



Random aliovalent cations induce synergistic bonding for high thermoelectric performance in NaSn2SbSe4
 0
0 Abstract
The dimensionless figure of merit ZT is the key metric for quantifying thermoelectric performance; however, its optimization is inherently limited by the intrinsic coupling between electrical and thermal transport properties. Herein, we perform first-principles calculations using the temperature-dependent effective potential method to investigate cation alloying in SnSe, where Sn2+ ions were randomly substituted by aliovalent cations - specifically Na+ and Sb3+ - a strategy that induces chemical bond synergy in NaSn2SbSe4, effectively decoupling electronic and phonon transport behaviors. Random cation occupation induces a mixed covalent-ionic bonding character, generating local bond-strength fluctuations that act as phonon-scattering centers. Furthermore, localized Sb-Se and Sn-Se antibonding states below the Fermi level correlate with the softening of low-frequency optical phonon branches. Combined with pronounced lattice anharmonicity, this phonon softening significantly enhances four-phonon scattering rates and suppresses the lattice thermal conductivity. Concurrently, aliovalent cation incorporation promotes electron delocalization between Sn-5p and Se-4p orbitals and strengthens covalent bonding character. This modified orbital hybridization alters band dispersion, reducing the hole effective mass while preserving high electrical conductivity in NaSn2SbSe4. The optimal balance between low lattice thermal conductivity and high power factor yields superior thermoelectric performance for NaSn2SbSe4 relative to SnSe across the entire temperature range, with a peak ZT of 0.88 at 800 K. This study establishes chemical bonding engineering as a promising strategy for enhancing thermoelectric performance and provides guidance for exploring high-performance high-entropy thermoelectric materials.
Keywords
INTRODUCTION
The escalating global energy crisis has motivated the recent growing interest in thermoelectric energy conversion, which is widely recognized as one of the most promising approaches for recovering waste heat from fossil fuel combustion. Consequently, the development of high-efficiency thermoelectric materials has emerged as the research focus in the pursuit of sustainable and efficient energy conversion solutions[1-3]. The thermoelectric conversion efficiency is commonly quantified in terms of the dimensionless figure of merit ZT, defined as ZT = S2σT/ (κe + κL), where S, σ, T, κL, and κe are the Seebeck coefficient, electrical conductivity, absolute temperature, lattice thermal conductivity, and electron thermal conductivity, respectively[4,5]. Achieving a high ZT value requires either enhancing the power factor (PF = S2σ) or suppressing the sum of thermal conductivities (κe + κL). Among the characteristic variables that control ZT - S, σ, κL, and κe - the lattice thermal conductivity κL is unique in that it can be tuned independently of thermal transport properties. Therefore, reducing κL has emerged as a key strategy for improving thermoelectric performance[6,7].
Conventional approaches to suppress κL include the introduction of point defects[8] and the engineering of nanostructures[9]. Although these methods effectively enhance phonon scattering, they often degrade charge-carrier transport due to increased carrier scattering, thereby limiting overall improvements in thermoelectric efficiency. Recently, entropy engineering has emerged as a promising alternative strategy, enabling an optimal balance between low lattice thermal conductivity and high electrical transport performance through the deliberate introduction of atomic-scale disorder[10-12]. This approach exploits configurational entropy to stabilize single-phase solid solutions composed of multiple principal elements, which leads to enhanced thermoelectric properties[13-16]. For instance, the high-entropy chalcogenides, (SnSe)1-x(NaSbSe2)x alloy remains phase stable in the cubic structure at ambient temperature, and its thermoelectric ZT value reaches 0.5 due to improved electrical transport arising from increased carrier concentration[17]. Similarly, Sn0.4(AgSb)0.3Se, designed via cationic alloying, attains a ZT of 0.82 at 842 K[18]. These advancements highlight the critical influence of local atomic structure - particularly cation disorder at the Sn site - on thermoelectric performance[19]. However, due to inherent limitations in experimental characterization techniques, direct observation of local structural details remains challenging, resulting in an incomplete understanding of how local disorder and configurational entropy affect thermoelectric behavior in entropically stabilized chalcogenides. This knowledge gap significantly impedes the establishment of clear structure-property relationships.
In this study, first-principles simulations are employed to investigate the relationship between local structural disorder and thermoelectric performance in entropy-stabilized chalcogenides. A cation-disordered NaSn2SbSe4 structure was constructed within a SnSe-based framework using the Monte Carlo special quasi-random structure (SQS) method[20]. In NaSn2SbSe4, the simultaneous occupation of the same Wyckoff site by two aliovalent cations - Na+ and Sb3+ - induces significant local structural heterogeneity. The disordered distribution of cations enhances asymmetry at the Sn sites, with a transition from uniform covalent bonding in pristine SnSe to a mixed covalent-ionic bonding character in NaSn2SbSe4. This evolution toward a heterogeneous bonding environment is expected to strengthen lattice anharmonicity and suppress phonon-mediated heat transport. Moreover, substituting Sn sites with cations of different ionic radii
METHODS
All first-principles calculations employed density functional theory (DFT) implemented with the projected augmented wave (PAW) pseudopotentials via the Vienna Ab initio Simulation Package (VASP)[21]. Quasi-random structures of NaSn2SbSe4 were generated via the Monte Carlo SQS method in the alloy theoretical automated toolkit (ATAT)[20], guided by the experimentally confirmed single-phase cubic structure of the (SnSe)1-x(NaSbSe2)x solid solution. The final selected configuration achieves the Perfect_match criterion in the mcsqs.log file by accurately reproducing all targeted pair and multi-body correlation functions. The corresponding bestsqs.out file contains the resulting optimal structure.
Owing to computational resource limitations, the structure of NaSn2SbSe4 used to evaluate thermal transport properties was constructed based on the SnSe unit cell. In this unit cell, the atomic fractions of Na, Sb, and Sn occupying the Sn Wyckoff positions were 25%, 25%, and 50%, respectively. These values were obtained from the SQS construction based on the target stoichiometry, consistent with the experimentally reported composition[17]. Although denoted by the nominal stoichiometry NaSn2SbSe4, the compound is modeled as a solid solution where the Sn and Sb atoms are randomly distributed on the cation sublattice. To account for this configurational disorder in our first-principles calculations, we employ the SQS method to generate supercells that best mimic the statistics of a perfectly random alloy. The plane-wave cutoff energy was set to 520 eV, and the Monkhorst-Pack k-mesh was configured as 9 × 9 × 9. To fully relax the lattice structure, the convergence thresholds for the total energy and Hellmann-Feynman force were set to 10-7 eV and 10-5 eV/Å, respectively. A 3 × 3 × 3 supercell comprising 216 atoms was employed for Ab Initio molecular dynamics (AIMD) simulation. The trajectories were propagated for 10 ps with a 1 fs integration interval under the canonical (NVT) ensemble. Second- to fourth-order interatomic force constants (IFCs) were obtained from the AIMD-derived potential energy surface within the framework of the temperature-dependent effective potential (TDEP) method[22,23]. By combining the TDEP method with the FourPhonon package[24], we solved the phonon Boltzmann transport equation (BTE) by incorporating four-phonon (4ph) scattering. The lattice thermal conductivity κL, phonon scattering rates, the Grüneisen parameter γ, and other relevant properties were computed on a 14 × 14 × 14 q-mesh in the irreducible Brillouin zone. After performing convergence testing, the Gaussian smearing parameter was chosen as 0.5. Four-phonon scattering rates were rapidly estimated under the relaxation time approximation (RTA) using 1 × 105 sampled processes within the maximum likelihood estimation method[25]. Bond orders were calculated using the integrated crystal orbital bond index (ICOBI) in the LOBSTER package[26].
