


TiO2-based photoreforming of alcohols as an alternative pathway for low-carbon H2 evolution
 0
0 
Abstract
Photocatalytic H2 production is a key reaction aiming at efficient solar energy conversion towards green fuels. While water splitting would be the ideal reaction for sustainable H2 production, thermodynamic and kinetic constraints have so far limited its applicability. Alternatively, photoreforming of organic derivatives, particularly those derived from renewable sources, can be an economic and technical solution to promote H2 evolution in parallel with the production of added-value oxidation products. Among different organic substrates, alcohol photoreforming stands out due to its large availability, cost-effectiveness, and the possibility to produce key chemical feedstocks. Such reactions have been studied since the early 70s, starting with primary alcohols such as methanol and, more recently, involving polyols such as glycerol. Despite different semiconductors having been employed as photocatalysts, fundamental understanding of the reaction mechanism is a necessary step towards the development of more efficient systems. In this manuscript, the fundamental aspects of alcohol photoreforming are reviewed, focusing on TiO2-based systems in which a vast literature is available. A systematic comparison of the light-driven reforming pathways of C1-C6 alcohols on TiO2-based photocatalysts is presented, extending from methanol to more complex substrates such as glycerol and biomass-derived compounds. The aim is to highlight how molecular complexity influences oxidative pathways, intermediate formation, hydrogen evolution, and catalyst requirements. The main findings employing different characterization techniques are summarized and corelated with surface modifications of the oxide aiming at improved H2 evolution rates and selectivity for different oxidation products. The discussion can work as a tool to promote the rational development of more efficient sunlight-driven photocatalysts for photoreforming.
Keywords
INTRODUCTION
The growing global energy demand, driven by population growth and industrial development, remains largely dependent on non-renewable sources for fuel and electricity. Molecular hydrogen (H2) production technologies are pointed as key vectors for net-zero CO2 emissions. It can be used as fuel due to its high energy density, producing only water as a by-product. Moreover, it also plays a vital role in the chemical industry, especially in fertilizer production and oil refining, with annual global demand reaching approximately 100 million tons in 2024[1,2]. Photoreforming of alcohols, plastic residues and other organic compounds such as sugars and biomass-derived molecules has gained attention as a promising pathway for the so-called low-carbon hydrogen production[3-6]. Moreover, photoreforming from agro-industrial waste streams couples solar energy conversion with the generation of fuels and chemicals, offering the dual benefit of hydrogen production and waste valorization[7-9]. During the photocatalytic process, organic compounds, such as alcohols derived from biomass, industrial residues, or by-products like glycerol, are converted into hydrogen and valuable oxidation products[7], including aldehydes, ketones, and carboxylic acids[10-17].
In heterogeneous photoreforming, the process relies on a semiconductor material that absorbs light to yield electron-hole pairs. In general, a heterogeneous photocatalyst is an inorganic semiconductor whose electronic structure is defined by two main energy bands (valence and conduction bands), separated by an energy gap (bandgap). When the photocatalyst absorbs a photon with energy equal to or greater than the bandgap, an electron-hole pair is generated, which can either migrate to the surface and participate in redox reactions with adsorbed species, or recombine, dissipating the energy without producing chemical change[18]. In the absence of molecular oxygen, the photogenerated electrons can drive proton reduction to produce hydrogen (H2), while the holes promote the oxidation of the organic substrate, Figure 1A. In the absence of an organic substrate, the electron/hole pair can potentially promote water splitting, Figure 1B, which is kinetically and thermodynamically less favorable than photoreforming. If both O2 and organic species are present, reactive oxygen species (ROS) are produced and can be used for environmental remediation purposes, Figure 1C.
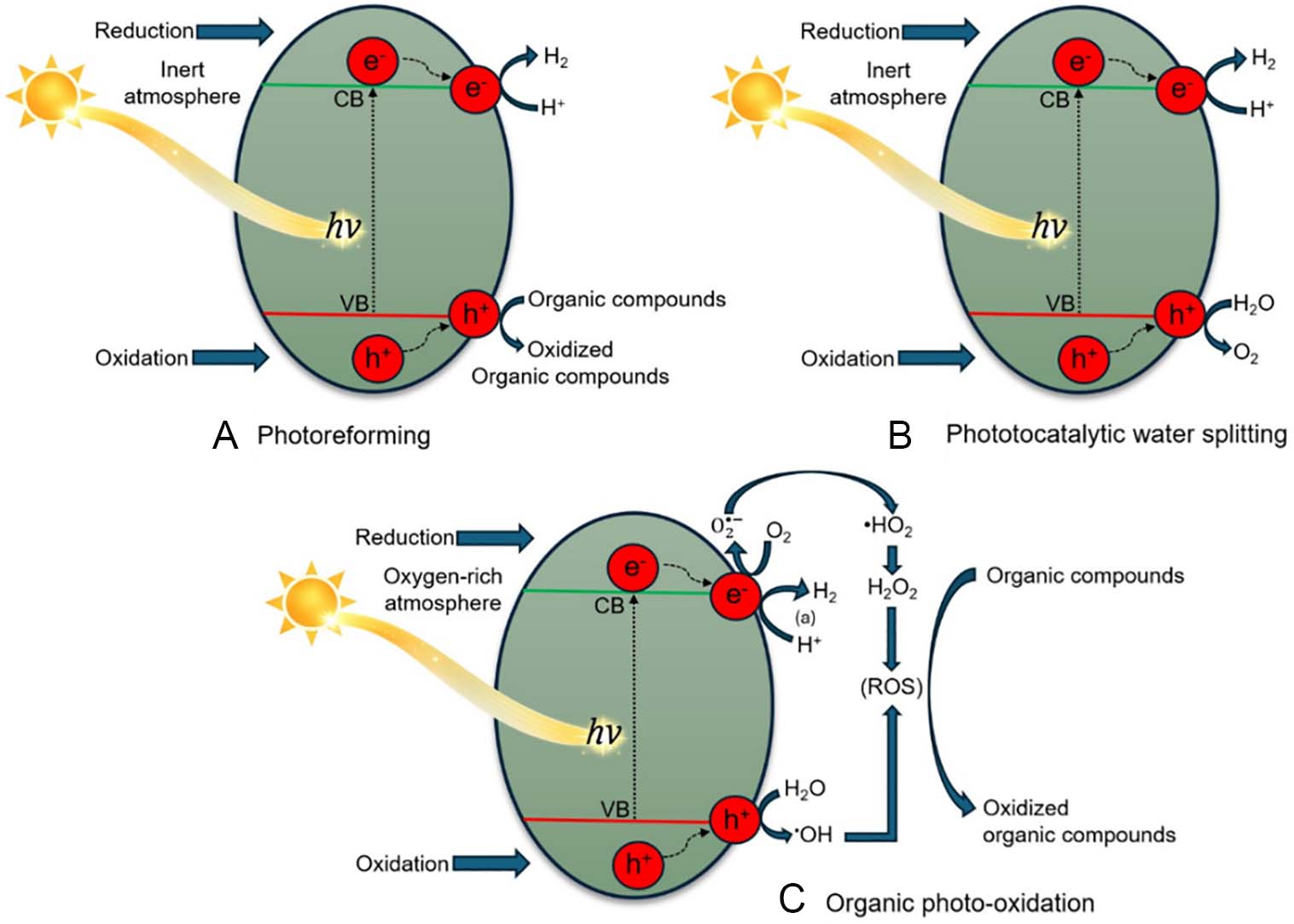
Figure 1. (A) Organic photoreforming, (B) photocatalytic water splitting in anaerobic conditions, and (C) organic photo-oxidation promoted by a heterogeneous photocatalyst.
Since most organic oxidation reactions are thermodynamically favorable, photoreforming uses light energy to drive charge generation and overcome the kinetic barriers associated with hydrogen production and substrate oxidation. Thus, the main challenge in the process is actually to control the oxidation step in order to increase selectivity for more valuable products, while simultaneously achieving efficient sunlight harvesting and suppressing charge recombination. Therefore, the photocatalyst is crucial in photoreforming, as its properties directly influence reaction efficiency, selectivity, and stability. Key characteristics include suitable band edge potential positions, a narrow bandgap for effective light absorption, high surface area, efficient charge separation, and chemical stability[19,20]. Commonly reported photocatalysts for photoreforming include CdS[21-24], graphitic carbon nitride (g-C3N4)[25-28], TiO2[9,29-34], and perovskite-type metal oxides and halides[35-37]. Among them, TiO2 remains one of the most widely used due to its chemical stability, non-toxicity, low cost, and favorable band edge positions for both water and alcohol oxidation and H2 evolution[20,38]. Nevertheless, despite its potential, TiO2-based photoreforming still faces important practical limitations, including the wide bandgap of TiO2 (~3.2 eV), which restricts light absorption predominantly to the UV region, the fast recombination of photogenerated charge carriers, challenges associated with carbon efficiency and product separation, as well as scalability issues for large-scale hydrogen production[4,5,10]. Moreover, most research in this field has focused on semiconductor materials development, while few studies are devoted to the systematic investigation of the influence of the organic species on the photoreforming process.
For instance, TiO2 photocatalytic performance is also strongly influenced by its crystalline phase and structural design. Among its three crystalline forms (anatase, rutile, and brookite), anatase and rutile are the most widely employed in photocatalysis, while brookite is less explored due to its lower stability and more complex synthesis routes[39]. Notably, mixed-phase TiO2 systems, such as the commercial P25 photocatalyst, often exhibit superior performance because the anatase/rutile heterojunction promotes more efficient charge separation and interfacial electron transfer, while also improving light absorption[39,40]. In addition, to overcome intrinsic drawbacks as UV-light dependence and rapid charge recombination, several modification strategies have been extensively investigated, such as doping[41,42], modification with other
In this context, this review offers a comprehensive overview of research conducted on the photoreforming of alcohols and biomass derivatives using TiO2-based photocatalysts. TiO2, despite its low sunlight activity, was chosen as a benchmark due to the extensive mechanistic and photocatalytic data available in the literature, while also serving as an important model platform for understanding fundamental principles that can guide the design of more advanced photocatalysts[20]. While previous reviews in the literature have mainly focused on TiO2 photocatalytic properties or hydrogen evolution systems, the present work provides an updated perspective centred on photoreforming mechanisms and catalyst-engineering strategies. The fundamental aspects of the photoreforming mechanism are discussed in detail, with methanol used as a model to illustrate key reaction pathways and the main spectroscopic techniques used to evaluate them. The review also explores the influence of TiO2 modification and the usage of co-catalysts on both hydrogen evolution and oxidative mechanisms. Furthermore, this review systematically compares the light-driven reforming pathways of C1-C6 alcohols, extending from methanol to more complex substrates such as glycerol and biomass-derived compounds, while discussing how molecular complexity influences oxidative pathways, intermediate formation, hydrogen evolution, and catalyst requirements. Therefore, Section "METHANOL PHOTOREFORMING AS A MODEL REACTION" initiates on methanol photoreforming as a model reaction to establish key mechanistic concepts and spectroscopic approaches. Subsequently, Section "PHOTOREFORMING OF C2+ ALCOHOLS" extends the analysis to C2+ alcohols, highlighting how increasing molecular complexity influences reaction pathways and intermediate formation. Finally, Section "GLYCEROL PHOTOREFORMING" addresses glycerol as a representative biomass-derived polyol, while Section "PHOTOREFORMING OF OTHER BIOMASS DERIVATIVES" covers other biomass derivatives.
