


Myocardial energy metabolism in aging
 0
0 Abstract
The natural process of aging in humans often increases one’s risk for a number of chronic diseases, including type 2 diabetes (T2D), dyslipidemia, hypertension, and cardiovascular disease (CVD). It is increasingly recognized that aging-related CVD in the absence of other confounding risk factors, such as obesity and T2D, has unique features. Although aging is accompanied by various molecular and physiological changes ultimately affecting whole-body homeostasis, alterations in myocardial energy metabolism are a common hallmark of CVD in elderly people. Under normal physiological conditions, the hearts of healthy individuals oxidize fatty acids, glucose, ketones, and amino acids to meet their energy demand. However, the relative contribution of these fuels for myocardial energy production changes during aging, including a decrease in fatty acid oxidation and an increase in overall glucose utilization (glucose uptake and glycolysis in particular). The heart is also associated with mitochondrial structural and functional abnormalities, resulting in the accumulation of reactive oxygen species and redox-regulated signaling that can exacerbate damage to oxidative phosphorylation capacity and aggravate cardiac dysfunction. We herein discuss the primary changes in myocardial energy metabolism and mitochondrial structure and function, as well as alterations in key molecular mediators that ensue during the physiological process of aging, while considering their potential impact on cardiac function. We have also highlighted the need for comprehensive clinical trials of potential lifestyle or established pharmacological interventions to attenuate myocardial energy metabolism and improve cardiac health in the setting of aging, which may lead to a healthy lifespan.
Keywords
INTRODUCTION
Aging is a natural process, accompanied by structural and functional changes in various organs including the heart[1]. Although recent developments in pharmacological and surgical treatments have improved life expectancy, the incidence of age-associated cardiovascular diseases (CVDs) has increased dramatically with high hospitalization rates and elevated mortality rates in elderly individuals[2,3]. Even without disease, aging is accompanied by cardiac cell damage aggravated by abnormal energy metabolism, alterations in mitochondrial structure and function, and oxidative stress resulting in cardiac fibrosis, hypertrophy, vascular remodeling, insufficient coronary perfusion/oxygenation, and declining cardiac function[4-7]. However, conventional suggestions of increased left ventricular mass during aging have been contradicted by considerable evidence of reduced cardiac mass in older adults in the absence of hypertension[8]. The association of cardiac complications with alterations in myocardial energy metabolic pathways has been extensively interrogated by researchers over the past century, where it is clear that in the context of other chronic pathologies such as obesity or type 2 diabetes (T2D), these alterations contribute to CVD risk[9,10]. Because aging also increases risk for many of these chronic pathologies, it presents a challenge in determining whether alterations in myocardial energy metabolism can directly precipitate CVD. Furthermore, aging-associated frailty, sarcopenia, inactivity, malnutrition, and poor sleeping patterns are also confounding factors that promote cardiometabolic abnormalities that increase the risk of CVD in elderly people[11].
To sustain continuous contractile activity, the heart relies heavily on the continuous production of adenosine triphosphate (ATP) by mitochondrial oxidative phosphorylation (OXPHOS). In elderly individuals, it is reasonable to presume that altered energy metabolism resulting in an energy deficit in the heart could contribute to their increased risk of CVD[12]. The compromised myocardial energy production results from several factors, including impaired mitochondrial OXPHOS and structural dynamics, decreased activity of electron transport chain (ETC) complexes, and alterations in energy substrate preference by the heart[13]. As every substrate for the heart matters due to its omnivore-like nature, the metabolic inflexibility that manifests in the heart during aging precipitates redox alterations that activate pathways that may contribute to ventricular dysfunction[14,15]. In this review, we aim to address the underlying mitochondrial structural and functional changes that develop in the aged heart. Furthermore, we will interrogate current knowledge of the specific perturbations of myocardial energy metabolism in aging as it relates to fatty acid, glucose, ketone, and amino acid utilization.
MITOCHONDRIAL ABNORMALITIES IN THE AGED MYOCARDIUM
Due to its enormous energy demands, the heart is highly susceptible to mitochondrial anomalies. Several preclinical and clinical studies have reported larger mitochondria with disrupted structure and function in the heart during aging (extensively reviewed in[13]), while cardiac mitochondria from aged rats (100 weeks of age) showed a significant increase in superoxide radical production with enhanced lipid peroxidation[16]. Furthermore, manganese superoxide dismutase (MnSOD), which transforms toxic superoxide to hydrogen peroxide to prevent accumulation of mitochondrial reactive oxygen species (ROS), is reduced in the hearts of aged (20-months of age) mice[17]. Sirtuin 3 (SIRT3), a deacetylase primarily localized in the mitochondria involved in regulating several physiological and pathophysiological processes, is downregulated in the heart during aging, resulting in suppressed MnSOD activity leading to increased ROS levels (extensively reviewed in[18]). Similarly, previous studies observed an increased expression of acetylated-MnSOD levels, which correlated with decreased SIRT3 activity in the hearts of aged (15-18 months of age) compared to young (2-4 months of age) male and female mice[19]. Marked alterations in mitochondrial ultrastructure, observed by a decreased cristae density, disturbed arrangement in the myofibrillar spaces, and enlarged size, were also observed in the hearts of aged male and female mice[19]. In addition, a recent mitochondrial multiscale 3D analysis at micro- and nano-resolution reported decreased expression of the cristae-remodeling protein optic atrophy 1 and linked remodeled cristae to reduced OXPHOS capacity in hearts of aged mice[20]. Thus, the reduced expression of optic atrophy 1 may serve as an early marker of overt mitochondrial remodeling in the heart during aging. Moreover, the removal capacity of dysfunctional and distorted mitochondria by mitophagy and the dynamic equilibrium of mitochondrial fusion and fission are also altered in aging[13]. Increases in cardiac mitochondrial protein carbonylation have also been observed in mice at the age of 24 months, which aggravates mitochondrial oxidative damage[7]. However, others have reported increased protein carbonylation levels in the cardiac tissues of aged (15-18 months of age) male but not female mice[19]. As mitochondrial DNA lacks protective histones, it is also vulnerable to mutations leading to oxidative damage, telomere shortening, and necrosis[21]. Telomere shortening, epigenetic modifications, mitochondrial dysfunction, and oxidative stress are known contributors to cellular senescence in the heart, a condition characterized by stable, irreversible cell cycle arrest linked to aging (extensively reviewed in[22,23]).
Paralleling observations in humans with heart failure, mitochondrial biogenesis is also compromised in the aged heart. Myocardial protein expression of peroxisome proliferator-activated receptor γ (PPARγ) coactivator 1-α (PGC1α), which stimulates mitochondrial biogenesis, is lower in aged animals[24]. The loss of telomeres represents an intrinsic driver of aging-associated multi-organ dysfunction and is considered a reliable model of aging that mimics the human aging process[25]. Accordingly, the telomerase-deficient mouse model exhibits declined PGC1α expression and compromised mitochondrial function when compared to their wild-type littermates at 12 months of age[26]. Intriguingly, cardiac-specific PGC1α overexpression in telomerase-deficient mice restored mitochondrial homeostasis, which alleviated aging-related declines in cardiac function. However, cardiac-specific PGC1α overexpression in wild-type mice accelerated aging with mitochondrial damage and ROS accumulation, resulting in shortened life span[26]. In other studies, moderate PGC1α activation inhibits age-related cardiac remodeling with increases in gene expression of several markers involved in mitochondrial biogenesis, metabolism, and myocardial contractility[27]. Hence, there appears to be a delicate balance between myocardial PGC1α activity and mitochondrial homeostasis during aging.
The interplay between mitophagy and biogenesis plays a key role in maintaining cardiac mitochondrial homeostasis, and during aging, impaired fission, fusion, and lysosomal degradation can further aggravate mitochondrial function in the heart[13]. Mechanistically, accumulation of dysfunctional mitochondria in the heart during aging is thought to be a result of decreased capacity for mitophagy[28]. In support of this, deletion of Pten-inducible kinase 1, a crucial mitochondrial kinase for maintaining quality control of mitochondria by mitophagy, leads to an age-dependent accumulation of damaged and dysfunctional mitochondria in mice, resulting in cardiac dysfunction[29]. In addition, mitochondrial ubiquitination decreases during aging, which leads to impaired mitophagy and removal of abnormal mitochondria in the heart, leading to increased cell damage[30]. Thus, accumulation of dysfunctional or distorted mitochondria due to ROS, mitochondrial DNA mutations, and defective ATP synthesis may contribute to cardiac dysfunction during aging.
