


The crucial role of alveolar fibroblasts in pulmonary fibrosis
 0
0 
Abstract
This study, “Alveolar fibroblast lineage orchestrates lung inflammation and fibrosis”, provides important new insights into the varied functions of alveolar fibroblasts. It highlights their key involvement in maintaining the stability of pulmonary alveoli and in regulating the prolonged tissue response to injury. Using a Scube2-creER system, the investigators achieved precise labeling of alveolar fibroblasts, thereby overcoming the limitations of previous methodologies that lacked the specificity to distinguish fibroblast subpopulations in different anatomical niches. By integrating lineage tracing, single-cell RNA sequencing, and targeted functional ablation assays, the study delineated the transition of inflammatory fibroblasts into a pro-fibrotic phenotype. These approaches further revealed the pivotal role of TGFβ in driving this fibrogenic conversion. This commentary will critically analyze these findings, discuss their significance, and explore future prospects.
Keywords
INTRODUCTION
Fibroblasts support each organ by maintaining the structure of the extracellular matrix (ECM)[1]. Following tissue injury, new subsets of fibroblasts, including those that produce large amounts of ECM, emerge at injury sites. Studies have shown that in chronic diseases, these fibroblasts may contribute to pathological fibrosis[2,3]. However, the cellular origin of fibrotic pathology remains controversial. Some reports suggest that these cells may transdifferentiate from other lineages, such as hematopoietic cells, epithelial cells, pericytes, or endothelial cells[4,5]. In contrast, a previous computational cell atlas analysis from this group indicated that pro-fibrotic fibroblasts responding to alveolar injury originate from alveolar fibroblasts[6]. This concept-that a specific cellular lineage can be activated by microenvironmental cues to
TREATMENT OF HUMAN IDIOPATHIC PULMONARY FIBROSIS
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive interstitial lung disease characterized by persistent fibrotic remodeling. Following diagnosis, the median survival is approximately 2.8 years, and mortality exceeds that of many cancers, earning IPF the designation of a “cancer-like” disorder. Currently, there is no cure for IPF. Nevertheless, comprehensive treatment strategies can help slow disease progression and alleviate symptoms. The current first-line therapies are pirfenidone and nintedanib. Long-term use of corticosteroids should be avoided - unless clear autoimmune features are present - as they provide no proven benefit and increase the risk of infection[10]. Nintedanib, a tyrosine kinase inhibitor, exerts therapeutic effects by blocking pro-fibrotic signaling pathways such as Platelet-Derived Growth Factor Receptor (PDGFR), Fibroblast Growth Factor Receptor (FGFR), and Vascular Endothelial Growth Factor Receptor (VEGFR)[11]. Its common side effects include diarrhea, elevated liver enzymes, and nausea. Pirfenidone is thought to exert anti-fibrotic effects by inhibiting Transforming Growth Factor Beta (TGFβ)[12]. It also has anti-inflammatory properties, which are
MARKING TOOLS USED IN THE STUDY
Under normal conditions, the lungs contain multiple fibroblast subpopulations, each defined by distinct anatomical locations within the alveolar interstitium and peribronchial regions[6]. Commonly used fibroblast markers, such as Pdgfra and Collagen, type I, alpha 1-Green Fluorescent Protein (Col1a1-GFP), cannot fully distinguish these subpopulations. Pdgfra is widely expressed in lung fibroblasts and can enrich the overall fibroblast population[6,13]; however, it labels all fibroblast subtypes indiscriminately and thus lacks the specificity needed to separate alveolar, interstitial, or perivascular fibroblasts. Similarly, the Col1a1-GFP reporter marks all collagen I-producing fibroblasts, offering broad coverage, but its signal is predominantly concentrated around bronchovascular
To overcome this limitation and enable precise labeling of alveolar fibroblasts, the experimental group generated and utilized Scube2-creER transgenic mice. In these mice, Scube2 is highly expressed in alveolar fibroblasts but not in other subpopulations (e.g., perivascular or peribronchial fibroblasts)[6]. Moreover, unlike conventional surface markers that reflect only transient cell states, Scube2-creER allows for
THE RESPONSE OF ALVEOLAR FIBROBLASTS TO INJURY
Having confirmed the specificity of Scube2-creER, the research team next investigated how alveolar fibroblasts respond to injury. To this end, they performed single-cell RNA sequencing using Scube2-creER; Rosa26-tdTomato mice. Lung tissues were harvested at days 0, 7, 14, and 21 post-injury, with three biologically independent replicate groups included in the experimental design. In addition to previously described mesenchymal subtypes, the researchers identified four distinct fibroblast clusters at the injury site, which they termed fibrotic fibroblasts, inflammatory fibroblasts, stress-activated fibroblasts, and proliferative fibroblasts[9].
