



Exploring the functional application of polymeric materials on photo- and electrocatalytic CO2 reduction reaction
 0
0 
Abstract
The electrochemical and photochemical conversion of CO2 into value-added chemicals and fuels presents a viable pathway toward carbon neutrality and sustainable energy solutions. Electrocatalysts and photocatalysts play a central role in this conversion by lowering activation barriers and steering reaction pathways toward desired products under ambient conditions. Among various types of catalytic materials, polymeric materials have emerged as a promising class of catalysts and functional supports due to their structural versatility, tunable functionality, and capacity to tailor the local reaction environment. This review provides a comprehensive overview of recent progress in polymer-based systems for photocatalytic and electrocatalytic CO2 reduction reactions (pCO2RR and
Keywords
INTRODUCTION
The extensive reliance on carbon-rich fossil fuels over the past two centuries has significantly elevated atmospheric CO2 concentrations, disrupting the global carbon cycle and accelerating environmental challenges such as global warming, ocean acidification, and other extreme weather events[1-5]. Mitigating these adverse impacts requires a multifaceted approach that includes reducing fossil fuel consumption, transitioning to renewable energy sources, and actively removing CO2 from the atmosphere[6-10].
In this context, the catalytic conversion of CO2 into value-added chemicals such as carbon monoxide (CO)[11,12], formic acid (HCOOH)[13,14], formate (HCOO-)[15,16], methanol (CH3OH)[17,18], methane (CH4)[19,20], ethylene (C2H4)[21,22], and ethanol (C2H5OH)[23,24] has emerged as a promising strategy for achieving carbon neutrality. Among the various strategies for CO2 utilization, photocatalytic and electrocatalytic CO2 reduction reactions (pCO2RR and eCO2RR) have garnered significant interest, as they offer sustainable routes for converting CO2 from an environmental pollutant into value-added chemical feedstocks, thereby contributing to a closed-loop carbon cycle[25-29].
Photocatalysts have gained attention for their ability to drive chemical reactions under mild conditions using solar energy, without requiring high temperatures, pressures, or external power input[30]. They have been widely applied in environmental remediation, where they degrade organic pollutants such as dyes[31], pharmaceuticals[32], and industrial residues without the need for harsh reagents, offering sustainable solutions for air and water purification[33]. In the field of renewable energy, photocatalytic water splitting provides a clean route to hydrogen production[34]. Of particular relevance, the pCO2RR enables the direct transformation of CO2 into fuels and chemical feedstocks, thereby contributing to carbon recycling and offering a pathway toward carbon neutrality.
In contrast, eCO2RR relies on an applied external potential to drive the reduction of CO2 at the cathode, typically coupled with oxidation reactions at the anode[35]. This process demands catalysts with high electrical conductivity, abundant active sites, and electrochemical stability[36-38]. Compared to pCO2RR,
Polymeric materials have recently emerged as promising candidates for both pCO2RR and eCO2RR, offering distinct advantages over conventional metal-based catalysts. Unlike noble metals such as Ag, Au, and Pd, which are expensive and pose environmental concerns, polymer-based systems are more suitable and
In this review, to the best of our knowledge, we present the first comprehensive summary that integrates the application of polymeric materials in both pCO2RR and eCO2RR. We focus on how polymeric materials contribute to performance enhancement through their diverse roles in structural design, charge transport, and interfacial modulation [Figure 1]. We begin by outlining the fundamental mechanisms of pCO2RR and eCO2RR, respectively. Then, we categorize recent advances in polymer-based photocatalysts over the past five years according to their functional roles, followed by a similar classification for electrocatalytic systems. Finally, we discuss the current challenges and propose future research directions in this emerging field.
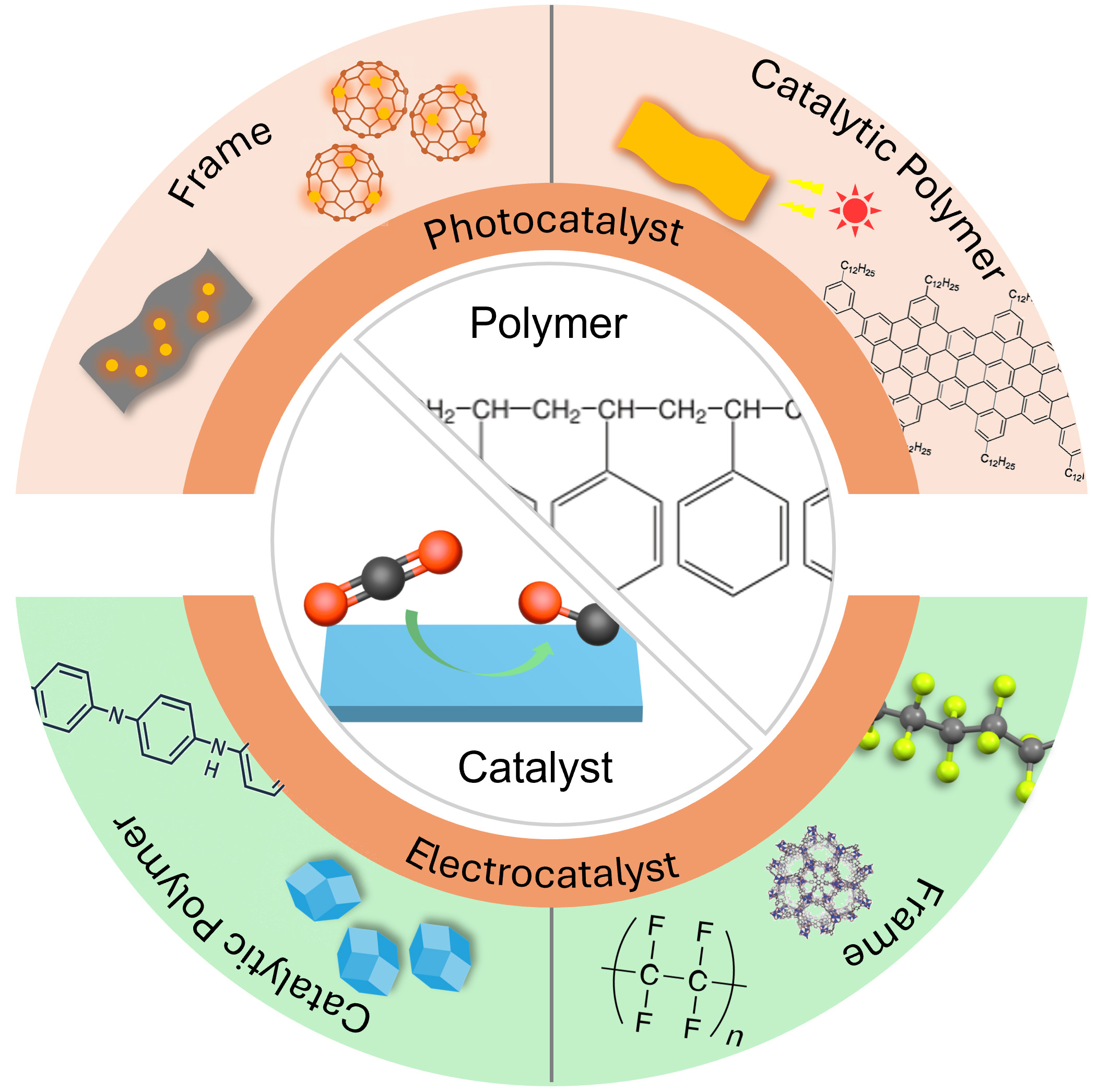
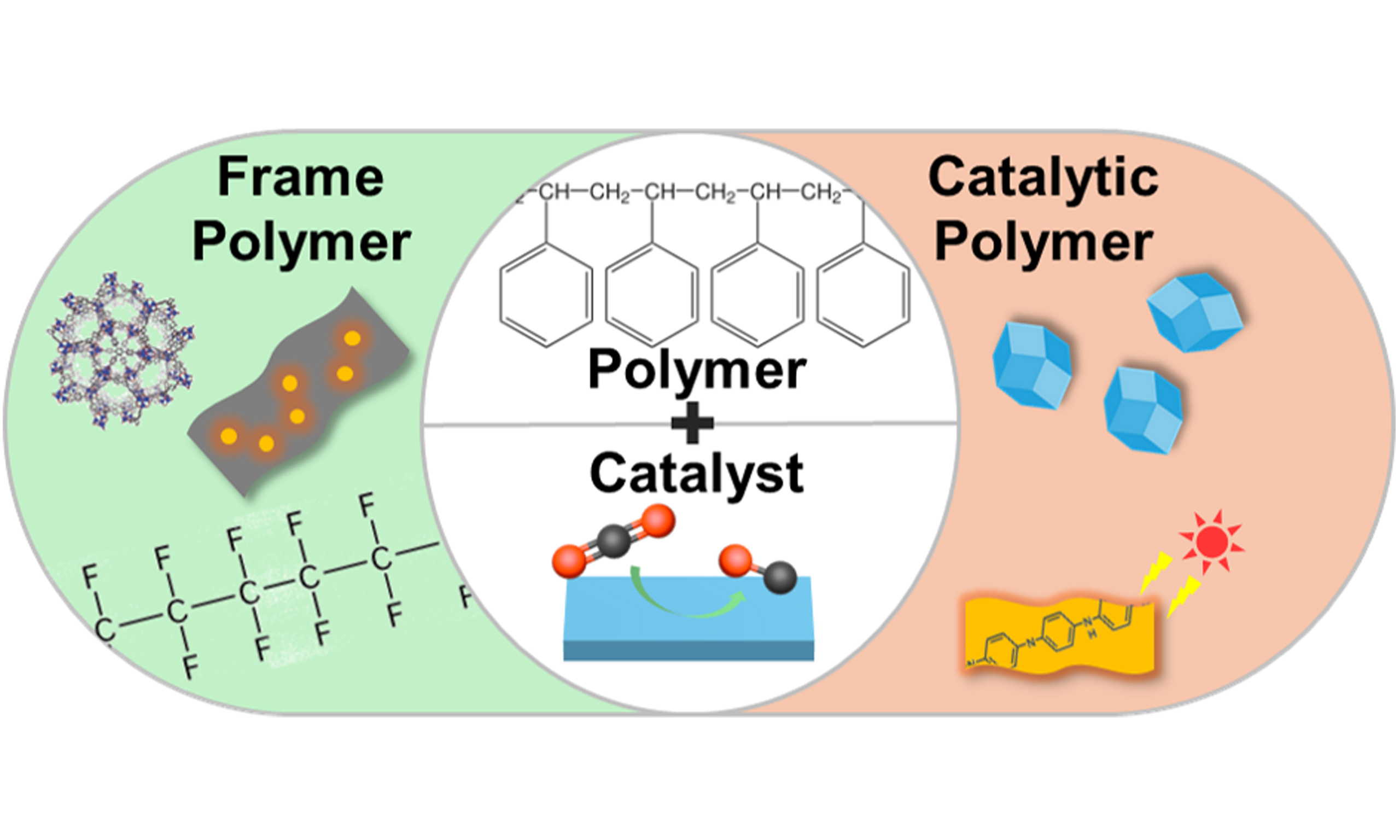
Figure 1. Overview of the functional application of polymeric materials on pCO2RR and eCO2RR. pCO2RR: Photocatalytic CO2 reduction reactions; eCO2RR: electrocatalytic CO2 reduction reactions.
As a continuation of this perspective, polymers play an increasingly important role in advancing photo- and electrocatalytic systems for CO2 reduction by enabling precise control over structural and electronic properties. Beyond merely forming porous networks, polymer materials provide versatile platforms to engineer catalytic environments through their tunable molecular architectures. The ability to design polymers with tailored functional groups and backbones enables the modulation of surface chemistry, facilitating uniform dispersion of catalytic sites and enhancing CO2 adsorption and activation.
Additionally, extended π-conjugation within polymer frameworks enhances visible-light absorption and promotes efficient charge separation and transport, reducing electron-hole recombination and supporting multi-electron transfer processes essential for complex CO2 reduction reactions.
The inherent flexibility and processability of polymers enable their integration into diverse device architectures, including thin films, coatings, and hybrid composites. Unlike rigid inorganic materials, polymers can be processed via scalable solution-based techniques, facilitating their application in practical devices. Furthermore, the dynamic nature of polymer chains can impart structural adaptability under reaction conditions, potentially stabilizing active sites and maintaining catalytic performance over time.
By leveraging these structural and functional advantages, ranging from molecular design and electronic tuning to device integration, polymer-based catalysts demonstrate a unique ability to bridge fundamental materials chemistry with enhanced catalytic function. This comprehensive approach highlights the importance of polymeric systems in developing efficient, stable, and selective CO2 photoreduction and electroreduction technologies and addresses the need for a more systematic analysis of structure-property-performance relationships.
FUNDAMENTAL PRINCIPLE FOR CO2 REDUCTION
PCO2RR mechanism
Photocatalytic reactions are complex processes in which light energy is absorbed to drive chemical transformations. These reactions are generally described by the following three key steps [Figure 2A]. First, when a photocatalyst absorbs photons with energy equal to or greater than its bandgap, electrons in the valence band (VB) are excited to the conduction band (CB). This excitation process leaves behind holes (h+) in the VB and generates mobile electrons (e-) in the CB. The pair formed by these charge carriers is referred to as an electron-hole pair. The bandgap energy (Eg) of the photocatalyst determines the minimum energy of light that can be absorbed and follows the relation Eg = hν, where h is Planck’s constant and ν is the frequency of the incident light.