MYOCARDIAL ENERGY METABOLISM DURING AGING
As previously stated, to maintain continuous contractile function, the adult healthy heart generates enormous quantities of ATP primarily from mitochondrial OXPHOS and aerobic glycolysis (see review[15]). The majority of myocardial ATP requirements (~95%) are fulfilled by mitochondrial OXPHOS and the remaining is met via glycolysis (~5%). In terms of fuel contribution, myocardial ATP production primarily relies on the oxidation of fatty acids (~40%-60%), with the remainder derived from the oxidation of glucose (~20%-40%), ketones (~10%-15%), and amino acids (~1%-2%) [Figure 1]. Moreover, the healthy heart is “metabolically flexible” in its ability to readily utilize the different substrates continuously provided to it via the coronary circulation to maintain ATP production throughout various physiological states (e.g., fasting, nutrient ingestion)[31]. Aging is accompanied by several degenerative changes in the myocardium with reduced organelle function in cardiomyocytes, which leads to a gradual decline in normal physiological function[13,14]. Although the effect of aging on myocardial metabolic flexibility is incompletely understood, a key alteration is the shift in the preference of fuel substrate for myocardial energy production. Below, we outline the major alterations in myocardial energy metabolism and their potential contribution(s) to the increased risk of CVD in elderly people [Figure 1]. Wherever relevant we will state the ages of the animals or humans studied, unless those details have not been clearly stated within the studies referenced herein.
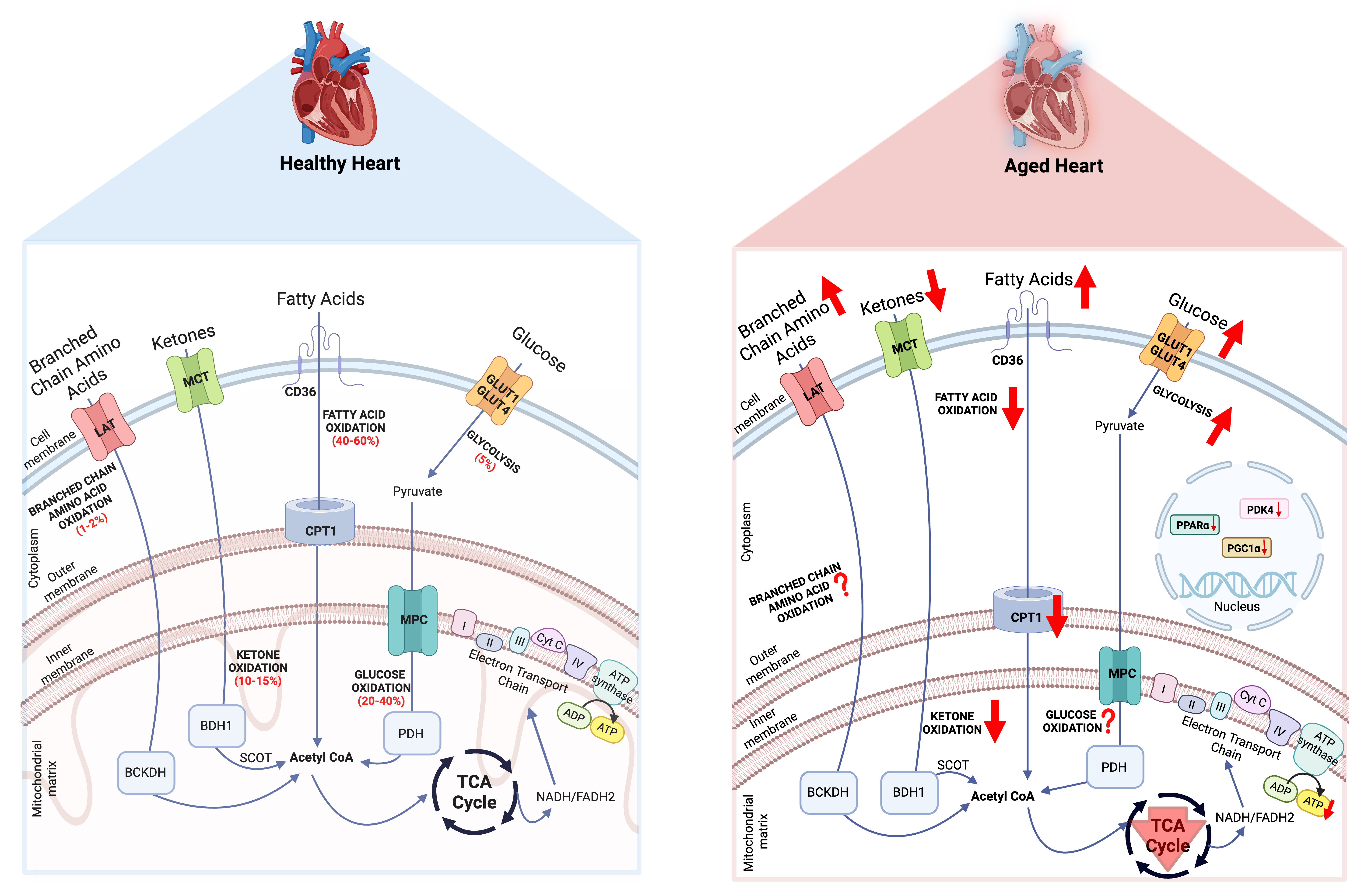
Figure 1. Energy metabolism in the healthy and aged heart. The red arrow facing up indicates an increase, and the arrow facing down indicates a decrease. PDK4: Pyruvate dehydrogenase kinase 4; PGC1α: PPAR gamma coactivator 1-alpha; PPARα: peroxisome proliferator-activated receptor α; PDH: pyruvate dehydrogenase; CPT1: carnitine palmitoyl transferase 1; BDH1: β-hydroxybutyrate dehydrogenase 1; LAT: L-type amino acid transporters; MPC: mitochondrial pyruvate carrier; MCT: monocarboxylate transporter; BCKDH: branched-chain alpha-keto acid dehydrogenase; NADH: nicotinamide adenine dinucleotide; SCOT: succinyl-CoA:3-ketoacid-CoA transferase. Created in BioRender. Seubert, J. (2026) https://BioRender.com/2piydk5.
Glucose is transported into cardiomyocytes via glucose transporter (GLUT) 1 or 4, followed by glycolysis to produce pyruvate. Pyruvate is transported into the mitochondria via the mitochondrial pyruvate carrier (MPC) and is converted by pyruvate dehydrogenase (PDH) to acetyl-coenzyme A (CoA). Fatty acids are transported into the cells via fatty acid transporter (CD36), esterified to CoA and then shuttled to mitochondria by carnitine palmitoyl transferase 1 (CPT1), which can then undergo β-oxidation to produce acetyl-CoA. Ketones are transported into the cell via monocarboxylate transporter, where β-hydroxybutyrate dehydrogenase 1 and succinyl-CoA:3-ketoacid-CoA transferase (SCOT) catalyze the oxidation of ketones to produce acetyl-CoA. Branched-chain amino acids are transported into the cell by L-type amino acid transporters and are catalyzed by branched-chain alpha-keto acid dehydrogenase to acetyl-CoA. The acetyl-CoA generated by oxidation of glucose, fatty acids, ketones, and branched-chain amino acids goes to the tricarboxylic acid cycle to produce nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2), which then enter the ETC to generate ATP. During aging, alterations in circulating glucose, fatty acids, ketones, and branched-chain amino acids lead to alterations in their oxidation with changes in expression of their transporters and key regulators.
