


Targeting proprotein convertase subtilisin/kexin type 9 in aging-associated cardiometabolic and neurological disorders
 0
0 Abstract
Proprotein convertase subtilisin/kexin type 9 (PCSK9), a crucial regulator of cholesterol metabolism, is gaining recognition for its broader involvement in aging-related disorders. Emerging evidence indicates that PCSK9 is closely linked to cardiovascular aging, neurodegenerative disorders, and metabolic dysfunction. This review synthesizes up-to-date understanding on PCSK9’s multifaceted contributions, emphasizing its role in oxidative stress, inflammation, and cellular senescence, alongside potential therapeutic implications. PCSK9 inhibition not only confers cardiovascular benefits via low-density lipoprotein-dependent and independent pathways but also exhibits broader therapeutic potential. For instance, PCSK9 inhibitors show promise in cancer immunotherapy by enhancing major histocompatibility complex class I-mediated tumor antigen presentation and in attenuating inflammation through suppressing the Toll-like receptor 4/nuclear factor kappa B pathway. However, clinical translation remains limited by tissue-specific responses and the incomplete understanding of long-term systemic effects. Overall, this review underscores the complex role of PCSK9 in the aging network and therefore highlights the need for further mechanistic studies to guide its therapeutic application.
Keywords
INTRODUCTION
With substantial medical breakthroughs and extension of human lifespan, aging has become a global health concern and has received increasing attention in recent years. Advanced aging itself serves as an independent risk factor for various pathological conditions, such as cardiovascular illnesses, neurodegenerative disorders, and impaired liver function[1-4]. Cellular senescence constitutes a key component of the aging phenotype. This state is characterized by the accumulation of senescent cells (SC) and their active secretion of a complex mixture of inflammatory signals and tissue-remodeling factors, commonly termed the senescence-associated secretory phenotype (SASP). It is closely linked to multi-organ aging, particularly through its role in shaping the systemic aging milieu[5,6]. Among these, proprotein convertase subtilisin/kexin type 9 (PCSK9) has gained increasing scientific focus for its potential functions within the systemic aging network. A population-based study conducted by Arif et al. revealed remarkably elevated plasma PCSK9 levels with aging, indicating a possible role of elevated PCSK9 levels in aging-related biological processes[7]. A large-scale network analysis conducted using iNetModels (iNetModels: an interactive database and visualization platform of Multi-Omics Biological Networks, MOBNs) reinforced this finding, demonstrating a robust positive link between aging and the upregulation of PCSK9 expression[7,8].
PCSK9, a serine protease mainly released by hepatocytes, is a crucial component in maintaining cholesterol balance through its modulation of low-density lipoprotein (LDL) receptor degradation[9,10]. This protease was first identified in 2003 by Seidah et al., characterizing its molecular structure[11]. Concurrently, a separate independent study subsequently noted its functional mutations in autosomal dominant hypercholesterolemia (ADH), exploring its clinical significance[12].
As a distinctive enzyme within the proteinase K subfamily, PCSK9 features a multi-domain structure including an N-terminal propeptide, a central catalytic domain, and a C-terminal domain rich in cysteine and histidine residues. Initially produced as an inactive zymogen, PCSK9 undergoes autocatalytic cleavage to generate its mature form PCSK9_62, prior to its secretion into circulation. Following secretion, 25%-40% of PCSK9_62 undergoes furin-mediated cleavage to generate PCSK9_55, a less active form of circulating PCSK9[13]. The C-terminal domain of mature PCSK9 engages with the LDL receptor (LDLR), promoting receptor degradation in endosomes/lysosomes[11,14]. This process regulates apolipoprotein B (apoB), lipoprotein absorption and LDL-cholesterol (LDL-C) metabolism in the liver[15]. Other targets include LDLR-related protein 1 (LRP1) and apolipoprotein E receptor 2 (ApoER2)[16]. PCSK9 is highly conserved across species, with critical structural residues in the catalytic and LDLR-binding domains maintained from humans to pufferfish[11]. These structural characteristics underlie the fundamental role of PCSK9 in cholesterol homeostasis and its therapeutic targeting for age-related diseases.
Previous research has established that PCSK9 exerts a pivotal role in cardiovascular and hepatic diseases[17]. However, its functions and mechanisms in multi-organ aging remain insufficiently explored, and most existing research has largely overlooked its roles beyond lipid-lowering effects. This review offers a comprehensive and integrative overview of PCSK9's involvement in diverse age-related diseases and proposes a conceptual framework integrating recent advances. Particular attention is given to the connection between PCSK9 and core aging phenotypes, alongside its systemic function in aging. By elucidating these connections, we aim to shed light on the links between metabolic regulation and chronic disease pathogenesis, while highlighting potential therapeutic opportunities. Based on a comprehensive survey of the existing literature, this review appears to be the first work that focuses on the function of PCSK9 in multi-organ aging.
PCSK9 IN AGE-RELATED CARDIOVASCULAR DISEASE
Extensive research has demonstrated a cardinal role of PCSK9 in age-related cardiovascular diseases (CVD), with emerging evidence uncovering multiple mechanisms. Previous studies have identified elevated serum PCSK9 concentrations in older adults versus their young counterparts, accompanied by parallel cardiac dysfunction[7,9]. A factorial Mendelian randomization (MR) study including 425,354 participants has demonstrated that genetically reduced PCSK9 is associated with lower LDL-C, apoB levels and a diminished likelihood of developing coronary artery disease (CAD)[18]. PCSK9 inhibitors (PCSK9i) are capable of mitigating pathologies evoked by elevated plasma cholesterol and LDL-C levels in natural aging, hyperactive PCSK9 state or proprotein convertase subtilisin/kexin (PCSK) knock-in models[19,20]. These findings collectively highlight the close link between PCSK9 dysregulation, aging processes, and the pathogenesis of cardiovascular dysfunction.
PCSK9 and atherosclerosis
PCSK9 has been established as a critical factor in atherosclerosis through its effects on lipid metabolism, vascular inflammation, and plaque stability. Both circulating PCSK9 and vascular smooth muscle cell (VSMC)-derived PCSK9 contribute to plaque development through distinct mechanisms. Circulating PCSK9 enhances platelet activation, leukocyte recruitment, and thrombus formation[21,22], whereas vascular PCSK9 promotes local vascular remodeling. Importantly, PCSK9i confer benefits that extend beyond LDL-C reduction, such as suppressing oxidized low-density lipoprotein (ox-LDL)- or high-fat diet (HFD)-induced VSMC proliferation and foam cell formation via small nucleolar RNA host gene 16 (SNHG16) downregulation[23].
The primary pathway by which PCSK9 regulates atherosclerosis is through LDL-C regulation. By promoting LDLR degradation, PCSK9 increases circulating LDL-C levels, facilitating subendothelial lipid deposition. Excess LDL-C subsequently undergoes oxidative modification by reactive oxygen species (ROS), triggering a cascade of endothelial dysfunction, inflammation, and plaque formation[9]. In contrast, PCSK9 loss-of-function (LOF) variants provide robust protection against atherosclerotic CVD, largely through LDL-C lowering[24]. Clinically, PCSK9i reduce LDL-C concentrations by up to 60%, translating into a marked decline in the incidence of major adverse cardiovascular events among individuals with CAD[25]. Additionally, Sirtuin 2 (SIRT2) regulates LDL metabolism through PCSK9 suppression, proving the effect of sirtuins in improving atherosclerosis[26]. Intriguingly, beyond cardiovascular outcomes, PCSK9-targeting variants have been associated with lifespan extension, highlighting LDLR as a genetic determinant of human longevity[27].
PCSK9 contributes to atherosclerosis through mechanisms beyond its classical lipid-regulatory function. Atherosclerosis progression is characterized by inflammatory responses, ranging from low-grade inflammation in early lesions to robust inflammatory activity in advanced plaques[28]. Recent evidence has identified LDL-independent pathways by which PCSK9 amplifies inflammation: adenylate cyclase-associated protein 1 (CAP1)-mediated activation of spleen tyrosine kinase (Syk)/protein kinase C delta (PKCδ) signaling, leading to downstream Toll-like receptor 4 (TLR4)/nuclear factor kappa B (NF-κB) activation, and phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR)-driven VSMC phenotypic modulation[29]. The identification of distinct associations between mature/furin-cleaved PCSK9 and CAD-related factors further supports the inflammation theory. Furin-cleaved PCSK9, in addition to its positive correlation with non-high-density lipoprotein (non-HDL) cholesterol and systolic blood pressure, is specifically linked to high-sensitivity C-reactive protein (hs-CRP), a key inflammation biomarker, implying its unique predictive potential for coronary events[30]. These findings indicate that PCSK9 serves not only as a lipid regulator but also as a pro-inflammatory mediator, integrating metabolic and immune pathways to drive atherosclerosis [Figure 1]. Clarifying these contributions will be crucial to optimizing therapeutic strategies, particularly in combination with antithrombotic agents. This may be especially beneficial for high-risk individuals who are statin-intolerant or have not achieved their LDL-C targets (e.g., familial hypercholesterolemia)[31].