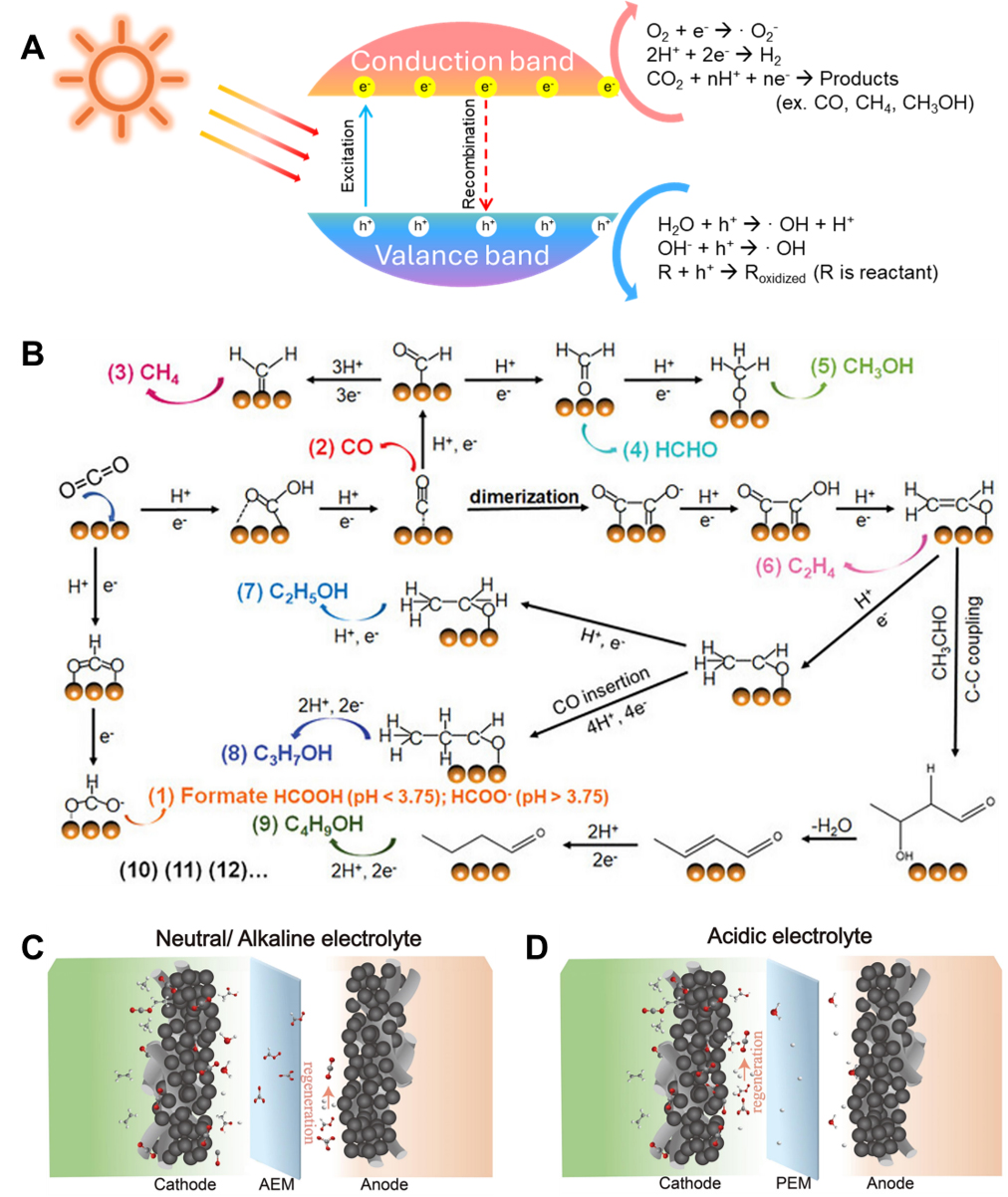
Figure 2. (A) Schematic illustration showing the pCO2RR mechanism; (B) Illustration of typical eCO2RR mechanism[47]. Copyright 2023 John Wiley & Sons; Schematic showing the eCO2RR mechanism in (C) Neutral/alkaline electrolyte and (D) Acidic electrolyte[53]. Copyright 2023 Elsevier. pCO2RR: Photocatalytic CO2 reduction reactions; eCO2RR: electrocatalytic CO2 reduction reactions.
Second, the photogenerated electron-hole pairs begin to migrate within the photocatalyst. In an ideal photocatalyst, these charge carriers efficiently travel to the surface where they can interact with adsorbed reactants. However, in practice, a significant portion of electron-hole pairs recombine before reaching the surface, releasing energy as heat or light. Such recombination is a major factor that limits photocatalytic efficiency. Therefore, suppressing charge recombination and promoting effective charge separation and transport are crucial in photocatalyst design. This can be achieved through strategies such as optimizing the crystal structure, surface modification, heterojunction engineering, and the introduction of cocatalysts.
Third, once electrons and holes reach the photocatalyst surface, they engage in redox reactions with the adsorbed reactants. Holes (h+) in the valence band possess strong oxidative power and can react with water (H2O) or hydroxide ions (OH-) adsorbed on the catalyst surface to produce hydroxyl radicals (•OH), which are highly reactive oxidizing agents. Additionally, the holes can directly oxidize or degrade organic pollutants through surface reactions.
Electrons in the conduction band possess reductive potential and can participate in various reduction reactions by interacting with adsorbed species such as oxygen (O2), hydrogen ions (H+), or carbon dioxide (CO2) on the catalyst surface. For instance, molecular oxygen can be reduced to form superoxide radicals (•O2-), which may further engage in subsequent redox processes.
To achieve the formation of multicarbon (C2+) products, the adsorbed intermediates should remain on the catalyst surface to undergo further transformation. After the formation of a CO2 radical anion, if it does not become desorbed, a new CO2 radical anion can be generated. The coupling of two CO2 radical anions leads to C-C bond formation, producing C2 products such as C2H4, C2H5OH, ethane (C2H6), and acetic acid
ECO2RR mechanism
CO2 is a highly stable molecule, requiring an activation energy of ~750 kJ mol-1 to cleave the C=O bond[47]. The eCO2RR involves sequential steps: CO2 adsorption, formation of a CO2•-, proton-coupled electron transfer (PCET), and product formation. These steps entail significant energy barriers, necessitating an external potential. As shown in Figure 2B, CO2 adsorbs onto the electrocatalyst surface and receives an electron to form a CO2- intermediate. This species undergoes PCET to generate either *OCHO or *COOH intermediates. In the *OCHO pathway, oxygen binds to the surface and the carbon is protonated, forming HCOOH or HCOO- depending on pH. In the *COOH pathway, carbon binds to the surface, leading to *CO formation. Weak *CO binding favors CO release, while strong binding drives further reduction to monocarbon (C1) products like CH4 and CH3OH or to C2+ products such as C2H4 and C2H5OH via *CO dimerization and successive PCET steps. Among these routes, the formation of C2+ products is critically governed by the C-C coupling process. A commonly accepted mechanism involves the dimerization of two *CO intermediates under strong *CO binding conditions to yield *CHCO species. From these intermediates, multiple pathways may proceed. Conventionally, *CHCO can be reduced to *OCCH2, which either continues along a pathway involving five PCET steps to produce C2H4, or undergoes desorption and reacts with OH- in solution to form acetate. Alternatively, less conventional but recently explored pathways suggest that *CHCO can be further reduced to *CH2CO and then to *OCHCH2, a key branching intermediate. This species may then be reduced to acetaldehyde or C2H5OH, or it can undergo additional coupling with *CO or *COH species, eventually leading to the formation of higher-value C3 products such as n-propanol through continued PCET steps[48].
A conventional three-electrode H-type cell, consisting of a working electrode, counter electrode, and reference electrode, is commonly employed in fundamental studies of eCO2RR. However, due to its limited mass transport and low current density, this configuration is unsuitable for practical applications. To overcome these limitations and achieve higher current densities, the electrocatalyst is typically supported on a gas diffusion electrode (GDE) and operated within a flow cell system, which enables CO2 delivery to the catalyst surface. To ensure uniform catalyst distribution on the GDE, spray-coating methods are commonly employed during catalyst deposition. The catalyst composition also critically influences the selectivity of CO2 reduction products. Metal-based catalysts are widely used, with Au, Ag, and Zn predominantly yielding CO[49], while Sn, In, and Bi mainly produce HCOOH or HCOO-[50]. Among various metal catalysts, Cu is uniquely capable of facilitating the formation of C2+ products with relatively high activity and selectivity, making it the most promising candidate for industrial-scale eCO2RR[51].
Aqueous electrolytes are widely employed in eCO2RR systems owing to their low cost, ease of handling, and tunable physicochemical properties[52]. These media inherently contain H+ and OH- species, with the solution pH governed by the balance between H+ and OH- concentrations. The pH not only modulates the equilibrium between dissolved CO2 and bicarbonate species but also plays a critical role in determining reaction pathways by affecting the stabilization of key intermediates. Since protonation steps are essential in both CO2 reduction and the competing hydrogen evolution reaction (HER), the local proton concentration exerts a significant influence on product selectivity during the process.
Neutral/alkaline environment
Currently, most eCO2RRs are conducted in neutral or alkaline electrolytes, where each system offers distinct benefits and limitations[53,54]. Alkaline media, for instance, provide higher CO2 solubility, lower overpotentials, and ample OH- availability, which can enhance catalytic performance. However, both alkaline and neutral electrolytes suffer from significant CO2 losses due to parasitic reactions that lead to carbonate formation (2OH- + CO2 → CO32- + H2O). This reaction occurs between CO2 and OH- ions present in the bulk solution or generated locally during the reduction process, thereby depleting the available carbon efficiency.
Although neutral electrolytes partially mitigate carbon loss via reduced carbonate regeneration, they face other performance challenges. In particular, during the anodic oxygen evolution reaction (OER), rapid consumption of OH- near the electrode surface hampers ion transport, severely impairing reaction kinetics. As a result, neutral eCO2RR systems typically exhibit substantial energy losses at the anode, representing a major bottleneck to their industrial application. Taken together, CO2 loss due to carbonate formation and anodic energy inefficiencies are key limitations that must be addressed to improve the overall efficiency and scalability of eCO2RR systems operating in neutral or alkaline environments [Figure 2C].
To address these limitations, three-electrode cell systems often adopt asymmetric electrolyte configurations, wherein different electrolytes are employed in the anode and cathode compartments. For example, alkaline electrolytes such as KOH are typically used at the anode to facilitate OER kinetics, while neutral electrolytes like KHCO3 are used at the cathode to minimize carbonate formation and maintain CO2 availability. This asymmetric design enables the optimization of individual half-cell environments, thereby improving overall energy efficiency and carbon utilization in eCO2RR systems.
Acidic environment
In contrast to neutral and alkaline systems, CO2 loss via carbonate formation can be effectively circumvented under acidic conditions. The eCO2RR in acidic media is a promising pathway to enhance carbon utilization efficiency by eliminating carbonate formation and minimizing CO2 crossover[54]. This is typically achieved through the use of a proton exchange membrane, which physically separates the cathode and anode compartments, thereby suppressing the migration of carbonate ions and other eCO2RR intermediates or products.
In principle, if H3O+ ions serve as the primary proton source, OH- ions are not generated, enabling eCO2RR to proceed without the formation of carbonate species. However, when H2O acts as the H+ donor, local generation of OH- becomes unavoidable, particularly at the cathode surface, potentially leading to carbonate formation. Under such conditions, the carbonate formed near the electrode may be reconverted to CO2 through reaction with H+ from the acidic bulk electrolyte, effectively confining carbonate cycling to the cathode interface [Figure 2D].
Despite these advantages, eCO2RR in acidic media faces significant challenges due to the dominant HER, which competes with eCO2RR by utilizing H+ more readily. This competition lowers the catalytic performance, especially for C2+ products that require multiple proton-electron transfers. Therefore, to enhance eCO2RR performance under acidic conditions, it is essential to develop catalysts and systems capable of modulating the local pH near the cathode through the rational design of the catalyst, electrolyte, and operating parameters.
POLYMER-BASED PHOTOCATALYST FOR PCO2RR
This section reviews recent advances, underlying principles, performance enhancements, and potential applications associated with these two polymer-based strategies. The incorporation of polymers into photocatalytic systems has opened new avenues for overcoming the limitations of conventional inorganic photocatalysts and for achieving improved efficiency. Polymers not only possess intrinsic photoactivity in some cases but can also be combined with other photocatalytic materials in various forms to generate synergistic effects. Here, we explore in detail the two primary roles of polymers in photocatalytic systems: as structural scaffolds and as active or directly functioning as the photocatalyst. This role-based classification provides insight into design strategies and performance enhancement mechanisms for polymer-based photocatalysts.
Polymer-supported architectures for photocatalysts
Commonly used inorganic photocatalysts such as TiO2[55-57], ZnO[58-60], and BiVO4[61-63] exhibit excellent photoactivity; however, they often suffer from aggregation when used in nanoparticle form, and their recovery and reuse remain challenging. These issues can be effectively addressed through the use of polymer supports. Polymers, with their large surface area, porous architecture, flexible processability, and tunable functionalization, provide an ideal platform for immobilizing and uniformly dispersing inorganic photocatalyst particles. By anchoring photocatalysts onto polymer supports, the stability of the photocatalytic system can be enhanced, the reaction efficiency maximized, and the recyclability significantly improved. Moreover, specific functional groups present in the polymer matrix can strengthen interactions with reactants or facilitate charge transport pathways, thereby further boosting the overall photocatalytic performance.
