


Cluster-model-embedded first-principles study of thermodynamic stability and elastic properties in (Zr, Ti)Cx carbides
 0
0 Abstract
Multi-component carbide ceramics have garnered significant attention as ultra-high-temperature structural materials due to their exceptionally high melting points and excellent mechanical properties. In this work, we systematically investigate the synergistic effects of C vacancies and Ti alloying on the thermodynamic stability and elastic behavior of (Zr, Ti)Cx carbides using first-principles calculations. Specific cluster structural models of [C-M6](C,□)5 (M = Zr/Ti, □ = vacancy) were constructed by considering the local chemical short-range orders of elemental distribution and the ordering of vacancies on C sublattice, which were then employed as inputs for first-principles calculations. The results reveal that the introduction of C vacancies decreases the free energy at high temperatures and enhances the thermodynamic stability, whereas Ti substitution for Zr tends to reduce stability. Notably, the ternary carbide Zr5Ti1C5 ([C-Zr5Ti1](C,□)5) with an equimolar ratio of Ti-to-vacancy exhibits superior high-temperature thermodynamic stability. Analysis of entropy contributions indicates that both vacancies and Ti addition primarily alter the free energy by modifying the lattice vibration modes, an effect dominated by the vibrational entropy. These two types of defects weaken the M-C bond strength, resulting in reduced binding energy and Young’s modulus. Furthermore, this synergistic effect considerably lowers the critical temperature required to stabilize the single-phase solid solution structure in multi-component carbides, which is attributed to a decrease in mixing enthalpy and an increase in configurational entropy caused by vacancies. The cluster-model-embedded first-principles approach offers valuable insight for designing high-performance carbides in complex ceramic systems.
Keywords
INTRODUCTION
Carbide ceramics composed of transition metals (TMs) have shown broad application prospects in aerospace and high-temperature (HT) structural materials due to their ultra-high melting points (> 3,273 K), and excellent chemical stability[1-4]. Among them, ZrC (face-centered cubic (FCC) NaCl type) has been widely used in wear-resistant coatings, high-speed cutting tools, and HT ceramic components owing to its high Young's modulus
Although experimental studies have demonstrated the advantages of vacancies and multi-element alloying in MC-type carbides, the intrinsic mechanisms governing thermodynamic stability are still unclear, largely due to the challenges in experimental characterization with sufficient accuracy. In recent years, first-principles calculations based on density functional theory (DFT) have become essential for atomic-level design and mechanistic exploration of material properties[18-20]. For instance, the Cluster Expansion Method (CEM) studies have revealed that the formation energy of ordered carbon vacancies in ZrCx is lower than that of disordered ones at 0 K, indicating superior stability of ordered configurations[21]. Research on (Ti1-yNiy)Cx ternary carbides using special quasi-random structure (SQS) method shows that the thermodynamic stability increases with C-vacancy concentration, and that high Ni content necessitates the presence of vacancies[22,23]. However, computational investigations into the ordered vacancies and chemical short-range orders (CSROs) in multi-component carbides are still limited. Conventional modeling approaches, such as SQS and supercell methods, are employed to construct the solid-solution structures of multi-component carbides, but often struggle to accurately capture ordered vacancy distribution and local chemical environments[24,25]. These methods also require large supercells for multi-component systems[26,27], leading to high computational costs that impede efficient screening and analysis. Therefore, there is a critical need for computational models capable of effectively representing ordered C-vacancies and incorporating CSROs to enable accurate theoretical insights into the thermodynamic stability and property modulation mechanisms of multi-component carbides.
In previous work, we developed a cluster-plus-glue-atom model to describe the CSROs in multi-component solid-solution alloys[28-30], providing a reliable approach to consider the interactions among multiple elements. Herein, we extend this model to the MC-type carbides. A series of structural models, encompassing binary (ZrC, Zr6C5, Zr3C2) and ternary (Zr5Ti1C6, Zr5Ti1C5, Zr5Ti1C4, Zr4Ti2C6, Zr4Ti2C5) carbides, will be constructed by systematically varying metal species (M = Zr, Ti) and C vacancy concentrations, with the ordered arrangement of C vacancies explicitly incorporated. These periodic cluster-based models will serve as input for first-principles calculations to determine equilibrium lattice constants, binding energy, formation energy, free energy, and HT elastic modulus, thereby elucidating the cooperative effect of vacancy concentration and Ti alloying on thermodynamic stability and elastic properties. Further analysis of entropy, electronic density of states, and phonon density of states will reveal the dominant factors governing free energy, bonding characteristics, and dynamical stabilities. Finally, the key factors affecting the formation of single-phase solid solution will be identified by evaluating the free energy change associated with the formation of (Zr, Ti)Cx from binary ZrCx and TiCx.
MATERIALS AND METHODS
First principles calculations
Construction of cluster structural models
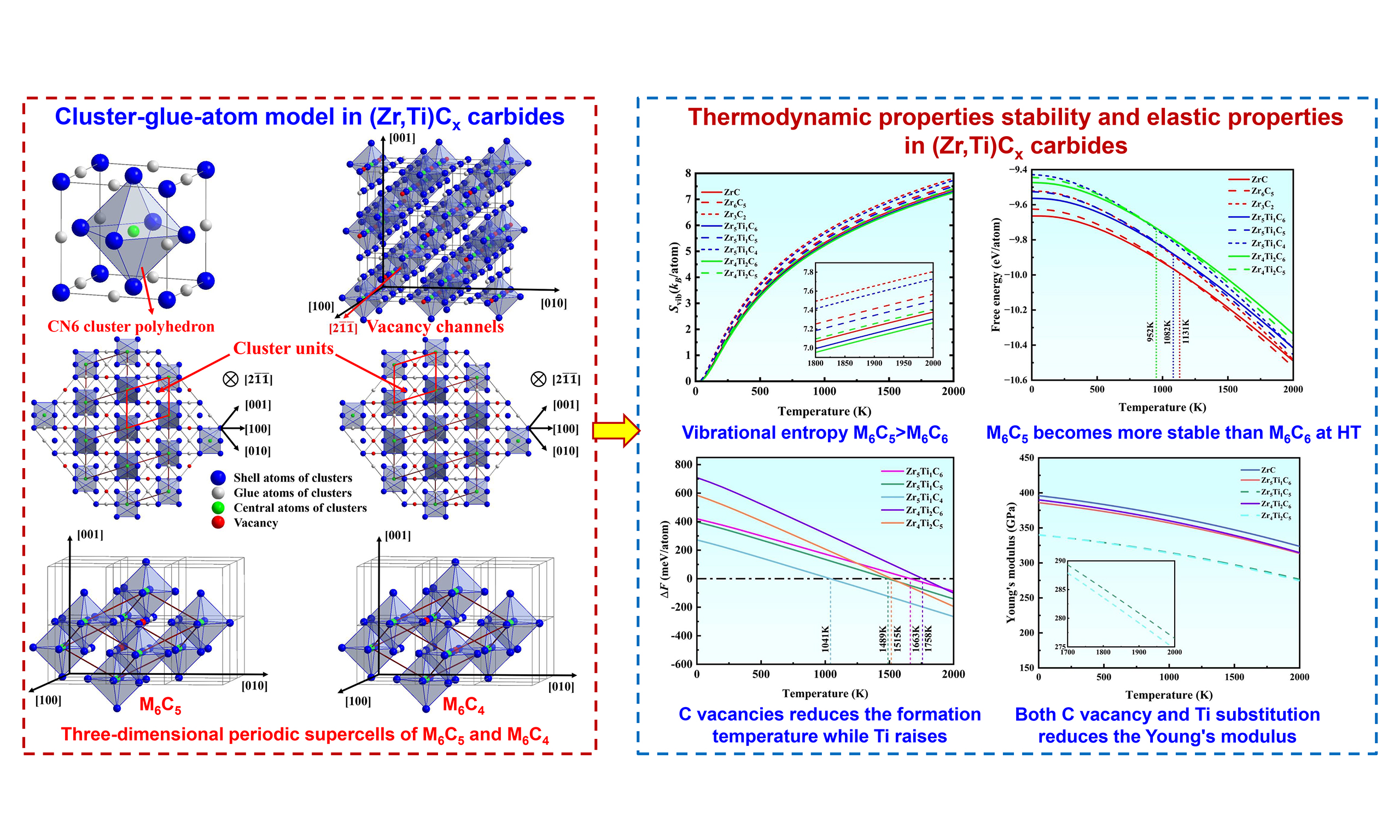
It is well known that vacancies in non-stoichiometric carbides are not randomly distributed[31]. Experimentally confirmed in systems such as ZrCx, TiCx, and NbCx (x < 1), the ordered arrangement of vacancies on the C sublattice is considered thermodynamically stable at room temperature (RT)[32-35]. Computational studies further reveal that chemical bonding in multi-component carbides is highly localized, occurring mainly between central atoms and their first- and second-nearest neighbors[36]. Therefore, these systems can be effectively represented by a cluster-plus-glue-atom structural model that reflects the coordination environment of the central atom over two nearest neighbors[28]. In this model, the cluster is defined as a coordination polyhedron comprising a central solute atom surrounded by its nearest-neighbor solvent atoms, where the strong interaction between them gives rise to the strongest CSRO. And other solute atoms having weaker interactions with the base serve as the glue atoms, occupying the inter-cluster sites to mediate atomic packing. The resulting structural unit is expressed as [cluster](glue atom)m, where m denotes the number of glue atoms per cluster. For MC-type solid solution carbides, the vacancies are located at the glue atom sites to ensure the integrity of the cluster structural unit since vacancies can generally be originated on the C side. Then, the cluster structural unit centered on C, with metal M atoms as shell atoms and the remaining C atoms and vacancies (C/□) serving as glue atoms are constructed. The cluster usually adopts an octahedron with a coordination number of CN = 6 (coordination number = 6), composed of a central C atom (marked by green sphere in Figure 1A) and six M atoms (blue spheres) in the first coordination shell. The second-nearest-neighbor sites around the central C atom (gray spheres in Figure 1A) serve as the glue atom sites, which are occupied by C or □. To preserve the crystal periodicity and stoichiometry of the ideal MC structure (M:C = 1:1), each [C-M6] cluster has five such sites, resulting in the cluster formula [C-M6](C,□)5, marked by red sphere in Figure 1B-F).
