


Gene therapy for inborn errors of immunity: past progress, current status and future directions
 0
0 
Abstract
Inborn errors of immunity (IEIs), also known as primary immunodeficiencies, are a group of rare inherited disorders that affect the immune system. They result in severe, opportunistic infections, severe autoimmune manifestations and a predisposition to malignancy. The only curative treatment for many years has been allogenic haematopoietic stem cell transplantation (alloHSCT). However, this requires the availability of a suitable donor and has risks of morbidity and mortality. Autologous gene therapy (GT) abrogates the immunological complications of alloHSCT and uses the patient’s own cells, removing the need for a donor. Preclinical proof-of-concept and clinical trials in humans have demonstrated that GT is safe and effective and can be used to correct a variety of IEIs. In this review, we outline the progress in developing GT for IEIs over the last four decades. We describe the gene editing technologies available to correct IEIs and their current applications. We also examine advances in GT development, the challenges to its application, and discuss future developments in the field, including emerging in vivo approaches.
Keywords
INTRODUCTION
Inborn errors of immunity (IEIs) are a group of rare inherited disorders that affect the immune system. Over 550 have been described to date[1,2]. Whilst individually rare, collectively IEIs affect approximately 1 in every 1,200 individuals[2]. In addition to increased susceptibility to infection, IEIs may present with autoimmunity, autoinflammation, and malignancy. Some IEIs, such as severe combined immunodeficiencies (SCID), require definitive treatment [e.g., allogeneic haematopoietic stem cell transplantation (alloHSCT) or gene therapy] early in life to prevent death from catastrophic infection.
For the majority of IEIs, alloHSCT is the only curative therapy, and, in many cases, is now the standard of care if a matched donor is available[3]. Since the first alloHSCT for IEIs more than 50 years ago, advances have been made in understanding the natural disease course of many indications[4,5]. This has enabled the publication of clear guidelines regarding when and how transplant should take place[3]. For example, the risk/benefit of alloHSCT is clear in diseases such as SCID, where without transplant the disease is universally fatal[6]. With current transplant practices, the survival rate is close to 95% in SCID patients transplanted under 3 months of age. Outcomes are expected to improve further with the expansion of universal newborn screening which will enable infants to undergo alloHSCT prior to the development of any infectious complications[7,8].
However, decisions about when to proceed with alloHSCT in rarer IEIs are challenging, as the natural course of the disease is less certain[9]. Risks of alloHSCT in many patients with IEIs are further increased due to infectious complications and/or significant organ dysfunction prior to or peri-transplant which increases the risks of transplant-related mortality[10]. Furthermore, the risks of graft-versus-host disease (GVHD), poor graft function or graft failure are increased in some IEIs, particularly those with uncontrolled autoinflammation and autoimmunity prior to transplant[10]. Other longer-term risks following transplant are well characterized and include endocrinopathies, chronic GVHD, autoimmune conditions, secondary malignancies and impaired growth[11].
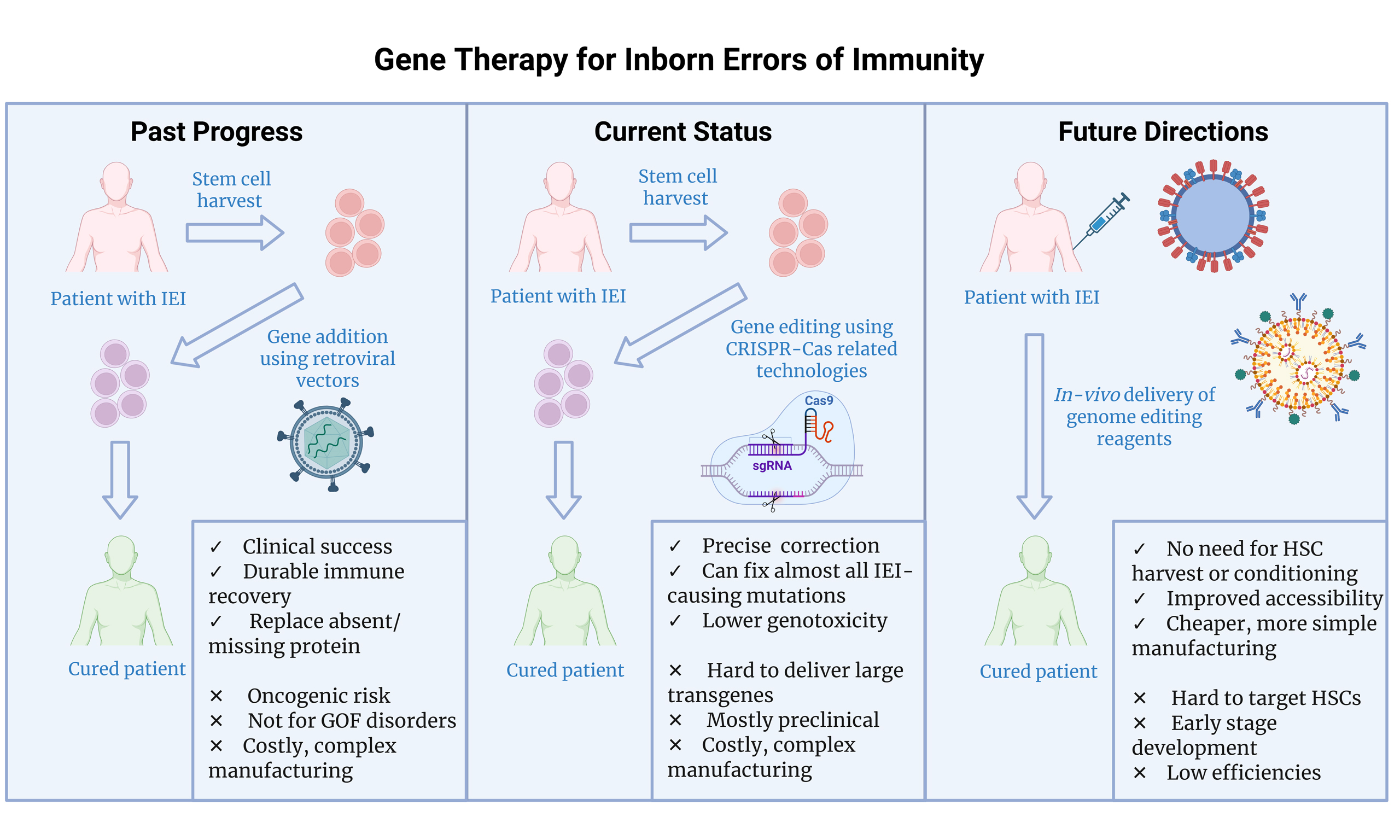
Autologous haematopoietic stem cell (HSC) gene therapies (GT) (HSC-GT) completely overcome the issues of donor availability and immunological complications (GVHD, immunosuppression-related infections, graft failure) associated with alloHSCT. HSC-GT involves harvesting the haematopoietic stem and progenitor cells from patients, typically via the apheresis of mobilized stem cells from the peripheral blood. Stem cells are then modified using gene addition/editing and returned to the patient following conditioning chemotherapy [Figure 1]. Modified patient HSCs home to the bone marrow and can repopulate all cell lineages with corrected progeny. HSC-GT does not require a donor and there is no risk of GVHD. Given the autologous nature, there is less risk of graft failure and a lack of requirement for immunosuppression following HSC infusion[12]. IEIs represent the ideal application for HSC-GT, given the clear link between genotype and disease phenotype and the defect being limited to cells that can be repopulated from corrected HSCs.
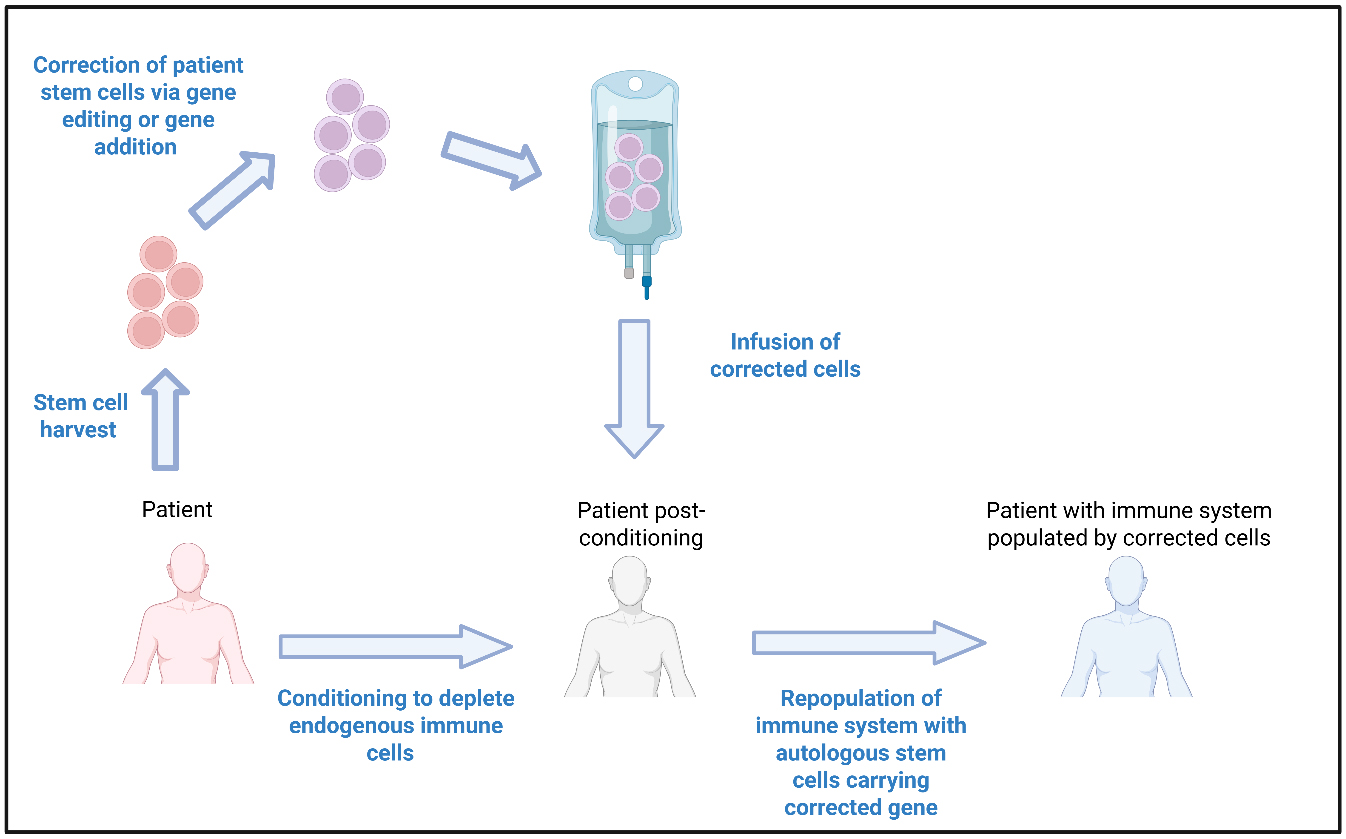
Figure 1. Autologous Haematopoietic Stem Cell Gene Therapies. Autologous HSC-GT involves harvesting the stem cells directly from the patient, which are then modified by gene addition or gene editing. Modified patient stem cells are then re-infused back into the patient, who in the meantime has undergone a conditioning regimen to deplete their endogenous immune cells. Following engraftment of the modified stem cells into the bone marrow of the patient, the newly introduced cells can repopulate the immune system with
In this review, we discuss the development of HSC-GT for IEIs, from gene addition to gene editing and give a perspective on advances and future directions.
