


Beyond point defects: a first-principles investigation of topological line defects in graphene as continuous catalytic highways for Li-S batteries
 0
0 Abstract
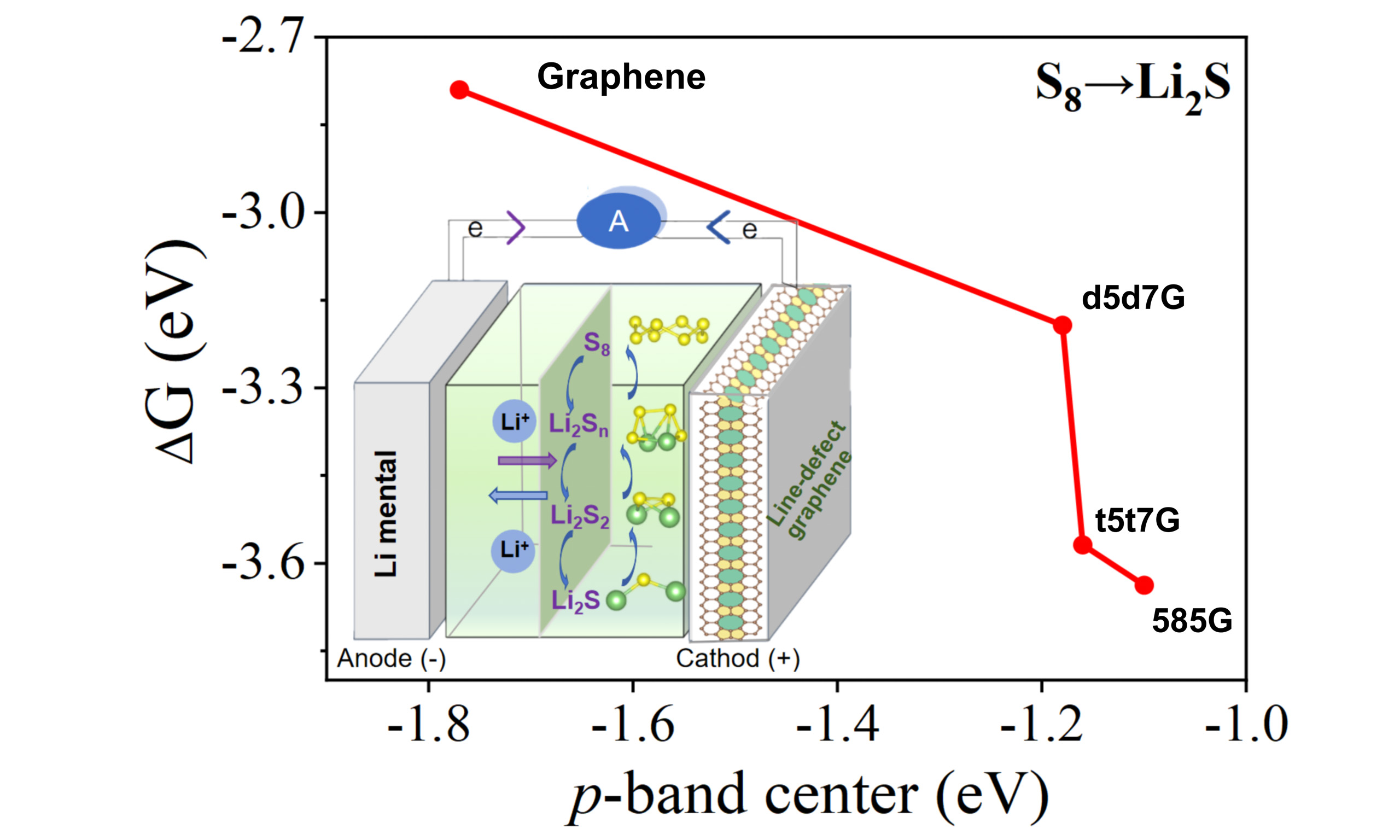
Lithium-sulfur (Li-S) batteries have long been plagued by the shuttle effect and sluggish sulfur conversion, which limit their practical application. To address these issues, we propose and investigate several line-defect graphene configurations (including d5d7G, t5t7G, and 585G) as sulfur cathode host materials in Li-S batteries and evaluate their adsorption and catalytic potential using first-principles calculations. The results demonstrate that the continuous high-density linear defects are rich in active sites, which not only enhance the adsorption and the catalytic activity for lithium polysulfides (LiPSs), but also maintain a low barrier for Li+ diffusion. A qualitative correlation between the p-band center and the Gibbs free energy barrier is established, directly linking the defect topology to the catalytic activity. Comprehensive screening identifies the 585G configuration as the most promising candidate, as it exhibits the strongest polysulfide adsorption (Ea = -0.90 eV to -1.86 eV), the highest p-band center (-1.10 eV), and the lowest Gibbs free energy barrier (2.35 eV). This work provides insights into how the line-defect architectures govern the catalytic activity in graphene, offering a new design paradigm for the carbon-based sulfur hosts.
Keywords
INTRODUCTION
To meet the world’s growing demand for sustainable and renewable energy, developing rechargeable batteries that combine high energy density with long cycle life is critical[1-4]. In recent years, lithium-sulfur (Li-S) batteries have attracted intense attention as an ideal successor to conventional lithium-ion technologies because of their exceptional theoretical capacity (1,675 mAh g-1) and energy density
To address these issues, different conductive encapsulation materials are introduced into the positive electrode system as sulfur carriers[12-16]. Carbon materials, especially graphene, are favored for their high conductivity, large surface area and two-dimensional honeycomb lattice[17,18]. However, the interaction between the pristine graphene and polysulfides is weak, so the discharged Li2S product readily detaches and becomes “dead sulfur”, while soluble higher-order polysulfides migrate to the lithium anode and generate the shuttle effect, leading to rapid capacity fade and low coulombic efficiency[19,20]. Notably, during the preparation and processing of graphene, structural defects, including the point and the line defects, inevitably are formed[21-23]. The defects expose additional active facets and edges sites, reshape atomic and electronic structures via local electron redistribution and band-gap tuning, and greatly boost the kinetics of the adsorption and the catalytic conversion[24-27]. Prior studies have successively examined a series of single-vacancy point-defect graphene structures as cathode hosts for Li-S batteries and found that the presence of the point-defect enhances the adsorption and catalytic conversion of polysulfides[28-30]. Concurrently, line-defect graphene has emerged and attracted much attention in recent years[31-33]. Characterized by continuously exposed, high-density carbon atoms, the line-defect graphene has been experimentally synthesized and verified to offer ample adsorption sites and induce significant local charge redistribution, showing great application potential in energy storage and electrocatalysis[34-37], thereby presenting a promising host candidate. Botello-Méndez et al. mapped the three lowest-energy reconstruction pathways of the divacancy defect graphene (d5d7G, t5t7G, and 585G) and demonstrated that charge is strongly localized along the defect axis with the line defects effectively behaving as “embedded metallic nanowires”[38]. Previous studies have applied the line-defect graphene as the electrode materials in batteries, and it has been believed that the line defects can provide a wider active area compared to the point defects, effectively regulating the pathways for charge transfer and ion diffusion, thereby improving the electrochemical performance[39]. However, their exploitation as the cathode hosts for Li-S batteries has yet to be explored. Experimentally synthesizing a well-defined series of the line-defect graphene is extremely challenging, limiting systematic investigations into their performance as sulfur hosts.
Here, using first-principles calculations, we systematically investigate the application potential of the line-defect graphene (d5d7G, t5t7G, and 585G) as cathode materials for Li-S batteries. The presence of line defects in graphene can simultaneously enhance the adsorption and the catalytic activity towards polysulfides. Through the partial density of states (PDOS) and charge density calculations, we elucidate how defect structure governs electrochemical performance and establish a qualitative relationship between the p-band center and the barrier of Gibbs free energy, enabling the correlation of defect topology with redox kinetics. Our screening reveals that the large-sized defects in the 585G create an optimal electronic environment at the active sites, endowing this structure with the strongest polysulfide adsorption
COMPUTATIONAL METHOD
All of the spin-polarized density functional theory (DFT) calculations are performed using the Vienna ab initio Simulation Package (VASP) program[40]. The exchange-correlation interaction is treated within the generalized gradient approximation (GGA) using the Perdew-Burke-Ernzerhof (PBE) functional[41-44]. The projector augmented wave (PAW) method is employed to describe the ion-electron interactions[45]. The plane-wave energy cutoff is set to 500 eV. For structural optimization and self-consistent field (SCF) calculations, gamma-centered k-point meshes of 3 × 3 × 1 and 8 × 8 × 1 are used for Brillouin zone sampling, respectively. The ab initio molecular dynamics (AIMD) calculations are carried out in the canonical (NVT) ensemble to assess the thermal stability of the line-defect graphene at high temperature, with a time step of
where H, T, S, EDFT, ZPE, and EH represent the reaction enthalpy, reaction temperature, entropy, the total energy of the system, zero-point energy, and the enthalpy function contribution, respectively. The reference energy of Li was obtained from bulk Li metal and normalized by the number of Li atoms. All thermodynamic parameters including entropy and enthalpy at 298.15 K (room temperature) are extracted from vibrational frequency calculations based on statistical thermodynamics. The Gibbs free energy calculation was performed at an applied potential of U = 0 V.