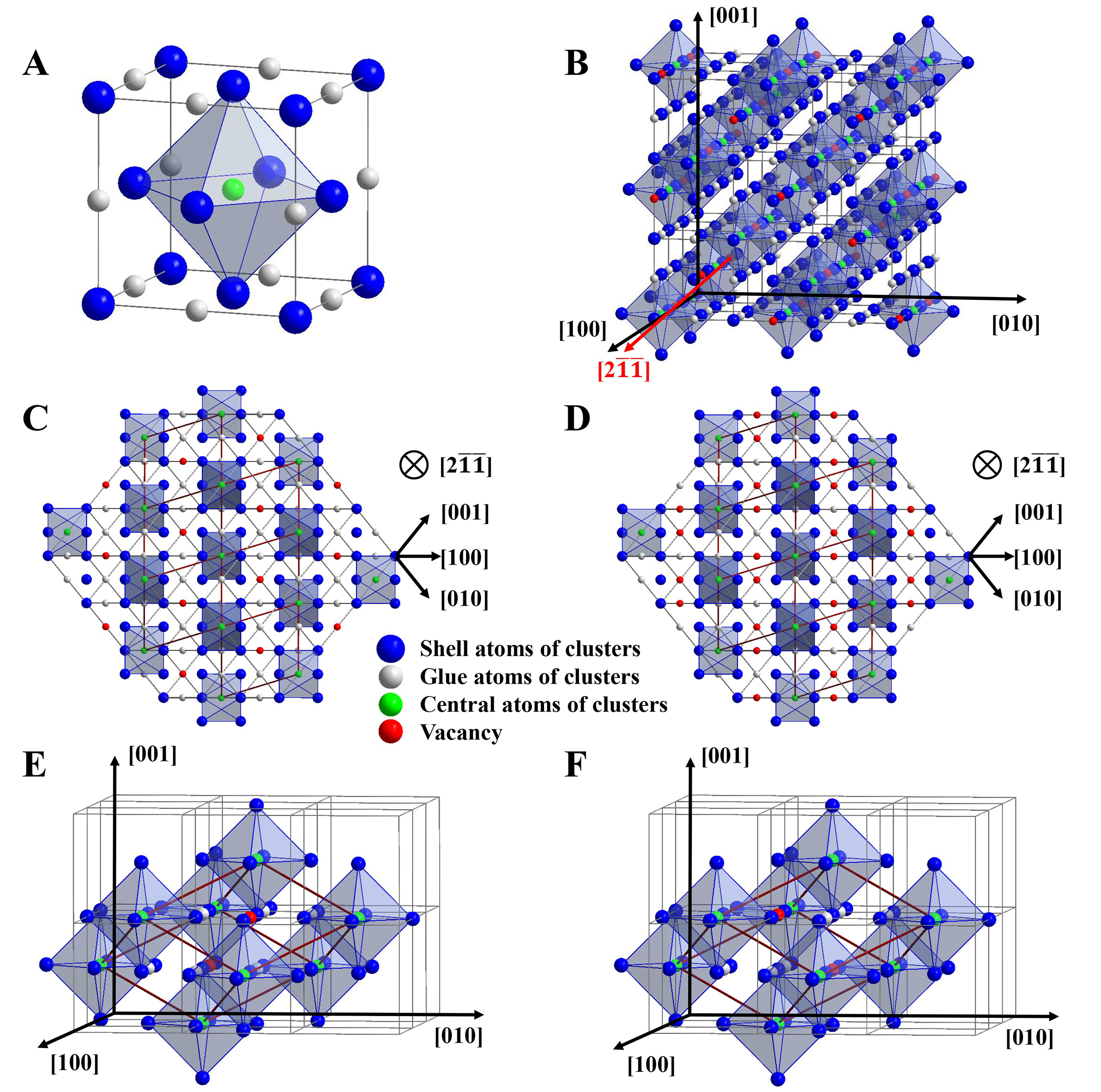
Figure 1. Cluster-glue-atom model in MC-type carbides. (A) Atomic structure of ZrC structure, showing that the CN6 cluster polyhedron is centered by a C atom (green sphere) and surrounded by six metal atoms (blue spheres); (B) Vacancy-ordered structure extracted from a 3 × 3 × 3 supercell of MC, showing the vacancy channels along the [211] direction; (C and D) Derived cluster units for M6C5 and M6C4, illustrating the arrangement of vacancies within the channel framework; (E and F) Three-dimensional periodic supercells of M6C5 and M6C4, constructed by the stacking of cluster units shown in (C and D). The cluster models were generated using the Visualization for Electronic and Structural Analysis (VESTA) software.
Since the introduction of specific vacancy sequences in the C sublattice has been shown to improve the structural stability[15,32], this study focuses on two C-deficient compositions, M6C5 and M6C4 (= M3C2). Guided by the ordered vacancy-channel distribution reported in the literature[37], the atomic structures of these carbides were reconstructed with a 3 × 3 × 3 supercell [Figure 1B]. Vacancy channels are clearly visible along the [211] direction of the MC lattice, where the vacancies occupy the second-nearest neighbor sites around the central C atom. Figure 1C and D show the vacancy channel distributions for M6C5 and M3C2, respectively, along this direction. In the M6C5 structure [Figure 1C], the C sublattice consists of two alternating layer types: one with two-thirds of sites occupied by C atoms and one-third forming vacancy rows, creating a channel-like structure, and the other fully occupied by C. In M3C2 containing a higher vacancy concentration [Figure 1D], the vacancy channel is more prominent: the first layer contains only one-third C rows and two-thirds vacancy rows, while the second layer is fully C-occupied.
Then, a periodic structural unit containing 12 sites (including vacancies), which fully incorporates the cluster model information, can be identified from the brown boxes in Figure 1C and D. Its composition corresponds exactly to the cluster formula [C-M6](C,□)5. The extracted structural units in three-dimension space are displayed in Figure 1E and F. The basis vectors (X, Y, Z) of the supercell are aligned with the [211], [121], and [211] directions of the original FCC lattice, respectively. And the lattice parameters are a = b = c =
It is noted that in the localized cluster model for MC metal carbides, both the M and C sublattices still maintain a complete NaCl-type lattice structure. Moreover, C occupies the center site of the cluster model, thereby guaranteeing that the glue atom site is occupied only by C/□ rather than metal atoms. Based on this model and previous evidence that Ti interacts more strongly with vacancies than Zr[38], Ti atoms were placed near C vacancies in the current model. To systematically evaluate the influence of Ti content and vacancy concentration on the structure and properties of carbides, the supercell structures corresponding to these designed carbide compositions are illustrated in Supplementary Figure 1.