GENE ADDITION FOR INBORN ERRORS OF IMMUNITY
The concept of genetic modification of human cells as a therapeutic option originated in the early 1970s, when it was hypothesized that the introduction of exogenous DNA into cells bearing genetic defects could be a viable therapeutic strategy for patients with genetic disease[13]. At the time, attempts at exogenous gene transfer in human cells showed limited efficiency. The use of more efficient transformation methods, such as engineered retroviruses, revolutionized the field[14-18]. Genetic disorders caused by a loss of function in a given protein could now be corrected using viral vectors to introduce a new, functional copy of the gene into patient cells [Figure 2].
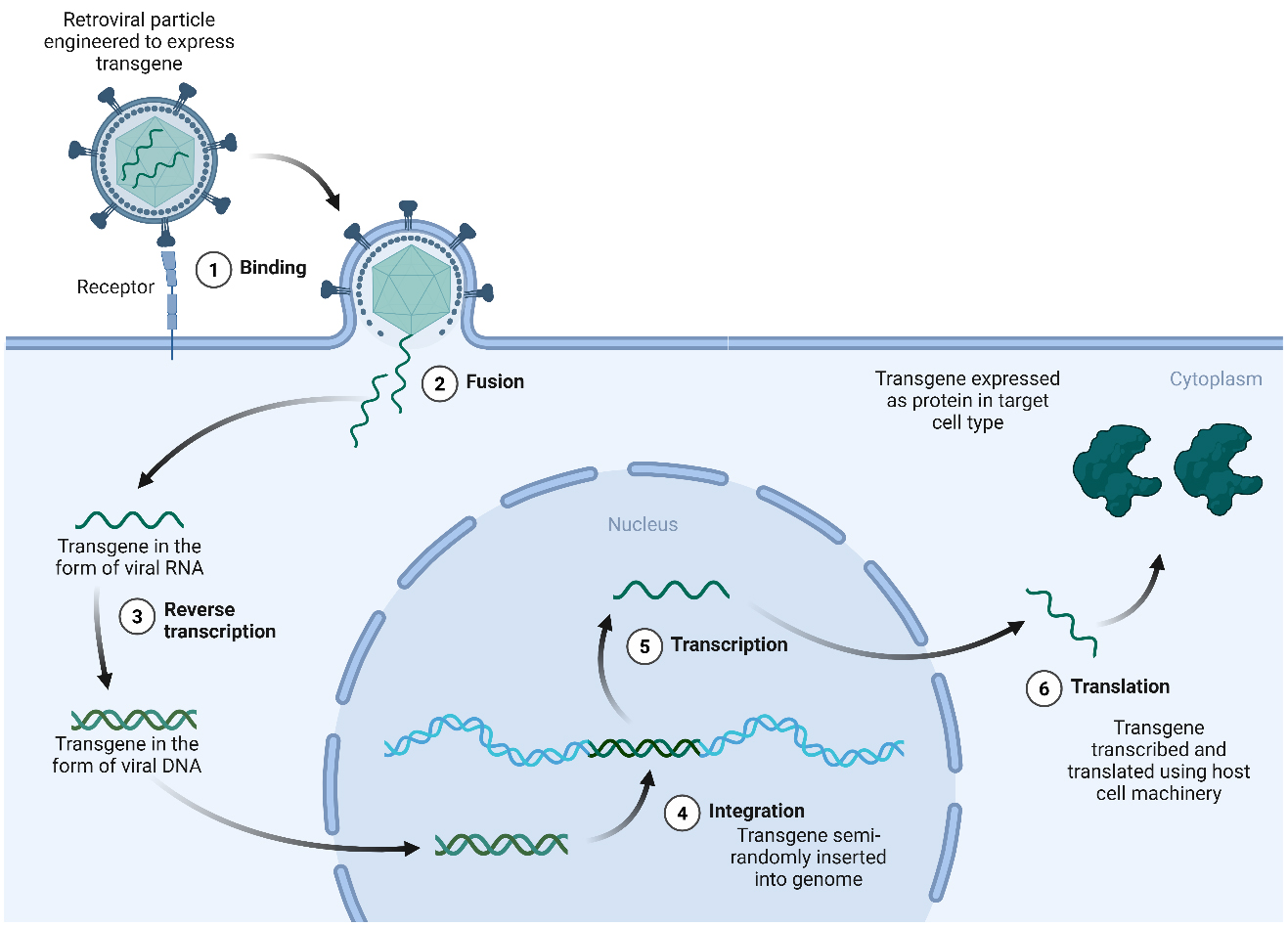
Figure 2. Gene addition for gene therapies using retroviral vectors. Gene transduction using retroviruses such as HIV-derived lentivirus or γ-retroviruses begins when virus particles enter the cell either by endocytosis or following direct fusion of the viral envelope protein with its cognate receptor on the membrane (1,2). Upon entry, the retroviral RNA genome is released into the cytoplasm where it is reverse transcribed by a viral reverse transcriptase (3) to produce cDNA which integrates semi-randomly into the host genome using a virally expressed integrase (4). While lentiviruses can transduce dividing and non-dividing cells, simple γ-retroviruses require the disappearance of the nuclear membrane during cell division, meaning that they are only able to successfully infect dividing cell types. Integrated sequences are then expressed using the host transcriptional (5) and translational (6) machinery. Promoter sequences either in the long-terminal repeat region of the virus, or more commonly for gene therapy approaches, from a mammalian promoter sequence incorporated into the genomic cargo, are used to regulate gene expression. Created in BioRender. Torrance R (2025) https://BioRender.com/thj9v37. cDNA: Complementary DNA; HIV: human immunodeficiency virus.
By 1990, γ-retroviral vectors had been successfully engineered to deliver complementary DNA (cDNA) encoding the adenosine deaminase gene (ADA) into the lymphocytes and bone marrow of patients with ADA-SCID. This provided the first proof-of-concept that GT could be a safe and effective therapeutic strategy for IEIs[14,15]. Following this success, over the next twenty years, a number of clinical trials examining safety and efficacy of GT using γ-retroviral vectors for IEIs were initiated, with promising results in
However, despite improvement of clinical phenotypes in many cases, the use of γ-retroviral vectors led to insertional oncogenesis in a number of patients. For example, despite sustained clinical benefit in 17/20 patients initially treated in a GT trial for X-SCID, 6/20 patients developed T cell leukemia linked to preferential vector integration in close proximity to protooncogenes such as the LMI domain only protein 2 (LMO2)[20,21,26,27]. Similar serious adverse events were also seen in the WAS trial where 7/10 enrolled patients developed leukemia[23], as well as in the Chronic Granulomatous Disease (CGD) trial where vector integration close to EVI1 was linked to the development of myelodysplasia with monosomy 7, a precursor to Acute Myeloid Leukaemia (AML)[25].
Interestingly, the rates of oncogenesis in ADA-SCID patients following γ-retroviral vector GT have been much lower than in other primary immunodeficiency disorders (PIDs), with only one case of vector-related leukemia in over 75 patients treated to date[28]. The exact reasons for this remain unclear, but it is thought to be related to intrinsic ADA deficiency in epithelial cells restricting thymopoiesis, which may in turn reduce the risk of leukemogenesis through a decreased rate of precursor T-cell expansion[29].
Due to the development of oncogenesis with γ-retroviral therapies, newer and safer self-inactivating (SIN)
Gene therapy clinical trials in progress for IEIs
| Disease | Target gene | Vector | Clinical trials (Interventional) | Reference |
| γ-retroviral, SIN γ-retroviral, adenoviral and lentiviral | ||||
| ADA-SCID | ADA | • γ-retroviral • Lentiviral (ex-vivo) • Lentiviral (in-vivo) | NCT00599781*; NCT00598481*; NCT00018018*; NCT01279720*; NCT00794508* NCT01380990*; NCT01852071*; NCT02022696*; NCT02999984*; NCT03765632*; NCT05432310 NCT03645460 | [16,37-41] [36] |
| X-linked SCID | IL2RG | • γ-retroviral • SIN γ-retroviral • Lentiviral (ex-vivo) • Lentiviral (in-vivo) | NCT00028236* NCT01410019*; NCT01175239; NCT01129544* NCT01512888; NCT01306019*; NCT03315078; NCT03311503; NCT03601286; NCT04286815 NCT03217617 | [42,43] [29] [31] |
| Wiskott Aldrich syndrome | WAS | • γ-retroviral • Lentiviral (ex-vivo) | NCT01410825 NCT01515462*; NCT01347242*; NCT01347346*; NCT03837483 NCT02333760 | [23] [32,33,44] |
| X-linked Chronic Granulomatous Disease | CYBB | • γ-retroviral • SIN γ-retroviral • Lentiviral (ex-vivo) • Adenoviral (in-vivo) | NCT00564759; NCT00927134*; NCT00394316; NCT00778882 NCT01906541 NCT01855685; NCT02234934*; NCT02757911; NCT03645486; NCT01381003 NCT06876363 | [24,25,45] [35] |
| Lentiviral only | ||||
| P47phox deficient Chronic Granulomatous Disease | NCF1 | • Lentiviral (ex-vivo) | NCT06253507; NCT05207657 | [46] |
| IPEX Syndrome | FOXP3 | • Lentiviral (ex-vivo) | NCT05241444 | [47] |
| Hemophagocytic lymphohistiocytosis | UNC13D PRF1 | • Lentiviral (ex-vivo) • Lentiviral (ex-vivo) | NCT06736080 Preclinical | [48] [49] |
| Deficiency of Adenosine Deaminase 2 | ADA2 | • Lentiviral (ex-vivo) | Preclinical | [50] |
| RAG2-SCID | RAG2 | • Lentiviral (ex-vivo) | Preclinical | [51] |
| X-linked lymphoproliferative disease | SH2D1A | • Lentiviral (ex-vivo) | Preclinical | [52] |
| X-linked agammaglobulinemia | BTK | • Lentiviral (ex-vivo) | Preclinical | [53] |
| AR-IFN-γR1 deficiency | IFN-γR1 | • Lentiviral (ex-vivo) | Preclinical | [54] |
| RAG1-SCID | RAG1 | • Lentiviral (ex-vivo) | NCT04797260 | [55] |
| Artemis-SCID | DCLRE1C | • Lentiviral (ex-vivo) | NCT05071222; NCT03538899 | [56] |
| Leukocyte adhesion deficiency | ITGB2 | • Lentiviral (ex-vivo) | NCT03812263*; NCT06282432; NCT03825783 | [57,58] |
However, lentiviral gene addition is not suitable to correct all IEIs as it can only be used when the genetic mutation results in a missing or absent protein or where overexpression is tolerated or advantageous (e.g., ADA-SCID). Furthermore, the semi-random nature of transgene integration that results from lentivirus transduction does not typically result in physiological expression in all cells. This is particularly important for IEIs where tightly regulated gene expression is required, such as for CTLA-4 insufficiency[59] or X-linked hyper IgM syndrome, where constitutive expression of the implicated gene CD40 ligand (CD40L) has been linked to lymphoproliferative disorders[60,61]. Furthermore, simple gene addition is not suitable for IEIs caused by dominant mutations, such as gain-of-function or dominant negative disorders, which make up approximately 27% of all IEIs[1]. For these indications, novel gene editing technologies may enable correction of the associated genetic defects while preserving the endogenous control machinery and gene expression profile[26,62,63].
GENE EDITING FOR INBORN ERRORS OF IMMUNITY
To date, several gene editing technologies have been used for the precise alteration of DNA sequences in IEIs. These include nuclease-based gene editors, base editors (BEs), and prime editors (PEs). The class of editor most suitable for a given indication depends on several factors, including the type of desired edit, the properties of the targeted DNA sequence, and the disease context.