To accurately simulate cation disorder, a 2 × 2 × 2 supercell (containing 64 atoms) based on the SnSe unit cell was constructed to model the structure of NaSn2SbSe4 for electrical transport calculations. The identical atomic alloying ratios were used in both electrical and thermal transport calculations. A plane-wave cutoff energy of 450 eV was employed, along with a Monkhorst-Pack k-mesh of 3 × 3 × 3. The convergence thresholds were set to 10-6 eV for the total energy and 10-2 eV/Å for the Hellmann-Feynman force, respectively. The electronic band structure was obtained using the Heyd-Scuseria-Ernzerhof (HSE06) functional in conjunction with spin-orbit coupling (SOC). Based on the electronic band structure obtained using the HSE06 functional with SOC, the electronic transport properties were computed by solving the semiclassical BTE under the relaxation time approximation as implemented in the BoltzTraP2 package[27].
RESULTS AND DISCUSSION
Disordered structure and chemical bond characterizations
To investigate cationic disorder arising from the entropy stabilization effect, appropriate cations were incorporated into the SnSe lattice, and quasi-random structures of NaSn2SbSe4 were constructed via the SQS method, as illustrated in Figure 1. The optimized lattice parameters of NaSn2SbSe4 are a = 5.954 Å and b = c = 5.931 Å, with α = β = γ = 90°, consistent with a typical tetragonal phase structure. Compared with cubic-phase SnSe, the lattice parameters along the a and b/c axes of NaSn2SbSe4 exhibit changes of 1.636% and 2.016%, respectively [Supplementary Table 1]. These variations are primarily attributed to the significantly greater contraction of Sb-Se, Na-Se, and Sn-Se bond lengths along the b/c-axes relative to those along the a-axis [Supplementary Table 2]. Analysis of bond lengths reveals that the random distribution of Na and Sb atoms, together with their distinct local bonding environments, results in significant structural deviations from pristine SnSe, with structural distortion degrees of 1.619% and 2.016% along the a and b/c-axes, respectively. This degree of distortion effectively mitigates excessive lattice strain while preserving the structural stability of the tetragonal phase, and is anticipated to maintain favorable electronic transport properties. As shown in Supplementary Table 3, the calculated optical dielectric constants of NaSn2SbSe4 along the a and b/c directions are approximately 4.17 and 3.70 times those of SnSe, respectively, confirming its significantly enhanced electronic polarizability. Higher electronic polarizability is closely associated with band structure features favorable for electron transport, predicting that NaSn2SbSe4 exhibits excellent electrical transport properties.
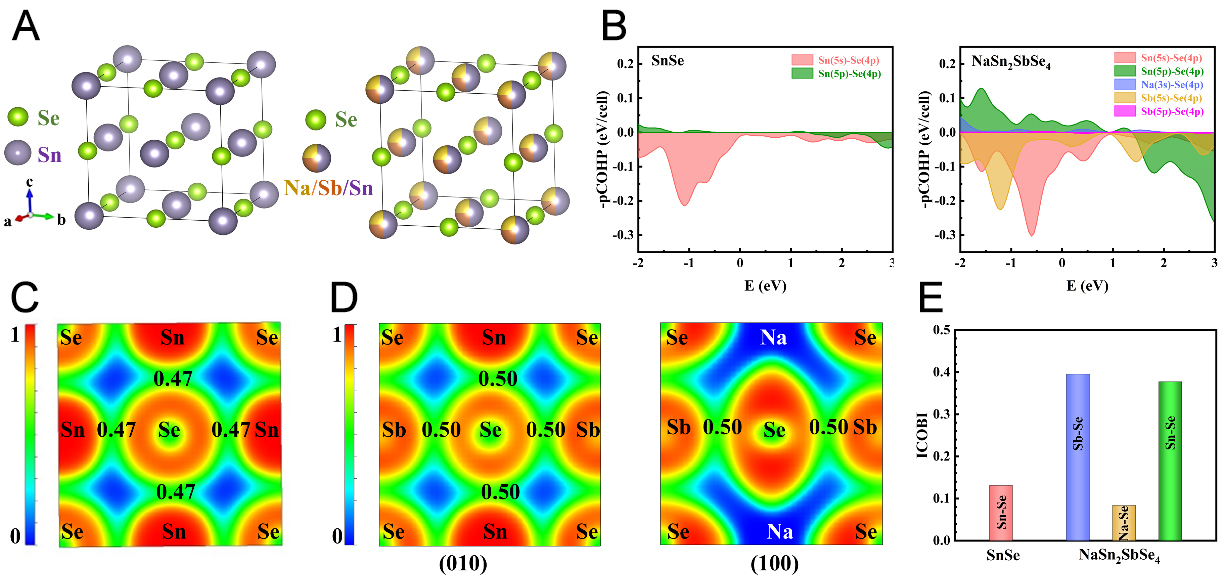
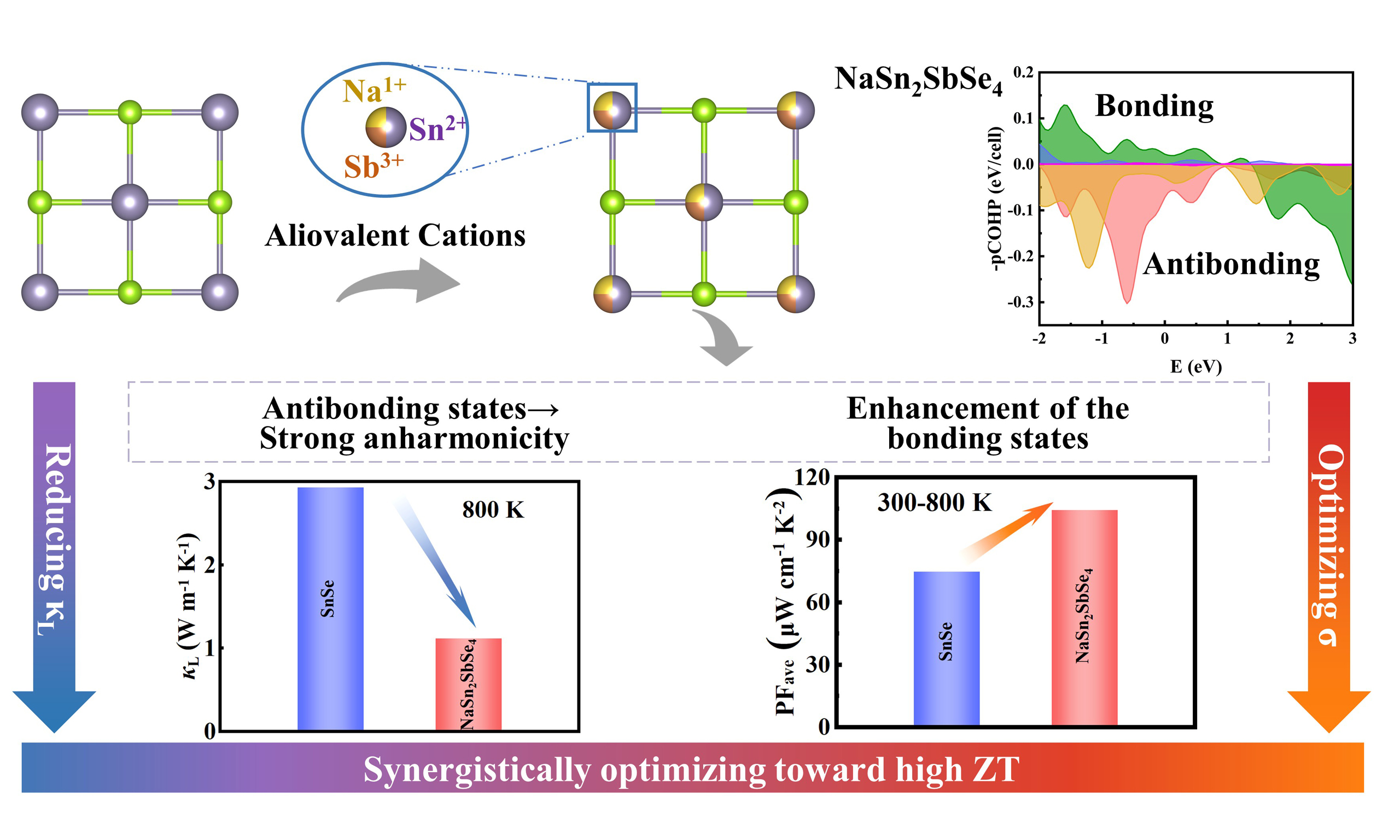
Figure 1. (A) The structures and (B) projected Crystal Orbital Hamilton Population (-pCOHP) of SnSe and NaSn2SbSe4. The Electron Localization Function (ELF) analysis of (C) SnSe and (D) NaSn2SbSe4, respectively; (E) The calculated integrated crystal orbital bond index (ICOBI) for the Sn-Se bond in SnSe and the Sb/Na/Sn-Se bonds in NaSn2SbSe4.
Furthermore, the structural distortion in NaSn2SbSe4 is strongly correlated with significant changes in its bonding characteristics[28]. To systematically investigate this effect, we employed projected Crystal Orbital Hamilton Population (-pCOHP) and Electron Localization Function (ELF) analyses. As shown in Figure 1B, the -pCOHP spectra of SnSe and NaSn2SbSe4 provide detailed insights into orbital interactions and bond strength, with negative and positive regions corresponding to antibonding and bonding states, respectively. In SnSe, the Sn-Se bonding is primarily attributed to the hybridization between Sn(5p) and Se(4p) orbitals, whereas the antibonding states arise mainly from the coupling of Sn(5s) and Se(4p) orbitals. In NaSn2SbSe4, the introduction of Na and Sb enhances the bonding interactions between Sn (5p) and Se (4p), which is expected to influence the electrical transport properties via band structure modification. Additionally, it introduces significant orbital interactions predominantly originating from Sb, where Sb(5s) forms a distinct antibonding state with Se(4p) and further enhances the existing Sn(5s)-Se(4p) antibonding interaction. It has been established in prior work that the formation of s-p antibonding states below the Fermi level plays a decisive role in shaping the phonon dynamics[29]. Consequently, the pronounced intensification of Sn(5s)-Se(4p) antibonding states from -1 eV to 0 eV in NaSn2SbSe4 relative to SnSe is expected to induce significantly stronger lattice anharmonicity, thereby contributing to a reduction in lattice thermal conductivity. We further investigated the influence of Sb and Na incorporation on electronic localization [Figure 