


SMN2 as a therapeutic target in spinal muscular atrophy: advances in gene expression modulation
 0
0  , , ...
, , ... Abstract
Spinal muscular atrophy (SMA) is a progressive neuromuscular degenerative disorder caused by mutations in the survival motor neuron 1 (SMN1) gene, leading to insufficient production of the survival motor neuron (SMN) protein. The nearly identical SMN2 gene modifies disease severity but generates only limited amounts of functional SMN protein due to a C-to-T transition in exon 7 that disrupts proper splicing. This review summarizes advances in understanding SMN2 splicing regulation and post-transcriptional modification in SMA pathogenesis. It discusses the roles of cis- and trans-acting elements in exon 7 inclusion, as well as the impact of epigenetic mechanisms such as histone acetylation and DNA methylation on SMN2 expression. This review also examines available therapeutic strategies, including antisense oligonucleotides (Nusinersen), small-molecule splicing modulators (Risdiplam and Branaplam), and gene therapy (Onasemnogene abeparvovec). Emerging approaches such as CRISPR/Cas9 genome editing and nanotechnology-based delivery systems are also highlighted. In addition, this review explores translational research using animal models, iPSC-derived neurons, and multi-omics approaches. Finally, it emphasizes the need for integrated therapeutic strategies that address both SMN-dependent and -independent pathways to improve treatment outcomes.
Keywords
INTRODUCTION
Spinal muscular atrophy (SMA) is a severe lower motor neuron disease inherited in an autosomal recessive manner and is among the most common genetic causes of infant mortality[1]. The first cases were described by Werdnig in 1891[2]. SMA type I alone accounts for around 60% of all cases[3]. The overall prevalence is estimated at 1-2 per 100,000 individuals, with an incidence of approximately 1 in 10,000 live births. Without treatment, the disease often leads to progressive motor decline and early death. Given its substantial health burden and severe clinical course, especially in infants and children, understanding SMA and improving therapeutic outcomes are of urgent medical importance. SMA is caused by biallelic mutations or deletions in the SMN1 gene on chromosome 5q13[4,5]. These genetic changes reduce the production of the survival motor neuron (SMN) protein.
Disease severity varies widely and is largely influenced by the copy number of SMN2, a nearly identical paralog that can partially compensate for SMN1 loss[6]. SMA is traditionally classified into five types (0-4) based on the age of onset and the highest motor milestones achieved. Type 0 presents prenatally with respiratory failure[7]. Type I (Werdnig-Hoffman disease) manifests before 6 months of age, with hypotonia and rapid progression[8]. Type II (Dubowitz disease) typically appears between 6 and 12 months; affected infants can sit but cannot walk[9]. Type III (Kugelberg-Welander disease) develops after 18 months, with patients initially able to walk but gradually losing ambulation[10]. Type IV is adult-onset and generally mild[11]. Although it is well established that SMN1 loss leads to motor neuron degeneration and SMN2 copy number influences severity, important questions remain. For example, the molecular basis of differential therapeutic responses in patients with similar SMN2 copy numbers is not fully understood [Table 1].
Clinical classification of spinal muscular atrophy by type, SMN2 copy number, and functional outcomes (adapted from Talbot & Tizzano, 2017)[12]
| Type | SMN2 copy number | Age of onset | Requires respiratory support at birth | Able to sit without support | Able to stand | Able to walk | Life expectancy |
| 0 | 1 | In utero | Yes | No | No | No | < 6 months |
| 1 | 1-2 | Birth - 6 months | No | No | No | No | < 2 years |
| 2 | 3 | 6-12 months | No | Yes | No | No | 10-40 years |
| 3 | 3-4 | 18 months- adulthood | No | Yes | Yes | Assisted | Adult |
| 4 | 4-8 | > 18 years of age | No | Yes | Yes | Yes | Adult |
Current therapies aim to modify SMN2 splicing and thereby increase the production of functional SMN protein. However, the precise mechanisms of action and long-term effects of these treatments remain under investigation. Additional factors such as alternative splicing regulation, epigenetic modifications, and the role of noncoding regions or other genetic modifiers require further exploration[13,14]. The SMN protein plays a central role in snRNP assembly, an important step in pre-mRNA splicing, which is a requirement for motor neuron survival[15]. While the SMN1 gene produces full-length SMN, SMN2 - which differs by a single silent C-to-T transition in exon 7 - predominantly generates transcripts lacking exon 7. As a result, SMN2 primarily produces a truncated, unstable SMN protein, with only a small fraction of transcripts yielding full-length, functional SMN[16]. Despite its limited output, this small amount of full-length SMN is clinically significant. Enhancing its production through targeted molecular therapies is promising. To optimize existing treatments and develop more durable, precise approaches, a deeper understanding of SMN2 regulation is essential. Investigating new strategies for modulating gene expression, such as RNA-based therapies, small molecules, and gene editing, could further optimize SMA management. This review examines the SMN2-targeted therapies currently available, focusing on their molecular mechanisms, therapeutic basis, and emerging strategies for enhancing gene expression modulation. By integrating insights from genetics, molecular biology, and clinical research, we aim to clarify how SMN2 modulation reshapes SMA management and identify directions for future therapeutic innovation.
POST-TRANSCRIPTIONAL REGULATION AND SMN2 SPLICING IN SMA PATHOGENESIS
Role of SMN2 in the pathogenesis of SMA
In eukaryotes, pre-mRNA transcribed by RNA polymerase II undergoes several maturation steps, including 5′ capping and 3′ polyadenylation. The most critical step is splicing, in which introns are removed and exons are joined to generate translatable mRNA[17-19]. Splicing can occur co-transcriptionally and is mediated by the spliceosome, a ribonucleoprotein complex that recognizes conserved splice sites[20,21]. Alternative splicing further expands proteomic diversity and regulates gene expression[22].
The pathogenesis of SMA is tightly linked to the splicing of SMN transcripts. The SMN protein is required for snRNP assembly and, therefore, for efficient pre-mRNA splicing. Reduced SMN impairs this process and compromises motor-neuron survival[23]. SMN2 differs from SMN1 by a single nucleotide substitution in exon 7, which promotes exon skipping and results in a truncated, unstable SMN protein [Figure 1][15].