METHANOL PHOTOREFORMING AS A MODEL REACTION
Methanol is often used as a model compound to study the photocatalytic activity over different semiconductor surfaces[73-76]. It is a simple molecule with a lower oxidation potential than water (0.103 V vs. standard hydrogen electrode for the methanol to formate oxidation)[77]. It readily donates electrons during photocatalysis, suppressing the recombination of charge carriers. The first study on photocatalytic oxidation of methanol on TiO2 dates back to the late 1970s and early 1980s[78-81]. The photoinduced process starts with light absorption by the semiconducting material through band gap excitation (3.2 eV, the case of Anatase TiO2), yielding an electron-hole pair or exciton. Once generated, these mobile charge carriers face a critical competition between productive and non-productive pathways, i.e., the effective promotion of redox reactions or the charge carrier recombination.
The non-productive electron-hole recombination occurs in a wide timescale range, from picoseconds to milliseconds depending on the structural properties of the semiconductor and dissipates the absorbed photon energy by radiative and/or nonradiative pathways. In TiO2, an n-type semiconductor, the photoexcited electrons on the conduction band can be trapped in shallow or deep trap states in the oxide surface. Such trap states are typically oxygen vacancies (Ov), which can be understood as unpaired electrons, located initially in an O2p orbital and then transferred, by the removal of the corresponding oxygen atom, into the conduction band formed by Ti3d orbitals[82,83]. Similarly, hole trapping can also occur at a bridging O2- or surface-bound OH- anions, resulting in the formation of O•- or/and OH• centres, respectively. Charge trapping and recombination dynamics are critical to the efficiency of the photocatalytic process and have been investigated by a myriad of time-resolved techniques.
Among them, Transient absorption spectroscopy (TAS) stands out because it provides direct, temporally resolved access to the dynamics of electrons, holes, excitons, and short-lived intermediates over an exceptionally broad temporal window. By tracking these species from femtoseconds to seconds, TAS uncovers mechanistic events that dictate photocatalytic performance, including exciton formation, hot-carrier cooling, charge separation, recombination pathways, carrier trapping, and interfacial charge transfer[84-89].
Fundamental photophysics studies on TiO2 nanoparticles revealed that electrons and holes are trapped onto the oxide surface within 50-150 fs following excitation. Electrons in the conduction band can be deeply trapped at the bulk within 50 ps, while holes are trapped in deep states at the surface within 200 fs[90-93]. The overall lifetime of trapped holes and electrons will depend on the crystal structure of the photocatalyst, its surface composition, defect concentration, and the presence of adsorbed species.
Tamaki et al.[90,94] investigated the reaction dynamics of the trapped holes by means of TAS in the presence of methanol and found that the transient absorption of the trapped holes decayed more rapidly, clearly indicating that trapped holes can react with the adsorbed alcohol on the surface. The lifetime of the trapped holes was around 300 ps in the presence of methanol. This ultrafast charge-transfer rate can be related to the adsorption behavior of methanol on the TiO2 surface, which is predominantly dissociative, resulting in the formation of surface alkoxide and hydroxyl groups[95-97]. Under UV irradiation, these surface alkoxide groups serve as effective hole trapping centers.
Generally, it is assumed that the hole or •OH-induced alcohol oxidation proceeds via two reaction steps
Continuous irradiation of oxygen-free methanol aqueous solutions in the presence of TiO2 nanoparticles will ultimately lead to the formation of CO2 as an oxidation product. However, if an excess of methanol is maintained in the course of the reaction, valuable oxidative products such as formaldehyde, formic acid, and even methyl formate can be obtained[73]. The following steps after the initial photoinduced hole transfer from TiO2 to adsorbed methanol molecules have been investigated by different groups.
Zhou and Guo have compared the oxidation of methanol and water on pristine Rutile TiO2(110) surfaces by means of advanced surface characterization techniques such as atom-resolved scanning tunning microscopy, temperature-programmed desorption (TPD)/time-of-flight spectroscopy, and photoelectron spectroscopy in combination with theoretical calculations[102,103]. Methanol adsorption on Ti4+ sites through its hydroxyl group lead to the formation of 5-fold coordinated Ti sites (Ti5cs) onTiO2 surface or, alternatively on bridging Ov. Both molecular and dissociative (proton transfer to the neighbouring oxygen on the surface) adsorption mechanism have been proposed. The energy difference between the two adsorption states is relatively small, so it is generally accepted that at higher temperatures they can easily interconvert between themselves. Under bandgap excitation, the adsorbed methanol molecules are photooxidized into formaldehyde by transferring both the hydroxyl and methyl hydrogen atoms to oxygen sites, reducing Ti4+ into Ti3+ ions. In fact, continuous irradiation of TiO2 nanoparticles under oxygen-free methanol vapor leads to the build-up of bluish color ascribed to the Ti3+ d-d transition. Density functional theory (DFT) calculations reveal that the effective interfacial hole transfer is thermodynamically favored by the previous dissociation of the hydroxyl hydrogen to yield methoxy anions adsorbed on the surface (H3CO-). Very similar conclusions were drawn by Diebold and Seleni for Anatase (101) TiO2[104]. In this case, the proposed overall mechanism for formaldehyde formation is summarized in Figure 2.
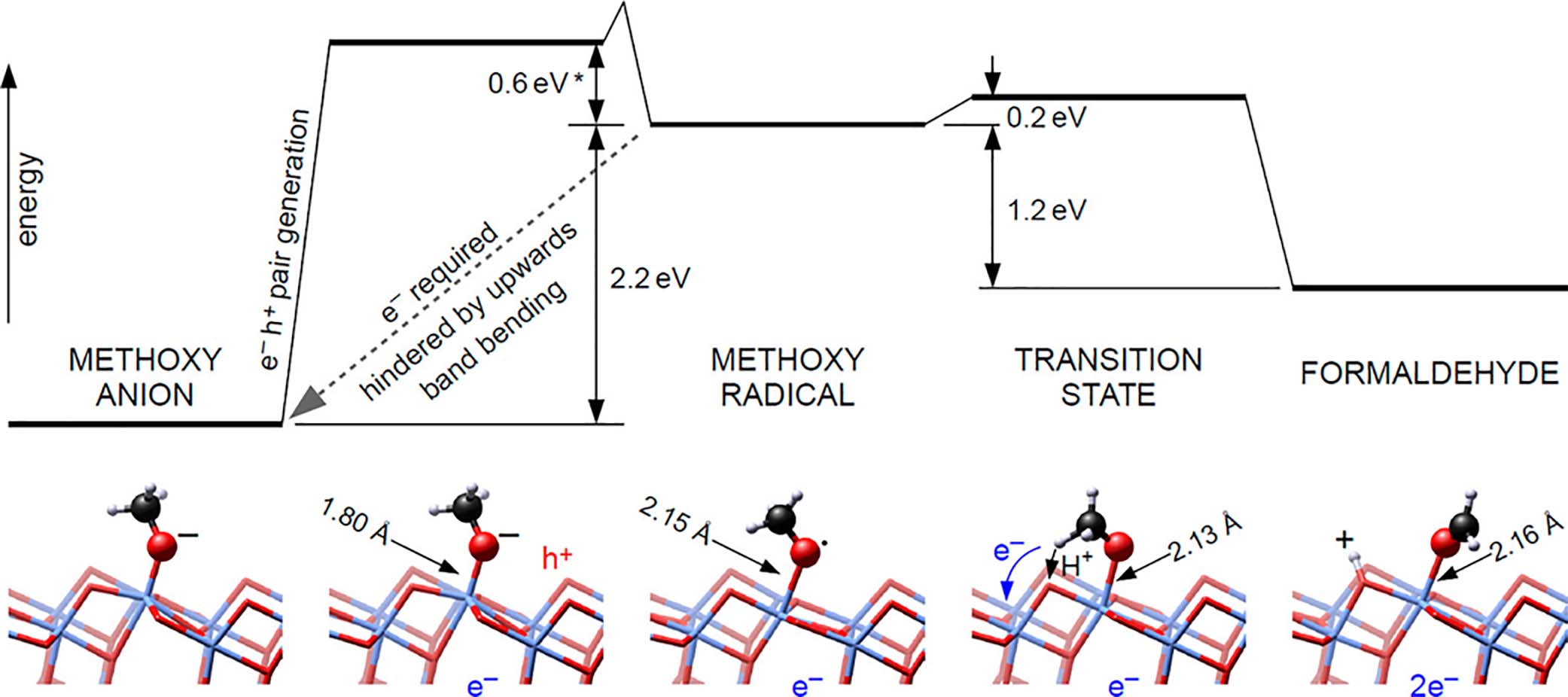
Figure 2. Calculated energy barriers for photoconversion of CH3O- (left) into formaldehyde (right) in Anatase TiO2(101). Reproduced from ref.[104] under the CC BY 4.0 license.
The presence of metallic cocatalysts, such as Pt, not only allows the formation of molecular H2 but also changes the energetics of the of hole transfer as the enhanced electron-hole separation intensifies the band bending and accelerate the photocatalyzed oxidation step[105]. In fact, TAS measurements employing TiO2 nanoparticles of different particle sizes, morphologies and crystalline phases reveal that Pt clusters on the oxide surface can efficiently scavenger the photoexcited electrons in the TiO2 conduction band, improving considerably the lifetime of the charge carriers[105,106]. Moreover, Kandiel et al. have provided evidence through isotopic studies that the molecular H2 is mainly produced from protons originating from water[107].
Al-Madanat and coauthors studied the influence of the platinum loading method on nanosized anatase TiO2 on both reductive and oxidative half-reactions[62]. Pt was added to TiO2 either by physical mixing with laser-ablated Pt nanoparticles (PtLA-TiO2) or via photodeposition (PtPD-TiO2). Using sealed reactors under argon at 25 °C and under simulated solar irradiation (1000 W Xenon lamp), methanol and naphthalene were tested as substrates. For methanol, both platinized TiO2 samples produced significantly higher amounts of acetaldehyde and formic acid compared to pristine TiO2, with PtPD-TiO2 showing about three times faster oxidation kinetics than PtLA-TiO2. CO2 formation was minimal but still higher for platinized samples, with PtPD-TiO2 outperforming PtLA-TiO2[62]. Hydrogen production was negligible for pristine TiO2 but substantially higher for the platinized catalysts, with PtPD-TiO2 achieving a photonic efficiency of 15.6% vs. 6.1% for PtLA-TiO2 after 1h of irradiation.
The superior performance of PtPD-TiO2 is attributed to better Pt dispersion and smaller Pt islands, enhancing catalytic activity and charge separation. Increasing Pt loading in PtLA-TiO2 only slightly improved activity but remained below that of PtPD-TiO2[62]. Overall, photodeposition of Pt provided a more effective and stable modification, especially enhancing hydrogen production and oxidation rates compared to physical mixing. Electron paramagnetic resonance (EPR) and TAS analyses has confirmed improved charge separation in PtPD, which was attributed to the preferential Pt deposition on the reductive {101} facets, enhancing electron transport. Interestingly, the enhancement achieved by Pt photodeposition strongly depends on the organic substrate. While the activity difference between PtPD-TiO2 and PtLA-TiO2 was not substantial for naphthalene in terms of photonic efficiency, methanol showed greater improvements. This was attributed to the current-doubling effect of methanol[98,108,109], which increases the average number of photogenerated electrons per particle, thereby benefiting more from the superior electron-capturing ability of the photodeposited Pt clusters.