RESULTS AND DISCUSSION
Structure and electronic properties of S8/Li2Sn clusters and the line-defect graphene
During discharge, the sulfur cathode successively generates a series of sulfur-containing species via the SRR. Figure 1A depicts the optimized molecular structures of S8 and Li2Sn (n = 1, 2, 4, 6, 8) clusters, which are in good agreement with the previous reports[49]. As the sulfur index n increases, Li2Sn evolves from short-chain motifs into intricate three-dimensional clusters. The long-chain LiPSs (Li2S8, Li2S6 and Li2S4) exhibit elongated S-S bond lengths and diminished bond energies, which facilitate their dissolution in the electrolyte and contribute to the shuttle effect.
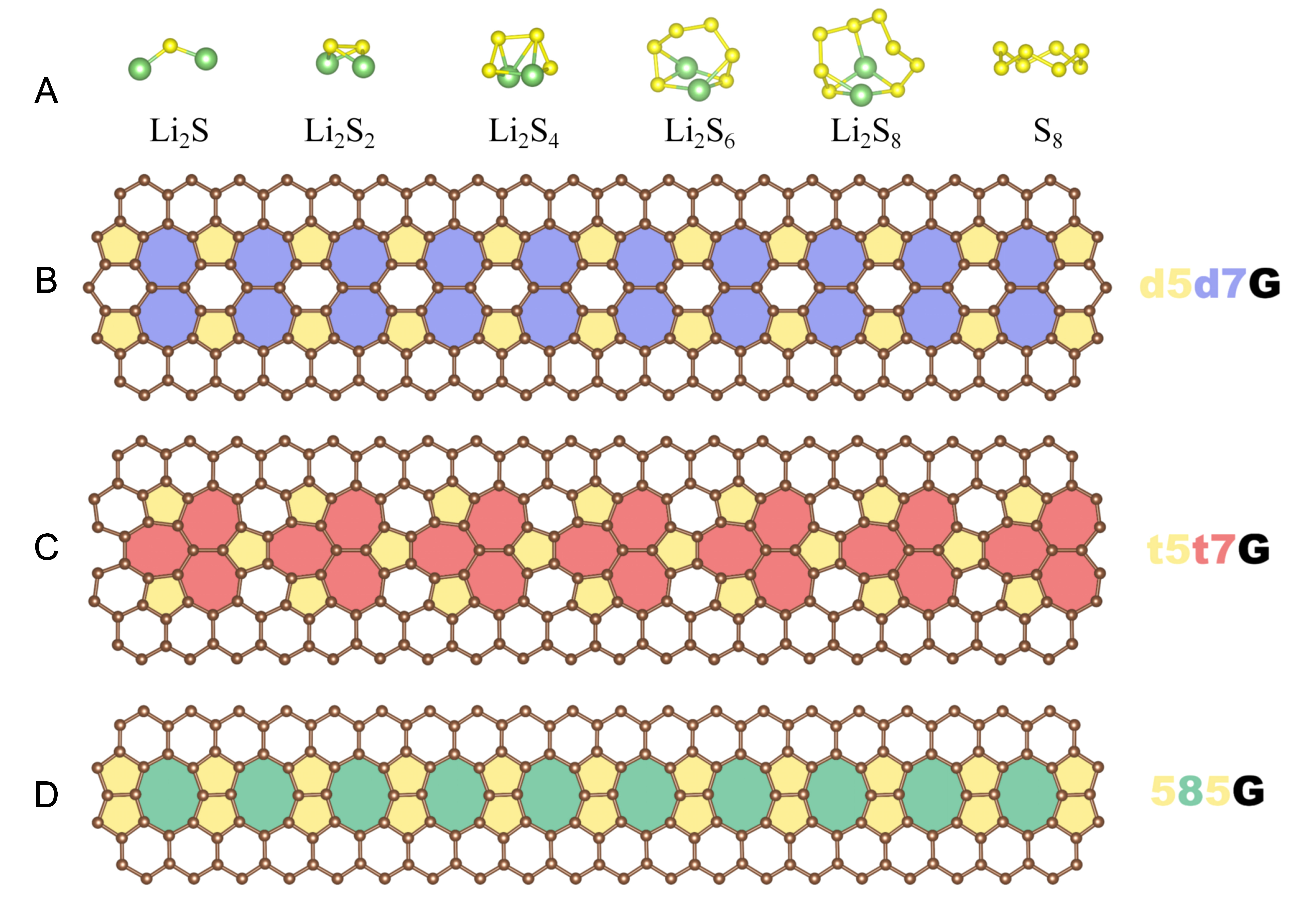
Figure 1. Atomic structures of (A) isolated S8 and Li2Sn (n = 1, 2, 4, 6, 8) clusters, and graphene with (B) d5d7, (C) t5t7, and (D) 585 line-defects.
To investigate the line-defect graphene as cathode materials for Li-S batteries, we construct three common-used line-defect graphene: double-pentagon-double-heptagon graphene (d5d7G), triple-pentagon-triple-heptagon graphene (t5t7G), and pentagon-octagon-pentagon graphene (585G) as shown in Figure 1B-D. A
To evaluate the stability of the line-defect graphene, the formation energy per unit length Ec(α) is calculated using the following equation[52]:
where α indicates the nature of the line-defect: d5d7G, t5t7G, and 585G, and the ET(α) is the total energy of a graphene plane containing an α-type line-defect in the supercells. Nα, Egr, and dα correspond to the number of the C atoms of the defective graphene in the supercells, the energy of one C atom in the pristine graphene, and the cell parameter in the direction of the defect extension in the supercells, respectively. The Ec(α) of d5d7G, t5t7G, and 585G are 0.82 eV Å-1, 0.79 eV Å-1, and 0.97 eV Å-1, respectively. Although the 585G possesses the highest formation energy, it has been successfully synthesized in experiments[53]. The lower Ec(α) of the other defects suggest that they are also synthetically feasible. It is reported that the t5t7G is a topological structure obtained through laser irradiation exhibiting excellent stability[38], which is consistent with our theoretical prediction. Experimental studies have demonstrated that well-defined graphene line defects can be successfully synthesized and manipulated through increasingly mature fabrication strategies[54,55], while line-defect-rich graphene structures have also been integrated into rechargeable battery electrodes and exhibited promising electrochemical performance[56,57]. Combined with the favorable formation energies by DFT conclusion in this work, these findings support the structural feasibility and practical relevance of line-defect graphene as sulfur-host materials.
To further validate the thermal stability, we perform AIMD simulations of these line-defect graphene at a temperature of 500 K. As shown in Figure 2, after a 2,000 fs time interval, these structures exhibit almost no significant changes when compared to their initial structures. The relatively minor energy fluctuations of the line-defect graphene demonstrate their structural integrity at high temperatures.
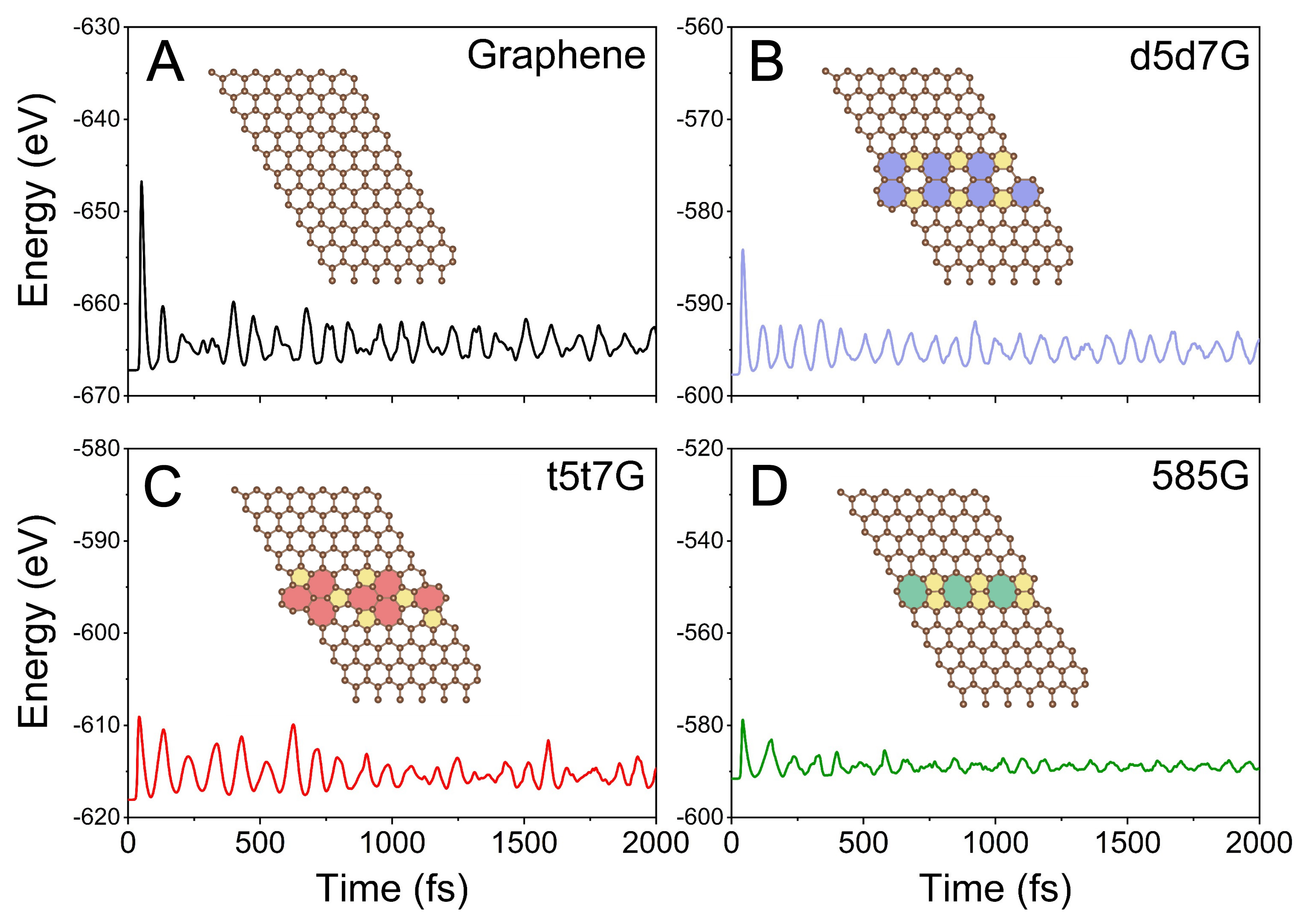
Figure 2. Energy fluctuations of (A) graphene, (B) d5d7G, (C) t5t7G, and (D) 585G with time progress of AIMD simulations at 500 K. Insets show the corresponding atomic snapshots at 2,000 fs. Different colors are used only to visually distinguish the structural motifs and do not represent different atomic species. AIMD: Ab initio molecular dynamics.