1C and D]. The results reveal that the ELF distribution in SnSe exhibits weakly covalent bonding characteristics, whereas NaSn2SbSe4 displays covalency and ionicity: the Sb atom significantly strengthens the covalent character of both Sn-Se and Sb-Se bonds, while the introduction of Na induces a partial transition of certain Sn-Se bonds from covalent to Na-Se ionic bonds[30]. We further employ the ICOBI to quantify bonding heterogeneity in NaSn2SbSe4. This index reflects differences in bond strength and allows direct comparison among bonds within a structure, making it a reliable indicator of bond non-uniformity[31]. As shown in Figure 1E, the calculated ICOBI values for the Sb-Se and Sn-Se bonds in NaSn2SbSe4 are 0.40 and 0.38, respectively - both notably higher than that of the Sn-Se bond in SnSe (0.13) - whereas the ICOBI value for the Na-Se bond is 0.08, which is lower than that of the Sn-Se bond in SnSe. This provides additional evidence that NaSn2SbSe4 exhibits mixed covalent-ionic bonding character, with the heterogeneous nature of strong and weak bonds acting to enhance lattice anharmonicity, strengthen phonon scattering, and consequently reduce lattice thermal conductivity[32]. Notably, structural distortion induces charge polarization, resulting in an inhomogeneous charge density distribution, with pronounced electron delocalization observed between Sn-5p and Se-4p orbitals. This modification is expected to further influence the band dispersion, thereby helping to preserve high electrical conductivity. These findings collectively demonstrate that the introduction of aliovalent cations and the resultant structural distortion profoundly influence the chemical bonding nature of the material. In this configuration, the synergistic interaction between Sb and Na fosters the formation of a complex chemical bonding network, giving rise to a unique electronic environment characterized by heterogeneous bonding interactions with varying orbital overlap strengths and bond polarizations. Such heterogeneous bonding characteristics are expected to suppress lattice thermal conductivity and concurrently maintain favorable electronic transport, thereby offering new avenues for the synergistic optimization of thermoelectric performance[33,34].
Thermal transport properties
To investigate the influence of chemical bonding variations on thermal transport properties, we calculate the phonon dispersion curves and phonon density of states (PhDOS) for both SnSe and NaSn2SbSe4. To enable a systematic comparison, the high-symmetry q-point path along Γ-X-M-Γ-R-M was employed. Within the harmonic approximation, the phonon dispersion of NaSn2SbSe4 was calculated at 0 K using Phonopy, and imaginary frequencies are observed in the resulting spectrum [Supplementary Figure 1]. Previous studies have shown that such behavior commonly occurs in strongly anharmonic materials[35], indicating that the reference structure of NaSn2SbSe4 does not correspond to a stable local minimum of the effective potential energy surface within the harmonic approximation. Instead, this behavior is consistent with a complex, multi-well potential energy surface, which is a characteristic feature of strongly anharmonic systems. In contrast, as shown in Figure 2A and B, the finite-temperature phonon spectra obtained using the TDEP method at 300 K show that these harmonic instabilities are removed through anharmonic renormalization, demonstrating that both SnSe and NaSn2SbSe4 are dynamically stable at 300 K[36-37]. Meanwhile, NaSn2SbSe4 shows a calculated formation energy of approximately -4 eV, indicating the thermodynamic stability of the corresponding structures. To accurately assess the phonon transport properties, the correction of the nonanalytic term and the Γ point splitting between transverse and longitudinal optical (TO-LO) phonon branches were incorporated into the analysis[38,39]. Notably, NaSn2SbSe4 shows a splitting of the optical branches at the Γ point. Compared to SnSe, NaSn2SbSe4 also demonstrates more pronounced softening of the optical phonon branches, which primarily originate from the vibrational contributions of the Sb, Sn, and Se atoms. For example, at the high-symmetry Γ point and in the frequency range of 0-50 cm-1, the phonon dispersion of NaSn2SbSe4 reveals low-frequency optical phonon branches, which are associated with lattice anharmonicity[23,40]. This observation is consistent with our initial prediction that the Sb/Na-induced disorder perturbs the local bonding environment and disrupts the periodicity required for coherent phonon propagation, thus strengthening the lattice anharmonicity. Concurrently, the phonon dispersion of NaSn2SbSe4 displays significant coupling between the acoustic and low-frequency optical modes - a feature that enhances phonon scattering and will reduce lattice thermal conductivity[39,41].