The role of Pt clusters in methanol photoreforming was also investigated by Zhou’s research group and compared to the pristine TiO2 employing ultrahigh vacuum techniques and well-ordered single crystals[110]. The authors conclude that Pt loading can favor the dissociation of methanol into methoxy and lower the desorption barrier of molecular hydrogen, which may work cooperatively for separating photoexcited charges to enhance the photocatalytic efficiency.
As expected, the main oxidative reaction pathway in the Pt/TiO2 and pristine TiO2 systems is similar, although with different kinetics. However, the presence of Pt clusters also leads to thermal dehydrogenation of methanol to CO species on the metal surface. Similar results were found by Walenta et al. using techniques such as Auger electron spectroscopy, TPD/reaction, isotopic labeling, and isothermal photoreactions[111]. The photo-oxidation of methoxy species is identified as the rate-determining step in this process. Key findings include an initial "conditioning" of the photocatalyst. Platinum (Pt) serves as a cocatalyst, primarily facilitating the evolution of molecular hydrogen by promoting the dimerization of surface hydrogen atoms. The selectivity for formaldehyde formation is basically due to easier thermal desorption of the molecule in comparison to methanol[111,112]. At temperatures below 250 K, the consecutive photooxidation of de aldehyde can be observed, leading to the formation of methyl formate, Equation 3.
At room temperature and in aqueous solution, the consecutive oxidation of formaldehyde leads to formic acid (Eqs. 4 and 5) and ultimately CO2. The methanol concentration, irradiance, pH, and photocatalyst load can be used to tune the selectivity as reported by Karimi Estahbanati and coauthors using TiO2 P25 modified with 1% Pt[10]. Hydrogen production from methanol significantly increased up to 10% concentration, remaining almost constant until 99%, which is consistent with the previous conclusion that the photooxidation of the adsorbed methoxy species is the rate-limiting step.
Kandiel et al. focused on the influence of physical and chemical parameters on the H2 evolution rates and oxidation products distribution on methanol photoreforming[113]. Different light intensities, pHs, and initial methanol concentrations were evaluated. The rate of photocatalytic H2 evolution is found to strongly depend on the methanol concentration initially present in the suspension. It increases with alcohol concentration and levels off, reaching a plateau. Furthermore, higher photonic efficiencies are observed for H2 evolution at low photon fluxes. Stochiometric amounts of formaldehyde are observed at short irradiation times independent of pH and methanol concentration. At longer irradiation times, formic acid and CO2 appear as minor products, being slightly favored at basic pHs.
Eder et al. expanded the understanding of heterogeneous photocatalytic alcohol oxidation by emphasizing the critical role of surface chemistry and thermal reactions in determining catalytic activity[114]. They found that the interaction of alcohols with TiO2(110) surfaces can guide the development of new photocatalysts and challenge the conventional electrochemical model. Their approach was based on surface investigations under ultra-high vacuum (UHV) conditions, employing post-illumination TPD technique to distinguish thermal from photochemical reactions. Their study also observed the photochemical oxidation of tertiary alcohols, leading to the formation of ketones and alkanes even in the absence of cocatalysts. They argued that the electrochemical model, where only a set of redox reactions accounts for the overall outcome, does not adequately explain alcohol photoreforming in the gas phase, as exemplified in Figure 3. They proposed a hole-mediated disproportionation mechanism, involving the homolytic cleavage of C-H bonds and the formation of H2 as a single-photon process. The authors highlighted the need for a holistic approach that integrates thermal surface chemistry and charge carrier dynamics for the rational design of photocatalysts. Although the study exhibits limitations such as the difficulty of replicating practical environments, it expands the scope of possible reactions and provides new guidelines aimed at innovative reactor concepts and selective oxidation under reductive conditions.
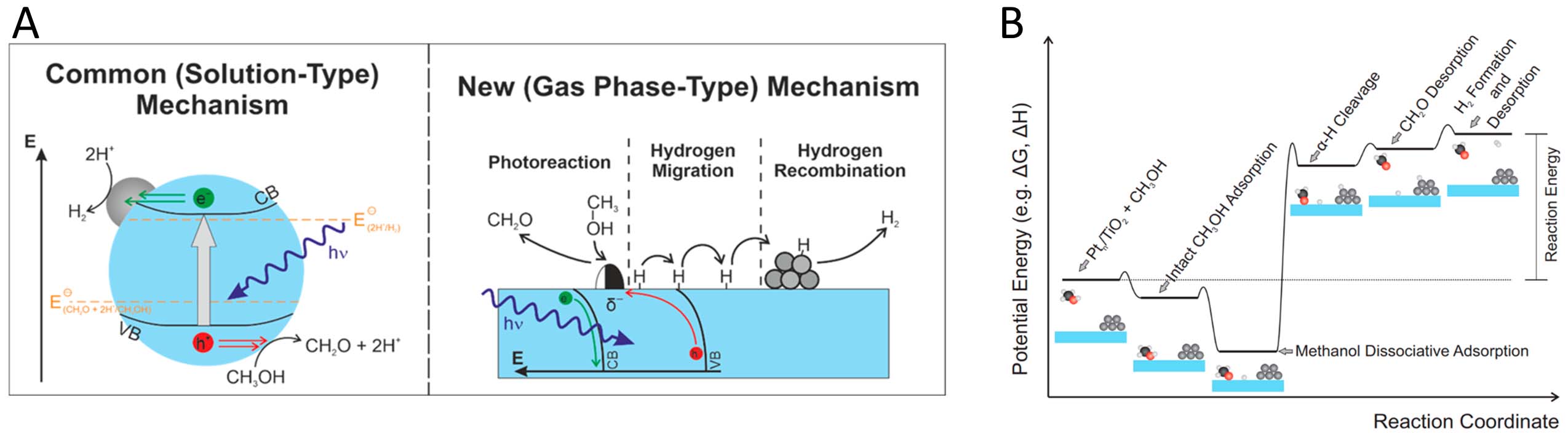
Figure 3. (A) Comparison of the conventional electrochemistry-type mechanism of methanol photoreforming (left) and the new mechanism based on surface science studies with gaseous reactants (right); (B) Hypothetical potential energy surface of the proceeding photooxidation of methanol adsorbed on TiO2 yielding formaldehyde and molecular hydrogen. Reproduced with permission from ref.[114]. Copyright 2023 American Chemical Society.
Interestingly, Courtois and collaborators investigated the origin of catalyst deactivation (“poisoning”) during the photoreforming of primary alcohols, such as methanol, on reduced rutile TiO2(110) under ultrahigh vacuum[115]. Contrary to the common attribution to increased trap states for charge carriers[116], the loss of activity was linked to thermal back-reactions, particularly between surface hydroxyls and aldehyde intermediates formed during methanol photo-oxidation[115]. These reactions regenerate methoxy species, reversing the desired oxidation step and progressively deactivating the surface. The absence of hydrogen evolution and the buildup of hydroxyl groups confirmed surface hydroxylation as the main cause of poisoning at room temperature. In contrast, tertiary alcohols like 2-methyl-2-pentanol, which undergo C-C bond cleavage instead of C-H oxidation, did not exhibit this deactivation, indicating that the effect arises from alcohol-specific back reactions rather than electronic or site-blocking factors[115]. Overall, the study identifies thermal back-reactions as the key mechanism limiting primary alcohol photoreforming on TiO2 surfaces.
Building upon the understanding of methanol photoreforming reaction steps, research efforts have increasingly focused on optimizing the activity and selectivity of photocatalysts. Material engineering has been a strategy to enhance the efficiency and selectivity of methanol photoreforming. By precisely tailoring the exposed crystal facets and introducing defects, the catalyst structure favored specific reaction pathways while improving charge separation. Yamakazi et al. explored rutile TiO2 nanorods modified with Pt, aiming to maximize photocatalytic activity for H2 production under visible light irradiation[117]. Additionally, defect introduction was performed via hydrogen spillover during Pt deposition, enabling controlled formation of Ti3+ and Ov. Pt-loaded TiO2 nanorods (TNR) reduced at 200 °C exhibited superior photocatalytic performance, producing hydrogen at rates approximately four times higher than conventional TiO2 powders reduced at 600 °C under visible light irradiation (λ > 420 nm, 500 W Xe lamp) in methanol solution (50 vol%). This enhancement was attributed to the controlled introduction of Ti3+ species without excessive Ov and effective spatial charge separation facilitated by the exposure of both {110} and {111} facets[117]. The aspect ratio of the nanorods, along with the balance between reductive {110} and oxidative {111} facets, played a critical role in catalytic efficiency. DFT calculations further revealed that oxygen vacancy formation is thermodynamically more favorable on the {111} facets, supporting the observed facet-dependent behavior.
Similarly, Hossein Hejazi and coauthors reported the influence of defect engineering in anisotropic brookite TiO2 nanorods, considering also the substrate specificity[118]. The nanorods were synthesized using hydrothermal methods, predominantly exposing the (210) facet. Defects were introduced through hydrogen reduction at 700 °C, enabling controlled formation of subsurface Ov and lattice distortions. Photocatalytic experiments using 1 wt% Pt as a cocatalyst showed that the methanol photoreforming rate was up to 11 times higher for the reduced anisotropic brookite in comparison to untreated brookite, achieving a specific methanol consumption of 99 mmol h-1 m-2 and hydrogen production of 88 mmol h-1 m-2. The catalyst showed clear substrate specificity, with methanol reforming being significantly more efficient than ethanol or isopropanol (50 vol%). The apparent quantum yield (AQY) for reduced brookite reached 33.5% at 334 nm but dropped to nearly zero in the visible range (≥ 380 nm). Experimental and DFT studies revealed that subsurface Ov, coupled with large electronic polarons, serve as active sites specifically for methanol oxidation. A direct correlation was established between the alcohol oxidation rate and hydrogen evolution, where methanol dissociation is favored by the combination of structural defects and exposed crystal facets in brookite[118]. Comparisons with isotropic brookite and reduced anatase further confirmed that crystal morphology and orientation play a critical role in the formation of catalytic sites.