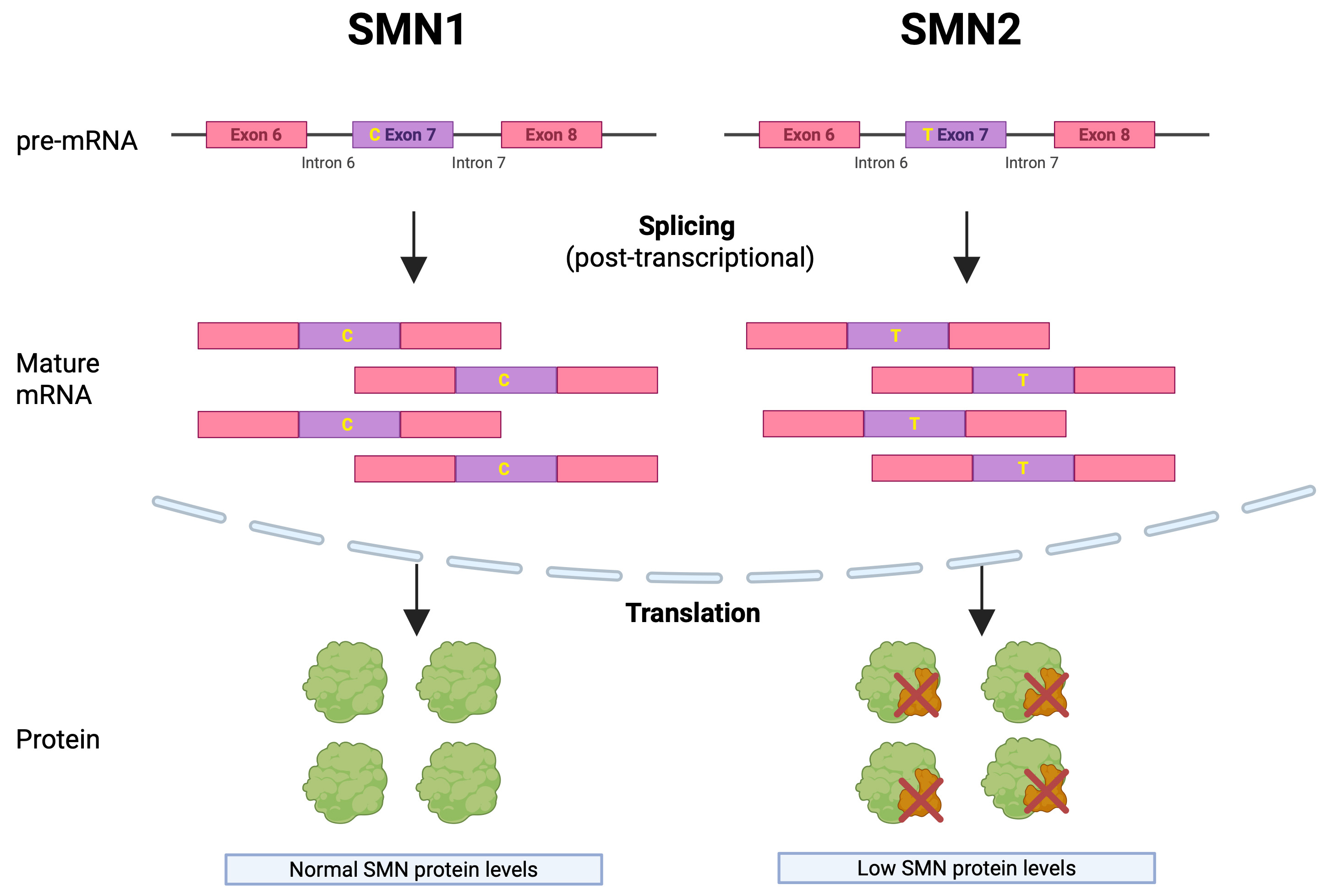
Figure 1. Differential Splicing of SMN1 and SMN2 Genes and Its Impact on SMN Protein Levels. Although nearly identical to SMN1, the SMN2 gene contains a single C-to-T substitution at codon 280 in exon 7. This silent mutation disrupts splicing, leading to exon 7 skipping in most transcripts and reduced production of full-length, functional SMN protein. Created in BioRender. Virata MCA, (2025) https://BioRender.com/6u6sj1t
Molecular modulators of SMN2 expression
The presence of a functional SMN1 gene largely determines whether an individual develops SMA. However, it was observed that in one case, two families carrying the same SMN1 mutation can exhibit markedly different clinical features[24]. This suggested that the molecular regulation of another gene may contribute to the clinical presentation of SMA. This gene was later identified as SMN2, which acts as a key disease modifier.
In patients lacking functional SMN1, the clinical phenotype depends on the number of SMN2 gene copies. Due to the C-to-T mutation in exon 7, SMN2 predominantly produces a truncated (and therefore unstable) protein, though a small fraction of transcripts generate full-length SMN. Consequently, SMA severity is inversely correlated with SMN2 copy number: the higher the SMN2 copy number, the milder the clinical presentation[25].
Several factors influence whether SMN2 produces truncated or full-length SMN protein, acting at different stages from transcription to protein synthesis. Prior to transcription, the degree of histone acetylation affects SMN2 expression. Kernochan et al. (2005) demonstrated that the SMN gene exhibits a distinct histone acetylation pattern, with high acetylation near the transcriptional start site and hypoacetylation approximately 1 kb upstream and downstream within intron 1[26]. The use of histone deacetylase inhibitors activated the SMN2 promoter, increased acetylation, and enhanced SMN2 expression. In addition, differential CpG methylation is associated with SMA severity[27]. After transcription, further modifications, including capping, splicing, and polyadenylation, are required before the mRNA can be translated into proteins[28].
During splicing, introns are removed while exons are retained. Since the SMN protein itself participates in spliceosome formation[28], reduced SMN leads to splicing abnormalities. Beyond the spliceosome, this process also involves a well-coordinated interplay between multiple factors, known as regulatory elements. Specifically, for SMN2, several regulatory elements determine whether exon 7 is excluded or not[16]. These elements can be broadly classified as exonic or intronic, and as enhancers or silencers, depending on their location and effect.
Exonic splicing silencers (ESSs) promote exon 7 skipping. For example, hnRNP A1/A2 binds ESS motifs, displacing U1 snRNP from the 5′ splice site (5′SS) and preventing spliceosome assembly[29]. An additional inhibitory terminal stem-loop structure (TSL1) forms near the C6U mutation that differentiates SMN1 from SMN2[16]. Conversely, exonic splicing enhancers (ESEs) stabilize exon 7 and promote its inclusion. Proteins such as SF2/ASF (SRSF1) and Tra2-B1 bind ESEs, enhance U1 snRNP recruitment to the weak exon 7 splice site, and counteract hnRNP-mediated inhibition[29,30]. Intronic regulatory elements play a similar role. Intronic splicing silencers (ISSs), such as ISS-N1, promote exon skipping and represent promising targets for antisense oligonucleotide (ASO)-based drugs currently undergoing clinical trials. In contrast, intronic splicing enhancers (ISEs) promote exon 7 retention. For example, TIA1 enhances exon 7 retention by recruiting U1 snRNP to the 5′ splice site[31,32]. Table 2 summarizes these regulatory elements.
Modifiers affecting SMN2 mRNA splicing (adapted from Singh et al., 2017)[102]
| Modifier | Effect on splicing |
| Within exon 7 | |
| hnRNP A1 | Negative |
| Extended inhibitory context (Exinct) | Negative |
| Sam68 | Negative |
| GAAGGA motif (at 22nd to 27th positions) | Negative |
| hnRNP Q1 | Positive |
| hnRNP G | Positive |
| SF2/ASF | Positive |
| Tra2-B1 | Positive |
| TDP-43 | Positive |
| SRp-30c | Positive |
| PSF + hnRNP M | Positive |
| Within intron 7 | |
| ISS-N1 | Negative |
| TIA1 | Positive |
| Element 2 | Positive |
| Outside exon 7 | |
| Element 1 | Negative |
| hnRNP C | Positive |
Structural constraints also influence less well-defined cis-acting factor binding sites, such as ESEs or ISEs, as well as ESSs or ISSs[33,34]. Miyaso et al. (2003) identified a conserved 24-nucleotide stem-loop structure within intron 7 that functions as an ISE. Deletion of this structure abolished the binding of an as-yet-unidentified trans-acting protein detected by a gel shift assay, thereby disrupting splicing. This effect occurs only when a C-to-T transition is present at position 6 of exon 7[35]. Trans-acting proteins also regulate SMN2 splicing through interactions with spliceosomal components. For example, serine/arginine-rich splicing factor 1 (SRSF1) generally promotes exon inclusion through the induction of SR protein-dependent CaMKIIδ exon skipping, while hnRNP A1 promotes exon 7 skipping by creating an ESS[36,37]. Miyajima et al. (2002) identified a 45-bp cis-acting element within intron 6 that regulates exon 7 splicing; mutation or ASO treatment targeting this element increased exon 7 inclusion[38].
Deletion analysis of SMN1 pre-mRNA revealed two intronic regions critical for exon 7 splicing: one within intron 6 (-112 to -68 bp, element 1) and one within intron 7 (+59 to +124 bp, element 2). In the presence of the C-to-T transition, element 1 promotes exon 7 skipping, whereas element 2 supports exon 7 inclusion. Deletion of these elements alone does not affect exon 7 splicing in wild-type SMN1. Thus, both the intronic deletions and the C-to-T mutation are required to change splicing patterns[38].
Polyadenylation, the addition of a polyadenosine [poly(A)] tail to the 3′ end of the mRNA, is essential for transcript stability and nuclear export prior to translation[39]. Abnormal SMN2 splicing, owing to SMN’s essential role in spliceosome assembly, is associated with nuclear accumulation of polyadenylated RNA granules (PARGs). These PARGs are detectable in motor neurons at presymptomatic stages of SMA and increase in number as the disease progresses[28].
THERAPEUTIC STRATEGIES TARGETING SMN2: NUSINERSEN, RISDIPLAM, AND BRANAPLAM
The main therapeutic agents currently targeting SMN2 are Nusinersen, Risdiplam, and Branaplam. This section reviews their mechanisms of action, modes of delivery, tissue distribution, and clinical efficacy, as summarized in Table 3.