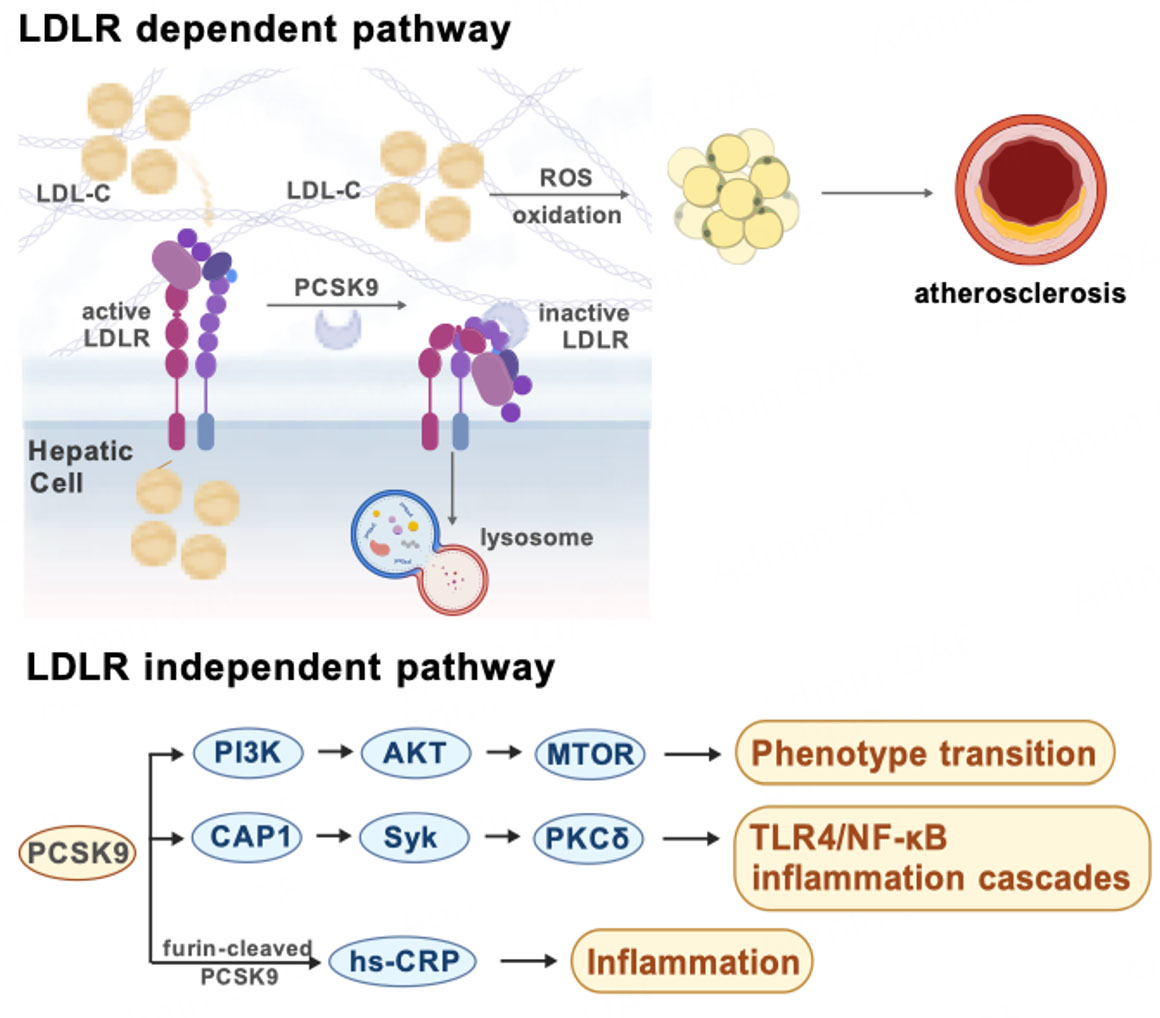
Figure 1. LDLR-dependent and independent mechanisms of PCSK9 in atherosclerosis. Created with BioGDP.com[94].
PCSK9 and cardiovascular aging
Age-related elevations in PCSK9 expression appear to establish a self-reinforcing cycle of cardiovascular deterioration. A critical distinction lies in the sources of PCSK9: circulating or systemic PCSK9 (primarily hepatic in origin) and vascular-derived PCSK9 (produced locally by vascular cells), each contributing uniquely to age-related pathology.
The impact of PCSK9 on aging exhibits spatial specificity. Systemic or hepatic PCSK9, functioning as an endocrine factor, promotes ectopic lipid deposition in vascular wall and increases thrombotic risk via platelet activation[9,21,22]. A progressive, age-dependent rise in circulating PCSK9 has been documented by large cohort studies, correlating with cardiac dysfunction and myocardial hypertrophy in elderly populations[10,32]. These associations seem to be driven by chronic inflammation, fibrotic remodeling, oxidative stress, and lipid accumulation, which collectively stimulate PCSK9 overexpression while being exacerbated by PCSK9 activity, thereby creating a vicious cycle. Apart from its recognized role in elevating LDL and cholesterol levels, PCSK9 also contributes to the thrombotic prevalence through platelet activation. Circulating PCSK9 binds to platelet CD36, triggering downstream signaling cascades that enhance platelet activation and thrombosis. This prothrombotic effect extends to the context of myocardial infarction (MI), where PCSK9 exacerbates microvascular obstruction and myocardial dilation[33]. These effects collectively foster a pro-inflammatory and pro-atherogenic state, contributing to aging phenotypes such as chronic inflammation and cardiac remodeling[34].
In contrast, vascular-derived PCSK9 mainly acts as a paracrine/autocrine factor, exerting more localized effects on vascular aging. At the cellular level, PCSK9 functions as a determinant factor on VSMC fate decision during aging. Under inflammatory conditions induced by stimuli such as lipopolysaccharide (LPS), oxidized LDL, and tumor necrosis factor-alpha (TNF-α), both VSMCs and endothelial cells (ECs) markedly increase PCSK9 expression. The outcomes vary according to the intensity and duration of exposure: moderate stimulation triggers adaptive autophagy, while sustained activation promotes senescence or apoptosis[16]. A recent investigation further demonstrated that increased PCSK9 expression suppresses ApoER2 levels in aortic smooth muscle cells, thereby inhibiting proliferation while promoting polyploidization, senescence, and apoptosis[35]. This graded response underscores PCSK9's central role in orchestrating VSMC life cycle decisions during vascular aging. Moreover, PCSK9 overexpression activates multiple oxidative stress pathways and upregulates senescence-associated genes, including p16, p21, and p53[36]. Conversely, PCSK9 inhibition attenuates vascular senescence via sirtuin 1-mediated downregulation of oxidative stress and senescence-related proteins[37]. These findings demonstrate that PCSK9 functions in normal cell-cycle suppression and oxidative stress activation, correlating with common hallmarks of cardiovascular aging including mitochondrial dysfunction and genomic alterations[34,38]. It also supports PCSK9 inhibition as a promising approach to mitigate age-related vascular damage.
Nevertheless, key questions remain regarding the relative significance of systemic versus vascular-derived PCSK9 in aging. Systemic or hepatic PCSK9 broadly influences metabolism and thrombosis, while vascular PCSK9 directly modulates local cell fate. Moreover, these two pools may engage in reciprocal regulation [Figure 2].