Fatty acid metabolism during aging
With aging, the heart undergoes significant metabolic remodeling, particularly in fatty acid metabolism, which is the primary energy source in the adult heart. Myocardial fatty acid oxidation rates decrease with the progression of age. For example, studies in isolated working hearts perfused at normal (50 mm Hg) and higher (80 mm Hg) workloads demonstrate reductions in myocardial palmitate oxidation in older mice (52-54 weeks of age) with systolic dysfunction and cardiac hypertrophy compared to young mice
The heart receives its fatty acid supply either as non-esterified fatty acids (NEFA) bound to albumin or from the hydrolysis of triacylglycerol (TAG)-containing lipoproteins, mediated by lipoprotein lipase[39]. Fatty acid metabolism in the myocardium involves fatty acid uptake into the cardiomyocyte, followed by esterification to CoA and transport of the fatty acid into the mitochondria for subsequent β-oxidation, though fatty acyl CoAs may also be stored as TAGs[39,43]. Finally, reducing equivalents (NADH, FADH2) produced from both β-oxidation and the Krebs cycle from fatty acid oxidation-derived acetyl-CoA feed into OXPHOS for ATP production[39,43].
During aging, elevated concentrations of circulating NEFAs were found in starved 60-week-old male fasted Wistar rats compared to younger rats at 7 and 35 weeks of age[44]. In addition, studies utilizing larger animal models such as male dogs at the age of 10-12 years also reported elevated concentrations of circulating NEFAs versus younger male dogs at 3-4 years of age[45]. In aged humans, circulating NEFA levels generally tend to be higher compared to younger individuals[46], though it can vary based on overall health. Some studies report a strong correlation between circulatory NEFA levels and sudden death in middle-aged men (42-53 years)[47], whereas others show no association between circulating NEFAs and the risk of sudden cardiac death in older men and women (mean age, 75 years)[48]. Nevertheless, circulating NEFA levels are highly associated with frailty, disability, and mobility limitation among men and women aged ≥ 65 years[46]. Similarly, elevated circulating TAG levels have been reported in older male and female rats (1 year of age) compared to younger rats (3 months of age)[49]. Interestingly, studies utilizing 52-54 week old mice reported no major differences in circulating TAG levels compared to 10-12 weeks old mice[32]. Moreover, 22-month-old mice appear to have slightly lower circulating TAG levels compared to 3-month-old mice[50]. In the healthy adult heart, 80% of the fatty acids taken up by the myocardium are oxidized to CO2 in the mitochondria and the remaining 20% are presumably converted to TAG[51]. In accordance, higher intramyocardial TAG levels have been strongly correlated with aging-associated cardiac diastolic
Cardiomyocyte fatty acid uptake is primarily dependent on sarcolemmal membrane transporters such as CD36 and fatty acid transport proteins (FATP)[39,54], of which the former may contribute to elevations in intramyocardial TAG content during aging[32]. In accordance, increased CD36 protein levels have been observed in cardiac tissues of C57BL/6 male mice at the age of 52-54 weeks compared to young mice at the age of 10-12 weeks[32]. Furthermore, CD36-deficient mice (52-54 weeks of age) have lower intramyocardial TAG, higher mitochondrial ATP production, and improved cardiac function versus their age-matched wild-type littermates. However, myocardial tissues collected during mitral valve replacement or heart transplant from middle-aged/aged individuals (> 40 years; median age, 61) exhibited a reduction in CD36 protein levels[55]. These findings suggest that the severity of CVD with comorbidities in aged individuals could affect cellular fatty acid transport by either up- or down-regulation of CD36 protein levels. Nevertheless, CD36 upregulation and elevated myocardial fatty acid uptake seem to be detrimental for cardiac health during aging. Moreover, there is consensus that the activity of CPT1, the rate-limiting enzyme for long-chain fatty acid transport into mitochondria for β-oxidation, decreases with aging in the heart[56-58]. The reduction in cardiac CPT1 activity for palmitoyl-CoA utilization has been observed to be specifically localized to the interfibrillar (prominently involved in ATP production) but not subsarcolemmal fraction of mitochondria in older Fischer 344 rats (24-28 months of age) compared to young rats (3-4 months of age)[58]. These observations suggest that even with elevations in myocardial fatty acid uptake, mitochondrial fatty acid uptake and subsequent oxidation reduce with aging.
The age-dependent decline in myocardial fatty acid utilization and ATP production also correlates with a lower expression of myocardial peroxisome proliferator-activated receptor α (PPARα), a key transcription factor regulating myocardial energy metabolism[59,60]. The importance of PPARα-associated reductions of myocardial fatty acid oxidation in the acceleration of cardiac abnormalities such as myocardial damage, cardiac fibrosis, inflammation, and abnormal myocardial cell growth has been demonstrated in PPARα-null mice[61]. Additionally, cardiac abnormalities along with decreased ATP production, abnormal mitochondrial cristae, and fibrosis were observed in the myocardium of PPARα knockout (KO) mice at the age of 16 weeks, which progressed further with aging[61] and strongly correlated with decreased longevity[62]. Of interest, cardiomyocyte-specific human aldose reductase (αMHC-hAR) overexpressing transgenic mice at the age of 12 months exhibit reduced cardiac function, which was associated with reduced myocardial expression of PPARα and increased PPARγ signaling[63,64]. This corresponded with decreased myocardial fatty acid oxidation and lipid accumulation, the latter of which was characterized by increased TAG, ceramide, and acylcarnitine levels[63,64]. Furthermore, genetic elimination of PPARα in αMHC-hAR mice resulted in the earlier onset of cardiac dysfunction at the age of 7 months[63]. Although reduced cardiac PPARα expression has been associated with aging-related cardiac dysfunction and reduced myocardial fatty acid oxidation, the potential beneficial actions of these changes have not been fully elucidated. Nevertheless, the imbalance between fatty acid uptake and oxidation leads to excess intracellular toxic lipid species (including ceramides and diacylglycerols) and eventually causes myocardial lipotoxicity during aging[36,65,66]. Although extensive evaluation is still needed to strengthen these conclusions, the alterations in myocardial fatty acid oxidation along with lipotoxicity may precipitate myocardial metabolic inflexibility during aging.
Glucose metabolism during aging
Metabolic pathways for different substrates are highly interconnected, with extensive intracellular cross-talk that coordinates fuel oxidation to ensure that the myocardium’s enormous energetic demand is met to support contractile function. This metabolic cross-talk underlies the “Glucose-Fatty Acid cycle”, in the heart, muscle, and adipose tissue, now frequently referred to as the “Randle cycle”[67]. Although absolute rates of myocardial glucose utilization in aged individuals do not increase following the decline in myocardial fatty acid oxidation[36], there may be an increase in the relative contribution of glucose to overall energy metabolism.
Unlike the young adult heart, the heart of an aged individual experiences higher glucose utilization towards energy production with a robust increase in glycolysis and similar rates of glucose oxidation[68,69]. Hearts from male Fisher 344 rats at the age of 24 months showed elevated myocardial glycolytic rates estimated by measuring 3H2O production from [5-3H]glucose during isolated working heart perfusion compared to hearts from younger rats at the age of 5 months[70]. Although the activity of enzymes involved in metabolic pathways generally decreases during aging, phosphofructokinase, the rate-limiting enzyme of glycolysis, was not significantly affected by age in male Wistar-Furth rats at the age of 31 months[71]. However, the activity of lactate dehydrogenase, another key enzyme in anaerobic metabolism, decreases in the hearts of rats aged 26 and 31 months[71]. Moreover, hearts from 52-54 week old male C57BL/6 mice demonstrated reduced myocardial glucose oxidation rates as estimated by 14CO2 production from [14C] glucose during isolated working heart perfusions[32]. Conversely, PET imaging using [11C]glucose in 14 older healthy individuals (mean age, 69 ± 4 years; 9 men and 6 women) under resting conditions showed higher myocardial glucose utilization compared to 16 younger healthy individuals (mean age, 26 ± 5 years; 5 men and 11 women)[72]. However, decreases in circulating estradiol levels are associated with cardiovascular events in women during postmenopause (median age, 50 years)[73], and myocardial glucose utilization was found to be similar in postmenopausal older women (mean age, 68 ± 4 years) compared to age-matched older men[72]. Notably, due to the limitation of PET imaging with [11C]glucose, it is challenging to differentiate the involvement of glycolysis or glucose oxidation in the observed increase in myocardial glucose utilization in older individuals. Moreover, dobutamine infusion, a well-characterized modulator of myocardial substrate metabolism[74,75], showed higher myocardial glucose utilization in younger healthy individuals but a blunted response in older healthy individuals[72]. When considering the fact that the cardiac contractile response to dobutamine diminishes with age[76], the metabolic response to dobutamine infusion observed in older individuals may reflect lower cardiac work. In the context of myocardial ATP production, it is unlikely that glycolysis can compensate for impaired glucose and fatty acid oxidation, thereby resulting in a persistent energy deficit and aberrant cardiac contraction during aging. Furthermore, a recent study utilizing transgenic mice expressing a mutated form of the phosphofructokinase-2/fructose bisphosphatase-2 that lacks fructose bisphosphatase-2 activity concluded that despite enhanced cardiac glycolysis, there was no acceleration of the cardiac aging phenotype[77]. Thus, increases in myocardial glycolytic capacity could be an adaptive rather than maladaptive metabolic alteration in the heart during aging.