Comparison of therapeutic agents targeting SMN2 splicing in spinal muscular atrophy (SMA): mechanisms, delivery, distribution, and clinical trial outcomes
| Drug | Mechanism of action | Mode of delivery | Tissue distribution | Efficacy and trials |
| Nusinersen[44,45] | Antisense oligonucleotide; promotes exon 7 inclusion in SMN2 mRNA | Intrathecal injection | Primarily CNS | ENDEAR (NCT02193074): infantile-onset SMA - Significantly improved motor milestone response and event-free survival CHERISH (NCT02292537): later-onset SMA - Significant motor function improvements; ≥ 3-point increase in HFMSE |
| Risdiplam[49,50] | Oral small molecule; binds two sites on SMN2 pre-mRNA to promote exon 7 inclusion | Oral (once daily) | Widely distributed in CNS and peripheral tissues | FIREFISH (NCT02913482): infantile-onset SMA - Significant improvements in motor milestones SUNFISH (NCT02908685): later-onset SMA - Significant improvements in motor function |
| Branaplam[53] | Small molecule; stabilizes spliceosome-pre-mRNA interaction to promote exon 7 inclusion | Oral (once weekly) | Widely distributed in CNS and peripheral tissues | MOONFISH (NCT02268552): SMA - Increased SMN2 mRNA/protein but unproven clinical benefit; trial discontinued due to peripheral neurotoxicity |
Nusinersen is a 2'-O-methoxyethyl phosphorothioate-modified antisense oligonucleotide. It binds specifically to a sequence in intron 7 of SMN2 pre-mRNA, blocking a splicing silencer and thereby promoting exon 7 inclusion[40]. This mechanism increases the production of full-length SMN protein in patients with SMA caused by chromosome 5q mutations that result in SMN protein deficiency[41]. Nusinersen is administered intrathecally at a dose of 12 mg (5 mL) per injection. Treatment begins with four loading doses: three given at 14-day intervals and a fourth 30 days after the third, followed by maintenance doses every four months[42]. After intrathecal administration, the drug distributes from cerebrospinal fluid (CSF) into CNS tissues, with relatively low plasma concentrations[43]. Its therapeutic efficacy is demonstrated by increased SMN2 mRNA containing exon 7 in the thoracic spinal cord and improvements in motor function. The pivotal Phase 3 clinical trials ENDEAR and CHERISH evaluated its effectiveness in SMA[44,45].
The ENDEAR trial evaluated infants with infantile-onset (type 1) SMA (symptom onset ≤ 6 months). It was terminated early because of clear clinical benefits, showing significant improvements in motor milestone responses, event-free survival, and overall survival compared with controls. The safety profile was favorable, with adverse event rates comparable between treatment and control groups[44].
The CHERISH trial evaluated children with later-onset (type 2) SMA (symptom onset > 6 months) using the same intrathecal 12 mg regimen. It reported significant improvements in motor function, assessed by the Hammersmith Functional Motor Scale Expanded (HFMSE). More participants receiving Nusinersen achieved a ≥ 3-point increase in HFMSE scores at 15 months, with the greatest benefits observed in younger children treated soon after symptom onset[45].
Risdiplam, unlike Nusinersen, is a small molecule that binds to two regions of SMN2 pre-mRNA: the exonic splicing enhancer 2 in exon 7 and the 5′ splice site in intron 7[46]. By promoting exon 7 inclusion during splicing, it increases the production of full-length, functional SMN protein. Importantly, Risdiplam is the first orally administered SMA therapy. It crosses the blood-brain barrier and achieves distribution in both CNS and peripheral tissues[47,48]. Its efficacy has been established in the FIREFISH and SUNFISH trials[49].
The FIREFISH trial was a two-part, open-label, multicenter study in infants aged 1-7 months with type 1 SMA. At 24 months, 44% of participants were able to sit unsupported for at least 30 s - a significant improvement compared with the natural history of the disease[49]. The SUNFISH trial, a two-part, Phase 3, randomized, double-blind, placebo-controlled study, evaluated older patients with type 2 or non-ambulant type 3 SMA. At 12 months, Risdiplam-treated participants showed significant improvements on the 32-item Motor Function Measure compared with placebo. Gains observed at 12 months were maintained or further improved at 24 months[50].
Branaplam is another small-molecule splicing modulator. It stabilizes the spliceosome-pre-mRNA interaction, promoting exon 7 inclusion in SMN2 transcripts and thereby enhancing SMN protein production[51]. Administered orally once weekly, it distributes to both the CNS and peripheral tissues. However, its therapeutic efficacy in humans remains unproven[52].
The MOONFISH trial (NCT02268552) investigated Branaplam in type 1 SMA. Although the drug demonstrated good CNS penetration and increased full-length SMN2 mRNA and SMN protein levels in blood, the trial was discontinued before reaching primary clinical endpoints due to concerns about preclinical neurotoxicity. Juvenile dog studies showed mild to moderate peripheral nerve fiber degeneration accompanied by elevated serum neurofilament light chain, a biomarker of neurodegeneration[53]. Similar adverse effects were later observed in the VIBRANT-HD trial for Huntington’s disease.
PROSPECTIVE COMBINATION STRATEGIES AND GENE THERAPY APPROACHES
Rationale and available therapies
SMA leads to progressive muscle weakness, loss of mobility, and respiratory complications. In severe cases, untreated SMA can result in death within two years. As a multisystem disorder, SMA involves not only motor neuron loss due to SMN protein deficiency but also secondary effects on muscle, the neuromuscular junction, and possibly other organs, including the heart, kidneys, liver, pancreas, spleen, bones, connective tissues, and the immune system. This broader phenotype underscores the need for SMN-independent treatments in conjunction with SMN-targeted therapies, as no single therapy can fully cure SMA[54,55]. Among recent advancements, gene therapy is considered the most effective disease-modifying approach for pediatric patients with bi-allelic SMN1 mutations. In contrast, neuroprotective and stem cell therapies remain insufficiently established as effective adjuncts.
Zolgensma (onasemnogene abeparvovec) is an adeno-associated virus serotype 9 (AAV9)-based gene therapy approved for pediatric patients under 2 years of age with SMA and bi-allelic SMN1 mutations. The STR1VE-US and STR1VE-EU trials demonstrated significantly higher rates of independent sitting and survival without permanent ventilation at 14-18 months of age, compared with untreated natural history cohorts[56,57]. The SPRINT trial further showed that presymptomatic infants with two or three copies of SMN2 achieved age-appropriate motor milestones, such as independent walking, with a 100% survival rate[58]. The START trial confirmed these benefits and reported no safety concerns up to 5-6 years post-treatment[59].
In comparison, Olesoxime has not proven effective as a disease-modifying therapy for slowing functional decline in SMA. However, it might provide some benefit when combined with agents that target different disease mechanisms[60]. Olesoxime was generally well tolerated and demonstrated an acceptable safety profile at the studied doses, but the pivotal trial failed to meet its primary endpoint. Specifically, the change from baseline to 24 months was not statistically significant between the treatment groups[61].
Stem cell therapy has also been investigated as a potential treatment, based on the rationale of replacing lost motor neurons. However, clinical evidence is limited, and controlled trials remain scarce. While no major adverse events have been reported, stem cell therapy has not demonstrated sustained clinical benefit. In a small case series of three children with SMA type I, repeated intrathecal and intravenous allogeneic mesenchymal stem cell infusions temporarily improved motor function. These gains, however, diminished after therapy was discontinued[62].
Challenges and long-term effects of combination therapies
The challenges associated with combination therapies are multifaceted. These include unknown additive or synergistic effects, an increased risk of adverse events, and high costs. There is also a lack of data on optimal timing, sequencing, and patient selection. Moreover, the long-term effects of combination therapies remain unclear, as most agents are relatively new, and real-world or long-term safety data are limited.
Olesoxime, a neuroprotective agent, is not part of standard care. It failed to achieve its primary endpoint, and its combination with gene therapy has not demonstrated additive or synergistic benefits[61]. Stem cell therapy remains investigational because it has not yielded sustained benefits for patients and it is not recommended outside of clinical studies[62]. Gene therapy with Zolgensma, by contrast, is an established first-line disease-modifying treatment that improves survival and motor function, especially when administered presymptomatically. However, it does not fully reverse established motor neuron loss[56,57]. In theory, combining Zolgensma, which delivers a functional SMN1 gene, with an SMN2-targeted agent such as Nusinersen or Risdiplam could enhance SMN protein levels in motor neurons and peripheral tissues. Nevertheless, there is no clear evidence that combination therapy is superior to early monotherapy. While generally well-tolerated, combination therapy has not shown additional gains in motor function compared to monotherapy alone. Indeed, some studies report that early treatment is more beneficial than combination therapy[63].
