


Epigenetic and epitranscriptomic regulation of cardiac metabolism in aging and disease
 0
0 Abstract
The heart’s metabolic plasticity, crucial for adapting to energy demands, is governed by epigenetic and epitranscriptomic mechanisms. Aging and cardiovascular diseases disrupt this equilibrium, leading to metabolic inflexibility, mitochondrial dysfunction, and pathological remodeling. This review explores how DNA methylation, histone modifications, and RNA methylation (e.g., m6A, m5C) dynamically regulate cardiac metabolism. Key findings reveal that age-related declines in SIRT1/NAD+ activity and FTO-mediated RNA demethylation impair fatty acid oxidation, while METTL3-driven m6A hypermethylation promotes glycolytic dependency. Dysregulation of TET enzymes and α-ketoglutarate (α-KG) further disrupts DNA hydroxymethylation and RNA modification, exacerbating oxidative stress and mitochondrial inefficiency. These alterations create self-reinforcing cycles of metabolic rigidity, contributing to heart failure, arrhythmias, and ischemia-reperfusion injury. Emerging therapeutic strategies, including BET inhibitors and NAD+ repletion, show promise in restoring metabolic flexibility by targeting epigenetic and epitranscriptomic pathways. Integrating multi-omics approaches and spatial epitranscriptomics offers novel insights into cell-specific regulatory networks, paving the way for precision interventions to counteract cardiac aging and disease.
Keywords
INTRODUCTION
Cardiovascular disease (CVD) remains the leading cause of death and disability worldwide, with the elderly population bearing a disproportionate burden. As the global population ages, the prevalence of CVD in older adults is expected to rise, posing significant challenges to healthcare systems. For instance, in the United States, the age-adjusted prevalence of heart disease in adults aged 18 and over was 5.5% in 2019, with higher rates observed in older age groups[1,2]. Moreover, the burden of CVD in the elderly is compounded by the presence of multiple comorbidities, such as frailty, obesity, and diabetes, which exacerbate risks and complicate management[3,4]. The heart, an organ with unparalleled energy demands, sustains its physiological function through a tightly regulated metabolic equilibrium, where a predominant fraction (~60%-90%) of ATP production in adults is derived from fatty acid β-oxidation, complemented by glucose, ketone, and lactate metabolism[5]. This metabolic plasticity ensures adaptability to fluctuating energy requirements, underpinning contractile efficiency and electrophysiological stability. However, aging perturbs this balance, often leading to a shift characterized by diminished mitochondrial fatty acid oxidation and increased reliance on glycolysis, reminiscent of fetal metabolic patterns[6]. This transition is associated with altered CPT1 and PDK4 expression, impairing fatty acid oxidation and uncoupling glucose oxidation from ATP synthesis[7,8]. The ensuing metabolic inflexibility reduces energy yield, promotes the accumulation of metabolic byproducts (e.g., excess lactate and reactive oxygen species), and triggers mitochondrial dysfunction, contributing to cardiomyocyte stress, interstitial fibrosis, and a proarrhythmic environment. These alterations collectively drive the progression of age-associated cardiovascular pathologies, including heart failure with preserved ejection fraction (HFpEF) and atrial fibrillation, highlighting the central role of metabolic dysregulation in cardiac aging.
Despite advances in medical therapy and interventions, conventional treatments for CVD in the elderly often fall short due to several limitations. Older adults are frequently underrepresented in clinical trials, leading to a paucity of evidence-based guidelines tailored to this demographic. Additionally, age-related changes in pharmacokinetics and pharmacodynamics increase the risk of adverse drug events, necessitating careful medication management. Furthermore, the presence of multimorbidity and geriatric syndromes requires a holistic, patient-centered approach that considers not only the cardiovascular condition but also the patient’s overall health status, functional ability, and personal preferences. Epigenetic mechanisms, encompassing DNA methylation, histone post-translational modifications, and chromatin remodeling, govern transcriptional programs without altering the genetic code, serving as dynamic rheostats of cardiac metabolism[9]. DNA methylation, catalyzed by DNMTs at CpG islands, is implicated in the repression of fatty acid oxidation genes, including PPARα and potentially CPT1A[10]. Concurrently, histone acetylation dynamics, regulated by antagonistic HATs and HDACs, modulate chromatin accessibility of metabolic gene clusters[11,12]. Notably, SIRT1, an NAD+-dependent class III HDAC, safeguards mitochondrial biogenesis primarily by deacetylating transcription factors like PGC-1α[13]. Beyond chromatin-based regulation, the emerging field of epitranscriptomics reveals RNA modifications - including well-characterized m6A, as well as potential roles of m5C and pseudouridylation (Ψ) - as post-transcriptional regulators of cardiac metabolism[14-16].
In light of these challenges, there is a pressing need for comprehensive reviews that synthesize the latest research on cardiac metabolism and aging, with a focus on identifying novel therapeutic targets and strategies that address the unique needs of the elderly population. Despite significant progress, fundamental questions remain regarding how epigenetic and epitranscriptomic mechanisms orchestrate metabolic reprogramming during cardiac aging and disease. A key challenge lies in understanding the temporal dynamics of epigenetic modifiers such as DNMT3A and SIRT6 - how do their fluctuating activities across the lifespan influence stage-specific metabolic shifts, including the regulation of enzymes like PDK4 and succinate dehydrogenase? Additionally, the cell-type specificity of these regulatory layers is poorly defined. For example, do cardiac fibroblasts exhibit distinct DNA hydroxymethylation patterns compared to cardiomyocytes? Furthermore, how do macrophage-derived extracellular vesicles deliver epitranscriptomic modifiers to influence myocardial metabolism? Another critical frontier is the bidirectional interplay between metabolic intermediates and the epigenetic landscape. For instance, age-related declines in α-ketoglutarate - a crucial cofactor for TET enzymes and RNA demethylases[17] - may contribute to metabolic rigidity, but whether this creates a self-reinforcing cycle remains to be determined. Moreover, the therapeutic potential of targeting these pathways is still underexplored. Could pharmacological inhibition of BET bromodomains or activation of FTO help mitigate age-associated metabolic inflexibility? If so, what are the implications for diastolic dysfunction or mitochondrial ROS accumulation? Recent advances in single-nucleus ATAC-seq have provided unprecedented insights into cell-specific chromatin accessibility changes in aged hearts[18], while spatial epitranscriptomic mapping of m6A modifications has revealed alterations in failing myocardium[19]. Integrating these high-resolution techniques with metabolomic flux analysis and CRISPR-based epigenome editing will be crucial for constructing a comprehensive multiscale model of cardiac aging. Such a model could ultimately enable the development of precision interventions aimed at restoring metabolic plasticity and counteracting the deleterious effects of aging on cardiovascular health.
EPIGENETIC CONTROL OF CARDIAC METABOLISM
In the physiological regulation of cardiac metabolism, epigenetic mechanisms orchestrate a dynamic equilibrium between fatty acid oxidation and glucose utilization, ensuring metabolic flexibility to meet the heart’s fluctuating energy demands. DNA methylation, mediated by DNA methyltransferases (DNMTs), regulates fatty acid oxidation pathways by modifying CpG islands within gene promoters[20]. The peroxisome proliferator-activated receptor alpha (PPARα), a master regulator of lipid metabolism, remains transcriptionally active under basal conditions due to hypomethylation at its promoter and enhancer regions[21]. This permissive methylation landscape enables PPARα to drive the expression of key metabolic genes, such as CPT1A and ACADL, which facilitate mitochondrial fatty acid uptake and β-oxidation[22,23]. DNMT3A, in coordination with methyl-CpG-binding protein 2 (MeCP2), modulates this system by repressing specific loci to fine-tune lipid metabolism in response to nutritional states, although the precise regulatory targets require further elucidation[24]. In parallel, histone acetylation-deacetylation cycles, governed by histone acetyltransferases (HATs) and histone deacetylases (HDACs), dynamically regulate glycolytic and glycogenolytic gene expression[25]. Under normoxic conditions, class II HDACs (e.g., HDAC4/5) suppress excessive glycolytic activation, in part by modulating transcription factors such as MEF2, which control glucose transporter (GLUT1, GLUT4) and hexokinase 2 (HK2) expression[26-28]. SIRT1 further refines this balance by deacetylating and activating PGC-1α, promoting mitochondrial biogenesis and coupling glucose-derived pyruvate to the TCA cycle[29]. Additionally, chromatin remodeling by the SWI/SNF complex modulates nucleosome positioning, facilitating dynamic transcriptional responses to metabolic cues[30]. This ensures that key transcription factors - such as PGC-1α and FOXO1 - can access regulatory elements in a substrate-dependent manner, allowing rapid metabolic adaptations. Collectively, this epigenetically governed regulatory network enables the heart to efficiently transition between energy substrates while preventing toxic metabolite accumulation, highlighting the intricate interplay between chromatin architecture and metabolic demand sensing.
The aging heart exhibits profound epigenetic dysregulation that disrupts metabolic plasticity, exemplified by the decline in SIRT1 deacetylase activity and aberrant DNA methylation landscapes, both converging to impair mitochondrial energetics and fuel substrate utilization[31,32]. Age-related NAD+ depletion, driven by reduced NAMPT expression and CD38 upregulation, diminishes SIRT1 activity, leading to histone hyperacetylation at glycolytic gene promoters (e.g., HK2) and dysregulation of fatty acid oxidation (FAO) pathways[33-36]. This shift locks the heart into a glucose-dependent state, characterized by downregulation of PPARα target genes such as CPT1A, which restricts mitochondrial fatty acid uptake, and a compensatory increase in GLUT1-mediated glucose transport - hallmarks of metabolic inflexibility[37]. Concomitantly, reduced SIRT1 activity impairs the deacetylation of key mitochondrial proteins, exacerbating oxidative stress[38]. Additionally, SIRT1-mediated regulation of PGC-1α-dependent transcriptional programs is disrupted, compromising NRF1-driven mitochondrial biogenesis and further impairing electron transport chain (ETC) efficiency[39]. These interrelated defects culminate in ROS-mediated damage to mtDNA and metabolic enzymes, reinforcing the decline in mitochondrial function and accelerating cardiac aging.
Parallel to SIRT1 dysfunction, aging remodels the cardiac methylome through altered DNMT3A and TET enzyme activity, establishing repressive methylation signatures at loci critical for mitochondrial homeostasis[40,41]. Age-related increases in DNMT3A expression, observed in certain cardiac cell types, promote hypermethylation of the TFAM promoter, encoding a key mtDNA packaging factor, thereby impairing mitochondrial transcription and replication[42]. Meanwhile, global hypomethylation at retrotransposon elements (e.g., LINE-1) has been implicated in genomic instability and heightened inflammatory signaling, though its precise role in cardiac aging warrants further investigation[43]. Notably, PPARα and CPT1B enhancers acquire age-dependent 5mC modifications via DNMT3A, repressing their transcription and diminishing FAO capacity[44-47]. Conversely, the TET2-mediated oxidation of 5mC to 5-hydroxymethylcytosine (5hmC) - a process requiring α-ketoglutarate (α-KG) - declines with age, likely due to reduced IDH2 activity and broader TCA cycle dysregulation, locking pro-glycolytic genes like LDHA in a transcriptionally active state[48-50]. This imbalance shifts cardiac metabolism toward a glycolysis-dependent phenotype, reinforcing metabolic inflexibility. Further compounding these changes, circadian epigenetic regulation becomes disrupted, as age-related blunting of BMAL1/CLOCK oscillations diminishes rhythmic SIRT1 expression, impairing its role in coordinating metabolic phase transitions[51]. Additionally, DNMT3A exhibits time-of-day-dependent rhythmic expression, with its activity peaking nocturnally, suggesting a role in regulating substrate utilization rhythms[52,53]. For instance, DNMT3A-mediated methylation of metabolic genes like PPARα and CPT1B, which control fatty acid oxidation, may exhibit circadian variation, influencing the daily switch between glucose and fatty acid utilization[54]. However, aging disrupts these rhythms, leading to dysregulated DNMT3A activity and loss of precise control over substrate utilization[55]. The resultant metabolic rigidity manifests as reduced ATP synthesis, lipid droplet accumulation, and NADH/NAD+ imbalance, which, in turn, disrupts epigenetic homeostasis by altering sirtuin activity and methyl donor (SAM/SAH) availability[56]. These interwoven defects in epigenetic-metabolic crosstalk contribute to mitochondrial dysfunction, diastolic dysfunction, and increased susceptibility to ischemic injury and arrhythmogenesis, highlighting the profound impact of epigenetic remodeling on cardiac senescence.