Another approach to increasing the efficiency of TiO2-based photocatalytic methanol oxidation is surface-level modifications without altering the underlying crystal structure. Monocrystalline rutile TiO2 with an exposed (110) surface offers an ideal model system for such investigations, particularly when coupled with the deposition of metal clusters as co-catalysts. The influence of their size and surface coverage remains insufficiently understood. By employing different cluster sources for size-selected deposition of Ni and Pt on TiO2(110) under ultrahigh vacuum, Eder and coauthors evaluated distinct size- and coverage-dependent effects on the photocatalytic reforming of alcohols[119]. Size-selected Pt and Ni clusters, ranging from single atoms to aggregates of over 47 atoms, were deposited on TiO2(110) under UHV via laser ablation, achieving precise control over both cluster size and surface coverage from 0.1% to 2% of a monolayer. A detailed study of methanol photoreforming on TiO2(110) under UHV
Furthermore, methanol reforming over Pd/P25 TiO2 was investigated and compared with Au- and Cu-based catalysts, as well as with the photocatalytic water-gas shift (WGS) reaction (CO + H2O → CO2 + H2)[120]. First, methanol reforming was significantly faster than the photocatalytic WGS reaction. Since both CO and H2O are produced during methanol reforming, the WGS reaction may contribute to overall H2 generation, though it proceeds more slowly and is less frequently reported. Nanoparticles were deposited on TiO2 via incipient wetness impregnation (IWI) (0.5 wt.%) and calcined at 400 °C, with Pd particles estimated at ~2.5 nm. Using a xenon arc lamp, photocatalytic methanol reforming and WGS reactions were tested in both liquid and gas phases. Pd/TiO2 exhibited the highest H2 production rate in the liquid phase (3.7 mL h-1), and this rate approximately doubled in the gas phase, where small amounts of CO were also detected, suggesting partial desorption of CO intermediates, identified as a rate-limiting step. Notably, the observed H2/CO2 ratio exceeded the theoretical 3:1, due to significant CO2 dissolution in water, as confirmed by pH monitoring, and CO2 release upon heating. Au/TiO2 was less effective than Pd for methanol reforming but showed superior performance in the photocatalytic WGS reaction, generating three times more H2 than
An extensive study of redox dynamics of Pt and Cu nanoparticles on TiO2 for methanol photoreforming was reported by Chiarello et al.[121] The study employed in situ modulated excitation X-ray absorption spectroscopy (ME-XAS) during the gas-phase photocatalytic oxidation of methanol, under both aerobic and anaerobic conditions, as compared in Figure 4. The photocatalysts were synthesized in a single step using flame spray pyrolysis (FSP) and were tested for their ability to oxidize methanol to CO2 in the presence (CH3OH + 3/2 O2 → CO2 + 2H2O) or absence (CH3OH + H2O → CO2 + 3H2) of oxygen under irradiation (300 W xenon lamp, 120 mW/cm2). Under anaerobic conditions, product formation included H2, CO2, formaldehyde, formic acid, and CO, with Pt/TiO2 achieving the highest H2 production rate
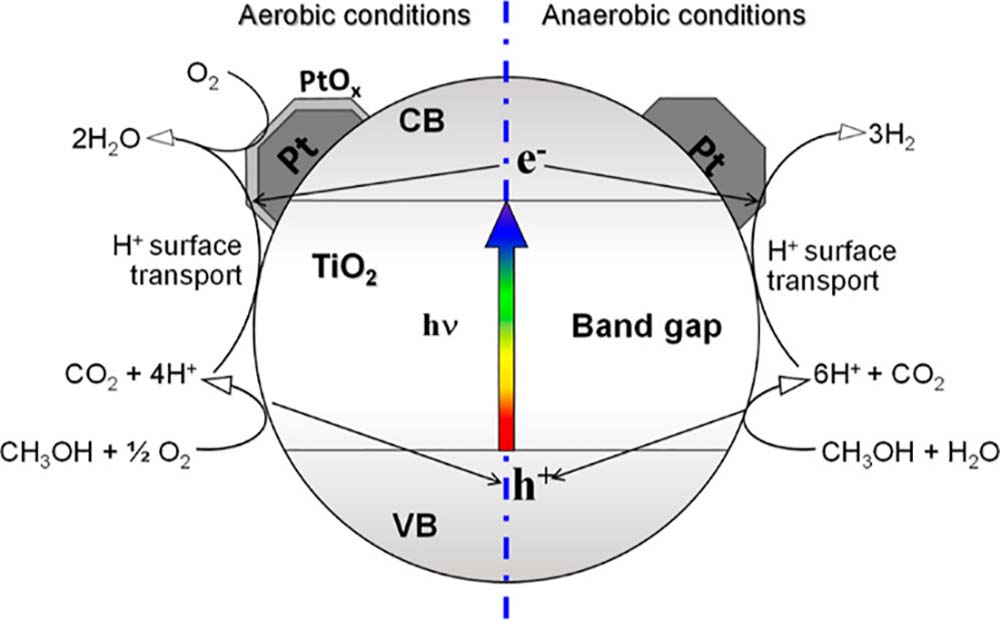
Figure 4. Proposed mechanism of photocatalytic methanol oxidation comparing aerobic (CH3OH + O2) and anaerobic (CH3OH + H2O) conditions over Pt/TiO2. Reproduced from ref.[121] under the CC BY 4.0 license.
In contrast, other study shows that supported CuO could be reduced to 1-2 nm Cu0 nanoparticles, forming active species (≈1 wt.% Cu/TiO2), which acted as efficient co-catalysts for hydrogen evolution[63]. The study also evaluated the influence of alcohol concentration, and correlate H2 production with alcohol physicochemical properties. Under UV irradiation (365 nm, 6.5mW cm-2), Cu2+ reduction was driven by α-hydroxy radicals generated during alcohol oxidation. These radicals, strong electron donors, promote Cu+/0 formation and enhance electron transfer to TiO2, boosting charge separation and hydrogen evolution. The reduction rate and H2 production followed the trend glycerol > ethylene glycol > methanol > ethanol, increasing with alcohol polarity, hydroxyl content, and lower oxidation potential. A similar trend was also observed for hydrogen generation by combining Cu2O and TiO2[122].
In fact, copper modification has been extensively explored as an effective approach to enhance TiO2 performance in methanol photoreforming without using precious metals[123-125]. A particularly effective strategy reported by Cwieka et al. involves introducing Cu as atomically dispersed Cu+ and Cu0 species on the TiO2 surface, without forming large Cu or Cu oxide nanoparticles, offering enhanced control over catalytic behavior and zero carbon emissions (CO or CO2) during methanol photoreforming[64]. Moreover, Cu-modified TiO2 (1 wt%) enabled a zero-carbon methanol photoreforming process, as only H2 was detected in the gas phase, with no trace of CO or CO2. In this case, TiO2 P90 was modified with highly dispersed copper via wet impregnation, followed by IR treatment (Vitalux 300 W) and calcination at 450 °C in a fritted glass reactor. In the liquid phase, carbon was fully retained as formaldehyde (~0.40 mmol) and formic acid (~0.41 mmol), confirming selective oxidation. The catalyst achieved an H2 production rate of
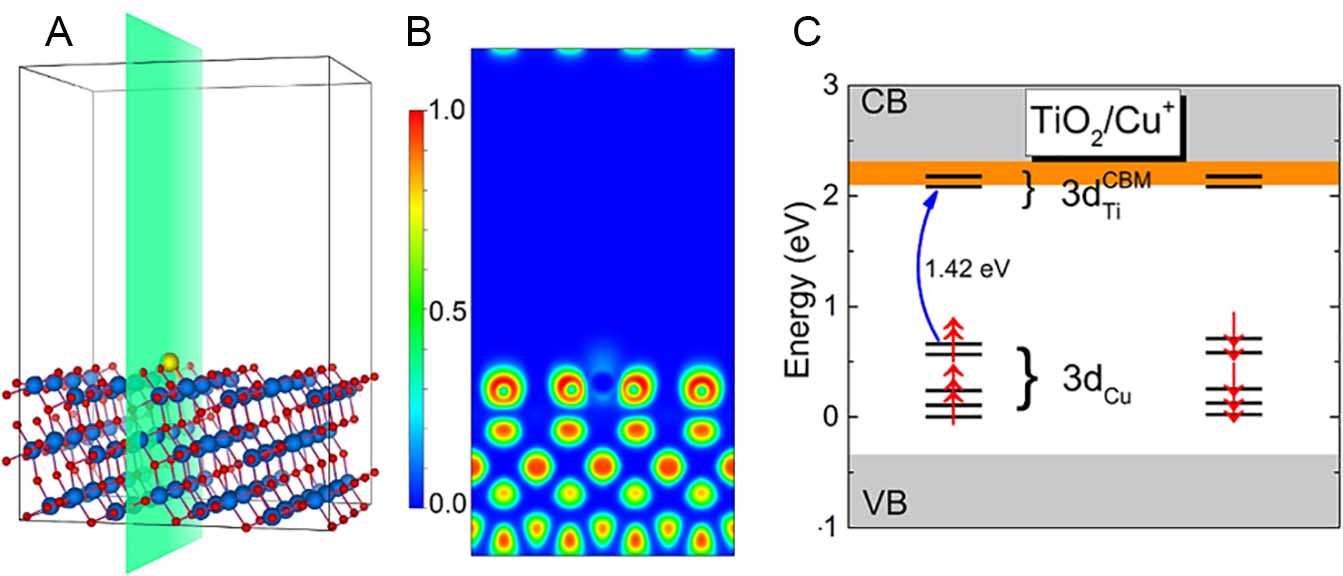
Figure 5. (A) Supercell of anatase TiO2(101) with an adsorbed Cu atom. Yellow, blue, and red spheres represent Cu, Ti, and O atoms, respectively. The electron localization function (ELF) is shown in green, (B) its projection on the (100) plane. (C) The Kohn-Sham eigenvalue spectrum with Cu⁺ 3d levels near the TiO2 bandgap; the gray and orange areas represent the TiO2 valence/conduction bands and the perturbed CBM, respectively. The blue arrow indicates a d-d transition between the localized Cu⁺ 3d state and the perturbed CBM. (A-C) is reproduced from ref.[64] under the CC BY 4.0 license.
The study from Plascencia-Hernández et al. employed P25-type TiO2 modified with Cu2O/CuO (CuxO)[126]. Firstly, CuxO polyhedral particles were synthesized at room temperature via chemical reduction with PVP and L-ascorbic acid, and then, CuxO and P25 were mixed in an ethanolic solution. The introduction of CuxO enabled the formation of a p-n heterojunction with TiO2, enhancing charge separation. Notably, under light irradiation, in situ self-reduction led to the generation of metallic Cu0, highlighting a dynamic and adaptive catalytic system. The Cu2O/CuO-modified TiO2 catalyst exhibited absorption in both UV and visible regions (up to ~650 nm), with enhanced activity under 470 nm excitation. Using a methanol-water mixture (1:10) at 1 g·L-1 catalyst concentration, the study showed that Cu oxides undergo self-reduction to Cu0 during photoreforming, forming a Schottky barrier that improves charge separation. Color changes in the solution also reflected the cyclic redox of copper species. The best catalysts (0.05% and 0.1% CuxO) achieved H2 production rates up to 2.86 mmol·g-1·h-1, about eight times higher than pure TiO2, alongside CO2 production, indicating complete methanol oxidation. The Cu2O/CuO-TiO2 p-n junction and in situ Cu0 formation both contribute to enhanced selectivity and stability, with performance maintained over multiple cycles.
A recent report showed an alternative approach to combine copper with TiO2, using a copper-based metal-organic framework (MOF), which offered additional advantages due to their high specific surface area, tuneable porous structure, and structural stability[127]. Composite materials based on P25 TiO2 and copper-benzene-1,3,5 tricarboxylate (HKUST-1) were synthesized via a simple physical mixing method in methanol overnight. Right after, water was added to reach 25 vol% of methanol and photocatalytic experiments for hydrogen were conducted. Similarly to the previous report from
Taking methanol as model alcohol, the mechanistic understanding of alcohol photoreforming can be extended to other alcohols with longer carbon chains. Moreover, photoreforming of diols and triols that could yield to value oxidation products and are derived from renewable sources, being therefore of great interest for sustainable low carbon hydrogen production. In the following sections, recent studies on photoreforming of C2+ alcohols are reviewed.
PHOTOREFORMING OF C2+ ALCOHOLS
As previous discussed, the photoreforming proceeds through pre-adsorption of the target alcohol on the photocatalyst surface, either at metal-oxygen bonds or Ov, leading to the formation of alkoxy intermediates. These species are subsequently oxidized by photogenerated holes, generating alkoxy radicals that undergo α-H abstraction. The resulting aldehydes can either desorb as products or remain adsorbed, undergoing further oxidation to acids or even complete mineralization to CO2[73,128,129]. In parallel, the release of protons is coupled to their reduction by photogenerated electrons at the metal co-catalyst, driving hydrogen evolution. For ethanol, for example, Coronado et al. have shown that the alcohol is adsorbed on the TiO2 surface by monodentate or bridged modes (Eqs. 6 and 7) to yield ethoxide groups[130].