Clustered regularly interspaced short palindromic repeats/Cas endonucleases
Three main classes of nucleases have been described for targeted genome engineering, each of which uses a targeting molecule (either protein or RNA) to guide a DNA nuclease to a specific region of the genome, where a double-stranded (ds) DNA break is created[64,65]. Historically, zinc-finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) which use protein moieties for targeting were utilized and demonstrated proof-of-principle of gene editing in human cells[65-67]. However, in recent years, the field has largely shifted towards using the Clustered Regularly Interspaced Short Palindromic Repeats associated with the Cas endonuclease (CRISPR-Cas) system for gene editing due to its ease of programmability[68]. CRISPR-Cas gene editing can be easily programmed by altering the guide RNA (gRNA) sequence, allowing targeting of the Cas endonuclease to different genomic loci based on complementary RNA:DNA base pairing. Indeed, following the first use of the CRISPR-Cas system to induce targeted double-stranded DNA (dsDNA) breaks in vitro in bacteria in 2011[68], there has been an exponential rise in applications of CRISPR-Cas technology from bacteria to human cells[69].
Regardless of the nuclease used, following the formation of a dsDNA break, the primary repair mechanism in human cells is non-homologous end joining (NHEJ) [Figure 3][70]. This results in semi-random insertions and deletions (indels) of bases at the site of the dsDNA break, often leading to frameshifts, an alteration in the coding sequence and thus gene knockout[71]. However, in the presence of a suitable “donor” template with homology to the DNA either side of the cut site, homology-directed repair (HDR) can take place, leading to the insertion of new sequences at the location of the break[72-74] [Figure 3]. Whereas NHEJ can occur at any time during the cell cycle, HDR only occurs during the S or G2 phases of the cell cycle, and thus typically happens at a lower frequency[75,76]. This is in contrast to base and prime editing (discussed below), which utilize repair mechanisms that are not cell cycle-dependent, allowing efficient gene editing of cycling and non-cycling cells.
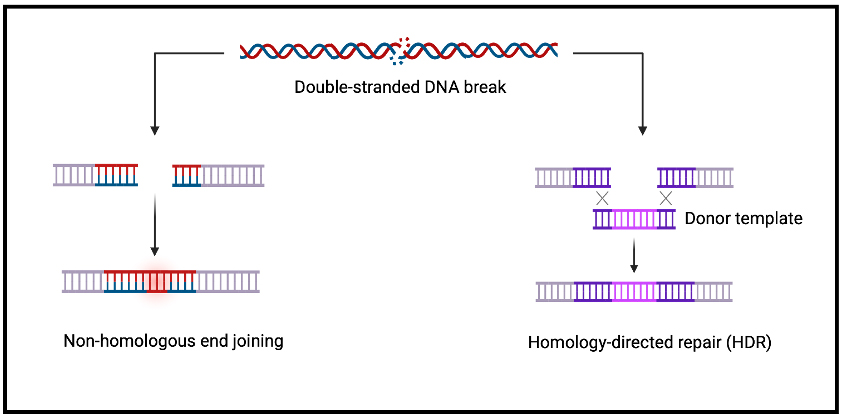
Figure 3. Mechanisms of repair of a dsDNA break. Following generation of a DSB by a CRISPR/Cas nuclease, the lesion is typically either repaired by either non-homologous end joining, which is highly error prone and often leads to gene disruption, or by
Several different types of repair templates can be utilized for HDR-based gene editing. For example, Adeno-associated virus type 6 (AAV6) and
Base editing
Base editing is a further development of the CRISPR/Cas technology that allows all four possible transition point mutations (A- > G, T- > C, C- > T, G- > A) to be performed, without the creation of double-strand DNA breaks (DSBs). BEs consist of a Cas nickase (nCas) fused to an adenine/cytosine deaminase domain[82]. As in the CRISPR-Cas system described above, the gRNA directs the nCas to the target sequence by binding to the target DNA strand, and producing a single-stranded DNA (ssDNA) substrate for the deaminase which deaminates specific nucleotides within this sequence [Figure 4][82]. The precise editing window varies depending on the BE but typically spans positions 4 to 8 of the protospacer sequence for BEs based on SpCas9, where the protospacer adjacent motif (PAM) is located at position 21 onwards[82]. After conversion of target bases in the editing window, the nCas creates a single-stranded break on the
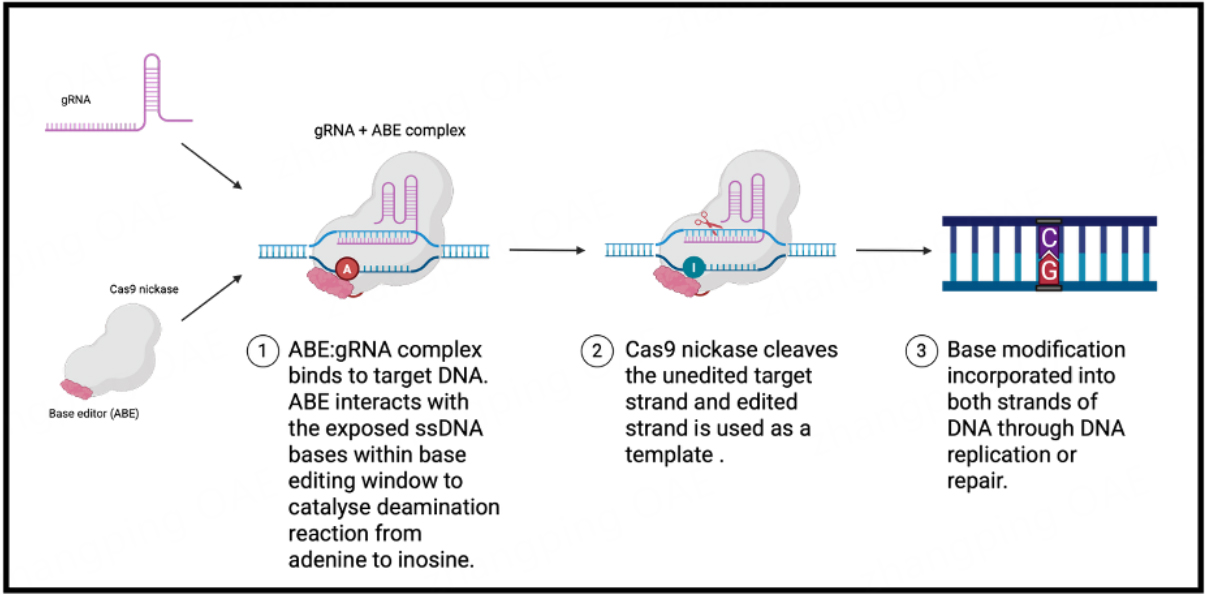
Figure 4. Schematic diagram showing the mechanism of adenine base editing. A gRNA targets a Cas nickase tethered to an adenine/cytosine deaminase to a specific sequence within the genome. There, specific deamination of target bases within a base editing window takes place, resulting in A:T - > G:C or C:G - > T:A transition mutations. Created in BioRender. Orf K (2025) https://BioRender.com/udxwt5j. ABEs: Adenine base editors; gRNA: guide RNA.
One of the major requirements for efficient base editing is the availability of a suitable PAM sequence that places the target base in an appropriate base editing window. Early BEs were exclusively based on SpCas9 (PAM=NGG) which theoretically permits the correction of just 26% and 28% of annotated, pathogenic transition mutations in ClinVar with cytosine base editors (CBEs) and adenine base editors (ABEs), respectively[82]. However, with the evolution of SpCas9 variants to accommodate different PAM sequences, the use of deaminase domains with alternative sequence specificities and editing window widths, and the use of alternative Cas enzymes (e.g., SaCas9, LbCas12a), around 95% of all pathogenic transition mutations in ClinVar are now targetable by base editing[82-87] [Table 2]. Selection of a particular BE/nCas combination depends on several factors, including the required base conversion efficacy, PAM availabilities and the presence of possibly deleterious bystander edits. Relative editing efficiencies and potential advantages/disadvantages of the different editors are largely dependent on the target sequence and indication.
Overview of different base editors with their respective PAM sequences
| Editing window | PAM sequence preference | Reference | |
| Adenine deaminase | |||
| ABE8e NGG | 4-8 | NGG | [85] |
| ABE8e SPRY | 4-8 | NRN, NYN | [83] |
| ABE8e NG | 4-8 | NG | [85] |
| ABEmax NG | 4-8 | NG | [88] |
| ABEmax SPRY-GFP | 4-8 | NRN, NYN | [89] |
| Cytosine deaminase | |||
| BE4max NRRH | 4-8 | NRRH | [90] |
| evoCDA BE4max NG | 3-12 | NG | [84] |
| evoFERNY BE4max NG | 3-8 | NG | [84] |
Prime editing
Although BEs can correct most pathogenic single nucleotide polymorphisms (SNPs), they cannot correct all types of mutation (e.g., substitutions, indels)[91]. PEs are able to mediate all single-nucleotide conversions, as well as small insertions and deletions without creating DSBs[91]. PEs consist of reverse transcriptase (RT) enzyme fused to a nCas domain [Figure 5]. A prime editing gRNA (pegRNA) can both direct the PE to the genomic target locus and encode the desired edit. As with nuclease-mediated gene editing and base editing, prime editing initiates with base pairing of the protospacer sequence to the target site in the genome and subsequent nicking of the non-protospacer bound strand[92]. Following nicking, the primer binding site within the pegRNA anneals to the nicked strand forming an RNA/DNA duplex that can be used to prime reverse transcription, with the reverse transcription template (RTT; also within the pegRNA) serving as a template. As reverse transcription occurs, the flap is extended, and the edit is incorporated. The edited
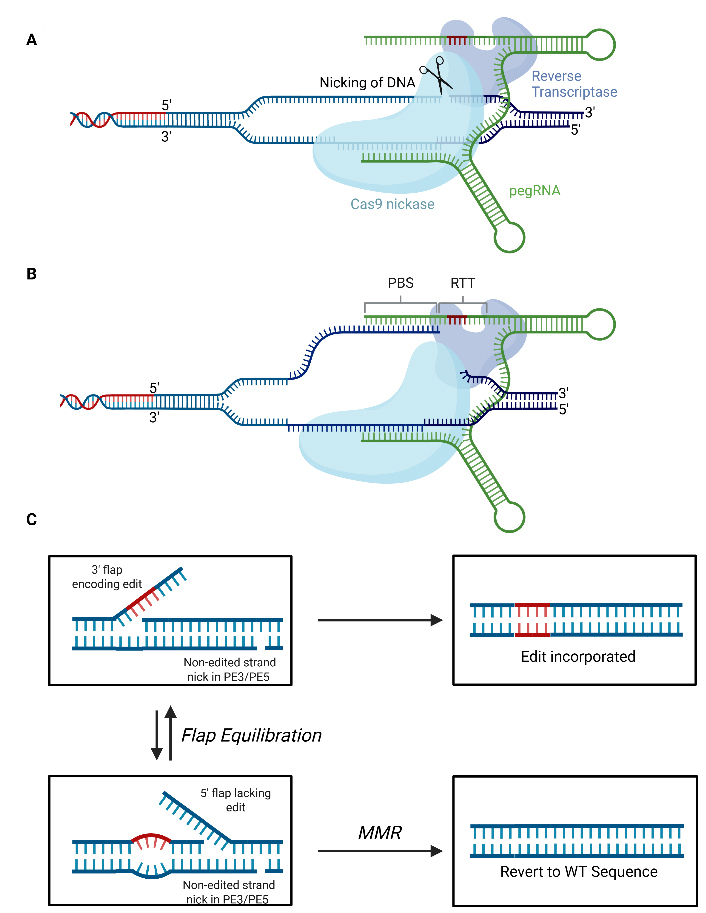
Figure 5. Mechanism of Prime Editing. (A) Prime editing initiates when the protospacer of the pegRNA binds to its cognate site within the genome before a nick in the phosphodiester backbone is created on the target strand; (B) Following nicking, the PBS anneals to the DNA flap with the resulting double-stranded DNA duplex acting to prime reverse transcription using the RTT as a template; (C) This produces a 3’ flap containing the desired edit which then undergoes flap equilibration with the 5’ flap lacking the edit. Degradation of the 5’ flap and ligation of the 3’ flap into the genome will result in the incorporation of the edit. Recognition of the mismatch between the 3’ flap containing the edit and the unedited by the MMR DNA repair machinery can prevent incorporation of the edit. A nick to the non-edited strand in the PE3 and PE5 systems and/or expression of a dominant negative mismatch repair protein MLH1 in PE4 and PE5 systems can bias DNA repair in favour of incorporation of the edit. RTT: Reverse transcription template; pegRNA: prime editing guide RNA; PBS: primer binding site; MMR: mismatch repair; WT: wildtype. Adapted from Doman et al.[92], Created in BioRender. Torrance R (2025) https://BioRender.com/06c7els.