During aging, blood glucose levels increase due in part to compromised GLUT mediated glucose uptake. For example, studies have reported decreased GLUT4 expression in the myocardium of rats at the age of 25 months[78,79], whereas other studies have observed an increase in myocardial GLUT4 protein expression in aged female C57BL/6 mice (22.5-months, 25-months, and 29-months [senescent]) compared to young female (5-month-old) mice[80]. Similarly, an elevation of GLUT4 protein expression was also observed in the hearts of senescence-accelerated male and female mice, a murine model of accelerated aging with short life span, deterioration in skin glossiness, learning, and memory[81]. However, the expression of GLUT1 protein was unaffected in myocardial extracts of aged (25 months) and senescent (4-8 weeks and 29 months) mice[80,81]. Of interest, Luptak and colleagues reported sustained elevations of glucose uptake and utilization in the hearts of 2-year-old transgenic mice with cardiac-specific overexpression of GLUT1 (GLUT1-TG)[82]. The left ventricular (LV) pressure-volume relationship for end-diastolic pressure suggests an impaired diastolic function in wild-type but not in GLUT1-TG hearts at the age of 22 months. Moreover, tolerance to ischemia-reperfusion injury was markedly improved in hearts from 22-month-old GLUT1-TG mice. Thus, lifelong overexpression of GLUT1 and sustained glucose uptake and utilization may confer cardioprotective properties in the aged heart.
Glycolytically-derived pyruvate has several metabolic fates, with 2 of the most important in the heart being conversion into lactate via lactate dehydrogenase, or transport into the mitochondrial matrix by the MPC for oxidation. In the hearts of healthy adult individuals where oxygen is not limiting, pyruvate oxidation via the mitochondrial pathway predominates, a process primarily controlled by PDH, the rate-limiting enzyme of glucose oxidation[83]. PDH is part of a multienzyme complex that decarboxylates pyruvate into acetyl-CoA, which enters the Krebs Cycle to generate reducing equivalents for OXPHOS in the mitochondrial ETC[43]. PDH activity is regulated by 4 isoforms of PDH kinase (PDK1-4) that respond to metabolic intermediates from glycolysis and fatty acid oxidation. Increases in NADH and acetyl-CoA derived from fatty acid oxidation stimulate PDKs, resulting in phosphorylation-mediated inhibition of PDH, whereas increased pyruvate from glycolysis deactivates PDKs, thereby activating PDH[84].
The reduction of myocardial fatty acid oxidation during aging could trigger a lowering of PDK expression in the heart. Indeed, PDK4 mRNA levels decrease by 57% and correlate with a 45% decrease in PDH phosphorylation in the hearts of 28-month-old F344 rats vs. young rats at the age of 4 months[85]. Furthermore, enzymatic kinetics of the PDH complex depicted a 60% increase in Vmax and a 1.6-fold decrease in the Michaelis constant (Km) with no change in PDH complex expression in the older F344 rats. The higher Vmax and lower Km indicate improved PDH catalytic efficiency without compromising PDH expression in the rat heart with aging. Similarly, another study utilizing aged mice (22-24 months) also reported lower PDK4 expression in hearts compared with young mice (4-6 months)[35]. In contrast, other studies have reported lower PDH activity in the fed state, even with a slight but significant decrease in PDK activity in the hearts of old rats at the age of 60 weeks compared with younger rats at the age of 7 weeks[44]. Likewise, PDH enzymatic activity measured without modulating PDKs also showed a reduced activity in the hearts of old rats at the age of 510 days compared to younger rats at the age of 75 days[86].
Although the mechanistic findings of glucose oxidation are not consistent among preclinical studies and clinically myocardial glucose utilization does not change in elderly healthy individuals[36], the decline in myocardial fatty acid oxidation may cause an increase in the relative contribution of myocardial glucose utilization to OXPHOS via the Randle Cycle effect. Thus, the shift from fatty acid oxidation to glucose oxidation may represent a compensatory response by cardiomyocytes to counteract the energy deficit arising from reduced fatty acid oxidation with aging. These compensatory metabolic adaptations, along with lower PDH activity and inconsistent glucose uptake, may take a heavy toll on the metabolic flexibility of the aged myocardium. Thus, further studies are required with careful consideration for the selection of models and methods to assess glucose uptake and oxidation during aging.
Ketone metabolism during aging
Ketones (β-hydroxybutyrate, acetoacetate, and acetone) are an alternate fuel source generated from hepatic fatty acid oxidation during prolonged fasting, while adherence to a very high-fat and low-carbohydrate ketogenic diet can lead to nutritional ketosis[87,88]. In preclinical animal models and heart failure patients, ketone oxidation-related enzymes and intermediates of ketone metabolism increase, implying that ketones are a critical alternative fuel source during cardiac dysfunction[89,90]. In the context of aging, with the impaired fatty acid oxidation and unchanged glucose oxidation, ketones could serve as an essential compensatory fuel during aging-related cardiac dysfunction.
In general, the ketogenic response in older individuals is diminished in response to fasting or adherence to ketogenic diets[91]. Similarly, aged rats (50-week-old) also exhibit decreased ketogenesis when compared to young rats at the age of 8 weeks in response to glucagon stimulation or fasting[92]. Moreover, aged rats (20-month-old) supplemented with ketogenic diets comprising 76% fat, 20% protein, and 4% carbohydrate for 12 weeks took longer to reach stable levels of β-hydroxybutyrate compared to young rats (4-month-
Of interest, β-hydroxybutyrate supplementation extends the lifespan of Caenorhabditis elegans by about 20%, primarily through activation of signaling pathways downstream of DAF-16, a forkhead box O homolog[96], which modulates metabolic homeostasis, stress resistance, and other longevity-associated processes[96]. In addition, 14-month-old mice maintained on a ketogenic diet significantly extended their lifespan, which was associated with preservation of physical and motor function, as well as improved cognitive function[97]. Long-term adherence to ketogenic diets in aged mice (20 months) also led to protection against aging-associated cardiac abnormalities, possibly through improving mitochondrial function[98]. Despite these salutary actions attributed to ketogenic dietary patterns, their metabolic effects with prolonged adherence remain controversial, as they may reduce insulin sensitivity, impair glucose tolerance, and induce cellular senescence in multiple organs, including the heart[99-101].
Isolated working heart perfusions in 22-24-month-old mice using 13C-labeled acetoacetate, free fatty acids, lactate, and glucose demonstrated lower myocardial ketone metabolism[35]. This may be attributed to a reduction in myocardial expression of SCOT, the rate-limiting enzyme of ketone oxidation, which is decreased in 8-month-old mice[102]. Moreover, cardiac-specific SCOT KO mice develop cardiac dysfunction and adverse remodeling, indicated by significantly decreased LV ejection fraction and posterior wall thickness compared to their cre expressing littermates at 30 weeks of age. Of note, ketogenic diet supplementation partially rescued the contractile dysfunction in cardiac-specific SCOT KO mice[102], suggesting that ketones may also work through oxidation-independent mechanisms in cardiomyopathy (for an extensive review of ketone-regulated signaling, please refer to the following reviews[88,103]). Contrary to the abovementioned studies, Rebrin et al. reported a significant increase in SCOT activity with no change in SCOT protein expression in the hearts of 24-month-old rats versus that of 4-month-old rats[104]. Thus, it remains inconclusive whether ketone oxidation or their actions beyond metabolism play a key role in aging-related metabolic inflexibility and cardiac dysfunction, which remains an area of active investigation.