Inflammatory fibroblasts were characterized by elevated expression of chemokines-such as CXCL1, CXCL2, and SAA3-as well as interferon-response genes, and played a key role in mediating early inflammatory cell recruitment. Fibrotic fibroblasts expressed high levels of collagen (COL1A1) and fibrosis markers (CTHRC1) and drove extracellular matrix deposition. Stress-activated fibroblasts expressed cell cycle arrest genes (e.g., CDKN1A/p21) and may represent a senescent state. Proliferative fibroblasts emerged transiently during the early stages of injury and contributed to tissue repair. The study mainly focused on inflammatory, fibrotic, and stress-activated fibroblasts[14]. Pseudo-temporal analysis indicated that
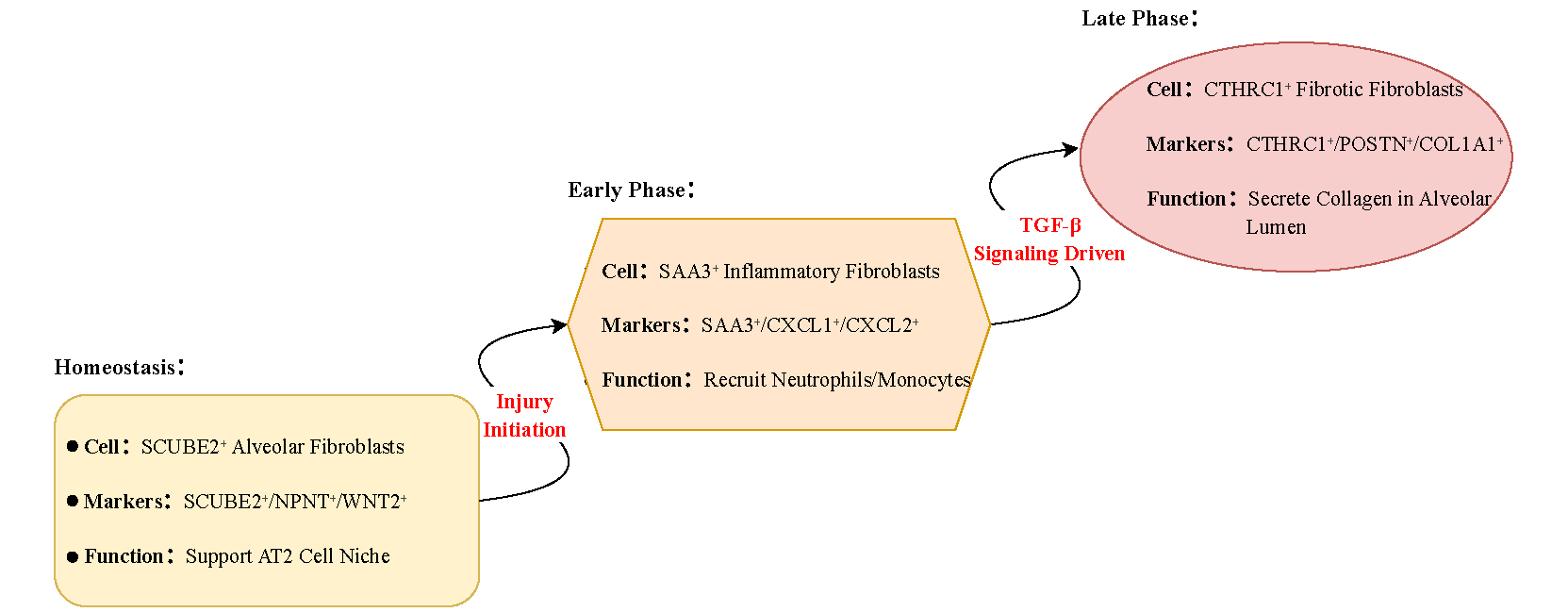
Figure 1. Transformation pathways of fibroblast subpopulations during pulmonary fibrosis. In homeostasis, SCUBE2+ alveolar fibroblasts support alveolar type II (AT2) cells. Under fibrotic stimulation, mediated by TGF-β signaling, these fibroblasts shift into an SAA3+ inflammatory state that recruits neutrophils and monocytes, and subsequently differentiate into CTHRC1+ fibrotic fibroblasts, which deposit collagen within the alveolar lumen.
EXPLORATION OF CTHRC1+ FIBROTIC FIBROBLASTS
To elucidate the functional role of CTHRC1+ fibrotic fibroblasts in pulmonary fibrosis-particularly their potential to directly drive collagen deposition-the experimental group generated a Cthrc1-creER knock-in mouse model by inserting a P2A-creERT2-T2A-GFP