Zhao et al. employed Ru(bpy)32+ (RuC) as a chromophore and a [Ru(tpy)(6-mbpy)(CH3CN)]2+ (RuCat) derivative as the CO2 reduction catalyst[64]. Polystyrene (PS) was utilized as a mechanical framework to support the catalysts, leading to the formation of a PS-RuC/RuCat hybrid structure [Figure 3A]. Photophysical analyses revealed that the metal-to-ligand charge transfer (MLCT) excited state of the RuC unit was significantly quenched in the presence of RuCat [Figure 3B]. Transient absorption spectroscopy further confirmed that the luminescence lifetime was shortened due to energy transfer from RuC to RuCat, indicating the activation of non-radiative decay channels and enhanced charge mobility, both of which contribute to improved catalytic efficiency. Photocatalytic experiments were conducted under visible light irradiation (λ > 505 nm, Xe lamp, 100 mW/cm2) in N,N-Dimethylformamide (DMF)/triethanolamine (TEOA) (4:1) containing 0.1 M
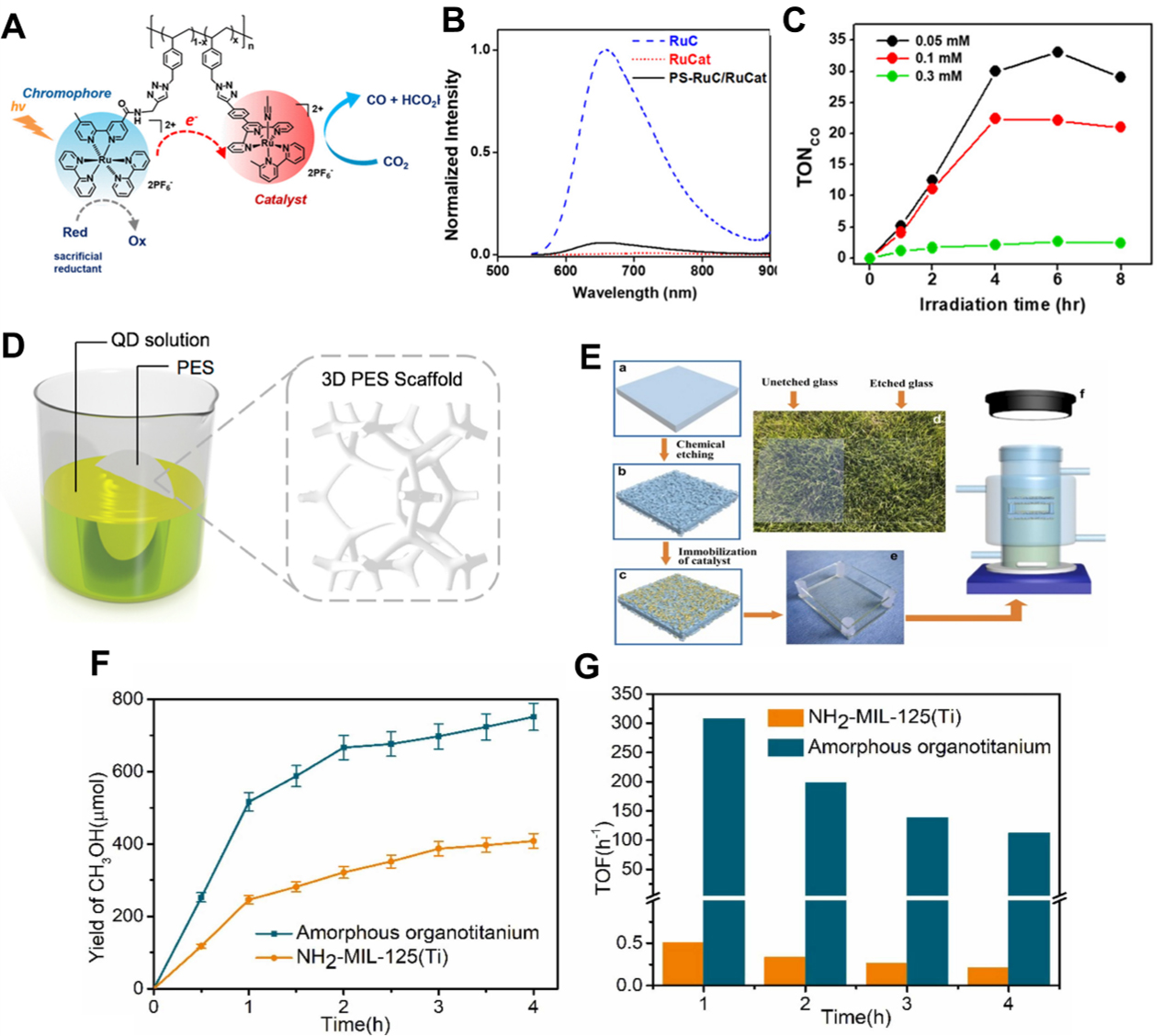
Figure 3. (A) Schematic illustration of PS-RuC/RuCat; (B) Emission spectra of RuC, RuCat, and Ps-RuC/RuCat; (C) TON of PS-RuCat as a function of irradiation time using a 300 W Xe arc light source (100 mW/cm2 at the sample) with a 505 nm long-pass filter[64]. Copyright 2021 American Chemical Society; (D) Schematic illustration of PQD/PES by self-attaching method[66]. Copyright 2021 Elsevier; (E) Etching process and glass slides before and after etching; (F) Yield of methanol of etched glass slides and NH2-MIL-125(Ti); (G) TOF of catalysts by reaction time under 300 W Xe lamp[67]. Copyright 2022 Elsevier. PQD: Perovskite quantum dot; PES: polyethersulfone; TON: turnover number; TOF: turnover frequency.
Izu et al. proposed a heterogeneous CO2 photoreduction system utilizing a polymer-based matrix, in which a transparent coordination polymer glass membrane embedded with metalloporphyrins was developed[65]. The [Zn(HPO4)(H2PO4)2]2 matrix served not only as a mechanical support for the Fe(TPP)Cl catalyst, but also promoted chemical interactions during synthesis and enabled uniform dispersion of the catalyst within the polymer network. Glass membranes with thicknesses of 3, 5, and 9 μm containing 0.5 wt% Fe(TPP)Cl were fabricated using a melt-quenching technique. The Soret band absorption peak at 435 nm indicated that the porphyrin retained its monomeric characteristics and remained stably dispersed within the dense polymer glass structure. Notably, the polymer glass exhibited high optical transparency, and its internal void network allowed CO2 and the sacrificial electron donor (BIH) to penetrate the embedded catalytic sites, enabling the entire membrane to participate in the reaction. Under 430 nm light irradiation for 48 h, the system produced 3.7 μmol of CO with a turnover frequency (TOF) of 1.2 h-1 and a CO selectivity exceeding 99%. Compared to conventional drop casting or adsorption methods, this polymer glass approach demonstrated superior catalyst dispersion, optical transmittance, and structural stability. These findings highlight the effectiveness of amorphous, polymer-based photocatalytic platforms for CO2 reduction.
Cheng et al. developed a monolithic perovskite quantum dots (PQDs) with Polyethersulfone (PES) film by uniformly immobilizing PQDs onto a three-dimensional polymeric support, PES, via electrostatic
Zhao et al. developed an amorphous organotitanium polymer immobilized on chemically etched glass surfaces to overcome the charge transport limitations and low photocatalytic activity typically associated with Ti-based metal-organic frameworks (MOFs)[67]. This polymeric catalyst was firmly anchored onto the etched glass substrate, enabling enhanced light transmittance by minimizing particle-induced scattering and promoting high immobilization efficiency through infiltration of the reactive polymer into the nanopores of the silica surface. As a result, the transmittance of the glass substrate increased from 89.9% to 98.1% after etching [Figure 3E]. The amorphous polymer catalyst exhibited a narrower bandgap of 2.21 eV compared to 2.54 eV for NH2-MIL-125(Ti), and its average excited-state electron lifetime extended to 5.23 ns, which is approximately 3.6 times longer than the 1.44 ns measured for the MOF counterpart. These enhanced photophysical properties translated into significantly improved catalytic performance. Under 300 W Xe lamp irradiation for 4 h in an aqueous solution of 0.2 M ethylenediamine (EDA), the glass-supported polymer catalyst achieved a methanol yield of 751.4 μmol with a TOF of 112 h-1 [Figure 3F and G]. In contrast, NH2-MIL-125(Ti) powder produced only 408.2 μmol of methanol with a TOF of 0.21 h-1 under identical conditions that represent an approximately 533-fold enhancement in TOF. Thermal and aqueous stability tests further highlighted the robustness of the polymer system. Even after heat treatment at 300 °C for
Thus, the strategy of utilizing polymers as inert frameworks offers an effective approach to improving both the efficiency and stability of photocatalysts by maintaining their well-dispersed state throughout the reaction process.
Polymer-assisted or polymer-based photocatalysts
Conventional inorganic photocatalysts exhibit excellent activity, yet they suffer from inherent limitations such as restricted light absorption confined to the UV region, rapid electron-hole recombination, low stability, and difficulties in recovery and reuse. As part of ongoing efforts to overcome these challenges and achieve significant enhancements in photocatalytic performance, polymeric materials have been strategically integrated into photocatalytic systems. Beyond serving as mere support, polymers can function as essential active components within the photocatalytic architecture. This section delves into the two principal roles of polymers in photocatalytic systems: as auxiliary agents that support light absorption and charge transport, and as photoactive materials that directly engage in photocatalytic reactions. By analyzing the underlying mechanisms and reviewing recent advancements, this section aims to provide a comprehensive understanding of the design principles and application potential of polymer-based photocatalysts.
Photocatalytic assistance by polymers
Photocatalytic materials generate electron-hole pairs upon light absorption; however, a significant portion of these charge carriers may recombine before participating in catalytic reactions, thereby reducing the overall efficiency of the photocatalyst. To address this limitation, polymeric materials can be introduced to suppress charge recombination and facilitate the separation and transport of electrons and holes, ensuring that more of the generated charges contribute effectively to the catalytic process. In addition, when polymers serve as structural frameworks supporting photoactive particles, their relatively larger surface area compared to the active particles can be leveraged. In such configurations, the polymer itself may absorb light and generate excited charge carriers, which are then transferred to the photoactive particles to drive the catalytic reaction. Conversely, the polymer can also accept charge carriers from the photoactive material and participate directly in the degradation of target compounds, further enhancing photocatalytic performance.
Skorjanc et al. developed a sulfur-rich thiacalix[4]arene-based porous organic polymer (SCX4+) and utilized it to fabricate a composite photocatalyst [gold nanoparticles (AuNPs)@SCX4+] by uniformly immobilizing AuNPs onto the polymer matrix[68]. The use of the polymer scaffold played a crucial role in preventing aggregation of the metal nanoparticles, while also facilitating gas diffusion and electron transfer, thereby enhancing the overall catalytic performance. The sulfur atoms in the SCX4+ framework form strong bonds with Au, effectively suppressing nanoparticles from leaching during the reaction [Figure 4A]. In photocatalytic CO2 reduction tests, the composite catalyst produced 6.74 μmol·g-1 of CO and 0.90 μmol·g-1 of CH4 over 4 h [Figure 4B]. The total consumed electrons corresponded to a rate of 5.24 μmol·g-1·h-1, which is comparable to that of more complex post-treated porous organic polymer (POP)-metal systems. Additionally, SCX4+ itself possesses inherent light absorption properties, allowing the polymer-metal composite to efficiently mediate light harvesting, charge separation, and electron transfer. This work demonstrates how a multifunctional polymer scaffold not only stabilizes metal nanoparticles but also contributes actively to photocatalytic processes, offering a synergistic platform for efficient CO2 photoreduction.
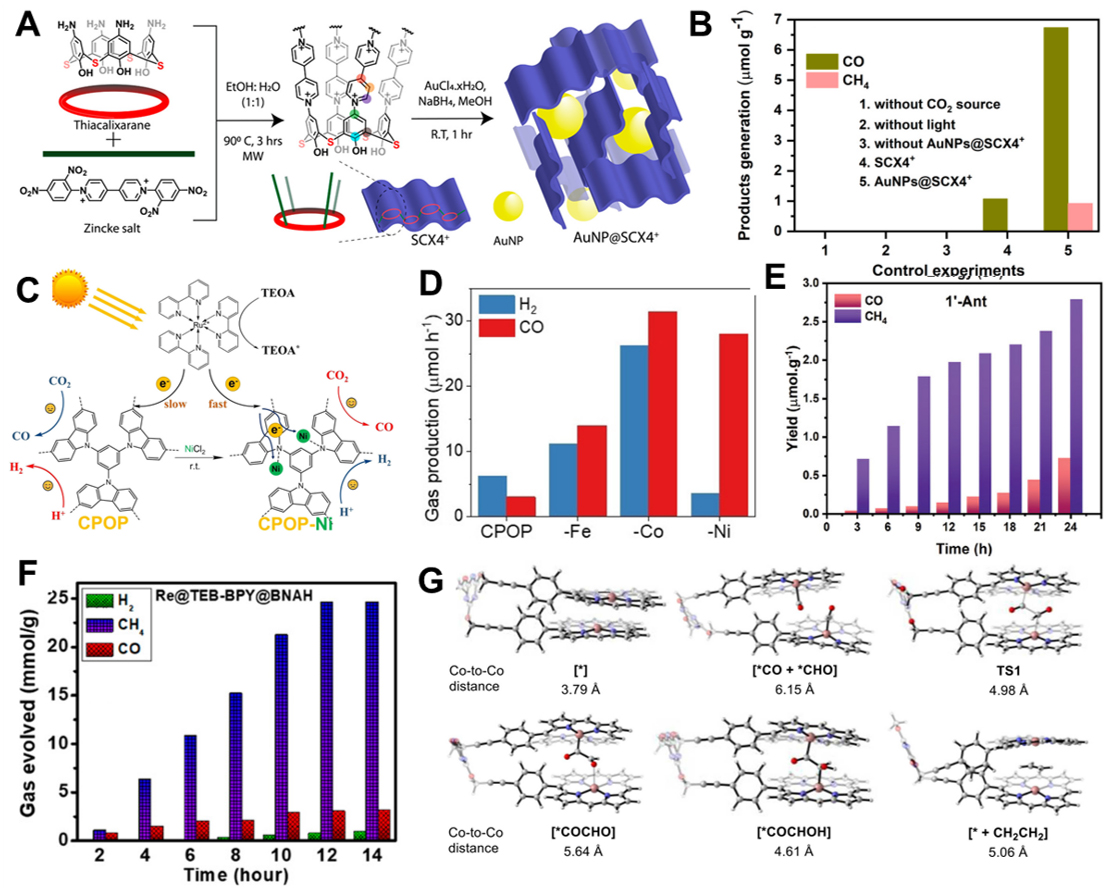
Figure 4. (A) Schematic illustration and synthesis method of AuNPs@SCX4+; (B) Products generation under visible light on various reaction conditions[68]. Copyright 2022 American Chemical Society; (C) Illustration of Ni incorporated conjugated porous polymers (CPOP-Ni) and its CO2 conversion; (D) Gas production of CO2 of CPOP and CPOP-M under visible light[71]. Copyright 2022 John Wiley & Sons; (E) Photocatalytic CO2 reduction activity of 1’-Ant with TEA as a sacrificial agent under visible light irradiation[72]. Copyright 2024 John Wiley & Sons; (F) Photocatalytic activity of Re@TEB-BPY@BNAH under visible light irradiation[74]. Copyright 2023 American Chemical Society; (G) Co distance change with various conditions[75]. Copyright 2024 Elsevier.