While this mechanism is broadly similar for different alcohols, the nature of the substrate strongly influences the reaction pathway and product distribution. For instance, the replacement of methanol by ethanol introduces C-C bond cleavage and additional α-hydrogens, leading to products such as acetaldehyde, acetic acid, and CO2[74]. Secondary alcohols, such as isopropanol, tend to undergo dehydrogenation more readily, with acetone as the main product, accompanied by small amounts of CO2, formic acid, or acetic acid from over-oxidation. In some cases, dehydration by-products like propylene can also appear[73,74]. Thus, compared to methanol, higher alcohols not only expand the spectrum of possible oxidation products but also reveal the complexity of competing pathways that influence the overall efficiency of hydrogen generation. Moreover, the balance between these transformations can be significantly affected by photocatalyst modifications, such as metal loading, defect engineering, or heterojunction design, which alter charge separation dynamics and surface reactivity.
Zhang and coauthors reported that induced defects in TiO2 can control product selectivity of ethanol photoreforming[131]. The defects were chemically formed into bare P25 TiO2 using NaBH4. Enhanced photoactivity was observed as the “defected” TiO2 exhibited improved light absorption, charge separation, and better product selectivity, resulting in both higher hydrogen purity and a better liquid-to-gaseous carbon ratio. Ov were created in the TiO2 lattice, forming Ti3+ sites, which were detected by EPR spectroscopy, with the signal increasing with increasing NaBH4 amounts. The dominant reaction pathway was determined through active species trapping experiments, which showed that the preferred ethanol oxidation pathway was related to direct hole transfer, indicating that selectivity relies on surface chemisorption. In this case, surface defects decreased the acetaldehyde adsorption energy, promoting its rapid desorption and suppressing overoxidation into CO2, thereby improving selectivity towards liquid hydrocarbon products. As represented in Figure 6, the oxidation mechanism involves C-H activation as the initial step of ethanol photoreforming on defected TiO2, with the abstraction of an α-H from the ethanol molecule and the formation of a 1-hydroxyethyl radical. Then, acetaldehyde was formed as the 1-hydroxyethyl radical underwent further oxidation by photogenerated holes. In this case, the so-called current doubling effect described for methanol reforming was not investigated. Liquid product production reached 0.08 mmol/h of liquid acetaldehyde (from 10 v/v % ethanol), and the total organic production (acetaldehyde and acetic acid) was 0.47 mmol in 6 h compared to 0.14 mmol for the pristine P25[131]. In relation to the H2 production, an optimal rate of 0.08 mmol/h, with purity greater than 99%, much higher than the 93.4% purity from bare P25 TiO2, was achieved.
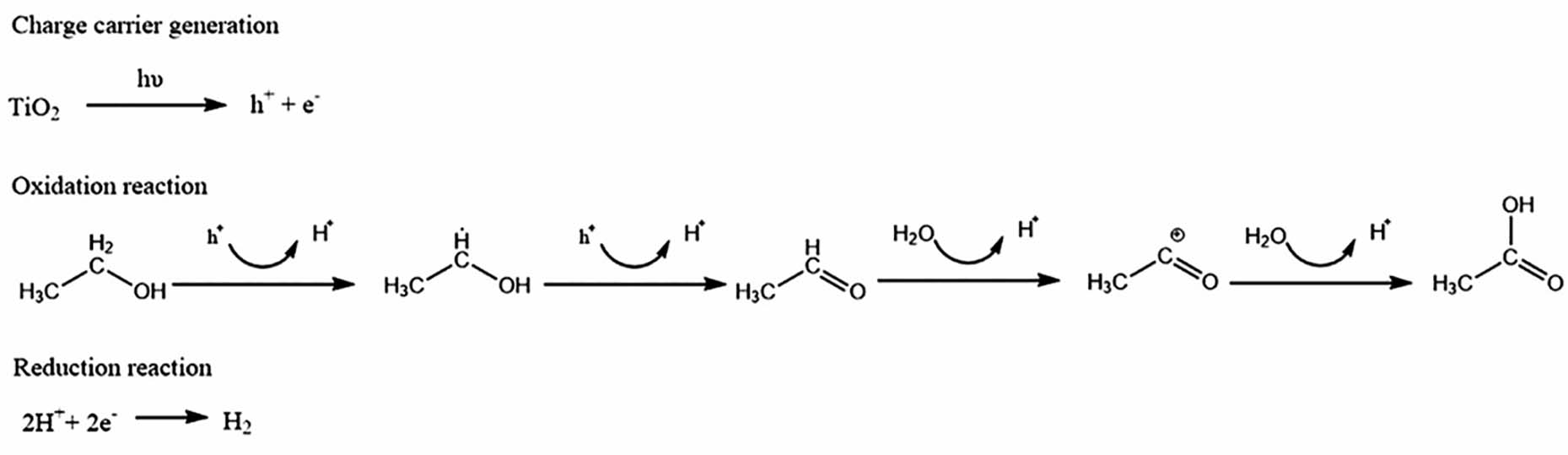
Figure 6. Proposed mechanism for the ethanol photoreforming reaction. Reproduced from ref.[131] under the CC BY 4.0 license.
Ethanol photoreforming was also evaluated using hierarchical Au/TiO2 nanoflower (TNF) photocatalysts under UV irradiation[132]. The effect of different calcination treatments on the physicochemical properties of the material were also evaluated, to benchmark their performance against selected reference photocatalysts. The Au/TNF-600-1 (600 °C, 1 h) sample exhibited the highest hydrogen production rate with 10 vol.% ethanol (23.8 mmol g-1h-1). This performance was superior to Au/P25 TiO2 and other Au/anatase reference photocatalysts. The hydrogen production rates increased with calcination temperature up to 600 °C but then decreased sharply at higher temperatures, reflecting the interplay among surface area, crystallite size, and polymorphic composition. The Au/TNF-600-1 photocatalyst also demonstrated excellent stability, with no loss in activity over five successive 3-h test cycles. Additionally, Piedra-Lopez and coauthors have shown that smaller and more uniformly distributed Au nanoparticles can yield higher performance on H2 production[65]. Their study compared the effects of two reduction methods, thermal reduction (TR) and photocatalytic reduction (PR), on Au/TiO2 (P25) photocatalysts prepared via deposition-precipitation (DP) with urea. The reduction route was found to critically influence nanoparticle size and distribution. The photocatalytically reduced sample achieved superior properties, with 94% of Au nanoparticles smaller than
Esteves et al. conducted a comprehensive investigation of TiO2-reduced graphene oxide (rGO)-Pt nanocomposites for photocatalytic hydrogen production from ethanol in both liquid and gas phases[134]. The catalysts were synthesized via a two-step procedure where TiO2-rGO nanocomposites were first prepared using a hydrothermal method at 125 °C. Subsequently, Pt decoration was achieved through photodeposition using a 450 W mercury-vapor lamp under N2 purging. The resulting nanocomposites had Pt loadings of 1.5%, 2.5%, and 3.8%, with rGO content optimized at 3 wt%. The TiO2-rGO3%-Pt3.8% composite achieved the highest initial hydrogen production rate of 560 mmol h-1g-1 with > 95% H2 selectivity. Gas-phase measurements confirmed a consistent H2/COx molar ratio of approximately 3, indicating near-complete selectivity with minimal byproducts, including CH4, CO, CO2, C2H6, and C2H4[134]. Platinum loading strongly influenced activity, with initial hydrogen production rates (slope 1) of 361, 356, and 567 mmol h-1 g-1 for Pt loadings of 1.5%, 2.5%, and 3.8%, respectively, followed by significantly lower rates (second slope) of 24, 34, and 66 mmol h-1 g-1, indicating a change in reaction regime over time. Stability tests in the liquid phase revealed a pronounced loss of activity upon cycling, attributed to platinum poisoning by adsorbed intermediates, notably acetaldehyde, and to nanoparticle agglomeration. In contrast, gas-phase experiments showed a much lower activity loss (ca. 8%), particularly for the TiO2-rGO-Pt system, highlighting the role of rGO in mitigating deactivation[134]. A rapid decrease in pH (from 7 to 3.5) was observed during liquid-phase reactions prior to stabilization, and this decline correlated with a decrease in the hydrogen production rate. Mechanistic analyses indicated a strong synergistic effect between rGO and Pt:rGO acts as an electron acceptor, suppressing charge recombination, while Pt significantly enhances hydrogen evolution activity. Voltametric studies further evidenced progressive catalyst poisoning, with oxidation peaks at 0.68 and 1.06 V vs. Ag/AgCl that gradually diminished over repeated cycles, supporting the role of reaction intermediates, particularly acetaldehyde, in activity loss. The dual-phase operation demonstrates that these nanocomposites are active in both liquid and gas phases, offering flexibility for hydrogen production processes. Although high hydrogen selectivity and the possibility of catalyst regeneration after treatment were observed, significant activity loss upon reuse and the requirement for relatively high Pt loadings (3.8%) may limit large-scale application[134]. Additional challenges include the need for UV irradiation, accumulation of intermediates, pH variations, and stability issues under continuous operation. Therefore, the development of flow reactor systems enabling the continuous removal of oxidation products may improve process stability and scalability.
TiO2 can also be modified with non-noble and earth-abundant materials to enhance its photocatalytic performance for ethanol photoreforming. In this context, the p-type semiconductor NiO was employed as a co-catalyst to form NiO-TiO2 heterojunctions synthesized via the sol-gel process[71]. The incorporation of NiO significantly improved hydrogen generation efficiency, doubling the H2 production rate compared to pure TiO2 and maintaining long-term stability. This enhancement arises from the formation of a p-n junction between NiO and TiO2, which facilitates charge separation by directing photogenerated electrons to the conduction band of TiO2 and holes to the valence band of NiO, thereby minimizing recombination. In situ DRIFTS analyses provided valuable mechanistic insights into ethanol adsorption and reaction pathways[71]. On both catalysts, ethanol adsorbed in molecular and dissociative forms, but the proportion of surface ethoxy species (CH3CH2OTi) was higher on NiO-TiO2. Upon illumination, pure TiO2 underwent a multistep oxidation via acetaldehyde and acetic acid intermediates, resulting in greater aldehyde accumulation and partial deactivation. In contrast, NiO-TiO2 favored direct dehydrogenation of ethanol through CH2CHO-type intermediates, accompanied by higher H2 evolution (20 mmol g-1 h-1) and increased oxidation reaction rates without surface poisoning by primary oxidation products.
Shahpal et al. evaluated the H2 evolution during aqueous-phase photo-oxidation of isopropanol (IPA) on Pt-decorated g-C3N4/TiO2 hybrid photocatalysts[135]. According to the hybrid band structure, the photo-generated electrons migrate from g-C3N4 to TiO2, while the photogenerated holes migrate from TiO2 to g-C3N4, reducing the likelihood of recombination. Furthermore, photodeposition of Pt nanoparticles localized the photogenerated electrons at the metal nanoparticles. In the proposed mechanism, IPA adsorbs on the surface of g-C3N4 or TiO2, where it is oxidized to acetone upon reacting with photo-generated holes, simultaneously forming H+ ions. In a parallel reaction, the adsorbed water molecules react with the photo-generated holes, producing additional H+ ions along with OH-. The hydronium ions then combine with the electrons accumulated on the Pt nanoparticles to form adsorbed H•, leading to evolved H2 gas.