In the pathological landscape of heart failure, dysregulated DNA hydroxymethylation emerges as a key contributor to maladaptive metabolic reprogramming, disrupting the balance between fatty acid oxidation (FAO) and glycolytic flux[57]. TET2-mediated conversion of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC) at FAO gene enhancers (e.g., CPT1A, PPARα) is markedly suppressed in failing hearts, likely due to reduced α-ketoglutarate (α-KG) availability, accumulation of succinate, and chronic inflammatory signaling - all hallmarks of mitochondrial dysfunction[48]. This hydroxymethylation deficit reinforces a transcriptionally repressive state, promoting a metabolic shift toward glucose dependency reminiscent of fetal cardiac metabolism. Concurrently, TET3 expression has been reported to increase in cardiac fibroblasts under pathological stress, potentially altering 5hmC patterns at pro-inflammatory gene loci (e.g., IL-6, TNF-α)[58]. This epigenetic reprogramming, in concert with ROS-mediated inhibition of electron transport chain (ETC) complexes, exacerbates mitochondrial dysfunction and impairs energy production. Further compounding these metabolic deficits, DNMT3A-mediated hypermethylation of PGC-1α regulatory regions contributes to suppressed mitochondrial biogenesis and oxidative metabolism[59]. Given that PGC-1α is also regulated by histone modifications and post-translational factors, DNMT3A may act in concert with other epigenetic regulators to create a self-reinforcing cycle of metabolic insufficiency. These interconnected pathways underscore the critical role of epigenetic remodeling in heart failure pathogenesis and highlight potential targets for therapeutic intervention.
Ischemia-reperfusion (I/R) injury establishes a persistent "metabolic memory" through histone modification-driven chromatin remodeling, perpetuating maladaptive responses even after blood flow is restored[60]. During ischemia, hypoxia-inducible factor 1α (HIF-1α) recruits histone acetyltransferases (HATs) such as p300 (EP300) to promoters of glycolytic and stress-response genes (e.g., HK2, PDK1, VEGFA), depositing H3K27ac marks that sustain transcription beyond the acute hypoxic phase[61]. This epigenetic priming, reinforced by ischemia-induced NAD+ depletion and subsequent suppression of SIRT1 deacetylase activity, promotes a metabolic shift favoring glycolysis at the expense of oxidative phosphorylation, impairing post-reperfusion recovery. Additionally, I/R injury induces H3K4me3 deposition at inflammatory gene loci, including NLRP3 inflammasome components, via the histone methyltransferase MLL1 (KMT2A), priming the myocardium for heightened immune activation upon subsequent ischemic insults[62]. While the precise cellular sources of this epigenetic memory remain under investigation, emerging evidence suggests that macrophages and cardiac fibroblasts may serve as reservoirs of maladaptive histone modifications, propagating chronic inflammation and metabolic inflexibility[63,64]. Pharmacological intervention targeting these chromatin modifications - such as HDAC inhibition to restore histone acetylation equilibrium or BET bromodomain blockade to disrupt transcriptional activation at dysregulated loci - has demonstrated potential in preclinical models, enhancing FAO and mitigating infarct expansion[65,66]. These findings underscore the centrality of epigenetic-metabolic interactions in I/R pathology and highlight novel therapeutic opportunities to reprogram cardiac metabolism by leveraging chromatin plasticity under pathological stress.
Sex differences in cardiac epigenetic aging significantly influence cardiovascular health, with evidence suggesting distinct patterns in DNMT3A and TET2 regulation. UK Biobank data indicate that DNMT3A mutations, linked to clonal hematopoiesis, are more prevalent in females (adjusted OR = 1.483,
EPITRANSCRIPTOMIC REGULATION OF METABOLIC FLUX
The epitranscriptomic regulation of metabolic flux is profoundly mediated by dynamic RNA modifications, with N6-methyladenosine (m6A) and 5-methylcytosine (m5C) serving as key modulators of mRNA stability and translational efficiency[70]. m6A, the most abundant internal mRNA modification, fine-tunes metabolic gene expression via a "write-erase-read" system that responds to nutrient availability[71]. In cardiomyocytes, METTL3-mediated m6A deposition on GLUT4 mRNA facilitates its recognition by YTHDF2, promoting its degradation through the CCR4-NOT deadenylase complex[72,73]. This process is dynamically regulated by insulin signaling: under hyperglycemic conditions, FTO-mediated demethylation of metabolic transcripts, including GLUT4, counteracts METTL3 activity, potentially stabilizing GLUT4 mRNA to enhance glucose uptake[74]. Conversely, fasting-induced m6A hypermethylation represses GLUT4 expression, shifting substrate utilization toward fatty acids[75]. This methylation-dependent regulatory cycle is essential for maintaining metabolic flexibility. Notably, in diabetic cardiomyopathy models, perturbations in the m6A/YTHDF2 axis have been associated with dysregulated GLUT4 expression and disrupted metabolic homeostasis, potentially contributing to altered glucose handling and oxidative stress[76]. These findings highlight the intricate epitranscriptomic control of cardiac metabolism and its implications for metabolic disorders.
In parallel with mRNA modifications, tRNA methylation fine-tunes the translational efficiency of metabolic enzymes, enabling rapid adaptation to nutrient fluctuations. NSUN2 catalyzes 5-methylcytosine (m5C) formation in both cytoplasmic and mitochondrial tRNAs, enhancing codon-anticodon pairing stability and optimizing translation of metabolic transcripts[77]. In mitochondria, NSUN3 specifically modifies the anticodon loop of mt-tRNAMet at cytosine-34, facilitating its conversion to 5-formylcytosine (f5C) - a modification essential for the efficient translation of oxidative phosphorylation (OXPHOS) proteins[78]. Loss of NSUN3 impairs mitochondrial protein synthesis, resulting in respiratory chain dysfunction and reduced metabolic flexibility, particularly in aged cardiomyocytes[79]. In pressure-overloaded hearts, NSUN2 deficiency leads to hypomethylation of cytosolic tRNALeu(CUN), reducing its stability and impairing the translation of fatty acid oxidation (FAO) enzymes such as CPT1A and ACADL. This translation bottleneck disrupts OXPHOS and shifts cardiac metabolism toward glycolysis[80]. Additionally, ALKBH1 mediates demethylation of N1-methyladenosine (m1A) in tRNAs under stress, potentially altering tRNA structure and redistributing ribosomal occupancy toward transcripts involved in adaptive responses, including HIF-1α[81].
Beyond tRNA, mitochondrial RNA modifications are critical for mitochondrial function and electron transport chain (ETC) efficiency. Pseudouridine (Ψ) modifications in mtDNA-encoded mRNAs - such as COXI, COXIII, and ATP6/ATP8 - are introduced by enzymes like RPUSD3 and TRUB2[82]. RPUSD3 is essential for COXI and COXIII translation and complex IV assembly, while TRUB2 mainly influences ATP6/ATP8 expression. These modifications support the structural integrity and translation of mitochondrial mRNAs, directly impacting ETC performance. Furthermore, mitochondria-encoded circular RNAs (mecciRNAs) add an additional layer of regulatory complexity. Although the presence and role of Ψ modifications in mecciRNAs remain poorly characterized, they may influence RNA stability and function, indirectly modulating ETC activity[83].
While NSUN2 and NSUN3 are established writers of m5C in tRNAs, the enzymes responsible for m5C modification in mtDNA-encoded mRNAs remain less defined. Emerging evidence suggests that METTL4 may participate in RNA methylation, but its direct involvement in mitochondrial mRNA modification is unclear[84]. Nonetheless, m5C modifications in mitochondrial RNAs, particularly in tRNAs, are known to enhance RNA stability and translational accuracy, which are critical for the efficient expression of OXPHOS proteins[85].
The interplay between m6A and tRNA epitranscriptomes establishes a hierarchical regulatory network, where m6A fine-tunes the expression of tRNA-modifying enzymes (e.g., NSUN2, ALKBH1), while tRNA modifications modulate the translational efficiency of m6A-modified mRNAs[86,87]. This multilayered regulatory system dynamically synchronizes metabolic gene expression with cellular bioenergetic demands, while its dysregulation contributes to metabolic disorders such as diabetic cardiomyopathy and heart failure. Emerging technologies, including tRNA-seq coupled with ribosome profiling, provide codon-resolution insights into how epitranscriptomic modifications influence metabolic protein synthesis, offering novel therapeutic targets to restore metabolic homeostasis in diseased hearts.
Aging disrupts the delicate equilibrium of epitranscriptomic regulation by progressively impairing RNA-modifying enzymes[87], with declining FTO demethylase activity and dysregulated writer proteins emerging as central contributors to metabolic inflexibility and oxidative stress[88]. FTO, a key m6A eraser required for maintaining insulin signaling homeostasis, exhibits age-associated activity loss, potentially due to Fe(II) cofactor depletion, NAD+ scarcity, or increased ubiquitin-mediated degradation under oxidative stress. This decline elevates m6A modification on insulin signaling transcripts such as INSR and IRS1, potentially altering their translation efficiency or stability[89]. Paradoxically, this dysregulation may contribute to impaired receptor recycling and downstream desensitization, leading to GLUT4 trafficking defects - a phenomenon termed "epitranscriptomic insulin resistance"[90]. Concurrently, FTO deficiency enhances m6A methylation on PPARδ mRNA, potentially altering its translation efficiency or stability, thereby impairing fatty acid oxidation (FAO) and shifting cardiomyocytes toward glucose dependency[91]. This shift exacerbates age-related lipotoxicity, mitochondrial dysfunction, and oxidative stress. In murine models, cardiac-specific FTO depletion recapitulates features of diabetic cardiomyopathy, including ectopic lipid accumulation, mitochondrial fragmentation, and metabolic inflexibility, underscoring its role as a crucial metabolic gatekeeper[92].
Parallel to FTO decline, the aging heart suffers from writer enzyme attrition, particularly METTL3/METTL14 complex destabilization, which disrupts the m6A landscape of antioxidant defense transcripts[72]. METTL14 downregulation reduces m6A deposition on regulatory regions of SOD2 and GPX4 mRNAs, impairing their recognition by the nuclear export machinery and subsequent cytoplasmic translation[93]. This leads to diminished synthesis of these critical ROS-scavenging enzymes, permitting H2O2 accumulation and peroxidation of mitochondrial cardiolipin - a key event in apoptosis initiation. Similarly, NSUN2-mediated m5C methylation of tRNA(Cys), essential for accurate translation of selenocysteine-containing antioxidants like thioredoxin reductase, declines with age due to oxidative inactivation of NSUN2’s catalytic domain[94]. The resultant mistranslation generates truncated, nonfunctional proteins that further compromise the glutathione redox cycle. These writer deficiencies create a self-amplifying loop: ROS accumulation oxidizes and inactivates additional RNA-modifying enzymes (e.g., ALKBH5), while impaired antioxidant synthesis permits persistent oxidative damage to rRNA and mtDNA-encoded transcripts. In human failing hearts, spatial transcriptomics reveals colocalization of METTL3-depleted zones with lipid peroxide hotspots and necroptotic markers, underscoring the spatial consequences of epitranscriptomic collapse. Therapeutic strategies targeting this axis, such as mitochondrially targeted METTL3 mRNA delivery or FTO-stabilizing chaperones, are now being explored to break this vicious cycle and restore redox-metabolic coherence in aged myocardium.
Disease-specific epitranscriptomic signatures are increasingly recognized as pivotal drivers of pathological metabolic and electrophysiological remodeling, with METTL3-mediated m6A hypermethylation in cardiac hypertrophy and pseudouridylation (Ψ) of arrhythmogenic circRNAs representing paradigmatic examples[95]. In pressure-overload-induced myocardial hypertrophy, METTL3 upregulation orchestrates a pro-glycolytic switch through m6A-dependent stabilization of HIF-1α and c-Myc mRNAs, which encode master regulators of anaerobic metabolism[96]. METTL3 deposits m6A marks at regulatory regions of these transcripts, enhancing their interaction with translation initiation factors to drive ribosome loading, thereby amplifying glycolytic enzyme production (e.g., HK2, LDHA) despite normoxic conditions. Concurrently, METTL3-mediated m6A methylation of PPARα mRNA facilitates YTHDF2-dependent decay, repressing fatty acid oxidation and fostering lipid droplet accumulation - a hallmark of metabolic immaturity[97]. This epitranscriptomic hijacking creates a self-reinforcing loop: HIF-1α activation and subsequent lactate accumulation disrupt METTL14 homeostasis, further skewing the m6A landscape toward pro-hypertrophic targets. Single-nucleus m6A-CLIP-seq in human hypertrophic hearts reveals METTL3 enrichment at splice junctions of TNNT2 transcripts, inducing isoform switching toward fetal troponin T variants that impair calcium sensitivity and diastolic relaxation.