Yu et al. proposed a CO2 photoreduction system in which the photocatalytic function is directly performed by a π-conjugated polymer incorporating metal porphyrin units[69]. They synthesized two cobalt(II) porphyrin-based conjugated polymers, cobalt(II) tetrakis(4-ethynylphenyl)porphyrin (CoPor)- 4,4’-dibromobiphenyl (DBBP) and CoPor-BBPA, by polymerizing CoPor with either DBBP or bis(4-bromophenyl)acetylene (BBPA). The conjugated polymer serves as the light-harvesting and charge-transporting framework, enabling CO2 reduction to occur at the cobalt-centered CoPor sites. Under UV light irradiation, CoPor-DBBP exhibited excellent photocatalytic performance with a CO production rate of 286.7 μmol·g-1·h-1, a CO selectivity of 90.4%, and an H2 production rate of 30.3 μmol·g-1·h-1. The quantum efficiency at 420 nm was measured at 0.41%, demonstrating competitive light-to-fuel conversion performance. In this system, the polymer played a dual role as both the light-harvesting antenna and electron transport framework. CoPor-DBBP possessed favorable structural and electronic characteristics, including a high specific surface area of 646 m2·g-1, a microporous structure centered around 1.0 nm, a bandgap of
Nandi et al. developed a photocatalyst system by anchoring nickel nanoparticles onto a triazine-based porous organic polymer (TrzPOP)[70]. Although the Ni nanoparticles serve as the catalytic active sites, the
Wang et al. synthesized metal-incorporated CPOPs by introducing transition metal ions (Fe, Co, and Ni) into a porous polymer matrix, aiming to develop selective CO2 reduction photocatalysts[71] [Figure 4C]. The CPOP framework exhibited a high BET surface area of 1,335 m2·g-1 prior to metal incorporation, providing abundant sites for metal coordination. Even after metal loading, the materials maintained high surface areas: 548 m2·g-1 for Fe, 732 m2·g-1 for Co, and 853 m2·g-1 for Ni. The presence and dispersion of metal ions were further confirmed through Transmission Electron Microscopy (TEM) and X-ray Photoelectron Spectroscopy (XPS) analyses. Photocatalytic experiments were conducted using Ru(bpy)32+ as the photosensitizer and TEOA as the sacrificial electron donor. Among the metal-incorporated systems, CPOP-Ni achieved a CO production rate of 28 μmol·h-1·mg-1 with 89% selectivity, while CPOP-Co exhibited an even higher CO production rate of 31 μmol·h-1·mg-1 [Figure 4D]. CPOP-Ni possessed an optical bandgap of 1.75 eV and a conduction band reduction potential of -0.67 V vs. NHE, making it suitable for CO2 photoreduction. Enhanced electron-hole separation in this system was demonstrated by increased photocurrent response, photoluminescence (PL) quenching, and reduced charge transfer resistance [electrochemical impedance spectroscopy (EIS) measurements]. This study confirms that porous polymer frameworks can not only serve as support but also enhance the catalytic performance and selectivity of transition metal centers through favorable electronic environments and structural synergy.
Jena et al. developed a CO2 reduction photocatalyst system by incorporating π-electron-rich polycyclic aromatic guest molecules into a donor-acceptor-type porous coordination polymer (PCP), effectively tuning the light-harvesting and charge separation properties[72]. In their design, anthracene (1-Ant) and pyrene (1-Pyr) were used as guest molecules in the PCP structure {[Zn(o-phen)(ndc)]·(guest)}n, forming non-covalent donor-acceptor charge-transfer (CT) complexes within the polymer matrix. To further enhance photocatalytic performance, they synthesized a multivariate PCP (1’-Ant) by partially substituting 45% of the redox-inactive Zn(II) centers into the 1-Ant structure with redox-active Co(II). This strategic modification resulted in a catalyst with significantly improved CO2 reduction activity. Under visible-light irradiation with triethylamine (TEA) as the sacrificial electron donor, the 1-Ant system achieved a CH4 yield of 1.24 mmol·g-1 with ~94% selectivity. The Co-doped 1’-Ant variant produced 2.79 mmol·g-1 of CH4 with the same high selectivity [Figure 4E], achieving a production rate of 116 μmol·g-1·h-1 and a TON of 3.54, which is more than four times that of the Zn-based system (TON = 0.80). This work demonstrates how polymeric coordination frameworks can serve as finely tunable platforms that precisely modulate electronic interactions with guest molecules, enabling enhanced photocatalytic CO2 conversion through synergistic charge transfer and catalytic activation.
Jiang et al. developed a conjugated porous polymer (CPP) photocatalyst by integrating a zinc porphyrin (ZnPor) as an electron donor and a rhenium complex as an electron acceptor, enabling efficient CO2 photoreduction under visible light[73]. In this system, the photoactive ZnPor units and catalytically active Re complexes were covalently embedded within a rigid polymer framework, providing a stable and selective platform for long-term CO2 conversion. The resulting ZnPor@Re composite exhibited a broad light absorption range from 200 nm to 1,000 nm, indicating strong photoresponse across the entire visible spectrum. PL measurements revealed that the emission peaks observed in the ZnPor precursor (ZnTBPP) were almost completely quenched in the ZnPor@Re system, suggesting efficient charge separation and suppression of electron-hole recombination due to the introduction of the Re complex. In photocatalytic performance tests, the ZnPor@Re system achieved a CO production yield of 66.2 mmol·g-1 with an exceptionally high CO selectivity of 99.8%. The quantum efficiency at 420 nm was measured to be 1.3%, highlighting the effectiveness of this covalently integrated donor-acceptor polymer in visible-light-driven CO2 reduction. This study showcases how conjugated porous polymers with integrated electron
Rahimi et al. synthesized a Re-based composite photocatalyst (Re@TEB-BPY) by post-metallating a conjugated microporous polymer (CMP) constructed from 1,3,5-triethynylbenzene (TEB) and
Wang et al. reported a flexible polymer-based catalyst, F-TotPp(Co), capable of selectively reducing
Polymers as active photocatalysts
Traditional photocatalyst research has primarily focused on inorganic semiconductors and metal complexes. However, growing interest has recently emerged in the intrinsic photocatalytic activity of polymers. Polymer-based photocatalysts offer significant advantages due to their chemically flexible structures and ease of functionalization, which enable precise tuning of light absorption properties, charge separation efficiency, and energy levels. In particular, CPPs have garnered attention for their broad
Chi et al. investigated a series of D-A conjugated polymers designed to optimize intramolecular electron transport and enhance intrinsic photocatalytic performance through precise molecular engineering of charge transmission pathways[76]. Using the same water-soluble electron-accepting unit, terephthalonitrile, and varying the electron-donating units (benzene, spirobifluorene, and pyrene), three polymers (B-2CN,
Rahimi et al. synthesized donor-acceptor conjugated polymers incorporating a (Z)-4-(2-hydroxy-3,5-diiodobenzylidene)-1-(4-iodophenyl)-2-methyl-1H-imidazol-5(4H)-one (o-HBDI-I3) (GFP) chromophore analogue[77]. Tris(4-ethynylphenyl)amine (TPA) and TEB were coupled with GFP to synthesize TPA-GFP and TEB-GFP [Figure 5A]. These polymers were evaluated as metal-free photocatalysts for visible-light-driven CO2 reduction. Photocatalytic experiments were conducted using 0.5 mg of catalyst in an acetonitrile:water (3:1) solvent system with TEA as the sacrificial electron donor, under 400-750 nm visible-light irradiation. TPA-GFP exhibited a remarkable CO production yield of 1,666 μmol·g-1 after 12 h, with a generation rate of 139 μmol·g-1·h-1 and a CO selectivity of 95% [Figure 5B]. In contrast, TEB-GFP showed significantly lower performance, producing 593 μmol·g-1 of CO with only 54% selectivity [Figure 5C]. Optical and electronic structure analyses revealed that TPA-GFP had an absorption peak at 545 nm and a narrow bandgap of 1.81 eV, indicating strong visible-light responsiveness. BET surface area measurements based on the Langmuir model showed that TPA-GFP had a surface area of 477.3 m2·g-1 compared to
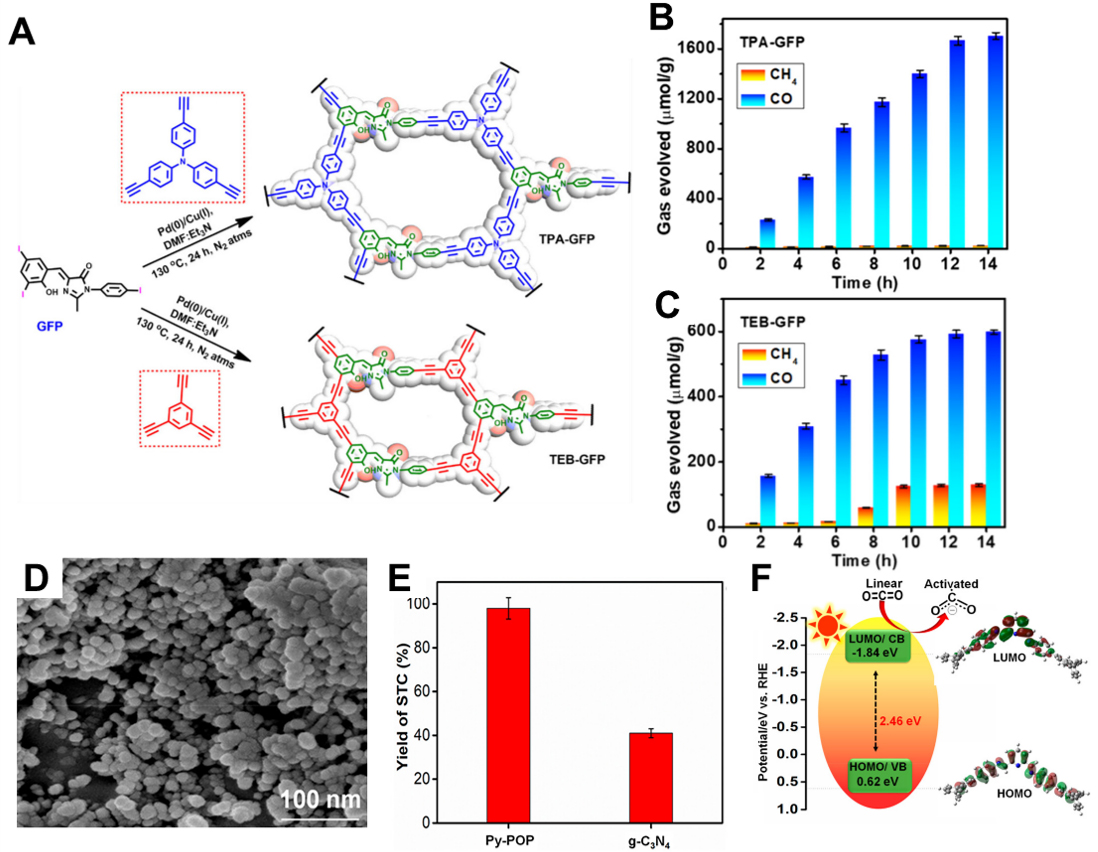
Figure 5. (A) Schematic illustration of TPA-GFP and TEB-GFP CMPs; (B) Photocatalytic activity of TPB-GFP and (C) TEB-GFP[77]. Copyright 2024 American Chemical Society; (D) FESEM images of Py-POP; (E) Performance comparison of Py-POP and g-C3N4; (F) Energy level of HOMO-LUMO of Py-POP[79]. Copyright 2023 American Chemical Society. TPA: Tris(4-ethynylphenyl)amine; CMP: conjugated microporous polymer; TEB: 1,3,5-triethynylbenzene; HOMO: highest occupied molecular orbital; LUMO: lowest unoccupied molecular orbital; POP: porous organic polymer; GFP: (Z)-4-(2-hydroxy-3,5-diiodobenzylidene)-1-(4-iodophenyl)-2-methyl-1H-imidazol-5(4H)-one (o-HBDI-I3); FESEM: field emission scanning electron microscopy.
Hong et al. designed a POP system incorporating two functional monomers: 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (4CzIPN) and 9-{4-[(2,2’:6’,2’’-terpyridin)-4’-yl]phenyl}-9H-carbazole (CzTPY), resulting in the formation of PolyIPN-TPY[78]. The resulting amorphous porous polymer exhibited a BET surface area of 160 m2·g-1 with an average pore size of 4.28 nm. Notably, PolyIPN-TPY displayed a high CO2 adsorption capacity of
Das et al. developed a photocatalyst based on a pyridine-functionalized polymer, Py-POP, synthesized through a Schiff-base condensation between an aromatic amine donor [1,3,5-tris(4-aminophenyl)benzene] and an electron-accepting unit (2,6-pyridinedicarboxaldehyde)[79]. This structure provides both a porous architecture and a π-conjugated framework, enabling effective light absorption and charge transport [Figure 5D]. Under photocatalytic conditions, Py-POP was employed to catalyze the conversion of styrene oxide and CO2 into styrene carbonate (STC). When irradiated with a 250 W Xe lamp for 10 h, the system achieved a high conversion efficiency of 94%. In comparison, a benchmark catalyst, g-C3N4, reached only 40% conversion under the same conditions, demonstrating the superior performance of Py-POP [Figure 5E]. Key indicators of photoactivity included a bandgap of 2.46 eV [Figure 5F], a conduction band position of
Summary of the comprehensive evaluation of polymer-based photocatalysts for pCO2RR
| Strategy | Photocatalyst | Sacrificial electron donor (SED) | Product | Activity | Selectivity (%) | Light source | Ref. |
| Polymer frame | PS-RuC/RuCat | BNAH | CO | TON = 41, TOF = 41 h-1 | - | Xe 300 W | [64] |
| Fe(TPP)Cl | BIH | CO | TOF = 1.2 | > 99 | - | [65] | |
| CsPbBr3 | - | CO | 32.45 μmol g-1 h-1 | - | - | [66] | |
| Glass-Ti-polymer | - | CH3OH | TOF = 112 | - | Xe 300 W | [67] | |
| Polymer-assisted photocatalysis | AuNPs | EtOH | CO, CH4 | TON = 5.24 | - | Xe 300 W | [68] |
| CoPor-DBBP | TEA | CO | 286.7 μmol g-1 h-1 | 90.4 | Xe 300W | [69] | |
| TrzPOP | TEOA | CH3OH | TON = 270 | LED 20 W | [70] | ||
| Ni | TEOA | CO | 28 μmol h-1 | 89 | Xe 300 W | [71] | |
| 1-Ant, 1-Pyr, 1’-Ant | TEA | CH4 | TON = 3.54 | 94 | Xe 300 W | [72] | |
| ZnPor@Re | BIH, TEOA | CO | 66.2 mmol g-1 | 99.8 | LED 90 mW/cm2 | [73] | |
| Re@TEB-BPY | TEA, BNAH | CH4 | TON = 71.1 | 96 | Xe 300 W | [74] | |
| Polymer catalyst | P-2CN | - | CO | 32 μmol h-1 | 61.5 | Xe 300 W | [76] |
| TPA-GFP, TEB-GFP | TEA | CO | 1,666 μmol h-1 | 95 | Xe 300 W | [77] | |
| PolyIPN-TPY | TEOA | CO | 265.7 μmol h-1 | 94.7 | LED 3 W | [78] | |
| Py-POP | - | STC | 94% conversion in 10 h | 99 | Xe 300 W | [79] |
POLYMER-BASED ELECTROCATALYST FOR ECO2RR
Polymer-based electrocatalysts have recently emerged as a promising class of materials for eCO2RR, offering exceptional structural tunability, interfacial control, and electronic modulation capabilities that enable precise tailoring of reaction pathways and product activity and selectivity.