Interestingly, Šalipur et al. illustrated the deactivation mechanisms of Pt-decorated g-C3N4/TiO2 photocatalysts for hydrogen production using IPA[136]. Under solar irradiation, the photocatalyst exhibited high initial activity (2.3 mmol g-1·h-1) but gradually deactivated to ~1.4 mmol g-1·h-1 due to the formation of inhibitory intermediates, such as propionic acid and pinacol, while Ti3+ surface defects were partially preserved[136]. Under visible light, activity was lower (1.1 mmol g-1·h-1) but stable, with complete oxidation to Ti4+ and absence of critical intermediates, highlighting a trade-off between initial activity and durability. Cross-deactivation tests confirmed that solar-light-generated intermediates caused H2 inhibition. The study revealed the dual role of Ov, the importance of TiO2-carbon-Pt charge transport, and the kinetics of intermediate formation vs. decomposition. These findings provide guidelines for spectral optimization, defect control, and prevention of inhibitory products, supporting the design of more efficient and durable photocatalysts.
Local structural features on the photocatalytic oxidation of IPA to acetone were examined using a systematic series of titania materials anchored on mesoporous silica (SBA-15)[60]. Titanium surface loading with monomeric and oligomeric Ti4+ species was prepared by grafting Ti4+ species onto SBA-15, with surface densities ranging from isolated low-loaded sites (0.02-0.35 Ti nm-2) to higher loadings (up to 2 Ti nm-2). The results showed that supported Ti4+ species exhibit superior selectivity compared to P25, particularly at low surface loadings where isolated tetrahedral TiO4 sites are present. These single-site catalysts limit the availability of charge carriers, preventing overoxidation to acetate, whereas P25 suffers from active site blocking due to acetate formation. Photocatalytic performance normalized to Ti content indicated that supported catalysts achieved comparable or superior activity per Ti atom. Additionally, band gap measurements indicated that quantum confinement effects in low-loaded catalysts influence charge carrier dynamics and reactivity. Structural analysis also revealed that increasing Ti loading promotes oligomeric Ti4+ species formation, which correlates with slightly reduced selectivity. Moreover, the presence of isolated Ti4+ sites restricted sequential oxidation steps, while the silica support provided additional adsorption sites that prevented active-site deactivation. In-situ DRIFTS confirmed that low-loaded catalysts maintain surface integrity and allow efficient acetone desorption. In summary, the study established clear structure-activity relationships: isolated tetrahedral Ti4+ sites provide the highest selectivity, whereas increasing Ti content favors oligomer formation, which triggers acetate formation and losses in photocatalytic activity.
Ethylene glycol photoreforming was recently reported by Roebuck et al., with particular emphasis on how the crystalline phase of titanium dioxide affects the photo-oxidation mechanism[137]. This substrate was selected because of its prevalence in bioderived compounds and its potential relevance as a waste or pollutant material. Its adsorption behavior and hole transfer mechanisms were analyzed across different TiO2 crystalline phases, including anatase, rutile, and P25. Among the studied samples, Pt/anatase and Pt/P25 photocatalysts exhibited similar hydrogen production activities, quantum efficiencies of 15.9% and 11.7%, respectively. In contrast, Pt/rutile showed the lowest performance with a quantum efficiency of only 4.3%. Over time, the hydrogen production rate for all catalysts slightly decreased, while CO2 evolution increased. The molar ratio of H2 to CO2 for Pt/P25 was approximately 2.5 after 4 h of irradiation, agreeing with the stoichiometric expectation for ethylene glycol photooxidation. Pt/anatase and Pt/rutile, however, displayed higher H2 to CO2 molar ratios of 3.7 and 3.1, respectively, suggesting additional H2 production from partial water oxidation for Pt/anatase and limited CO2 generation for Pt/rutile. With respect to oxidation pathways, Pt/anatase and Pt/P25 primarily oxidized ethylene glycol to glycolaldehyde, following an indirect oxidation pathway via hydroxyl radicals, leading to full mineralization, whereas Pt/rutile favored direct C-C bond cleavage to formaldehyde, which failed to undergo further conversion to formic acid and thereby limited hydrogen evolution. The inability of Pt/rutile to effectively oxidize formaldehyde was identified as a key factor for its lower hydrogen output. Overall, an indirect hydroxyl radical-driven mechanism predominated on anatase-containing catalysts, while the direct hole transfer mechanism governed on rutile. Therefore, the choice of TiO2 polymorph is a critical strategy for enhancing hydrogen production, as it governs the oxidation mechanism. Although P25 exhibited lower overall H2 production than pure anatase, it showed a more efficient conversion of ethylene glycol per unit surface area, indicating that the mixed-phase advantage may lie in its additive ability to convert both ethylene glycol and its intermediate species. This dual functionality of mixed-phase TiO2 helps to avoid the accumulation of specific intermediates and consequent surface poisoning.
Some recent works have considered the photoreforming of alcohols with longer and more complex chains. The influence of butanol isomer on photothermal hydrogen production using Ti@TiO2 core-shell photocatalysts was studied by El Hakim et al.[138] Structural differences among 1-butanol (1-BuOH), 2-butanol (2-BuOH), and tert-butanol (t-BuOH) were evaluated with respect to hydrogen evolution kinetics, surface adsorption, and reaction mechanisms. A clear performance hierarchy for hydrogen production was observed: 1-BuOH significantly outperformed 2-BuOH, which in turn exceeded t-BuOH across all tested temperatures (up to 69 °C). This trend was attributed to stronger adsorption of primary alcohols on the TiO2 surface, facilitating more effective interaction with photogenerated holes. Photothermal effects were pronounced, with reaction rates increasing with temperature under illumination; however, no hydrogen formation occurred in dark conditions, confirming the photonic origin of the reaction.
Product analysis showed that 1-BuOH and 2-BuOH produced H2 exclusively as a reduction product, whereas t-BuOH generated a mixture of H2, CH4, and C4H6, indicating a C-C bond scission in tertiary alcohols[138]. Activation energies for 1-BuOH and 2-BuOH were similar (20-21 kJ·mol-1), comparable to glycerol (25 ± 5 kJ·mol-1), while t-BuOH exhibited a lower activation energy of 13 kJ·mol-1, reflecting a different rate-limiting mechanism. Mechanistically, primary and secondary alcohols followed a photothermal pathway involving photogeneration of electron-hole pairs in TiO2, thermal activation of shallowly trapped holes, transfer to highly reactive free holes at the surface, and subsequent hole-mediated O-H bond cleavage leading to hydrogen formation. In contrast, t-BuOH displayed a distinct mechanism characterized by lower activation energy, weaker surface adsorption, and C-C bond scission, producing multiple gaseous products. Therefore, for hydrogen production, primary alcohols were preferred due to their higher yields and exclusive H2 formation.
Courtois and coauthors investigated the photochemical transformations of isobutanol on rutile TiO2(110), as a model system to understand temperature-dependent photocatalytic processes of biomass-derived chemicals[66]. A strong temperature dependence in the reaction selectivity was observed and would arise from the competition between thermal desorption of initial photoproducts from the surface and secondary photochemical reactions of surface-bound intermediates. They revealed that isobutanol desorbs from
Mechanistic analysis evidence that isobutoxy species are the key photoactive intermediates, supported by the complete suppression of disproportionation after UV exposure, and that oxidized TiO2(110) surfaces enhanced alkoxy formation and photo-oxidation efficiency compared to reduced surfaces, with hydroxylated surfaces exhibiting intermediate behavior[66]. The primary photochemical pathway involved isobutanol conversion to isobutoxy with release of protons and electrons, followed by photo-oxidation of isobutoxy to isobutanal, while at low temperatures secondary photo-oxidation occurred, in which isobutanal photochemically split into propyl and formyl radicals, the former recombining with surface hydrogen to yield propane and the latter reacting with lattice oxygen to form formate that decomposed into CO. Quantitative analysis showed that higher surface coverages of isobutanol increase photochemical efficiency. A significant fraction of surface isobutoxy was converted under UV illumination, and the thermal desorption processes exhibited Arrhenius-type temperature dependence.
The same group also investigated the photocatalytic conversion of tertiary alcohols on rutile TiO2(110) surfaces under highly defined vacuum conditions[139]. The study shows that tertiary alcohols, typically regarded as inert toward oxidation, undergo diverse reaction pathways under photocatalytic conditions, particularly because of the absence of an α-hydrogen, which normally facilitates C-H bond cleavage in primary and secondary alcohols. Moreover, the addition of co-catalysts, specifically platinum, influences the reaction rates and the emergence of new reaction channels, including the formation of molecular hydrogen and long-chain alkanes. The experiments were conducted under UHV conditions to ensure a highly controlled environment and to minimize interference from oxygen and water.
The tested photocatalyst was a rutile TiO2(110) single crystal, and the studied tertiary alcohols were 3-methyl-3-hexanol, 2-methyl-2-pentanol, and 2-methyl-2-butanol[139]. Platinum clusters were deposited on the TiO2(110) crystal at well-defined coverages, ranging from 0.1% of a monolayer up to Pt25 clusters, in order to assess their co-catalytic effects. Under UV illumination, contrary to typical oxidation pathways observed for primary and secondary alcohols, tertiary alcohols underwent a photocatalytic disproportionation reaction, yielding an alkane and the corresponding ketone. For instance, as represented in Figure 7, 3-methyl-3-hexanol produced 2-pentanone and ethane, as well as 2-butanone and propane, as well as 2-butanone and propane. Instead of the common C-H bond cleavage, the reaction proceeded via C-C bond cleavage; however, bonds to methyl groups were not cleaved. Instead, the longer alkyl chains in the α-position to the hydroxyl group were abstracted. This process exhibited 100% selectivity for the abstraction of the longer carbon chain in tertiary alcohols with two methyl groups at the α-C position, such as 2-methyl-2-butanol and 2-methyl-2-pentanol, which yielded acetone and the respective alkane exclusively. The introduction of platinum loadings significantly increased the overall reaction rate and also opened a new reaction channel, involving the formation of H2 and a long-chain alkane, resulting from the recombination of two alkyl moieties. In the case of 2-methyl-2-pentanol on Pt-loaded TiO2, hexane was detected (from propyl radical recombination), along with H2, which were not observed on bare TiO2.