In arrhythmogenic contexts, Ψ modifications deposited by dyskerin (DKC1) on circular RNAs (circRNAs) emerge as critical regulators of cardiac excitability[98]. The arrhythmia-associated circRNA CDR1as undergoes DKC1-mediated Ψ modification at conserved sites within its miRNA interaction domains, which stabilizes its interaction with argonaute 2 (AGO2) and enhances sequestration of miR-7 - a microRNA involved in ion channel regulation[99]. This Ψ-driven sponging indirectly modulates potassium channel transcripts (KCNH2, KCNQ1), prolonging action potential duration and promoting early afterdepolarizations in ventricular myocytes[100]. Concurrently, Ψ-modified circRNA HRCR (heart-related circRNA) adopts a unique topology that recruits YBX1 to stabilize SCN5A mRNA, increasing sodium channel density and contributing to conduction velocity heterogeneity[101]. Intriguingly, hypoxia triggers NSUN5-dependent Ψ deposition on mitochondrial circRNA SCAR (senescence-associated circRNA), which interacts with POLG and is implicated in mtDNA replication fidelity - a process disrupted in atrial fibrillation, where SCAR Ψ levels inversely correlate with mitochondrial ROS bursts and conduction block[102]. Spatial Ψ mapping in infarct border zones reveals Ψ hotspots on miR-328-binding circRNAs, which coordinate focal adhesion kinase (FAK) translation to promote fibrotic substrate formation[103]. These findings position RNA pseudouridylation as a regulatory nexus linking epitranscriptomic dysregulation to arrhythmogenic metabolic-electrical coupling, with potential therapeutic interventions aimed at modulating Ψ writer enzymes such as DKC1 to restore ionic homeostasis in diseased hearts.
CROSSTALK BETWEEN EPIGENETIC AND EPITRANSCRIPTOMIC LAYERS
The intricate crosstalk between epigenetic and epitranscriptomic layers is exemplified by the functional coupling of histone acetyltransferase p300 and the m6A reader YTHDF2, a synergistic interaction that bridges chromatin remodeling and post-transcriptional regulation[104]. p300, a master regulator of histone acetylation, enhances transcriptional activation of metabolic genes such as PDK4 and GLUT1 by acetylating histones H3K27 and H3K9, thereby promoting transcription factor recruitment and chromatin accessibility[105]. Concurrently, YTHDF2, a cytoplasmic m6A-binding protein, recognizes m6A marks on transcripts of these same genes, accelerating their degradation via recruitment of deadenylase complexes[106]. This paradoxical coordination creates a regulatory circuit wherein p300-driven transcriptional activation is counterbalanced by YTHDF2-mediated mRNA turnover, fine-tuning metabolic gene expression in response to energetic stress. Recent studies suggest that p300-mediated acetylation of YTHDF2 at lysine 267 may enhance its binding affinity for m6A-modified RNAs, potentially linking nuclear histone modifications with cytoplasmic mRNA stability[107]. This post-translational modification of YTHDF2 by p300 establishes a molecular axis integrating histone acetylation landscapes with epitranscriptomic dynamics, particularly evident in hypertrophied cardiomyocytes, where hyperacetylation of both histones and YTHDF2 drives pathological glycolytic switching[108].
Equally compelling is the role of non-coding RNAs, such as miRNAs and lncRNAs, as interfaces between epigenetic and epitranscriptomic regulations[109,110]. These molecules serve as critical connectors, linking DNA methylation (an epigenetic mark) with RNA modifications (epitranscriptomic marks)[111,112]. For instance, the lncRNA NEAT1 acts as a scaffold for DNMT1, a DNA methyltransferase, thereby influencing DNA methylation patterns that can indirectly affect the expression of genes involved in RNA modification[113,114]. Similarly, miRNAs like miR-29 are regulated by DNA methylation; DNMT3B-mediated methylation of CpG islands within the miR-29 promoter suppresses its transcription, while, when expressed, miR-29b targets METTL3, a key m6A writer, modulating m6A deposition on transcripts involved in fatty acid oxidation[115,116]. Another example is miR-133a, whose promoter hypermethylation in aging hearts leads to decreased miR-133a levels, resulting in derepression of ALKBH5, an m6A eraser, thereby altering m6A levels on target transcripts like CPT1A and impacting metabolic function[117,118]. Conversely, TET2-driven demethylation at the miR-145 locus promotes its expression, which downregulates FTO, stabilizing m6A-modified SIRT1 mRNA and enhancing mitochondrial function[109,119]. This bidirectional circuitry, where DNA methylation status dictates miRNA and lncRNA profiles, which in turn modulate the epitranscriptomic machinery, shapes the cardiac metabolic epigenome[120,121]. In aging hearts, hypermethylation of the miR-133a promoter correlates with diminished levels of this miRNA, relieving its repression of ALKBH5 and thereby reducing m6A levels on CPT1A mRNA, exacerbating lipid metabolic dysfunction[122,123]. Such multilayered integration of DNA methylation, miRNA networks, and RNA modifications underscores the hierarchical complexity of metabolic regulation, offering novel therapeutic nodes to disrupt pathological feedback loops in age-related cardiovascular diseases.
The bidirectional regulation of metabolic intermediates serves as a critical nexus linking epigenetic and epitranscriptomic mechanisms, with α-ketoglutarate (α-KG) and NAD+ emerging as central players in this dynamic interplay. α-KG, TCA cycle intermediate, functions as an essential cofactor for TET DNA demethylases and AlkB homolog RNA demethylases, thereby synchronizing nuclear and cytoplasmic regulatory layers[124]. Concurrently, α-KG serves as a co-substrate for ALKBH5 and FTO, which mediate oxidative demethylation of m6A-modified RNAs, positioning α-KG as a metabolic regulator that integrates DNA hydroxymethylation with RNA methylation dynamics[125]. In aged cardiomyocytes, reduced α-KG availability - due to mitochondrial dysfunction or IDH2 downregulation - impairs both TET2 activity and ALKBH5-mediated RNA demethylation, resulting in concurrent hypermethylation of DNA regulatory elements and increased m6A deposition[126]. This imbalance exacerbates metabolic inflexibility, as hypermethylation-associated transcriptional repression of lipid metabolism genes limits fatty acid utilization, while m6A accumulation on PDK4 mRNA stabilizes its transcripts, further inhibiting glucose oxidation. These interwoven metabolic-epigenetic circuits underscore the pivotal role of α-KG in maintaining transcriptional and post-transcriptional homeostasis.
NAD+, a pivotal coenzyme in redox reactions, exerts pleiotropic effects on both histone deacetylases and RNA modification machinery, creating a unified regulatory axis. SIRT1, an NAD+-dependent class III histone deacetylase, deacetylates histones (e.g., H3K56ac) and non-histone targets like PGC-1α, thereby promoting mitochondrial biogenesis and oxidative metabolism[127]. Concurrently, NAD+ levels modulate the activity of RNA demethylases such as FTO, potentially through indirect metabolic interactions[128]. Age-related NAD+ depletion disrupts this coordination: diminished SIRT1 activity leads to hyperacetylation of histones at glycolytic gene promoters (e.g., HK2, LDHA), while impaired FTO function is associated with increased m6A methylation on transcripts encoding antioxidant enzymes like SOD2, potentially affecting their stability and translation efficiency[129]. This dual impairment manifests as a metabolic crisis - exacerbated glycolysis, mitochondrial ROS overproduction, and defective redox homeostasis - hallmarks of cardiac aging. Notably, NAD+ repletion via precursors like NMN restores SIRT1-mediated deacetylation of FOXO1, facilitating its transcriptional activity on oxidative metabolism genes, while concurrently supporting FTO activity to regulate m6A modifications on Nrf2 mRNA, contributing to antioxidant defense restoration[130]. These findings reveal NAD+ as a metabolic integrator that synchronizes chromatin-based and RNA-centric regulatory networks, offering a unified therapeutic target to counteract age-associated metabolic decline. The interdependency of α-KG and NAD+ in bridging epigenetic and epitranscriptomic layers underscores the sensitivity of cardiac metabolism to metabolic intermediate flux, with perturbations in these molecules triggering cascading failures that accelerate cardiovascular aging. Targeting these regulatory nodes through pharmacological or nutritional interventions may thus restore metabolic-epigenetic crosstalk coherence, optimizing energy substrate utilization and enhancing myocardial resilience.
Defective mitophagy, driven by PARK2 promoter hypermethylation or m6A-modified PINK1 mRNA, leads to the accumulation of damaged mitochondria, which produce excessive reactive oxygen species (ROS) that cause epigenetic and epitranscriptomic damage[131-133]. Hypermethylation of the PARK2 promoter reduces parkin expression, while m6A modification of PINK1 mRNA promotes its degradation, impairing the recruitment of parkin to damaged mitochondria and disrupting their clearance[134,135]. This results in elevated ROS, which oxidize DNA, histones, and RNA, altering DNA methylation, histone acetylation, and m6A RNA modifications, thus disrupting gene regulation and contributing to cellular dysfunction[136,137]. Concurrently, damaged mitochondria release mitochondrial DNA (mtDNA) into the cytosol, activating the cGAS-STING pathway, which triggers inflammation and further increases ROS production[138,139]. This creates a vicious cycle where inflammation amplifies oxidative stress, exacerbating epigenetic and epitranscriptomic damage, potentially worsening conditions like neurodegeneration or cancer.
We summarized and mapped the epigenetics-epitranscriptome-metabolism axis of healthy versus aging hearts, as detailed in Figure 1.
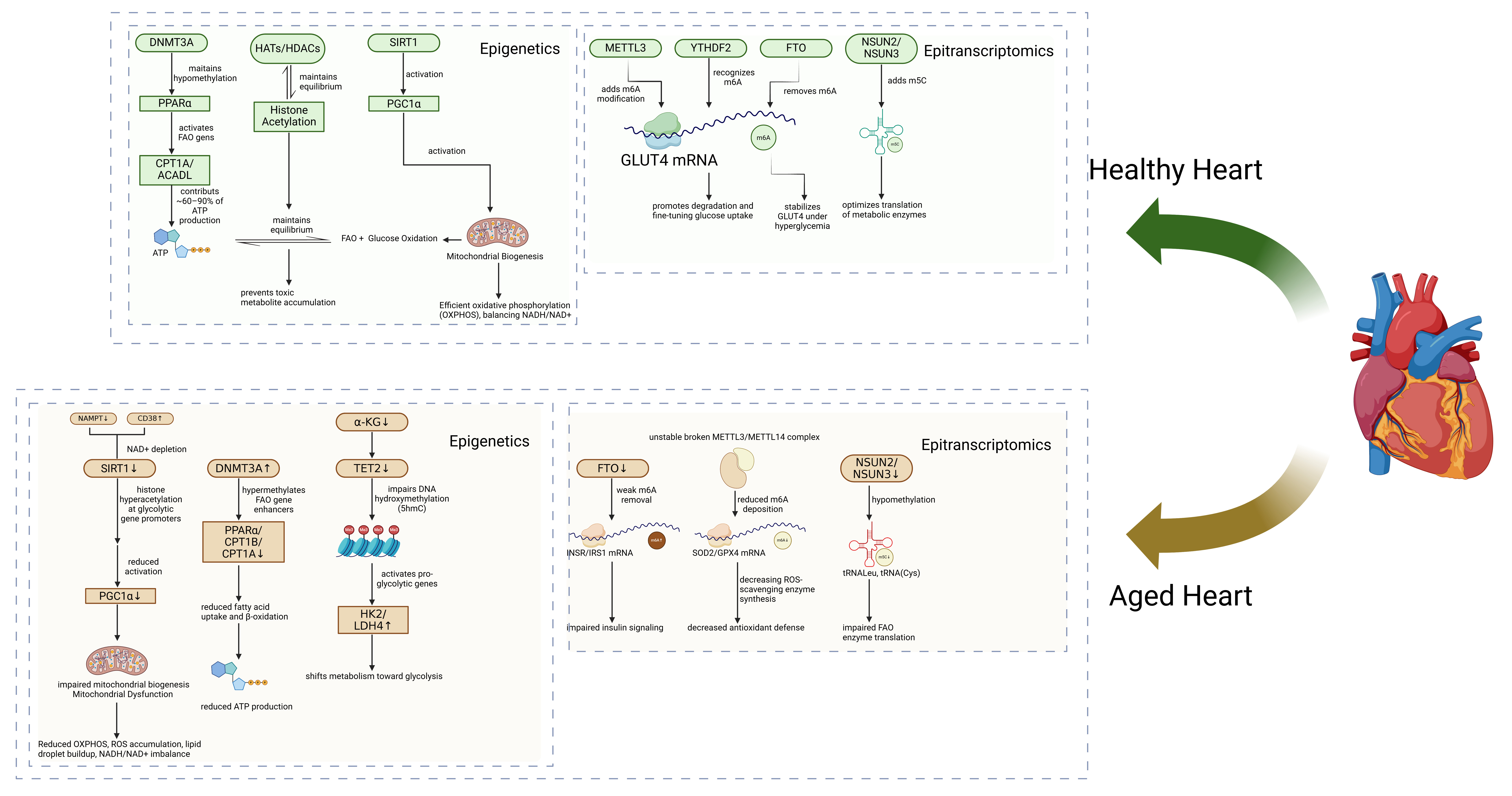
Figure 1. The epigenetic-epitranscriptomic-metabolic axis in healthy vs. aged hearts. Created in BioRender.com. Johnzy L., 2025 https://BioRender.com/fh4fk4u.