Active site engineering in polymeric catalysts for eCO2RR
Coordination polymer
Coordination polymers have been attracted as tunable electrocatalysts for eCO2RR, where the metal centers act as active sites and govern product selectivity. By varying the metal species and coordination environment, these materials enable the formation of different products such as HCOO-, CO, CH4, and C2+ compounds. The following examples demonstrate how tailoring metal-ligand interactions in coordination polymers can effectively steer eCO2RR pathways. Tin is regarded as one of the most effective metal centers for eCO2RR to HCOO-. For example, Geng et al.[80] proposed a framework functionalization approach to enhance the catalytic performance of tin-based coordination polymers for eCO2RR by introducing amino groups. Among the evaluated ligands, p-phthalic acid, 2-aminoterephthalic acid, and 2,5-diaminoterephthalic acid, the tin 2,5-diaminoterephthalic acid (Sn-DaPTA) catalyst exhibited the highest surface Sn2+ content and the densest distribution of -NH2 groups within the coordination framework[80]. As a result, Sn-DaPTA recorded a superior Faradaic efficiency (FE) for HCOO- (FEHCOO-) of 85% at a current density of 11 mA/cm2. This enhanced activity is primarily attributed to the abundant -NH2 functionalities, which promote stronger CO2 adsorption and facilitate subsequent reduction steps. Cu stands out not only for its ability to promote C-C coupling toward C2+ products, but also for its high selectivity toward CH4 under specific coordination environments. For instance, Chen et al.[81] reported the synthesis of a layered coordination polymer catalyst (CuPEDOT) comprising Cu2+ ions and 3,4-ethylenedioxythiophene (EDOT), via a wet-chemical route [Figure 6A]. Notably, attempts to produce this material through the direct reaction of pre-polymerized PEDOT with copper chloride dihydrate were unsuccessful, likely due to the inherent insolubility of PEDOT in common solvents. In contrast, the employed method facilitated the in situ formation of CuPEDOT, where the limited solubility of EDOT and the mild oxidative conditions provided by Cu2+ contributed to a slow and controlled coordination process. This gradual reaction enabled the assembly of an ordered, layered structure through the direct coordination between Cu2+ centers and EDOT ligands [Figure 6B]. The resulting CuPEDOT indicated remarkable selectivity toward CH4 production, achieving a FE of 62.7% at a current density of
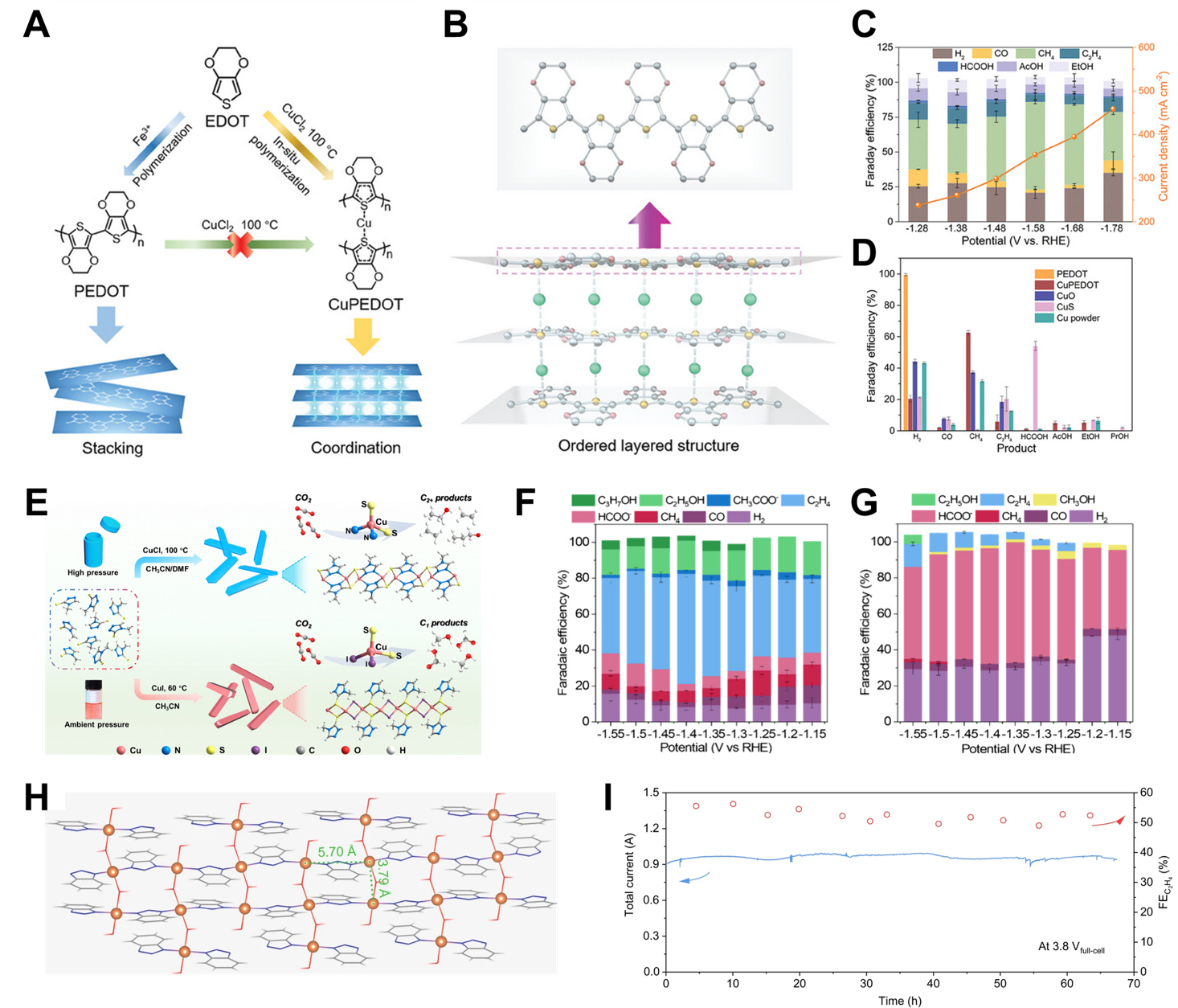
Figure 6. Active site engineering in coordination polymer catalysts. (A) Schematic illustration of the synthesis process of CuPEDOT; (B) Structural configuration of CuPEDOT; (C) Selectivity and activity of CuPEDOT; (D) FE for various products of other conventional Cu-based catalysts[81]. Copyright 2023 John Wiley & Sons; (E) Scheme for fabrication process of Cu-N2S2 and Cu-I2S2; (F) FE of Cu-N2S2; (G) FE of Cu-I2S2[82]. Copyright 2024 American Chemical Society; (H) Structural configuration of Cu(OH)BTA; (I) Operational durability test for Cu(OH)BTA[83]. Copyright 2023 Springer Nature. FE: Faradaic efficiency; CuPEDOT: Cu coordination polymer 3,4-ethylenedioxythiophene.
Cu is also one of the most extensively investigated metals for the production of C2+ compounds during
Metal-organic frameworks
As a subclass of polymers, MOFs exhibit exceptional structural tunability, high surface area, and
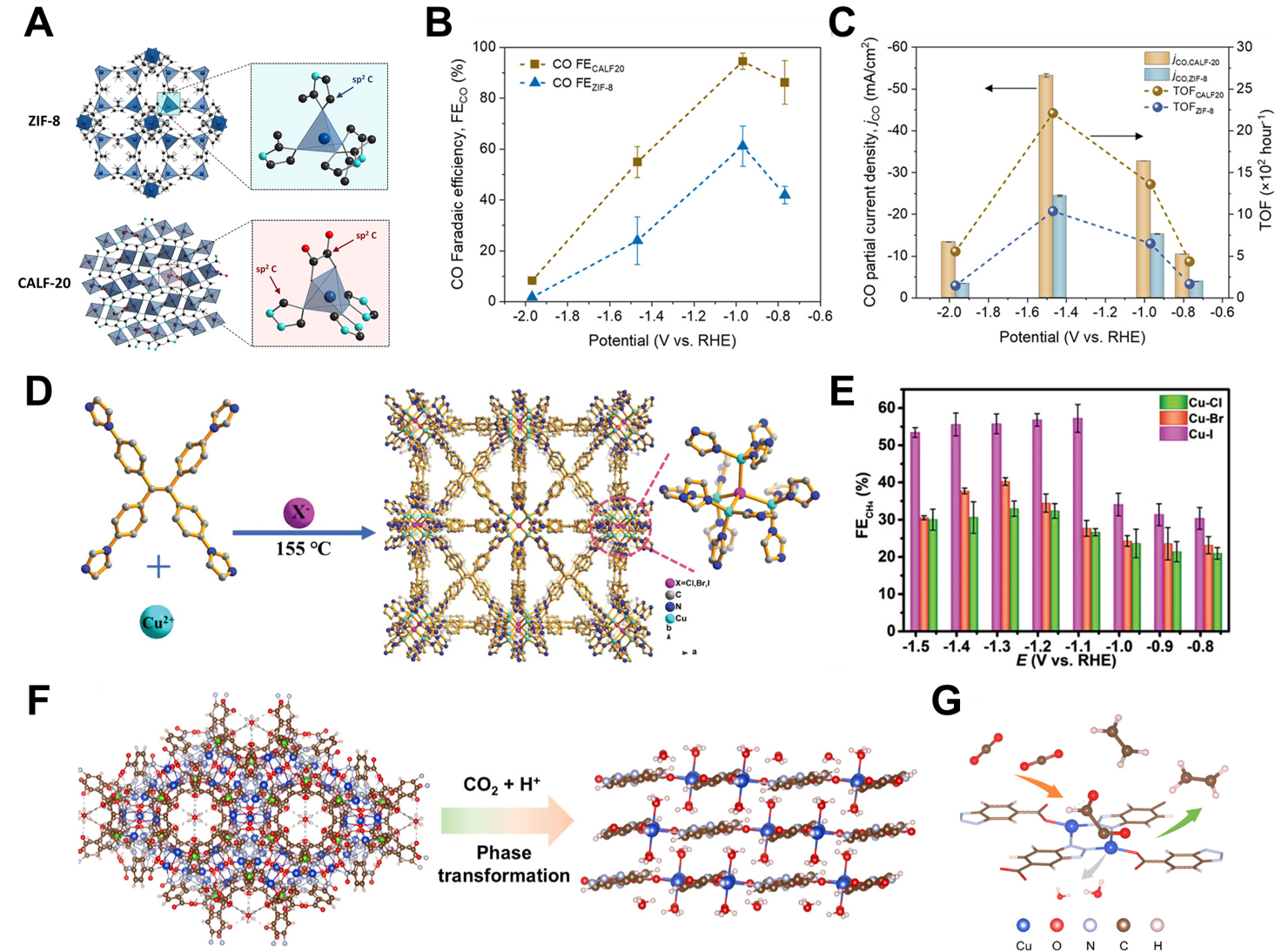
Figure 7. Active site engineering in MOF catalysts. (A) Molecular frameworks of ZIF-8 and CALF20; (B) FECO of ZIF-8 and CALF20; (C) Catalytic activity of ZIF-8 and CALF20 for CO[84]. Copyright 2021 American Chemical Society; (D) Schematic depicting the synthesis process and molecular structure of Cu-X MOFs; (E) FECH4 of various Cu-X MOFs[85]. Copyright 2022 John Wiley & Sons; (F) Illustration of structural transformation of Cu-btca nanocages to nanosheets; (G) Scheme for Cu-btca nanosheets during eCO2RR[88]. Copyright 2024 American Chemical Society. MOFs: Metal-organic frameworks; FE: faradaic efficiency.