Calculation methods
The first-principles DFT calculations were performed using the plane-wave (PW) basis set and projector-augmented wave (PAW) method, as implemented in the Vienna ab initio Simulation Package (VASP)[39-41]. The electronic exchange-correlation potential was described within the generalized-gradient approximation (GGA) using the Perdew-Burke-Ernzerhof (PBE) parameterization[42]. The C-2s22p2, Ti-3p63d24s2, and Zr-4s24p65s24d2 were treated as the valence states. The kinetic energy cutoff of 800 eV was employed for the plane-wave basis. Ionic positions and lattice parameters of cluster structural models were fully relaxed during the geometrical optimization, with convergence criteria set to a Hellmann-Feynman force tolerance below 0.02 eV Å-1 and an energy tolerance of 1 × 10-8 eV. Γ-centered k-point meshes with a spacing of 0.02 Å-1 were generated according to the Monkhorst-Pack scheme for all calculations. All structural models were built and visualized using the Visualization for Electronic and Structural Analysis (VESTA) software[43], and the electronic density of states was calculated using VASPKIT toolkit[44].
The formation energy (Ef) and binding energy (Eb) of each carbide were calculated using the fundamental physical expressions of Equation (1) and (2):
where Ef represents the energy required to form the carbide structure, defined as the difference between the total energy of the optimized structure (Etot) and the sum of the energies of individual atoms in their standard elemental states (EM), n is the total number of atoms. For Zr and Ti, their standard states are the hexagonal close-packed (HCP) structure with the space group of P63/mmc, while the carbon adopts the graphite structure with the same space group. The DFT-D3 dispersion correction[45] is applied to accurately describe the interlayer van der Waals interactions in graphite. Eb reflects the energy associated with atomic bonding relative to isolated states (Esingle). The ideal single-atom energy (Esingle), i.e., the energy of an isolated atom, is calculated by placing a single atom in a large supercell with a lattice constant of 15 Å to minimize the interactions among periodic images.
To assess the temperature-dependent stability, the Helmholtz free energy F(T) of carbide was computed as[46]:
where Fvib(T), Fel(T), Fmag(T), and Sconf represent the vibrational, electronic, and magnetic free energies, and configurational entropy, respectively. As the system is non-magnetic, Fmag = 0. Lattice dynamics calculations and related thermodynamic properties (free energy and entropy) were performed using the finite displacement method. Supercells with lattice constants larger than 9 Å were built to avoid spurious self-interactions, where each atom was displaced by 0.01 Å along the three Cartesian directions. Force constants were obtained via finite differences, and phonon frequencies were Fourier-interpolated to a 25 × 25 × 25 q-point grid using the phonopy package[47]. The resulting force-constant matrices were subsequently utilized to compute the thermodynamic properties of carbides. Thermodynamic quantities were evaluated within the harmonic approximation up to 2,000 K. The vibrational free energy and vibrational entropy were derived from phonon calculations, as follows[48]:
where kB is Boltzmann constant, q is the wave vector, ν denotes the phonon mode index, ωq,ν represents the phonon frequency at the wave vector modes q and ν, T is the absolute temperature, and
where n(ε) is the electronic density of states, εf is the Fermi energy, and f = f(ε, T) is the Fermi-Dirac distribution function. The configurational entropy Sconf is calculated using Equation (9)[50]:
where the sublattice h contains X sites and sublattice k contains Y sites; R is the ideal gas constant; Nh and Nk are the number of element types in sublattices h and k, respectively.
The elastic modulus of carbide at 0 K was computed from the stress-strain relationship[51,52]. For the crystal symmetries, VASP generated the necessary strain configurations, where strains with magnitudes of ±1% and ±2% were applied along the relevant Cartesian directions in both tensile and compressive sense for each component. Its high-temperature behavior was approximated using the empirical function proposed by Zakarian et al.[53]:
where E0 is the elastic modulus at 0 K from DFT, Tmax is the melting temperature calculated from the Calculation of Phase Diagrams (CALPHAD) method[54,55]. Thus, E(T) can be determined for any temperature given E0 and Tmax.
RESULTS AND DISCUSSION
Formation and binding energies of (Zr, Ti)-C carbides
To validate the selected computational parameters and the accuracy of the cluster model, the calculated lattice constants of Zr-based carbides were compared with available experimental data. The DFT-calculated lattice constant of stoichiometric ZrC is a = 4.710 Å, showing excellent agreement with the experimental values of 4.692-4.705 Å[56,57]. With the introduction of C vacancies, the lattice constant of Zr6C5 (C/Zr = 0.83) increases to a = 4.719 Å, consistent with the reported value of 4.702 Å[58]. This expansion is attributed to the Coulomb repulsion between excess electrons localized at vacancy sites[59], leading to a local lattice distortion and an overall increase in the lattice constant. Upon Ti doping, the lattice constant decreases to a = 4.664 Å for Zr5Ti1C5, due to the smaller atomic radius of Ti (1.47 Å) compared to Zr (1.60 Å). This result agrees well with the reported value (~4.633 Å) of (Zr0.8Ti0.2)C0.8[60].
The structural stability of designed carbides was evaluated by calculating their formation energy Ef and binding energy Eb, as shown in Figure 2. ZrC exhibits the most negative formation energy
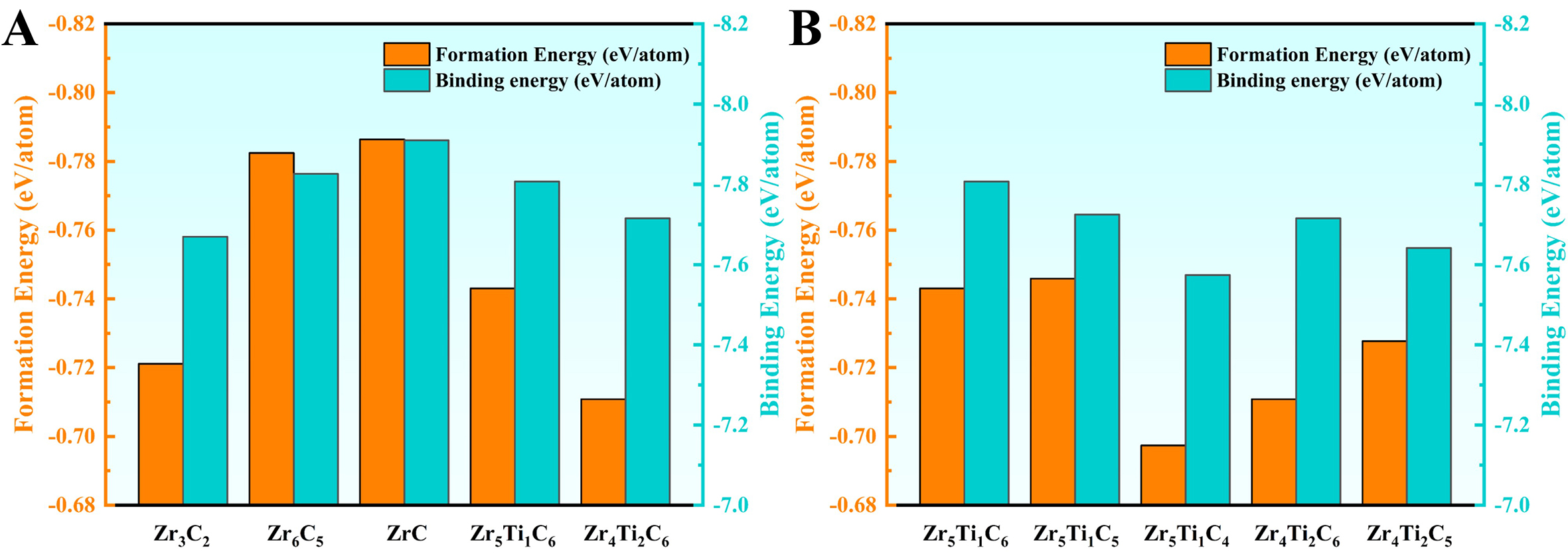
Figure 2. Formation energy (orange y-axis) and binding energy (cyan y-axis) of (Zr, Ti)-C carbides. (A) Zr3C2, Zr6C5, ZrC, Zr5Ti1C6, and Zr4Ti2C6 with different stoichiometric ratios; (B) Zr5Ti1C6, Zr5Ti1C5, Zr5Ti1C4, Zr4Ti2C6, and Zr4Ti2C5 by varying the concentrations of Ti and vacancies.