Amino acid metabolism during aging
There is growing recognition that cardiovascular pathologies are also associated with perturbations in amino acid metabolism[105], though this element of myocardial metabolism in the context of aging has been understudied. Of relevance, the essential amino acids referred to as branched-chain amino acids (BCAAs), which include leucine, isoleucine, and valine, have important actions in the myocardium[106]. While comprehensive studies of myocardial BCAA metabolism during aging are lacking, BCAA supplementation increases the average life span of mice and is associated with increased mitochondrial biogenesis in cardiomyocytes at 21 months of age[107]. However, recent longitudinal studies involving the assessment of amino acid metabolites in serum samples collected from older individuals (mean age, 73 years) living without T2D in comparison to archived samples of the same individuals over the previous 15-year period, observed a strong correlation for elevated serum BCAA levels with cardiac dysfunction in older individuals[108]. Although participants were not controlled for post-prandial and fasting states, which may affect BCAA measurements, it cannot be ruled out that post-prandial rises in circulating BCAA levels might be independent of observed cardiac dysfunction in older individuals[108]. Conversely, studies have found an association between aging-related frailty and increased risk of death with decreasing serum BCAA levels, while other studies observed decreased serum BCAAs levels in healthy aged individuals[109,110]. Nevertheless, accumulating evidence that high circulating BCAA levels are linked to contractile dysfunction and different forms of heart failure[10] suggests that alterations in BCAA metabolism might be a predictor of age-related cardiovascular risk. Moreover, high circulating BCAA levels with disrupted BCAA metabolism elicit insulin resistance and metabolic inflexibility in heart failure[111], which may imply that these perturbations are also associated with myocardial metabolic inflexibility during aging.
POTENTIAL INTERVENTIONS TO IMPROVE CARDIOVASCULAR HEALTH IN AGING
Although aging is an inevitable process, lifestyle changes and pharmacological interventions can slow or attenuate aging-associated cardiovascular complications. There are a few well characterized interventions, such as calorie restriction, dietary modifications, exercise, and pharmacological compounds [Table 1] that promote healthy aging with lower cardiovascular complications and/or lifespan extension (extensively reviewed in[112,113]).
Pharmacological interventions targeting myocardial energy metabolism
| Interventions | Experimental models/subjects | Effects | References | |
| SGLT2 Inhibitors (e.g., empagliflozin, dapagliflozin) | Diabetic heart | ↑ Myocardial ATP production ↑ Ketone metabolism | [124,125] | |
| GLP-1 receptor agonists (e.g., liraglutide) | Diabetic heart | ↑ Insulin secretion ↑ Myocardial glucose metabolism ↑ PDH activity | [126,127] | |
| Metformin | Diabetic heart | ↑ AMPK-mediated glucose uptake ↑ Myocardial glucose metabolism | [128] | |
| PDK inhibitors (e.g., dichloroacetate) | Obesity & hypertension | ↑ Myocardial glucose oxidation ↑ PDH activity | [129] | |
Besides several pre-clinical studies (extensively reviewed in[114]), a randomized controlled trial of caloric restriction designed to achieve 25% calorie restriction showed significant decreases in body weight, serum cholesterol, TAGs, and mean blood pressure without adverse cardiovascular events in nonobese individuals aged 21-51 years[115,116]. Although the molecular mechanisms underlying the protective cardiovascular effects of calorie restriction are still not completely understood, it has beneficial effects on metabolism, mitochondrial activity, oxidative stress, and inflammation[117]. In addition, dietary interventions, such as omega-3 fatty acids[118], unsaturated fatty acids[119], ketone supplements[120], and dietary inorganic nitrate from beetroot[121], are associated with improved cardiometabolic health in clinical trials involving adults with or without cardiovascular complications. However, there is a need for longer and more targeted trials to evaluate the impact of dietary interventions in elderly populations. Moreover, clinical trials with endurance training or walking consistently demonstrate improved exercise capacity, physical function, and quality of life in older individuals with cardiovascular complications[122,123].
Accumulating pre-clinical and clinical evidence supported by large, randomized trials demonstrates the cardiovascular therapeutic potential of sodium-glucose cotransporter 2 inhibitors (e.g., empagliflozin, dapagliflozin)[124,125] and glucagon-like peptide-1 receptor agonists (e.g., semaglutide, liraglutide, dulaglutide)[126,127] through weight loss and anti-inflammatory effects, particularly in cohorts of obese individuals (extensively reviewed in[126]). In addition, metformin, a commonly prescribed drug to treat T2D, leads to improvement in cardiometabolic health and lifespan in mice by reducing oxidative stress and inflammation via increased adenosine monophosphate-activated protein kinase activity and antioxidant actions[128]. Moreover, pharmacological PDH activation via PDK inhibition restores glucose oxidation, improves cardiac function, and attenuates hypertrophy in aged female mice[129]. However, the translational potential of these pharmacological interventions to improve cardiovascular outcomes, healthspan, or lifespan in elderly individuals is yet to be evaluated.
CONCLUSIONS
The aged heart is energy-deficient, primarily due to a decrease in myocardial fatty acid oxidation, increased glycolysis uncoupled from glucose oxidation, and deterioration in mitochondrial health and oxidative capacity. The former combined with increased fatty acid availability to the heart during aging leads to cardiac lipotoxicity, while alterations in glucose utilization also contribute to the pathology of age-related cardiac dysfunction. Although it remains inconclusive whether the metabolism of ketones and amino acids, or their signaling actions, play a key role in cardiac dysfunction during aging, the perturbations in fatty acid metabolism may be a physiological adaptation for the less energy-efficient aged heart. Moreover, deteriorations in mitochondrial structure and oxidative metabolism in the aged heart are due to transcriptional changes, mutations in mitochondrial DNA, oxidative stress, and epigenetic changes in genes encoding metabolic enzymes. As aging itself is a heterogeneous natural process, with our current knowledge, it remains inconclusive whether the overall aging-related alterations in myocardial energy metabolism are adaptive or maladaptive. This is further construed by the fact that aging is associated with increased risk for several chronic disorders (e.g., obesity, T2D) that will impact myocardial energy metabolism, making this an extremely difficult question to answer. As such, both preclinical and clinical research studies involving healthy aging in the absence of obesity and/or T2D are necessary. In addition, emerging developments in the cardioprotective actions of calorie restriction, dietary supplements, exercise, and pharmacological interventions of metabolism (e.g., PDK inhibition, ketone supplementation, etc.) warrant future clinical studies to investigate their effects on cardiac health in aging. Despite accumulating evidence of phenotypic differences between men and women in cardiac energy metabolism and cardiovascular risk factors, the mechanistic understanding of sex differences still requires comprehensive evaluation during aging, as a decrease in estrogen levels has been strongly correlated with cardiovascular complications at the menopausal age in women. Nonetheless, pharmacological or lifestyle changes to optimize myocardial energy metabolism and metabolic inflexibility after considering sex differences may serve as a protective approach to improve cardiac health while supporting a healthy aging process.
DECLARATIONS
Authors’ contributions
Writing the first version of the manuscript: Gopal K, Ussher JR
Commenting, editing, and approving all versions of the manuscript: Gopal K, Heidari M, Seubert JM, Ussher JR
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This manuscript was supported by a Project Grant from the Canadian Institutes of Health Research to Ussher JR. Ussher JR is supported by a Tier 2 Canada Research Chair in Pharmacotherapy of Energy Metabolism in Obesity.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Kovacic JC, Moreno P, Nabel EG, Hachinski V, Fuster V. Cellular senescence, vascular disease, and aging: part 2 of a 2-part review: clinical vascular disease in the elderly. Circulation. 2011;123:1900-10.