CURRENT CLINICAL AND PRECLINICAL APPLICATIONS OF GENE EDITING FOR IEI
Several approaches utilizing the genome editing technologies described above have been developed for the treatment of different IEIs. The vast majority of these applications have been for those disorders caused by homozygous or heterozygous loss-of-function (LOF) mutations where gene editing offers a rival approach to gene addition, with the additional benefit of allowing tightly regulated, physiological expression due to the preservation of the endogenous control machinery[59-61]. These approaches for IEIs caused by LOF mutations typically involve the use of CRISPR/Cas9 nucleases to induce a dsDNA break within the endogenous gene, before a therapeutic (full or partial) transgene is introduced at the cut site by HDR. Indeed, such an approach has been successfully applied in multiple IEIs including SCID-X1, X-linked hyper IgM, recombinase activating gene 1 (RAG1) SCID, recombinase activating gene 2 (RAG2) SCID, WAS,
Gene editing strategies for IEIs and current trial status if applicable
| Disease | Gene | Approach | Stage of development | Reference |
| HDR | ||||
| X-linked SCID | IL2RG | - HDR knock-in of full length IL2RG cDNA at translation start site using CRISPR/Cas & AAV6 in HSCs | Preclinical | [78] |
| X-linked hyper IgM syndrome | CD40L | - HDR knock-in of CD40L cDNA in 5’UTR with TALEN or CRISPR/Cas + AAV6 in HSCs & T cells - HDR knock-in of CD40L cDNA in 1st intron using CRISPR/Cas + AAV6 in HSCs and T cells | Preclinical Preclinical | [77] [94] |
| RAG1-SCID | RAG1 | - HDR knock-in of RAG1 cDNA in-frame in exon 2 using CRISPR/Cas + AAV6 or IDLV in HSCs | Preclinical | [95] |
| RAG2-SCID | RAG2 | - HDR knock-in of RAG2 cDNA upstream of START codon using CRISPR/Cas + AAV6 in iPSCs - HDR knock-in of RAG2 cDNA upstream of START codon using CRISPR/Cas + AAV6 in HSCs - HDR knock-in of RAG2 cDNA downstream of START codon using CRISPR/Cas + AAV6 in HSCs | Preclinical Preclinical Preclinical | [96] [78] [97] |
| Wiskott Aldrich syndrome | WAS | - HDR knock-in of WAS cDNA at translation start site using CRISPR/Cas + AAV6 in HSCs | Preclinical | [79] |
| X-linked agammaglobulinemia | BTK | - HDR knock-in of Btk cDNA + 300bp of terminal intron into exon 2 using CRISPR/Cas + AAV6 in HSCs | Preclinical | [98] |
| IL7Rα SCID | IL7RA | - HDR knock-in of promoterless IL7RA cDNA in exon 1 using CRISPR/Cas + AAV6 in T cells & HSCs | Preclinical | [99] |
| IPEX syndrome | FOXP3 | - HDR knock-in of FOXP3 cDNA at translation start site + tNGFR selectable marker using CRISPR/Cas + AAV6 in T cells & HSCs | Preclinical | [80] |
| X-linked chronic granulomatous disease | CYBB | - HDR knock-in of CYBB into AAVS1 safe harbour using ZFN + AAV6 in HSCs - HDR knock-in of CYBB at endogenous exon 2 using CRISPR/Cas + AAV6 in HSCs - HDR: Mutation-specific correction of c.676C > T using CRISPR/Cas + ssODN in HSCs | Preclinical Preclinical Preclinical | [100] [101] [81] |
| XMEN syndrome | MAGT1 | - HDR knock-in of MAGT1 in the endogenous exon 1 using CRISPR/Cas + AAV6 in HSCs & T cells | Preclinical | [102] |
| X-linked lymphoproliferative disease | SH2D1A | - HDR knock-in of SH2D1A cDNA within the first exon using TALEN + AAV6 or CRISPR/Cas + AAV6 in T cells | Preclinical | [103] |
| CTLA-4 Insufficiency | CTLA4 | - HDR knock-in of CTLA-4 cDNA in the first intron using CRISPR/Cas + AAV6 in T cells | Preclinical | [59] |
| Sting-associated vasculopathy with onset in infancy | STING1 | - HDR knock-in of coSTING1 cDNA (exon 5-8) downstream of endogenous intron 4 in patient-derived iPSCs and healthy-donor derived HSCs | Preclinical | [104] |
| Base editing | ||||
| X-linked SCID | IL2RG | - Base editing of the c.444C > T, p.Q144X mutation in IL2RG | Clinical NCT06851767 | Not published |
| CD3δ SCID | CD3D | - Mutation specific correction of c.202C > T using adenine base editing in HSCs | Preclinical | [105] |
| X-linked chronic granulomatous disease | - Mutation specific correction of c.676C > T and c.1075G > A using adenine base editing in HSCs | Clinical NCT06325709 | [106] | |
| Prime editing | ||||
| P47phox deficient chronic granulomatous disease | NCF1 | - Prime editing to restore the 2 nucleotide GT deletion in HSCs | Clinical NCT06559176 | [107] |
| Other | ||||
| Hemophagocytic lymphohistiocytosis | UNC13D | - CRISPR/Cas9-mediated excision of an intronic cryptic splice donor site using two gRNAs in murine HSCs | Preclinical | [108] |
| Severe congenital neutropenia | ELANE | - Disruption of ELANE promoter TATA box using dual CRISPR-Cas9D10A nickases operating on opposite strands | Preclinical | [109] |
However, in some cases, a knock-in approach, such as the one described above, is not suitable or feasible, and a mutation-specific approach is more appropriate. This may be true, for example, when high levels of editing are required to correct the disease phenotype. Given that CRISPR/Cas9 HDR is cell
In recent years, several mutation-specific strategies have been developed for IEIs. For example,
Increasingly, newer gene editing technologies such as base and prime editing are also being applied to permit mutation specific correction without the production of DSBs, thus preventing mixtures of uncontrollable indel byproducts from forming, reducing p53 activation and reducing the risk of chromosomal translocations and other gross genomic aberrations[110]. Indeed, recently, base editing of the same prototypical c.676C > T X-CGD mutation described above was successfully achieved at efficiencies 3.5-fold greater than the CRISPR/Cas + ssODN approach that had been previously trialled, with no large chromosomal rearrangements detected and an improved reported genotoxicity profile[106]. Similarly, base editing has recently been used to correct the founder c.202C > T mutation in the CD3D gene, which is causative of CD3δ SCID, leading to the production of mature, functional T cells in artificial thymic organoids (ATO)[105]. Finally, Prime Medicine has recently announced its work demonstrating that prime editing is able to successfully correct defects in long-term hematopoietic stem cells (LT-HSCs) derived from p47phox-deficient CGD patients[107].
T CELL GENE THERAPY FOR IEI
For most IEIs, correction of the HSC compartment is needed for multilineage expression and robust immune reconstitution. HSCs have the advantage of their inherent property of self-renewal, thus potentially providing a source of corrected immune cells for the lifetime of the recipient[36,79,111]. However, in several IEIs, the defect is limited to the lymphoid compartment. In diseases such as IPEX syndrome, CD40 ligand deficiency, CTLA-4 insufficiency and X-linked lymphoproliferative syndrome (XLP), it is defects in T cells (or T cell subsets) that result in the disease phenotype[59,80,94,112,113]. In diseases such as these, correction of the T cell compartment alone may improve the disease phenotype whilst offering advantages over HSC GT[59].
There are several advantages of a T cell GT approach over autologous CD34+ cells as the starting material. Firstly, the conditioning needed to engraft engineered CD34+ cells is more toxic than the lymphodepletion required prior to a T cell infusion[114,115]. We also know from studies exploring the role of alloHSCT in some T cell mediated IEIs such as CTLA-4 insufficiency that there is a high risk of stem cell engraftment failure due to the inflammatory environment present in patients with this disease[116].
Secondly, as T cells are terminally differentiated cells, the genotoxic risks from gene editing procedures may be less than in CD34+ cells[94,117,118]. CRISPR/Cas9 gene editing is a new technology, and whilst Advanced Therapy Investigational Medicinal Products (ATIMPs) using this are in clinical trials or approved for use in humans [e.g., Casgevy (exagamglogene autotemcel)], there are potential risks of genotoxic events[114]. There have been no concerns regarding CRISPR/Cas9 mediated homology directed repair in T cells and several trials of T cells engineered using this technology are currently recruiting (e.g., NCT05643742).
Whilst the long-term persistence of autologous T cell GT for IEIs is not yet known, extrapolating from experience with autologous chimeric antigen receptor (CAR)-T therapies and early T cell GT studies (in ADA-SCID), it is likely to be long-term[119]. Experience from T cell cellular therapy suggests that genetically modified T cells can persist for > 10 years if enough T stem cell memory cells are included in the therapeutic product[119].
Robust preclinical proof-of-principle of T cell GT approaches (both viral gene addition and gene editing) have been published for IPEX syndrome, CD40 ligand deficiency, XLP and CTLA-4 insufficiency[52,59,80,94,120,121]. Proof of principle in humans is awaited but a trial of the lentiviral CD4+ T cells approach for IPEX syndrome is being assessed in a phase I clinical trial (NCT05241444) and a trial of a lentiviral T cell GT approach for XLP is expected to open in the UK later in 2025[52]. Whilst it remains to be seen whether T cell correction can offer lasting improvement in disease phenotype in humans, in select IEIs, preclinical data suggests that this is a promising and potentially less toxic approach than HSC correction. While T cell-based GTs hold considerable promise, long-term risks remain incompletely defined. In particular, uncertainties exist regarding persistence and the potential for functional exhaustion, which can only be fully assessed in human clinical trials. Several such studies are expected to open soon and will be critical in addressing these unknowns.
GENE THERAPY FOR ADOLESCENTS AND ADULTS WITH IEI
As data for efficacy of alloHSCT has accumulated in the non-SCID IEI setting, successful GT approaches for non-SCID IEIs have been published[35,44,122,123]. In recent years, increasing numbers of adolescent and adult patients with non-SCID IEIs have been treated with alloHSCT, raising the question of whether GT could be applied to older patients[124]. As they have lived with their IEIs for several years, these individuals often have more comorbidities, making them higher-risk transplant candidates[10,125]. Patients with uncontrolled autoinflammation prior to transplant have an increased risk of graft failure and GVHD[10]. Older patients may therefore stand to benefit most from less toxic, autologous GT approaches.