Sun et al.[86] developed a strategy to systematically regulate product selectivity in eCO2RR to CH4 and C2H4 by tuning the electronic structure of Cu active sites through ligand functionalization of UiO-66-based MOF composites. Specifically, they reported a series of Cu/UiO-66-L (M) catalysts, where M denotes the metal nodes (Zr, Hf, Ce) and L refers to the substituents on 1,4-benzenedicarboxylic acid linkers (H, F, NH2). By selecting appropriate combinations of metal nodes and functional groups, they achieved significant suppression of the HER while simultaneously promoting the formation of either CH4 or C2H4. Notably,
Organic polymer modifiers for eCO2RR interfaces
Polymeric materials have emerged as versatile modifiers capable of tailoring the local microenvironment surrounding electrocatalysts, thereby enhancing both activity and selectivity in eCO2RR. By influencing factors such as interfacial ion transport, CO2 adsorption, and intermediate stabilization, polymers play a critical role in optimizing reaction pathways and suppressing competing processes. For example, Li et al.[89] presented that modifying Ag-based catalysts with electro-activated CO2-binding organic molecules, specifically indigo derivatives, can create synergistic interfacial sites that substantially enhance both the selectivity and catalytic activity for CO production during eCO2RR. The dynamic coordination between these organic modifiers and CO2 facilitates its activation and promotes the enrichment of key *CO2- and *COOH intermediates in the vicinity of the Ag surface. A systematic evaluation of various indigo derivatives bearing different substituents revealed that indigo-6,6’-dicarboxylic acid (DCId) possesses the most favorable CO2- adsorption energy for CO formation. Despite its high performance, DCId suffers from limited operational stability due to its dissolution into the electrolyte under high current conditions. To overcome this limitation, the active CO2-binding moieties of DCId were covalently incorporated into a polymeric matrix via amidation with 2,2’-(ethylenedioxy)diethylamine, resulting in an indigo-based polymer (P-Id). This macromolecular anchoring strategy significantly improved the stability of the interface, enabling the P-Id-modified Ag catalyst to achieve over 90% FECO across a wide current density range of 100~1,200 mA cm-2 and delivering a high Ag mass activity of 174 A mg-1 toward CO production[89]. Xu et al.[90] developed a precursor system comprising oxygen vacancy-rich Bi2O3 nanosheets coated with a polypyrrole (PPy) layer (Bi2O3@PPY NSs) for highly selective eCO2RR to HCOO-. The Bi2O3 nanosheets were initially synthesized via a hydrothermal method, followed by controlled room-temperature polymerization of pyrrole monomers on their surface to form a conformal PPY coating [Figure 8A]. Upon exposure to electrochemical conditions, the Bi2O3 phase underwent an in situ transformation into a
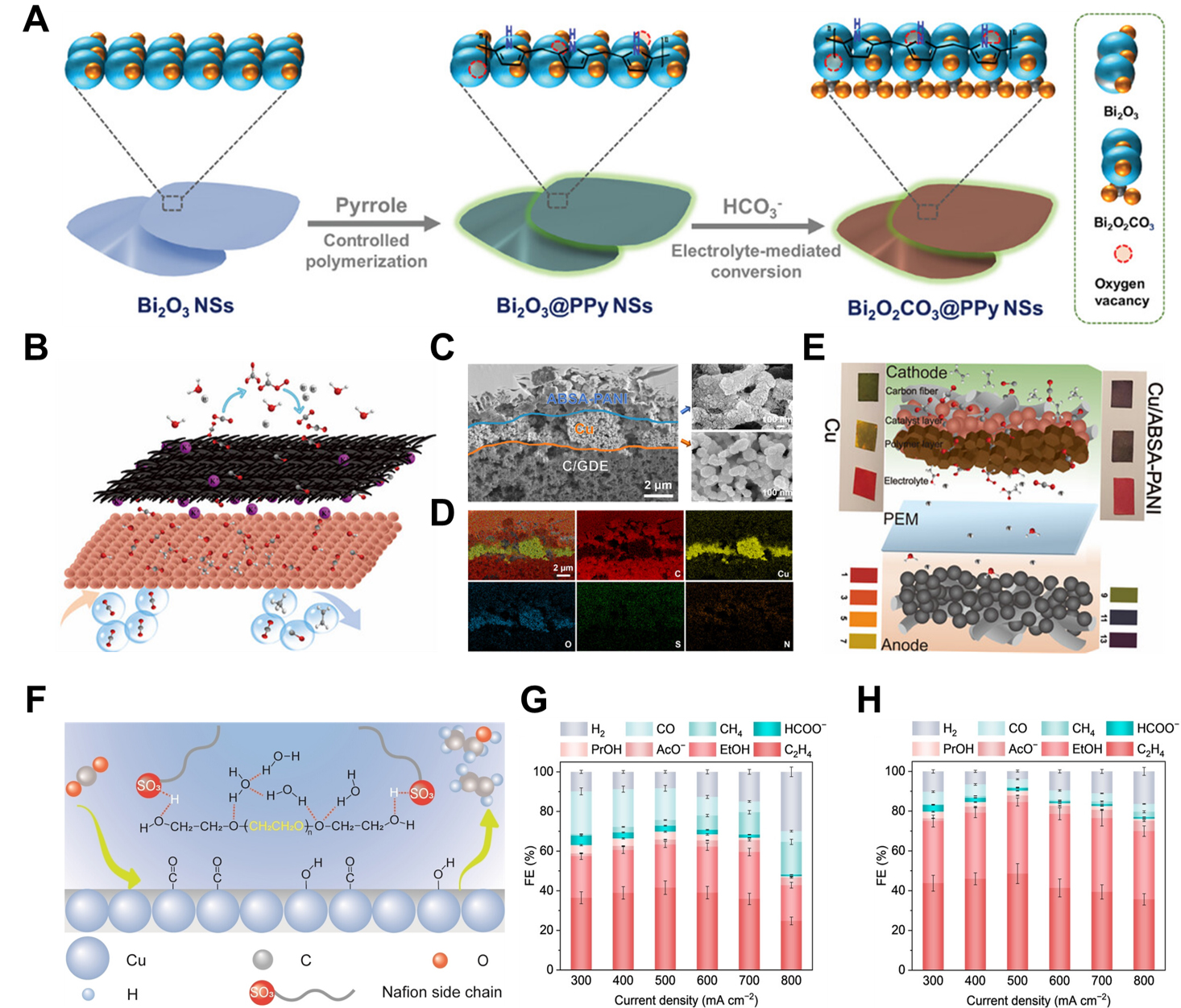
Figure 8. Organic polymer modifiers. (A) Illustration of synthesis procedure for Bi2O2CO3@PPY NSs[90]. Copyright 2023 John Wiley & Sons; (B) Schematic of ABSA-PANI/Cu/C electrode; (C) SEM images and (D) EDX mapping of ABSA-PANI/Cu/C electrode; (E) Schematic of ABSA-PANI/Cu/C electrode’s reaction mechanism in acidic environment[91]. Copyright 2025 John Wiley & Sons; (F) Schematic depicting the influence of PEG at the Cu-Nafion interface; FE and current density for (G) unmodified Cu and (H) CuPEG[92]. Copyright 2025 John Wiley & Sons. FE: Faradaic efficiency; EDX: energy-dispersive X-ray; PEG: polyethylene glycol; SEM: scanning electron microscopy.
As illustrated in Figure 8B, Su et al.[91] introduced a multifunctional interfacial layer composed of a conductive polymer, polyaniline (PANI) modified by p-aminobenzenesulfonic acid (p-ABSA), positioned between the Cu nanoparticle catalyst layer and the electrolyte. This polymer interface was designed to establish a favorable local microenvironment for eCO2RR, while simultaneously optimizing ion transport and electronic conductivity, thus promoting the reaction kinetics under acidic conditions. The successful and homogeneous deposition of the ABSA-PANI layer was confirmed by cross-sectional SEM and
In addition to organic polymer modifiers, ionomers such as Nafion and Sustainion have been widely employed in eCO2RR systems not only as ionic conductors and binders but also as regulators of the
Polymer-supported architectures for eCO2RR
Beyond modulating the microenvironment at the catalyst-electrolyte interface, polymers also play a crucial role in engineering the interfacial properties between the catalyst and the electrode substrate, thereby further optimizing the reaction conditions for efficient eCO2RR. For instance, Zhang et al.[95] fabricated a self-supporting electrode by anchoring Ag single atom (AgSA) catalysts onto a PANI-modified GDE, referred to as GDE-PANI-AgSA. As presented in Figure 9A, PANI was vertically polymerized on the GDE surface via in situ oxidative polymerization of aniline monomers, followed by mild aqueous-phase reduction of Ag+ precursors to immobilize AgSA onto the PANI matrix. This hierarchical architecture ensures high dispersion and full exposure of AgSA active sites. Notably, the electron-donating amino groups in the PANI framework facilitate CO2 adsorption and activation, while the N-coordinated Ag centers enhance *COOH intermediate stabilization. As a result, the GDE-PANI-AgSA electrode achieves nearly 100% FECO in 1 M KOH under flow cell conditions, clearly demonstrating its superior catalytic performance [Figure 9B]. Xu et al.[96] developed a Cu-based catalyst by magnetron sputtering Cu onto a PANI-coated polytetrafluoroethylene (PTFE) membrane, resulting in Cu-PANI composites. As depicted in Figure 9C and D, the sputtered Cu retained the nanoparticulate microstructure of the underlying PANI layer, and elemental mapping confirmed the uniform distribution of Cu, N, and C throughout the catalyst layer. Compared to bare Cu, the Cu-PANI composite exhibited a substantial enhancement in hydrocarbon selectivity, increasing from 22.2% to 71.8%. This improved performance was attributed to the nitrogen-containing sites in PANI, which facilitate CO2 adsorption and the formation of *CO intermediates. Furthermore, the Cu/PANI interface effectively suppressed the diffusion and premature desorption of *CO, thereby promoting further hydrogenation and C-C coupling pathways that favor hydrocarbon production [Figure 9E].
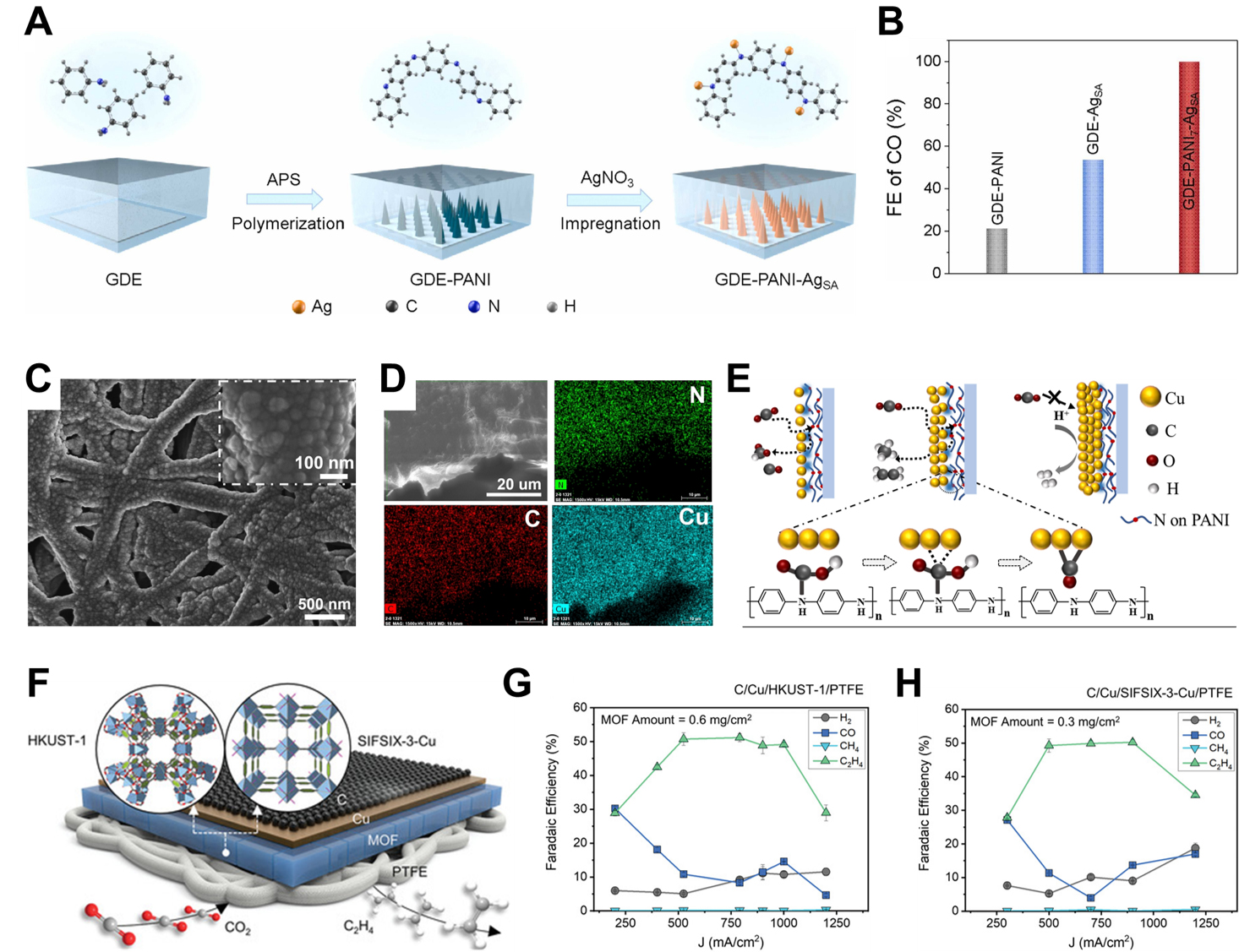
Figure 9. Polymer-supported designs. (A) Schematic of the fabrication process of GDE-PANI-AgSA; (B) FECO of GDE-PANI-AgSA[95]. Copyright 2024 Elsevier; (C) SEM image and (D) Elemental mapping of Cu-PANI; (E) Schematic representation of the interaction mechanism between CO2 and Cu in Cu-PANI during eCO2RR[96]. Copyright 2024 John Wiley & Sons; (F) Schematic of MOF-functionalized GDEs; FE and total current density of (G) C/Cu/HKUST-1/PTFE and (H) C/Cu/SIFSIX-3-Cu/PTFE[98]. Copyright 2022 John Wiley & Sons. FE: Faradaic efficiency; GDE: gas diffusion electrode; MOF: metal-organic framework; SEM: scanning electron microscopy.