Free energies of (Zr, Ti)-C carbides
Since the formation energy reflects the structural stability at 0 K alone, the Helmholtz free energy (F) was computed to evaluate the thermodynamic stability of carbides at elevated temperatures. Figure 3 shows the temperature dependence of free energy from 0 to 2,000 K, indicating a decrease in free energy with rising the temperature for all carbides. For the binary Zr-C carbides, the introduction of C vacancies reduces the stability at low temperatures, as evidenced by the fact that ZrC has a lower free energy than Zr6C5 below
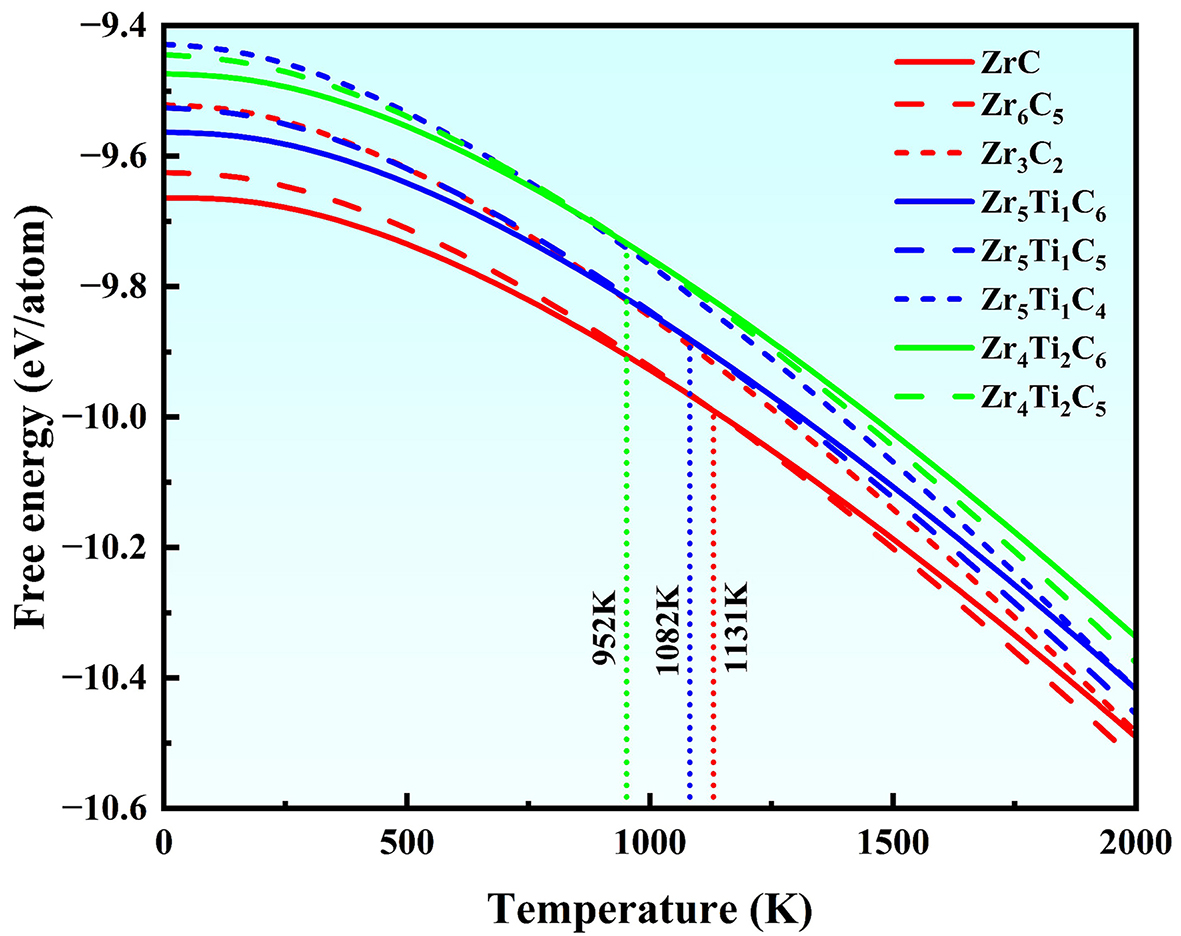
Figure 3. Helmholtz free energy as a function of temperature for (Zr, Ti)-C carbides.
In ternary (Zr, Ti)-C carbides, the free energy increases with Ti content (solid lines in Figure 3), indicating that the substitution of Ti for Zr reduces the thermodynamic stability. C vacancies also induce a stability inversion in these carbides, but the transition temperature decreases with higher Ti content: from 1,131 K for ZrC-Zr6C5 pair to 1,082 K for Zr5Ti1C6-Zr5Ti1C5 and 952 K for Zr4Ti2C6-Zr4Ti2C5 (blue and cyan curves in Figure 3). This indicates that Ti addition preferentially stabilizes the M6C5 structure over the M6C6 at high temperatures. However, excessive C vacancies are detrimental, as Zr5Ti1C4 exhibits higher free energy than Zr5Ti1C5 across the entire temperature range. Therefore, Zr5Ti1C5, with a specific combination of one C vacancy and one Ti atom, demonstrates an optimal HT stability due to the synergistic effect of vacancy and metal alloying. It should be noted that the present free energy trends are evaluated from 0 to 2,000 K. Extrapolation to ultra-high temperatures (> 2,500 K) may involve additional contributions such as anharmonic lattice softening, phase transitions, or decomposition[8], which are beyond the scope of the current model and should be considered in further high-temperature assessments.
Elastic properties of (Zr, Ti)-C carbides
Elastic properties, particularly Young's modulus, are critical indicators for ultra-high-temperature structural materials, as they reflect the bonding strength and structural integrity. The temperature-dependent Young's moduli of carbides were calculated using first-principles results combined with the empirical model proposed by Zakarian et al.[53], as shown in Figure 4. It is found that the modulus decreases with the temperature due to the weakening of interatomic bonds by thermal vibrations. To validate the model, compositions close to the experimentally-studied ZrC0.96 and ZrC0.85 were selected due to the scarcity of HT elastic data for carbides. The calculated moduli of ZrC and Zr6C5 agree well with available experimental and theoretical data[63,64]. Both calculations and experiments confirm that increasing C vacancy concentration significantly reduces the Young's modulus. Although vacancies may enhance the local M-M bonding, the overall reduction in bond density due to missing C atoms diminishes the material’s resistance to elastic deformation. Ti-containing Zr5Ti1C6 and Zr4Ti2C6 exhibit lower Young's modulus than pure ZrC across the temperature range, which is attributed to the weaker bonding strength of Ti-C compared to Zr-C, leading to reduced stiffness. Notably, Zr5Ti1C5 maintains a slightly higher modulus than Zr4Ti2C5 at HTs, indicating superior HT structural stability, consistent with the free energy trends. Within the studied temperature range (below 2,000 K), the stable-lattice approximation provides a solid basis for interpreting the elastic properties, while beyond this range would necessitate accounting for additional factors such as lattice softening and carbon evaporation.
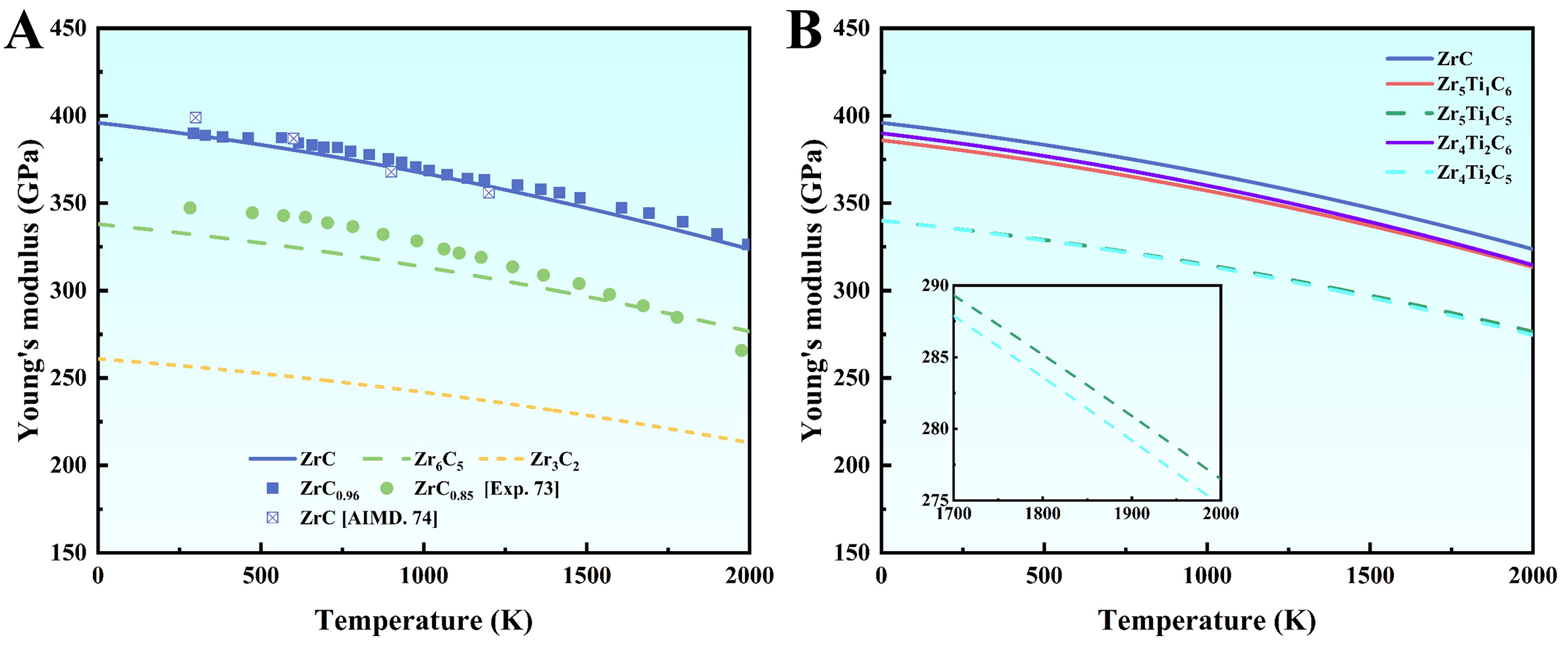
Figure 4. Young's modulus as a function of temperature for ZrCx in (A) and (Zr, Ti)Cx series of carbides in (B), where scattered points represent experimental data, and solid lines denote corresponding theoretical fittings.