2. Heidenreich PA, Trogdon JG, Khavjou OA, et al. Forecasting the future of cardiovascular disease in the United States: a policy statement from the American Heart Association. Circulation. 2011;123:933-44.
3. Lutz W, Sanderson W, Scherbov S. The coming acceleration of global population ageing. Nature. 2008;451:716-9.
4. Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: part II: the aging heart in health: links to heart disease. Circulation. 2003;107:346-54.
5. Gorbunova V, Seluanov A, Mita P, et al. The role of retrotransposable elements in ageing and age-associated diseases. Nature. 2021;596:43-53.
6. Peverill RE. Changes in left ventricular size, geometry, pump function and left heart pressures during healthy aging. Rev Cardiovasc Med. 2021;22:717.
7. Dai D, Chen T, Johnson SC, Szeto H, Rabinovitch PS. Cardiac aging: from molecular mechanisms to significance in human health and disease. Antioxid Redox Signal. 2012;16:1492-526.
8. Khouri MG, Maurer MS, Rumbarger LEK, King DL. Assessment of age‐related changes in left ventricular structure and function by freehand three‐dimensional echocardiography. Am J Geri Cardiol. 2007;14:118-25.
9. Lopaschuk GD, Folmes CD, Stanley WC. Cardiac energy metabolism in obesity. Circ Res. 2007;101:335-47.
10. Lopaschuk GD, Karwi QG, Tian R, Wende AR, Abel ED. Cardiac energy metabolism in heart failure. Circ Res. 2021;128:1487-513.
11. Benjamin EJ, Muntner P, Alonso A, et al. Heart disease and stroke statistics—2019 update: a report from the American Heart Association. Circulation. 2019;139:e56-528.
13. Lesnefsky EJ, Chen Q, Hoppel CL. Mitochondrial metabolism in aging heart. Circ Res. 2016;118:1593-611.
15. Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005;85:1093-129.
16. Sawada M, Carlson JC. Changes in superoxide radical and lipid peroxide formation in the brain, heart and liver during the lifetime of the rat. Mech Ageing Dev. 1987;41:125-37.
17. Li Y, Ma Y, Song L, et al. SIRT3 deficiency exacerbates p53/Parkin-mediated mitophagy inhibition and promotes mitochondrial dysfunction: implication for aged hearts. Int J Mol Med. 2018;41:3517-26.
18. Kincaid B, Bossy-Wetzel E. Forever young: SIRT3 a shield against mitochondrial meltdown, aging, and neurodegeneration. Front Aging Neurosci. 2013;5:48.
19. Jamieson KL, Keshavarz-Bahaghighat H, Darwesh AM, Sosnowski DK, Seubert JM. Age and sex differences in hearts of soluble epoxide hydrolase null mice. Front Physiol. 2020;11:48.
20. Molina-Riquelme I, Barrientos G, Breitsprecher L, et al. Multiscale mitochondrial cristae remodeling links Opa1 downregulation to reduced OXPHOS capacity in aged hearts. Proc Natl Acad Sci USA. 2025;123:e2508911123.
21. Larsson N. Somatic mitochondrial DNA mutations in mammalian aging. Annu Rev Biochem. 2010;79:683-706.
22. Hu C, Zhang X, Teng T, Ma Z, Tang Q. Cellular senescence in cardiovascular diseases: a systematic review. Aging Dis. 2022;13:103.
23. Chen MS, Lee RT, Garbern JC. Senescence mechanisms and targets in the heart. Cardiovasc Res. 2022;118:1173-87.
24. Viña J, Gomez-cabrera MC, Borras C, et al. Mitochondrial biogenesis in exercise and in ageing. Adv Drug Delivery Rev. 2009;61:1369-74.
25. López-otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194-217.
26. Zhu X, Shen W, Yao K, et al. Fine-tuning of PGC1α expression regulates cardiac function and longevity. Circ Res. 2019;125:707-19.
27. Whitehead N, Gill JF, Brink M, Handschin C. Moderate modulation of cardiac PGC-1α expression partially affects age-associated transcriptional remodeling of the heart. Front Physiol. 2018;9:242.
28. Nàger M, Calvoli M, Larsen KB, Birgisdottir AB. The multifaceted role of autophagy and mitophagy in cardiovascular health and disease. Autophagy Rep. 2025;4:2572511.
29. Billia F, Hauck L, Konecny F, Rao V, Shen J, Mak TW. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc Natl Acad Sci USA. 2011;108:9572-7.
30. Ma L, Zhu J, Gao Q, Rebecchi MJ, Wang Q, Liu L. Restoring pharmacologic preconditioning in the aging heart: role of mitophagy/autophagy. J Gerontol Ser A. 2017;72:489-98.
31. Karwi QG, Uddin GM, Ho KL, Lopaschuk GD. Loss of metabolic flexibility in the failing heart. Front Cardiovasc Med. 2018;5:68.
32. Koonen DP, Febbraio M, Bonnet S, et al. CD36 expression contributes to age-induced cardiomyopathy in mice. Circulation. 2007;116:2139-47.
33. Ledee D, Portman MA, Kajimoto M, Isern N, Olson AK. Thyroid hormone reverses aging-induced myocardial fatty acid oxidation defects and improves the response to acutely increased afterload. PLoS ONE. 2013;8:e65532.
34. Sample J, Cleland JG, Seymour AL. Metabolic remodeling in the aging heart. J Mol Cell Cardiol. 2006;40:56-63.
35. Hyyti OM, Ledee D, Ning X, Ge M, Portman MA. Aging impairs myocardial fatty acid and ketone oxidation and modifies cardiac functional and metabolic responses to insulin in mice. Am J Physiol Heart Circ Physiol. 2010;299:H868-75.
36. Kates AM, Herrero P, Dence C, et al. Impact of aging on substrate metabolism by the human heart. J Am Coll Cardiol. 2003;41:293-9.
37. Herrero P, Soto PF, Dence CS, et al. Impact of hormone replacement on myocardial fatty acid metabolism: potential role of estrogen. J Nucl Cardiol. 2005;12:574-81.
38. Barger PM, Kelly DP. Fatty acid utilization in the hypertrophied and failing heart: molecular regulatory mechanisms. Am J Med Sci. 1999;318:36.
39. Lopaschuk GD, Ussher JR, Folmes CDL, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. 2010;90:207-58.
40. Coughlin SS, Tefft MC, Rice JC, Gerone JL, Baughman KL. Epidemiology of idiopathic dilated cardiomyopathy in the elderly: pooled results from two case-control studies. Am J Epidemiol. 1996;143:881-8.
41. Dávila-Román VG, Vedala G, Herrero P, et al. Altered myocardial fatty acid and glucose metabolism in idiopathic dilated cardiomyopathy. J Am Coll Cardiol. 2002;40:271-7.
42. Funada J, Betts TR, Hodson L, et al. Substrate utilization by the failing human heart by direct quantification using arterio-venous blood sampling. PLoS ONE. 2009;4:e7533.
43. Chen L, Chen M, Yang X, et al. Energy metabolism in cardiovascular diseases: unlocking the hidden powerhouse of cardiac pathophysiology. Front Endocrinol. 2025;16:1617305.
44. Nakai N, Sato Y, Oshida Y, et al. Effects of aging on the activities of pyruvate dehydrogenase complex and its kinase in rat heart. Life Sci. 1997;60:2309-14.
45. Bhashyam S, Parikh P, Bolukoglu H, et al. Aging is associated with myocardial insulin resistance and mitochondrial dysfunction. Am J Physiol Heart Circ Physiol. 2007;293:H3063-71.
46. Ahiawodzi PD, Buzkova P, Djousse L, Ix JH, Kizer JR, Mukamal KJ. Nonesterified fatty acids and hospitalizations among older adults: the cardiovascular health study. J Gerontol Ser A. 2021;76:1326-32.
47. Jouven X, Charles M, Desnos M, Ducimetière P. Circulating nonesterified fatty acid level as a predictive risk factor for sudden death in the population. Circulation. 2001;104:756-61.
48. Djoussé L, Biggs ML, Ix JH, et al. Nonesterified fatty acids and risk of sudden cardiac death in older adults. Circ Arrhythm Electrophysiol. 2012;5:273-8.