Jia et al.[97] presented the fabrication of a series of 3D hierarchical metal/PANI-carbon paper (M/PANI-CP) electrodes through in situ electropolymerization. This strategy enables the integration of various metals such as Cu, Pd, Zn, Sn and PANI onto a carbon paper scaffold, resulting in hybrid electrodes with tunable electrocatalytic properties toward eCO2RR. The product selectivity was found to depend strongly on the choice of metal, with the formation of C2H4, CO, or HCOOH observed accordingly. Among the different combinations, Cu/PANI exhibited notable performance, achieving a FEC2H4 of 59.4% at 30.2 mA cm-2[97]. This hierarchical 3D architecture offers a high surface area and abundant defect sites, which are beneficial for enhancing both activity and selectivity. Furthermore, the in situ polymerization process ensures minimal interfacial resistance at both the metal/PANI and PANI/CP interfaces, contributing to a lower onset potential and reduced overpotential during operation. Improved selectivity was also attributed to the prolonged residence time of key intermediates within the catalyst matrix, which facilitates C-C coupling into C2+ products. Additionally, the 3D structure supports uniform metal dispersion, effectively suppressing metal agglomeration and thereby improving long-term catalytic stability. Nam et al.[98] engineered a
Summary of the comprehensive evaluation of polymer-based electrocatalysts for eCO2RR
| Strategy | Electrocatalysts | Electrolyte | Performance about activity or selectivity | Ref. |
| Active site engineering | Sn-DaPTA | 0.5 M KHCO3 | FEHCOO- of 85% at 11 mA cm-2 | [80] |
| CuPEDOT | 1 M KOH | FECH4 of 62.7% at 354 mA cm-2 | [81] | |
| Cu-N2S2 | 0.1 M KHCO3 | FEC2H4 of 61.2% and FEC2+ of 82.2% | [82] | |
| Cu-I2S2 | FEHCOO- of 66.9% | |||
| Cu(OH)BTA | 1 M KOH | FEC2H4 of 57% at 500 mA cm-2 | [83] | |
| CALF20 | 1 M KOH | FECO of 94.8% with partial current density of 32.8 mA cm-2 | [84] | |
| Cu-I MOF | 1 M KOH | FECH4 of 57.2% | [85] | |
| Cu/UiO-66-H (Ce) | 1 M KOH | FECH4 of 58% | [86] | |
| Cu/UiO-66-F (Ce) | FEC2H4 of 44% | |||
| S-HKUST-1 | 1 M KOH | FEC2H4 of 57.2% at 400 mA cm-2 | [87] | |
| Cu-btca MOFs | 3 M KCl + 0.5 M H2SO4 | FEC2H4 of 51.2% and FEC2+ of 81.9% at 300 mA cm-2 | [88] | |
| Organic polymer modifiers | P-Id-modified Ag catalyst | 1 M KOH | FECO over 90% across 100 ~ 1200 mA cm-2 | [89] |
| Bi2O2CO3@PPY NSs | 0.5 M KHCO3 | FEHCOO- of 95.8% | [90] | |
| GDE/Cu/ABSA-PANI | 1 M KCl + 0.5 M H2SO4 | FEC2+ of 81% at 600 mA cm-2 | [91] | |
| CuPEG | 1 M KOH | FEC2+ of 90.3% at 500 mA cm-2 | [92] | |
| G3-NH2/Cu | 1 M KOH | FE for acetate of 47% with partial current density of 202 mA cm-2 | [93] | |
| G3-OCH3/Cu | FECH4 of 73.2% | |||
| Naf850/Sus/Cu | 0.1 M CsHCO3 | FEC2+ over 90% with total current density for 12.1 mA cm-2 | [94] | |
| Polymer-supported architectures | GDE-PANI-AgSA | 1 M KOH | nearly 100% FECO | [95] |
| Cu-PANI composites | 0.1 M KHCO3 | FE for hydrocarbons of 71.8% | [96] | |
| Cu/PANI-CP | 0.1 M KCl | FEC2H4 of 59.4% at 30.2 mA cm-2 | [97] | |
| HKUST-1-functionalized GDE | 1 M KOH | FEC2H4 above 48% with partial current density of 491 mA cm-2 | [98] |
CONCLUSION AND FUTURE PROSPECTS
This paper explores the applications of polymeric materials in pCO2RR and eCO2RR, emphasizing their role in achieving carbon neutrality and sustainable energy solutions. We highlight how polymers, with their structural tunability and chemical stability, enhance catalytic systems by improving CO2 adsorption, charge separation, and light harvesting. In photocatalysis, polymers act as support for inorganic catalysts, form hybrid architectures with metal complexes, or function directly as photoactive materials. Conjugated polymer systems with donor-acceptor structures are particularly effective for charge transport and light absorption. For electrocatalysis, polymers contribute to active site engineering in coordination polymers and MOFs, enabling precise control over electronic environments and product selectivity. They also serve as interfacial modifiers and support, improving catalyst activity and stability and suppressing undesired side reactions.
While significant progress has been made, challenges remain, including limited light absorption, charge recombination, and poor long-term stability. Further direction emphasizes optimizing polymer architectures, developing metal-free systems, and employing advanced design strategies to achieve high performance and scalability in CO2 reduction technologies.
Optimizing polymer architecture is crucial for enhancing the efficiency of light harvesting and charge transport. Tailoring the conjugation length, introducing donor-acceptor structures, and designing porous networks can facilitate stronger light absorption and more efficient exciton dissociation. Additionally, layered structures and nanoscale ordering can provide efficient pathways for charge carriers, reducing recombination losses and improving overall catalytic activity.
Developing metal-free systems provides a sustainable and eco-friendly alternative to noble metal catalysts. Polymer-based systems reduce material costs and environmental risks while offering structural versatility. However, their catalytic performance still lags behind noble metals, highlighting the need for continued research to enhance efficiency and long-term stability. Despite their environmental advantages over
Advanced design strategies such as machine learning-assisted polymer discovery, tandem systems, and high-throughput screening are emerging approaches for developing polymers. These tools can accelerate the identification of polymer configurations and predict structure-property relationships, thereby overcoming current limitations and enabling scalable, high-performance CO2 reduction technologies.
DECLARATIONS
Authors’ contributions
Proposed the topic of this review: Kim, S. Y.
Prepared the manuscript: Kim, J. H.; Jeong, J.
Data curation: Kim, J. H.; Jeong, J.; Jo, H. J.; Han, S. M.; Kim, Y. J.
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This research was supported in part by the NRF funded by the Korean government [RS-2022-NR068133, RS-2025-00558945] and in part by Korea Institute of Marine Science & Technology Promotion (KIMST) funded by the Ministry of Oceans and Fisheries (RS-2024-00406639).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Zhou, P.; Navid, I. A.; Ma, Y.; et al. Solar-to-hydrogen efficiency of more than 9% in photocatalytic water splitting. Nature 2023, 613, 66-70.
2. Lim, J. W.; Cho, W. S.; Lee, J. Kinetic Investigation of CuSx Formation on Cu substrates for enhanced electrochemical CO2 reduction to HCOOH. Electron. Mater. Lett. 2025, 21, 245-51.
3. Kim, J. H.; Jo, H. J.; Han, S. M.; Kim, Y. J.; Kim, S. Y. Recent advances in electrocatalysts for anion exchange membrane water electrolysis: design strategies and characterization approaches. Energy. Mater. 2025, 5, 500099.
4. Prasanna, M.; Logeshwaran, N.; Ramakrishnan, S.; Yoo, D. J. Metallic 1T-N-WS2/WO3 heterojunctions featuring interface-engineered Cu-S configuration for selective electrochemical CO2 reduction reaction. Small 2024, 20, 2306165.
5. Zhang, Z.; Li, Y.; Zhong, Y.; et al. sp2/sp3-Hybridized nitrogen-mediated electrochemical CO2 capture and utilization. Sci. Adv. 2025, 11, eadw6592.
6. Ma, J.; Cho, J. H.; Lee, C.; et al. Unraveling the harmonious coexistence of ruthenium states on a self-standing electrode for enhanced hydrogen evolution reaction. Energy. Environ. Mater. 2024, 7, e12766.
7. Srivastava, V.; Chauhan, R. K.; Lohia, P.; Yadav, S. Investigation of Eco-friendly perovskite solar cell employing niobium pentoxide as electron transport material using SCAPS-1D. Trans. Electr. Electron. Mater. 2024, 25, 294-303.
8. Dhankar, U.; Dahiya, S.; Chawla, R. Mitigating the solar PV landscape: a comprehensive review of challenges and innovations. Trans. Electr. Electron. Mater. 2025, 26, 429-56.
9. Ma, J.; Kim, S. Y. Development of catalysts and reactor designs for CO2 electroreduction towards C2+ products. Energy. Mater. 2025, 5, 500052.
10. Kim, J. H.; Hong, S. H.; Ahn, S. H.; Kim, S. Y. Recent progress in photocathode interface engineering for photoelectrochemical CO2 reduction reaction to C1 or C2+ products. Exploration 2025, 5, 70010.
11. Kim, J. H.; Kim, J.; Ma, J.; et al. Spontaneous metal-chelation strategy for highly dense Ni single-atom catalysts with asymmetric coordination in CO2 electroreduction. Small 2025, 21, 2409481.
12. Ma, J.; Lee, W.; Kim, J. H.; et al. Leveraging the intermetal distance in dual-atom catalysts: revealing optimized synergistic interactions for CO2 electroreduction. ACS. Nano. 2025, 19, 18698-707.
13. Lu, P.; Tan, X.; Zhao, H.; et al. Atomically dispersed indium sites for selective CO2 electroreduction to formic acid. ACS. Nano. 2021, 15, 5671-8.
14. Shen, H.; Jin, H.; Li, H.; et al. Acidic CO2-to-HCOOH electrolysis with industrial-level current on phase engineered tin sulfide. Nat. Commun. 2023, 14, 2843.
15. Chi, L. P.; Niu, Z. Z.; Zhang, X. L.; et al. Stabilizing indium sulfide for CO2 electroreduction to formate at high rate by zinc incorporation. Nat. Commun. 2021, 12, 5835.
16. El-Nagar, G. A.; Yang, F.; Stojkovikj, S.; et al. Comparative spectroscopic study revealing why the CO2 electroreduction selectivity switches from CO to HCOO- at Cu-Sn- and Cu-in-based catalysts. ACS. Catal. 2022, 12, 15576-89.
17. Zhang, G.; Wang, T.; Zhang, M.; et al. Selective CO2 electroreduction to methanol via enhanced oxygen bonding. Nat. Commun. 2022, 13, 7768.
18. Wu, Y.; Jiang, Z.; Lu, X.; Liang, Y.; Wang, H. Domino electroreduction of CO2 to methanol on a molecular catalyst. Nature 2019, 575, 639-42.
19. Wu, H.; Tian, B.; Xu, W.; et al. Pressure-dependent CO2 electroreduction to methane over asymmetric Cu-N2 single-atom sites. J. Am. Chem. Soc. 2024, 146, 22266-75.
20. Zhou, X.; Shan, J.; Chen, L.; et al. Stabilizing Cu2+ ions by solid solutions to promote CO2 electroreduction to methane. J. Am. Chem. Soc. 2022, 144, 2079-84.
21. Kim, J.; Lee, T.; Jung, H. D.; et al. Vitamin C-induced CO2 capture enables high-rate ethylene production in CO2 electroreduction. Nat. Commun. 2024, 15, 192.
22. Fan, D.; Zhang, S.; Li, Y.; et al. High selective electrocatalytic reduction of carbon dioxide to ethylene enabled by regulating the microenvironment over Cu-Ag nanowires. J. Colloid. Interface. Sci. 2024, 662, 786-95.
23. Liu, Z.; Song, L.; Lv, X.; et al. Switching CO2 electroreduction toward ethanol by delocalization state-tuned bond cleavage. J. Am. Chem. Soc. 2024, 146, 14260-6.
24. Chang, S.; Gao, J.; Xuan, Y.; Li, D.; Wang, K. Amine modification enables selective CO2 electroreduction to ethanol via coupling of carbon-containing intermediates. Chem. Catal. 2025, 101383.
25. Li, Q.; Zheng, X.; Zhu, Z.; et al. Regulating organic modifiers on metal-based catalysts for enhanced electrocatalytic CO2 reduction. Chem. Asian. J. 2025, 20, e202401345.
26. Birdja, Y. Y.; Pérez-gallent, E.; Figueiredo, M. C.; Göttle, A. J.; Calle-vallejo, F.; Koper, M. T. M. Advances and challenges in understanding the electrocatalytic conversion of carbon dioxide to fuels. Nat. Energy. 2019, 4, 732-45.
27. Fan, L.; Liu, C.; Zhu, P.; et al. Proton sponge promotion of electrochemical CO2 reduction to multi-carbon products. Joule 2022, 6, 205-20.
28. Du, Z. Y.; Li, S. B.; Liang, G. H.; et al. Promoting water activation via molecular engineering enables efficient asymmetric C-C coupling during CO2 electroreduction. J. Am. Chem. Soc. 2024, 146, 32870-9.
29. Guo, F.; Zhao, Y.; Li, R.; Xu, H.; Sun, W. Photo/electrocatalytic reduction of CO2 to C2+ products on MOF-based catalysts. ChemNanoMat 2023, 9, e202300313.
30. Uekert, T.; Bajada, M. A.; Schubert, T.; Pichler, C. M.; Reisner, E. Scalable photocatalyst panels for photoreforming of plastic, biomass and mixed waste in flow. ChemSusChem 2021, 14, 4190-7.
31. Kumari, H.; Sonia; Suman; et al. A review on photocatalysis used for wastewater treatment: dye degradation. Water. Air. Soil. Pollut. 2023, 234, 349.
32. Ruziwa, D. T.; Oluwalana, A. E.; Mupa, M.; et al. Pharmaceuticals in wastewater and their photocatalytic degradation using nano-enabled photocatalysts. J. Water. Process. Eng. 2023, 54, 103880.
33. Sonu; Sharma, S.; Dutta, V.; et al. An overview of heterojunctioned ZnFe2O4 photocatalyst for enhanced oxidative water purification. J. Environ. Chem. Eng. 2021, 9, 105812.
34. Hu, H.; Ye, F.; Wang, T.; Xu, R.; Zhu, Y.; Deng, C. Co-deposition of amorphous carbon and CdS with the Host NiO HMs for superior photocatalytic H2 production via water splitting. Electron. Mater. Lett. 2024, 20, 627-38.
35. Kibria, M. G.; Edwards, J. P.; Gabardo, C. M.; et al. Electrochemical CO2 reduction into chemical feedstocks: from mechanistic electrocatalysis models to system design. Adv. Mater. 2019, 31, 1807166.