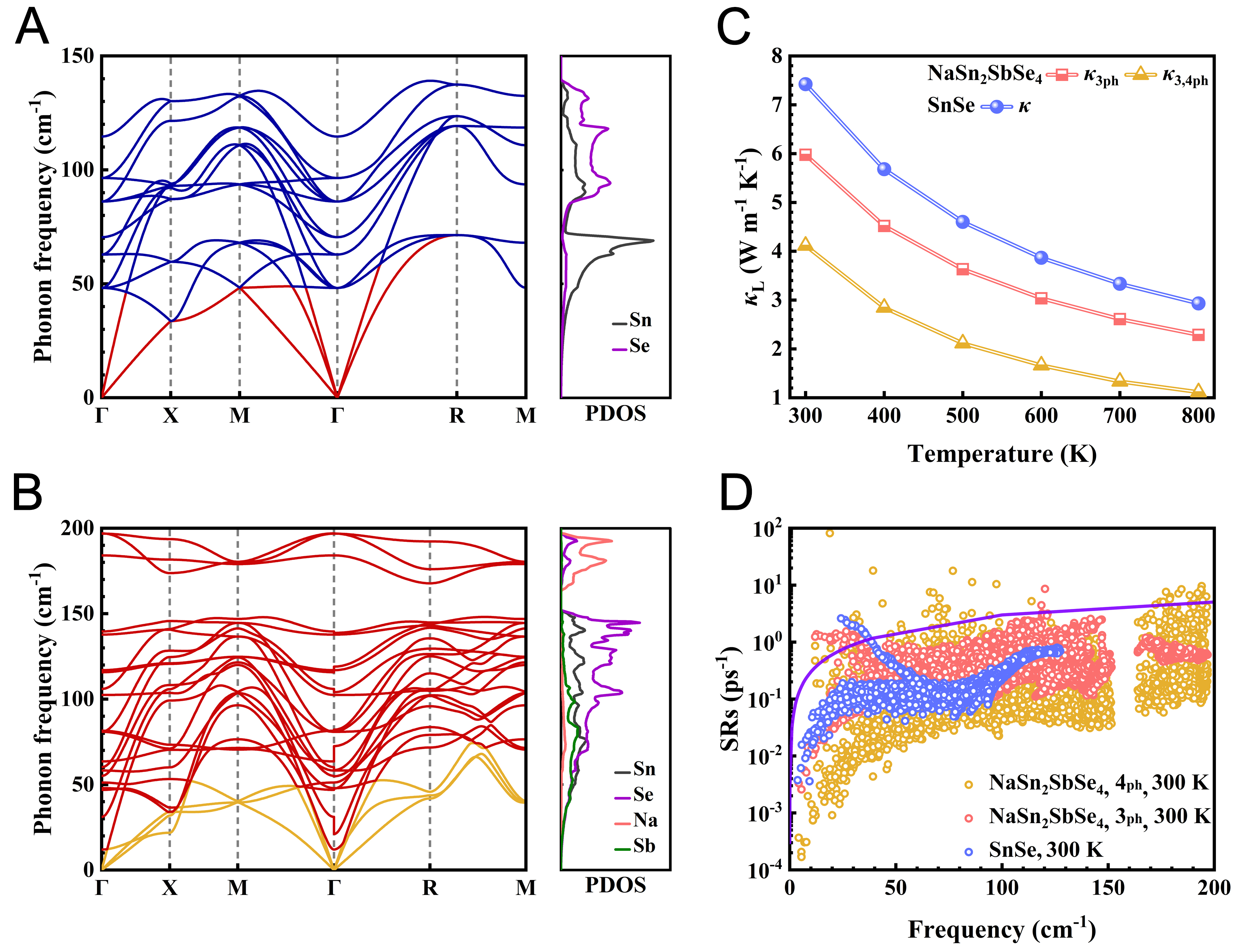
Figure 2. Calculated phonon dispersion curves and projected phonon density of states (PDOS) at T = 300 K for (A) SnSe and (B) NaSn2SbSe4. The optical branches are shown in blue in (A) and red in (B), and the acoustic branches are shown in red in (A) and yellow in (B). The grey, purple, red, and green solid lines in subfigures (A) and (B) represent the PDOS of Sn, Se, Na, and Sb atoms, respectively; (C) κL calculated from the three-phonon (3ph) and combined three- and four-phonon (3,4ph) scattering models over the temperature range from 300 K to 800 K, and (D) the calculated 3ph and 4ph scattering rates (SRs) as a function of phonon frequency at 300 K for SnSe and NaSn2SbSe4.
Lattice thermal conductivity κL quantifies the phonon contribution to thermal transport. For materials with pronounced anharmonicity, such as NaSn2SbSe4, it is essential to consider quartic anharmonicity effects to predict κL values accurately[42,43]. Therefore, we investigated κL using three-phonon (3ph) and combined three- and four-phonon (3,4ph) scattering models, with results presented in Figure 2C. For both SnSe and NaSn2SbSe4, the κL exhibits a steady decline, approximately inversely proportional to temperature in the range of 300-800 K. This phenomenon arises primarily from enhanced lattice vibrations at elevated temperatures, which facilitate phonon interactions and strengthen phonon scattering processes[39,42]. Notably, NaSn2SbSe4 exhibits an approximately 50% reduction in κL relative to SnSe, aligned with the observations extracted from the phonon dispersion assessment. Additionally, for NaSn2SbSe4, κ3,4ph is significantly reduced relative to κ3ph due to the inclusion of the 4ph scattering mechanism, highlighting the significant impact of 4ph scattering processes on phonon thermal transport. Specifically, the theoretically obtained κ3ph and κ3,4ph values are 3.0 and 1.6 W m-1 K-1 at 600 K for NaSn2SbSe4, respectively. At 800 K, the κ3,4ph of NaSn2SbSe4 is
Phonon scattering rates are primarily determined by two factors: the anharmonic scattering strength, typically characterized by the Grüneisen parameters γ[48], and the number of available scattering channels, quantified by the phonon scattering phase space W. The phonon scattering phase space is defined as the set of all accessible final states in momentum space, thereby quantifying the number of scattering channels permitted by simultaneous energy and momentum conservation[41,49]. To clarify the underlying origin of the low κL, both the Grüneisen parameter γ and the scattering phase space W were analyzed. As shown in Figure 3A, the magnitude of γ in NaSn2SbSe4 within the 0-25 cm-1 frequency range is significantly larger than that in SnSe, thereby indicating its stronger anharmonicity. This finding further supports that a high Grüneisen parameter γ plays an important role in enhancing phonon scattering in NaSn2SbSe4. Figure 3B presents the calculated 3ph scattering phase space (W3ph). Under the conservation of energy and momentum, low-frequency phonons mainly participate in combination processes that up-convert population into high-frequency phonons, while high-frequency phonons primarily evolve via intrinsic 3ph scattering. Therefore, within the 3ph phase space, phonon scattering is predominantly governed by splitting processes (λ → λ′ + λ″) in the 50-200 cm-1 range, and by combination processes (λ + λ′ → λ″) at frequencies below
Figure 3. (A) Calculated Grüneisen parameter γ of SnSe and NaSn2SbSe4 at 300 K; (B) Decomposition of 3ph scattering phase space (W3ph) of NaSn2SbSe4 at 300 K into combination (λ + λ′ → λ″) and splitting (λ → λ′ + λ″) processes; (C) Decomposition of the 4ph scattering phase space (W4ph) of NaSn2SbSe4 at 300 K into redistribution (λ + λ′ → λ″+ λ‴), recombination (λ + λ′+ λ″ → λ‴), and splitting (λ → λ′ + λ″+ λ‴) processes; (D) Decomposition of 4ph scattering rates of NaSn2SbSe4 at 300 K into the normal and Umklapp processes.