Thermodynamic stability of designed (Zr, Ti)-C carbides
The above results reveal that the introduction of vacancies leads to an intersection in the free energy curves at HTs, resulting in a stability transition, whereas the substitution of Ti for the base Zr raises the free energy and reduces the thermodynamic stability of (Zr,Ti)-C ternary carbides [Figure 3]. To further elucidate the influence of vacancies and Ti addition on free energy, Figure 5 and Supplementary Figure 2A and B present the entropy contributions in the designed different carbides as a function of temperature. The results show that the vibrational entropy (Svib = 0-8 kB/atom) dominates the free energy at elevated temperatures, compared to the electronic entropy (Sel = 0-0.6 kB/atom) and configurational entropy (Sconf = 0-0.28 kB/atom). Moreover, the C vacancies increase all entropy components [Figure 5], thereby lowering the free energy and enhancing the thermodynamic stability. This is the primary reason for the stability transition observed in vacancy-containing carbides at HTs. In contrast, the Ti addition reduces the vibrational entropy while increasing the configurational entropy, with a negligible impact on electronic entropy [Figure 5B]. Given the dominant role of vibrational entropy, the net effect of Ti alloying is an increase in the overall free energy, consistent with the trends shown in Figure 3.
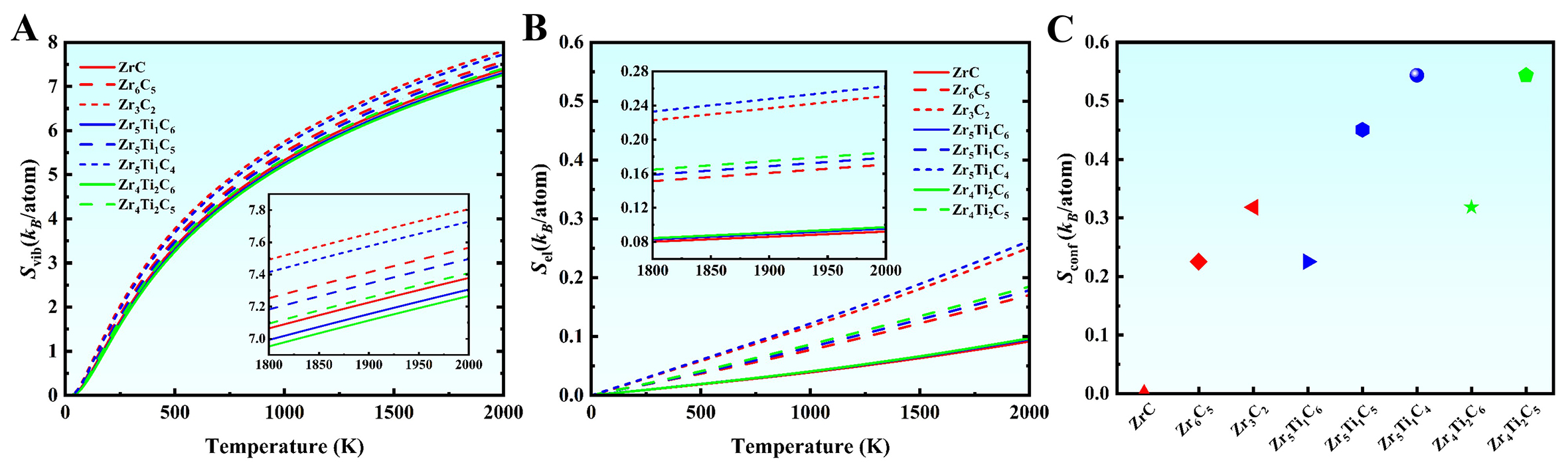
Figure 5. Contributions to the total entropy as a function of temperature for (Zr, Ti)Cx carbides. (A) Vibrational entropy; (B) Electronic entropy; (C) Configurational entropy.
For the dominant vibrational entropy, it originates from lattice vibrations, and its magnitude is directly governed by the phonon density of states (PhDOS), as presented in Figure 6A-C and
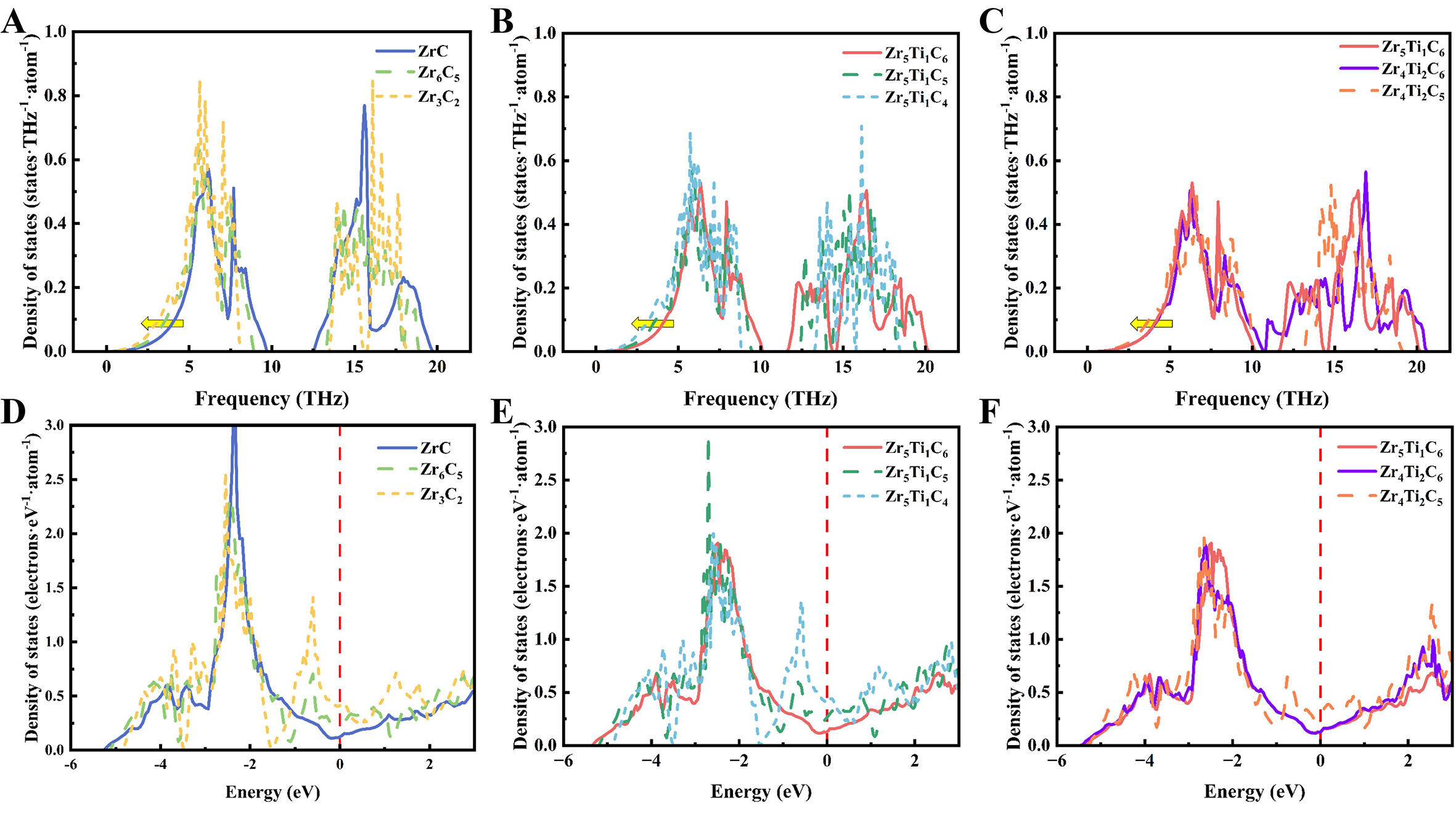
Figure 6. Density of states analysis for (Zr, Ti)Cx carbides. (A-C) Phonon density of states probing lattice dynamics; (D-F) Electronic density of states revealing electronic structure.