As shown in Figure 3A-D, to explore the effect of line defects on the electronic structure of graphene, we calculate the PDOS of the intrinsic/defective graphene. Compared with those of the pristine graphene, the electron states around the Fermi level are enhanced in the line-defect graphene, especially in the 585G. The increased electronic-state occupation near the Fermi level indicates a stronger metallic character and a higher density of electronically accessible states, which can facilitate electron transport and accelerate interfacial charge-transfer processes during the electrochemical reactions, thereby potentially improving the electronic conductivity of the electrode. In the Figure 3E-H, due to its non-polar nature, the pristine graphene exhibits a symmetric distribution of electron gain and loss among carbon atoms. In contrast, the line defects break the intrinsic lattice symmetry, leading to a substantial redistribution of Bader charges along the defect zones. This symmetry-breaking induces an extended local polarization across the defect region rather than isolated polar spots, which may facilitate the adsorption of S8 and LiPSs[39].
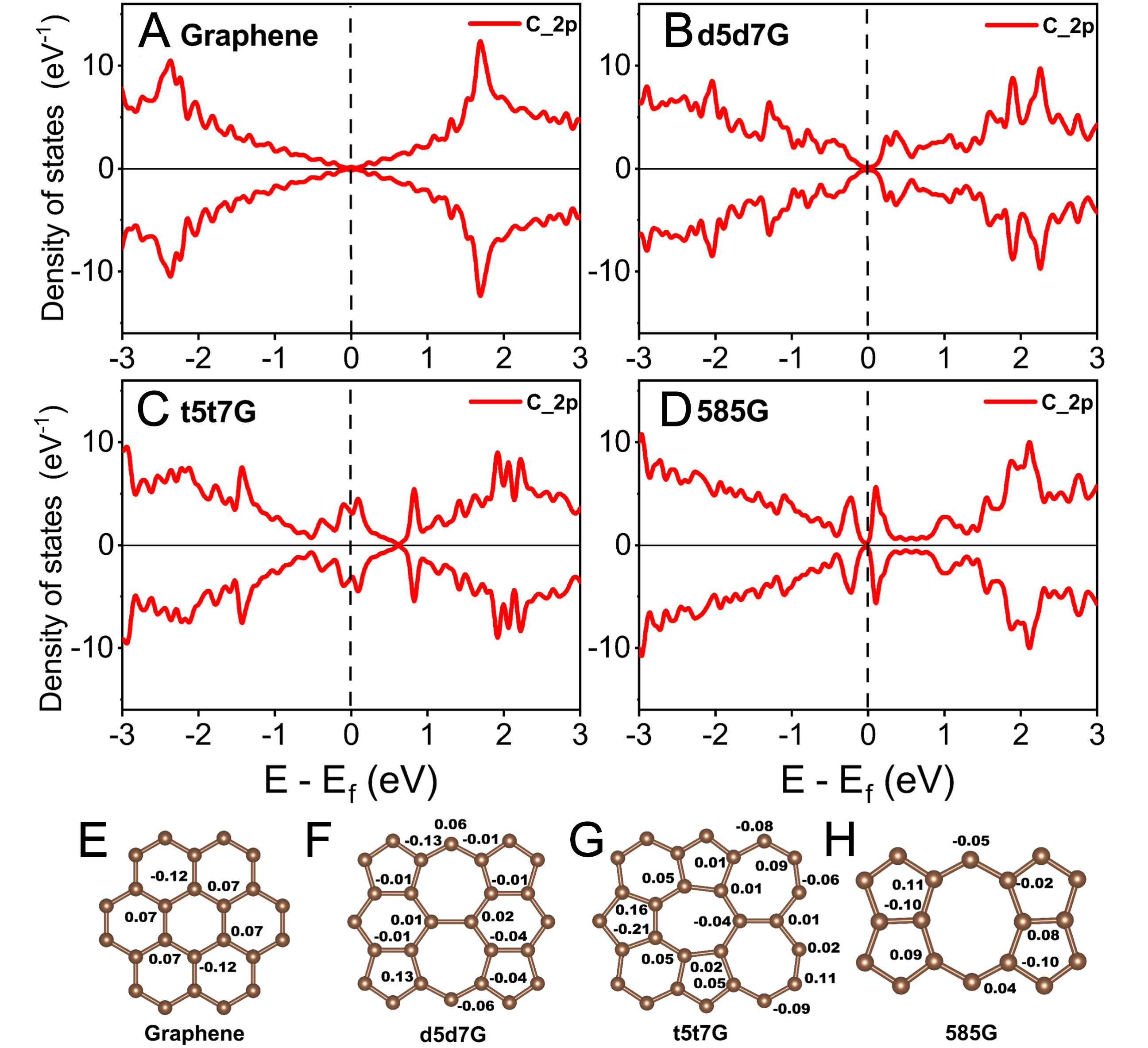
Figure 3. Partial density of states (PDOS) for (A) graphene, (B) d5d7G, (C) t5t7G, and (D) 585G. Bader charge distributions of (E) graphene, (F) d5d7G, (G) t5t7G, and (H) 585G. The numbers in panels (E-H) denote the Bader charge (in units of e) on the key atoms.
Adsorption of S8 and LiPSs on the line-defect graphene
The adsorption strength of S8 and LiPSs on cathode materials is crucial in mitigating the shuttle effect. The adsorption structures of S8 and LiPSs on the intrinsic and the line-defect graphene are relaxed and illustrated in Figure 4. The S8 molecules adsorb parallel to both pristine and line-defect graphene surfaces. Similarly, the short-chain LiPSs (Li2S, Li2S2) adsorb with the Li-Li axis parallel to the cathode surface. In contrast, the long-chain LiPSs (Li2S4, Li2S6, and Li2S8) adsorb with the Li-Li axis perpendicular to the cathode surface, except that the Li-Li axis of Li2S4 tends to align parallel to the substrate in t5t7G. From a top-view perspective, S8 and LiPSs are preferentially adsorbed directly above the center of the defects in the line-defect graphene. There is a large octagon defect in the 585G, and S8 and LiPSs preferentially adsorb atop the octagon defect.
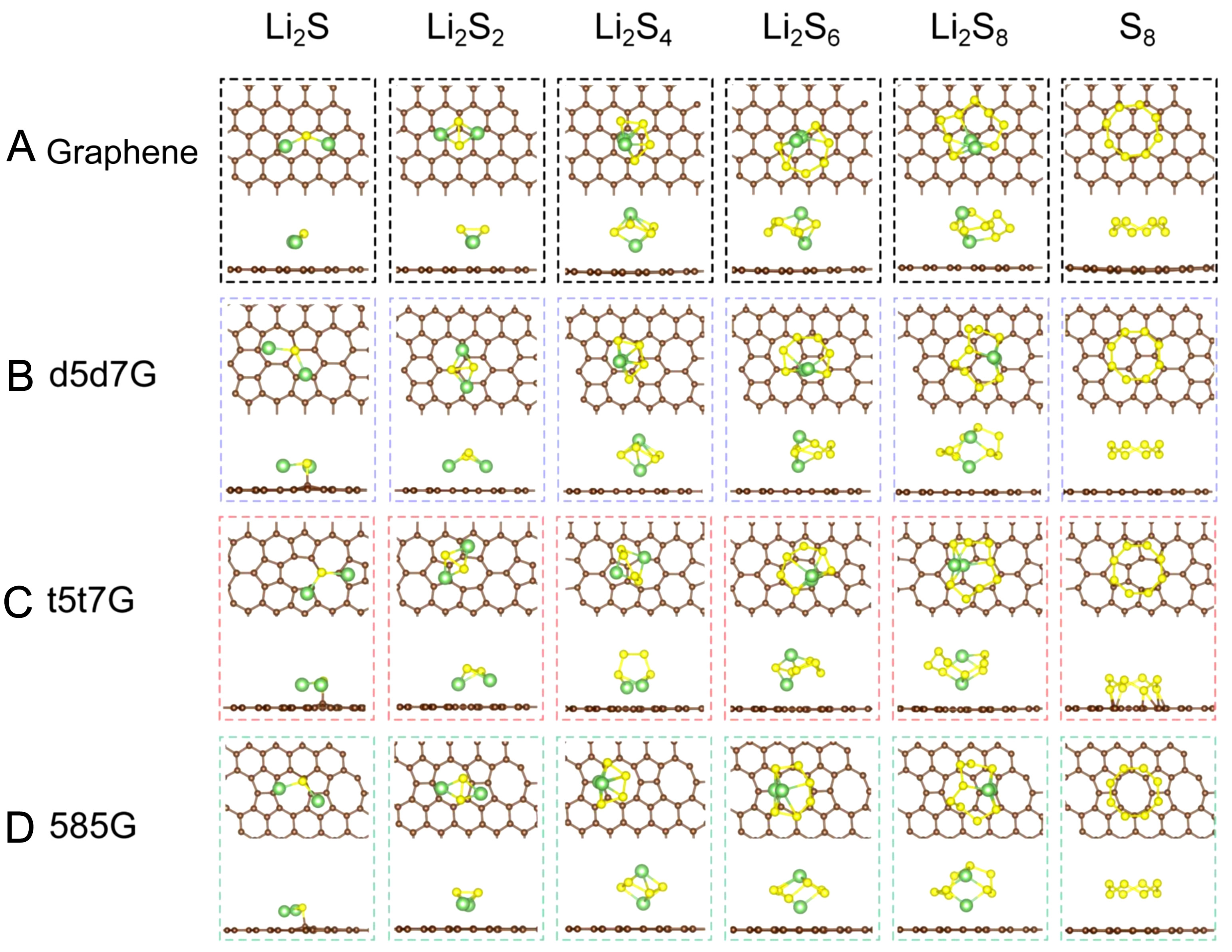
Figure 4. Stable adsorption configuration of polysulfides on the (A) graphene, (B) d5d7G, (C) t5t7G, and (D) 585G (top and side views).