CRITICAL EVALUATION OF METTL3 AND FTO IN CARDIAC PATHOLOGY
The roles of METTL3 and FTO in cardiac metabolism are pivotal. This section evaluates conflicting findings, methodological limitations, and gaps in knowledge to provide a more nuanced understanding of their contributions to cardiac pathology and guide future research.
Research suggests that METTL3, an m6A methyltransferase, promotes hypertrophic cardiomyopathy by enhancing m6A modifications on transcripts involved in protein kinase signaling and intracellular pathways, such as those in the MAPK cascade[140]. However, some studies indicate METTL3 may have protective effects, potentially reducing pathological cardiac hypertrophy and myocardial fibrosis in specific contexts[141]. These discrepancies suggest that METTL3’s effects may depend on factors such as disease stage, stress conditions, or specific cellular environments, which require further clarification to reconcile these findings.
Similarly, FTO, an m6A demethylase, is implicated in maintaining cardiac contractile function and metabolic flexibility, with its deficiency linked to metabolic inflexibility and diabetic cardiomyopathy in murine models[142]. Yet, in other contexts, such as cancer, FTO inhibition is beneficial, highlighting a context-dependent role that complicates its therapeutic application in cardiac diseases. For instance, FTO’s protective role in suppressing cardiac fibrosis post-myocardial infarction contrasts with its potential to exacerbate arrhythmias in FTO-deficient mice, indicating varied effects across different cardiac conditions (FTO Cardiac Fibrosis; FTO Deficiency). These conflicting findings underscore the need for a deeper understanding of the conditions under which METTL3 and FTO exert their effects.
A significant limitation in the current article is the heavy reliance on preclinical models, particularly mice, which may not fully recapitulate human cardiac physiology. Differences in metabolic profiles between mice and humans could affect the translatability of findings related to METTL3 and FTO. For example, murine models of FTO depletion show impaired cardiac function, but human cardiomyocytes may respond differently due to variations in metabolic pathways or cofactor availability, such as NAD+ or Fe(II). Additionally, many studies utilize bulk RNA sequencing or m6A-CLIP-seq, which lack the resolution to provide cell-type-specific insights. Given the heart’s composition of multiple cell types (e.g., cardiomyocytes, fibroblasts, endothelial cells), understanding how METTL3 and FTO function in each is crucial for developing targeted therapies. Techniques like m6A-CLIP-seq are also sensitive to RNA degradation and sample quality variability, which can introduce biases and affect data accuracy.
Several knowledge gaps persist in the field. First, the cell-type specificity of METTL3 and FTO in the heart remains underexplored. While their roles in cardiomyocytes are relatively well-studied, their functions in other cardiac cell types, such as fibroblasts or immune cells, are less clear. For instance, METTL3’s role in stabilizing fetal isoforms of troponin T in cardiomyocytes may not apply to fibroblasts, which could have distinct epitranscriptomic profiles (IGFBP3 Fibrosis). Second, the temporal dynamics of METTL3 and FTO activity during disease progression are not well-characterized. Determining whether their dysregulation is an early event or a consequence of cardiac pathology is essential for therapeutic timing. Third, the potential off-target effects of modulating METTL3 and FTO are a concern. For example, global activation of FTO to restore metabolic homeostasis might inadvertently stabilize oncogenic transcripts, posing risks that need careful evaluation. Finally, there is a lack of human studies, as most research relies on preclinical models, limiting the applicability of findings to human cardiac pathology.
We summarized the SIRT1-NAD+ and METTL3-m6A-PPARα crosstalk and plotted a figure as detailed in Figure 2.
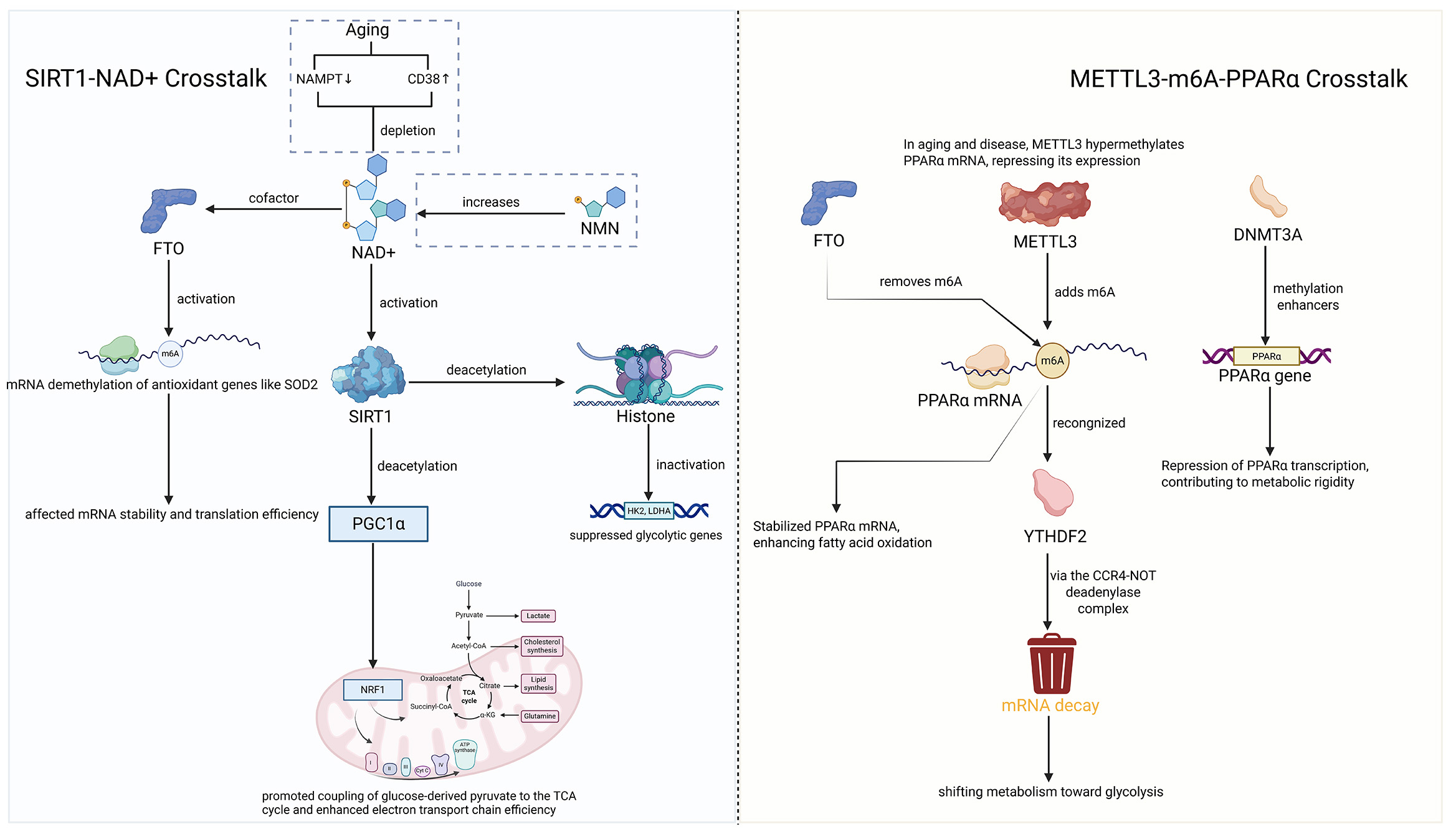
Figure 2. SIRT1-NAD+ and METTL3-m6A-PPARα crosstalk. Created in BioRender.com. Johnzy L., 2025 https://BioRender.com/vhiidg1.
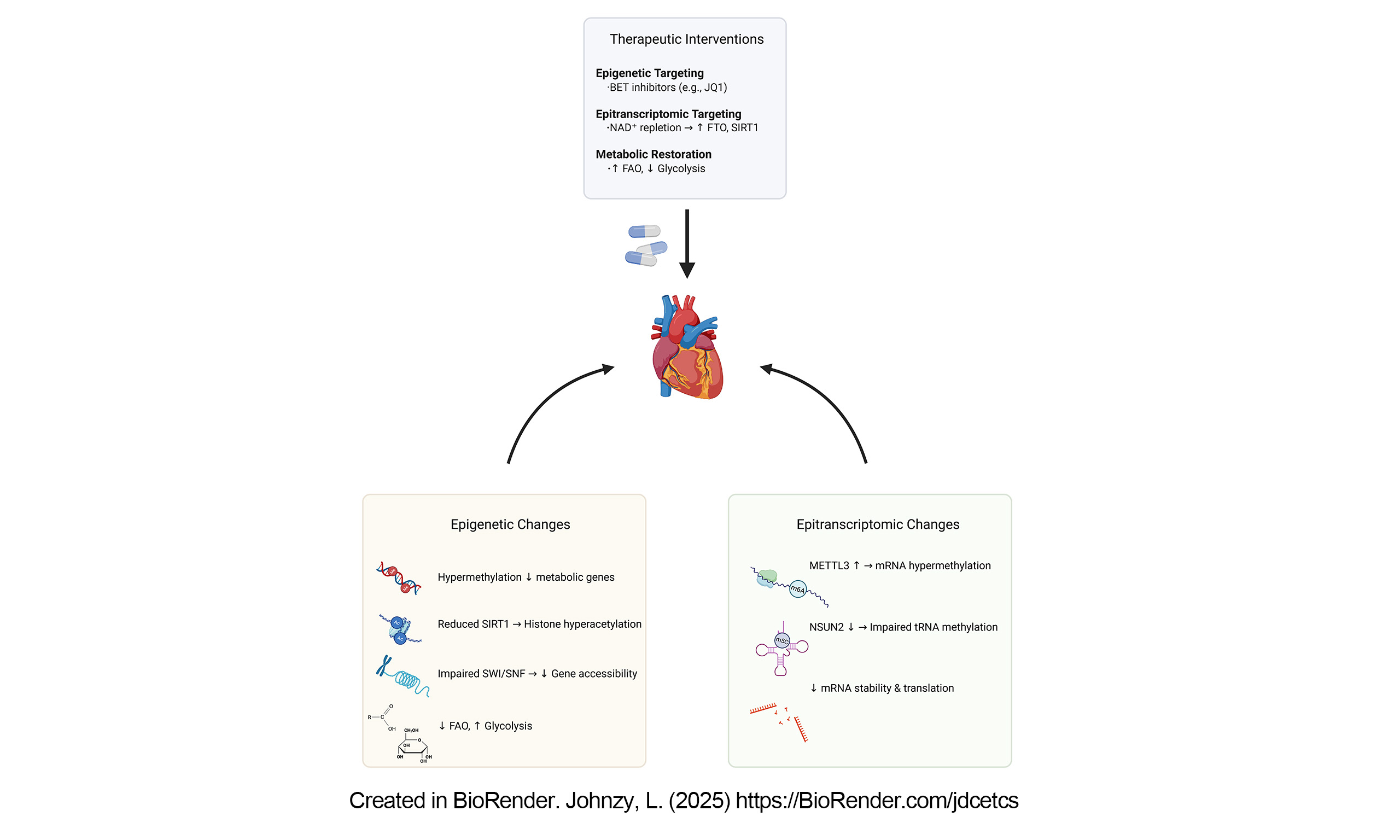
THERAPEUTIC IMPLICATIONS AND TECHNOLOGICAL FRONTIERS
Emerging therapeutic strategies targeting epigenetic and epitranscriptomic regulators, such as BET inhibitors and FTO modulators, hold transformative potential for metabolic cardiomyopathy and age-related cardiac dysfunction. BET bromodomain inhibitors, exemplified by JQ1, have demonstrated pleiotropic benefits beyond their original oncological applications. JQ1 selectively inhibits BRD4, a transcriptional coactivator that promotes pro-inflammatory and hypertrophic gene expression by recognizing acetylated histones. In metabolic cardiomyopathy, where chronic lipotoxicity and mitochondrial dysfunction contribute to maladaptive remodeling, JQ1 modulates metabolic substrate preference by suppressing BRD4-mediated transcriptional activation of glycolytic genes (e.g., HK2, LDHA) while influencing pathways involved in fatty acid oxidation, potentially through indirect effects on PPARα signaling[143]. Preclinical studies indicate that JQ1 administration is associated with reduced myocardial FDG uptake - a marker of metabolic remodeling - and improved diastolic function in obesity-induced cardiomyopathy models, effects potentially mediated by alterations in chromatin accessibility and autophagy activation[144]. Additionally, its ability to suppress MYC-regulated transcriptional networks may mitigate profibrotic signaling, suggesting potential synergy with conventional metabolic therapies targeting insulin resistance and lipid overload[143].