Immunologic and developmental timing in gene therapy
Zolgensma is an AAV9-based gene therapy, and its use raises immunologic concerns. Patients with pre-existing anti-AAV9 antibodies can neutralize the therapy, reducing its efficacy. Zhang et al. (2005), analyzing the U.S. Food and Drug Administration’s Adverse Event Reporting System (FAERS), identified 281 Zolgensma-related adverse events, the most common being elevated liver enzymes, fever, vomiting, and thrombocytopenia[64]. Nevertheless, the therapy has a favorable safety profile when thorough screening and post-therapy management and monitoring are performed[65]. The steroids used to manage elevated liver transaminases may interfere with vaccination schedules, particularly in countries where live tuberculosis (TB) vaccination is administered. Consequently, the Polish Vaccinology Association has provided recommendations for administering gene therapy in newborns who have received the live TB vaccine[66].
SMA causes irreversible motor neuron loss prenatally, which accelerates rapidly after birth, especially in severe cases. Developmental timing is therefore critical, as the therapeutic window for optimal efficacy is narrow regardless of disease severity. Studies have shown that patients treated presymptomatically or at an early symptomatic stage are more likely to achieve the best outcomes. The START trial highlighted both the long-term benefits of gene therapy and the importance of early identification of affected infants for maximizing therapeutic efficacy[59].
CURRENT MODELS AND APPROACHES OF SPINAL MUSCULAR ATROPHY
Over the past decade, various models have been developed that closely reproduce different forms of SMA and their respective phenotypes, enabling a deeper understanding of disease mechanisms and the identification of potential treatments, as summarized in Table 4.
Preclinical and experimental models for studying spinal muscular atrophy (SMA): key features, advantages, limitations, and applications
| Model/Approach | Key features | Advantages | Limitations |
| Mouse[15,69,70] | Genetically tractable; shares neuromuscular similarity with humans | Models SMA via human SMN2/SMNΔ7 transgenes; suitable for drug efficacy & pathogenesis studies | Native mouse has only one SMN gene; full knockout is lethal, heterozygous mice show no phenotype |
| Drosophila[73] | Single SMN gene; rapid lifecycle; low cost | Models severe and late-onset SMA; suitable for peripheral neuromuscular studies | Null mutations lethal at the larval stage; limited phenotype diversity |
| Zebrafish[74] | Transparent larvae; sequenced genome; high-throughput capacity | Cost-effective; well-defined motor circuits; multiple genetic manipulation tools | Full SMN deletion is lethal |
| iPSCs[77-79] | Pluripotent; differentiate into motor neurons | Human-derived; models motor neuron pathogenesis; enables testing of SMN-independent therapies | Challenges with reprogramming efficiency and reproducibility |
| Omics approaches[80] | Includes proteomics, genomics, metabolomics, etc. | Identifies molecular targets (e.g., SMN2 splicing modifiers) | Complexity of data integration |
| Epigenetic regulators[81,82] | DNA methylation profiling of SMA patients | Potential modifiers of phenotype severity | Inconclusive evidence in some large cohorts |
| CRISPR/Cas9[83,84] | Genome editing with sgRNA and Cas9 endonuclease | Correct SMN2 splicing; increases full-length SMN production | Clinical safety and efficacy still under investigation |
Preclinical models
Mouse
One of the most widely used SMA models is the mouse. Mice are popular for exploring disease pathogenesis and assessing therapeutic efficacy because of their anatomical and physiological similarities to the human neuromuscular system. Additionally, their suitability for genetic manipulation makes them ideal for studying various neuromuscular disorders[67]. A limitation of mouse models is that mice have only a single SMN gene. Complete knockout (Smn-/-) mice are embryonically lethal, while heterozygous (Smn+/-) mice do not exhibit a typical SMA phenotype[15]. To overcome this, genetic modifications were introduced by adding one or both of the human SMN2 transgene and SMNΔ7 cDNA into Smn-/- mice, thereby extending survival. Recent studies have shown that dual SMN administration of optimal doses of exon 7 inclusion-promoting ISS-N1 antisense oligonucleotides to human SMN2-carrying SMA mice produces longer-lived, symptomatic SMN∆7 mouse models, which are valuable for assessing long-term therapeutic effects[68]. For example, Zhao et al. (2016) demonstrated the pharmacokinetics, pharmacodynamics, and efficacy of SMN-C1 at varying doses in transgenic SMA mouse models[69]. SMN-C1 is a small-molecule SMN2 splicing modifier that promotes exon 7 inclusion, thereby increasing SMN protein production. Consequently, these mouse models provide a useful platform for testing low-molecular-weight compounds that correct alternative splicing defects of SMN2 exon 7. Mouse models have also been instrumental in elucidating SMA pathogenesis. In a study by Seo et al. (2016), oxidative stress was induced in transgenic mice, triggering the production of reactive oxygen species. This led to multiple co-skipping events of SMN2 exons 5 and 7, as well as increased skipping of exon 3 in all tissues except the testis[70].
Drosophila
Drosophila melanogaster, commonly known as the fruit fly, has also been used to model SMA because it is inexpensive, easy to maintain, and genetically tractable. Its short generation time, high fecundity, and well-characterized organ systems and cellular pathways make it particularly suitable for studying peripheral neuromuscular defects in SMA[71]. Like mice, Drosophila possess only a single SMN gene. A null SMN mutation is lethal at the larval stage, limiting fly models to severe SMA phenotypes, as evidenced by defects in larval synapses and musculature[72]. In a study by Spring et al. (2019), 14 Drosophila lines expressing SMA patient-derived missense mutations in the SMN gene were examined for organism viability, lifespan, locomotor function, neuromuscular junction structure, and muscle health. Remarkably, the fly model could also replicate late-onset (Type IV) SMA, which is difficult to reproduce in other systems such as mice[73]. This study demonstrated that Drosophila can model SMA-related declines in locomotor function and can also be used to study secondary neuromuscular symptoms in peripheral systems of patients undergoing antisense oligonucleotide therapy.
Zebrafish
Like other animal models discussed, zebrafish have a single SMN gene, and complete deletion of this gene leads to embryonic lethality. Nevertheless, zebrafish remain a valuable model for SMA. Their well-defined motor neuron circuits and relatively simple neuromuscular structure are advantageous. Additional benefits include their low-cost genetic manipulation, small size, and rapid reproduction. Furthermore, features unique to zebrafish, such as optical transparency during larval stages, a fully sequenced genome, and suitability for high-throughput screening - advantages not available in mouse models- further enhance their utility[74]. The zebrafish model is particularly useful for studying the effects of reduced SMN levels on motor neuron development in vivo. By 24 h post-fertilization, each spinal cord segment contains three primary motor neurons that target specific myotome regions. Within the next few days, 20-30 secondary motor axons join these neurons to form three distinct motor nerves. These axons can be easily visualized in living embryos, allowing researchers to directly observe and quantify disruptions in motor neuron structure and development[75].
Various strategies have been developed to mimic human SMA in zebrafish, and numerous models have been developed over the past two decades to study disease mechanisms and identify therapeutic targets. These models have led to significant advances, including the discovery of potential genetic modifiers such as U snRNPs, Etv5b, PLS3, CORO1C, Pgrn, Cpg15, Uba1, Necdin, and Pgk1[74]. Collectively, these findings provide insight into SMA pathogenesis across different phenotypes and support the development of diverse therapeutic approaches.
Induced pluripotent stem cells
Induced pluripotent stem cells (iPSCs) represent a promising model because they can self-renew and differentiate into various cell types, including motor neurons[76]. Using specific reprogramming methods, iPSCs can be used to model SMA. A major challenge of this approach lies in the reprogramming method itself, which requires efficient and consistent differentiation to produce reliable iPSC models. To address this, researchers have developed advanced protocols incorporating small molecules, lineage-specific transcription factors, 3D culture systems, and organoids[77]. SMN replacement strategies in SMA treatment still face limitations, such as late-onset peripheral neuromuscular dysfunction, highlighting the need for complementary therapies that act independently of SMN.
For example, a 2024 study by Pagliari et al. investigated Stathmin-2 (STMN2), a neuronal microtubule regulator implicated in neurodegenerative diseases[78]. The study showed that STMN2 overexpression restored axonal growth and corrected outgrowth defects in iPSC-derived motor neurons from an SMA patient[78]. Similarly, a 2016 study by Ohuchi et al. generated iPSCs from fibroblasts of a patient with SMA Type III and used the model to test a TRH analog - a potential therapeutic agent that activates the SMN2 gene and inhibits glycogen synthase kinase-3β[79]. Together, SMA-iPSC models serve as powerful tools for studying neuromuscular pathogenesis and for efficiently screening drugs that target pathways beyond the SMN protein.