Subsequently, we calculate the adsorption energy Ea for the line-defect graphene to the isolated S8 and LiPSs by the following equation[58]:
where Et, ELD, and EPS represent the energy of the adsorbed system, the free-standing line-defect graphene, and the isolated S8 and LiPSs, respectively. A more negative adsorption energy indicates stronger binding and a more stable structure after adsorption. Figure 5 depicts the adsorption energies of the stable structures, with corresponding values listed in Supplementary Table 1. The adsorption energies are weakest with the range from -0.45 eV to -0.88 eV for S8 and LiPSs in the pristine graphene. Both d5d7G and t5t7G show moderate adsorption enhancement, with adsorption energies ranging from -0.84 eV to -1.72 eV and -0.89 eV to -1.74 eV, respectively. Notably, 585G exhibits the strongest adsorption among all samples, with values between -0.89 eV and -1.86 eV. Meanwhile, the adsorption energy of Li2S on 585G is distinctly higher than that on d5d7G and t5t7G. Notably, the adsorption energies from Li2S4 to Li2S are significantly enhanced in the line-defect graphene relative to the pristine graphene. Compared to the pristine graphene, the line-defect graphene exhibits polarity due to the extended local polarization induced by symmetry-breaking, which enhances the electronic interaction with the polysulfides. Meanwhile, the adsorption energies are generally higher than those reported for representative point-defect graphene systems in previous studies (-0.58 eV to -1.06 eV)[28,59], indicating that the line-defect graphene provides stronger polysulfide anchoring capability. In contrast to the point-defect graphene, the line-defect graphene provides a continuous and high-density array of active sites, which significantly enhances the polysulfides adsorption and effectively suppresses the shuttle effect. Among them, the t5t7G and the 585G, with relatively large and concentrated defect structures, show prominent adsorption advantages.
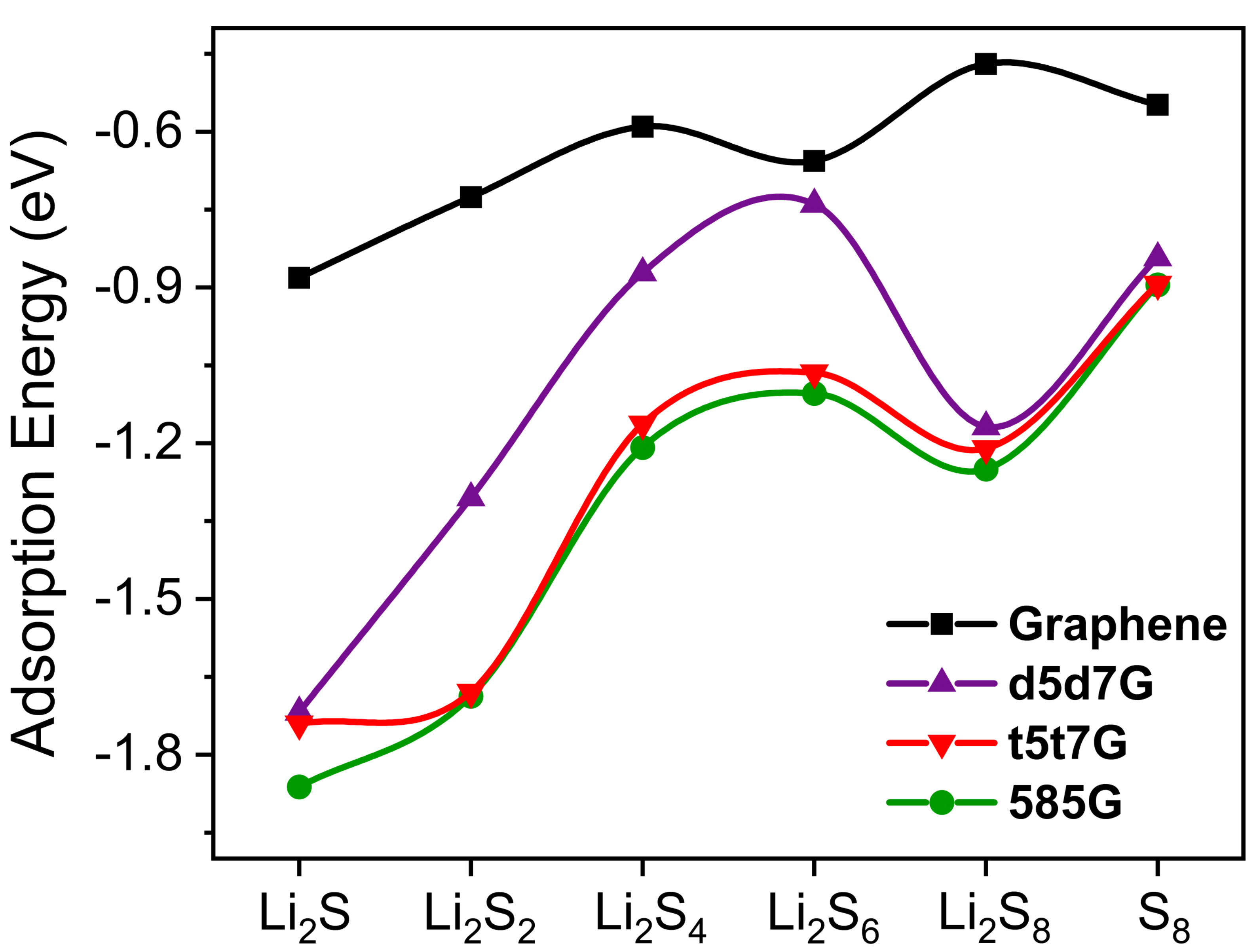
Figure 5. The adsorption energies of S8 and LiPSs on graphene and the line-defect graphene.
Li2S adsorption constitutes the first step of the entire nucleation process and significantly influences subsequent Li2S nucleation and growth[60,61]. Owing to the similar bonding character of Li2S and other LiPSs, a substrate that strongly binds Li2S usually exhibits high affinity toward other LiPSs as well[62]. Therefore, we choose Li2S as a representative model to further investigate the interaction between LiPSs and these materials. In the Supplementary Figure 1, to further elucidate the macroscopic crystallization behavior of Li2S and evaluate whether it induces active-site passivation, AIMD simulations were performed using a (Li2S)5 cluster at 500 K for 10,000 fs. Characteristically, even when the (Li2S)5 cluster was initially positioned away from the line defect, the molecules spontaneously rearranged and anchored onto the 585G framework during the dynamic relaxation. More importantly, the optimized snapshot exhibits a distinct 2D surface-spreading growth mode, where Li2S species wet the flat 1D periodic atomic boundary uniformly, instead of rolling up into spherical 3D insulating clusters as typically observed on pristine graphene[29]. This prominent structural alignment confirms that the 585G serves as a low-barrier, continuous catalytic highway that may enable orderly Li2S film deposition without blocking the active electrochemical interfaces.
Furthermore, we systematically analyzed the average variations in Li-S bond lengths and Li-S-Li bond angles, as well as the charge transfer upon Li2S adsorption, as listed in Supplementary Table 2. For the weakest adsorption in the pristine graphene, the charge transfer (-0.31 e) is minimal, the Li-S bond-length change (0.01 Å) is negligible, and the bond-angle variation (1.60°) is slight. The line-defect graphene exhibit significantly enhanced Li-S bond-length changes, bond-angle variations, charge transfer numbers and adsorption energies. Among them, the 585G with large-sized octagonal defects exhibits the highest adsorption energy, corresponding to the largest charge transfer number (-0.99 e), as well as the maximum bond-length change (0.24 Å) and bond-angle variation (13.80°). The large-sized defect structure of the 585G provides a favorable environment for the electron interactions.