Parallel advances in epitranscriptomic therapeutics highlight the potential of FTO agonists to counteract age-related metabolic decline. While FTO inhibitors demonstrate efficacy in cancer by destabilizing oncogenic m6A-modified transcripts, FTO activation may help restore cardiac metabolic homeostasis by reversing aberrant m6A marks that accumulate during aging[145]. Notably, age-associated FTO downregulation correlates with hypermethylation of SIRT1 and CPT1A mRNAs, impairing mitochondrial biogenesis and fatty acid oxidation[146]. Pharmacological FTO activation, potentially through allosteric modulators or NAD+-boosting compounds, could restore m6A equilibrium, stabilizing transcripts encoding oxidative phosphorylation components and antioxidant enzymes[147]. Intriguingly, FTO’s enzymatic activity is dependent on α-ketoglutarate and NAD+, linking its function to mitochondrial TCA cycle flux and cellular redox status, thus providing a rationale for metabolic co-therapies. Novel therapeutic platforms leveraging bioactive peptides and growth factors (e.g., VEGF, FGF) are being explored to enhance intracellular NAD+ levels and reactivate endogenous FTO, thereby improving mitochondrial function and extracellular matrix remodeling in aged tissues. Such strategies may synergize with senolytics to eliminate pro-inflammatory senescent cells, addressing both metabolic inflexibility and tissue dysfunction in aging hearts.
These interventions, however, face significant translational challenges, including cell-type specificity and off-target effects. The broad chromatin-modifying activity of JQ1 may inadvertently disrupt homeostatic gene regulatory networks, highlighting the need for cardiac-targeted delivery systems or isoform-specific BET inhibitors. Likewise, systemic FTO activation could stabilize oncogenic transcripts, necessitating strategies for spatiotemporal control, such as nanoparticle-based delivery or inducible gene editing systems. Advances in single-cell multi-omics and spatial epitranscriptomics will be essential for mapping context-specific vulnerabilities, refining therapeutic windows, and optimizing dosing regimens. By integrating these precision-medicine approaches, next-generation therapies may enable targeted metabolic reprogramming, reversing the epigenetically driven decline in cardiac plasticity associated with aging and metabolic disease.
We summarized some representative BET inhibitors, FTO activators, and NAD+ boosters, and the relevant comparative analyses are detailed in Table 1.
Therapeutic challenges table comparing BET inhibitors, FTO activators and NAD+ boosters
| Drug name | Type | Metabolic effects | Cell-type specificity challenges | Clinical trial status | Off-target risks | References |
| Apabetalone (RVX-208) | BET inhibitor | Enhances HDL metabolism, inhibits BRD4, improves lipid metabolism and reduces cardiovascular inflammation | Variable response in vascular vs. myocardial cells Primarily studied in hepatocytes and monocytes; limited cardiomyocyte data | Completed Phase III trial (NCT02586155) | Lower off-target risks compared to pan-BET inhibitors like JQ1 May affect circadian gene expression | [1] |
| JQ1 | BET inhibitor | Suppresses BRD4 and super-enhancer activity, downregulates pro-inflammatory and cardiac hypertrophy genes. Promotes FAO, improves mitochondrial | Affects multiple cell types, broad systemic influence | Early-phase and preclinical studies | Disrupts circadian rhythm and reproductive axis | [2] |
| PLX51107 | BET inhibitor | Modulates inflammatory transcription networks | Studies report cell-type selectivity in immune cells but limited in cardiac data | Ongoing (e.g., NCT02683395) | Hematological toxicity possible | [3] |
| I-BET151 | BET inhibitor | Induces structural and functional alterations in cardiac mitochondria | Cardiac mitochondria show significant impairment in mice | Early-phase oncology studies | Cardiotoxicity potential evident in animal models | [4] |
| CPI-0610 | BET inhibitor | Regulates inflammatory and metabolic gene networks | Well-studied in hematologic malignancies, limited cardiac context | Phase II MANIFEST trial ongoing | Potential hematopoietic system effects | [5] |
| OTX015 | BET inhibitor | Downregulates metabolism and inflammation-related gene expression | Primarily acts on lymphoma cells; cardiometabolic data lacking | Phase I (Amorim et al., 2016)[161] | Possible mitochondrial and epigenetic perturbation | [6] |
| ABBV-744 | BET inhibitor | Selective inhibition of BRD4-BD2 reduces inflammatory gene expression | Lower systemic impact; cardiomyocyte specificity pending validation | Ongoing trials (e.g., NCT03360006) | Fewer off-target effects due to BD2 selectivity | [3] |
| FB23-2 | FTO activator | binds to the catalytic pocket of FTO, elevating m6A levels and suppressing expression of metabolic regulators | Good selectivity toward FTO, but potential impact on hematopoietic and hepatic systems noted | Preclinical stage, studied in models of leukemia and metabolic disorders | Minor effect on ALKBH5 and other 2-oxoglutarate-dependent dioxygenases | [7,8] |
| Rhein | FTO activator | suppresses FTO activity by directly binding to its active pocket, thereby increasing m6A methylation and reducing gluconeogenesis and lipid accumulation. Improves insulin sensitivity and mitigates hepatic steatosis in obese mice | Primarily studied in liver cells; direct cardiomyocyte-specific modulation is underexplored | Preclinical; no direct cardiovascular trial data available | Exhibits polypharmacology affecting multiple pathways, including NF-κB and AMPK | [9] |
| Entacapone | FTO activator | Enhances FTO demethylase activity, which in turn reduces m6A methylation levels on key transcripts involved in insulin signaling and lipid metabolism. This has been associated with improved insulin sensitivity and glucose uptake | Cell-type dependent effects are prominent in adipocytes and hepatocytes, where demethylation of mRNA modulates transcriptional programs specific to metabolic regulation | Not specifically trialed in cardiometabolic disease; mostly used as a COMT inhibitor for Parkinson’s disease | May affect other iron-dependent dioxygenases | [7] |
| Meclofenamic Acid (MA) | FTO activator | Inhibits FTO demethylase without affecting ALKBH5, altering m6A patterns and downstream gene expression related to metabolism. May reduce adipogenesis and insulin resistance | In vitro evidence mainly; in vivo cardiac-specific responses require further validation | No clinical trials for FTO-targeted use; originally an NSAID | Known to inhibit cyclooxygenase enzymes (COX-1/2), hence affecting inflammatory pathways | [8] |
| NR | NAD+ booster | Restores NAD⁺ levels and improves mitochondrial function, enhancing oxidative metabolism in cardiac tissue. Relates to the function of left ventricular and inflammation | Effective in cardiomyocytes, but its tissue-specific absorption and downstream pathway modulation may vary across individuals | Multiple clinical trials are underway or completed. Safe dosage and favorable outcomes were reported in Phase I/II studies | Potential to interfere with methylation pathways or NAD⁺ consumers like CD38, especially at high doses | [10] |
| NMN | NAD+ booster | Increases NAD⁺ biosynthesis, enhances mitochondrial activity, and improves cardiac glucose utilization and oxidative phosphorylation | High uptake in skeletal muscle and liver; bioavailability and efficacy in heart tissue are promising but require further validation | Human clinical trials show good tolerance and improved biomarkers, but long-term cardiac effects still under investigation | Can influence insulin signaling, liver fat metabolism, and renal function; long-term safety data limited | [10] |
| NAM | NAD+ booster | Enhances NAD⁺ biosynthesis but can inhibit sirtuin activity at high concentrations, leading to reduced mitochondrial biogenesis and fatty acid oxidation | Sirtuin inhibition creates complex effects depending on concentration | Used in several metabolic studies; low doses beneficial, while high doses may disrupt epigenetic regulation | Inhibits SIRT1 and other NAD⁺-dependent enzymes at pharmacologic levels | [10] |
Recent breakthroughs in single-cell multi-omics technologies have revolutionized our ability to explore metabolic diversity and develop precise interventions for aging and cardiovascular diseases[148]. Spatial epitranscriptomics, powered by platforms like 10x Visium, enables high-resolution mapping of RNA modification landscapes, such as N6-methyladenosine (m6A) and 5-methylcytosine (m5C), within the complex architecture of cardiac tissue[149,150]. While direct applications of 10x Visium for m6A mapping in cardiac infarct border zones are still emerging, studies leveraging spatial transcriptomics have already demonstrated significant region-specific gene expression changes in these critical areas[151]. For example, research has identified heightened expression of mechanical stress-response genes, such as Csrp3, in the border zone following myocardial infarction, underscoring the spatial heterogeneity of cardiac remodeling[152]. Integrating spatialATAC-seq, which maps chromatin accessibility, with spatial epitranscriptomics could further illuminate how epigenetic regulation and RNA modifications like m6A orchestrate gene expression in a spatially dependent manner[153]. In the context of aging, this integrated approach could pinpoint senescent cell niches - regions of cellular aging - marked by m6A hypermethylation of key metabolic genes, such as PDK4 and LDHA[154]. These modifications are associated with localized lactate accumulation and fibrosis, contributing to impaired heart function. Complementary spatial metabolomic profiling enhances these insights by capturing zonated fluctuations in metabolites like α-ketoglutarate and NAD+ across myocardial layers, revealing how metabolic and epigenetic states are coordinated spatially. By combining these advanced spatial omics technologies, researchers can unravel the intricate interplay between metabolism, epigenetics, and spatial organization in the aging heart, paving the way for targeted therapies to mitigate age-related cardiac pathologies. However, spatial transcriptomics, such as 10x Visium, is limited by its resolution (approximately 55 micrometers), which may not capture single-cell details in dense cardiac tissue. It also struggles with detecting low-abundance transcripts, potentially missing rare cell types or subtle RNA modifications like m6A. Single-cell omics faces challenges from tissue dissociation, which can alter gene expression or exclude fragile cells, introducing biases. Both technologies generate complex datasets requiring advanced computational tools, and their high costs limit scalability for large-scale studies.
While the potential of BET inhibitors and NAD+ supplementation as therapeutic strategies for restoring cardiac metabolic flexibility has been highlighted, it is crucial to consider the translational challenges and clinical trial data to provide a balanced perspective on their feasibility and safety in human patients. For BET inhibitors, although they are primarily being tested in clinical trials for cancer, their application in heart failure is still largely preclinical[155]. Studies have demonstrated that BET inhibitors, such as JQ1, can suppress innate inflammatory and profibrotic transcriptional networks in heart failure models, suggesting potential benefits in modulating cardiac metabolism[156]. However, the lack of specific clinical trials for heart failure means that their efficacy and safety in this context remain to be fully established. Challenges include potential off-target effects, the need for cardiac-specific delivery systems, and ensuring isoform-specific inhibition to avoid disrupting homeostatic gene networks. On the other hand, NAD+ supplementation has shown more promising results in both preclinical and early clinical studies. For instance, nicotinamide administration has been shown to reduce cardiac injury markers, such as troponin T, in patients undergoing cardiac surgery, and observational studies link NAD+ precursor-rich diets to lower cardiac-specific mortality[157]. Additionally, initial human studies with nicotinamide riboside (NR) have demonstrated that it is safe and effective in increasing NAD+ levels without adverse effects[158]. Despite these encouraging findings, large-scale clinical trials specifically designed for heart failure patients are still needed to confirm the therapeutic potential and long-term safety of NAD+ supplementation in this population. Therefore, while both BET inhibitors and NAD+ supplementation hold promise as strategies to target epigenetic and epitranscriptomic pathways in cardiac metabolism disorders, further research and clinical trials are essential to translate these preclinical findings into effective and safe treatments for human patients.