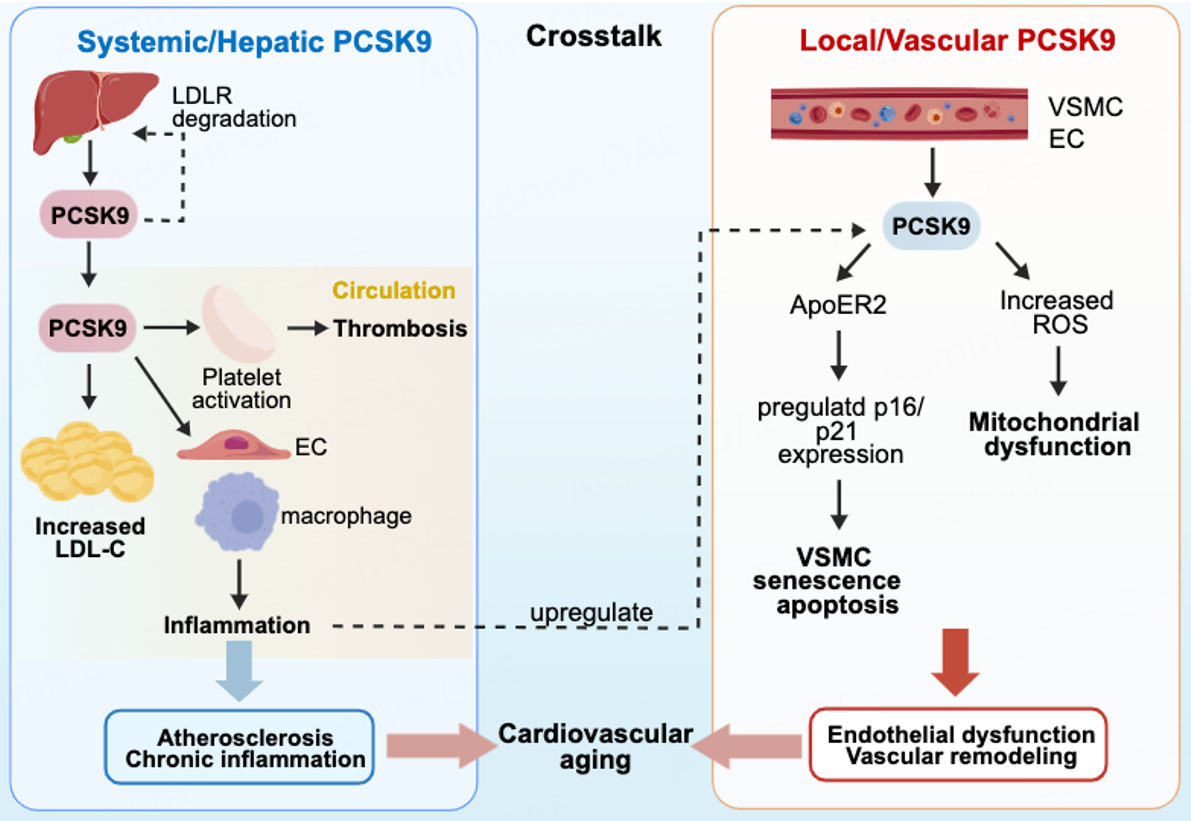
Figure 2. Distinct and interacting roles of systemic versus vascular-derived PCSK9 in cardiovascular aging created with BioGDP.com[94].
We believe that circulating PCSK9 heightens the baseline cardiovascular risk by promoting LDL-C production, systemic inflammation, and thrombotic propensity, increasing susceptibility to atherosclerotic plaque formation and other complications. Vascular-derived PCSK9, however, acts as a direct cellular mediator, governing local processes such as vascular cell senescence, death, dysfunction, and gene expression. Notably, crosstalk between these two pools has been noted where systemic inflammation can upregulate vascular PCSK9 expression[16]. A recent study reported hepatocyte-specific inhibition of PCSK9 reduces circulating PCSK9 by approximately 83%[39]. Combined with the evidence for vascular-derived PCSK9 secretion, these findings indicate local PCSK9 may modulate hepatic PCSK9 synthesis in return[15]. Animal studies indicate that PCSK9i ameliorate age-related cardiac dysfunction and vascular remodeling, yet it remains uncertain whether these benefits arise predominantly from improved systemic metabolism (via reduced hepatic PCSK9) or direct vascular effects[10,32]. Despite these uncertainties, current evidence strongly supports PCSK9 as both a biomarker and a drug target in cardiovascular aging.
PCSK9 AND NEURODEGENERATIVE DISORDERS
Growing evidence underscores the significant impact of PCSK9 on brain physiology and neurodegeneration. The effects of PCSK9 in the brain differ depending on whether it originates from the peripheral circulation or is synthesized locally within the central nervous system (CNS). The condition of the blood-brain barrier (BBB) is a key determinant in modulating these site-specific actions.
Circulating PCSK9 and blood-brain barrier dysfunction
The structural integrity of BBB is essential for protecting the CNS from neurotoxic substances and maintaining normal neural function[40]. Circulating PCSK9, which does not readily cross an intact BBB, primarily interacts with vascular ECs and astrocytes[41]. The pathological impact of circulating PCSK9 becomes particularly significant in BBB dysfunction. BBB breakdown, triggered by various factors, leads to a cascade of neurological deficits and has been implicated in the pathogenesis of numerous sporadic cases of Alzheimer’s disease (AD), amyotrophic lateral sclerosis (ALS) and Parkinson’s disease (PD)[40]. Previous studies have documented age-related BBB disruption in the normal human brain, characterized by progressive increases in the BBB permeability starting in the hippocampus[42]. This compromised vascular integrity may allow circulating PCSK9 to accumulate in brain tissues during aging, potentially contributing to neurodegeneration.
Locally produced PCSK9 in AD, ALS, and PD
In contrast, PCSK9 is also intrinsically expressed within the CNS by neurons, astrocytes, and other glial cells[43-45]. Unlike circulating PCSK9, this locally synthesized form acts on a broader range of neural cells through receptors such as LDLR, LRP1, apoER2 and CD36. These receptor-mediated interactions regulate crucial brain functions, including cholesterol homeostasis, neural development, cell survival, and inflammatory responses[41].
The role of local PCSK9 is evident across several neurodegenerative conditions. PCSK9’s role in AD pathophysiology is multifaceted, involving amyloid-β (Aβ) pathology, neuroinflammation, and altered cholesterol metabolism[44]. Hippocampal PCSK9 expression is elevated in hyperlipidemia-mediated neuronal apoptosis, and co-increases with apoptotic markers B-cell lymphoma 2 (Bcl-2), Bax, caspase-3 and beta-site amyloid precursor protein cleaving enzyme 1 (BACE1), suggesting both direct and indirect modes of action[46]. Both in vitro and in vivo studies have confirmed its pro-inflammatory activity, whereas PCSK9 ablation protects against cognitive decline in AD mouse models by enhancing Aβ clearance and reducing neuroinflammation[47]. Clinical observations further support this view, reporting higher cerebrospinal fluid (CSF) PCSK9 levels in AD patients and particularly among apolipoprotein E4 (APOE4) carriers. Notably, these increases appear in the early stages of AD without clear correlation to later disease progression, suggesting that PCSK9 may contribute to APOE4-associated lipid dysregulation in AD pathogenesis[48]. Additionally, APOE4 accelerates disruption of BBB, pericyte loss and early neurovascular dysfunction. These findings align with our earlier discussions of BBB and favor the mode of action for PCSK9 through both circulating and localized pathways[49].
Elevated CSF PCSK9 concentration is also reported in non-AD neural disorders, pointing to a broader contribution of PCSK9 in neurodegenerative processes[50]. Neurodegenerative diseases exhibit distinct pathologies, yet oxidative stress is a common hallmark across disorders including AD and PD[51]. PCSK9 induces oxidative stress in neurons through the activation of nicotinamide adenine dinucleotide phosphate oxidase 2. This activation subsequently leads to an elevated generation of ROS and facilitates oxidized LDL accumulation via LDLR degradation. It may also amplify oxidative injury indirectly by enhancing neuroinflammation. Although these mechanisms are not fully understood, preclinical evidence suggests that PCSK9 inhibition confers neuroprotective effects[52].
Neuroprotective potential of PCSK9 inhibition
The relationship between PCSK9 and neurodegeneration has also been investigated through drug-target MR studies, which assess the causal impacts of inhibiting PCSK9 on the onset or progression of neurodegenerative conditions. These studies, however, have produced inconsistent results, particularly in ALS and PD [Table 1][53-55].
Contradictory Mendelian randomization results of PCSK9
| Study | Population/Design | Key findings |
| Rosoff et al., 2022[53] | Large-scale genome-wide association studies (GWAS) | No adverse cognitive effects linked to PCSK9 inhibition; neutral cognitive impact |
| Huang et al., 2024[54] | Large-scale GWAS focusing on four diseases: ALS, PD, AD, and coronary heart disease (CHD) | PCSK9 inhibition associated with reduced ALS risk but increased PD susceptibility |
| Xia et al., 2025[55] | Cross-trait meta-analysis; bidirectional Mendelian randomization (MR) study | dyslipidemia and ALS within a single patient are caused in part by a shared genetic basis; PCSK9 inhibition and low LDL-C reduced ALS risk |
These divergent findings may stem from disease-specific mechanisms, while therefore altering PCSK9's role. In ALS, PCSK9 modulates disease progression through dual regulation of lipid metabolic and neuroprotective pathways. For instance, Xia and coworkers identified a positive genetic correlation between ALS and lipid metabolism. Their MR analysis further revealed that 55 specific single nucleotide polymorphisms (SNPs) in PCSK9 have been linked to decreased ALS susceptibility, underscoring PCSK9’s regulatory role in lipid metabolism during ALS pathogenesis[55]. Moreover, in vitro and in vivo studies have indicated that PCSK9 promotes neuronal apoptosis via mediating ApoER2 degradation[56,57], indicating that PCSK9 inhibition may confer neuroprotection by suppressing apoptotic pathways. In contrast, the role of PCSK9 in PD remains poorly understood. While Huang et al. reported no significant association[54], a recent case-control study found elevated PCSK9 levels in PD patients relative to those in the healthy control group[58], highlighting conflicting evidence in the field.