Supplementary Figure 2 further illustrates the positive relationship among adsorption energy, charge transfer, and Li-S bond/angle distortion. Upon adsorption, the greater Li-S bond/angle distortion corresponds to the higher adsorption energy and the larger charge transfer. This arises because the stronger binding stretches the Li-S bond, pulls the sulfur closer to the defective active sites, and thereby increases mutual charge transfer. Thus, the amount of charge transfer between Li2S and the defective substrate can serve as a descriptor for the adsorption strength of the host material. By calculating the charge density difference ∆ρ after Li2S adsorption, thus providing insight into the interaction mechanism between polysulfides and the substrates. As shown in Supplementary Figure 3, the yellow and cyan parts represent the charge accumulation and loss, respectively. In the line-defect graphene, charge accumulates between S and the defect-site C while depleting between Li and S, evidencing that Li2S adsorption is governed by S-C chemical bonding, which endows it with strong adsorption capability.
To gain deeper insight into the interaction between the line-defect graphene and polysulfides, the PDOS for Li2S4 adsorbed on the four systems are further analyzed as a representative model system [Figure 6]. Compared with the pristine graphene, there are pronounced electronic-state occupation near the Fermi level in the line-defect graphene, indicating their high electrochemical activity and conductivity, which is advantageous for the reversible charge/discharge reactions. What’s more, little appreciable overlap exists between the C2p orbital and the Li2s/S2p orbitals around the Fermi level in the pristine graphene and d5d7G, reflecting a weak chemical interaction. Meanwhile, the Li2s states remain relatively localized in these two systems, indicating limited orbital coupling and interfacial charge redistribution involving Li species. An obvious hybridization between C2p and S2p orbitals occurs in the 585G around the Fermi level, while it also occurs in the t5t7G at -0.5 eV to -1 eV. The orbital hybridization between C2p and S2p is highest in the 585G, which is consistent with the strongest adsorption. In addition, the enhanced overlap between the delocalized C2p states and Li2s states in the 585G suggests strengthened interfacial orbital coupling and charge-transfer interaction involving Li species, which further contributes to the improved adsorption and catalytic conversion behavior.
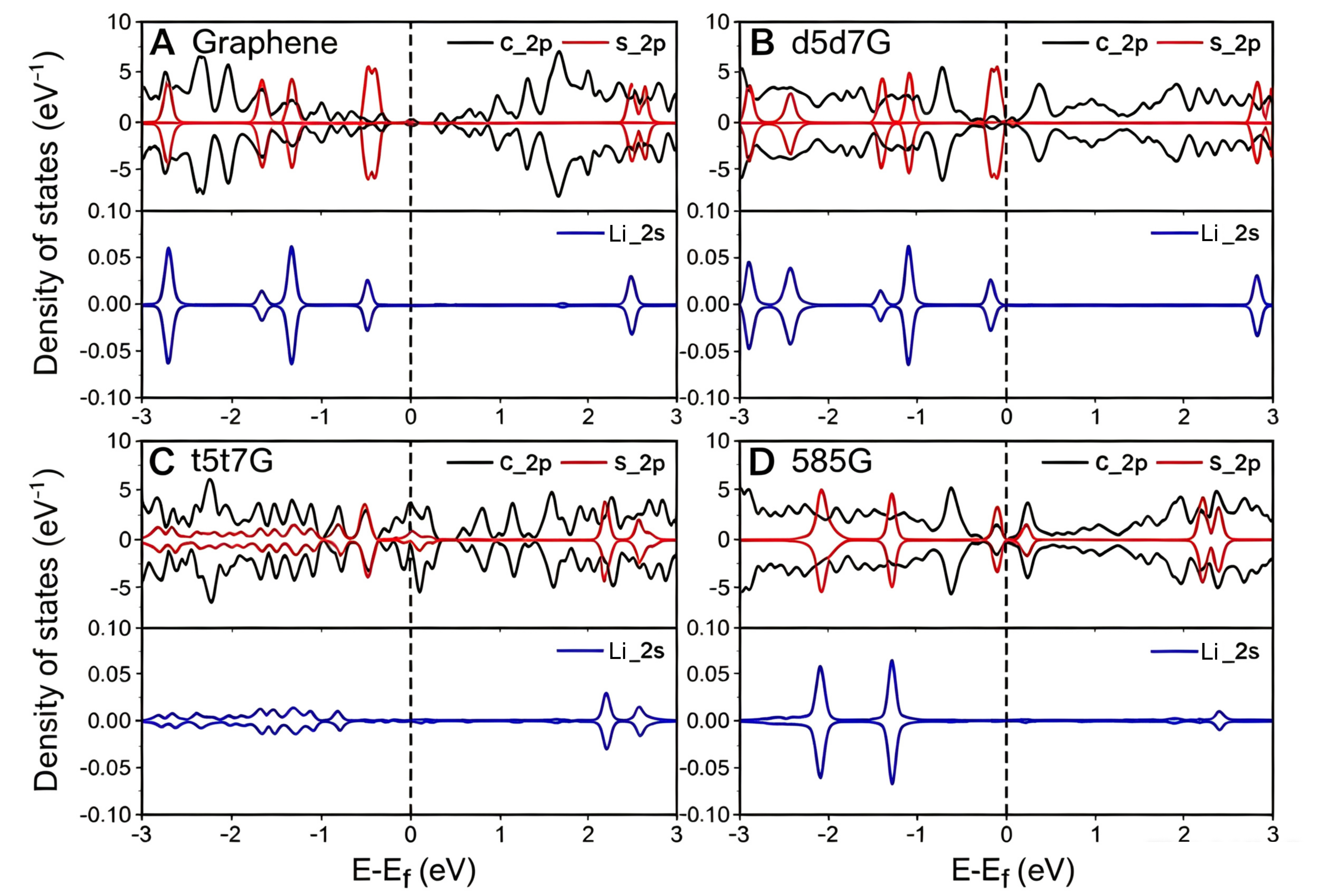
Figure 6. The PDOS of C2p, S2p and Li2s for Li2S4 absorbed on (A) graphene, (B) d5d7G, (C) t5t7G, and (D) 585G. PDOS: Partial density of states.
Catalytic and diffusion kinetic properties of the line-defect graphene
The conversion of polysulfides during the discharge process are crucial for the battery performance. We explore the role of line defects in graphene in the kinetics of LiPSs conversion through the Gibbs free energy barrier analysis. Figure 7 presents the Gibbs free energy barriers for the complete discharge process, which involves the reduction of S8 to Li2S. The Gibbs free energy barriers for the reaction steps from S8 to Li2S2 is negative, signifying the spontaneous reactions. The Gibbs free energy barriers for converting solid-phase Li2S2 to Li2S are positive, identifying this conversion as the rate-limiting stage, in agreement with the previous studies[63]. The barriers of the rate-limiting step for the pristine graphene, d5d7G, t5t7G, and 585G are 2.59, 2.46, 2.40, and 2.35 eV, respectively. Among them, the 585G exhibits the lowest energy barrier in the final step, demonstrating the highest catalytic efficiency and combining superior adsorption performance with catalytic advantages. Compared to the pristine graphene, the introduction of line defects lowers the barrier of the rate-limiting step for the Li-S battery. This indicates that line defects can not only effectively trap polysulfides, but also enhance the catalytic conversion performance of graphene.
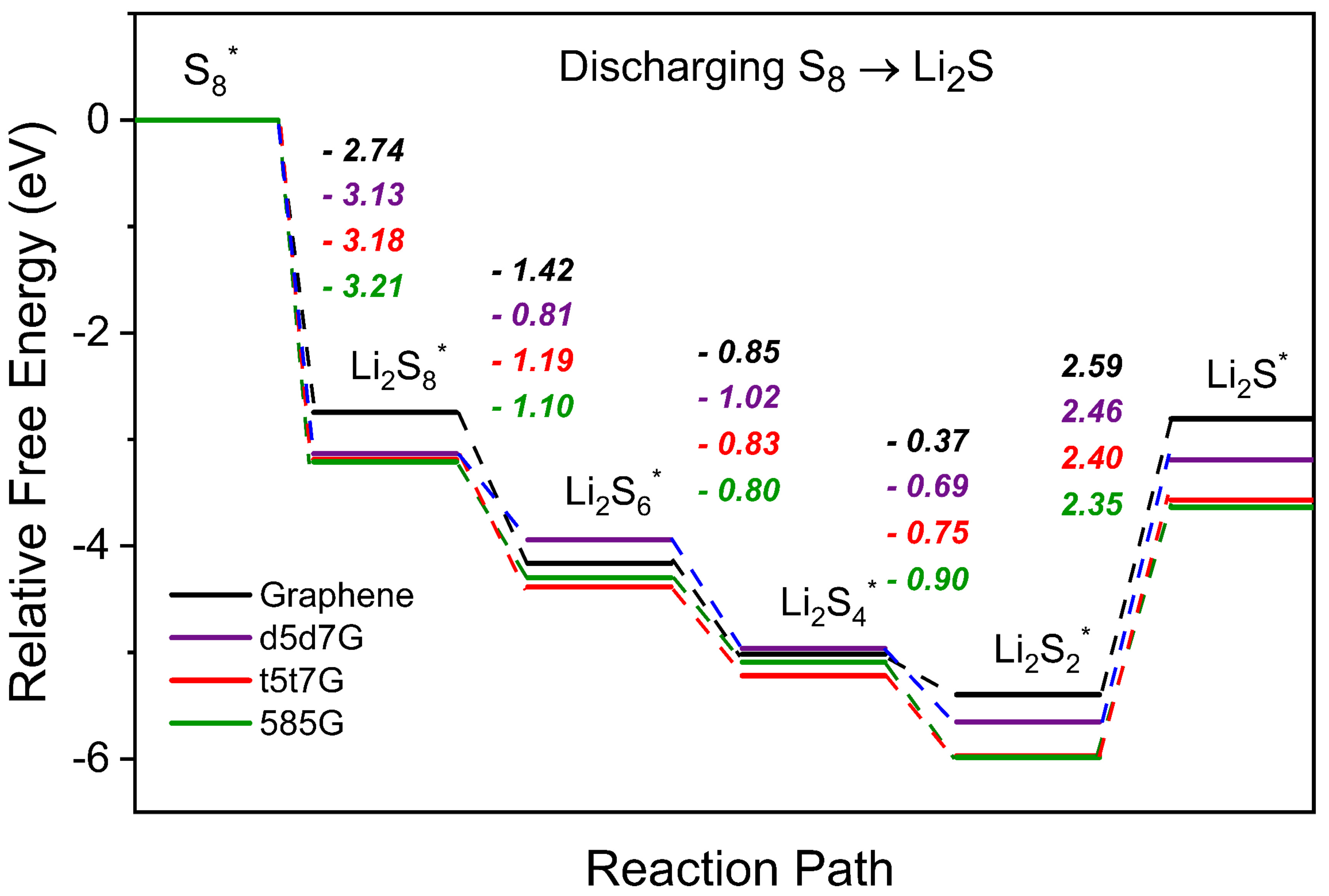
Figure 7. The free energy plots of sulfur reduction process during discharge reaction on graphene and the line-defect graphene, respectively.