49. Reaven GM. Effect of age and sex on triglyceride metabolism in the rat. J Gerontol. 1978;33:368-71.
50. Houtkooper RH, Argmann C, Houten SM, et al. The metabolic footprint of aging in mice. Sci Rep. 2011;1:134.
51. Wisneski JA, Gertz EW, Neese RA, Mayr M. Myocardial metabolism of free fatty acids. Studies with 14C-labeled substrates in humans. J Clin Investig. 1987;79:359-66.
52. Zhao L, Zou X, Feng Z, et al. Evidence for association of mitochondrial metabolism alteration with lipid accumulation in aging rats. Exp Gerontol. 2014;56:3-12.
53. Van Der Meer RW, Rijzewijk LJ, Diamant M, et al. The ageing male heart: myocardial triglyceride content as independent predictor of diastolic function. Eur Heart J. 2008;29:1516-22.
54. Luiken JJFP, Turcotte LP, Bonen A. Protein-mediated palmitate uptake and expression of fatty acid transport proteins in heart giant vesicles. J Lipid Res. 1999;40:1007-16.
55. Cai Y, Liu H, Song E, et al. Deficiency of telomere-associated repressor activator protein 1 precipitates cardiac aging in mice via p53/PPARα signaling. Theranostics. 2021;11:4710-27.
56. Mcmillin JB, Taffet GE, Taegtmeyer H, Hudson EK, Tate CA. Mitochondrial metabolism and substrate competition in the aging Fischer rat heart. Cardiovasc Res. 1993;27:2222-8.
57. Odiet JA, Boerrigter ME, Wei JY. Carnitine palmitoyl transferase-I activity in the aging mouse heart. Mech Ageing Dev. 1995;79:127-36.
58. Gómez LA, Heath SD, Hagen TM. Acetyl-l-carnitine supplementation reverses the age-related decline in carnitine palmitoyltransferase 1 (CPT1) activity in interfibrillar mitochondria without changing the l-carnitine content in the rat heart. Mech Ageing Dev. 2012;133:99-106.
59. Iemitsu M, Miyauchi T, Maeda S, et al. Aging-induced decrease in the PPAR-α level in hearts is improved by exercise training. Am J Physiol Heart Circ Physiol. 2002;283:H1750-60.
60. Poynter ME, Daynes RA. Peroxisome proliferator-activated receptor α activation modulates cellular redox status, represses nuclear factor-κB signaling, and reduces inflammatory cytokine production in aging. J Biol Chem. 1998;273:32833-41.
61. Watanabe K, Fujii H, Takahashi T, et al. Constitutive regulation of cardiac fatty acid metabolism through peroxisome proliferator-activated receptor α associated with age-dependent cardiac toxicity. J Biol Chem. 2000;275:22293-9.
62. Howroyd P, Swanson C, Dunn C, Cattley RC, Corton JC. Decreased longevity and enhancement of age-dependent lesions in mice lacking the nuclear receptor peroxisome proliferator-activated receptor α (PPARα). Toxicol Pathol. 2004;32:591-9.
63. Son N, Ananthakrishnan R, Yu S, et al. Cardiomyocyte aldose reductase causes heart failure and impairs recovery from ischemia. PLoS ONE. 2012;7:e46549.
64. Thiagarajan D, Ananthakrishnan R, Zhang J, et al. Aldose reductase acts as a selective derepressor of PPARγ and the retinoic acid receptor. Cell Rep. 2016;15:181-96.
65. Drosatos K, Schulze PC. Cardiac lipotoxicity: molecular pathways and therapeutic implications. Curr Heart Fail Rep. 2013;10:109-21.
66. Rodriguez-calvo R, Serrano L, Barroso E, et al. Peroxisome proliferator-activated receptor down-regulation is associated with enhanced ceramide levels in age-associated cardiac hypertrophy. J Gerontol Ser A Biol Sci Med Sci. 2007;62:1326-36.
67. Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963;281:785-9.
68. Nyberg M, Jones AM. Matching of O2 utilization and O2 delivery in contracting skeletal muscle in health, aging, and heart failure. Front Physiol. 2022;13:898395.
69. Lakatta EG, Yin FC. Myocardial aging: functional alterations and related cellular mechanisms. Am J Physiol Heart Circ Physiol. 1982;242:H927-41.
70. Abu-Erreish GM, Neely JR, Whitmer JT, Whitman V, Sanadi DR. Fatty acid oxidation by isolated perfused working hearts of aged rats. Am J Physiol Endocrinol Metab. 1977;232:E258.
71. Ji LL, Dillon D, Wu E. Myocardial aging: antioxidant enzyme systems and related biochemical properties. Am J Physiol Regul Integr Comp Physiol. 1991;261:R386-92.
72. Soto PF, Herrero P, Kates AM, et al. Impact of aging on myocardial metabolic response to dobutamine. Am J Physiol Heart Circ Physiol. 2003;285:H2158-64.
73. El Khoudary SR, Aggarwal B, Beckie TM, et al. Menopause transition and cardiovascular disease risk: implications for timing of early prevention: a scientific statement from the American Heart Association. Circulation. 2020;142:25.
74. Goodwin GW, Ahmad F, Doenst T, Taegtmeyer H. Energy provision from glycogen, glucose, and fatty acids on adrenergic stimulation of isolated working rat hearts. Am J Physiol Heart Circ Physiol. 1998;274:H1239-47.
75. Hall JL, Stanley WC, Lopaschuk GD, et al. Impaired pyruvate oxidation but normal glucose uptake in diabetic pig heart during dobutamine-induced work. Am J Physiol Heart Circ Physiol. 1996;271:H2320-9.
77. Faakye A, Harold KM, Matsuzaki S, et al. The effect of enhanced glycolysis on cardiac aging. GeroScience. 2025;47:6455-72.
78. Hall JL, Mazzeo RS, Podolin DA, Cartee GD, Stanley WC. Exercise training does not compensate for age-related decrease in myocardial GLUT-4 content. J Appl Physiol. 1994;76:328-32.
79. Mooradian AD, Chehade JM, Kim J. Age-related changes in thyroid hormone effects on glucose transporter isoforms of rat heart. Life Sci. 1999;65:981-9.
80. Martineau LC, Chadan SG, Parkhouse WS. Age-associated alterations in cardiac and skeletal muscle glucose transporters, insulin and IGF-1 receptors, and PI3-kinase protein contents in the C57BL/6 mouse. Mech Ageing Dev. 1999;106:217-32.
81. Ozaki N, Sato E, Kurokawa T, Ishibashi S. Early changes in the expression of GLUT4 protein in the heart of senescence-accelerated mouse. Mech Ageing Dev. 1996;88:149-58.
82. Luptak I, Yan J, Cui L, Jain M, Liao R, Tian R. Long-term effects of increased glucose entry on mouse hearts during normal aging and ischemic stress. Circulation. 2007;116:901-9.
83. Patel MS, Nemeria NS, Furey W, Jordan F. The pyruvate dehydrogenase complexes: structure-based function and regulation. J Biol Chem. 2014;289:16615-23.
84. Sugden MC, Holness MJ. Interactive regulation of the pyruvate dehydrogenase complex and the carnitine palmitoyltransferase system. FASEB J. 1994;8:54-61.
85. Moreau R, Heath SHD, Doneanu CE, Harris RA, Hagen TM. Age-related compensatory activation of pyruvate dehydrogenase complex in rat heart. Biochem Biophys Res Commun. 2004;325:48-58.
86. Chuffa LG, Seiva FR. Combined effects of age and diet-induced obesity on biochemical parameters and cardiac energy metabolism in rats. Ind J Biochem Biophys. 2013;50:40-7.
87. Puchalska P, Crawford PA. Multi-dimensional roles of Ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metab. 2017;25:262-84.
88. Cotter DG, Schugar RC, Crawford PA. Ketone body metabolism and cardiovascular disease. Am J Physiol Heart Circ Physiol. 2013;304:H1060-76.
89. Aubert G, Martin OJ, Horton JL, et al. The failing heart relies on ketone bodies as a fuel. Circulation. 2016;133:698-705.