Due to thymic involution, there were concerns that autologous HSC-GT may not result in robust multilineage immune reconstitution in older patients with IEIs. In 2017, a 30-year-old patient with
<i>IN VIVO</i> GENE THERAPY
Despite remarkable clinical results, ex vivo GT has major limitations. The need for a bespoke cellular product, harvesting of HSCs from the patient and ex vivo manufacturing all make GT expensive and complex to deliver [Figure 6]. In vivo GT has the potential to transform GT for IEIs from a unique cellular product to an off-the-shelf drug. This would dramatically reduce costs, complexity of delivery and toxicity associated with GT for IEIs.
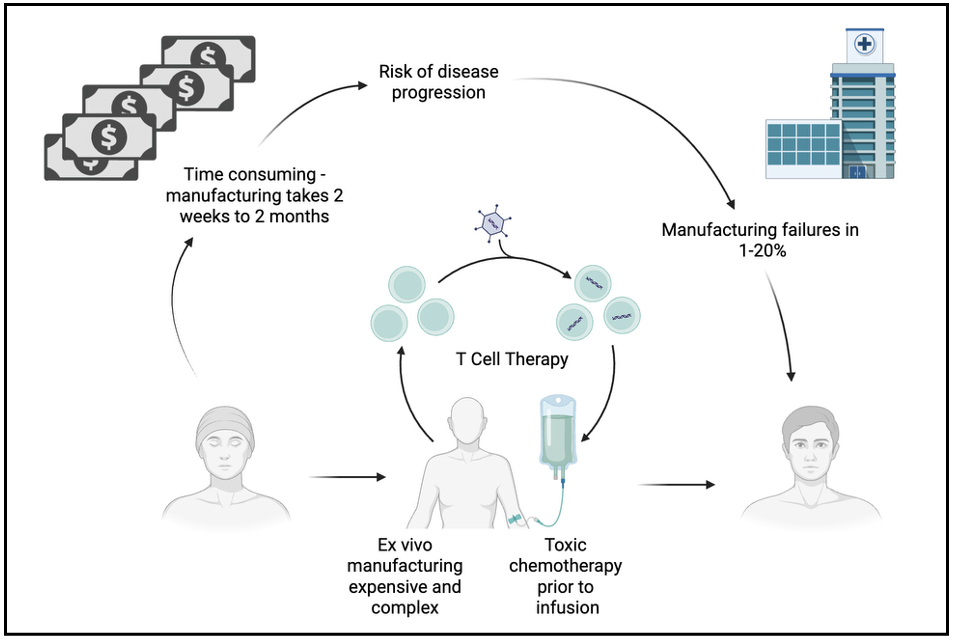
Figure 6. Potential limitations of ex vivo T cell therapy relative to in-vivo T cell therapy. Created in BioRender. Fox TA (2025) https://BioRender.com/kkpbc4l.
In vivo GT requires a delivery platform that is non-toxic, non-immunogenic and can target a specific tissue. The main delivery technologies can be divided into virus or virus-like vectors (also known as enveloped delivery vehicles) and lipid nanoparticles (LNPs). Both technologies are in use in human clinical trials of in vivo GTs for other diseases. Two notable examples, at the advanced stage of clinical development, include adeno-associated virus-based therapies for haemophilia[127,128] and lipid-nanoparticle CRISPR therapy for congenital amyloidosis[129]. Whilst these examples demonstrate that organ-specific transgene transduction and gene editing can be safely and effectively performed in vivo, both approaches target the liver. The liver contains 10%-15% of the body’s circulating blood volume making it an attractive and accessible target for in vivo gene delivery vectors administered intravenously[130].
Targeting HSCs in vivo is more challenging due to both the physical location of the cells and the immune microenvironment of the bone marrow niche. Compared to the liver, HSCs are a rare population of cells (50,000-200,000 cells are estimated to be actively involved in haematopoiesis)[131]. Whilst both viral and
One of the challenges of targeting HSCs in the peripheral blood is that vectors can be sequestered if the protein on the cell of interest is shared by other cell types. This phenomenon was observed in
Several groups are working on HSC-targeted LNPs and the last few years have seen some exciting progress in this field[136,137]. CD117-targeted LNPs have been demonstrated to deliver mRNA in vivo in mice[136]. Interestingly, the authors of this study delivered mRNA coding for pro-apoptotic PUMA (p53 up-regulated modulator of apoptosis) to deplete HSCs with the proposed clinical application of non-genotoxic conditioning[136]. Such technology (if the results can be replicated in non-human primates and humans) could have several applications to the IEI field.
In vivo HSC GT is set to first be attempted in humans to treat IEIs. Ensoma Therapeutics has developed a virus-like particle (VLP) platform based on the helper-dependent adenovirus 3/35++ technology previously described and is using it to deliver the CYBB gene to HSCs for the treatment of X-CGD[138]. Ensoma’s approach (a VLP product called EN-374) consists of two VLPs: one containing a Sleeping Beauty 100X transposase and Flp recombinase and another with a transposase, myeloid promoter and a copy of the CYBB gene. The second VLP also contains a methylguanine methyltransferase (MGMT) marker containing the P140K mutation which enables engineered HSCs to be enriched by the administration of temozolomide[138]. Ensoma Therapeutics is expected to open a trial of EN-374 in the United States and Europe in 2025 (NCT06876363). Patients will undergo mobilization of HSCs, followed by IV injection of the VLP product EN-374 with simultaneous immune prophylaxis. Patients will receive temozolomide following VLP infusion to enrich the engineered HSCs in vivo. The results of such a trial will be eagerly awaited, as if it is successful, similar approaches could be used for many other IEIs in which correction of the HSC compartment is desired.
In vivo GT represents the next frontier in the GT field. Given its transformational potential to reduce toxicity, costs, and complexity, it could ultimately reach many more patients than ex vivo approaches. By removing the need for bespoke cell harvesting, ex vivo manufacturing, cryostorage, and transplantation expertise, in vivo GT offers the potential to broaden access to patients in low- and middle-income countries. However, whether this promise can be realized will depend on parallel progress in addressing regulatory, infrastructural, and cost barriers that continue to limit equitable deployment of advanced therapies. Indeed, progressing in vivo GT worldwide is contingent on navigating stringent regulatory hurdles, including requirements for detailed biodistribution, long-term safety, and immunogenicity data. Platforms such as Ensoma’s VLP system are addressing these challenges through stepwise trial designs and close regulatory engagement, with forthcoming clinical studies expected to provide critical insights.
STATUS OF GENE THERAPY FOR IEI
Despite the promising preclinical development and clinical trials of GT approaches outlined in this review, these therapies remain experimental, and access to them is almost exclusively through clinical trials. In the United States, no GTs for IEIs are approved by the Federal Drug Administration (FDA). In the European Union a single GT, Strimvelis, a γ-retroviral-based HSC therapy for ADA-SCID is approved by the European Medicines Agency (EMA)[139]. In 2024, a patient treated with Strimvelis developed T cell leukemia 4.7 years after treatment because of retroviral integration at the LMO2 locus followed by acquisition of a complex set of somatic mutations[28]. The risk of mutagenesis is well characterized with γ-retroviral vectors but in ADA-SCID, it is mitigated in part by disease-related factors. After extensive investigation the EMA concluded that the risk/benefit analysis for Strimvelis remains favourable, and it remains approved in Europe albeit with robust monitoring arrangements[28]. The evidence for autologous GT is strongest for ADA-SCID with 100% overall survival and 90%-95% engraftment[111,140]. Recently updated treatment guidelines for the management of ADA-SCID recommend consideration of autologous GT for the condition when a matched family donor is not available[140]. This makes ADA-SCID, the only IEI for which GT is considered a standard of care. However, aside from Strimvelis which is a fresh cell product necessitating travel to the only treatment centre in Milan, GT can only be accessed through clinical trials. As of October 2025, there were trials of lentiviral HSC GT for ADA-SCID open and recruiting IN the United States (NCT05432310) and China (NCT03645460).
In other SCID IEIs, access is also limited to clinical trials. For X-SCID, there are trials of lentiviral vector approaches open and recruiting in the UK (NCT03601286), USA (NCT01306019, NCT03311503) and China (NCT03217617). Several trials are open and recruiting As of October 2025 for Artemis SCID (NCT03538899, NCT05071222) and RAG1-deficient SCID (NCT04797260). In the non-SCID context, there are trials open of GT for X-linked (NCT06559176 - prime editing of HSCs) and autosomal recessive (NCT05207657) CGD. A trial of a lentiviral vector modified T cell GT approach for IPEX syndrome is open and recruiting in the USA (NCT05241444). Several trials of GT (HSC and T cell) in other IEIs are expected to open soon (e.g., NCT06876363, NCT06876363).
The lack of availability to GT despite strong efficacy and safety data has now been recognized as a major issue for the field. The reasons are complex and are discussed in the next section along with potential solutions.
ADVANCES IN GENE THERAPY FOR IEI
Improving access to gene therapy
GT is not used in routine clinical practice. The reasons are twofold. Firstly, simultaneous advances in alloHSCT have reduced some of the impetus to develop and deliver autologous GTs. These advances, coupled with the current higher costs of GT compared to alloHSCT, have reduced the attractiveness of GT in financially constrained healthcare systems. The development of haploidentical transplant protocols has dramatically increased donor availability[141,142]. Reduced intensity conditioning protocols, coupled with targeted drug dosing, have reduced the toxicity associated with the procedure[143]. Advances in GVHD prophylaxis including graft manipulation have reduced GVHD and improved infection prophylaxis with reduced morbidity from infectious pathogens[144-147].
Despite the laudable advances in alloHSCT, autologous GT offers clear advantages, including universal applicability (no need for a donor) and the absence of GVHD risk. It also has a better safety and efficacy profile, although the number of patients treated remains small. The risks of mortality and morbidity associated with alloHSCT remain unacceptably high, particularly in the absence of a sibling donor[7,142,143,148]. Take ADA-SCID as an example. Five-year overall survival for alloHSCT preceded by enzyme replacement therapy was 79.6% compared to 100% for autologous GT[111]. Headline overall survival rates do not tell the whole story - reduced morbidity to both patient (and potentially an alloHSCT donor) due to lack of GVHD and reduced infectious complications due to a lack of immunosuppression all lead to improved quality of life in patients receiving GT versus alloHSCT[36]. As clinical experience with GT increases, the body of evidence to support the use of these more expensive autologous therapies will increase which will ultimately lower the costs for the health system.
However, even with increasing clinical data demonstrating improved survival and quality of life following GT compared to alloHSCT, a second reason explains the lack of widespread use of GT for IEIs. This reason is non-clinical and relates to the widespread commercial disinvestment in GTs for rare diseases that has occurred over the last decade[149-151]. In the rare and ultra-rare disease space, the costs of developing a novel GT product to the point of achieving market authorization are prohibitive and unattractive to for-profit pharmaceutical companies. There is now widespread recognition that in the rare disease space the current for-profit model of drug development is not fit for purpose.
Initiatives in both the United States and Europe are trying to address the challenge of access to GT for rare diseases. In Europe, the cross-border AGORA consortium (access to GT for rare diseases) was founded in 2022[149]. AGORA is working with key stakeholders from across the field to address current barriers to access. A similar taskforce has been established in the United States[152]. At present, international initiatives are largely region-specific, focused either in Europe or the United States due to differing regulatory frameworks; however, the issue of access is regularly discussed in forums such as the American Society of Gene and Cell Therapy (ASGCT) and European Society for Gene & Cell Therapy (ESGCT), which provide opportunities to share experience and align approaches across jurisdictions.