Catalytic activity is closely correlated with the band center position; systematically investigating the p-band centers of different line defects is key to uncovering the origin of their activity differences. To this end, we calculate the p-band centers of the pristine graphene and three line-defect graphene by analyzing their C2p orbital density of states [Supplementary Figure 4]. The p-band center of the pristine graphene is -1.77 eV. Introduction of the line defects modifies the electronic structure, which shifts the p-band center upward to the Fermi level. The p-band center of d5d7G, t5t7G, and 585G are -1.18 eV, -1.16 eV, and -1.10 eV, respectively. Notably, the 585G exhibits the highest p-band center, which aligns well with its superior adsorption and catalytic performance Consistent with the established theory, a p-band center closer to the Fermi level corresponds to higher energy of bonding-state electrons, which facilitates electron exchange[64,65].
To further probe the role of the p-band center in catalytic activity of these systems, we examine its correlation with the Gibbs free energy barrier during the discharge progress as depicted in Figure 8. A distinct trend is observed: a higher p-band center (closer to the Fermi level) correlates with a lower Gibbs free energy barrier for both the solid-solid conversion from Li2S2 to Li2S and the overall discharge reaction (from S8 to Li2S). A higher p-band center closer to the Fermi level indicates that the carbon p orbitals possess stronger electronic activity and are more readily involved in interfacial charge transfer during the sulfur-conversion reactions. Such enhanced electronic coupling can strengthen the orbital hybridization between the defected graphene and sulfur species, thereby facilitating electron-transfer kinetics and reducing the Gibbs free energy barrier of the catalytic conversion process. This supports the use of the p-band center as an effective descriptor for connecting defect geometry with catalytic efficacy. Insights from p-band center analysis thus offer a promising avenue for predicting and optimizing the catalytic behavior of related materials.
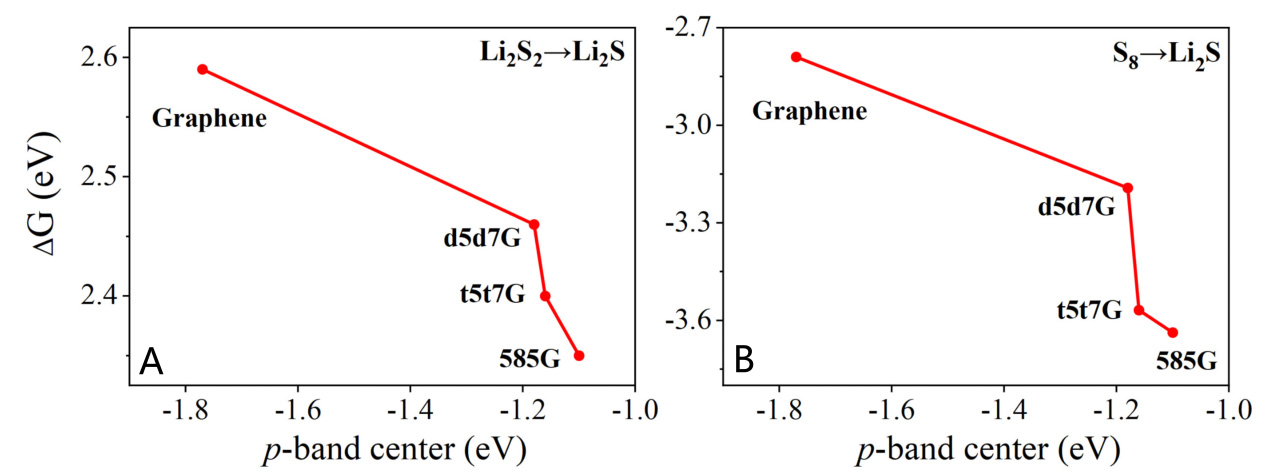
Figure 8. Relationship between the p-band center and Gibbs free energy barrier for (A) Li2S2-Li2S and (B) S8-Li2S on graphene, d5d7G, t5t7G, and 585G.
To elucidate the reaction kinetics in Li-S batteries, we investigate Li⁺ diffusion on the four material surfaces. As illustrated in Supplementary Figure 5, multiple diffusion paths are selected for each material to ensure the simulation accuracy, with the corresponding diffusion barriers listed in Figure 9. The diffusion barrier is highly path-dependent. For the pristine graphene, the Li+ diffusion barrier is 0.31 eV, comparable to the value reported in the previous study[66]. For the line-defect graphenes, the barrier varies due to differences in the line-defect structures and the electronic structure. For the d5d7G, the lowest diffusion barrier is 0.24 eV for the diffusion from the five-membered ring to the five-membered ring corresponding to the path 1. For the t5t7G, the lowest diffusion barrier is 0.31 eV for the diffusion from the five-membered ring to the seven-membered ring corresponding to the path 2. Meanwhile, for the 585G, the lowest diffusion barrier is 0.28 eV for the diffusion from the five-membered ring to the five-membered ring corresponding to the path 2. Compared with the reported point-defect graphene systems (0.69 eV)[66], the present line-defect graphene also exhibits relatively low Li⁺ diffusion barriers, suggesting favorable catalytic conversion kinetics. The line defects in graphene eliminate the issue of Li atoms being trapped at vacancies and unable to hop to adjacent sites. Furthermore, the abundance and continuity of defect sites enable sustained Li⁺ diffusion with low overall barriers, resulting in markedly smoother ion transport.
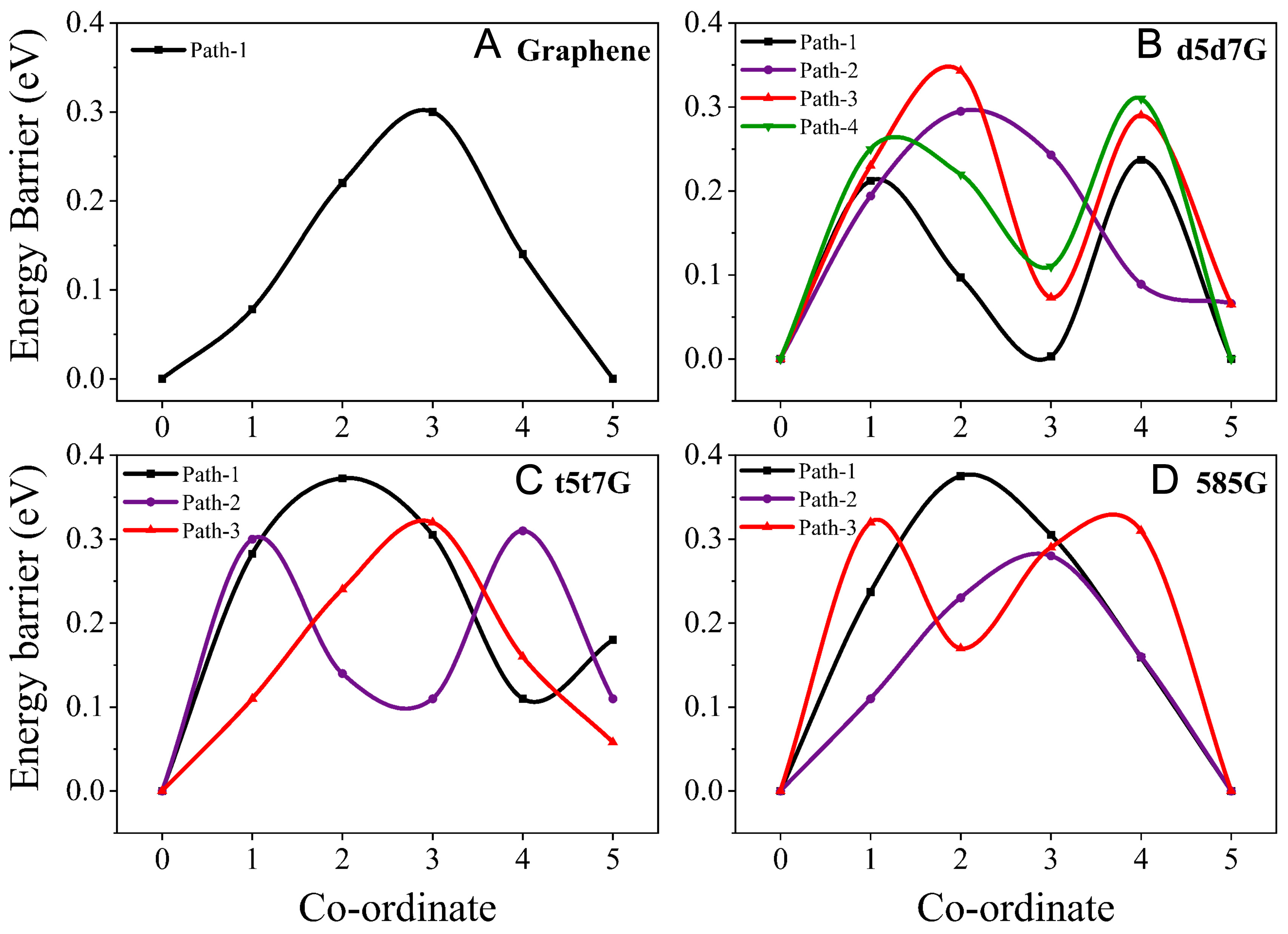
Figure 9. The Energy barriers for Li+ transport along different paths on (A) graphene, (B) d5d7G, (C) t5t7G and (D) 585G.