Moreover, the electronic density of states (DOS) in the designed carbides is also presented in Figure 6D-F to further clarify the effects of vacancies and Ti addition on the thermodynamic stability. Analysis of the total and partial density of states (TDOS and PDOS; see Figure 6D-F, Supplementary Figure 2D and Supplementary Figure 3) shows a non-zero DOS at the Fermi level (set to zero and marked by a vertical dashed line) for all carbides, confirming their metallic bonding character. Compared with the ZrC, the covalent-character-dominated TDOS peaks decrease considerably in Zr6C5 and Zr3C2 [Figure 6D], indicating weakened bonding interactions between C-p and Zr-d orbitals and a corresponding reduction in M-C bond strength. A new hybridization peak emerges near ~ -0.8 eV, attributed to the rearrangement of local electronic structure caused by C vacancies, which promotes Zr-Zr metallic bonding near vacancy sites. Furthermore, the increased DOS at the Fermi level implies enhanced metallicity and weaker bonding strength. Figure 6E and F further reveal that the intensity of C-p and Zr/Ti-d hybridization peaks decreases with higher Ti content, suggesting that the Ti substitution for Zr also weakens bonding strength and undermines structural stability. The evolution of the electronic structure elucidated by DOS analysis clarifies the origin of bond weakening due to both C vacancies and increased Ti content. This weakening leads to a higher binding energy and a decreased Young's modulus, consistent with the trends observed in
Formation of single-phase solid solution in multi-component carbides
The strong covalent bonding in carbides presents a formidable obstacle to the direct formation of single-phase FCC solid solution structure in multi-component systems from their binary counterparts. Therefore, the ability to form a single phase is a key indicator for evaluating the thermodynamic stability of multi-component carbides. The single-phase formability of (Zr, Ti)Cx was assessed by calculating the free energy change (∆F) associated with its formation from binary ZrCx and TiCx, as defined in Equation (11):
where y is the number of Ti atoms in the M6C6 metal sublattice, x is the number of C atoms, and T is the temperature. Figure 7A shows the temperature dependence of ∆F for multi-component carbides. At low temperatures, ∆F is positive for all carbides, suggesting that (Zr, Ti)Cx is unstable and difficult to form as a stable single-phase solid solution. As the temperature increases, ∆F decreases and eventually becomes negative, indicating the formation of a single-phase solid solution becomes feasible. For instance, Zr5Ti1C6 achieves the thermodynamic stability (∆F < 0) above 1,663 K. With one C vacancy, Zr5Ti1C5 stabilizes above 1,489 K, and with two vacancies, Zr5Ti1C4 stabilizes above 1,041 K, demonstrating that C vacancies significantly reduce the formation temperature. These computational results align well with existing experimental observations[60]. For instance, stoichiometric Zr0.8Ti0.2C1.0 failed to form a complete solid solution at 1,873 K, whereas non-stoichiometric (Zr, Ti)Cx (x = 0.7-0.9) readily forms a single-phase FCC structure, confirming the beneficial role of vacancies in promoting the formation of single-phase solid solution. Furthermore, a higher Ti content raises ∆F of multi-component carbides, necessitating increased synthesis temperature. Compared to low-Ti Zr5Ti1C6, the synthesis temperature for Zr4Ti2C6 increases to 1,758 K, indicating greater synthesis difficulty. This trend is consistent with experimental reports that higher Ti content complicates the synthesis[67,68]. However, introducing C vacancies substantially lowers the synthesis temperature, as in Zr4Ti2C5, where it drops to 1,515 K, thereby facilitating the synthesis process.
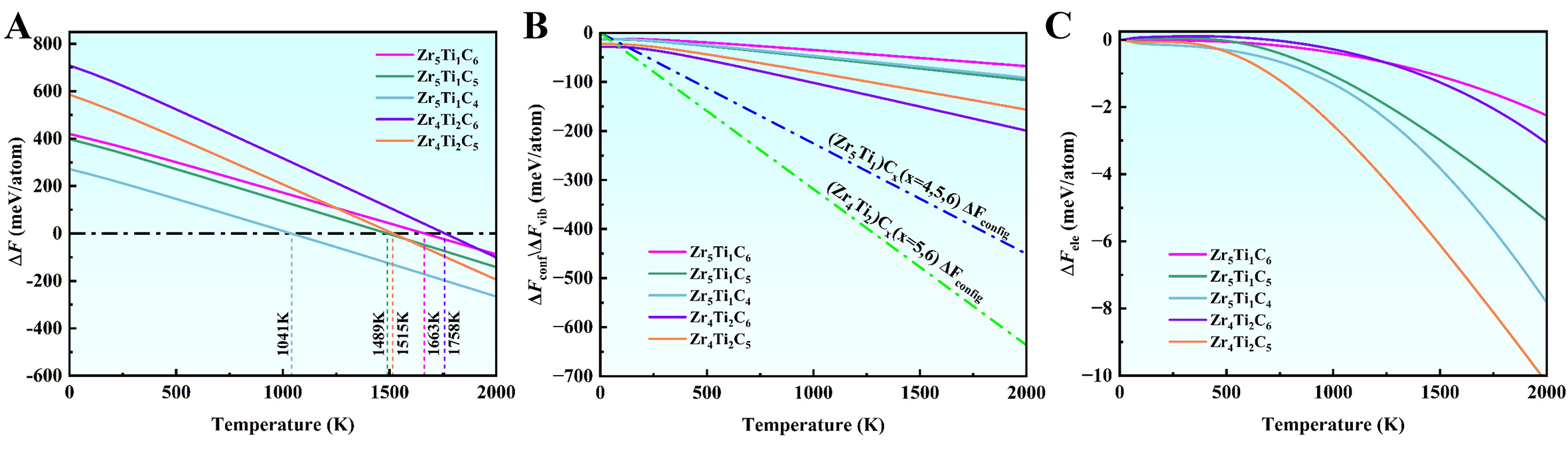
Figure 7. Free energy changes associated with the formation of single-phase (Zr, Ti)Cx solid solution from binary ZrCx and TiCx. (A) Helmholtz free energy change (ΔF); (B) Contributions from vibrational (∆Fvib) and configurational (∆Fconf); (C) Electronic free energy change (∆Fele).
Intrinsically, the single-phase formation ability of multi-component carbides is governed by the mixing enthalpy (∆H) between their constituent binary carbides (i.e., the total energy difference at 0 K), along with three free energy contributions: configurational (∆Fconf), vibrational (∆Fvib), and electronic (∆Fele) free energy changes. To further elucidate the single-phase formability of multi-component carbides, Figure 7 presents the individual contributions to the free energy change. C vacancies significantly reduce the ∆H, but the Ti addition increases it. Meanwhile, increased Ti content raises ∆Fconf, ∆Fvib, and ∆Fele [Figure 7B and C], thereby favoring a reduction in the total free energy [Figure 7A]. Comparative analysis shows that ∆H and ∆Fconf dominate, with contributions far exceeding those of ∆Fele and ∆Fvib. These results indicate that in multi-component carbide systems, the competition between mixing enthalpy and entropy (∆Fconf, ∆Fvib, and ∆Fele) collectively determines the temperature range for forming single-phase solid solution and the magnitude of thermodynamic driving force. This conclusion not only validates the reliability of computational approaches but also provides a theoretical basis for the rational design and optimization of non-stoichiometric multi-component carbides.