Recent advancements in multi-omics databases and consortia, such as the Genotype-Tissue Expression (GTEx) project and the Cardiac Aging Atlas, provide valuable resources for understanding the molecular mechanisms underlying cardiac aging and heart failure. These databases offer comprehensive data on gene expression, including that of key regulators like SIRT1 and FTO, across various tissues, including cardiac tissue[150]. Analysis of such data reveals correlations between the expression levels of SIRT1 and FTO and metabolic gene signatures, which are crucial for maintaining cardiac metabolic homeostasis. For instance, studies indicate that SIRT1 downregulation in heart failure is associated with reduced expression of metabolic genes like Mn-superoxide dismutase and thioredoxin 1, impacting mitochondrial function and cardiomyocyte survival[159,160]. Similarly, FTO’s role in m6A RNA demethylation suggests its influence on metabolic gene expression, potentially linked to mitochondrial biogenesis and fatty acid oxidation, as seen in preclinical models. Furthermore, spatial transcriptomics and epitranscriptomics techniques have been employed to map RNA modifications, such as m6A and m5C, in explanted hearts compared to donor controls, providing insights into the spatial distribution of these modifications in the context of heart failure. These findings underscore the importance of integrating data from large-scale consortia to advance our understanding of the epitranscriptomic landscape in cardiac diseases and to identify potential therapeutic targets.
CONCLUSION
The intricate interplay between epigenetic and epitranscriptomic mechanisms plays a central role in regulating cardiac metabolic homeostasis, integrating hierarchical layers of DNA methylation, histone modifications, chromatin remodeling, and RNA modifications to dynamically coordinate substrate utilization and energy production. DNA hydroxymethylation by TET enzymes and m6A-mediated mRNA decay via YTHDF2 exemplify how these regulatory networks modulate fatty acid oxidation and glycolytic flux, ensuring metabolic flexibility under physiological conditions. However, aging and disease disrupt this equilibrium, as evidenced by SIRT1/NAD+ axis depletion, FTO dysfunction, and METTL3-driven m6A hypermethylation, which collectively induce metabolic rigidity, mitochondrial inefficiency, and ROS accumulation. These findings highlight the essential roles of epigenetic modifiers and RNA-modifying enzymes in preserving metabolic plasticity, positioning them as key targets for therapeutic intervention.
Advancing cross-scale investigations that connect molecular mechanisms, cellular behavior, and organ-level pathophysiology is essential for understanding cardiac aging and disease. While single-cell multi-omics has revealed cell-type-specific epigenetic landscapes in aging cardiomyocytes and fibroblasts, integrating spatial epitranscriptomics, metabolomic imaging, and organ-on-chip systems is crucial to elucidate how localized metabolic-epigenetic disruptions - such as senescent cell niches with aberrant m6A signatures - propagate dysfunction across myocardial tissue. For instance, mitochondrial α-ketoglutarate depletion in aged hearts not only compromises TET2-mediated DNA demethylation but also impairs ALKBH5-dependent RNA modifications, highlighting the necessity of multiscale models that simultaneously capture metabolite flux, chromatin dynamics, and RNA epitranscriptomic alterations.
Therapeutic innovation is poised to leverage these insights through synergistic combinations of epigenetic-targeting drugs and metabolic co-interventions. BET inhibitors like JQ1, which suppress BRD4-driven glycolytic addiction, may be complemented by NAD+ precursors (e.g., NMN) to concurrently reactivate SIRT1 and FTO, restoring both histone deacetylation and m6A homeostasis. Nutritional strategies - such as ketogenic diets to promote β-hydroxybutyrate-mediated histone deacetylase activation or selenium-rich regimens to stabilize NSUN2-dependent tRNA methylation - could further potentiate pharmacological interventions. However, translating these approaches into clinical practice requires precision delivery platforms, such as lipid nanoparticles conjugated with cardiac-tropic peptides, to minimize off-target effects. As spatial multi-omics and federated learning algorithms advance, the vision of dynamically adjustable, metabolism-aware epigenome editing is moving closer to clinical realization, offering promising strategies to counteract the rigid metabolic trajectories underlying cardiac aging and disease.
DECLARATIONS
Authors’ contributions
Conceived and designed research, synthesized and interpreted data, drafted, edited, revised, and approved the final manuscript: Liu J, Yang M, Zhou B
Availability of data and materials
Not applicable.
Financial support and sponsorship
This work was supported by the National Natural Science Foundation of China (32470826), the Beijing Municipal Health Commission (2024-3-034), the Student Research Training program of Tsinghua University (2511T0682), and the National Undergraduate Training Program for Innovation and Entrepreneurship & Student Research Training Program (S202410003143).
Conflicts of interest
Zhou B is a Youth Editorial Board member of The Journal of Cardiovascular Aging. Zhou B was not involved in any steps of editorial processing, notably including reviewer selection, manuscript handling, or decision making, while the other authors have declared that they have no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
1. Yazdanyar A, Newman AB. The burden of cardiovascular disease in the elderly: morbidity, mortality, and costs. Clin Geriatr Med. 2009;25:563-77.
2. Rodgers JL, Jones J, Bolleddu SI, et al. Cardiovascular risks associated with gender and aging. J Cardiovasc Dev Dis. 2019;6:19.
3. Qu C, Liao S, Zhang J, et al. Burden of cardiovascular disease among elderly: based on the Global Burden of Disease Study 2019. Eur Heart J Qual Care Clin Outcomes. 2024;10:143-53.
4. Gadó K, Szabo A, Markovics D, Virág A. Most common cardiovascular diseases of the elderly - A review article. Dev Health Sci. 2022;4:27-32.
5. Wu D, Jian C, Peng Q, et al. Prohibitin 2 deficiency impairs cardiac fatty acid oxidation and causes heart failure. Cell Death Dis. 2020;11:181.
6. Slawik M, Vidal-Puig AJ. Lipotoxicity, overnutrition and energy metabolism in aging. Ageing Res Rev. 2006;5:144-64.
7. White PJ, McGarrah RW, Grimsrud PA, et al. The BCKDH kinase and phosphatase integrate BCAA and lipid metabolism via regulation of ATP-citrate lyase. Cell Metab. 2018;27:1281-93.e7.
8. Chen X, Kang R, Kroemer G, Tang D. Organelle-specific regulation of ferroptosis. Cell Death Differ. 2021;28:2843-56.
10. Sun Z, Wu Y, Ordog T, et al. Aberrant signature methylome by DNMT1 hot spot mutation in hereditary sensory and autonomic neuropathy 1E. Epigenetics. 2014;9:1184-93.
11. Wu Y, Chen J, Sun Y, et al. PGN and LTA from staphylococcus aureus induced inflammation and decreased lactation through regulating DNA methylation and histone H3 acetylation in bovine mammary epithelial cells. Toxins. 2020;12:238.
12. Fang WF, Chen YM, Lin CY, et al. Histone deacetylase 2 (HDAC2) attenuates lipopolysaccharide (LPS)-induced inflammation by regulating PAI-1 expression. J Inflamm. 2018;15:3.
14. Ghazi T, Nagiah S, Chuturgoon AA. Fusaric acid decreases p53 expression by altering promoter methylation and m6A RNA methylation in human hepatocellular carcinoma (HepG2) cells. Epigenetics. 2021;16:79-91.
15. Yang X, Gao Y, Ji Z, et al. Dual functional molecular imprinted polymer-modified organometal lead halide perovskite: synthesis and application for photoelectrochemical sensing of salicylic acid. Anal Chem. 2019;91:9356-60.
16. Yang H, Sun Y, Li Q, Jin F, Dai Y. Diverse epigenetic regulations of macrophages in atherosclerosis. Front Cardiovasc Med. 2022;9:868788.
17. Namba-Fukuyo H, Funata S, Matsusaka K, et al. TET2 functions as a resistance factor against DNA methylation acquisition during Epstein-Barr virus infection. Oncotarget. 2016;7:81512-26.
18. Choi J, Diao H, Faliti CE, et al. Bcl-6 is the nexus transcription factor of T follicular helper cells via repressor-of-repressor circuits. Nat Immunol. 2020;21:777-89.
19. Paramasivam A, Priyadharsini JV. m6A RNA methylation in heart development, regeneration and disease. Hypertens Res. 2021;44:1236-7.
20. Piersanti RL, Santos JEP, Sheldon IM, Bromfield JJ. Lipopolysaccharide and tumor necrosis factor-alpha alter gene expression of oocytes and cumulus cells during bovine in vitro maturation. Mol Reprod Dev. 2019;86:1909-20.
21. Zeybel M, Hardy T, Wong YK, et al. Multigenerational epigenetic adaptation of the hepatic wound-healing response. Nat Med. 2012;18:1369-77.
22. Li R, Li X, Zhao J, et al. Mitochondrial STAT3 exacerbates LPS-induced sepsis by driving CPT1a-mediated fatty acid oxidation. Theranostics. 2022;12:976-98.
23. Wang H, Zhong J, Zhang C, et al. The whole-transcriptome landscape of muscle and adipose tissues reveals the ceRNA regulation network related to intramuscular fat deposition in yak. BMC Genomics. 2020;21:347.
24. Liao CG, Liang XH, Ke Y, et al. Active demethylation upregulates CD147 expression promoting non-small cell lung cancer invasion and metastasis. Oncogene. 2022;41:1780-94.
25. Leung YT, Shi L, Maurer K, et al. Interferon regulatory factor 1 and histone H4 acetylation in systemic lupus erythematosus. Epigenetics. 2015;10:191-9.
26. Wang Z, Zhang Y, Zhu S, et al. A small molecular compound CC1007 induces cross-lineage differentiation by inhibiting HDAC7 expression and HDAC7/MEF2C interaction in BCR-ABL1(-) pre-B-ALL. Cell Death Dis. 2020;11:738.
27. Liu B, Ou WC, Fang L, Tian CW, Xiong Y. Myocyte enhancer factor 2A plays a central role in the regulatory networks of cellular physiopathology. Aging Dis. 2023;14:331-49.
28. Tobin SW, Hashemi S, Dadson K, et al. Heart failure and MEF2 Transcriptome dynamics in response to β-blockers. Sci Rep. 2017;7:4476.
29. Yu X, Zhang L, Wen G, et al. Upregulated sirtuin 1 by miRNA-34a is required for smooth muscle cell differentiation from pluripotent stem cells. Cell Death Differ. 2015;22:1170-80.
30. Wu S, Fatkhutdinov N, Fukumoto T, et al. SWI/SNF catalytic subunits' switch drives resistance to EZH2 inhibitors in ARID1A-mutated cells. Nat Commun. 2018;9:4116.
31. Ronnebaum SM, Patterson C. The FoxO family in cardiac function and dysfunction. Annu Rev Physiol. 2010;72:81-94.
32. Koczor CA, Ludlow I, Fields E, et al. Mitochondrial polymerase gamma dysfunction and aging cause cardiac nuclear DNA methylation changes. Physiol Genomics. 2016;48:274-80.
33. Hoernle K, Abt DL, Fischer KM, et al. Arc-parallel flow in the mantle wedge beneath Costa Rica and Nicaragua. Nature. 2008;451:1094-7.
34. Tarragó MG, Chini CCS, Kanamori KS, et al. A potent and specific CD38 inhibitor ameliorates age-related metabolic dysfunction by reversing tissue NAD+ decline. Cell Metab. 2018;27:1081-95.e10.
35. Paneni F, Diaz Cañestro C, Libby P, Lüscher TF, Camici GG. The aging cardiovascular system: understanding it at the cellular and clinical levels. J Am Coll Cardiol. 2017;69:1952-67.