All results in this work are obtained via first-principles calculations. It should be noted that our theoretical simulations only focus on the electrode material itself, while a practical lithium-sulfur battery consists of electrodes, electrolyte and other components. Theoretical simulations confirm the high thermodynamic stability of line-defect graphene. This material can retain stable structures throughout adsorption and catalysis, thereby benefiting its long-term cycling stability. In fact, previous experimental studies have demonstrated that line-defect graphene can be synthesized in a controllable manner[54-57]. Therefore, the electrochemical performance of the line-defect graphene can be further verified by assembling full batteries in follow-up experimental investigations.
CONCLUSION
Using first-principles calculations, we systematically evaluated three line-defect graphenes (d5d7G, t5t7G, and 585G) as cathode hosts for Li-S batteries. With the adsorption energies of -0.8 eV to -1.9 eV for S8/LiPSs, the line-defect graphene offers stronger chemical anchoring than that of the point-defect graphene with the low Li⁺ diffusion barrier. The p-band center exhibits a clear negative correlation with the Gibbs free energy barrier. A higher p-band center, corresponding to a position closer to the Fermi level, is associated with a lower Gibbs free energy barrier and thus superior catalytic performance. This correlation indicates that modulating the p-orbital valence electron population can shift the p-band center and thereby regulate the interfacial redox kinetics. Through comprehensive screening, the 585G is identified with the strongest adsorption, the highest p-band center, and consequently the most promising electrochemical performance as a cathode host for Li-S battery. This study elucidates the mechanism by which line-defect governs catalytic activity and provides a new design paradigm for the high-performance Li-S cathodes.
DECLARATIONS
Authors’ contributions
Conceptualization, investigation, methodology, writing - original draft. Zhao, Y.
Data curation, software,validation: Sun, H.
Data curation, writing - original draft: Zhu, Y.
Formal analysis, methodology, validation: Tian, J.
Methodology, validation: Tang, M.
Formal analysis, data curation, validation: Sun, J.
Visualization: Zhang, H.
Resources, investigation, writing - review and editing: Pan, Y.
Investigation, funding acquisition: He, Y.
Conceptualization, investigation, funding acquisition, project administration, supervision, writing - review and editing: Li, Q.
Availability of data and materials
The original contributions presented in this study are included in the article and its Supplementary Materials. Requests for further information may be directed to the corresponding authors.
AI and AI-assisted tools statement
During the preparation of this manuscript, the AI tool DeepSeek-V3 (version V3.2, released 2025-12-01) was used solely for language editing. The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship
This work is supported by the National Natural Science Foundation of China (Nos. 52336003, 22179066, 92372127, 11904409 and 12004210), Youth Innovation Technology Project of Higher School in Shandong Province (2022KJ139), and the Natural Science Foundation of Shandong, China(ZR202210060028, ZR2024ZD34).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
Supplementary Materials
REFERENCES
1. Li, Q.; Li, H.; Xia, Q.; et al. Extra storage capacity in transition metal oxide lithium-ion batteries revealed by in situ magnetometry. Nat. Mater. 2021, 20, 76-83.
2. Xu, Z.; Zhang, X.; Yang, J.; Cui, X.; Nuli, Y.; Wang, J. High-voltage and intrinsically safe electrolytes for Li metal batteries. Nat. Commun. 2024, 15, 9856.
3. Gui, H.; Wang, Z.; Cao, Y.; Xu, F. Hybridization-tuned dual-chain conjugated polythioether quinones for high-energy rechargeable magnesium batteries. Chem. Sci. 2026, 17, 325-34.
4. Sun, J.; Liu, S.; Chen, L.; et al. Accelerating charging and elevating capacity of TiO2 by interface space charge storage. Rare. Met. 2025, 44, 5404-11.
5. Zhao, Y.; Xi, Y.; Zhu, Y.; et al. The synergistic effect of heteronuclear diatom enhances the catalytic and adsorption ability of lithium-sulfur batteries. Appl. Surf. Sci. 2025, 706, 163600.
6. Zhou, S.; Shi, J.; Liu, S.; et al. Visualizing interfacial collective reaction behaviour of Li-S batteries. Nature 2023, 621, 75-81.
7. Song, H.; Münch, K.; Liu, X.; et al. All-solid-state Li-S batteries with fast solid-solid sulfur reaction. Nature 2025, 637, 846-53.
8. Zhou, J.; Holekevi Chandrappa, M. L.; Tan, S.; et al. Healable and conductive sulfur iodide for solid-state Li-S batteries. Nature 2024, 627, 301-5.
9. Li, J.; Gao, L.; Pan, F.; et al. Engineering strategies for suppressing the shuttle effect in lithium-sulfur batteries. NanoMicro. Lett. 2023, 16, 12.
10. Wang, T.; Tan, X.; Ma, Y.; Ma, Y.; Wu, Y. High-entropy materials regulating lithium polysulfides for advanced lithium-sulfur batteries. Chem. Eng. J. 2025, 519, 165580.
11. Zhang, H.; Wan, F.; Li, X.; et al. Atomically dispersed Co-Ru dimer catalyst boosts conversion of polysulfides toward high-performance lithium-sulfur batteries. Adv. Mater. 2025, 37, e2500950.
12. Zhang, H.; Zhang, M.; Liu, R.; et al. Fe3O4-doped mesoporous carbon cathode with a plumber’s nightmare structure for high-performance Li-S batteries. Nat. Commun. 2024, 15, 5451.
13. Liu, R.; Wei, Z.; Peng, L.; et al. Establishing reaction networks in the 16-electron sulfur reduction reaction. Nature 2024, 626, 98-104.
14. Cheng, Q.; Zhao, J.; Chen, Z.; et al. Unveiling the capacity enhancement mechanism of carbon interlayers in lithium-sulfur batteries. Adv. Energy. Mater. 2025, 15, 2404757.
15. Saroha, R.; Lee, J. S.; Cho, S. W.; Cho, C.; Park, J.; Cho, J. S. Polysulfide barrier comprising bismuth selenide nanocrystals well anchored within N-doped carbon microspheres for stable Li-S batteries. Energy. Mater. 2025, 5, 500089.
16. Lu, W.; Xu, Y.; Dazon, C.; Macaulay, F.; Pimenta, V.; Serre, C. MOF and related composites as selective functional separators and interlayers for Li-S batteries. Energy. Mater. 2025, 5, 500075.
17. Xu, Y.; Ma, Y. B.; Gu, F.; Yang, S. S.; Tian, C. S. Structure evolution at the gate-tunable suspended graphene-water interface. Nature 2023, 621, 506-10.
18. Zhang, J.; Zhao, B.; Qian, M.; et al. Optimizing electrocatalysis and domain effects of graphene on Li2S cathodes for high-efficient-stable Li-S batteries. Small 2025, 21, e2409172.
19. Huangfu, J.; Feng, P.; Di, X.; et al. Controllable construction of active sites for catalytic conversion and spatial constraints applied to high‐performance lithium-sulfur batteries. Adv. Energy. Mater. 2025, 15, 2502210.
20. Tian, J.; Xing, F.; Gao, Q. Graphene-based nanomaterials as the cathode for lithium-sulfur batteries. Molecules 2021, 26, 2507.
21. Klein, B. P.; Stoodley, M. A.; Deyerling, J.; et al. One-step synthesis of graphene containing topological defects. Chem. Sci. 2025, 16, 19403-13.
22. Zhang, Y.; Liu, J.; Xu, Y.; Xie, C.; Wang, S.; Yao, X. Design and regulation of defective electrocatalysts. Chem. Soc. Rev. 2024, 53, 10620-59.
23. Tiwari, S. K.; Pandey, S. K.; Pandey, R.; et al. Stone-wales defect in graphene. Small 2023, 19, e2303340.
24. Guo, R.; Lv, C.; Xu, W.; et al. Effect of intrinsic defects of carbon materials on the sodium storage performance. Adv. Energy. Mater. 2020, 10, 1903652.