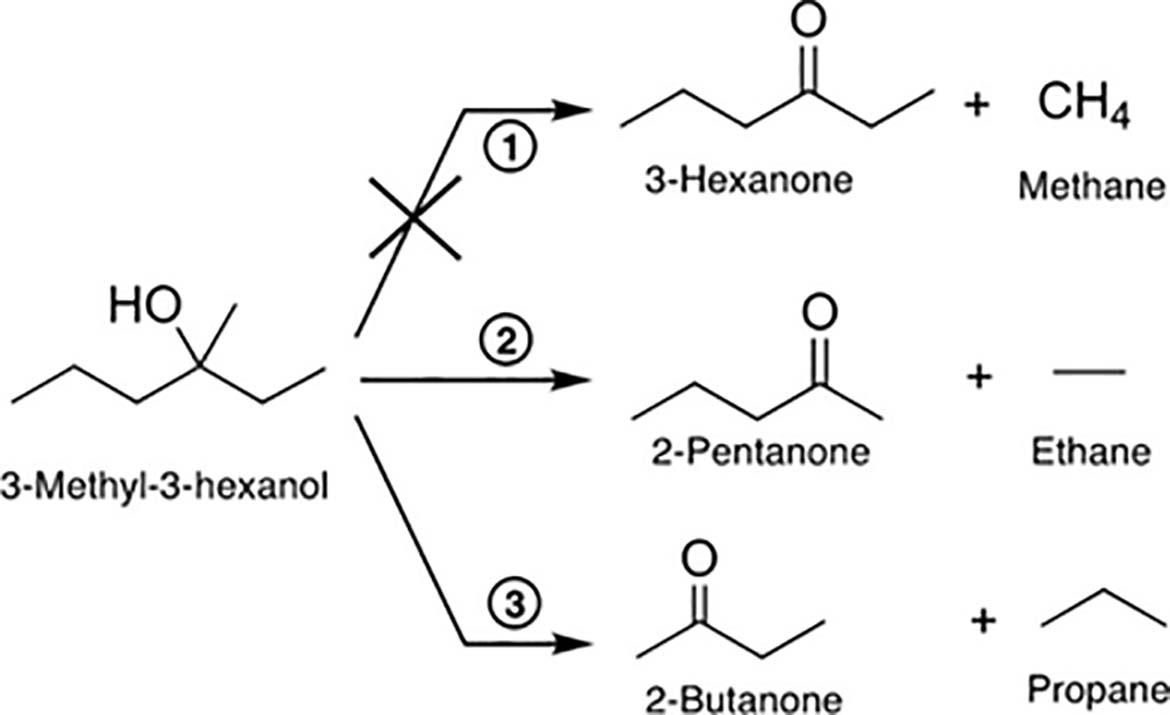
Figure 7. Reaction scheme for the photoreforming of 3-methyl-3-hexanol on Ptx/TiO2(110) and on TiO2(110) under UV illumination. Reproduced from ref.[139] under the CC BY 4.0 license.
The study confirmed that the reaction is truly catalytic under illumination, with no evidence of catalyst poisoning by unwanted surface species[139]. Selectivity patterns, such as the pentanone-to-butanone ratio of about 2:1 for 3-methyl-3-hexanol, remained largely unaffected by variations in Pt coverage or temperature in the range of 230-360 K. The observed preference for ethyl over propyl cleavage, along with the consistent absence of methyl cleavage, is in agreement with thermochemical principles, as methyl radical formation requires significantly higher energy. The proposed reaction mechanism involves photoactive alkoxy species generated upon surface adsorption undergoing a hole-mediated oxidation process, leading to homolytic C-C bond scission, with longer alkyl chains such as ethyl and propyl interacting more strongly with the TiO2 surface and remaining adsorbed, subsequently recombining in a consecutive thermal reaction step with hydrogen atoms derived from dissociative alcohol adsorption. In summary, the study demonstrated novel and unexpected photocatalytic reactions of tertiary alcohols on rutile TiO2(110), characterized by disproportionation into an alkane and a ketone via C-C bond cleavage [Figure 8].
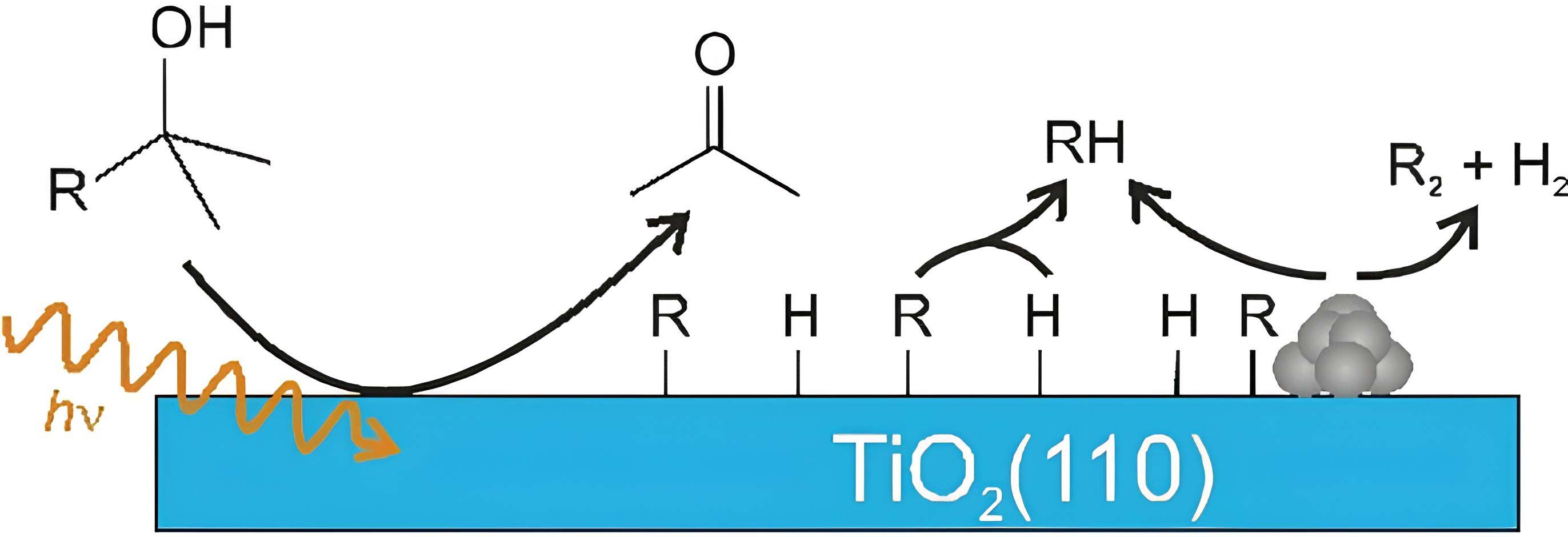
Figure 8. Photocatalytic disproportionation of tertiary alcohols on rutile TiO2(110) under UV irradiation, yielding an alkane and a ketone via C-C bond cleavage. Reproduced from ref.[139] under the CC BY 4.0 license.
GLYCEROL PHOTOREFORMING
Glycerol is widely used as a sacrificial agent in photocatalytic reforming due to its unique structural and chemical properties[140-142]. As a triol, glycerol contains three hydroxyl groups, which makes it highly reactive and easily oxidizable under photocatalytic conditions. Its low redox potential facilitates electron transfer to the photocatalyst, enhancing charge separation and promoting hydrogen evolution. Moreover, as it shares chemical features with polyols and sugars present in lignocellulosic or agricultural residues, glycerol also serves as a model substrate for biomass photoreforming. Additionally, it is a byproduct of biodiesel production, making it abundant, inexpensive, and renewable, which increases its appeal for sustainable energy applications[143-147]. Compared to other alcohols, glycerol often leads to higher hydrogen production rates and the formation of valuable intermediates such as dihydroxyacetone (DHA), formic acid, and glyceraldehyde (GA), providing opportunities for combined energy and chemical valorization[148]. These characteristics make glycerol a model substrate for investigating photocatalytic hydrogen generation and designing efficient photocatalyst systems. Selected results on photoreforming of glycerol using different TiO2-based photocatalysts are summarized in Table 1, while key aspects of the process and materials development are discussed in the following paragraphs.
Performance of TiO2-based systems on glycerol photoreforming
| Photocatalyst | Irradiation conditions | Glycerol conc. | H2 evolution rate | AQE | Main oxidation products (selectivity) | Ref. |
| Pt/TiO2 | 30 W LED lamp, 380 nm | 0.06 M | 1.35 mmol·g-1·h-1 | - | - | [149] |
| Pt/TiO2@0.3% (plates) | 300 W Xe lamp, visible light | 10 vol.% | 316 mmol·h-1·m-2 | - | - | [142] |
| Pt/TiO2 | 300 W Xe lamp | 1 M | 4.93 mmol·g-1·h-1 | - | GA, GA | [150] |
| Pt/TiO2 brookite | UV irradiation | 2 mM | 9.3 mM (after 4 h) | - | DHA (6.8%); GA (8.1%) | [151] |
| Pt/TiO2-PyCF | 125 W Hg lamp, 365 nm | 10 vol.% | 10.5 mmol·g-1·h-1 | - | DHA, GA | [152] |
| Zeolite (USY62)-Ti-Pt | 125 W Hg lamp | 10 vol.% | 11.7 mmol·g-1·h-1 | - | CO2 | [153] |
| Pt-g-C3N4-TiO2 | 125 W LED lamp, 365 nm | 10 vol.% | 27.5 mmol·g-1·h-1 | - | GA, DHA | [141] |
| Pt/N-TiO2 | UV lamp (125 W, 360 nm) | 0.92 wt% | 0.345 mmol·g_cat-1·h-1 (avg; 8 h) | - | DHA | [42] |
| Pt/TiO2-MCH | UV irradiation | 10 vol.% | 26.1 mmol·g -1·h-1 | - | CO2 | [154] |
| 2 wt% Au/P25 TiO2 | UV 365 nm, 6.5 mW/cm2 | 15 vol.% | 32.7 mmol·g-1·h-1 | - | - | [155] |
| Au/TiO2@n - octadecane | 300W Xe lamp | 10 vol.% | 5.44 mmol·g-1·h-1 | - | - | [156] |
| TiO2-rGO-Au | 365 nm LED lamp | 10 vol.% | 64 mmol·g-1·h-1 | - | CO2 | [157] |
| 0.1 wt% Ag, 0.3 wt% Pd/TiO2 (anatase) | UV lamp 100 W (360 nm cutoff) | 0.5 vol.% | 13.9 mmol·g-1·h-1 | - | formaldehyde, hydroxyacetone, ethylene glycol | [14] |
| Pd/rGO/TiO2 | UV lamp 100 W, 365 nm | 5 vol.% | 41.0 mmol·g-1·h-1 | - | - | [158] |
| Cu-TiO2 | UV: 125 W Hg lamp | 10 vol.% | 3.57 mmol·g-1·h-1 | - | CO2 | [159] |
| Cu-TiO2 | 365 nm LED irradiation (18 W, ~55 mW cm-2) | 0.11 M | 1.83 mmol·g-1·h-1 | 3.4% | methanol (40%) CO2 (31%), CO (4%) | [160] |
| 1 wt.% Cu/TiO2 | UV, 365 nm, 6.5 mW cm-2 | 20 vol.% | 20.5 mmol·g·h-1 | - | - | [63] |
| Cu/P25 | 125 W Hg lamp, 365 nm | 10 vol.% | 4.5 mmol·g-1·h-1 | - | - | [161] |
| Cu-TiO2/MCH (carbonaceous support) | Hg lamp (125 W) | 10 vol.% | 4.6 mmol·g-1·h-1 | - | Glyceric acid and glycolic acid | [162] |
| TiO2-CuO | UV LED, 365 nm | 10 vol.% | 3.65 mmol·g -1·h-1 | - | - | [163] |
| CuO-TiO2 | Solar (CPC, 47 kJ L-1) | 0.075 M (≈ 6.9 g L-1) | 12 mmol h-1 | STH = 1.42% | CO2, formate and glycolate | [164] |
| CuO:TiO2 | 125 W Hg lamp, 365 nm | 10 vol.% | 17.6 mmol·g-1·h-1 | - | Formic acid, glycolaldehyde | [165] |
| 3% Cu2O-P25 | 125 W Hg, 365 nm | 2 mM | 0.17 mmol·g-1·h-1 | - | DHA (10%), GA (5.4%) | [72] |
| 3% Cu2O-P25 | 30 W LED, 380 nm | 0.075 M | 18.8 mmol·g-1·h-1 | STH = 1.71% | Formic acid, CO2 | [30] |
| Cu2O/TiO2 | 400 W metal halide lamp | 5 vol.% | 0.67 mmol·g-1·h-1 | - | - | [166] |
| CuO-NiO-TiO2 (TiO2:MeO = 10:1; Cu:Ni = 1:2) | Solar (CPC, 2.10 m2) | 0.075 M | 0.062 mmol·kJ UVA-1 | - | - | [167] |
| NiO-TiO2 (7.5% Ni) | Xenon lamp 300W | 10 vol.% | 8.0 mmol·g-1·h-1 | - | GA, DHA | [168] |
| NiO-Ni-TiO2 (10% Ni loading) | 300 W Xe lamp | 10 vol.% | 24.5 mmol·g-1·h-1 | 6.86% (AQY at 365 nm) | DHA (21 %) and GA | [169] |
| O-Ni2/TiO2 | 365 nm LED irradiation (60 W), | ≈2.6 vol.% | 2.54mmol·g-1·h-1 | - | Syngas (H2 + CO) 96.1% | [170] |
| 3% Carbon dot/TiO2 | 300 W Xe lamp, 1-sun | 10 vol.% | 0.33 mmol·g-1·h-1 | - | CO2 | [171] |
| TiO2/C | UV Hg lamp 125 W | 10 vol.% | - | - | CO2 | [172] |
| Zr-RuCP6-Zr-RuP6@Pt-TiO2 + TEMPO | 460 ± 15 nm, 80 mW | 0.10 M | 2.67 mmol·g-1·h-1 | 0.516% | Glyceric acid | [173] |
In the context of photocatalytic applications using TiO2, the kinetics and mechanistic pathways of glycerol transformation can be influenced by diverse factors, as reported by Panagiotopoulou and coauthors[174]. Under photo-oxidation conditions, TiO2 (P25) promotes the complete oxidation of glycerol to CO2, with reaction rates increasing considerably upon the addition of Pt. Under photoreforming conditions (in the absence of O2), TiO2 alone exhibits very low activity. In contrast, when Pt (0.5 wt.%) is added as a cocatalyst, H2 and CO2 production rates increased by about 25 and 60 times, respectively. Analysis of liquid-phase products revealed a series of common intermediates formed during both oxidation and reforming reactions, including acetol, acetaldehyde, ethanol, methanol, GA, glycoaldehyde, acetone, and acrolein. Their formation, first increasing and then decreasing with irradiation time, indicates sequential oxidation and reforming steps leading ultimately to CO2. In the presence of oxygen, adsorbed intermediates were more rapidly oxidised, resulting in a cleaner surface compared with the oxygen-free conditions of photo-reforming, in which persistent surface residues were observed. In general, the reaction mechanism involves two main initial routes: hydrogenolysis of glycerol into propylene glycol and dehydrogenation into GA, followed by decarbonylation and dehydration steps yielding oxygenated species such as acetaldehyde, ethanol, and acetone (see scheme in Figure 9). In both oxidation and reforming conditions, these intermediates are eventually transformed into CO2, where in the latter, protons from water are reduced to generate H2. Consequently, both processes share the same overall pathway, differing mainly in the terminal electron acceptor and the presence of oxygen.