Omics and systems approaches
Small molecules have become a major focus of research because of their ability to selectively bind RNA and modulate pre-mRNA splicing, an approach used in the treatment of human diseases. Wang et al. (2018) analyzed SMN-C2 and C3, close analogs of RG-7916, which act as selective RNA-binding ligands that modulate pre-mRNA splicing[80]. To understand how SMN-C2 and C3 affect exon 7 splicing, they identified cellular targets through proteomic and genomic studies. These studies showed that the small molecules bind directly to the AGGAAG motif on exon 7 of SMN2 pre-mRNA, inducing conformational changes in two or three unpaired nucleotides at the intron 6/exon 7 junction. This structural alteration enhances the binding of splicing modulators to the SMN-C2/C3 pre-mRNA complex, thereby promoting SMN2 splicing[80]. The application of omic approaches enabled investigators to uncover modifying factors that may support the development of more targeted and specific SMA therapies.
Another potential modifying factor in the molecular mechanisms underlying SMA pathogenesis is epigenetic regulation, which can alter patterns of gene expression. Zheleznyakova et al. (2013) performed the first genome-wide methylation profiling of patients with mild and severe forms of SMA, aiming to correlate DNA methylation status with phenotype severity[81]. They identified 10 CpG sites with significantly different methylation levels between SMA patients and healthy controls, located near genes associated with the activity of Rab and Rho GTPases, which are crucial regulators of actin dynamics, vesicle formation, and axogenesis[81]. These findings may encourage further studies examining how Rab and Rho GTPase activity influences SMA severity. In contrast, Zwartkruis et al. (2025) analyzed DNA methylation across 365 patients with SMA and found no association between blood DNA methylation and disease severity or treatment. However, their results did show age-related DNA methylation changes in intron 1 and 3′ UTR, highlighting DNA methylation as a possible modifier of SMN expression during development and aging[82].
Genomic engineering
The Clustered Regularly Interspaced Short Palindromic Repeats-Cas9 (CRISPR/Cas9) system is a powerful genome-editing tool composed of two key components. The first is a single-guide RNA (sgRNA) designed to match a target gene. It consists of two elements: CRISPR RNA (crRNA), which is complementary to the target sequence, and trans-activating crRNA (tracrRNA), which binds to Cas9. The second is CRISPR-associated protein 9 (Cas9), an endonuclease that introduces double-stranded DNA breaks, thereby enabling targeted modifications[83]. Li et al. (2019) successfully used Cas9 with sgRNA to disrupt intronic splicing silencers (ISS-N1 and ISS+100) in SMN2, rescuing SMA-associated phenotypes in human iPSCs and transgenic mice. Their approach significantly increased exon 7 inclusion and enhanced the production of full-length SMN protein in SMA-iPSCs and motor neurons, resulting in markedly improved survival rates[84]. These findings demonstrate the potential of CRISPR/Cas9 as a therapeutic strategy in humans and provide a foundation for developing efficient interventions for SMA and other diseases.
BIOMARKERS AND CLINICAL OUTCOMES
SMA research is shifting its focus from understanding disease pathophysiology to developing therapeutic interventions. As a result, the identification of biomarkers has become a rapidly growing area of investigation. Biomarkers may expedite diagnosis, help predict responses to experimental treatments, and confirm therapeutic efficacy.
Genetic biomarkers
Plastin 3, a protein that binds to F-actin - an important component of the axonal cytoskeleton - acts as a positive and protective modifier. Its overexpression delays axonal pruning. Coronin 1C, a protein that interacts with Plastin 3, functions as a genetic enhancer by increasing F-actin levels and restoring vesicular pools, thereby supporting normal functioning of the neuromuscular junction[85].
Proteomic biomarkers
Two proteins under consideration as possible biomarkers for SMA are the Survival Motor Neuron (SMN) protein and Neurofilaments.
The SMN protein is directly produced by transcription and translation of the SMN1 and SMN2 genes. Reduced SMN levels constitute the central pathophysiology of SMA[86]. Although most studies indicate that SMN protein levels correlate with gene copy number, they are also affected by degradation pathways such as the ubiquitin-proteasome system (UPS). Dysfunction in these pathways may result in elevated SMN levels[85].
Another emerging diagnostic biomarker for SMA and other neurodegenerative conditions is the measurement of neurofilament levels in CSF or serum[87]. Neurofilaments are structural proteins in neurons that, when damaged, can leak into the CSF and eventually into the blood, particularly the light chain component (NfL)[88]. Beyond serving as a diagnostic biomarker, NfL is also under investigation as a prognostic and treatment-response marker for neurodegenerative disorders[88] and potentially for SMA. Notably, pre-treatment NfL levels have been found to inversely correlate with motor function scores in patients with SMA type I[85]. However, caution is warranted when interpreting these results, as elevated NfL can also occur in conditions such as traumatic brain injury, ALS, and other neurodegenerative diseases[88].
Electrophysiologic biomarkers
Electrophysiologic biomarkers apply principles of muscle physiology to assess the presence and severity of SMA at the organ level. One well-established measure is the Compound Muscle Action Potential (CMAP), which indirectly reflects the number of intact motor neurons. CMAP values are typically lower in more severe SMA types and depend on both the number of functional motor units and the average amplitude generated by each unit[85]. These factors can be quantitatively assessed using traditional Motor Unit Number Estimation (MUNE) techniques and by measuring the size of the average single motor unit potential (SMUP).
Another promising tool is Electrical Impedance Myography (EIM). This technique measures the resistance of intra- and extracellular fluids to electrical stimulation as well as the reactance of the muscle cell membrane. In a longitudinal study, Rutkove et al. (2012) demonstrated that EIM effectively tracked disease progression in SMA patients. Importantly, the study concluded that muscle reactance, rather than phase, is the preferred parameter when using a handheld electrode array, as phase measurements are more affected by subcutaneous fat[89].
Imaging modalities
Because SMA alters muscle properties, imaging techniques such as MRI and ultrasound have been investigated as alternative biomarkers. These modalities reveal characteristic findings including muscle atrophy and fat infiltration[90]. Muscle fat fraction has been shown to correlate with functional motor scores and disease progression[85]. To ensure reproducibility and reliability, protocols and validation studies are necessary. Moreover, results must be interpreted in light of normal age-dependent changes, which remain to be fully defined.
Clinical outcome assessment
Assessing clinical outcomes in patients with SMA is crucial for evaluating therapeutic efficacy. Several assessment tools have been developed or adapted to capture the full spectrum of SMA (types I-IV), with a primary focus on motor function. A summary of these tools is provided in Table 5. According to the systematic review by Erdos and Wild (2022), baseline motor endpoints in patients treated with Nusinersen or the gene therapy onasemnogene abeparvovec were measured using these assessment tools. For patients with SMA type I, CHOP-INTEND and HINE-2 were most commonly applied[98]. For SMA types II-IV, a wider range of instruments was used: while CHOP-INTEND remained frequently employed, other studies utilized HINE-2, MFM, HFSME, RULM, 6MWT, or a combination of CHOP-INTEND and HFSME. These findings highlight the importance of routine clinical evaluation, as current evidence does not yet support the use of the aforementioned biomarkers as reliable substitutes for the comprehensive clinical assessment of SMA patients.