cassette into the terminal exon of the endogenous Cthrc1 locus. This model specifically labels CTHRC1-expressing fibrotic fibroblasts after injury upon tamoxifen induction. However, because the GFP signal generated by this construct was too weak to be detected by flow cytometry or microscopy, the tdTomato reporter gene was used for cell tracking instead. To achieve this, the researchers crossed Cthrc1-creER mice with Rosa26-tdTomato reporter mice, replacing GFP with tdTomato (red fluorescence) for visualization and lineage tracing. Using this system, they monitored the spatiotemporal dynamics of CTHRC1+ fibroblasts during the progression of lung fibrosis. Similar to the Scube2-creER system, both models rely on tamoxifen induction; however, they target distinct fibroblast populations: Scube2-creER specifically labels alveolar fibroblasts, whereas Cthrc1-creER labels fibrotic fibroblasts [Figure 2]. Following tamoxifen induction, approximately 50% of tdTomato-labeled cells were ablated, accompanied by a significant reduction in pulmonary fibrosis, as evidenced by decreased hydroxyproline content and reduced collagen I deposition. These findings establish CTHRC1+ fibroblasts as key effector cells in driving fibrotic progression. Accordingly, therapeutic approaches could be developed by targeting Cthrc1 with antibodies or small-molecule inhibitors. Moreover, since CTHRC1+ fibroblasts are modulated by TGFβ signaling, inhibition of the TGFβ pathway also represents a promising therapeutic strategy.