CONCLUSIONS
The present work systematically investigated the thermodynamic stability and elastic properties of binary (ZrC, Zr6C5, Zr3C2) and ternary (Zr5Ti1C6, Zr5Ti1C5, Zr5Ti1C4, Zr4Ti2C6, Zr4Ti2C5) carbides using first principles calculations. The main findings are summarized as follows:
(i) Based on the cluster-plus-glue-atom model representing the local CSROs of elemental distribution, the cluster structural unit of MC-type carbides was identified as [C-M6](C,□)5 (M = Zr/Ti, □ = vacancy), where vacancies occupy the glue atom sites along with C atoms. A 12-atom periodic supercell structure was then used as the input for the first principles calculations. The results demonstrate that introducing C vacancies reduces both the formation energy and free energy at HTs, thereby enhancing thermodynamic stability of FCC carbides. In contrast, Ti substitution for base Zr increases both energy terms, reducing stability. The variation in free energy is mainly governed by entropy contributions: C vacancies significantly raise the vibrational, electronic, and configurational entropies, lowering the free energy. Ti alloying, however, reduces the dominant vibrational entropy, leading to a net increase in free energy. Owing to the synergistic effect of C vacancies and Ti alloying, the [C-Zr5Ti1](C,□)5 (=Zr5Ti1C5) carbide exhibits the lowest formation energy and free energy among all compositions studied, indicating exceptional thermodynamic stability.
(ii) Analysis of the electronic and vibrational density of states was performed to elucidate the factors governing carbide stability. The phonon spectra confirm the dynamic stability of all modeled carbides, as no imaginary frequencies are present. C vacancies enhance the low-frequency phonon contribution, whereas Ti substitution shifts phonon states to higher frequencies due to its lower atomic mass. Changes in elastic properties are closely linked to electronic structure: both vacancies and Ti addition weaken the covalent-type TDOS peaks in the total density of states, reducing M-C bond strength, which in turn leads to increased binding energy and decreased Young’s modulus.
(iii) The single-phase formability of multi-component carbides was also evaluated. Results indicate that C vacancies significantly lower the synthesis temperature required to form single-phase FCC solid solution in (Zr, Ti)-C carbides from binary precursors, thereby improving the single-phase formability. This is attributed to the reduced mixing enthalpy in vacancy-containing carbides. Conversely, increasing Ti content raises the mixing enthalpy and synthesis temperature, making it more challenging to achieve single-phase solid solutions in multi-component (Zr, Ti)-C systems. Therefore, in (Zr, Ti)-C carbides, introducing an appropriate concentration of carbon vacancies while maintaining a higher content of Zr rather than Ti is of great importance for the high-temperature stability of materials in extremely high-temperature applications.
DECLARATIONS
Authors’ contributions
Made substantial contributions to conception and design of the study and performed data analysis and interpretation: Zhang, Q.; Niu, B.; Wang, Q.; Zhang, Z.
Performed data acquisition, as well as provided administrative, technical, and material support: Li, Z.;
Availability of data and materials
The data that support the findings of this study are available from the corresponding author upon reasonable request.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
Dong, C. is an Associate Editor of the journal Journal of Materials Informatics, but was not involved in any steps of editorial processing, notably including reviewer selection, manuscript handling, and decision making, while the other authors have declared that they have no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
Supplementary Materials
REFERENCES
1. Wyatt, B. C.; Nemani, S. K.; Hilmas, G. E.; Opila, E. J.; Anasori, B. Ultra-high temperature ceramics for extreme environments. Nat. Rev. Mater. 2023, 9, 773-89.
2. Xiang, H.; Xing, Y.; Dai, F.; et al. High-entropy ceramics: present status, challenges, and a look forward. J. Adv. Ceram. 2021, 10, 385-441.
3. Shojaie-Bahaabad, M.; Bozorg, M.; Najafizadeh, M.; Cavaliere, P. Ultra high temperature ceramic coatings in thermal protection systems (TPS). Ceram. Int. 2024, 50, 9937-51.
4. Lun, H.; Zeng, Y.; Xiong, X.; et al. Oxidation behavior of non-stoichiometric (Zr,Hf,Ti)Cx carbide solid solution powders in air. J. Adv. Ceram. 2021, 10, 741-57.
5. Harrington, T. J.; Gild, J.; Sarker, P.; et al. Phase stability and mechanical properties of novel high entropy transition metal carbides. Acta. Mater. 2019, 166, 271-80.
6. Wang, X. G.; Guo, W. M.; Kan, Y. M.; Zhang, G. J.; Wang, P. L. Densification behavior and properties of hot-pressed ZrC ceramics with Zr and graphite additives. J. Eur. Ceram. Soc. 2011, 31, 1103-11.
7. Yang, Q.; Wang, X.; Bao, W.; et al. Influence of equiatomic Zr/(Ti,Nb) substitution on microstructure and ultra-high strength of (Ti,Zr,Nb)C medium-entropy ceramics at 1900 °C. J. Adv. Ceram. 2022, 11, 1457-65.
8. Huang, S.; Dai, F.; Xiang, X.; et al. Strengthening or softening: on the impact of off-stoichiometry on the mechanical properties of ZrC. Acta. Mater. 2025, 289, 120892.
9. Dai, F.; Sun, Y.; Ren, Y.; Xiang, H.; Zhou, Y. Segregation of solute atoms in ZrC grain boundaries and their effects on grain boundary strengths. J. Mater. Sci. Technol. 2022, 101, 234-41.
10. Hossain, M. D.; Borman, T.; Oses, C.; et al. Entropy landscaping of high-entropy carbides. Adv. Mater. 2021, 33, e2102904.
11. He, L.; Liu, L.; Peng, F.; et al. Host lattice and solid solution formation in an octal-cation (NbTaZrTiHfVWMo)C high entropy carbide ceramic. J. Eur. Ceram. Soc. 2023, 43, 5792-801.
12. Deadmore, D. L. Vaporization of tantalum carbide-hafnium carbide solid solutions. J. Am. Ceram. Soc. 2006, 48, 357-9.
13. Han, X.; Girman, V.; Sedlak, R.; et al. Improved creep resistance of high entropy transition metal carbides. J. Eur. Ceram. Soc. 2020, 40, 2709-15.
14. Ye, B.; Wen, T.; Huang, K.; Wang, C. Z.; Chu, Y. First‐principles study, fabrication, and characterization of (Hf0.2Zr0.2Ta0.2Nb0.2Ti0.2)C high‐entropy ceramic. J. Am. Ceram. Soc. 2019, 102, 4344-52.
15. Zeng, Y.; Wang, D.; Xiong, X.; et al. Ablation-resistant carbide Zr0.8Ti0.2C0.74B0.26 for oxidizing environments up to 3,000 °C. Nat. Commun. 2017, 8, 15836.
16. Sheindlin, M.; Falyakhov, T.; Petukhov, S.; Valyano, G.; Vasin, A. Recent advances in the study of high-temperature behaviour of non-stoichiometric TaCx, HfCx and ZrCx carbides in the domain of their congruent melting point. Adv. Appl. Ceram. 2018, 117, s48-55.
17. Liu, D.; Hou, Y.; Meng, J.; Zhang, A.; Han, J.; Zhang, J. The significant influence of carbon content on mechanical and thermal properties of (VNbTaMoW)0.5Cx high entropy carbides. J. Eur. Ceram. Soc. 2022, 42, 5262-72.
18. Schmid, B.; Koutná, N.; Ntemou, E.; et al. Mechanical properties of VC/ZrC and VC/HfC superlattices. Acta. Mater. 2024, 270, 119852.
19. Wang, Z.; Yu, Z.; Zhang, L.; et al. Tailoring structural, mechanical, and electronic properties of (Ti0.25Ta0.25Zr0.25Nb0.25-La )C high-entropy carbide ceramics via rare-earth La incorporation: a comprehensive first-principles study. Ceram. Int. 2025, 51, 33721-30.
20. Chen, L.; Kong, Q.; Liu, Q.; et al. Covalent bonds enhancing and microstructure evolution induced by carbon content in multi-component (TiZrNbMo)C ceramics. J. Materiomics. 2025, 11, 101048.
21. Zhang, Y.; Liu, B.; Wang, J.; Wang, J. Theoretical investigations of the effects of ordered carbon vacancies in ZrC1-x on phase stability and thermo-mechanical properties. Acta. Mater. 2016, 111, 232-41.
22. Zunger, A.; Wei, S.; Ferreira, L. G.; Bernard, J. E. Special quasirandom structures. Phys. Rev. Lett. 1990, 65, 353-6.
23. Kim, J. First-principles investigation of the elastic properties and phase stability of (Ti1-xNix)C1-y ternary metastable carbides. J. Alloys. Compd. 2021, 853, 157349.