Assessment tools for motor endpoints in patients with SMA
| Clinical outcome assessment tool | Components | Utility/Limitations |
| Children’s Hospital of Philadelphia Infant Test for Neuromuscular Disorders (CHOP INTEND)[91,92] | • 16-item observational test scored based on predetermined graded responses | • Developed for SMA type I patients • Limited use in older patients due to positional requirements and reliance on primitive reflexes |
| CHOP ATEND (modified CHOP INTEND scale)[92] | • Assesses head and trunk control, antigravity limb movements, rolling, reaching, and resistance to movement using a 0-4 scale across 16 items to monitor motor function progression or therapeutic response | • Designed to measure motor function in non-sitters in their wheelchair |
| Hammersmith Infant Neurological Examination (HINE)[93] | • Evaluates motor milestones (head control, sitting, voluntary grasp, kicking, rolling, crawling, standing, walking) | • Useful for determining developmental milestones in infantile-onset SMA (2-24 months) • Also used in conditions such as cerebral palsy |
| Hammersmith Functional Motor Scale (HFMS)[94,95] | • 20-item scale assessing ability to perform activities (maximum score: 40) | • Requires frequent positional changes • May cause fatigue • Used for SMA types II and III with limited ambulation |
| Hammersmith Functional Motor Scale Expanded (HFMSE)[95,96] | • 33-item ordinal scale (maximum score: 66) | • Limited by psychometric properties • Correlates with SMN2 copy number, CMAP, Forced Vital Capacity (FVC), and muscle strength |
| Modified Hammersmith Functional Motor Scale (MHFMS)[95,97] | • 20-item scale assessing gross motor function (rolling, sitting, crawling, kneeling, standing, transitions), each scored 0-2 | • Designed to minimize fatigue and reduce frequent positional changes required in HFMS • Developed for SMA type II |
| Modified Hammersmith Functional Motor Scale Extended (MHFMS-EXTEND)[95] | • Incorporates 8 additional gross motor items and timed tests | • May have a ceiling effect, limiting sensitivity to clinical improvement especially in stronger patients |
| Revised Hammersmith Scale (RHS)[95,96] | • 36-item scale (maximum score: 69) including two timed tests; modification of HFMSE to reduce floor and ceiling effects | • Applied in SMA types II and III |
| Motor Function Measure (MFM)[92] | • Assesses motor function across three domains: standing/transfers, axial/proximal function, and distal function | • Also validated in other neuromuscular disorders |
| Revised Upper Limb Module (RULM)[92,96] | • 20-item scale (maximum score: 37) with an entry item identifying the participant’s functional level | • Assesses upper extremity function in non-ambulatory SMA patients |
FUTURE DIRECTIONS
Currently approved therapeutic drugs for patients with SMA are primarily designed for the severe phenotypes. The timing of treatment is critical: initiating therapy before symptom onset offers significantly better outcomes. This underscores the importance of early detection, which can be achieved through neonatal or prenatal genetic screening. Supporting this, recent evidence suggests that presymptomatic developmental processes contribute to SMA pathogenesis in prenatal mouse models[99].
Extensive studies have proven that SMN functions ubiquitously, not only in the central nervous system but also in peripheral organs. Although many therapeutic targets have been identified, increasing SMN levels alone is insufficient to control or cure peripheral symptoms, particularly in milder disease forms. Therefore, future research should explore potential combinatorial therapies, including those targeting SMN-independent pathways and genetic modifications. For example, Hatanaka et al. (2024) reported that combining gene supplementation with genome editing in an SMA mouse model produced encouraging results. Using a CRISPR-Cas9-based homology-independent targeted integration (HITI) strategy alongside SMN1 cDNA supplementation, they observed improved cellular and tissue phenotypes, elevated SMN1 expression, and markedly prolonged survival in SMA mice[100]. Building on such findings, it is anticipated that combinatorial gene therapies will receive growing attention as potential curative strategies for SMA and a broad spectrum of inherited genetic disorders, both in vitro and in vivo. Another promising direction lies in targeted therapy and drug delivery. Nanomedicine has emerged as an efficient drug delivery platform capable of directing therapies to specific cellular and molecular targets. Notably, nanotechnology-based systems have successfully delivered drugs across the blood-brain barrier and blood-cerebrospinal fluid barrier, reaching precise brain cells and signaling pathways[101]. Ongoing research into novel nanocarriers and drug targets is expected to significantly influence the future of SMA therapy. Their synergistic application will be particularly important for addressing pathology in both the central and peripheral nervous systems, especially within organ systems affected by spinal muscular atrophy.
CONCLUSION
This review highlights the critical role of SMN2 splicing regulation and post-transcriptional modification in spinal muscular atrophy (SMA). A single point mutation - a C-to-T transition - alters the inclusion of exon 7 in SMN2 transcripts, functioning as a molecular switch that determines whether functional SMN protein is produced. This process is regulated by cis-acting elements and trans-acting splicing factors, many of which are promising drug targets for enhancing full-length SMN protein production. Other post-transcriptional mechanisms, including alternative splicing, polyadenylation, and epigenetic modifications such as histone acetylation and CpG methylation, also affect SMN2 expression and are crucial considerations for therapy. Current drugs, including Nusinersen and Risdiplam, show that enhancing exon 7 inclusion through splicing modulation provides clear clinical benefits, underscoring the therapeutic potential of targeting post-transcriptional regulation in SMA.
Looking forward, more effective SMA treatments will depend on deeper and more precise insights into SMN2 splicing regulation. Promising strategies include RNA-based drugs, small-molecule splicing modulators, and gene-editing tools such as CRISPR/Cas9 to boost the production of functional SMN protein. Additional therapeutic opportunities lie in targeting epigenetic modifiers and splicing regulators beyond exon 7, such as ISS-N1, ESEs, and ISEs. The development of improved disease models and the identification of reliable biomarkers will be essential for evaluating therapeutic efficacy and tailoring treatment plans. Ultimately, the central goal remains the restoration of normal SMN2 splicing through precise post-transcriptional interventions, a cornerstone of current and future SMA research and treatment.
DECLARATIONS
Authors’ contributions
Conceptualization, writing - original draft: Virata MCA, Abello RMR, Brillante EMB, Corneta MB
Visualization, writing - review and editing: Virata M
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
1. National Center for Biotechnology Information. Spinal muscular atrophy; 1998. Available from: https://www.ncbi.nlm.nih.gov/books/NBK22213/ [Last accessed on 22 Sep 2025].
2. Nishio H, Niba ETE, Saito T, Okamoto K, Takeshima Y, Awano H. Spinal muscular atrophy: the past, present, and future of diagnosis and treatment. Int J Mol Sci. 2023;24:11939.
3. Verhaart IEC, Robertson A, Wilson IJ, et al. Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy - a literature review. Orphanet J Rare Dis. 2017;12:124.
4. Lefebvre S, Bürglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155-65.
5. Prior TW, Leach ME, Finanger EL. Spinal muscular atrophy; 2000. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1352/ [Last accessed on 22 Sep 2025].
6. Butchbach ME. Copy number variations in the survival motor neuron genes: implications for spinal muscular atrophy and other neurodegenerative diseases. Front Mol Biosci. 2016;3:7.
7. Dubowitz V. Very severe spinal muscular atrophy (SMA type 0): an expanding clinical phenotype. Eur J Paediatr Neurol. 1999;3:49-51.
9. Prior TW, Leach ME, Finanger EL. Spinal muscular atrophy, type II. Available from: https://www.ncbi.nlm.nih.gov/medgen/95975 [Last accessed on 22 Sep 2025].
10. Salort-Campana E, Quijano-Roy S. Clinical features of spinal muscular atrophy (SMA) type 3 (Kugelberg-Welander disease). Arch Pediatr. 2020;27:7S23-8.
11. Wijngaarde CA, Stam M, Otto LAM, et al. Muscle strength and motor function in adolescents and adults with spinal muscular atrophy. Neurology. 2020;95:e1988-98.
12. Talbot K, Tizzano EF. The clinical landscape for SMA in a new therapeutic era. Gene Ther. 2017;24:529-33.
13. Calder AN, Androphy EJ, Hodgetts KJ. Small molecules in development for the treatment of spinal muscular atrophy. J Med Chem. 2016;59:10067-83.
14. Dhuri K, Bechtold C, Quijano E, et al. Antisense oligonucleotides: an emerging area in drug discovery and development. J Clin Med. 2020;9:2004.
15. Bowerman M, Becker CG, Yáñez-Muñoz RJ, et al. Therapeutic strategies for spinal muscular atrophy: SMN and beyond. Dis Model Mech. 2017;10:943-54.
16. Singh RN, Singh NN. Mechanism of splicing regulation of spinal muscular atrophy genes. In: Sattler R, Donnelly CJ, editors. RNA Metabolism in Neurodegenerative Diseases. Cham: Springer International Publishing; 2018. pp. 31-61.
17. Bruce A, Alexander J, Julian L, Martin R, Keith R, Peter W. Proteins are made on polyribosomes; 2002. Available from: https://www.ncbi.nlm.nih.gov/books/NBK26829/ [Last accessed on 22 Sep 2025].
18. Kiss T. Small nucleolar RNA-guided post-transcriptional modification of cellular RNAs. EMBO J. 2001;20:3617-22.
19. Wise JA, Lou H. Transcription | messenger RNA processing in eukaryotes. Encyclopedia of Biological Chemistry III. Elsevier; 2021. pp. 411-9.