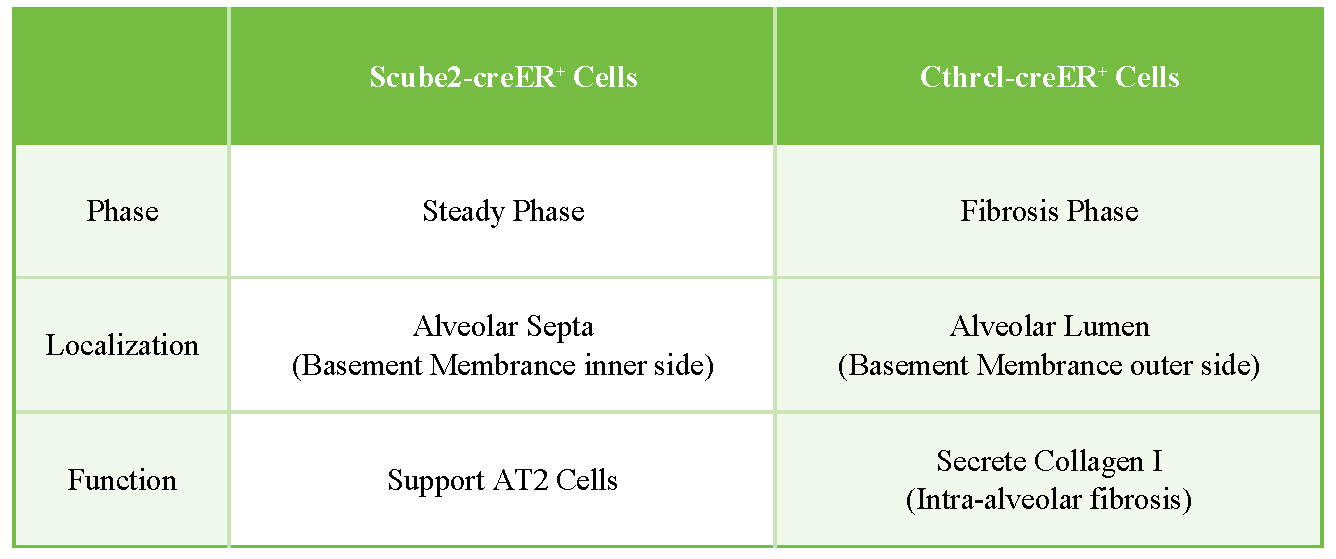
Figure 2. Spatial distribution of Scube2-creER- and Cthrc1-creER-labeled fibroblasts. In homeostasis, Scube2-creER-labeled fibroblasts are located within the alveolar septa, where they support AT2 cells. During fibrosis, Cthrc1-creER‑labeled fibroblasts localize to the alveolar lumen, where they secrete collagen I and drive intra-alveolar fibrosis.
DISADVANTAGES
Although this study investigated the role of alveolar fibroblasts in pulmonary fibrosis, it did not fully rule out potential contributions from other fibroblast subpopulations to disease pathogenesis. Thus, the findings cannot be generalized to all fibroblasts under all pathological conditions. Moreover, mouse models do not replicate the chronicity, microenvironment complexity (e.g., aging, comorbidities), or heterogeneous disease progression observed in human IPF. In addition, the number of IPF samples used for comparison was limited (n = 3), which may have led to biased conclusions. Although the study partially addressed this limitation by integrating two large databases containing over 16 IPF samples[16,17], further expansion of the dataset is required to verify cross-species consistency. A larger sample size would improve both the statistical power and experimental robustness of the study findings. Furthermore, TGFβ-targeted therapy should be evaluated in large animal models to enhance clinical relevance. While the study focused on the TGFβ signaling pathway, it did not identify specific downstream effectors or clarify its synergistic or antagonistic interactions with other signaling pathways (such as IL-1β). Notably, simultaneous knockout of the tgfbr2 gene to inhibit TGFβ signaling significantly reduced pulmonary fibrosis, lowering collagen deposition by approximately 50%. However, this intervention also appeared to exacerbate inflammatory responses, indicating the need for concurrent administration of anti-inflammatory drugs.
CONCLUSION
This study employed an ingeniously designed Scube2-creER system to achieve precise labeling of alveolar fibroblasts. Through a cross-species, multi-omics systematic investigation, it revealed the critical functions of alveolar fibroblasts and the TGFβ signaling pathway in pulmonary fibrosis pathogenesis, thereby providing an important foundation for understanding IPF mechanisms and facilitating the development of targeted therapies. These findings identify TGFβ or CTHRC1 as potential therapeutic targets, although validation in large animal IPF models remains necessary before clinical translation. Moreover, the study may also offer valuable insights into fibrotic mechanisms in other organs.