Some proposed solutions being explored include harmonised infrastructure for production, platform technology approvals and recognition of approvals across jurisdictions, standardized regulatory approach across different countries and data sharing to streamline safety assays[150]. Ultimately, it is anticipated that the use of non-traditional manufacturing and distribution models will be needed, for example, where an academic medical centre delivers a therapy on a per-patient basis under the European Union’s Hospital Exemption policy[149,152]. Many leading centers in the GT field, such as Great Ormond Street Hospital in London and San Raffaele in Milan, already operate Good Manufacturing Practices (GMP) manufacturing facilities, which could serve as prototypes for producing therapies at reduced cost within these emerging non-traditional models.
Non-genotoxic conditioning and epitope editing
Two major advances in the cell and GT field are expected to further increase access to ex vivo GT for IEIs by reducing the toxicity, complexity and costs of treatment: the development of non-genotoxic conditioning and the use of epitope editing to enable selective enrichment of engineered cells[153-155]. There are two main targets being explored for targeted immunotherapeutic conditioning approaches, c-Kit (CD117) and CD45[155-157]. Both antigens are broadly expressed on HSCs (CD45 30 times more than c-kit[155]). Approaches for both targets have demonstrated promising preclinical efficacy[157,158]. IEIs are a desirable first target indication for these approaches as in the non-malignant setting the anti-neoplastic effect of genotoxic agents is unwarranted. Both CD45 and c-Kit targeted approaches have been assessed in clinical trials in the alloHSCT setting for IEI. A total of 16 IEI patients were treated with a CD45 targeted antibody (in combination with fludarabine, cyclophosphamide and alemtuzumab for immunosuppression); 15 of these engrafted (94%) and 11 (69%) achieved full or high level chimerism in the myeloid and lymphoid lineages, suggesting that the approach can facilitate engraftment and warrants further investigation[159]. Another trial of single-agent c-kit targeted antibody JSP191 is under investigation in SCID patients (NCT0293064)[160]. Whilst successful engraftment has been observed, several patients did not achieve robust multilineage engraftment and as such, it is not yet clear whether this approach will be successful[160]. Anti c-kit or
Whilst non-genotoxic conditioning has shown mixed results in clinical trials a parallel advance has the potential to improve the efficacy of non-genotoxic conditioning is epitope editing. Epitope editing refers to the use of base or prime editing to change several amino acids in the antibody-binding epitope of a protein. This creates “stealth receptors” which have preserved function but shield the cell from targeting with an antibody or CAR. Proof-of-principle of epitope editing of HSCs at several loci including CD45 and c-kit has now been demonstrated[153,162]. Non-genotoxic conditioning in combination with epitope editing may enable selection of an engineered population of cells facilitating engraftment. Such approaches are currently being investigated.
CONCLUSIONS
Autologous GT has the potential to be an effective, safe, durable curative therapy for many IEIs, with less toxicity than alloHSCT. Both viral gene addition and gene editing can be used to correct or engineer autologous HSCs or T cells for therapeutic benefit. The choice of engineering technology and starting material depends on the target disease but we now have a versatile GT toolbox which can be applied to many IEIs. Autologous GT is now recommended as a standard of care for patients with ADA-SCID who lack a family donor. Despite this remarkable progress and recognition of clinical efficacy in treatment guidelines, access to GT for IEIs is almost exclusively restricted to clinical trials, in just a few treatment centres worldwide. Due to the rare nature of IEIs, the commercial development of GT approaches is not attractive to for-profit companies, which limits patient access to effective autologous therapies. Developments in GT, particularly in vivo delivery, may improve commercial interest, but issues surrounding marketing authorisation, costs, and regulatory hurdles need to be addressed before GT can emerge as a major therapeutic option. As the field advances, ensuring that the benefits of GT extend beyond early adopters and high-income settings is an ethical imperative, so that transformative treatments are accessible to all patients who need them.
DECLARATIONS
Author contributions
Conceptualization and manuscript drafting, manuscript revision: Torrance R, Orf K, Fox TA
All authors reviewed and approved the final manuscript.
Availability of data and materials
Not applicable.
Financial support and sponsorship
Torrance R was supported by the Medical Research Council (MR/N013867/1). Orf K was supported
Conflicts of interest
All authors declare that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
1. Poli MC, Aksentijevich I, Bousfiha AA, et al. Human inborn errors of immunity: 2024 update on the classification from the International Union of Immunological Societies Expert Committee. J Hum Immun. 2025;1:e20250003.
2. Boyle JM, Buckley RH. Population prevalence of diagnosed primary immunodeficiency diseases in the United States. J Clin Immunol. 2007;27:497-502.
3. Lankester AC, Albert MH, Booth C, et al; Inborn Errors Working Party of the European Society for Blood and Marrow Transplantation and the European Society for Immune Deficiencies. EBMT/ESID inborn errors working party guidelines for hematopoietic stem cell transplantation for inborn errors of immunity. Bone Marrow Transplant. 2021;56:2052-62.
4. Bach FH, Albertini RJ, Joo P, Anderson JL, Bortin MM. Bone-marrow transplantation in a patient with the Wiskott-Aldrich syndrome. Lancet. 1968;2:1364-6.
5. Castagnoli R, Delmonte OM, Calzoni E, Notarangelo LD. Hematopoietic stem cell transplantation in primary immunodeficiency diseases: current status and future perspectives. Front Pediatr. 2019;7:295.
6. Gennery AR, Lankester A; Inborn Errors Working Party (IEWP) of the European Society for Blood and Marrow Transplantation (EBMT). Long term outcome and immune function after hematopoietic stem cell transplantation for primary immunodeficiency. Front Pediatr. 2019;7:381.
7. Pai SY, Logan BR, Griffith LM, et al. Transplantation outcomes for severe combined immunodeficiency, 2000-2009. N Engl J Med. 2014;371:434-46.
8. Therrell BL, Padilla CD, Borrajo GJC, et al. Current status of newborn bloodspot screening worldwide 2024: a comprehensive review of recent activities (2020-2023). Int J Neonatal Screen. 2024;10:38.
9. Gratwohl A, Brand R, Frassoni F, et al; Acute and Chronic Leukemia Working Parties. Cause of death after allogeneic haematopoietic stem cell transplantation (HSCT) in early leukaemias: an EBMT analysis of lethal infectious complications and changes over calendar time. Bone Marrow Transplant. 2005;36:757-69.
10. Fox TA, Massey V, Lever C, et al. Pre-transplant immune dysregulation predicts for poor outcome following allogeneic haematopoietic stem cell transplantation in adolescents and adults with inborn errors of immunity (IEI). J Clin Immunol. 2025;45:64.
11. Heimall J, Puck J, Buckley R, et al. Current knowledge and priorities for future research in late effects after hematopoietic stem cell transplantation (HCT) for severe combined immunodeficiency patients: a consensus statement from the second pediatric blood and marrow transplant consortium international conference on late effects after pediatric HCT. Biol Blood Marrow Transplant. 2017;23:379-87.
12. Albert MH, Hauck F, Wiebking V, et al. Allogeneic stem cell transplantation in adolescents and young adults with primary immunodeficiencies. J Allergy Clin Immunol Pract. 2018;6:298-301.e2.
13. Mercola KE, Cline MJ. Sounding boards. The potentials of inserting new genetic information. N Engl J Med. 1980;303:1297-300.
14. Bordignon C, Notarangelo LD, Nobili N, et al. Gene therapy in peripheral blood lymphocytes and bone marrow for ADA- immunodeficient patients. Science. 1995;270:470-5.
15. Blaese RM, Culver KW, Miller AD, et al. T lymphocyte-directed gene therapy for ADA- SCID: initial trial results after 4 years. Science. 1995;270:475-80.
16. Aiuti A, Slavin S, Aker M, et al. Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science. 2002;296:2410-3.
17. Gaspar HB, Bjorkegren E, Parsley K, et al. Successful reconstitution of immunity in ADA-SCID by stem cell gene therapy following cessation of PEG-ADA and use of mild preconditioning. Mol Ther. 2006;14:505-13.
18. Candotti F, Shaw KL, Muul L, et al. Gene therapy for adenosine deaminase-deficient severe combined immune deficiency: clinical comparison of retroviral vectors and treatment plans. Blood. 2012;120:3635-46.
19. Cavazzana-Calvo M, Hacein-Bey S, de Saint Basile G, et al. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science. 2000;288:669-72.
20. Gaspar HB, Parsley KL, Howe S, et al. Gene therapy of X-linked severe combined immunodeficiency by use of a pseudotyped gammaretroviral vector. Lancet. 2004;364:2181-7.
21. Hacein-Bey-Abina S, Le Deist F, Carlier F, et al. Sustained correction of X-linked severe combined immunodeficiency by ex vivo gene therapy. N Engl J Med. 2002;346:1185-93.
22. Boztug K, Schmidt M, Schwarzer A, et al. Stem-cell gene therapy for the Wiskott-Aldrich syndrome. N Engl J Med. 2010;363:1918-27.
23. Braun CJ, Boztug K, Paruzynski A, et al. Gene therapy for Wiskott-Aldrich syndrome-long-term efficacy and genotoxicity. Sci Transl Med. 2014;6:227ra33.
24. Kang EM, Choi U, Theobald N, et al. Retrovirus gene therapy for X-linked chronic granulomatous disease can achieve stable long-term correction of oxidase activity in peripheral blood neutrophils. Blood. 2010;115:783-91.
25. Stein S, Ott MG, Schultze-Strasser S, et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med. 2010;16:198-204.
26. Fischer A. Gene therapy for inborn errors of immunity: past, present and future. Nat Rev Immunol. 2023;23:397-408.
27. Hacein-Bey-Abina S, Von Kalle C, Schmidt M, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415-9.
28. Cesana D, Cicalese MP, Calabria A, et al. A case of T-cell acute lymphoblastic leukemia in retroviral gene therapy for ADA-SCID. Nat Commun. 2024;15:3662.
29. Hacein-Bey-Abina S, Pai SY, Gaspar HB, et al. A modified γ-retrovirus vector for X-linked severe combined immunodeficiency. N Engl J Med. 2014;371:1407-17.
30. Thornhill SI, Schambach A, Howe SJ, et al. Self-inactivating gammaretroviral vectors for gene therapy of X-linked severe combined immunodeficiency. Mol Ther. 2008;16:590-8.
31. Mamcarz E, Zhou S, Lockey T, et al. Lentiviral gene therapy combined with low-dose busulfan in infants with SCID-X1. N Engl J Med. 2019;380:1525-34.
32. Ferrua F, Cicalese MP, Galimberti S, et al. Lentiviral haemopoietic stem/progenitor cell gene therapy for treatment of Wiskott-Aldrich syndrome: interim results of a non-randomised, open-label, phase 1/2 clinical study. Lancet Haematol. 2019;6:e239-53.
33. Magnani A, Semeraro M, Adam F, et al. Long-term safety and efficacy of lentiviral hematopoietic stem/progenitor cell gene therapy for Wiskott-Aldrich syndrome. Nat Med. 2022;28:71-80.
34. Booth C, Sevilla J, Almarza E, et al. Lentiviral gene therapy for severe leukocyte adhesion deficiency type 1. N Engl J Med. 2025;392:1698-709.
35. Kohn DB, Booth C, Kang EM, et al; Net4CGD consortium. Lentiviral gene therapy for X-linked chronic granulomatous disease. Nat Med. 2020;26:200-6.
36. Kohn DB, Booth C, Shaw KL, et al. Autologous ex vivo lentiviral gene therapy for adenosine deaminase deficiency. N Engl J Med. 2021;384:2002-13.
37. Aiuti A, Cattaneo F, Galimberti S, et al. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N Engl J Med. 2009;360:447-58.
38. Shaw KL, Garabedian E, Mishra S, et al. Clinical efficacy of gene-modified stem cells in adenosine deaminase-deficient immunodeficiency. J Clin Invest. 2017;127:1689-99.