20. Lodish HF. Molecular cell biology, 6th ed. United States: W.H. Freeman; 2008. Available from: https://books.google.com/books/about/Molecular_Cell_Biology.html?id=K3JbjG1JiUMC [Last accessed on 24 Sep 2025].
21. Wahl MC, Will CL, Lührmann R. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136:701-18.
22. McManus CJ, Graveley BR. RNA structure and the mechanisms of alternative splicing. Curr Opin Genet Dev. 2011;21:373-9.
23. Pellizzoni L. Chaperoning ribonucleoprotein biogenesis in health and disease. EMBO Rep. 2007;8:340-5.
24. Ropper AH, Samuels MA, Klein JP, Prasad S. Adams and victor's principles of neurology, 11e. McGraw-Hill Education; 2019. Available from: https://neurology.mhmedical.com/content.aspx?bookid=1477§ionid=215135193 [Last accessed on 24 Sep 2025].
25. Hassan HA, Zaki MS, Issa MY, El-Bagoury NM, Essawi ML. Genetic pattern of SMN1, SMN2, and NAIP genes in prognosis of SMA patients. Egypt J Med Hum Genet. 2020;21:44.
26. Kernochan LE, Russo ML, Woodling NS, et al. The role of histone acetylation in SMN gene expression. Hum Mol Genet. 2005;14:1171-82.
27. Cao YY, Qu YJ, He SX, et al. Association between SMN2 methylation and disease severity in Chinese children with spinal muscular atrophy. J Zhejiang Univ Sci B. 2016;17:76-82.
28. Narcís JO, Tapia O, Tarabal O, et al. Accumulation of poly(A) RNA in nuclear granules enriched in Sam68 in motor neurons from the SMNΔ7 mouse model of SMA. Sci Rep. 2018;8:9646.
29. Eperon IC, Makarova OV, Mayeda A, et al. Selection of alternative 5' splice sites: role of U1 snRNP and models for the antagonistic effects of SF2/ASF and hnRNP A1. Mol Cell Biol. 2000;20:8303-18.
30. Xue J, Ma T, Zhang X. TRA2: the dominant power of alternative splicing in tumors. Heliyon. 2023;9:e15516.
31. Singh NN, Lee BM, DiDonato CJ, Singh RN. Mechanistic principles of antisense targets for the treatment of spinal muscular atrophy. Future Med Chem. 2015;7:1793-808.
32. Förch P, Puig O, Martínez C, Séraphin B, Valcárcel J. The splicing regulator TIA-1 interacts with U1-C to promote U1 snRNP recruitment to 5' splice sites. EMBO J. 2002;21:6882-92.
33. Blencowe BJ. Exonic splicing enhancers: mechanism of action, diversity and role in human genetic diseases. Trends Biochem Sci. 2000;25:106-10.
34. Goren A, Ram O, Amit M, et al. Comparative analysis identifies exonic splicing regulatory sequences-The complex definition of enhancers and silencers. Mol Cell. 2006;22:769-81.
35. Miyaso H, Okumura M, Kondo S, Higashide S, Miyajima H, Imaizumi K. An intronic splicing enhancer element in survival motor neuron (SMN) pre-mRNA. J Biol Chem. 2003;278:15825-31.
36. Han J, Ding JH, Byeon CW, et al. SR proteins induce alternative exon skipping through their activities on the flanking constitutive exons. Mol Cell Biol. 2011;31:793-802.
37. Kashima T, Rao N, David CJ, Manley JL. hnRNP A1 functions with specificity in repression of SMN2 exon 7 splicing. Hum Mol Genet. 2007;16:3149-59.
38. Miyajima H, Miyaso H, Okumura M, Kurisu J, Imaizumi K. Identification of a cis-acting element for the regulation of SMN exon 7 splicing. J Biol Chem. 2002;277:23271-7.
39. Passmore LA, Coller J. Roles of mRNA poly(A) tails in regulation of eukaryotic gene expression. Nat Rev Mol Cell Biol. 2022;23:93-106.
40. Li Q. Nusinersen as a therapeutic agent for spinal muscular atrophy. Yonsei Med J. 2020;61:273-83.
42. U.S. Food and Drug Administration. SPINRAZA (Nusinersen) injection, for intrathecal use: highlights of prescribing information; 2016. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/209531lbl.pdf [Last accessed on 22 Sep 2025].
43. Neil EE, Bisaccia EK. Nusinersen: a novel antisense oligonucleotide for the treatment of spinal muscular atrophy. J Pediatr Pharmacol Ther. 2019;24:194-203.
44. Finkel RS, Mercuri E, Darras BT, et al. Nusinersen versus Sham control in infantile-onset spinal muscular atrophy. N Engl J Med. 2017;377:1723-32.
45. Mercuri E, Darras BT, Chiriboga CA, et al. Nusinersen versus Sham control in later-onset spinal muscular atrophy. N Engl J Med. 2018;378:625-35.
46. Ratni H, Scalco RS, Stephan AH. Risdiplam, the first approved small molecule splicing modifier drug as a blueprint for future transformative medicines. ACS Med Chem Lett. 2021;12:874-7.
47. Baranello G, Darras BT, Day JW, et al. Risdiplam in type 1 spinal muscular atrophy. N Engl J Med. 2021;384:915-23.
48. Sturm S, Günther A, Jaber B, et al. A phase 1 healthy male volunteer single escalating dose study of the pharmacokinetics and pharmacodynamics of risdiplam (RG7916, RO7034067), a SMN2 splicing modifier. Br J Clin Pharmacol. 2019;85:181-93.
49. Masson R, Mazurkiewicz-Bełdzińska M, Rose K, et al. Safety and efficacy of risdiplam in patients with type 1 spinal muscular atrophy (FIREFISH part 2): secondary analyses from an open-label trial. Lancet Neurol. 2022;21:1110-9.
50. Oskoui M, Day JW, Deconinck N, et al. Correction to: Two-year efficacy and safety of risdiplam in patients with type 2 or non-ambulant type 3 spinal muscular atrophy (SMA). J Neurol. 2023;270:2547-9.
51. Cheung AK, Hurley B, Kerrigan R, et al. Discovery of small molecule splicing modulators of survival motor neuron-2 (SMN2) for the treatment of spinal muscular atrophy (SMA). J Med Chem. 2018;61:11021-36.
52. Keller CG, Shin Y, Monteys AM, et al. An orally available, brain penetrant, small molecule lowers huntingtin levels by enhancing pseudoexon inclusion. Nat Commun. 2022;13:1150.
53. Theil D, Kuhle J, Brees D, et al. Neurofilament light chain: a translational safety biomarker for drug-induced peripheral neurotoxicity. Toxicol Pathol. 2023;51:135-47.
54. Yeo CJJ, Darras BT. Overturning the Paradigm of spinal muscular atrophy as just a motor neuron disease. Pediatr Neurol. 2020;109:12-9.
55. Haque US, Yokota T. Recent progress in gene-targeting therapies for spinal muscular atrophy: promises and challenges. Genes. 2024;15:999.
56. Day JW, Finkel RS, Chiriboga CA, et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy in patients with two copies of SMN2 (STR1VE): an open-label, single-arm, multicentre, phase 3 trial. Lancet Neurol. 2021;20:284-93.
57. Mercuri E, Muntoni F, Baranello G, et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy type 1 (STR1VE-EU): an open-label, single-arm, multicentre, phase 3 trial. Lancet Neurol. 2021;20:832-41.
58. Strauss KA, Farrar MA, Muntoni F, et al. Onasemnogene abeparvovec for presymptomatic infants with three copies of SMN2 at risk for spinal muscular atrophy: the Phase III SPR1NT trial. Nat Med. 2022;28:1390-7.
59. Mendell JR, Al-Zaidy SA, Lehman KJ, et al. Five-year extension results of the phase 1 START trial of onasemnogene abeparvovec in spinal muscular atrophy. JAMA Neurol. 2021;78:834-41.
60. Muntoni F, Bertini E, Comi G, et al. Long-term follow-up of patients with type 2 and non-ambulant type 3 spinal muscular atrophy (SMA) treated with olesoxime in the OLEOS trial. Neuromuscul Disord. 2020;30:959-69.
61. Bertini E, Dessaud E, Mercuri E, et al. Safety and efficacy of olesoxime in patients with type 2 or non-ambulatory type 3 spinal muscular atrophy: a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol. 2017;16:513-22.