24. Liu, S.; Qin, L.; Zhang, H.; et al. Design of superhard high-entropy diborides via high-throughput DFT and thermodynamics calculations. Ceram. Int. 2024, 50, 17977-87.
25. Liu, S.; Zhang, S.; Liu, S.; et al. Stability and mechanical properties of single-phase quinary high-entropy metal carbides: first-principles theory and thermodynamics. J. Eur. Ceram. Soc. 2022, 42, 3089-98.
26. Liu, Y.; Lu, Y.; Wang, W. Y.; et al. Effects of solutes on thermodynamic properties of (TMZrU)C (TM = Ta, Y) medium-entropy carbides: a first-principles study. J. Mater. Inf. 2023, 3, 17.
27. Meng, H.; Chu, Y. Surface energies in high‐entropy carbides with variable carbon stoichiometry. J. Am. Ceram. Soc. 2022, 105, 5835-42.
28. Ma, Y.; Wang, Q.; Jiang, B.; et al. Controlled formation of coherent cuboidal nanoprecipitates in body-centered cubic high-entropy alloys based on Al2(Ni,Co,Fe,Cr)14 compositions. Acta. Mater. 2018, 147, 213-25.
29. Yuan, J.; Li, Z.; Yang, Y.; et al. Applications of machine learning method in high-performance materials design: a review. J. Mater. Inf. 2024, 4, 14.
30. Wang, W. Y.; Shang, S. L.; Wang, Y.; et al. Atomic and electronic basis for the serrations of refractory high-entropy alloys. NPJ. Comput. Mater. 2017, 3, 23.
31. Bie, X.; Hou, J.; Zhou, X.; Song, J. Sub-stoichiometry and vacancy structures in V/Nb carbide precipitates by cluster expansion and first-principles calculations. Acta. Mater. 2024, 269, 119806.
32. Hu, W.; Xiang, J.; Zhang, Y.; et al. Superstructural nanodomains of ordered carbon vacancies in nonstoichiometric ZrC0.61. J. Mater. Res. 2012, 27, 1230-6.
33. Gusev, A. I. Phase equilibria, phases and compounds in the Ti-C system. Russ. Chem. Rev. 2002, 71, 439-63.
34. Gusev, A. I.; Rempel, A. A. Order-disorder phase transition channel in niobium carbide. Phys. Stat. Sol. 1986, 93, 71-80.
35. Xiang, J.; Hu, W.; Liu, S.; et al. Spark plasma sintering of the nonstoichiometric ultrafine-grained titanium carbides with nano superstructural domains of the ordered carbon vacancies. Mater. Chem. Phys. 2011, 130, 352-60.
36. Hong, Q.; Van, De. Walle. A. Prediction of the material with highest known melting point fromab initiomolecular dynamics calculations. Phys. Rev. B. 2015, 92, 020104.
37. Gusev, A. I.; Rempel, A. A. Superstructures of non-stoichiometric interstitial compounds and the distribution functions of interstitial atoms. Phys. Stat. Sol. 1993, 135, 15-58.
38. Zeng, Y.; Xiong, X.; Li, G.; Chen, Z.; Sun, W.; Wang, D. Microstructure and ablation behavior of carbon/carbon composites infiltrated with Zr-Ti. Carbon 2013, 54, 300-9.
39. Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15-50.
40. Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B. Condens. Matter. 1996, 54, 11169-86.
41. Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B. 1999, 59, 1758-75.
42. Perdew, J. P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865-8.
43. Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272-6.
44. Wang, V.; Xu, N.; Liu, J.; Tang, G.; Geng, W. VASPKIT: a user-friendly interface facilitating high-throughput computing and analysis using VASP code. Comput. Phys. Commun. 2021, 267, 108033.
45. Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456-65.
46. Ma, D.; Grabowski, B.; Körmann, F.; Neugebauer, J.; Raabe, D. Ab initio thermodynamics of the CoCrFeMnNi high entropy alloy: importance of entropy contributions beyond the configurational one. Acta. Mater. 2015, 100, 90-7.
47. Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1-5.
48. Togo, A.; Chaput, L.; Tanaka, I.; Hug, G. First-principles phonon calculations of thermal expansion in Ti3SiC2,Ti3AlC2, and Ti3GeC2. Phys. Rev. B. 2010, 81, 174301.
49. Olsson, P.; Blomqvist, J.; Bjerkén, C.; Massih, A. Ab initio thermodynamics investigation of titanium hydrides. Comput. Mater. Sci. 2015, 97, 263-75.
50. Ye, B.; Wen, T.; Nguyen, M. C.; Hao, L.; Wang, C.; Chu, Y. First-principles study, fabrication and characterization of (Zr0.25Nb0.25Ti0.25V0.25)C high-entropy ceramics. Acta. Mater. 2019, 170, 15-23.
51. Page Y, Saxe P. Symmetry-general least-squares extraction of elastic data for strained materials fromab initio calculations of stress. Phys. Rev. B. 2002, 65, 104104.
52. Wu, X.; Vanderbilt, D.; Hamann, D. R. Systematic treatment of displacements, strains, and electric fields in density-functional perturbation theory. Phys. Rev. B. 2005, 72, 035105.
53. Zakarian, D.; Khachatrian, A.; Firstov, S. Universal temperature dependence of Young’s modulus. Metal. Powder. Rep. 2019, 74, 204-6.
54. Réjasse, F.; Trolliard, G.; Léchelle, J.; et al. Study of the TiC1-x - TiO2 reactive interface. Acta. Mater. 2018, 146, 225-36.
55. Luo, Z.; Du, Y.; Mao, H.; Tang, S.; Peng, Y.; Liu, Z. Phase field simulation of the lamellar precipitation in the TiC-ZrC system. Ceram. Int. 2018, 44, 22041-4.
56. Lei, J.; Okimura, H.; Brittain, J. O. The electrical resistance of the group IV transition metal monocarbides and mononitrides in the temperature range 20-1000 °C. Mater. Sci. Eng. A. 1990, 123, 129-40.
57. Mahday, A. A.; Sherif, El-Eskandarany. M.; Ahmed, H.; Amer, A. Mechanically induced solid state carburization for fabrication of nanocrystalline ZrC refractory material powders. J. Alloys. Compd. 2000, 299, 244-53.
59. Zhou, Y.; Watts, J.; Li, C.; Fahrenholtz, W. G.; Hilmas, G. E. Vacancy ordering in zirconium carbide with different carbon contents. J. Eur. Ceram. Soc. 2023, 43, 5814-21.
60. Lun, H.; Yuan, J.; Zeng, Y.; Xiong, X.; Wang, Q.; Ye, Z. Mechanisms responsible for enhancing low‐temperature oxidation resistance of nonstoichiometric (Zr,Ti)C. J. Am. Ceram. Soc. 2022, 105, 5309-24.
61. Lu, W.; Xu, J.; Huang, S.; et al. The coupling of carbon non-stoichiometry and short-range order in governing mechanical properties of high-entropy ceramics. NPJ. Comput. Mater. 2025, 11, 64.
62. Kim, H.; Kim, M.; Kim, J.; Kim, J. Enhancing the hydrogen storage properties of (Ti, M)C1-x materials (M = W, Mg, Ni, and Al) depends on the carbon vacancies: potential for the recycling of scraps. Int. J. Refract. Met. Hard. Mater. 2024, 118, 106475.
63. Baranov, V. M.; Knyazev, V. I.; Korostin, O. S. The temperature dependence of the elastic constants of nonstoichiometric zirconium carbides. Strength. Mater. 1973, 5, 1074-7.
64. Sangiovanni, D.; Tasnádi, F.; Harrington, T.; Odén, M.; Vecchio, K.; Abrikosov, I. Temperature-dependent elastic properties of binary and multicomponent high-entropy refractory carbides. Mater. Des. 2021, 204, 109634.
65. Evitts, L.; Middleburgh, S.; Kardoulaki, E.; Ipatova, I.; Rushton, M.; Lee, W. Influence of boron isotope ratio on the thermal conductivity of uranium diboride (UB2) and zirconium diboride (ZrB2). J. Nucl. Mater. 2020, 528, 151892.
66. Esters, M.; Oses, C.; Hicks, D.; et al. Settling the matter of the role of vibrations in the stability of high-entropy carbides. Nat. Commun. 2021, 12, 5747.
67. Markström, A.; Andersson, D.; Frisk, K. Combined ab-initio and experimental assessment of A1-xBxC mixed carbides. Calphad 2008, 32, 615-23.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].