These findings enhance our understanding of PCSK9 as a key modulator in neurodegeneration, yet its complex and sometimes contradictory roles, along with insufficient clinical validation, warrant cautious interpretation. Future studies should prioritize mechanistic investigations in human-derived models and stratified clinical trials to determine whether PCSK9 represents a viable therapeutic target or merely a biomarker of disrupted CNS homeostasis.
PCSK9 AND LIVER AGING
The liver undergoes progressive pathological alterations during aging, including mitochondrial dysfunction, ROS accumulation, and dysregulated lipid metabolism[7,59]. These changes drive chronic inflammation, cellular senescence and apoptosis, thereby accelerating age-related hepatic deterioration, such as steatotic liver disease, liver fibrosis and hepatocellular carcinoma (HCC)[4,60,61]. Central to this process, key pro-inflammatory signaling cascades such as NF-κB, TNF, and interferon-γ pathways exhibit remarkable upregulation in aging livers, amplifying tissue injury and dysfunction.
PCSK9 has emerged as a crucial mediator in exacerbating these age-related hepatic disorders. The interplay between PCSK9 and ROS production has been demonstrated using p47phox and gp91phox knockout mouse models, which revealed a direct link between PCSK9 expression and ROS generation[62,63]. Similarly, human ECs and SMCs treated with PCSK9 display concentration-dependent increases in ROS levels[62]. Elevated PCSK9 expression is further associated with increased apoptosis in VSMCs, coupled with mitochondrial morphological shifts toward fission and subsequent mitochondrial dysfunction[64].
In the liver, high PCSK9 levels are correlated with aggravated apoptosis, enhanced TNF signaling, and impaired cholesterol metabolism through suppression of LDLR activity. Transcriptomic analyses identified PCSK9 as a central differentially expressed gene (DEG) in aging-associated hepatic gene clusters across human and rat cohorts. Importantly, aging livers with elevated PCSK9 expression exhibit accelerated metabolic dysfunction and heightened oxidative stress compared to low-PCSK9 counterparts[7]. In summary, PCSK9 contributes to hepatic aging by amplifying oxidative stress, promoting apoptosis, and disrupting lipid and cholesterol homeostasis. Therapeutically, PCSK9 inhibition represents a promising strategy to attenuate liver aging by restoring metabolic balance and reducing inflammatory burden.
METABOLIC AND INFLAMMATORY AGING
PCSK9 and glucose metabolism
PCSK9 is abundantly produced in hepatocytes, neurons, and intestinal epithelial cells, where it participates in diverse physiological processes such as liver regeneration, programmed cell death, and insulin secretion. Its role in metabolic aging is complex and highly tissue-specific. Recent studies have highlighted novel functions of PCSK9 in pancreatic β-cells. Obesity- and hyperglycemia-induced ROS stimulate PCSK9 overexpression, which in turn exacerbates vascular damage, a process that can be reversed by PCSK9 inhibition[65]. The complexity of this relationship is further underscored by genetic evidence.
The role of PCSK9 in glucose regulation and the development of diabetes appears paradoxical. Elevated circulating PCSK9 levels are a common feature of both major forms of diabetes, including type 1 and type 2, suggesting its involvement in pancreatic β-cell dysfunction and insulin resistance[66,67]. Shi et al. further established a strong correlation between elevated PCSK9 levels and a greater likelihood of progressing to type 2 diabetes (T2DM) in prediabetic women[68], with this pathway potentially overlapping with statin-induced insulin resistance[69]. Intriguingly, short- and long-term treatment with the PCSK9i evolocumab does not increase diabetes risk, and may even reduce incidence compared to placebo[70]. However, genetic studies complicate this picture: PCSK9 variants linked to lower LDL-C levels and heightened T2DM susceptibility[71-73]. This apparent paradox may stem from the distinct role of locally produced PCSK9 in pancreatic islets, which operates independently of circulating PCSK9. Specifically, PCSK9 deficiency or LOF mutations in pancreatic δ-cells upregulate LDLR expression, leading to intracellular cholesterol accumulation, lipotoxicity, and ultimately impaired insulin secretion[74]. Collectively, these findings highlight the dual, tissue-specific regulatory mechanisms of PCSK9 in diabetes development.
Inflammaging
The role of PCSK9 in inflammaging and cytokine interactions constitutes a vital aspect of its age-related pathophysiology. A persistent, low-level inflammatory state is widely acknowledged as a fundamental driver of the aging process[75]. Within this framework, immunosenescence - the progressive aging of the immune system - functions as a key trigger in initiating and perpetuating inflammaging. The concept of inflammaging, originally proposed by Franceschi et al., refers to the characteristic age-associated upregulation of pro-inflammatory mediators that sustains a persistent, low-level inflammatory state[76]. This chronic inflammatory milieu is now understood to be a unifying mechanism underlying multiple age-related diseases, including T2DM, cardiovascular disorders, cognitive decline and cancer[75,77].
Beyond its metabolic functions, PCSK9 has also been implicated in skin aging. Ultraviolet B (UVB) radiation induces PCSK9 upregulation in macrophages, which activates interferon regulatory factor 3 (IRF3) and triggers stimulator of interferon genes (STING)-dependent inflammatory signaling[78]. This cascade ultimately promotes keratinocyte damage, accelerating skin aging processes.
PCSK9 in cancer
The involvement of PCSK9 in tumorigenesis and progression shows remarkable complexity, with its functions varying significantly across different cancer types, stages and even microenvironmental factors [Figure 3].
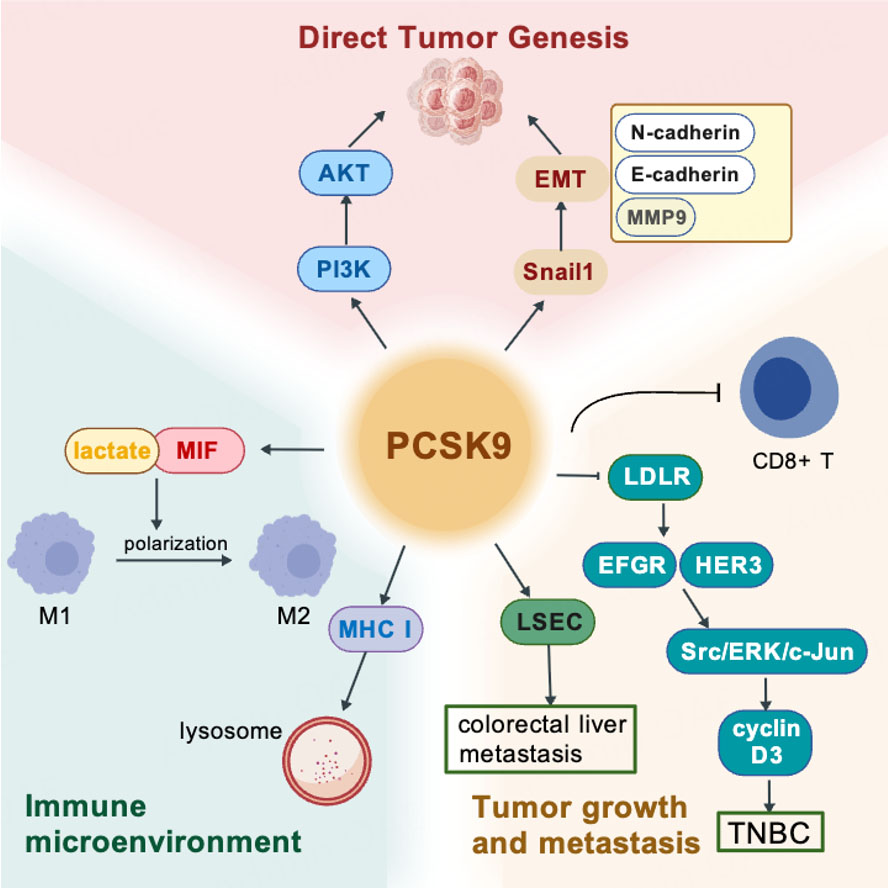
Figure 3. PCSK9's multifunction in cancer. Created with BioGDP.com[94].