62. Villanova M, Bach JR. Allogeneic mesenchymal stem cell therapy outcomes for three patients with spinal muscular atrophy type 1. Am J Phys Med Rehabil. 2015;94:410-5.
63. Mirea A, Shelby ES, Axente M, et al. Combination therapy with nusinersen and onasemnogene abeparvovec-xioi in spinal muscular atrophy type I. J Clin Med. 2021;10:5540.
64. Zhang W, Yin Y, Yang D, et al. Comprehensive analysis of adverse events associated with onasemnogene abeparvovec (Zolgensma) in spinal muscular atrophy patients: insights from FAERS database. Front Pharmacol. 2024;15:1475884.
65. Waldrop MA, Karingada C, Storey MA, et al. Gene therapy for spinal muscular atrophy: safety and early outcomes. Pediatrics. 2020;146:e20200729.
66. Kotulska K, Jozwiak S, Jedrzejowska M, et al. Newborn screening and gene therapy in SMA: challenges related to vaccinations. Front Neurol. 2022;13:890860.
67. Sleigh JN, Gillingwater TH, Talbot K. The contribution of mouse models to understanding the pathogenesis of spinal muscular atrophy. Dis Model Mech. 2011;4:457-67.
68. Kray KM, McGovern VL, Chugh D, Arnold WD, Burghes AHM. Dual SMN inducing therapies can rescue survival and motor unit function in symptomatic ∆7SMA mice. Neurobiol Dis. 2021;159:105488.
69. Zhao X, Feng Z, Ling KK, et al. Pharmacokinetics, pharmacodynamics, and efficacy of a small-molecule SMN2 splicing modifier in mouse models of spinal muscular atrophy. Hum Mol Genet. 2016;25:1885-99.
70. Seo J, Singh NN, Ottesen EW, Sivanesan S, Shishimorova M, Singh RN. Oxidative stress triggers body-wide skipping of multiple exons of the spinal muscular atrophy gene. PLoS One. 2016;11:e0154390.
71. Baenas N, Wagner AE. Drosophila melanogaster as an alternative model organism in nutrigenomics. Genes Nutr. 2019;14:14.
72. Praveen K, Wen Y, Matera AG. A Drosophila model of spinal muscular atrophy uncouples snRNP biogenesis functions of survival motor neuron from locomotion and viability defects. Cell Rep. 2012;1:624-31.
73. Spring AM, Raimer AC, Hamilton CD, Schillinger MJ, Matera AG. Comprehensive modeling of spinal muscular atrophy in drosophila melanogaster. Front Mol Neurosci. 2019;12:113.
74. Gonzalez D, Vásquez-Doorman C, Luna A, Allende ML. Modeling spinal muscular atrophy in zebrafish: current advances and future perspectives. Int J Mol Sci. 2024;25:1962.
75. McWhorter ML, Monani UR, Burghes AH, Beattie CE. Knockdown of the survival motor neuron (Smn) protein in zebrafish causes defects in motor axon outgrowth and pathfinding. J Cell Biol. 2003;162:919-31.
76. Cerneckis J, Cai H, Shi Y. Induced pluripotent stem cells (iPSCs): molecular mechanisms of induction and applications. Signal Transduct Target Ther. 2024;9:112.
77. Bautista CM, Sterneckert J. Progress and challenges in directing the differentiation of human iPSCs into spinal motor neurons. Front Cell Dev Biol. 2022;10:1089970.
78. Pagliari E, Taiana M, Manzini P, et al. Targeting STMN2 for neuroprotection and neuromuscular recovery in Spinal Muscular Atrophy: evidence from in vitro and in vivo SMA models. Cell Mol Life Sci. 2024;82:29.
79. Ohuchi K, Funato M, Kato Z, et al. Established stem cell model of spinal muscular atrophy is applicable in the evaluation of the efficacy of thyrotropin-releasing hormone analog. Stem Cells Transl Med. 2016;5:152-63.
80. Wang J, Schultz PG, Johnson KA. Mechanistic studies of a small-molecule modulator of SMN2 splicing. Proc Natl Acad Sci USA. 2018;115:E4604-12.
81. Zheleznyakova GY, Voisin S, Kiselev AV, et al. Genome-wide analysis shows association of epigenetic changes in regulators of Rab and Rho GTPases with spinal muscular atrophy severity. Eur J Hum Genet. 2013;21:988-93.
82. Zwartkruis MM, Kortooms JV, Gommers D, et al. Comprehensive analysis across SMN2 excludes DNA methylation as an epigenetic biomarker for spinal muscular atrophy. iScience. 2025;28:112461.
83. Redman M, King A, Watson C, King D. What is CRISPR/Cas9? Arch Dis Child Educ Pract Ed. 2016;101:213-5.
84. Li JJ, Lin X, Tang C, et al. Disruption of splicing-regulatory elements using CRISPR/Cas9 to rescue spinal muscular atrophy in human iPSCs and mice. Natl Sci Rev. 2020;7:92-101.
85. Kariyawasam DST, D'Silva A, Lin C, Ryan MM, Farrar MA. Biomarkers and the development of a personalized medicine approach in spinal muscular atrophy. Front Neurol. 2019;10:898.
86. Burghes AH, Beattie CE. Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat Rev Neurosci. 2009;10:597-609.
87. Darras BT, Crawford TO, Finkel RS, et al. Neurofilament as a potential biomarker for spinal muscular atrophy. Ann Clin Transl Neurol. 2019;6:932-44.
88. Virata MCA, Catahay JA, Lippi G, Henry BM. Neurofilament light chain: a biomarker at the crossroads of clarity and confusion for gene-directed therapies. Neurodegener Dis Manag. 2024;14:227-39.
89. Rutkove SB, Gregas MC, Darras BT. Electrical impedance myography in spinal muscular atrophy: a longitudinal study. Muscle Nerve. 2012;45:642-7.
90. Huang Y, Chen T, Hu Y, Li Z. Muscular MRI and magnetic resonance neurography in spinal muscular atrophy. Clin Radiol. 2024;79:673-80.
91. Glanzman AM, Mazzone E, Main M, et al. The children's hospital of philadelphia infant test of neuromuscular disorders (CHOP INTEND): test development and reliability. Neuromuscul Disord. 2010;20:155-61.
92. Nelson LL, Iannaccone ST. Clinical outcome assessments in Duchenne muscular dystrophy and spinal muscular atrophy: past, present and future. Neuromuscul Disord. 2021;31:1028-37.
93. Bishop KM, Montes J, Finkel RS. Motor milestone assessment of infants with spinal muscular atrophy using the hammersmith infant neurological exam-part 2: experience from a nusinersen clinical study. Muscle Nerve. 2018;57:142-6.
94. Main M, Kairon H, Mercuri E, Muntoni F. The Hammersmith functional motor scale for children with spinal muscular atrophy: a scale to test ability and monitor progress in children with limited ambulation. Eur J Paediatr Neurol. 2003;7:155-9.
95. Ramsey D, Scoto M, Mayhew A, et al. Revised hammersmith scale for spinal muscular atrophy: a SMA specific clinical outcome assessment tool. PLoS One. 2017;12:e0172346.
96. Wolfe A, Stimpson G, Ramsey D, et al. Disease trajectories in the revised hammersmith scale in a cohort of untreated patients with spinal muscular atrophy types 2 and 3. J Neuromuscul Dis. 2024;11:665-77.
97. Krosschell KJ, Scott CB, Maczulski JA, Lewelt AJ, Reyna SP, Swoboda KJ; Project Cure SMA. Reliability of the modified hammersmith functional motor scale in young children with spinal muscular atrophy. Muscle Nerve. 2011;44:246-51.
98. Erdos J, Wild C. Mid- and long-term (at least 12 months) follow-up of patients with spinal muscular atrophy (SMA) treated with nusinersen, onasemnogene abeparvovec, risdiplam or combination therapies: a systematic review of real-world study data. Eur J Paediatr Neurol. 2022;39:1-10.
99. Motyl AAL, Faller KME, Groen EJN, et al. Pre-natal manifestation of systemic developmental abnormalities in spinal muscular atrophy. Hum Mol Genet. 2020;29:2674-83.
100. Hatanaka F, Suzuki K, Shojima K, et al. Therapeutic strategy for spinal muscular atrophy by combining gene supplementation and genome editing. Nat Commun. 2024;15:6191.
101. Nguyen TT, Dung Nguyen TT, Vo TK, et al. Nanotechnology-based drug delivery for central nervous system disorders. Biomed Pharmacother. 2021;143:112117.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Issue
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].