Electronic transport properties
To systematically characterize the influence of structural disorder on electrical transport properties, we built a supercell and conducted a preliminary investigation of the electronic structure of NaSn2SbSe4, as shown in Supplementary Figures 5 and 6. Upon the incorporation of SOC in the calculations, NaSn2SbSe4 exhibits a relatively narrow band gap (~0.25 eV)[52]. Furthermore, as shown in Supplementary Figure 6C, the charge density at the valence band maximum (VBM) demonstrates significant delocalization and hole transport pathways between the Sn and Se atoms. This is further supported by Supplementary Figure 7, where the projected density of states confirms the hybridization of Sn-5p and Se-4p orbitals, also highlighting their contribution to the valence band edge. The above observation not only indicates enhanced hole transport capability in NaSn2SbSe4 but also corroborates the prior chemical bonding analysis, which revealed that strengthened hybridization between Sn-5p and Se-4p orbitals leads to greater delocalization and increased covalency. This effect of strengthened hybridization gives rise to an electronic environment that modulates the band structure, thereby creating favorable conditions for carrier transport and electrical conductivity in NaSn2SbSe4[33,53].
To comprehensively evaluate and compare the electronic transport properties of SnSe and NaSn2SbSe4, we employed Boltzmann transport theory to calculate and analyze how the Seebeck coefficient (S), electrical conductivity (σ), and PF vary with carrier concentration and temperature. As shown in Figure 4A, the Seebeck coefficients of SnSe and NaSn2SbSe4 display an inverse dependence on carrier concentration, initially increasing at low doping levels and then decreasing at higher concentrations. According to the Mott relation[54], the Seebeck coefficient varies with carrier concentration n as S ∝ n-2/3. At low p-type doping levels, the observed deviation from the expected trend can be attributed to the bipolar conduction effect induced by a small bandgap. This effect occurs because, although holes dominate charge transport at low carrier concentrations, intrinsically excited minority electrons also make a non-negligible contribution[55]. The opposite-sign contributions of these two types partially compensate each other, thereby markedly suppressing the Seebeck response in the low-doping regime. By contrast, at high carrier concentrations, the Seebeck coefficient is mainly determined by the majority hole carriers and thus follows the expected trend.
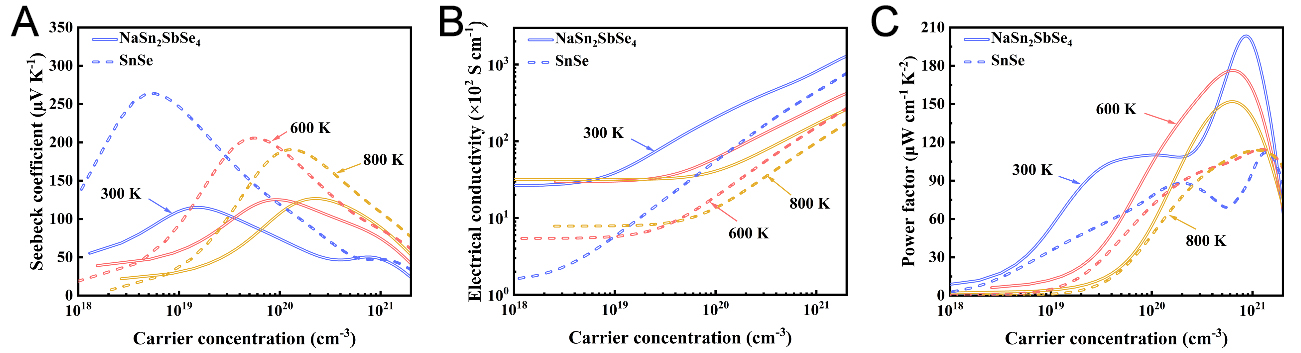
Figure 4. Calculated (A) Seebeck coefficient, (B) electrical conductivity, and (C) power factor of SnSe and NaSn2SbSe4 at 300, 600, and 800 K as a function of carrier concentration.
The carrier relaxation time (τ) was calculated for SnSe and NaSn2SbSe4 according to the deformation potential theory[56,57], with the results presented in Supporting Information Supplementary Table 4. NaSn2SbSe4 consistently demonstrates a longer relaxation time than SnSe at the same temperature. This behavior is primarily attributed to the reduction in the hole effective mass[52,58]. Electrical conductivity was calculated based on the obtained relaxation time, and the results are shown in Figure 4B. At a given temperature, the electrical conductivities of SnSe and NaSn2SbSe4 increase with increasing charge carrier concentration. At the same temperature and carrier concentration, NaSn2SbSe4 shows higher electrical conductivity than SnSe, mainly because its longer relaxation time reduces electron scattering[59]. For example, at a carrier concentration of 1021 cm-3 and 600 K, the electrical conductivity increases from 1.976 × 103 S cm-1 in SnSe to 6.063 × 103 S cm-1 in NaSn2SbSe4. Moreover, the electronic thermal conductivity (κe) quantifies the contribution of charge carriers to thermal transport within the material. Supplementary Figure 8 presents the calculated κe values for SnSe and NaSn2SbSe4, showing that κe is higher in NaSn2SbSe4 than in SnSe under identical temperature and hole concentration conditions, which reflects proportional dependence on electrical conductivity.
The power factor PF, expressed as PF = S2σ, measures the effectiveness of electronic transport in thermoelectric materials. To further explore the contradictory dependence between the Seebeck coefficient and electrical conductivity, the power factors of SnSe and NaSn2SbSe4 were analyzed with respect to carrier concentration, as shown in Figure 4C. Despite the reduction in the Seebeck coefficient, the significant enhancement in electrical conductivity ensures that NaSn2SbSe4 maintains a higher optimal power factor compared to SnSe. Specifically, at 300 K, the peak power factor of NaSn2SbSe4 reaches 203.42 μW cm-1 K-2 at a hole concentration of 8.69 × 1020 cm-3, which is twice as high as that of SnSe.