DECLARATIONS
Authors’ contributions
Have full access to all data in this study and take responsibility for the integrity and accuracy of the data analyses: Zhu H, Hong Z, Yi M
Conceived and designed the study, supervised the study: Zhou Y, Zhang H
Drafted the manuscript: Zhu H, Hong Z,
Collected the data: Song S, Qian L, Hong Y, Zhang X, Tan L
All authors read and approved the final manuscript.
Availability of data and materials
Not applicable.
Financial support and sponsorship
This work was supported by the National Natural Science Foundation of China (81401988), China Postdoctoral Science Foundation (2019M661907), Jiangsu Postdoctoral Science Foundation (2019K159, 2019Z153), General Project of Jiangsu Provincial Health Committee (H2023136), General Project of Nantong Municipal Health Committee (MS2023013), Jiangsu Provincial Research Hospital (YJXYY202204-YSB28), Beijing Municipal Public Welfare Development and Reform Pilot Project for Medical Research Institutes (JYY2023-14, JYY2023-15), and College Student Innovation Program (202210304128Y and 2023103041055).
Conflicts of interest
Zhang H is a Junior Editorial Board member of Journal of Translational Genetics and Genomics. He was not involved in any steps of editorial processing, notably including reviewer selection, manuscript handling, or decision making. The other authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
1. Plikus MV, Wang X, Sinha S, et al. Fibroblasts: origins, definitions, and functions in health and disease. Cell. 2021;184:3852-72.
2. Henderson NC, Rieder F, Wynn TA. Fibrosis: from mechanisms to medicines. Nature. 2020;587:555-66.
3. Hinz B, Lagares D. Evasion of apoptosis by myofibroblasts: a hallmark of fibrotic diseases. Nat Rev Rheumatol. 2020;16:11-31.
4. Friedman SL, Sheppard D, Duffield JS, Violette S. Therapy for fibrotic diseases: nearing the starting line. Sci Transl Med. 2013;5:167sr1.
5. Pakshir P, Noskovicova N, Lodyga M, et al. The myofibroblast at a glance. J Cell Sci. 2020;133:jcs227900.
6. Tsukui T, Sun KH, Wetter JB, et al. Collagen-producing lung cell atlas identifies multiple subsets with distinct localization and relevance to fibrosis. Nat Commun. 2020;11:1920.
7. Chen K, Qu C. Impact of occult hepatitis B virus infection and high-fat diet on hepatocellular carcinoma development. Hepatoma Res. 2024;10:38.
8. Qu DC, Neu D, Khawaja ZQ, et al. Epigenetic effects of high-fat diet on intestinal tumorigenesis in C57BL/6J-ApcMin/+ mice. J Transl Genet Genom. 2023;7:3-16.
9. Tsukui T, Wolters PJ, Sheppard D. Alveolar fibroblast lineage orchestrates lung inflammation and fibrosis. Nature. 2024;631:627-34.
10. Raghu G, Remy-Jardin M, Richeldi L, et al. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2022;205:e18-47.
11. Huang J, Beyer C, Palumbo-Zerr K, et al. Nintedanib inhibits fibroblast activation and ameliorates fibrosis in preclinical models of systemic sclerosis. Ann Rheum Dis. 2016;75:883-90.
12. Wollin L, Wex E, Pautsch A, et al. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur Respir J. 2015;45:1434-45.
13. Buechler MB, Pradhan RN, Krishnamurty AT, et al. Cross-tissue organization of the fibroblast lineage. Nature. 2021;593:575-9.
14. Cao J, Spielmann M, Qiu X, et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature. 2019;566:496-502.
15. Ding Z, Liu Y, Huang Q, et al. m6A- and immune-related lncRNA signature confers robust predictive power for immune efficacy in lung squamous cell carcinoma. VIEW. 2023;4:20220083.
16. Adams TS, Schupp JC, Poli S, et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci Adv. 2020;6:eaba1983.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].