Emerging literature presents contradictory findings regarding PCSK9 as a regulator of colorectal cancer (CRC) pathogenesis through both metabolic and direct oncogenic pathways. Clinical studies suggest a modest association between elevated LDL-C and CRC risk, independent of traditional metabolic factors such as HDL-C, triglycerides, body mess index (BMI) or diabetes[79]. However, genetic studies failed to demonstrate a clear link between reduced PCSK9 activity and CRC risk. In contrast, experimental evidence indicates that PCSK9 overexpression in colon cancer cells directly drives malignant progression through two synergistic mechanisms. First, it activates the PI3K/AKT signaling cascade, promoting epithelial-mesenchymal transition and thereby enhancing proliferation, migration and metastasis. Second, it reprograms the tumor microenvironment by promoting M2 macrophage phenotype polarization through lactate accumulation and regulating of macrophage migration inhibitory factor (MIF)[80]. These dual effects - direct promotion of tumor cell aggressiveness and remodeling of the immune microenvironment - position PCSK9 inhibition as a promising therapeutic strategy in CRC. Supporting this notion, clinical samples consistently show higher PCSK9 expression correlating with advanced tumor grade[80].
Distinct perspectives have emerged from studies of colorectal liver metastasis. PCSK9 expression was identified in human and murine liver sinusoidal endothelial cells (LSECs), and exposure to media conditioned by CRC stem cells resulted in a marked upregulation of its expression. Functionally, PCSK9 promoted LSEC activation, thereby creating a pro-metastatic microenvironment that facilitated colorectal liver metastasis[81]. This finding highlights a novel interaction between PCSK9 and CRC progression and suggests therapeutic potential in targeting the metastatic niche.
Beyond colorectal and liver cancers, PCSK9 has been implicated as a histology-dependent prognostic biomarker. RNA sequencing analyses revealed that elevated PCSK9 expression predicted improved overall patient survival in Luminal B breast cancer and ovarian cancer but forecasted a worse prognosis in bladder cancer, renal clear cell carcinoma, and pancreatic cancer[82]. However, PCSK9 exhibits heterogeneity in its role across cancers. In triple-negative breast cancer (TNBC), PCSK9 drives tumor progression by downregulating LDLR, leading to activation of epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor 3 (HER3) signaling, which promotes tumor cell growth and metastasis[83]. These divergent effects highlight variable and contingent functions of PCSK9 in cancer, likely influenced by intrinsic tumor characteristics such as histologic origin, basal oncogenic signaling pathways, lipid metabolic patterns and the immune landscape of the tumor microenvironment. Notably, tumors with constitutively active PI3K/AKT signaling and high LDLR expression (e.g., colorectal, pancreatic cancer) are particularly susceptible to PCSK9-mediated pro-tumorigenic effects. In contrast, hormone receptor-driven tumors (e.g., ovarian cancer) often lack these molecular prerequisites. These intrinsic tumor traits may serve as potential predictive biomarkers for PCSK9’s role in cancer progression, and could be critical for patient stratification in future PCSK9-targeted therapies.
PCSK9 also modulates immune surveillance within the tumor microenvironment. Mechanistically, PCSK9 binds to major histocompatibility complex class I (MHC I) proteins, diverting them to lysosomal degradation and thereby reducing surface expression. Both genetic deletion and antibody-mediated blockade of PCSK9 restore MHC I expression on the surface of cancer cells, which improves antigen presentation and promotes cytotoxic T lymphocyte (CTL) infiltration[84]. Similarly, in HCC, high-level PCSK9 expression correlates with poor overall survival and reduced presence of CD8+ T cells within tumors, while PCSK9 inhibition enhances elimination of tumor cells and synergizes with anti-programmed cell death protein 1 (PD-1) immunotherapy in vivo[85]. This antitumor efficacy of PCSK9 suppression requires the presence of CD8+ T-cells, and it acts through the LDLR-mammalian target of rapamycin complex 1 (mTORC1) signaling pathway.
Taken together, the evidence reveals the remarkable complexity of PCSK9’s role in tumorigenesis and cancer progression. Its effects vary not only across cancer types and disease stages but also within distinct microenvironmental contexts [Figure 2]. This variability provides both challenges and opportunities for developing PCSK9-targeted strategies, particularly in combination with immunotherapy.
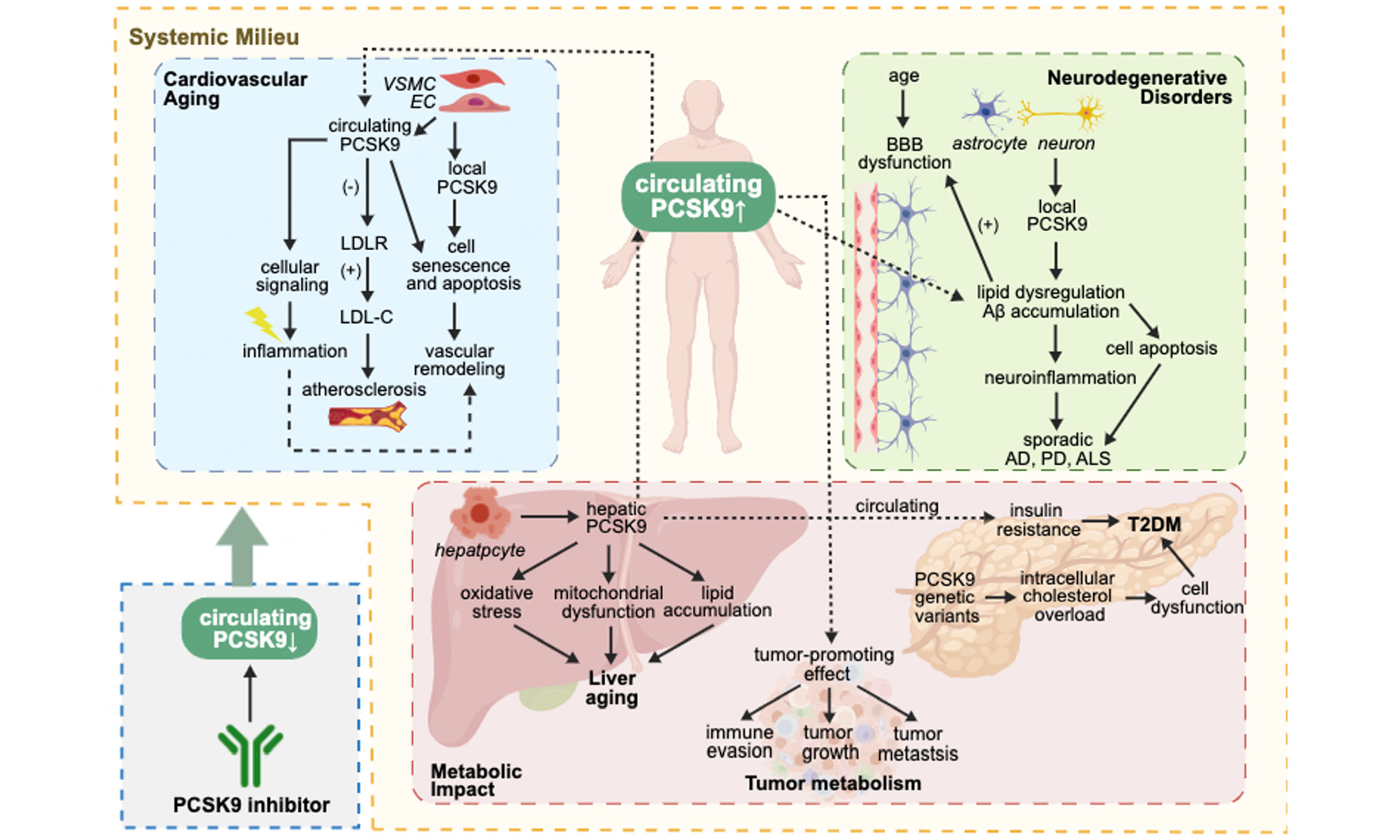
Systemic aging milieu and PCSK9 in multi-organ aging
Aging represents more than just the sum of organ dysfunction. It is a systemic process driven by intricate interactions across cells, and organs. Emerging research highlights the crucial role of systemic milieu changes in aging, characterized by inflammaging, metabolic dysregulation, and the accumulation of "pro-geronic" factors. These elements drive the gradual deterioration of function across multiple organ systems, particularly affecting vasculature and brain[86]. As a predominantly liver-derived secretory protein whose circulating levels rise with age, PCSK9 is well-positioned to serve as a key mediator within this systemic aging network.
Circulating factors from aged or senescent tissues can induce aging phenotypes in youth tissues. Transcriptomic studies revealed that aortic tissues and cerebromicrovascular ECs exhibit altered gene expression patterns during aging, which are partially reversed upon exposure to young blood[87,88]. Conversely, exposure to blood from aged individuals accelerates vascular aging[89], mediated by inflammatory signaling molecules, SASP components (e.g., eotaxin-1), and other circulating factors[86]. Within this paradigm, PCSK9’s role extends beyond its well-known hepatic LDLR regulation and may act synergistically with other humoral factors to exacerbate vascular and neuronal aging. For example, PCSK9 amplifies inflammation via the TLR4/NF-κB pathway - a mechanism also strongly activated by SASP factors such as follistatin-like protein 1 (FSTL1)[29,90,91]. Additionally, pro-geronic factors disrupt the BBB and impair cerebral microvascular function, facilitating the influx of circulating proteins including PCSK9. By converging with local inflammatory and oxidative stress pathways, PCSK9 exacerbates neuroinflammation, accelerates cognitive decline, and promotes age-related neurodegenerative diseases[40,92]. Thus, the systemic milieu acts as a critical conduit linking hepatic PCSK9 secretion to remote aging effects in the brain.