Thermoelectric figure of merit
Based on the analysis of structural features and chemical bonding characteristics, together with thermal and electrical transport properties, it is evident that, compared to SnSe, the incorporation of aliovalent cations in NaSn2SbSe4 induces pronounced local structural distortions and establishes a heterogeneous bonding environment. The Sb(5s)/Sn(5s)-Se(4p) antibonding states located below the Fermi level correlate with the softening of low-frequency optical phonon branches in NaSn2SbSe4. This softening, coupled with the strong lattice anharmonicity, significantly enhances four-phonon scattering rates and effectively suppresses the lattice thermal conductivity. Furthermore, the incorporation of aliovalent cations enhances the hybridization between Sn-5p and Se-4p orbitals, leading to greater electronic delocalization and increased covalency - factors that promote high carrier mobility and enable superior electrical conductivity. The synergistic optimization of low thermal conductivity and high electrical conductivity leads to a significant enhancement in thermoelectric performance. To assess its practical potential, we calculated the thermoelectric figure of merit, given by ZT = S2σT/ (κe + κL), with the results shown in Figure 5. The optimal ZT values of NaSn2SbSe4 surpass those of SnSe. Specifically, at 800 K, the maximum ZT value reaches 0.88 with an optimized carrier concentration of 2.39 × 1020 cm-3. These results demonstrate that the regulation of chemical bonding is a promising approach for enhancing the thermoelectric performance of SnSe-derived alloys such as NaSn2SbSe4, thereby validating its significant potential as a novel thermoelectric compound for practical applications. At the intermediate temperature of 600 K, the ZT value of NaSn2SbSe4 at the optimal carrier concentration is 0.75, surpassing the ZT value of 0.63 for SnSe. Furthermore, the ZT values calculated using κ3ph are also presented as lower bounds (see Supplementary Figure 9), confirming the favorable thermoelectric properties of NaSn2SbSe4. Moreover, the temperature-dependent optimized ZT values of SnSe and NaSn2SbSe4 are plotted in Figure 5B, where NaSn2SbSe4 consistently demonstrates superior ZT values across the entire temperature range. Notably, the constructed crystal structure introduces only a limited degree of disorder that affects thermal transport, resulting in an overestimation of the lattice thermal conductivity and underestimation of the ZT values. The excellent thermoelectric performance of NaSn2SbSe4 highlights its potential as a high-performance thermoelectric material.
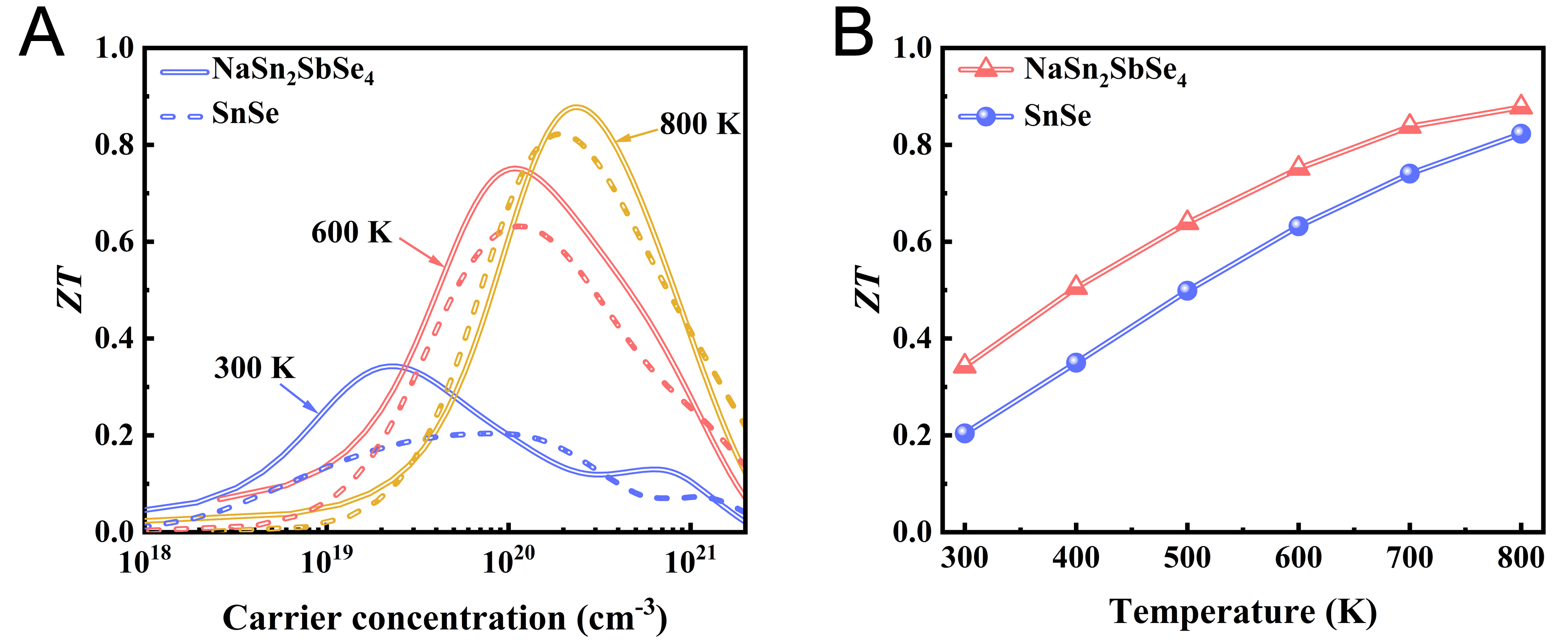
Figure 5. (A) Calculated ZT values of SnSe and NaSn2SbSe4 as a function of carrier concentration; (B) Optimized ZT values of SnSe and NaSn2SbSe4 at different temperatures.ZT: Dimensionless thermoelectric figure of merit.
CONCLUSIONS
In conclusion, we designed the NaSn2SbSe4 alloy through random substitution of Sn2+ with aliovalent cations, specifically Na+ and Sb3+. By combining first-principles, we systematically investigated the microscopic mechanism underlying the chemical bonding modulation and its impact on thermoelectric performance. For thermal transport, the Sb(5s)-Se(4p) and Sn(5s)-Se(4p) interactions in NaSn2SbSe4 exhibit enhanced antibonding states below the Fermi level, softening low-frequency optical phonons. Combined with pronounced lattice anharmonicity, phonon softening boosts four-phonon scattering and suppresses lattice thermal conductivity. Meanwhile, Sn-5p and Se-4p orbital hybridization improves electron delocalization and covalent bonding, sustaining high electrical conductivity. The low lattice thermal conductivity and high electrical conductivity indicate that chemical bonding regulation is a promising strategy to enhance the thermoelectric performance of SnSe-derived alloys such as NaSn2SbSe4, highlighting its potential for practical applications. Consequently, a deeper understanding of chemical bonding is essential for the discovery of new compounds and could accelerate the development of advanced high-performance thermoelectric materials. For instance, doping with ternary or quaternary cations could be investigated, potentially leveraging the high-entropy alloy strategy.
DECLARATIONS
Authors’ contributions
Conceived and designed the study: Li, L.; Li, Y.