36. Wu, Z.; Zhu, P.; Cullen, D. A.; et al. A general synthesis of single atom catalysts with controllable atomic and mesoporous structures. Nat. Synth. 2022, 1, 658-67.
37. Wang, Q.; Liu, K.; Hu, K.; et al. Attenuating metal-substrate conjugation in atomically dispersed nickel catalysts for electroreduction of CO2 to CO. Nat. Commun. 2022, 13, 6082.
38. Hao, Q.; Zhong, H.; Wang, J.; et al. Nickel dual-atom sites for electrochemical carbon dioxide reduction. Nat. Synth. 2022, 1, 719-28.
39. Chang, H.; Kwon, S.; Bae, G.; Jeon, S. Rational design of arbitrary topology in three-dimensional space via inverse calculation of phase modulation. Nanophotonics 2024, 13, 971-82.
41. Pelras, T.; Mahon, C. S.; Müllner, M. Synthesis and applications of compartmentalised molecular polymer brushes. Angew. Chem. Int. Ed. 2018, 57, 6982-94.
42. Park, J.; Chae, D.; Lim, H.; et al. Daytime radiative cooling sheet functionalized by Al2O3-assisted organic composite. Adv. Sci. 2025, 12, 2417584.
43. Mazur, F.; Pham, A.; Chandrawati, R. Polymer materials as catalysts for medical, environmental, and energy applications. Appl. Mater. Today. 2023, 35, 101937.
44. Nghiem, T. L.; Coban, D.; Tjaberings, S.; Gröschel, A. H. Recent advances in the synthesis and application of polymer compartments for catalysis. Polymers 2020, 12, 2190.
45. Wang, Q.; Astruc, D. State of the art and prospects in metal-organic framework (MOF)-based and MOF-derived nanocatalysis. Chem. Rev. 2020, 120, 1438-511.
46. Li, H.; Gao, Y.; Xiong, Z.; Liao, C.; Shih, K. Enhanced selective photocatalytic reduction of CO2 to CH4 over plasmonic Au modified g-C3N4 photocatalyst under UV-vis light irradiation. Appl. Surf. Sci. 2018, 439, 552-9.
47. Zhang, W.; Jin, Z.; Chen, Z. Rational-designed principles for electrochemical and photoelectrochemical upgrading of CO2 to value-added chemicals. Adv. Sci. 2022, 9, 2105204.
48. Zhan, C.; Dattila, F.; Rettenmaier, C.; et al. Key intermediates and Cu active sites for CO2 electroreduction to ethylene and ethanol. Nat. Energy. 2024, 9, 1485-96.
49. Yoo, S.; Kim, D.; Deng, G.; et al. Impact of heterocore atoms on CO2 electroreduction in atomically precise silver nanoclusters. J. Am. Chem. Soc. 2025, 147, 12546-54.
50. Weng, C.; Wang, C.; Song, Y.; et al. In-situ reconstruction of active bismuth for enhanced CO2 electroreduction to formate. Chem. Eng. J. 2025, 505, 159732.
51. Kim, I.; Lee, G.; Kim, S.; et al. Unveiling the reconstruction of copper bimetallic catalysts during CO2 electroreduction. Nat. Catal. 2025, 8, 697-713.
52. Dang, H.; Guan, B.; Zhu, L.; et al. A review on photocatalytic and electrocatalytic reduction of CO2 into C2+ products: recent advances and future perspectives. Energy. Fuels. 2025, 39, 10109-33.
53. Su, L.; Hua, Q.; Yang, Y.; et al. Regulating competing reaction pathways for efficient CO2 electroreduction in acidic conditions. J. Energy. Chem. 2025, 105, 326-51.
54. Ozden, A.; García de Arquer, F. P.; Huang, J. E.; et al. Carbon-efficient carbon dioxide electrolysers. Nat. Sustain. 2022, 5, 563-73.
55. Guo, Q.; Zhou, C.; Ma, Z.; Yang, X. Fundamentals of TiO2 photocatalysis: concepts, mechanisms, and challenges. Adv. Mater. 2019, 31, 1901997.
56. Gao, D.; Long, H.; Wang, X.; Yu, J.; Yu, H. Tailoring antibonding-orbital occupancy state of selenium in Se-enriched ReSe2+x cocatalyst for exceptional H2 evolution of TiO2 photocatalyst. Adv. Funct. Mater. 2023, 33, 2209994.
57. Li, L.; Chen, X.; Quan, X.; Qiu, F.; Zhang, X. Synthesis of CuOx/TiO2 photocatalysts with enhanced photocatalytic performance. ACS. Omega. 2023, 8, 2723-32.
58. Li, Z.; Li, R.; Jing, H.; et al. Blocking the reverse reactions of overall water splitting on a Rh/GaN-ZnO photocatalyst modified with Al2O3. Nat. Catal. 2023, 6, 80-8.
59. Bi, T.; Du, Z.; Chen, S.; He, H.; Shen, X.; Fu, Y. Preparation of flower-like ZnO photocatalyst with oxygen vacancy to enhance the photocatalytic degradation of methyl orange. Appl. Surf. Sci. 2023, 614, 156240.
60. Saeed, M.; Khan, I.; Adeel, M.; Akram, N.; Muneer, M. Synthesis of a CoO-ZnO photocatalyst for enhanced visible-light assisted photodegradation of methylene blue. New. J. Chem. 2022, 46, 2224-31.
61. Wang, S.; Li, C.; Qi, Y.; et al. Etched BiVO4 photocatalyst with charge separation efficiency exceeding 90%. Nat. Commun. 2025, 16, 3776.
62. Hunge, Y. M.; Uchida, A.; Tominaga, Y.; et al. Visible light-assisted photocatalysis using spherical-shaped BiVO4 photocatalyst. Catalysts 2021, 11, 460.
63. Akrami, S.; Murakami, Y.; Watanabe, M.; et al. Enhanced CO2 conversion on highly-strained and oxygen-deficient BiVO4 photocatalyst. Chem. Eng. J. 2022, 442, 136209.
64. Zhao, Y.; Kim, S.; Eom, Y. K.; Valandro, S. R.; Schanze, K. S. Polymer chromophore-catalyst assembly for photocatalytic CO2 reduction. ACS. Appl. Energy. Mater. 2021, 4, 7030-9.
65. Izu, H.; Tabe, H.; Namiki, Y.; Yamada, H.; Horike, S. Heterogenous CO2 reduction photocatalysis of transparent coordination polymer glass membranes containing metalloporphyrins. Inorg. Chem. 2023, 62, 11342-9.
66. Cheng, R.; Chung, C.; Wang, S.; et al. Three-dimensional self-attaching perovskite quantum dots/polymer platform for efficient solar-driven CO2 reduction. Mater. Today. Phys. 2021, 17, 100358.
67. Zhao, Y.; Ye, X.; Liu, Y.; et al. Fabrication of glass immobilized amorphous organotitanium polymer for enhancing catalytic turnover frequency and stabilities in photocatalytic reduction of CO2. Appl. Catal. A. Gen. 2022, 647, 118910.
68. Skorjanc, T.; Kamal, K. M.; Alkhoori, A.; et al. Polythiacalixarene-embedded gold nanoparticles for visible-light-driven photocatalytic CO2 reduction. ACS. Appl. Mater. Interfaces. 2022, 14, 30796-801.
69. Yu, Z.; Xiao, Y.; Guo, S.; et al. Visible light-driven selective reduction of CO2 by acetylene-bridged cobalt porphyrin conjugated polymers. ChemSusChem 2022, 15, e202200424.
70. Nandi, D. K.; Seth, J.; Chakrabortty, P.; et al. Ni Nanoparticles supported over triazine based porous organic polymer for selective CO2 photo-reduction to methanol. ChemCatChem 2023, 15, e202301018.
71. Wang, W.; Xi, Y.; Yang, C.; et al. Incorporation of metal active sites on porous polycarbazoles for photocatalytic CO2 reduction. ChemCatChem 2022, 14, e202101872.
72. Jena, R.; Rahimi, F. A.; Karmakar, S.; et al. Electron rich guest regulated enhanced CO2 reduction in a multivariate porous coordination polymer. Adv. Funct. Mater. 2024, 34, 2407721.
73. Jiang, J.; Chen, Y.; Ji, H. Zinc porphyrin and rhenium complex-based donor-acceptor conjugated porous polymer for visible-light-driven conversion of CO2 to CO. J. CO2. Util. 2022, 60, 101972.
74. Rahimi, F. A.; Dey, S.; Verma, P.; Maji, T. K. Photocatalytic CO2 reduction based on a Re(I)-integrated conjugated microporous polymer: role of a sacrificial electron donor in product selectivity and efficiency. ACS. Catal. 2023, 13, 5969-78.
75. Wang, K.; Li, Q.; Chen, X.; et al. Porous organic polymers with shiftable active Co(II) sites for photocatalytic reduction of CO2 to C2H4. Appl. Catal. B. Environ. Energy. 2025, 362, 124765.
76. Chi, X.; Lan, Z. A.; Chen, Q.; et al. Electronic transmission channels promoting charge separation of conjugated polymers for photocatalytic CO2 reduction with controllable selectivity. Angew. Chem. Int. Ed. 2023, 62, e202303785.
77. Rahimi, F. A.; Singh, A.; Jena, R.; Dey, A.; Maji, T. K. GFP chromophore integrated conjugated microporous polymers toward bioinspired photocatalytic CO2 reduction to CO. ACS. Appl. Mater. Interfaces. 2024, 16, 43171-9.
78. Hong, X.; Fang, Y.; Chao, D. A metal-free porous organic polymer with high CO2 adsorption for robust aqueous CO2 photoreduction via molecular regulation. Mol. Catal. 2023, 549, 113476.
79. Das, N.; Paul, R.; Biswas, S.; et al. Photo-responsive signatures in a porous organic polymer enable visible light-driven CO2 photofixation. ACS. Sustainable. Chem. Eng. 2023, 11, 2066-78.
80. Geng, W.; Wang, Q.; Zhu, C.; et al. Enhanced CO2 electroreduction to formate over tin coordination polymers via amino-functionalization. J. Power. Sources. 2022, 529, 231252.
81. Chen, X.; Jia, S.; Chen, C.; et al. Highly stable layered coordination polymer electrocatalyst toward efficient CO2-to-CH4 conversion. Adv. Mater. 2024, 36, 2310273.
82. Wang, J.; Sun, M.; Xu, H.; et al. Coordination environment engineering of metal centers in coordination polymers for selective carbon dioxide electroreduction toward multicarbon products. ACS. Nano. 2024, 18, 7192-203.
83. Liang, Y.; Zhao, J.; Yang, Y.; et al. Stabilizing copper sites in coordination polymers toward efficient electrochemical C-C coupling. Nat. Commun. 2023, 14, 474.
84. Al-attas, T. A.; Marei, N. N.; Yong, X.; et al. Ligand-engineered metal-organic frameworks for electrochemical reduction of carbon dioxide to carbon monoxide. ACS. Catal. 2021, 11, 7350-7.
85. Zhang, Y.; Zhou, Q.; Qiu, Z.; et al. Tailoring coordination microenvironment of Cu(I) in metal-organic frameworks for enhancing electroreduction of CO2 to CH4. Adv. Funct. Mater. 2022, 32, 2203677.
86. Sun, M.; Cheng, J.; Anzai, A.; Kobayashi, H.; Yamauchi, M. Modulating electronic states of Cu in metal-organic frameworks for emerging controllable CH4/C2H4 selectivity in CO2 electroreduction. Adv. Sci. 2024, 11, 2404931.
87. Wen, C. F.; Zhou, M.; Liu, P. F.; et al. Highly ethylene-selective electrocatalytic CO2 reduction enabled by isolated Cu-S motifs in metal-organic framework based precatalysts. Angew. Chem. Int. Ed. 2022, 61, e202111700.
88. Yu, J.; Xiao, J.; Guo, L.; et al. In situ phase transformation-enabled metal-organic frameworks for efficient CO2 electroreduction to multicarbon products in strong acidic media. ACS. Nano. 2024, 18, 33602-13.
89. Li, Z.; Li, X.; Wang, R.; et al. Electro-activated indigos intensify ampere-level CO2 reduction to CO on silver catalysts. Nat. Commun. 2025, 16, 3206.
90. Xu, Y.; Guo, Y.; Sheng, Y.; et al. Selective CO2 electroreduction to formate on polypyrrole-modified oxygen vacancy-rich Bi2O3 nanosheet precatalysts by local microenvironment modulation. Small 2023, 19, 2300001.
91. Su, L.; Hua, Q.; Feng, G.; et al. Multifunctional conductive polymer modification for efficient CO2 electroreduction in acidic electrolyte. Adv. Funct. Mater. , 2025, 2425636.
92. Wang, Y.; Cheng, Y.; Liu, S.; et al. Enhancing CO2 electroreduction to multicarbon products by modulating the surface microenvironment of electrode with polyethylene glycol. Angew. Chem. Int. Ed. 2025, 64, e202420661.
93. Yang, L.; Lv, X.; Peng, C.; et al. Promoting CO2 electroreduction to acetate by an amine-terminal, dendrimer-functionalized Cu catalyst. ACS. Cent. Sci. 2023, 9, 1905-12.
94. Kim, C.; Bui, J. C.; Luo, X.; et al. Tailored catalyst microenvironments for CO2 electroreduction to multicarbon products on copper using bilayer ionomer coatings. Nat. Energy. 2021, 6, 1026-34.
95. Zhang, T.; Lu, X.; Qi, W.; Qin, G.; Li, S. Efficient electroreduction of CO2 to CO on silver single-atom catalysts: activity enhancement through coordinated modulation of polyaniline. Appl. Catal. B. Environ. Energy. 2024, 349, 123896.
96. Xu, Y.; Zhao, Y.; Kochubei, A.; et al. Copper/polyaniline interfaces confined CO2 electroreduction for selective hydrocarbon production. ChemSusChem 2024, 17, e202400209.
97. Jia, S.; Zhu, Q.; Chu, M.; et al. Hierarchical metal-polymer hybrids for enhanced CO2 electroreduction. Angew. Chem. Int. Ed. 2021, 60, 10977-82.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].