90. Takahara S, Soni S, Maayah ZH, Ferdaoussi M, Dyck JRB. Ketone therapy for heart failure: current evidence for clinical use. Cardiovasc Res. 2022;118:977-87.
91. Hernandez A, Truckenbrod L, Federico Q, et al. Metabolic switching is impaired by aging and facilitated by ketosis independent of glycogen. Aging. 2020;12:7963-84.
92. Okuda Y, Kawai K, Yamashita K. Age-related change in ketone body metabolism: diminished glucagon effect on ketogenesis in adult rats. Endocrinology. 1987;120:2152-7.
93. Hernandez AR, Hernandez CM, Campos KT, et al. The antiepileptic ketogenic diet alters hippocampal transporter levels and reduces adiposity in aged rats. J Gerontol Ser A. 2018;73:450-8.
94. Eap B, Nomura M, Panda O, et al. Ketone body metabolism declines with age in mice in a sex-dependent manner. bioRxiv. 2022. [Preprint].
95. Niezen S, Connelly MA, Hirsch C, et al. Elevated plasma levels of ketone bodies are associated with all‐cause mortality and incidence of heart failure in older adults: the CHS. J Am Heart Assoc. 2023;12:e029960.
96. Edwards C, Canfield J, Copes N, Rehan M, Lipps D, Bradshaw PC. D-beta-hydroxybutyrate extends lifespan in C. elegans. Aging. 2014;6:621-44.
97. Roberts MN, Wallace MA, Tomilov AA, et al. A ketogenic diet extends longevity and healthspan in adult mice. Cell Metab. 2017;26:539-46.e5.
98. Yu Y, Wang F, Wang J, Zhang D, Zhao X. Ketogenic diet attenuates aging-associated myocardial remodeling and dysfunction in mice. Exp Gerontol. 2020;140:111058.
99. Han Y, Ramprasath T, Zou M. β-hydroxybutyrate and its metabolic effects on age-associated pathology. Exp Mol Med. 2020;52:548-55.
100. Wei S, Schell JR, Chocron ES, et al. Ketogenic diet induces p53-dependent cellular senescence in multiple organs. Sci Adv. 2024;10:eado1463.
101. Greenwell AA, Saed CT, Tabatabaei Dakhili SA, et al. An isoproteic cocoa butter-based ketogenic diet fails to improve glucose homeostasis and promote weight loss in obese mice. Am J Physiol Endocrinol Metab. 2022;323:E8-20.
102. Keller MA, Nakamura M. Ketone catabolism is essential for maintaining normal heart function during aging. bioRxiv. 2025. [Preprint].
103. Lopaschuk GD, Dyck JRB. Ketones and the cardiovascular system. Nat Cardiovasc Res. 2023;2:425-37.
104. Rebrin I, Brégère C, Kamzalov S, Gallaher TK, Sohal RS. Nitration of tryptophan 372 in succinyl-CoA:3-ketoacid CoA transferase during aging in rat heart mitochondria. Biochemistry. 2007;46:10130-44.
105. Huang Y, Zhou M, Sun H, Wang Y. Branched-chain amino acid metabolism in heart disease: an epiphenomenon or a real culprit? Cardiovasc Res. 2011;90:220-3.
106. Harper AE, Miller RH, Block KP. Branched-chain amino acid metabolism. Annu Rev Nutr. 1984;4:409-54.
107. D'antona G, Ragni M, Cardile A, et al. Branched-chain amino acid supplementation promotes survival and supports cardiac and skeletal muscle mitochondrial biogenesis in middle-aged mice. Cell Metab. 2010;12:362-72.
108. Kovalik J, Zhao X, Gao F, et al. Amino acid differences between diabetic older adults and non-diabetic older adults and their associations with cardiovascular function. J Mol Cell Cardiol. 2021;158:63-71.
109. Chaleckis R, Murakami I, Takada J, Kondoh H, Yanagida M. Individual variability in human blood metabolites identifies age-related differences. Proc Natl Acad Sci USA. 2016;113:4252-9.
110. Kouchiwa T, Wada K, Uchiyama M, et al. Age-related changes in serum amino acids concentrations in healthy individuals. Clin Chem Lab Med. 2012;50:861-70.
111. Xia J, Nong Y, Teng J, et al. Unlocking metabolic flexibility in heart failure with preserved ejection fraction: Bridging fundamental mechanisms to clinical innovation. iScience. 2025;28:113471.
112. Alfaras I, Di Germanio C, Bernier M, et al. Pharmacological strategies to retard cardiovascular aging. Circ Res. 2016;118:1626-42.
113. Ye M, Feng S, Xu Z, He W, Liu C, Zhu W. Aging and metabolism in HFpEF: pathophysiology and therapeutic implications. Metabolism. 2026;176:156460.
114. Weiss EP, Fontana L. Caloric restriction: powerful protection for the aging heart and vasculature. Am J Physiol Heart Circ Physiol. 2011;301:H1205-19.
115. Racette SB, Silver RE, Barry VG, et al. Diet quality and nutritional adequacy during a 2-year calorie restriction intervention: the comprehensive assessment of long-term effects of reducing intake of energy 2 trial. Am J Clin Nutr. 2026;123:101182.
116. Ravussin E, Redman LM, Rochon J, et al. A 2-year randomized controlled trial of human caloric restriction: feasibility and effects on predictors of health span and longevity. J Gerontol Ser A. 2015;70:1097-104.
117. Marzetti E, Wohlgemuth SE, Anton SD, Bernabei R, Carter CS, Leeuwenburgh C. Cellular mechanisms of cardioprotection by calorie restriction: state of the science and future perspectives. Clin Geriatr Med. 2009;25:715-32.
118. Lechner K, Scherr J, Lorenz E, et al. Omega-3 fatty acid blood levels are inversely associated with cardiometabolic risk factors in HFpEF patients: the Aldo-DHF randomized controlled trial. Clin Res Cardiol. 2021;111:308-21.
119. Carbone S, Billingsley HE, Canada JM, et al. Unsaturated fatty acids to improve cardiorespiratory fitness in patients with obesity and HFpEF. JACC. 2019;4:563-5.
120. Selvaraj S, Karaj A, Chirinos JA, et al. Crossover trial of exogenous ketones on cardiometabolic endpoints in heart failure with preserved ejection fraction. JACC. 2025;13:102435.
121. Eggebeen J, Kim-Shapiro DB, Haykowsky M, et al. One week of daily dosing with beetroot juice improves submaximal endurance and blood pressure in older patients with heart failure and preserved ejection fraction. JACC. 2016;4:428-37.
122. Brubaker PH, Avis T, Rejeski WJ, Mihalko SE, Tucker WJ, Kitzman DW. Exercise training effects on the relationship of physical function and health-related quality of life among older heart failure patients with preserved ejection fraction. J Cardiopulm Rehabil Prev. 2020;40:427-33.
123. Gary R, Lee SY. Physical function and quality of life in older women with diastolic heart failure: effects of a progressive walking program on sleep patterns. Prog Cardiovasc Nurs. 2007;22:72-80.
124. Lopaschuk GD, Verma S. Mechanisms of cardiovascular benefits of sodium glucose co-transporter 2 (SGLT2) inhibitors: a state-of-the-art review. JACC. 2020;5:632-44.
125. Verma S, Rawat S, Ho KL, et al. Empagliflozin Increases cardiac energy production in diabetes: novel translational insights into the heart failure benefits of SGLT2 inhibitors. JACC. 2018;3:575-87.
126. Marx N, Husain M, Lehrke M, Verma S, Sattar N. GLP-1 receptor agonists for the reduction of atherosclerotic cardiovascular risk in patients with type 2 diabetes. Circulation. 2022;146:1882-94.
127. Huang J, Kwok AJ, Li JCY, et al. Body-wide multi-omic counteraction of aging with GLP-1R agonism. Cell Metab. 2025;37:2362-80.e8.
128. Martin-montalvo A, Mercken EM, Mitchell SJ, et al. Metformin improves healthspan and lifespan in mice. Nat Commun. 2013;4:2192.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].