Performed simulations and data curation: Qin, Y.
Conducted software reliability testing: Song, Q.; Bai, L.; Bao, Q.
Provided valuable suggestions: Li, L.; Li, Y.; Cheng, X.; Zhao, X.
Drafted the manuscript: Qin, Y.; Li, L.
Availability of data and materials
The data supporting this article have been included as part of the Supplementary Materials. The DFT simulations and the crystal structure schematic of NaSn2SbSe4 are freely available. Further data are available from the corresponding authors upon request.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This work was supported by the “National Key R&D Program of China”, the National Science Foundation of China (Grant No. 22209201), Future Plans of Young Scholars, and Qilu Young Scholars Program of Shandong University.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
Supplementary Materials
REFERENCES
1. Qin, Y.; Qin, B.; Wang, D.; Chang, C.; Zhao, L. Solid-state cooling: thermoelectrics. Energy. Environ. Sci. 2022, 15, 4527-41.
2. Qin, B.; Wang, D.; Hong, T.; et al. High thermoelectric efficiency realized in SnSe crystals via structural modulation. Nat. Commun. 2023, 14, 1366.
3. Wei, J.; Yang, L.; Ma, Z.; et al. Review of current high-ZT thermoelectric materials. J. Mater. Sci. 2020, 55, 12642-704.
4. Ganesan, P.; Gantepogu, C. S.; Duraisamy, S.; et al. Maximizing thermoelectric performance in SnTe through strategic co-doping, nanostructuring, and topological insights. J. Mater. Chem. A. 2025, 13, 8559-70.
5. Jin, Y.; Qiu, Y.; Bai, S.; et al. Modifying roles of CuSbSe2 in realizing high thermoelectric performance of GeTe. Adv. Energy. Mater. 2024, 14, 2400623.
6. Ghosh, T.; Dutta, M.; Sarkar, D.; Biswas, K. Insights into low thermal conductivity in inorganic materials for thermoelectrics. J. Am. Chem. Soc. 2022, 144, 10099-118.
7. Song, X.; Zhao, Y.; Ni, J.; Meng, S.; Dai, Z. Thermal transport properties of anisotropic materials RbCaX (X = As, Sb) with strong anharmonicity. Comput. Mater. Sci. 2022, 213, 111618.
8. Yang, X.; Xie, C.; Sun, J.; et al. Extended phase homogeneity and improved out-of-plane charge transfer in Sb and Te co-alloyed n-type BiSe layered compound with extraordinary thermoelectric performance. Mater. Today. Phys. 2023, 33, 101047.
9. Kim, Y.; Zhao, L.; Kanatzidis, M. G.; Seidman, D. N. Analysis of nanoprecipitates in a Na-Doped PbTe-SrTe thermoelectric material with a high figure of merit. ACS. Appl. Mater. Interfaces. 2017, 9, 21791-7.
10. Lin, N.; Han, S.; Ghosh, T.; et al. Metavalent bonding in cubic SnSe alloys improves thermoelectric properties over a broad temperature range. Adv. Funct. Mater. 2024, 34, 2315652.
11. Xia, M.; Record, M.; Boulet, P. Investigation of PbSnTeSe high-entropy thermoelectric alloy: a DFT approach. Materials 2022, 16, 235.
12. Liu, R.; Chen, H.; Zhao, K.; et al. Entropy as a gene‐like performance indicator promoting thermoelectric materials. Adv. Mater. 2017, 29, 1702712.
13. Wang, W.; Liu, S.; Wang, Y.; et al. Tailoring local chemical fluctuation of high-entropy structures in thermoelectric materials. Sci. Adv. 2024, 10, eadp4372.
14. Li, S.; Hou, S.; Xue, W.; et al. Manipulation of phase structure and Se vacancy to enhance the average thermoelectric performance of AgBiSe2. Mater. Today. Phys. 2022, 27, 100837.
15. Dutta, M.; Pal, K.; Etter, M.; Waghmare, U. V.; Biswas, K. Emphanisis in cubic (SnSe)0.5(AgSbSe2)0.5: dynamical off-centering of anion leads to low thermal conductivity and high thermoelectric performance. J. Am. Chem. Soc. 2021, 143, 16839-48.
16. Arora, R.; Waghmare, U. V.; Rao, C. N. R. Metavalent bonding origins of unusual properties of group IV chalcogenides. Adv. Mater. 2022, 35, 2208724.
17. Wang, Y.; Qin, B.; Shi, H.; Su, L.; Wang, D.; Zhao, L. Contrasting thermoelectric properties in cubic SnSe-NaSbSe2 and SnSe-NaSbTe2: High performance achieved via increasing cation vacancies and charge densities. Acta. Mater. 2023, 247, 118754.
18. Wang, H.; Mao, L.; Tan, X.; et al. Nontrivial thermoelectric behavior in cubic SnSe driven by spin-orbit coupling. Nano. Energy. 2018, 51, 649-55.
19. Luo, Y.; Hao, S.; Cai, S.; et al. High thermoelectric performance in the new cubic semiconductor AgSnSbSe3 by high-entropy engineering. J. Am. Chem. Soc. 2020, 142, 15187-98.
20. Van De Walle, A.; Tiwary, P.; De Jong, M.; et al. Efficient stochastic generation of special quasirandom structures. Calphad 2013, 42, 13-8.
21. Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B. 1996, 54, 11169-86.
22. Kim, D. S.; Hellman, O.; Herriman, J.; et al. Nuclear quantum effect with pure anharmonicity and the anomalous thermal expansion of silicon. Proc. Natl. Acad. Sci. U.S.A. 2018, 115, 1992-7.
23. Li, C.; Guo, D.; Ren, X.; et al. Two-channel thermal transport and scattering channel of high-temperature phase SnSe using temperature-dependent effective potential. Mater. Today. Commun. 2023, 36, 106590.
24. Han, Z.; Yang, X.; Li, W.; Feng, T.; Ruan, X. FourPhonon: An extension module to ShengBTE for computing four-phonon scattering rates and thermal conductivity. Comput. Phys. Commun. 2022, 270, 108179.
25. Guo, Z.; Han, Z.; Feng, D.; Lin, G.; Ruan, X. Sampling-accelerated prediction of phonon scattering rates for converged thermal conductivity and radiative properties. npj. Comput. Mater. 2024, 10, 31.
26. Nelson, R.; Ertural, C.; George, J.; Deringer, V. L.; Hautier, G.; Dronskowski, R. LOBSTER: local orbital projections, atomic charges, and chemical‐bonding analysis fromprojector‐augmented‐wave‐based density‐functional theory. J. Comput. Chem. 2020, 41, 1931-40.
27. Madsen, G. K.; Carrete, J.; Verstraete, M. J. BoltzTraP2, a program for interpolating band structures and calculating semi-classical transport coefficients. Comput. Phys. Commun. 2018, 231, 140-5.
28. Jana, S. S.; Banerjee, R.; Maiti, T. Disorder by design: high-entropy oxides as next generation thermoelectric materials. J. Mater. Chem. A. 2025, 13, 27050-68.
29. Slade, T. J.; Pal, K.; Grovogui, J. A.; et al. Contrasting SnTe-NaSbTe2 and SnTe-NaBiTe2 thermoelectric alloys: high performance facilitated by increased cation vacancies and lattice softening. J. Am. Chem. Soc. 2020, 142, 12524-35.