36. Wei Z, Xia J, Li J, et al. SIRT1 promotes glucolipid metabolic conversion to facilitate tumor development in colorectal carcinoma. Int J Biol Sci. 2023;19:1925-40.
37. Lopaschuk GD, Karwi QG, Tian R, Wende AR, Abel ED. Cardiac energy metabolism in heart failure. Circ Res. 2021;128:1487-513.
38. Chun SK, Lee S, Flores-Toro J, et al. Loss of sirtuin 1 and mitofusin 2 contributes to enhanced ischemia/reperfusion injury in aged livers. Aging Cell. 2018;17:e12761.
39. Li X, Chen C, Zhan X, et al. R13 preserves motor performance in SOD1(G93A) mice by improving mitochondrial function. Theranostics. 2021;11:7294-307.
40. Chang Y, Sun L, Kokura K, et al. MPP8 mediates the interactions between DNA methyltransferase Dnmt3a and H3K9 methyltransferase GLP/G9a. Nat Commun. 2011;2:533.
41. Mohni KN, Wessel SR, Zhao R, et al. HMCES maintains genome integrity by shielding Abasic sites in single-strand DNA. Cell. 2019;176:144-53.e13.
42. Lesker TR, Durairaj AC, Gálvez EJC, et al. An integrated metagenome catalog reveals new insights into the murine gut microbiome. Cell Rep. 2020;30:2909-22.e6.
43. Delahaye F, Wijetunga NA, Heo HJ, et al. Sexual dimorphism in epigenomic responses of stem cells to extreme fetal growth. Nat Commun. 2014;5:5187.
44. Ghoneim HE, Fan Y, Moustaki A, et al. De novo epigenetic programs inhibit PD-1 blockade-mediated T cell rejuvenation. Cell. 2017;170:142-57.e19.
45. Zhang Q, Zhang Y, Sun S, et al. ACOX2 is a prognostic marker and impedes the progression of hepatocellular carcinoma via PPARα pathway. Cell Death Dis. 2021;12:15.
46. Pararasa C, Ikwuobe J, Shigdar S, et al. Age-associated changes in long-chain fatty acid profile during healthy aging promote pro-inflammatory monocyte polarization via PPARγ. Aging Cell. 2016;15:128-39.
47. Lim JH, Kang YJ, Lee BY, et al. Epigenome-wide base-resolution profiling of DNA methylation in chorionic villi of fetuses with Down syndrome by methyl-capture sequencing. Clin Epigenetics. 2019;11:180.
48. Mehta AP, Li H, Reed SA, Supekova L, Javahishvili T, Schultz PG. Replacement of 2'-Deoxycytidine by 2'-Deoxycytidine analogues IN THe E. coli genome. J Am Chem Soc. 2016;138:14230-3.
49. Hou X, Shi X, Zhang W, et al. LDHA induces EMT gene transcription and regulates autophagy to promote the metastasis and tumorigenesis of papillary thyroid carcinoma. Cell Death Dis. 2021;12:347.
50. Flores A, Sandoval-Gonzalez S, Takahashi R, et al. Increased lactate dehydrogenase activity is dispensable in squamous carcinoma cells of origin. Nat Commun. 2019;10:91.
51. Tan X, Ye J, Liu W, et al. Acrylamide aggravates cognitive deficits at night period via the gut-brain axis by reprogramming the brain circadian clock. Arch Toxicol. 2019;93:467-86.
52. Hudec M, Dankova P, Solc R, Bettazova N, Cerna M. Epigenetic regulation of circadian rhythm and its possible role in diabetes mellitus. Int J Mol Sci. 2020;21:3005.
53. Tatton-Brown K, Seal S, Ruark E, et al. Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nat Genet. 2014;46:385-8.
54. Bellet MM, Sassone-Corsi P. Mammalian circadian clock and metabolism - the epigenetic link. J Cell Sci. 2010;123:3837-48.
55. Kwapis JL, Alaghband Y, Kramár EA, et al. Epigenetic regulation of the circadian gene Per1 contributes to age-related changes in hippocampal memory. Nat Commun. 2018;9:3323.
56. Lamming DW, Ye L, Katajisto P, et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science. 2012;335:1638-43.
57. Hardy T, Mann DA. Epigenetics in liver disease: from biology to therapeutics. Gut. 2016;65:1895-905.
58. Hattori M, Yokoyama Y, Hattori T, et al. Global DNA hypomethylation and hypoxia-induced expression of the ten eleven translocation (TET) family, TET1, in scleroderma fibroblasts. Exp Dermatol. 2015;24:841-6.
59. Koh JH, Hancock CR, Terada S, Higashida K, Holloszy JO, Han DH. PPARβ is essential for maintaining normal levels of PGC-1α and mitochondria and for the increase in muscle mitochondria induced by exercise. Cell Metab. 2017;25:1176-85.e5.
60. Deng W, Wei X, Xie Z, et al. Inhibition of PLK3 attenuates tubular epithelial cell apoptosis after renal ischemia-reperfusion injury by blocking the ATM/P53-mediated DNA damage response. Oxid Med Cell Longev. 2022;2022:4201287.
61. Gao Q, Luo J, Pan J, et al. Integrative analyses identify HIF-1α as a potential protective role with immune cell infiltration in adamantinomatous craniopharyngioma. Front Immunol. 2022;13:949509.
62. Byrne NJ, Soni S, Takahara S, et al. Chronically elevating circulating ketones can reduce cardiac inflammation and blunt the development of heart failure. Circ Heart Fail. 2020;13:e006573.
63. Taylor MD, Sadhukhan S, Kottangada P, et al. Nuclear role of WASp in the pathogenesis of dysregulated TH1 immunity in human Wiskott-Aldrich syndrome. Sci Transl Med. 2010;2:37ra44.
64. Yang J, Wang B, Li N, Zhou Q, Zhou W, Zhan Z. Salvia miltiorrhiza and carthamus tinctorius extract prevents cardiac fibrosis and dysfunction after myocardial infarction by epigenetically inhibiting Smad3 expression. Evid Based Complement Alternat Med. 2019;2019:6479136.
65. Eichner LJ, Curtis SD, Brun SN, et al. HDAC3 is critical in tumor development and therapeutic resistance in Kras-mutant non-small cell lung cancer. Sci Adv. 2023;9:eadd3243.
66. Sullivan JM, Badimon A, Schaefer U, et al. Autism-like syndrome is induced by pharmacological suppression of BET proteins in young mice. J Exp Med. 2015;212:1771-81.
67. Stomper J, Niroula A, Belizaire R, McConkey M, Bandaru TS, Ebert BL. Sex differences in DNMT3A-mutant clonal hematopoiesis and the effects of estrogen. Cell Rep. 2025;44:115494.
68. Sano S, Oshima K, Wang Y, et al. Tet2-mediated clonal hematopoiesis accelerates heart failure through a mechanism involving the IL-1β/NLRP3 inflammasome. J Am Coll Cardiol. 2018;71:875-86.
69. Wang S, Hu S, Luo X, et al. Prevalence and prognostic significance of DNMT3A- and TET2- clonal haematopoiesis-driver mutations in patients presenting with ST-segment elevation myocardial infarction. EBioMedicine. 2022;78:103964.
70. Li X, Xiong X, Zhang M, et al. Base-resolution mapping reveals distinct m1A methylome in nuclear- and mitochondrial-encoded transcripts. Mol Cell. 2017;68:993-1005.e9.
71. Gokhale NS, McIntyre ABR, McFadden MJ, et al.
72. Zhao Z, Meng J, Su R, et al. Epitranscriptomics in liver disease: basic concepts and therapeutic potential. J Hepatol. 2020;73:664-79.
73. Wu C, Chen W, He J, et al. Interplay of m6A and H3K27 trimethylation restrains inflammation during bacterial infection. Sci Adv. 2020;6:eaba0647.
74. Berulava T, Buchholz E, Elerdashvili V, et al. Changes in m6A RNA methylation contribute to heart failure progression by modulating translation. Eur J Heart Fail. 2020;22:54-66.
75. Weems JC, Griesel BA, Olson AL. Class II histone deacetylases downregulate GLUT4 transcription in response to increased cAMP signaling in cultured adipocytes and fasting mice. Diabetes. 2012;61:1404-14.
76. Chen YG, Chen R, Ahmad S, et al. N6-Methyladenosine Modification Controls Circular RNA Immunity. Mol Cell. 2019;76:96-109.e9.
77. Van Haute L, Lee SY, McCann BJ, et al. NSUN2 introduces 5-methylcytosines in mammalian mitochondrial tRNAs. Nucleic Acids Res. 2019;47:8720-33.
78. Cui L, Ma R, Cai J, et al. RNA modifications: importance in immune cell biology and related diseases. Signal Transduct Target Ther. 2022;7:334.
79. Chen H, Yang H, Zhu X, et al. m5C modification of mRNA serves a DNA damage code to promote homologous recombination. Nat Commun. 2020;11:2834.
80. Antonicka H, Choquet K, Lin ZY, Gingras AC, Kleinman CL, Shoubridge EA. A pseudouridine synthase module is essential for mitochondrial protein synthesis and cell viability. EMBO Rep. 2017;18:28-38.
81. Hermon SJ, Sennikova A, Becker S. Quantitative detection of pseudouridine in RNA by mass spectrometry. Sci Rep. 2024;14:27564.
82. Goh YT, Koh CWQ, Sim DY, Roca X, Goh WSS. METTL4 catalyzes m6Am methylation in U2 snRNA to regulate pre-mRNA splicing. Nucleic Acids Res. 2020;48:9250-61.
84. Zheng L, Chen X, He X, et al. METTL4-mediated mitochondrial DNA N6-methyldeoxyadenosine promoting macrophage inflammation and atherosclerosis. Circulation. 2025;151:946-65.
85. Li Y, Liu Y, Chen Z, et al. Protopanaxadiol ameliorates NAFLD by regulating hepatocyte lipid metabolism through AMPK/SIRT1 signaling pathway. Biomed Pharmacother. 2023;160:114319.
86. Ohshiro T, Konno M, Asai A, et al. Single-molecule RNA sequencing for simultaneous detection of m6A and 5mC. Sci Rep. 2021;11:19304.
87. Shah P, Ding Y, Niemczyk M, Kudla G, Plotkin JB. Rate-limiting steps in yeast protein translation. Cell. 2013;153:1589-601.
88. Gan XT, Zhao G, Huang CX, Rowe AC, Purdham DM, Karmazyn M. Identification of fat mass and obesity associated (FTO) protein expression in cardiomyocytes: regulation by leptin and its contribution to leptin-induced hypertrophy. PLoS One. 2013;8:e74235.
89. Su R, Dong L, Li C, et al. R-2HG exhibits anti-tumor activity by targeting FTO/m6A/MYC/CEBPA signaling. Cell. 2018;172:90-105.e23.
90. Burghardt KJ, Goodrich JM, Dolinoy DC, Ellingrod VL. DNA methylation, insulin resistance and second-generation antipsychotics in bipolar disorder. Epigenomics. 2015;7:343-52.
91. Pitt B, Kober L, Ponikowski P, et al. Safety and tolerability of the novel non-steroidal mineralocorticoid receptor antagonist BAY 94-8862 in patients with chronic heart failure and mild or moderate chronic kidney disease: a randomized, double-blind trial. Eur Heart J. 2013;34:2453-63.
92. Saucedo R, Valencia J, Gutierrez C, et al. Gene variants in the FTO gene are associated with adiponectin and TNF-alpha levels in gestational diabetes mellitus. Diabetol Metab Syndr. 2017;9:32.
93. Gu C, Wang Z, Zhou N, et al. Mettl14 inhibits bladder TIC self-renewal and bladder tumorigenesis through N6-methyladenosine of Notch1. Mol Cancer. 2019;18:168.
94. Fang L, Wang W, Li G, et al. CIGAR-seq, a CRISPR/Cas-based method for unbiased screening of novel mRNA modification regulators. Mol Syst Biol. 2020;16:e10025.
95. Xu Z, Lv B, Qin Y, Zhang B. Emerging roles and mechanism of m6A methylation in cardiometabolic diseases. Cells. 2022;11:1101.
96. Liu C, Li C, Liu Y. The role of metabolic reprogramming in pancreatic cancer chemoresistance. Front Pharmacol. 2022;13:1108776.
97. Deng LJ, Deng WQ, Fan SR, et al. m6A modification: recent advances, anticancer targeted drug discovery and beyond. Mol Cancer. 2022;21:52.
