: Advances in Neurofibromatosis - The Future is Bright" of the journal Rare Disease and Orphan Drugs. Gurcharanjeet Kaur was not involved in any steps of the editorial process, notably including reviewers' selection, manuscript handling, or decision-making. The other authors declare that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Hirbe AC, Gutmann DH. Neurofibromatosis type 1: a multidisciplinary approach to care. Lancet Neurol. 2014;13:834-43.
2. Fisher MJ, Blakeley JO, Weiss BD, et al. Management of neurofibromatosis type 1-associated plexiform neurofibromas. Neuro Oncol. 2022;24:1827-44.
3. Na B, Shah SR, Vasudevan HN. Past, present, and future therapeutic strategies for NF-1-associated tumors. Curr Oncol Rep. 2024;26:706-13.
4. Weiss B, Widemann BC, Wolters P, et al. Sirolimus for progressive neurofibromatosis type 1-associated plexiform neurofibromas: a neurofibromatosis Clinical Trials Consortium phase II study. Neuro Oncol. 2015;17:596-603.
5. Robertson KA, Nalepa G, Yang FC, et al. Pilot phase II trial of imatinib mesylate in neurofibromatosis type 1 patients with plexiform neurofibromas. Lancet Oncol. 2012;13:1218-24.
6. Widemann BC, Babovic-Vuksanovic D, Dombi E, et al. Phase II trial of pirfenidone in children and young adults with neurofibromatosis type 1 and progressive plexiform neurofibromas. Pediatr Blood Cancer. 2014;61:1598-602.
7. Widemann BC, Dombi E, Gillespie A, et al. Phase 2 randomized, flexible crossover, double-blinded, placebo-controlled trial of the farnesyltransferase inhibitor tipifarnib in children and young adults with neurofibromatosis type 1 and progressive plexiform neurofibromas. Neuro Oncol. 2014;16:707-18.
8. Shih CS. Study of sutent®/sunitinib (SU11248) in subjects with NF-1 plexiform neurofibromas; 2018. Available from: https://clinicaltrials.gov/study/NCT01402817 [Last accessed on 11 Feb 2026].
9. Gorai S, Rathore G, Das K. Selumetinib-A comprehensive review of the new FDA-approved drug for neurofibromatosis. Indian Dermatol Online J. 2024;15:701-5.
10. Akbarzadeh M, Vaez-Gharamaleki Y, Hosseini M. Mirdametinib approval for neurofibromatosis type 1 with inoperable plexiform neurofibromas: the journey of MEK inhibitors. ESMO Rare Cancers. 2025;3:100015.
11. Solares I, Viñal D, Morales-Conejo M, Rodriguez-Salas N, Feliu J. Novel molecular targeted therapies for patients with neurofibromatosis type 1 with inoperable plexiform neurofibromas: a comprehensive review. ESMO Open. 2021;6:100223.
12. Gross AM, Wolters PL, Dombi E, et al. Selumetinib in children with inoperable plexiform neurofibromas. N Engl J Med. 2020;382:1430-42.
13. FDA. FDA approves selumetinib for adults with neurofibromatosis type 1 with symptomatic, inoperable plexiform neurofibromas. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-selumetinib-adults-neurofibromatosis-type-1-symptomatic-inoperable-plexiform [Last accessed on 11 Feb 2026].
14. Chen AP, Coyne GO, Wolters PL, et al. Efficacy and safety of selumetinib in adults with neurofibromatosis type 1 and symptomatic, inoperable plexiform neurofibromas (KOMET): a multicentre, international, randomised, placebo-controlled, parallel, double-blind, phase 3 study. Lancet. 2025;405:2217-30.
15. Moertel CL, Hirbe AC, Shuhaiber HH, et al. ReNeu: a pivotal, phase IIb trial of mirdametinib in adults and children with symptomatic neurofibromatosis type 1-associated plexiform neurofibroma. J Clin Oncol. 2025;43:716-29.
16. FDA. FDA approves mirdametinib for adult and pediatric patients with neurofibromatosis type 1 who have symptomatic plexiform neurofibromas not amenable to complete resection. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-mirdametinib-adult-and-pediatric-patients-neurofibromatosis-type-1-who-have-symptomatic [Last accessed on 11 Feb 2026].
17. Mol I, Bell T, Hu Y, Langseth A, Weber M, Moradian H. CO122 indirect treatment comparison (ITC) of mirdametinib and selumetinib for the treatment of children with neurofibromatosis type 1-associated plexiform neurofibromas (NF1-PN). Value Health. 2025;28:S45.
18. Gross AM, Dombi E, Wolters PL, et al. Long-term safety and efficacy of selumetinib in children with neurofibromatosis type 1 on a phase 1/2 trial for inoperable plexiform neurofibromas. Neuro Oncol. 2023;25:1883-94.
19. Healx Limited. A phase 2, open-label, single arm, non-controlled, single-stage study to evaluate the safety and effects of HLX-1502 in patients with plexiform neurofibroma and neurofibromatosis type 1 (INSPIRE-NF1). Available from: https://clinicaltrials.gov/study/NCT06541847 [Last accessed on 11 Feb 2026].
20. Korf B. Phase II study of binimetinib in children and adults with NF1 Plexiform neurofibromas (NF108-BINI). Available from: https://clinicaltrials.gov/study/NCT03231306 [Last accessed on 11 Feb 2026].
21. Fisher MJ, Shih CS, Rhodes SD, et al. Cabozantinib for neurofibromatosis type 1-related plexiform neurofibromas: a phase 2 trial. Nat Med. 2021;27:165-73.
22. Dhall G. Study of cabozantinib with selumetinib for plexiform neurofibromas (NF113). Available from: https://clinicaltrials.gov/study/NCT06502171 [Last accessed on 11 Feb 2026].
23. Sundby RT, Szymanski JJ, Pan AC, et al. Early detection of malignant and premalignant peripheral nerve tumors using cell-free DNA fragmentomics. Clin Cancer Res. 2024;30:4363-76.
24. Bai RY, Shi J, Liu J, et al. Development of an adeno-associated virus vector for gene replacement therapy of NF1-related tumors. Nat Commun. 2025;16:8594.
25. Mueller S, Reddy AT, Dombi E, et al. NFB-17. MEK inhibitor binimetinib shows clinical activity in children with neurofibromatosis type 1- associated plexiform neurofibromas: a report from pnoc and the nf clinical trials consortium. Neuro Oncol. 2020;22:iii420-1.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].