CONCLUSION
This study sets out to gain a better understanding of PCSK9 as a pivotal regulator of aging processes across multiple organ systems, extending far beyond its well-known role in LDL cholesterol metabolism [Figure 4]. In summary, PCSK9 serves as a multifunctional molecule within the aging organism, integrating metabolic, inflammatory, and senescent pathways across cardiovascular, neural, hepatic, and systemic compartments. In cardiovascular aging, PCSK9 drives atherosclerosis through both lipid-regulatory and non-lipid-regulatory mechanisms and exacerbates vascular senescence, while its inhibition demonstrates protective effects. These effects differ depending on the origin of PCSK9, with systemic and local PCSK9 contributing distinctly and interactively. In the CNS, PCSK9 contributes to Alzheimer's pathology through Aβ accumulation and neuroinflammation, yet exhibits complex, disease-specific effects in other neurodegenerative disorders. The protease further influences hepatic aging by accelerating metabolic dysfunction and oxidative stress. Furthermore, PCSK9 actively participates in systemic aging phenotype by synergizing with inflammatory cytokines and other systemic milieu factors. This intricate mechanism exacerbates metabolic dysfunction, closely linking to the progression of T2DM, inflammaging, and context-dependent roles in cancer progression. These results suggest that PCSK9 acts in various biological processes and reveals a mechanism of complexity, positioning it as a modulator of the systemic aging landscape.
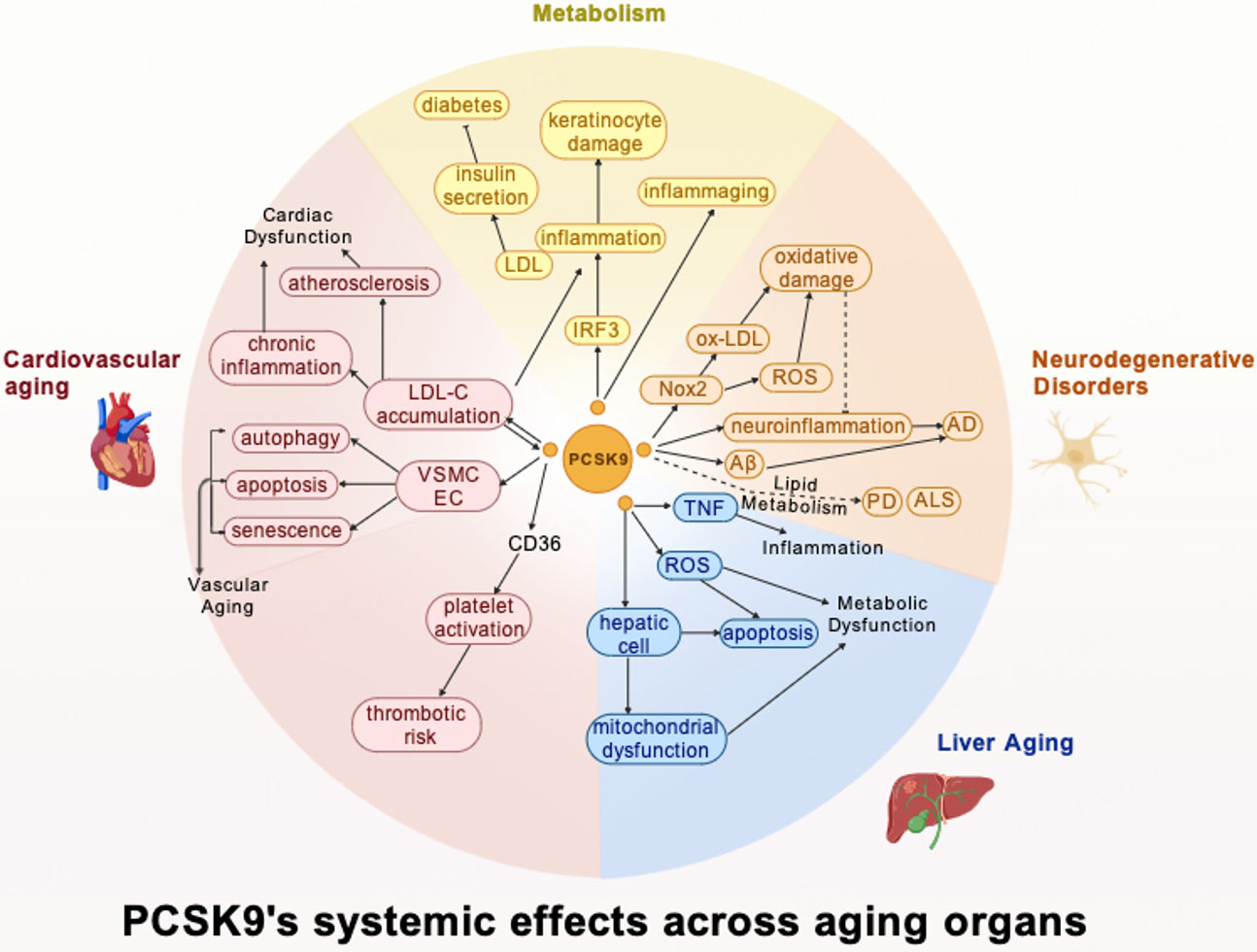
Figure 4. PCSK9's systemic effects across aging organs. Created with BioGDP.com[94].
Despite these advances, critical knowledge gaps remain in understanding PCSK9's systemic role in aging and disease. First, current research lacks comprehensive investigations of PCSK9's whole-body functions and multi-tissue interactions, requiring cautious interpretation of existing findings. Clinical observations reveal puzzling contradictions - such as its opposing effects on PD versus ALS risk and context-dependent impacts on tumor prognosis, highlighting the need for more sophisticated, human-relevant models and replicated experiments. Future studies should prioritize longitudinal clinical studies to clarify PCSK9's temporal dynamics in aging processes, alongside mechanistic work in metabolic signaling networks. Therapeutically, while PCSK9 inhibition shows promise for mitigating multiple age-related pathologies[93], personalized approaches must carefully balance benefits (e.g., cardiovascular protection) against potential risks (e.g., pancreatic β-cell dysfunction). Integrating PCSK9-targeted therapies with established anti-aging interventions could yield synergistic benefits, offering new avenues to address the complex challenges of aging-related diseases.
DECLARATIONS
Acknowledgment
The Graphical Abstract was created using BioGDP.com.
Authors’ contributions
Wrote the initial draft: Huang K
Involved in editing and revising the manuscript: Huang K, Ren J
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
Ren J is an Editorial Board Member of The Journal of Cardiovascular Aging. Ren J was not involved in any steps of the editorial process, notably including reviewers' selection, manuscript handling, or decision-making. Huang K declares that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Costantino S, Paneni F, Cosentino F. Ageing, metabolism and cardiovascular disease. J Physiol. 2016;594:2061-73.
2. Ferrucci L, Fabbri E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol. 2018;15:505-22.
4. Sanfeliu-Redondo D, Gibert-Ramos A, Gracia-Sancho J. Cell senescence in liver diseases: pathological mechanism and theranostic opportunity. Nat Rev Gastroenterol Hepatol. 2024;21:477-92.
5. Abavisani M, Faraji S, Ebadpour N, Karav S, Sahebkar A. Beyond the hayflick limit: how microbes influence cellular aging. Ageing Res Rev. 2025;104:102657.
6. Ajoolabady A, Pratico D, Bahijri S, et al. Hallmarks and mechanisms of cellular senescence in aging and disease. Cell Death Discov. 2025;11:364.
7. Arif M, Matyas C, Mukhopadhyay P, et al. Data-driven transcriptomics analysis identifies PCSK9 as a novel key regulator in liver aging. Geroscience. 2023;45:3059-77.
8. Arif M, Zhang C, Li X, et al. iNetModels 2.0: an interactive visualization and database of multi-omics data. Nucleic Acids Res. 2021;49:W271-6.
9. Tan D, Yang X, Yang J, Fan G, Xiong G. PCSK9 in vascular aging and age-related diseases. Aging Dis. 2025;17:691-711.
10. Csiszar A, Tarantini S, Yabluchanskiy A, Ungvari Z. PCSK9: an emerging player in cardiometabolic aging and its potential as a therapeutic target and biomarker. Geroscience. 2024;46:257-63.
11. Seidah NG, Benjannet S, Wickham L, et al. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proc Natl Acad Sci USA. 2003;100:928-33.