39. Cicalese MP, Ferrua F, Castagnaro L, et al. Gene therapy for adenosine deaminase deficiency: a comprehensive evaluation of short- and medium-term safety. Mol Ther. 2018;26:917-31.
40. Gaspar HB, Cooray S, Gilmour KC, et al. Long-term persistence of a polyclonal T cell repertoire after gene therapy for X-linked severe combined immunodeficiency. Sci Transl Med. 2011;3:97ra79.
41. Cicalese MP, Ferrua F, Castagnaro L, et al. Update on the safety and efficacy of retroviral gene therapy for immunodeficiency due to adenosine deaminase deficiency. Blood. 2016;128:45-54.
42. Hacein-Bey-Abina S, Garrigue A, Wang GP, et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008;118:3132-42.
43. Howe SJ, Mansour MR, Schwarzwaelder K, et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest. 2008;118:3143-50.
44. Aiuti A, Biasco L, Scaramuzza S, et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science. 2013;341:1233151.
45. Kang HJ, Bartholomae CC, Paruzynski A, et al. Retroviral gene therapy for X-linked chronic granulomatous disease: results from phase I/II trial. Mol Ther. 2011;19:2092-101.
46. Schejtman A, Aragão-Filho WC, Clare S, et al. Lentiviral gene therapy rescues p47phox chronic granulomatous disease and the ability to fight Salmonella infection in mice. Gene Ther. 2020;27:459-69.
47. Masiuk KE, Laborada J, Roncarolo MG, Hollis RP, Kohn DB. Lentiviral gene therapy in HSCs restores lineage-specific Foxp3 expression and suppresses autoimmunity in a mouse model of IPEX syndrome. Cell Stem Cell. 2019;24:309-317.e7.
48. Takushi SE, Paik NY, Fedanov A, et al. Lentiviral Gene therapy for familial hemophagocytic lymphohistiocytosis type 3, caused by UNC13D genetic defects. Hum Gene Ther. 2020;31:626-38.
49. Ghosh S, Carmo M, Calero-Garcia M, et al. T-cell gene therapy for perforin deficiency corrects cytotoxicity defects and prevents hemophagocytic lymphohistiocytosis manifestations. J Allergy Clin Immunol. 2018;142:904-913.e3.
50. Hong Y, Casimir M, Houghton BC, et al. Lentiviral mediated ADA2 gene transfer corrects the defects associated with deficiency of adenosine deaminase type 2. Front Immunol. 2022;13:852830.
51. van Til NP, de Boer H, Mashamba N, et al. Correction of murine Rag2 severe combined immunodeficiency by lentiviral gene therapy using a codon-optimized RAG2 therapeutic transgene. Mol Ther. 2012;20:1968-80.
52. Panchal N, Houghton B, Diez B, et al. Transfer of gene-corrected T cells corrects humoral and cytotoxic defects in patients with X-linked lymphoproliferative disease. J Allergy Clin Immunol. 2018;142:235-245.e6.
53. Seymour BJ, Singh S, Certo HM, et al. Effective, safe, and sustained correction of murine XLA using a UCOE-BTK promoter-based lentiviral vector. Mol Ther Methods Clin Dev. 2021;20:635-51.
54. Hahn K, Pollmann L, Nowak J, et al. Human lentiviral gene therapy restores the cellular phenotype of autosomal recessive complete IFN-γR1 deficiency. Mol Ther Methods Clin Dev. 2020;17:785-95.
55. Garcia-Perez L, van Eggermond M, van Roon L, et al. Successful preclinical development of gene therapy for recombinase-activating gene-1-deficient SCID. Mol Ther Methods Clin Dev. 2020;17:666-82.
56. Cowan MJ, Yu J, Facchino J, et al. Lentiviral gene therapy for artemis-deficient SCID. N Engl J Med. 2022;387:2344-55.
57. Booth C, Sevilla J, Lopez MC, et al. Severe leukocyte adhesion deficiency-I (LAD-I) lentiviral-mediated ex-vivo gene therapy: ongoing phase 1/2 study results. Clinical Immunology. 2023;250:109354.
58. Kohn DB, Rao GR, Almarza E, et al. A phase 1/2 study of lentiviral-mediated ex-vivo gene therapy for pediatric patients with severe leukocyte adhesion deficiency-I (LAD-I): results from phase 1. Blood. 2020;136:15.
59. Fox TA, Houghton BC, Petersone L, et al. Therapeutic gene editing of T cells to correct CTLA-4 insufficiency. Sci Transl Med. 2022;14:eabn5811.
60. Brown MP, Topham DJ, Sangster MY, et al. Thymic lymphoproliferative disease after successful correction of CD40 ligand deficiency by gene transfer in mice. Nat Med. 1998;4:1253-60.
61. Sacco MG, Ungari M, Catò EM, et al. Lymphoid abnormalities in CD40 ligand transgenic mice suggest the need for tight regulation in gene therapy approaches to hyper immunoglobulin M (IgM) syndrome. Cancer Gene Ther. 2000;7:1299-306.
62. Liu X, Li G, Liu Y, Zhou F, Huang X, Li K. Advances in CRISPR/Cas gene therapy for inborn errors of immunity. Front Immunol. 2023;14:1111777.
63. Mudde A, Booth C. Gene therapy for inborn error of immunity - current status and future perspectives. Curr Opin Allergy Clin Immunol. 2023;23:51-62.
64. Bibikova M, Golic M, Golic KG, Carroll D. Targeted chromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases. Genetics. 2002;161:1169-75.
65. Gaj T, Gersbach CA, Barbas CF 3rd. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31:397-405.
66. Durai S, Mani M, Kandavelou K, Wu J, Porteus MH, Chandrasegaran S. Zinc finger nucleases: custom-designed molecular scissors for genome engineering of plant and mammalian cells. Nucleic Acids Res. 2005;33:5978-90.
67. Christian M, Cermak T, Doyle EL, et al. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics. 2010;186:757-61.
68. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816-21.
69. Wang JY, Doudna JA. CRISPR technology: a decade of genome editing is only the beginning. Science. 2023;379:eadd8643.
70. Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181-211.
71. van Overbeek M, Capurso D, Carter MM, et al. DNA repair profiling reveals nonrandom outcomes at Cas9-mediated breaks. Mol Cell. 2016;63:633-46.
72. Rouet P, Smih F, Jasin M. Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol Cell Biol. 1994;14:8096-106.
73. Rouet P, Smih F, Jasin M. Expression of a site-specific endonuclease stimulates homologous recombination in mammalian cells. Proc Natl Acad Sci U S A. 1994;91:6064-8.
74. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281-308.
75. Bak RO, Dever DP, Porteus MH. CRISPR/Cas9 genome editing in human hematopoietic stem cells. Nat Protoc. 2018;13:358-76.
76. Heyer WD, Ehmsen KT, Liu J. Regulation of homologous recombination in eukaryotes. Annu Rev Genet. 2010;44:113-39.
77. Kuo CY, Long JD, Campo-Fernandez B, et al. Site-specific gene editing of human hematopoietic stem cells for X-linked hyper-IgM syndrome. Cell Rep. 2018;23:2606-16.
78. Pavel-Dinu M, Wiebking V, Dejene BT, et al. Gene correction for SCID-X1 in long-term hematopoietic stem cells. Nat Commun. 2019;10:1634.
79. Rai R, Romito M, Rivers E, et al. Targeted gene correction of human hematopoietic stem cells for the treatment of Wiskott -Aldrich Syndrome. Nat Commun. 2020;11:4034.
80. Goodwin M, Lee E, Lakshmanan U, et al. CRISPR-based gene editing enables FOXP3 gene repair in IPEX patient cells. Sci Adv. 2020;6:eaaz0571.
81. De Ravin SS, Brault J, Meis RJ, et al. Enhanced homology-directed repair for highly efficient gene editing in hematopoietic stem/progenitor cells. Blood. 2021;137:2598-608.
82. Anzalone AV, Koblan LW, Liu DR. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol. 2020;38:824-44.
83. Huang TP, Heins ZJ, Miller SM, et al. High-throughput continuous evolution of compact Cas9 variants targeting single-nucleotide-pyrimidine PAMs. Nat Biotechnol. 2023;41:96-107.
84. Thuronyi BW, Koblan LW, Levy JM, et al. Continuous evolution of base editors with expanded target compatibility and improved activity. Nat Biotechnol. 2019;37:1070-9.
85. Richter MF, Zhao KT, Eton E, et al. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat Biotechnol. 2020;38:883-91.
86. Kim YB, Komor AC, Levy JM, Packer MS, Zhao KT, Liu DR. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat Biotechnol. 2017;35:371-6.
87. Landrum MJ, Lee JM, Benson M, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46:D1062-7.
88. Huang TP, Zhao KT, Miller SM, et al. Circularly permuted and PAM-modified Cas9 variants broaden the targeting scope of base editors. Nat Biotechnol. 2019;37:626-31.
89. Walton RT, Christie KA, Whittaker MN, Kleinstiver BP. Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science. 2020;368:290-6.
90. Miller SM, Wang T, Randolph PB, et al. Continuous evolution of SpCas9 variants compatible with non-G PAMs. Nat Biotechnol. 2020;38:471-81.
91. Anzalone AV, Randolph PB, Davis JR, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149-57.
92. Doman JL, Sousa AA, Randolph PB, Chen PJ, Liu DR. Designing and executing prime editing experiments in mammalian cells. Nat Protoc. 2022;17:2431-68.
93. Pluciennik A, Dzantiev L, Iyer RR, Constantin N, Kadyrov FA, Modrich P. PCNA function in the activation and strand direction of MutLα endonuclease in mismatch repair. Proc Natl Acad Sci U S A. 2010;107:16066-71.
94. Vavassori V, Mercuri E, Marcovecchio GE, et al. Modeling, optimization, and comparable efficacy of T cell and hematopoietic stem cell gene editing for treating hyper-IgM syndrome. EMBO Mol Med. 2021;13:e13545.
95. Castiello MC, Brandas C, Ferrari S, et al. Exonic knockout and knockin gene editing in hematopoietic stem and progenitor cells rescues RAG1 immunodeficiency. Sci Transl Med. 2024;16:eadh8162.
96. Gardner CL, Pavel-Dinu M, Dobbs K, et al. Gene editing rescues in vitro T cell development of RAG2-deficient induced pluripotent stem cells in an artificial thymic organoid system. J Clin Immunol. 2021;41:852-62.
97. Iancu O, Allen D, Knop O, et al. Multiplex HDR for disease and correction modeling of SCID by CRISPR genome editing in human HSPCs. Mol Ther Nucleic Acids. 2023;31:105-21.
98. Bahal S, Zinicola M, Moula SE, et al. Hematopoietic stem cell gene editing rescues B-cell development in X-linked agammaglobulinemia. J Allergy Clin Immunol. 2024;154:195-208.e8.
99. Rai R, Steinberg Z, Romito M, et al. CRISPR/Cas9-based disease modeling and functional correction of interleukin 7 receptor alpha severe combined immunodeficiency in T-Lymphocytes and hematopoietic stem cells. Hum Gene Ther. 2024;35:269-83.
100. De Ravin SS, Reik A, Liu PQ, et al. Targeted gene addition in human CD34+ hematopoietic cells for correction of X-linked chronic granulomatous disease. Nat Biotechnol. 2016;34:424-9.
101. Sweeney CL, Pavel-Dinu M, Choi U, et al. Correction of X-CGD patient HSPCs by targeted CYBB cDNA insertion using CRISPR/Cas9 with 53BP1 inhibition for enhanced homology-directed repair. Gene Ther. 2021;28:373-90.
102. Brault J, Liu T, Bello E, et al. CRISPR-targeted MAGT1 insertion restores XMEN patient hematopoietic stem cells and lymphocytes. Blood. 2021;138:2768-80.