30. Li, J.; Ma, Z.; Wang, H.; et al. Boosting thermoelectric properties of high-entropy chalcogenides through local structural distortion and tailored chemical bonding. J. Am. Chem. Soc. 2025, 147, 41629-38.
31. Müller, P. C.; Ertural, C.; Hempelmann, J.; Dronskowski, R. Crystal orbital bond index: covalent bond orders in solids. J. Phys. Chem. C. 2021, 125, 7959-70.
32. Das, S. S.; Sadeghi, S. N.; Esfarjani, K.; Zebarjadi, M. The interplay of chemical bonding and thermoelectric properties in doped cubic GeTe. J. Mater. Chem. A. 2024, 12, 14072-86.
33. Wan, B.; Gao, Z.; Huang, X.; et al. Bonding heterogeneity inducing low lattice thermal conductivity and high thermoelectric performance in 2D CdTe2. ACS. Appl. Energy. Mater. 2022, 5, 9549-58.
34. Dutta, M.; Pal, K.; Waghmare, U. V.; Biswas, K. Bonding heterogeneity and lone pair induced anharmonicity resulted in ultralow thermal conductivity and promising thermoelectric properties in n-type AgPbBiSe3. Chem. Sci. 2019, 10, 4905-13.
35. Zacharias, M.; Volonakis, G.; Giustino, F.; Even, J. Anharmonic electron-phonon coupling in ultrasoft and locally disordered perovskites. npj. Comput. Mater. 2023, 9, 153.
36. Yue, T.; Sui, P.; Zhao, Y.; Ni, J.; Meng, S.; Dai, Z. Theoretical prediction of mechanics, transport, and thermoelectric properties of full Heusler compounds Na2KSb and X2CsSb (X=K,Rb). Phys. Rev. B. 2022, 105, 184304.
37. Zhou, J.; Hellman, O.; Bernardi, M. Electron-phonon scattering in the presence of soft modes and electron mobility in SrTiO3 perovskite from first principles. Phys. Rev. Lett. 2018, 121, 226603.
38. Yue, T.; Zhao, Y.; Ni, J.; Meng, S.; Dai, Z. Strong quartic anharmonicity, ultralow thermal conductivity, high band degeneracy and good thermoelectric performance in Na2TlSb. npj. Comput. Mater. 2023, 9, 17.
39. Ai, P.; Tang, S.; Wan, D.; et al. Synergistic effect of lone-pair electron and atomic distortion in introducing anomalous phonon transport in layered PbXSeF (X= Cu, Ag) compounds with low lattice thermal conductivity. Mater. Today. Phys. 2024, 48, 101572.
40. Sarkar, D.; Dolui, K.; Taneja, V.; et al. Chemical bonding tuned lattice anharmonicity leads to a high thermoelectric performance in cubic AgSnSbTe3. Angew. Chem. Int. Ed. 2023, 62, e202308515.
41. Bai, S.; Liu, D.; Shi, H.; et al. Revealing the origin of anisotropic rashba spin‐orbital splitting and four‐phonon scattering in strontium‐tin‐selenium thermoelectrics. Adv. Funct. Mater. 2024, 35, 2414288.
42. Sun, J.; Hu, M.; Zhang, C.; Bai, L.; Zhang, C.; Wang, Q. Ultralow thermal conductivity of layered Bi2O2Se induced by twisting. Adv. Funct. Mater. 2022, 32, 2209000.
43. Lin, S.; Yue, J.; Ren, W.; Shen, C.; Zhang, H. Strong anharmonicity and medium-temperature thermoelectric efficiency in antiperovskite Ca3XN (X = P, As, Sb, Bi) compounds. J. Mater. Chem. A. 2024, 12, 19567-79.
44. Wang, X.; Gao, Z.; Zhu, G.; et al. Role of high-order anharmonicity and off-diagonal terms in thermal conductivity: a case study of multiphase CsPbBr3. Phys. Rev. B. 2023, 107, 214308.
45. Song, X.; Wang, J.; Zhao, Y.; Ni, J.; Meng, S.; Dai, Z. Extremely strong four-phonon scattering and ultra-low lattice thermal conductivity due to quartic anharmonicity in fluoride perovskites XHgF3 (X = K, Rb). Phys. Lett. A. 2022, 456, 128550.
46. Yue, T.; Zhao, Y.; Ni, J.; Meng, S.; Dai, Z. Microscopic mechanism of low lattice thermal conductivity in natural superlattice materials BaXYF(X= Cu, Ag;Y= Se, Te) including fully quartic anharmonicity. Phys. Rev. B. 2023, 107, 024301.
47. Yuan, X.; Zhao, Y.; Sui, P.; Ni, J.; Dai, Z. Anomalous phonon transport and thermoelectric properties in honeycomb compounds ACuTe (A = Na, K, Rb). J. Mater. Chem. A. 2025, 13, 5106-18.
48. Zhang, T.; Yu, T.; Ning, S.; et al. Extremely low lattice thermal conductivity leading to superior thermoelectric performance in Cu4TiSe4. ACS. Appl. Mater. Interfaces. 2023, 15, 32453-62.
49. Li, Z.; Liu, F.; Cao, P.; et al. Chemical pressure‐driven three‐ and four‐phonon scattering in SnTe: Toward suppressed lattice thermal conductivity and enhanced thermoelectric performance. Adv. Funct. Mater. 2025, 35, 2507645.
50. Chen, C.; Feng, Z.; Yao, H.; et al. Intrinsic nanostructure induced ultralow thermal conductivity yields enhanced thermoelectric performance in Zintl phase Eu2ZnSb2. Nat. Commun. 2021, 12, 5718.
51. Song, X.; Zhao, Y.; Ni, J.; Meng, S.; Dai, Z. High thermoelectric performance in XAgSe2 (X = Sc, Y) from strong quartic anharmonicity and multi-valley band structure. J. Mater. Chem. A. 2023, 11, 17138-44.
52. Yu, Y.; Zhou, C.; Ghosh, T.; et al. Doping by Design: Enhanced thermoelectric performance of GeSe alloys through metavalent bonding. Adv. Mater. 2023, 35, 2300893.
53. Wang, T.; Duan, X.; Zhang, H.; et al. Origins of three‐dimensional charge and two‐dimensional phonon transports in Pnma phase PbSnSe2 thermoelectric crystal. InfoMat 2023, 5, e12481.
55. Xia, M.; Boulet, P.; Record, M. Influence of biaxial and isotropic strain on the thermoelectric performance of PbSnTeSe high-entropy alloy: a density-functional theory study. Mater. Today. Phys. 2024, 49, 101590.
56. Bardeen, J.; Shockley, W. Deformation potentials and mobilities in non-polar crystals. Phys. Rev. 1950, 80, 72-80.
57. Xi, J.; Long, M.; Tang, L.; Wang, D.; Shuai, Z. First-principles prediction of charge mobility in carbon and organic nanomaterials. Nanoscale 2012, 4, 4348.
58. Shi, H.; Su, L.; Bai, S.; et al. Realizing high in-plane carrier mobility in n-type SnSe crystals through deformation potential modification. Energy. Environ. Sci. 2023, 16, 3128-36.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].