12. Abifadel M, Varret M, Rabès JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34:154-6.
13. Oleaga C, Hay J, Gurcan E, et al. Insights into the kinetics and dynamics of the furin-cleaved form of PCSK9. J Lipid Res. 2021;62:100003.
15. Ferri N, Tibolla G, Pirillo A, et al. Proprotein convertase subtilisin kexin type 9 (PCSK9) secreted by cultured smooth muscle cells reduces macrophages LDLR levels. Atherosclerosis. 2012;220:381-6.
16. Wang X, Liu L, Zhai L, Palade P, Wang X, Mehta JL. Direct impact of PCSK9 on SMC senescence and apoptosis: a new focus in cardiovascular diseases. Arterioscler Thromb Vasc Biol. 2024;44:1491-6.
17. Ajoolabady A, Pratico D, Mazidi M, et al. PCSK9 in metabolism and diseases. Metabolism. 2025;163:156064.
18. Cupido AJ, Reeskamp LF, Hingorani AD, et al. Joint genetic inhibition of PCSK9 and CETP and the association with coronary artery disease: a factorial mendelian randomization study. JAMA Cardiol. 2022;7:955-64.
19. Carreras A, Pane LS, Nitsch R, et al. In vivo genome and base editing of a human PCSK9 knock-in hypercholesterolemic mouse model. BMC Biol. 2019;17:4.
20. Nicholls SJ, Puri R, Anderson T, et al. Effect of evolocumab on progression of coronary disease in statin-treated patients: the GLAGOV randomized clinical trial. JAMA. 2016;316:2373-84.
21. Punch E, Klein J, Diaba-Nuhoho P, Morawietz H, Garelnabi M. Effects of PCSK9 targeting: alleviating oxidation, inflammation, and atherosclerosis. J Am Heart Assoc. 2022;11:e023328.
22. Kong N, Xu Q, Cui W, Feng X, Gao H. PCSK9 inhibitor inclisiran for treating atherosclerosis via regulation of endothelial cell pyroptosis. Ann Transl Med. 2022;10:1205.
23. Liu Y, Zhao Y, Feng P, Jiang H. PCSK9 inhibitor attenuates atherosclerosis by regulating SNHG16/EZH2/TRAF5-mediated VSMC proliferation, migration, and foam cell formation. Cell Biol Int. 2023;47:1267-80.
24. Langsted A, Nordestgaard BG, Benn M, Tybjærg-Hansen A, Kamstrup PR. PCSK9 R46L Loss-of-function mutation reduces lipoprotein(a), LDL cholesterol, and risk of aortic valve stenosis. J Clin Endocrinol Metab. 2016;101:3281-7.
25. Ostadal P, Steg PG, Poulouin Y, et al. Metabolic risk factors and effect of alirocumab on cardiovascular events after acute coronary syndrome: a post-hoc analysis of the ODYSSEY OUTCOMES randomised controlled trial. Lancet Diabetes Endocrinol. 2022;10:330-40.
26. Toulassi IA, Al Saedi UA, Gutlapalli SD, et al. A paradigm shift in the management of atherosclerosis: protective role of sirtuins in atherosclerosis. Cureus. 2021;13:e12735.
27. Chen H, Zhou X, Hu J, et al. Genetic insights into the association of statin and newer nonstatin drug target genes with human longevity: a Mendelian randomization analysis. Lipids Health Dis. 2023;22:220.
28. Gusev E, Sarapultsev A. Atherosclerosis and inflammation: insights from the theory of general pathological processes. Int J Mol Sci. 2023;24:7910.
29. Shin D, Kim S, Lee H, et al. PCSK9 stimulates Syk, PKCδ, and NF-κB, leading to atherosclerosis progression independently of LDL receptor. Nat Commun. 2024;15:2789.
30. Kataoka Y, Harada-Shiba M, Hori M, et al. Circulating furin-cleaved proprotein convertase subtilisin/kexin type 9 concentration predicts future coronary events in Japanese subjects. JACC Asia. 2021;1:360-8.
31. College of Cardiovascular Physicians. Chinese expert consensus on anti-thrombotic therapy for pan-vascular diseases (2023 edition). Cardiol Plus. 2024;9:49-69.
32. Matyas C, Trojnar E, Zhao S, et al. PCSK9, A promising novel target for age-related cardiovascular dysfunction. JACC Basic Transl Sci. 2023;8:1334-53.
33. Qi Z, Hu L, Zhang J, et al. PCSK9 (proprotein convertase subtilisin/Kexin 9) enhances platelet activation, thrombosis, and myocardial infarct expansion by binding to platelet CD36. Circulation. 2021;143:45-61.
34. Abdellatif M, Schmid ST, Fuerlinger A, Kroemer G. Anti-ageing interventions for the treatment of cardiovascular disease. Cardiovasc Res. 2025;121:1524-36.
35. Guo Y, Tang Z, Yan B, et al. PCSK9 (proprotein convertase subtilisin/kexin type 9) triggers vascular smooth muscle cell senescence and apoptosis: implication of its direct role in degenerative vascular disease. Arterioscler, Thromb, Vasc Biol. 2022;42:67-86.
36. Ajoolabady A, Pratico D, Bahijri S, Tuomilehto J, Uversky VN, Ren J. Hallmarks of cellular senescence: biology, mechanisms, regulations. Exp Mol Med. 2025;57:1482-91.
37. Wang Y, Cao S, Wang Z, et al. PCSK9 affects vascular senescence through the SIRT1 pathway. Exp Gerontol. 2025;201:112701.
38. Ungvari Z, Tarantini S, Sorond F, Merkely B, Csiszar A. Mechanisms of vascular aging, a geroscience perspective: JACC focus seminar. J Am Coll Cardiol. 2020;75:931-41.
39. Lee RG, Mazzola AM, Braun MC, et al. Efficacy and safety of an investigational single-course CRISPR base-editing therapy targeting PCSK9 in nonhuman primate and mouse models. Circulation. 2023;147:242-53.
40. Sweeney MD, Zhao Z, Montagne A, Nelson AR, Zlokovic BV. Blood-brain barrier: from physiology to disease and back. Physiol Rev. 2019;99:21-78.
41. Jaafar AK, Techer R, Chemello K, Lambert G, Bourane S. PCSK9 and the nervous system: a no-brainer? J Lipid Res. 2023;64:100426.
42. Montagne A, Barnes SR, Sweeney MD, et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron. 2015;85:296-302.
43. Poirier S, Prat A, Marcinkiewicz E, et al. Implication of the proprotein convertase NARC-1/PCSK9 in the development of the nervous system. J Neurochem. 2006;98:838-50.
44. Papotti B, Adorni MP, Marchi C, et al. PCSK9 affects astrocyte cholesterol metabolism and reduces neuron cholesterol supplying in vitro: potential implications in Alzheimer's disease. Int J Mol Sci. 2022:23.
45. Piao MX, Bai JW, Zhang PF, Zhang YZ. PCSK9 regulates apoptosis in human neuroglioma u251 cells via mitochondrial signaling pathways. Int J Clin Exp Pathol. 2015;8:2787-94.
46. Zhao XS, Wu Q, Peng J, et al. Hyperlipidemia-induced apoptosis of hippocampal neurons in apoE-/- mice may be associated with increased PCSK9 expression. Mol Med Rep. 2017;15:712-8.
47. Vilella A, Bodria M, Papotti B, et al. PCSK9 ablation attenuates Aβ pathology, neuroinflammation and cognitive dysfunctions in 5XFAD mice. Brain Behav Immun. 2024;115:517-34.
48. Papotti B, Palumbo M, Adorni MP, et al. Influence of APOE4 genotype on PCSK9-lipids association in cerebrospinal fluid and serum of patients in the Alzheimer's disease continuum. J Alzheimers Dis. 2024;102:162-72.
49. Halliday MR, Rege SV, Ma Q, et al. Accelerated pericyte degeneration and blood-brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer's disease. J Cereb Blood Flow Metab. 2016;36:216-27.
50. Courtemanche H, Bigot E, Pichelin M, et al. PCSK9 concentrations in cerebrospinal fluid are not specifically increased in Alzheimer's disease. J Alzheimers Dis. 2018;62:1519-25.
51. Teleanu DM, Niculescu AG, Lungu II, et al. An overview of oxidative stress, neuroinflammation, and neurodegenerative diseases. Int J Mol Sci. 2022;23:5938.
52. Park LM, Pacher P, Lohoff FW. Targeting oxidative stress in neurodegenerative disorders: a novel role for PCSK9 inhibition? ACS Chem Neurosci. 2024;15:2662-4.
53. Rosoff DB, Bell AS, Jung J, Wagner J, Mavromatis LA, Lohoff FW. Mendelian randomization study of PCSK9 and HMG-CoA reductase inhibition and cognitive function. J Am Coll Cardiol. 2022;80:653-62.