103. Houghton BC, Panchal N, Haas SA, et al. Genome editing with TALEN, CRISPR-Cas9 and CRISPR-Cas12a in combination with AAV6 homology donor restores T cell function for XLP. Front Genome Ed. 2022;4:828489.
104. Pavel-Dinu M, Viel S, Selvaraj S, et al. Correcting autoinflammation in STING-associated vasculopathy with onset in infancy (SAVI) by human stem cell genome-editing. Research Square 2024.
105. McAuley GE, Yiu G, Chang PC, et al. Human T cell generation is restored in CD3δ severe combined immunodeficiency through adenine base editing. Cell. 2023;186:1398-1416.e23.
106. Bzhilyanskaya V, Ma L, Liu S, et al. High-fidelity PAMless base editing of hematopoietic stem cells to treat chronic granulomatous disease. Sci Transl Med. 2024;16:eadj6779.
107. Heath JM, Orenstein JS, Tedeschi JG, et al. Prime editing efficiently and precisely corrects causative mutation in chronic granulomatous disease, restoring myeloid function: toward development of a prime edited autologous hematopoietic stem cell therapy. Blood. 2023;142:7129.
108. Dettmer-Monaco V, Weißert K, Ammann S, et al. Gene editing of hematopoietic stem cells restores T-cell response in familial hemophagocytic lymphohistiocytosis. J Allergy Clin Immunol. 2024;153:243-255.e14.
109. Nasri M, Ritter MU, Mir P, et al. CRISPR-Cas9n-mediated ELANE promoter editing for gene therapy of severe congenital neutropenia. Mol Ther. 2024;32:1628-42.
110. Fiumara M, Ferrari S, Omer-Javed A, et al. Genotoxic effects of base and prime editing in human hematopoietic stem cells. Nat Biotechnol. 2024;42:877-91.
111. Cuvelier GDE, Logan BR, Prockop SE, et al. Outcomes following treatment for ADA-deficient severe combined immunodeficiency: a report from the PIDTC. Blood. 2022;140:685-705.
112. Egg D, Rump IC, Mitsuiki N, et al. Therapeutic options for CTLA-4 insufficiency. J Allergy Clin Immunol. 2022;149:736-46.
113. Panchal N, Ghosh S, Booth C. T cell gene therapy to treat immunodeficiency. Br J Haematol. 2021;192:433-43.
114. Frangoul H, Locatelli F, Sharma A, et al; CLIMB SCD-121 Study Group. Exagamglogene autotemcel for severe sickle cell disease. N Engl J Med. 2024;390:1649-62.
115. Albert MH, Slatter M, Gennery A, et al. Busulfan/fludarabine- or treosulfan/fludarabine-based conditioning regimen in patients with Wiskott-Aldrich syndrome given allogeneic hematopoietic cell transplantation - an EBMT inborn errors working party and scetide retrospective analysis. Blood. 2018;132:2175.
116. Tsilifis C, Speckmann C, Lum SH, et al. Hematopoietic stem cell transplantation for CTLA-4 insufficiency across Europe: a European Society for Blood and Marrow Transplantation Inborn Errors Working Party study. J Allergy Clin Immunol. 2024;154:1534-44.
117. Ferrari S, Jacob A, Cesana D, et al. Choice of template delivery mitigates the genotoxic risk and adverse impact of editing in human hematopoietic stem cells. Cell Stem Cell. 2022;29:1428-44.e9.
118. Asperti C, Canarutto D, Porcellini S, et al. Scalable GMP-compliant gene correction of CD4+ T cells with IDLV template functionally validated in vitro and in vivo. Mol Ther Methods Clin Dev. 2023;30:546-57.
119. Melenhorst JJ, Chen GM, Wang M, et al. Decade-long leukaemia remissions with persistence of CD4+ CAR T cells. Nature. 2022;602:503-9.
120. Passerini L, Mel ER, Sartirana C, et al. CD4+ T cells from IPEX patients convert into functional and stable regulatory T cells by FOXP3 gene transfer. Sci Transl Med. 2013;5:215ra174.
121. Hubbard N, Hagin D, Sommer K, et al. Targeted gene editing restores regulated CD40L function in X-linked hyper-IgM syndrome. Blood. 2016;127:2513-22.
122. Pai SY. Treatment of primary immunodeficiency with allogeneic transplant and gene therapy. Hematology Am Soc Hematol Educ Program. 2019;2019:457-65.
123. Pai SY, Cowan MJ. Stem cell transplantation for primary immunodeficiency diseases: the North American experience. Curr Opin Allergy Clin Immunol. 2014;14:521-6.
124. Albert MH, Sirait T, Eikema DJ, et al. Hematopoietic stem cell transplantation for adolescents and adults with inborn errors of immunity: an EBMT IEWP study. Blood. 2022;140:1635-49.
125. Burns SO, Morris EC. How I use allogeneic HSCT for adults with inborn errors of immunity. Blood. 2021;138:1666-76.
126. Morris EC, Fox T, Chakraverty R, et al. Gene therapy for Wiskott-Aldrich syndrome in a severely affected adult. Blood. 2017;130:1327-35.
127. Pipe SW, Leebeek FWG, Recht M, et al. Gene therapy with etranacogene dezaparvovec for hemophilia B. N Engl J Med. 2023;388:706-18.
128. Ozelo MC, Mahlangu J, Pasi KJ, et al; GENEr8-1 Trial Group. Valoctocogene roxaparvovec gene therapy for hemophilia A. N Engl J Med. 2022;386:1013-25.
129. Gillmore JD, Gane E, Taubel J, et al. CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N Engl J Med. 2021;385:493-502.
130. Maestro S, Weber ND, Zabaleta N, Aldabe R, Gonzalez-Aseguinolaza G. Novel vectors and approaches for gene therapy in liver diseases. JHEP Rep. 2021;3:100300.
131. Lee-Six H, Øbro NF, Shepherd MS, et al. Population dynamics of normal human blood inferred from somatic mutations. Nature. 2018;561:473-8.
132. Li C, Georgakopoulou A, Mishra A, et al. In vivo HSPC gene therapy with base editors allows for efficient reactivation of fetal γ-globin in β-YAC mice. Blood Adv. 2021;5:1122-35.
133. Li C, Georgakopoulou A, Newby GA, et al. In vivo base editing by a single i.v. vector injection for treatment of hemoglobinopathies. JCI Insight. 2022;7:e162939.
134. Li C, Wang H, Gil S, et al. Safe and efficient in vivo hematopoietic stem cell transduction in nonhuman primates using HDAd5/35++ vectors. Mol Ther Methods Clin Dev. 2022;24:127-41.
135. Wang H, Germond A, Li C, et al. In vivo HSC transduction in rhesus macaques with an HDAd5/3+ vector targeting desmoglein 2 and transiently overexpressing cxcr4. Blood Adv. 2022;6:4360-72.
136. Breda L, Papp TE, Triebwasser MP, et al. In vivo hematopoietic stem cell modification by mRNA delivery. Science. 2023;381:436-43.
137. Shi D, Toyonaga S, Anderson DG. In vivo RNA delivery to hematopoietic stem and progenitor cells via targeted lipid nanoparticles. Nano Lett. 2023;23:2938-44.
138. Kattula S, Rajani GM, Chandra V, et al.
139. Valsecchi MC. Rescue of an orphan drug points to a new model for therapies for rare diseases. Nat Italy. 2023.
140. Grunebaum E, Booth C, Cuvelier GDE, Loves R, Aiuti A, Kohn DB. Updated management guidelines for adenosine deaminase deficiency. J Allergy Clin Immunol Pract. 2023;11:1665-75.
141. Bertaina A, Merli P, Rutella S, et al. HLA-haploidentical stem cell transplantation after removal of αβ+ T and B cells in children with nonmalignant disorders. Blood. 2014;124:822-6.
142. Kurzay M, Hauck F, Schmid I, et al. T-cell replete haploidentical bone marrow transplantation and post-transplant cyclophosphamide for patients with inborn errors. Haematologica. 2019;104:e478-82.
143. Fox TA, Chakraverty R, Burns S, et al. Successful outcome following allogeneic hematopoietic stem cell transplantation in adults with primary immunodeficiency. Blood. 2018;131:917-31.
144. Balashov D, Shcherbina A, Maschan M, et al. Single-center experience of unrelated and haploidentical stem cell transplantation with TCRαβ and CD19 depletion in children with primary immunodeficiency syndromes. Biol Blood Marrow Transplant. 2015;21:1955-62.
145. Shah RM, Elfeky R, Nademi Z, et al. T-cell receptor αβ+ and CD19+ cell-depleted haploidentical and mismatched hematopoietic stem cell transplantation in primary immune deficiency. J Allergy Clin Immunol. 2018;141:1417-26.e1.
146. Slatter MA, Gennery AR. Hematopoietic cell transplantation in primary immunodeficiency - conventional and emerging indications. Expert Rev Clin Immunol. 2018;14:103-14.
147. Marty FM, Ljungman P, Chemaly RF, et al. Letermovir prophylaxis for cytomegalovirus in hematopoietic-cell transplantation. N Engl J Med. 2017;377:2433-44.
148. Chiesa R, Wang J, Blok HJ, et al. Hematopoietic cell transplantation in chronic granulomatous disease: a study of 712 children and adults. Blood. 2020;136:1201-11.
149. Fox T, Bueren J, Candotti F, et al; AGORA Initiative. Access to gene therapy for rare diseases when commercialization is not fit for purpose. Nat Med. 2023;29:518-9.
151. Aiuti A, Pasinelli F, Naldini L. Ensuring a future for gene therapy for rare diseases. Nat Med. 2022;28:1985-8.
152. Kliegman M, Zaghlula M, Abrahamson S, et al. A roadmap for affordable genetic medicines. Nature. 2024;634:307-14.
153. Casirati G, Cosentino A, Mucci A, et al. Epitope editing enables targeted immunotherapy of acute myeloid leukaemia. Nature. 2023;621:404-14.
154. Kwon HS, Logan AC, Chhabra A, et al. Anti-human CD117 antibody-mediated bone marrow niche clearance in nonhuman primates and humanized NSG mice. Blood. 2019;133:2104-8.
155. Cavazzana M, Calvo C. A new step toward non-genotoxic conditioning prior to hematopoietic stem cell transplantation. Mol Ther. 2024;32:1604-5.
156. Pang WW, Czechowicz A, Logan AC, et al. Anti-CD117 antibody depletes normal and myelodysplastic syndrome human hematopoietic stem cells in xenografted mice. Blood. 2019;133:2069-78.
157. Saha A, Hyzy S, Lamothe T, et al. A CD45-targeted antibody-drug conjugate successfully conditions for allogeneic hematopoietic stem cell transplantation in mice. Blood. 2022;139:1743-59.
158. Uchida N, Stasula U, Demirci S, et al. Fertility-preserving myeloablative conditioning using single-dose CD117 antibody-drug conjugate in a rhesus gene therapy model. Nat Commun. 2023;14:6291.
159. Straathof KC, Rao K, Eyrich M, et al. Haemopoietic stem-cell transplantation with antibody-based minimal-intensity conditioning: a phase 1/2 study. Lancet. 2009;374:912-20.
160. Agarwal R, Dvorak CC, Kwon H, et al. Non-genotoxic anti-CD117 antibody conditioning results in successful hematopoietic stem cell engraftment in patients with severe combined immunodeficiency. Blood. 2019;134:800-800.
161. Arai Y, Choi U, Corsino CI, et al. Myeloid conditioning with c-kit-Targeted CAR-T cells enables donor stem cell engraftment. Mol Ther. 2018;26:1181-97.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].