25. Lv, J.; Luo, X.; Liu, J.; Zhao, B.; Cheng, Y.; Liu, S. Interfacial charge transfer and defect engineering in the MoTe2/graphene heterostructure for tailored carrier kinetics and nonlinear absorption. Adv. Funct. Materials. 2025, 35, 2505740.
26. Lu, J.; Tian, R.; Sui, X.; et al. In-situ construction of impurity-free defective graphene nanosheets as high-performance lithium-ion battery anodes via electron beam irradiation. Energy 2026, 344, 139752.
27. Zhang, N.; Yang, Z.; Gao, C.; et al. Rapid electron/ion transport in a 3D holey graphene aerogel framework toward high-loading Na-Se batteries. J. Mater. Chem. A. 2026, 14, 18769-78.
28. Yi, Z.; Su, F.; Huo, L.; et al. New insights into Li2S2/Li2S adsorption on the graphene bearing single vacancy: a DFT study. Appl. Surf. Sci. 2020, 503, 144446.
29. Su, F.; Yi, Z.; Xie, L.; et al. Critical role of surface defects in the controllable deposition of Li2S on graphene: from molecule to crystallite. ACS. Appl. Mater. Interfaces. 2020, 12, 53435-45.
30. Jia, Y.; Zhang, L.; Du, A.; et al. Defect graphene as a trifunctional catalyst for electrochemical reactions. Adv. Mater. 2016, 28, 9532-8.
31. Santos, R. M. D.; Sousa, L. E.; Galvão, D. S.; Ribeiro, L. A. Tuning penta-graphene electronic properties through engineered line defects. Sci. Rep. 2020, 10, 8014.
32. Bhatt, M. D.; Kim, H.; Kim, G. Various defects in graphene: a review. RSC. Adv. 2022, 12, 21520-47.
33. Duman, Ş.; Schattauer, C.; Libisch, F. Line defects as valley filters in graphene. Phys. Status. Solidi. (b). 2025, 262, 2400418.
34. Zhang, W.; Yin, J.; Zhang, P.; Ding, Y.; Jiang, Y. Molecular dynamics study on the relaxation properties of bilayered graphene with defects. Bull. Mater. Sci. 2017, 40, 1255-61.
35. Gillen, R.; Maultzsch, J. Family behavior and Dirac bands in armchair nanoribbons with 4-8 defect lines. J. Phys. Condens. Matter. 2024, 36.
36. Wang, J.; Zhao, X.; Zou, G.; et al. Crystal-defect engineering of electrode materials for energy storage and conversion. Mater. Today. Nano. 2023, 22, 100336.
37. Wu, B.; Meng, H.; Chen, X.; et al. Structural modulation of nanographenes enabled by defects, size and doping for oxygen reduction reaction. Angew. Chem. Int. Ed. 2025, 64, e202415071.
38. Botello-Méndez, A. R.; Declerck, X.; Terrones, M.; Terrones, H.; Charlier, J. C. One-dimensional extended lines of divacancy defects in graphene. Nanoscale 2011, 3, 2868-72.
39. Khossossi, N.; Singh, D.; Ainane, A.; Ahuja, R. Recent progress of defect chemistry on 2D materials for advanced battery anodes. Chem. Asian. J. 2020, 15, 3390-404.
40. Wang, V.; Xu, N.; Liu, J.; Tang, G.; Geng, W. VASPKIT: a user-friendly interface facilitating high-throughput computing and analysis using VASP code. Comput. Phys. Commun. 2021, 267, 108033.
41. Rafiu, R.; Kriaa, K.; Saikot, M. S. H.; et al. Comprehensive first-principles analysis and device simulations of vacancy-ordered D2CeX6 double perovskites for high-efficiency lead-free solar cells. RSC. Adv. 2025, 15, 44711-48.
42. Cabrera-Tinoco, H.; Borja-Castro, L.; Valencia-Bedregal, R.; et al. Pyridinic-N Coordination effect on the adsorption and activation of CO2 by single vacancy iron-doped graphene. Langmuir 2024, 40, 6703-17.
43. Abbasi, R.; Horri, A.; Faez, R. The geometric structure and electronic properties of van der Waals C-borophane/h-BC6N/Graphene heterostructure: a first-principles study. Physica. B. 2025, 699, 416692.
44. Shi, X.; Liu, X.; Nie, C.; et al. SnHPO4: a layered Tin(II) phosphate with enhanced birefringence. Inorg. Chem. 2025, 64, 2127-32.
45. Varrassi, L.; Ellinger, F.; Flage-larsen, E.; et al. Automated workflow for accurate high-throughput GW calculations using plane waves. npj. Comput. Mater. 2025, 11, 1833.
46. González, J. E.; Besse, R.; Lima, M. P.; Da Silva, J. L. F. Decoding Van der Waals impact on chirality transfer in perovskite structures: density functional theory insights. J. Chem. Inf. Model. 2024, 64, 1306-18.
47. Tran, T. N.; Anh Duy, N. V.; Hieu, N. H.; et al. Electric field enhances the electronic and diffusion properties of penta-graphene nanoribbon anodes in lithium-ion batteries. RSC. Adv. 2024, 14, 33524-35.
48. He, Q.; Yu, B.; Wang, H.; Rana, M.; Liao, X.; Zhao, Y. Oxygen defects boost polysulfides immobilization and catalytic conversion: first-principles computational characterization and experimental design. Nano. Res. 2020, 13, 2299-307.
49. Zhang, H.; Wang, S.; Wang, Y.; Huang, B.; Dai, Y.; Wei, W. Borophosphene: a potential anchoring material for lithium-sulfur batteries. Appl. Surf. Sci. 2021, 562, 150157.
50. Podlivaev, A.; Openov, L. Out-of-plane path of the Stone-Wales transformation in graphene. Phys. Lett. A. 2015, 379, 1757-61.
51. Chen, J.; Sun, C.; Lin, D.; et al. Anisotropic growth mechanism of epitaxial graphene on 4H-SiC. Appl. Surf. Sci. 2025, 690, 162596.
52. Li, Y.; Pan, Y.; Cong, Y.; et al. Decoration of defective graphene with MoS2 enabling enhanced anchoring and catalytic conversion of polysulfides for lithium-sulfur batteries: a first-principles study. Phys. Chem. Chem. Phys. 2022, 24, 29214-22.
53. Lahiri, J.; Lin, Y.; Bozkurt, P.; Oleynik, I. I.; Batzill, M. An extended defect in graphene as a metallic wire. Nat. Nanotechnol. 2010, 5, 326-9.
54. Kim, K.; Lee, H. B.; Johnson, R. W.; et al. Selective metal deposition at graphene line defects by atomic layer deposition. Nat. Commun. 2014, 5, 4781.
55. Chen, J.; Autès, G.; Alem, N.; et al. Controlled growth of a line defect in graphene and implications for gate-tunable valley filtering. Phys. Rev. B. 2014, 89.
56. Guo, H.; Long, D.; Zheng, Z.; Chen, X.; Ng, A. M. C.; Lu, M. Defect-enhanced performance of a 3D graphene anode in a lithium-ion battery. Nanotechnology 2017, 28, 505402.
57. Dong, C.; Yang, L.; Lian, C.; et al. Scalable solid-phase synthesis of defect-rich graphene for oxygen reduction electrocatalysis. Green. Energy. Environ. 2023, 8, 224-32.
58. Sinthika, S.; Pushpa Selvi, M.; Nimma Elizabeth, R.; Gavali, D. S.; Thapa, R. Understanding the role of lithium bonds in doped graphene nanoribbons as cathode hosts for Li‐S batteries: a first‐principles study. Int. J. Energy. Res. 2022, 46, 4405-16.
59. Guan, L.; Hu, H.; Li, L.; et al. Intrinsic defect-rich hierarchically porous carbon architectures enabling enhanced capture and catalytic conversion of polysulfides. ACS. Nano. 2020, 14, 6222-31.
60. Chen, H.; Wu, Z.; Zheng, M.; et al. Catalytic materials for lithium-sulfur batteries: mechanisms, design strategies and future perspective. Mater. Today. 2022, 52, 364-88.
61. Ji, L.; Yang, D.; Xue, J.; et al. Flexible titanium nitride‐based membrane reactor for S8/Li2S and dendrite regulation in lithium‐sulfur batteries. Adv. Energy. Mater. 2025, 15, 2404738.
62. Jin, L.; Wang, H.; Zhao, H.; Ji, Y.; Li, Y. Unfolding the structure-property relationships of Li2S anchoring on two-dimensional materials with high-throughput calculations and machine learning. J. Energy. Chem. 2023, 82, 31-9.
63. Zhou, G.; Zhao, S.; Wang, T.; et al. Theoretical calculation guided design of single-atom catalysts toward fast kinetic and long-life Li-S batteries. Nano. Lett. 2020, 20, 1252-61.
64. Zhou, J.; Liu, X.; Zhu, L.; et al. Deciphering the modulation essence of p bands in Co-based compounds on Li-S chemistry. Joule 2018, 2, 2681-93.
65. Zhao, Z.; Yi, Z.; Li, H.; et al. Synergetic effect of spatially separated dual co-catalyst for accelerating multiple conversion reaction in advanced lithium sulfur batteries. Nano. Energy. 2021, 81, 105621.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].