54. Huang Q, Zhang Q, Cao B. Causal relationship between PCSK9 inhibitor and common neurodegenerative diseases: A drug target Mendelian randomization study. Brain Behav. 2024;14:e3543.
55. Xia K, Huang N, Wang Y, et al. Shared genetic link and causal inference between blood lipids, lipid-lowering drugs and amyotrophic lateral sclerosis. Neural Regen Res. 2025.
56. Kysenius K, Muggalla P, Mätlik K, Arumäe U, Huttunen HJ. PCSK9 regulates neuronal apoptosis by adjusting ApoER2 levels and signaling. Cell Mol Life Sci. 2012;69:1903-16.
57. Wang L, Wang Z, Shi J, et al. Inhibition of proprotein convertase subtilisin/kexin type 9 attenuates neuronal apoptosis following focal cerebral ischemia via apolipoprotein E receptor 2 downregulation in hyperlipidemic mice. Int J Mol Med. 2018;42:2098-106.
58. Jahed MR, Habibi SAH, Vaseghi G, Amiri H, Montazeri H, Eshraghi A. Association between plasma PCSK9 levels and lipid profile in patients with Parkinson's disease and comparison with healthy subjects. Curr J Neurol. 2022;21:236-43.
59. Xu X, Pang Y, Fan X. Mitochondria in oxidative stress, inflammation and aging: from mechanisms to therapeutic advances. Signal Transduct Target Ther. 2025;10:190.
60. Li X, Li C, Zhang W, Wang Y, Qian P, Huang H. Inflammation and aging: signaling pathways and intervention therapies. Signal Transduct Target Ther. 2023;8:239.
61. Mohammed S, Thadathil N, Selvarani R, et al. Necroptosis contributes to chronic inflammation and fibrosis in aging liver. Aging Cell. 2021;20:e13512.
62. Ding Z, Liu S, Wang X, et al. PCSK9 regulates expression of scavenger receptors and ox-LDL uptake in macrophages. Cardiovasc Res. 2018;114:1145-53.
63. Ding Z, Liu S, Wang X, et al. Hemodynamic shear stress via ROS modulates PCSK9 expression in human vascular endothelial and smooth muscle cells and along the mouse aorta. Antioxid Redox Signaling. 2015;22:760-71.
64. Xu R, Li T, Luo J, et al. PCSK9 increases vulnerability of carotid plaque by promoting mitochondrial dysfunction and apoptosis of vascular smooth muscle cells. CNS Neurosci Ther. 2024;30:e14640.
65. Barale C, Tempesta G, Melchionda E, et al. PCSK9 expression in vascular smooth muscle cells: role of insulin resistance and high glucose. Int J Mol Sci. 2025;26:1003.
66. Levenson AE, Shah AS, Khoury PR, et al. Obesity and type 2 diabetes are associated with elevated PCSK9 levels in young women. Pediatr Diabetes. 2017;18:755-60.
67. Levenson AE, Wadwa RP, Shah AS, et al. PCSK9 is increased in youth with type 1 diabetes. Diabetes Care. 2017;40:e85-7.
68. Shi J, Zhang W, Niu Y, et al. Association of circulating proprotein convertase subtilisin/kexin type 9 levels and the risk of incident type 2 diabetes in subjects with prediabetes: a population-based cohort study. Cardiovasc Diabetol. 2020;19:209.
69. Shu X, Wu J, Zhang T, et al. Statin-induced geranylgeranyl pyrophosphate depletion promotes PCSK9-dependent adipose insulin resistance. Nutrients. 2022;14:5314.
70. O'Donoghue ML, Giugliano RP, Wiviott SD, et al. Long-term evolocumab in patients with established atherosclerotic cardiovascular disease. Circulation. 2022;146:1109-19.
71. Schmidt AF, Swerdlow DI, Holmes MV, et al. PCSK9 genetic variants and risk of type 2 diabetes: a mendelian randomisation study. Lancet Diabetes Endocrinol. 2017;5:97-105.
72. Lotta LA, Sharp SJ, Burgess S, et al. Association between low-density lipoprotein cholesterol-lowering genetic variants and risk of type 2 diabetes: a meta-analysis. JAMA. 2016;316:1383-91.
73. Liu S, Wan J, Wang D, et al. Effect of the PCSK9 R46L genetic variant on plasma insulin and glucose levels, risk of diabetes mellitus and cardiovascular disease: a meta-analysis. Nutr Metab Cardiovasc Dis. 2024;34:1339-51.
74. Da Dalt L, Ruscica M, Bonacina F, et al. PCSK9 deficiency reduces insulin secretion and promotes glucose intolerance: the role of the low-density lipoprotein receptor. Eur Heart J. 2019;40:357-68.
75. Ajoolabady A, Pratico D, Vinciguerra M, Lip GYH, Franceschi C, Ren J. Inflammaging: mechanisms and role in the cardiac and vasculature. Trends Endocrinol Metab. 2023;34:373-87.
76. Franceschi C, Bonafè M, Valensin S, et al. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 2000;908:244-54.
77. Ajoolabady A, Pratico D, Tang D, Zhou S, Franceschi C, Ren J. Immunosenescence and inflammaging: mechanisms and role in diseases. Ageing Res Rev. 2024;101:102540.
78. Luan C, He Y, Liu W, et al. PCSK9 inhibition interrupts the cross-talk between keratinocytes and macrophages and prevents UVB-induced skin damage. J Biol Chem. 2023;299:104895.
79. Chan WC, Liu L, Bouras E, et al. Associations of blood lipids and LDL cholesterol lowering drug-targets with colorectal cancer risk: a Mendelian randomisation study. Br J Cancer. 2025;132:103-10.
80. Wang L, Li S, Luo H, Lu Q, Yu S. PCSK9 promotes the progression and metastasis of colon cancer cells through regulation of EMT and PI3K/AKT signaling in tumor cells and phenotypic polarization of macrophages. J Exp Clin Cancer Res. 2022;41:303.
81. Martin A, Gerovska D, Arauzo-Bravo MJ, et al. Inhibition of PCSK9 Attenuates liver endothelial cell activation induced by colorectal cancer stem cells during liver metastasis. Cancers. 2025;17:1977.
82. Ungvari Z, Menyhart O, Lehoczki A, Fekete M, Bianchini G, Győrffy B. PCSK9 expression and cancer survival: a prognostic biomarker at the intersection of oncology and geroscience. Geroscience. 2026;48:435-49.
83. Li T, Wu R, Luo KQ. PCSK9 promotes the malignancy of triple-negative breast cancer cells by reducing cholesterol levels at the plasma membrane to activate EGFR and HER3. Adv Sci. 2025;12:e2408514.
84. Liu X, Bao X, Hu M, et al. Inhibition of PCSK9 potentiates immune checkpoint therapy for cancer. Nature. 2020;588:693-8.
85. Xu W, Hu M, Lu X, et al. Inhibition of PCSK9 enhances the anti-hepatocellular carcinoma effects of TCR-T cells and anti-PD-1 immunotherapy. Int J Biol Sci. 2024;20:3942-55.
86. Gulej R, Patai R, Ungvari A, et al. Impacts of systemic milieu on cerebrovascular and brain aging: insights from heterochronic parabiosis, blood exchange, and plasma transfer experiments. Geroscience. 2025;47:6207-376.
87. Ximerakis M, Holton KM, Giadone RM, et al. Heterochronic parabiosis reprograms the mouse brain transcriptome by shifting aging signatures in multiple cell types. Nat Aging. 2023;3:327-45.
88. Kiss T, Tarantini S, Csipo T, et al. Circulating anti-geronic factors from heterochonic parabionts promote vascular rejuvenation in aged mice: transcriptional footprint of mitochondrial protection, attenuation of oxidative stress, and rescue of endothelial function by young blood. Geroscience. 2020;42:727-48.
89. Kiss T, Nyúl-Tóth Á, Gulej R, et al. Old blood from heterochronic parabionts accelerates vascular aging in young mice: transcriptomic signature of pathologic smooth muscle remodeling. Geroscience. 2022;44:953-81.
90. Tang ZH, Peng J, Ren Z, et al. New role of PCSK9 in atherosclerotic inflammation promotion involving the TLR4/NF-κB pathway. Atherosclerosis. 2017;262:113-22.
91. Yan X, Ding JY, Zhang RJ, et al. FSTL1 accelerates nucleus pulposus cell senescence and intervertebral disc degeneration through TLR4/NF-κB pathway. Inflammation. 2024;47:1229-47.
92. Sweeney MD, Sagare AP, Zlokovic BV. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. 2018;14:133-50.
93. Zhang G, Chang S, Zhao F, et al. The benefits of PCSK9 inhibitors in patients with acute coronary syndrome: a systematic review and meta-analysis. Emerg Crit Care Med. 2024;4:28-34.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].