98. Schwartz S, Bernstein DA, Mumbach MR, et al. Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell. 2014;159:148-62.
99. Azubel M, Koivisto J, Malola S, et al. Nanoparticle imaging. Electron microscopy of gold nanoparticles at atomic resolution. Science. 2014;345:909-12.
100. Glazer AM, Davogustto G, Shaffer CM, et al. Arrhythmia variant associations and reclassifications in the eMERGE-III sequencing study. Circulation. 2022;145:877-91.
101. Jiang L, Wang X, Zhan X, et al. Advance in circular RNA modulation effects of heart failure. Gene. 2020;763:100036.
102. Chen LY, Wang L, Ren YX, et al. The circular RNA circ-ERBIN promotes growth and metastasis of colorectal cancer by miR-125a-5p and miR-138-5p/4EBP-1 mediated cap-independent HIF-1α translation. Mol Cancer. 2020;19:164.
103. Mills RE, Walter K, Stewart C, et al. Mapping copy number variation by population-scale genome sequencing. Nature. 2011;470:59-65.
105. Guermah M, Palhan VB, Tackett AJ, Chait BT, Roeder RG. Synergistic functions of SII and p300 in productive activator-dependent transcription of chromatin templates. Cell. 2006;125:275-86.
106. Shi H, Wei J, He C. Where, When, and How: context-dependent functions of RNA methylation writers, readers, and erasers. Mol Cell. 2019;74:640-50.
107. Zawistowski JS, Bevill SM, Goulet DR, et al. Enhancer remodeling during adaptive bypass to MEK inhibition is attenuated by pharmacologic targeting of the P-TEFb complex. Cancer Discov. 2017;7:302-21.
108. Fang F, Li G, Jing M, et al. C646 modulates inflammatory response and antibacterial activity of macrophage. Int Immunopharmacol. 2019;74:105736.
109. Ma F, Lei YY, Ding MG, Luo LH, Xie YC, Liu XL. LncRNA NEAT1 interacted with DNMT1 to regulate malignant phenotype of cancer cell and cytotoxic T cell infiltration via epigenetic inhibition of p53, cGAS, and STING in lung cancer. Front Genet. 2020;11:250.
110. Fierro C, Gatti V, La Banca V, et al. The long non-coding RNA NEAT1 is a ΔNp63 target gene modulating epidermal differentiation. Nat Commun. 2023;14:3795.
111. Hirose T, Yamazaki T, Nakagawa S. Molecular anatomy of the architectural NEAT1 noncoding RNA: the domains, interactors, and biogenesis pathway required to build phase-separated nuclear paraspeckles. Wiley Interdiscip Rev RNA. 2019;10:e1545.
112. Dukatz M, Dittrich M, Stahl E, et al. DNA methyltransferase DNMT3A forms interaction networks with the CpG site and flanking sequence elements for efficient methylation. J Biol Chem. 2022;298:102462.
113. Skeparnias I, Zhang J. Structural basis of NEAT1 lncRNA maturation and menRNA instability. Nat Struct Mol Biol. 2024;31:1650-4.
114. Jacq A, Becquet D, Guillen S, et al. Direct RNA-RNA interaction between Neat1 and RNA targets, as a mechanism for RNAs paraspeckle retention. RNA Biol. 2021;18:2016-27.
115. Yamazaki T, Souquere S, Chujo T, et al. Functional domains of NEAT1 architectural lncRNA induce paraspeckle assembly through phase separation. Mol Cell. 2018;70:1038-53.e7.
116. Chen ZX, Mann JR, Hsieh CL, Riggs AD, Chédin F. Physical and functional interactions between the human DNMT3L protein and members of the de novo methyltransferase family. J Cell Biochem. 2005;95:902-17.
117. Naveed A, Cooper JA, Li R, et al. NEAT1 polyA-modulating antisense oligonucleotides reveal opposing functions for both long non-coding RNA isoforms in neuroblastoma. Cell Mol Life Sci. 2021;78:2213-30.
118. Gu T, Lin X, Cullen SM, et al. DNMT3A and TET1 cooperate to regulate promoter epigenetic landscapes in mouse embryonic stem cells. Genome Biol. 2018;19:88.
119. Dong P, Xiong Y, Yue J, et al. Long non-coding RNA NEAT1: a novel target for diagnosis and therapy in human tumors. Front Genet. 2018;9:471.
120. Jiang L, Shao C, Wu QJ, et al. NEAT1 scaffolds RNA-binding proteins and the Microprocessor to globally enhance pri-miRNA processing. Nat Struct Mol Biol. 2017;24:816-24.
121. Kotini AG, Mpakali A, Agalioti T. Dnmt3a1 upregulates transcription of distinct genes and targets chromosomal gene clusters for epigenetic silencing in mouse embryonic stem cells. Mol Cell Biol. 2011;31:1577-92.
122. Wang Z, Fan P, Zhao Y, et al. NEAT1 modulates herpes simplex virus-1 replication by regulating viral gene transcription. Cell Mol Life Sci. 2017;74:1117-31.
123. Smith AM, LaValle TA, Shinawi M, et al. Functional and epigenetic phenotypes of humans and mice with DNMT3A overgrowth syndrome. Nat Commun. 2021;12:4549.
124. Bosselut R. Control of intra-thymic αβ T cell selection and maturation by H3K27 methylation and demethylation. Front Immunol. 2019;10:688.
125. Bayliak M, Burdyliuk N, Lushchak V. Growth on alpha-ketoglutarate increases oxidative stress resistance in the yeast saccharomyces cerevisiae. Int J Microbiol. 2017;2017:5792192.
126. Lin AP, Abbas S, Kim SW, et al. D2HGDH regulates alpha-ketoglutarate levels and dioxygenase function by modulating IDH2. Nat Commun. 2015;6:7768.
127. Xu C, Wang L, Fozouni P, et al. SIRT1 is downregulated by autophagy in senescence and ageing. Nat Cell Biol. 2020;22:1170-9.
128. Wangpaichitr M, Wu C, Li YY, et al. Exploiting ROS and metabolic differences to kill cisplatin resistant lung cancer. Oncotarget. 2017;8:49275-92.
129. Afanas'ev I. New nucleophilic mechanisms of ros-dependent epigenetic modifications: comparison of aging and cancer. Aging Dis. 2014;5:52-62.
130. Chatterjee S, Daenthanasanmak A, Chakraborty P, et al. CD38-NAD+Axis regulates immunotherapeutic anti-tumor T cell response. Cell Metab. 2018;27:85-100.e8.
131. Jiménez-Loygorri JI, Villarejo-Zori B, Viedma-Poyatos Á, et al. Mitophagy curtails cytosolic mtDNA-dependent activation of cGAS/STING inflammation during aging. Nat Commun. 2024;15:830.
132. Liu J, Zhang C, Hu W, Feng Z. Parkinson's disease-associated protein Parkin: an unusual player in cancer. Cancer Commun. 2018;38:40.
133. Jiang X, Liu B, Nie Z, et al. The role of m6A modification in the biological functions and diseases. Signal Transduct Target Ther. 2021;6:74.
134. Harbauer AB, Hees JT, Wanderoy S, et al. Neuronal mitochondria transport Pink1 mRNA via synaptojanin 2 to support local mitophagy. Neuron. 2022;110:1516-31.e9.
135. Agirre X, Román-Gómez J, Vázquez I, et al. Abnormal methylation of the common PARK2 and PACRG promoter is associated with downregulation of gene expression in acute lymphoblastic leukemia and chronic myeloid leukemia. Int J Cancer. 2006;118:1945-53.
136. Kietzmann T, Petry A, Shvetsova A, Gerhold JM, Görlach A. The epigenetic landscape related to reactive oxygen species formation in the cardiovascular system. Br J Pharmacol. 2017;174:1533-54.
137. Lee Y, Choe J, Park OH, Kim YK. Molecular mechanisms driving mRNA degradation by m6A modification. Trends Genet. 2020;36:177-88.
138. Sun Y, Shen W, Hu S, et al. METTL3 promotes chemoresistance in small cell lung cancer by inducing mitophagy. J Exp Clin Cancer Res. 2023;42:65.
139. Wang X, Lu Z, Gomez A, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117-20.
140. Dorn LE, Lasman L, Chen J, et al. The N6-methyladenosine mRNA methylase METTL3 controls cardiac homeostasis and hypertrophy. Circulation. 2019;139:533-45.
141. Kmietczyk V, Riechert E, Kalinski L, et al. m6A-mRNA methylation regulates cardiac gene expression and cellular growth. Life Sci Alliance. 2019;2:e201800233.
142. Mathiyalagan P, Adamiak M, Mayourian J, et al. FTO-dependent N6-methyladenosine regulates cardiac function during remodeling and repair. Circulation. 2019;139:518-32.
143. Donato E, Croci O, Sabò A, et al. Compensatory RNA polymerase 2 loading determines the efficacy and transcriptional selectivity of JQ1 in Myc-driven tumors. Leukemia. 2017;31:479-90.
144. Mu J, Zhang D, Tian Y, Xie Z, Zou MH. BRD4 inhibition by JQ1 prevents high-fat diet-induced diabetic cardiomyopathy by activating PINK1/Parkin-mediated mitophagy in vivo. J Mol Cell Cardiol. 2020;149:1-14.
145. Wang JY, Chen LJ, Qiang P. The potential role of N6-methyladenosine (m6A) demethylase fat mass and obesity-associated gene (FTO) in human cancers. Onco Targets Ther. 2020;13:12845-56.
146. Elkashef SM, Lin AP, Myers J, et al. IDH mutation, competitive inhibition of FTO, and RNA methylation. Cancer Cell. 2017;31:619-20.
147. Shaaban Z, Khoradmehr A, Amiri-Yekta A, Jafarzadeh Shirazi MR, Tamadon A. Pathophysiologic mechanisms of obesity- and chronic inflammation-related genes in etiology of polycystic ovary syndrome. Iran J Basic Med Sci. 2019;22:1378-86.
148. Kanemaru K, Cranley J, Muraro D, et al. Spatially resolved multiomics of human cardiac niches. Nature. 2023;619:801-10.
149. Riahi R, Wang S, Long M, et al. Mapping photothermally induced gene expression in living cells and tissues by nanorod-locked nucleic acid complexes. ACS Nano. 2014;8:3597-605.
150. Kuppe C, Ramirez Flores RO, Li Z, et al. Spatial multi-omic map of human myocardial infarction. Nature. 2022;608:766-77.
151. Yamada S, Ko T, Hatsuse S, et al. Spatiotemporal transcriptome analysis reveals critical roles for mechano-sensing genes at the border zone in remodeling after myocardial infarction. Nat Cardiovasc Res. 2022;1:1072-83.
152. Longenecker JZ, Gilbert CJ, Golubeva VA, Martens CR, Accornero F. Epitranscriptomics in the heart: a focus on m6A. Curr Heart Fail Rep. 2020;17:205-12.
153. Lee AC, Lee Y, Choi A, et al. Spatial epitranscriptomics reveals A-to-I editome specific to cancer stem cell microniches. Nat Commun. 2022;13:2540.
154. Palmer JA, Rosenthal N, Teichmann SA, Litvinukova M. Revisiting cardiac biology in the era of single cell and spatial omics. Circ Res. 2024;134:1681-702.
155. Duan Q, McMahon S, Anand P, et al. BET bromodomain inhibition suppresses innate inflammatory and profibrotic transcriptional networks in heart failure. Sci Transl Med. 2017;9:eaah5084.
156. Mills RJ, Humphrey SJ, Fortuna PRJ, et al. BET inhibition blocks inflammation-induced cardiac dysfunction and SARS-CoV-2 infection. Cell. 2021;184:2167-82.e22.
157. Abdellatif M, Sedej S, Kroemer G. NAD+ metabolism in cardiac health, aging, and disease. Circulation. 2021;144:1795-817.
158. Lauritzen KH, Olsen MB, Ahmed MS, et al. Instability in NAD+ metabolism leads to impaired cardiac mitochondrial function and communication. Elife. 2021;10:e59828.
159. Lu TM, Tsai JY, Chen YC, et al. Downregulation of Sirt1 as aging change in advanced heart failure. J Biomed Sci. 2014;21:57.
160. Costantino S, Mengozzi A, Velagapudi S, et al. Treatment with recombinant Sirt1 rewires the cardiac lipidome and rescues diabetes-related metabolic cardiomyopathy. Cardiovasc Diabetol. 2023;22:312.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Issue
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].