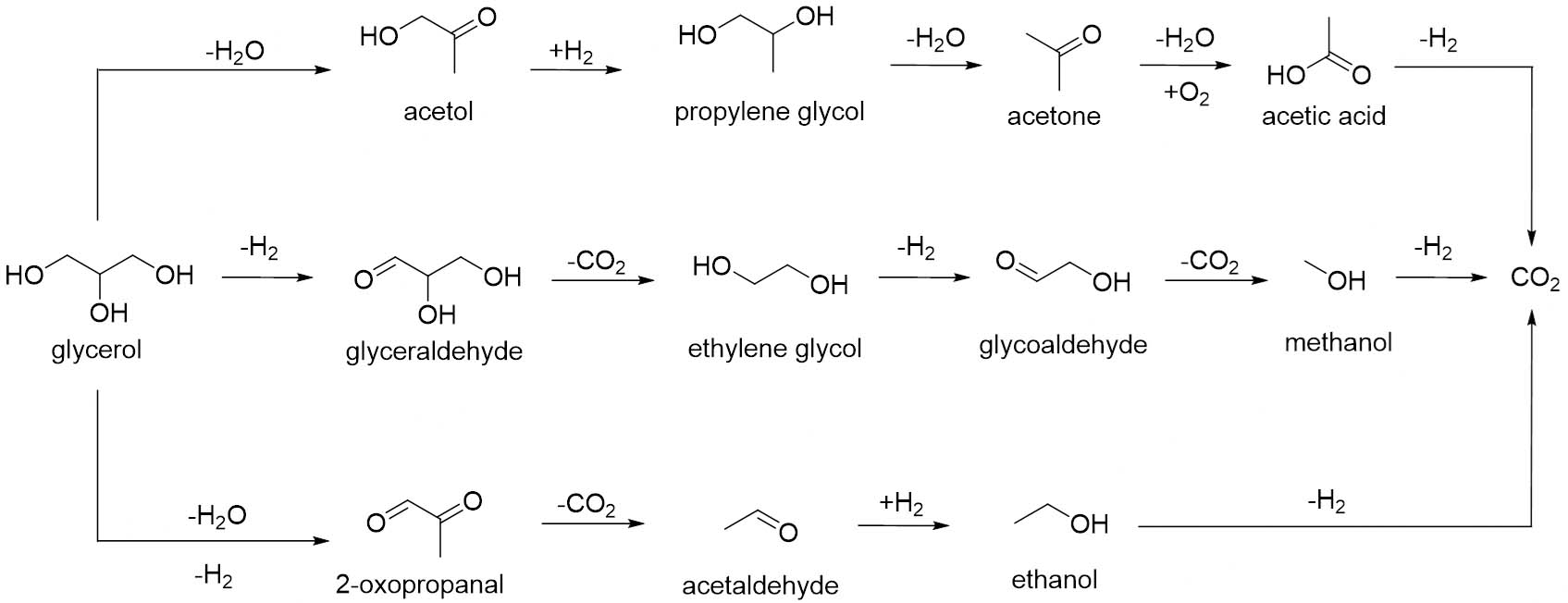
Figure 9. Schematic representation of the main reaction pathways involved in glycerol transformation.
Wahab and Idriss also investigated the photocatalytic reforming and oxidation pathways of glycerol using anatase TiO2 modified with silver (0.1 at.%) and palladium (0.3 at.%)[14]. Photocatalytic tests (100 W,
Additionally, Hu and coworkers investigated how different crystalline phases of TiO2 (anatase and rutile) affect the generation of ROS and the selective conversion of glycerol into formic acid[175]. In this case, rutile favors ROS formation via the sequential pathway O2 → O2•- → H2O2 → •OH, facilitated by its low oxygen vacancy formation energy (0.96 vs. 4.49 eV for anatase). Heterojunctions between anatase and rutile further lower this energy to -0.77 eV, thereby enhancing charge separation. The TiO2 sample containing 74% anatase achieved the highest glycerol photoreforming performance with 66% conversion and 42% selectivity toward formic acid. Key intermediates such as GA and glycolaldehyde were identified. Moreover, excess •OH causes over-oxidation to CO2, but adjusting the solution pH to 11 could suppresses this, increasing selectivity to 59%. The main pathway under neutral conditions was glycerol → GA → glycolaldehyde → formic acid, while excess •OH drives undesired CO2 formation. Further report by Pecoraro et al.[151] on glycerol photoreforming using the TiO2 polymorphs (anatase, brookite, and rutile) combined with Pt reveals that H2 generation arises from both glycerol dehydrogenation and, predominantly, from water dissociation. The observed superior performance of brookite was attributed to two distinctive surface properties. First, Fourier-transform infrared (FT-IR) spectroscopy revealed a higher hydrophilicity, leading to an enhanced ability to adsorb water molecules. Second, the distribution and nature of platinum sites were more favorable, with a predominance of Pt0 species located at edges and steps, which are known to be more catalytically active. Consequently, the following trend in hydrogen evolution efficiency was Pt-brookite > Pt-P25 ≈ Pt-anatase > Pt-rutile. Under UV irradiation, the Pt-brookite catalyst achieved an H2 yield 3.1 times higher than that of the reference Pt-P25. These results, further supported by the higher
In this regard, Maslova et al. demonstrated the importance of structural control over both the support and the metallic phase to maximize glycerol photoreforming using platinum-supported TiO2 catalysts[150]. TiO2 support was prepared via a microemulsion route, enabling controlled synthesis of nanoparticles with high uniformity, stability, and specific surface area. This process yielded a material predominantly in the anatase phase, with a small fraction of rutile, and exhibiting a large surface area favorable for metal dispersion. To optimize the anchoring sites, the TiO2 support underwent a dehydroxylation treatment under dynamic vacuum at various temperatures, with 500 °C selected as the optimal temperature. Platinum was then introduced using Pt(COD)Me2 (COD: 1,5-cyclooctadiene, Me: -CH3) as a precursor, via a surface organometallic chemistry (SOMC) approach for precise control over the formation and dispersion of metallic nanoparticles. Highly dispersed and ultrasmall platinum nanoparticles (≈1 nm) were obtained after reduction under H2 atmosphere at 300 °C (Pt-OM@TiO2-m-500). For comparison, photocatalysts were also prepared through conventional techniques, such as IWI and DP, using the same metal precursor. Different characterizations confirmed that the SOMC-prepared catalysts exhibited significantly higher platinum dispersion and smaller nanoparticles on the TiO2 surface. Photocatalytic tests were performed in a reactor irradiated with simulated sunlight
Escamilla-Mejía et al. presented a study focused on the preferential incorporation of platinum in TiO2-carbon composites[154]. The work highlights how the selective localization of Pt within specific regions of the TiO2/carbon matrix can positively influence glycerol photoreforming. For the studies, a commercial TiO2 P25 and a carbonaceous support (MSH) derived from olive pit biochar were used, where the TiO2-MCH composite was prepared via the sol-gel method, followed by photodeposition of platinum and a final calcination step. The carbonaceous supports enhanced intermediate adsorption, although excessive adsorption could potentially hinder light absorption and electron transfer if intermediates block TiO2 active sites. The results demonstrated that the composite exhibited a higher H2/CO2 ratio during glycerol photoreforming, indicating improved overall system efficiency and greater selectivity toward hydrogen generation compared to other catalysts tested in the study. This improvement was attributed to the strategic placement of Pt in regions that facilitate proton reduction, thereby minimizing charge recombination and preventing excessive oxidation of the substrate. The authors emphasized that the synergistic interaction of TiO2, the carbonaceous support, and Pt contributes to enhanced catalytic stability over multiple reaction cycles, maintaining consistent performance and preventing significant degradation of the active material. Overall, the study underscores the importance of targeted synthetic strategies that account for factors such as selective metal incorporation, functional support design, and light absorption dynamics to optimize photocatalytic efficiency.
Zhong et al. presented a multifunctional approach based on Au/TiO2@n-octadecane microcapsules with an organic core and a photothermal shell[156]. The study reports a multifunctional Au/TiO2 (AT) encapsulated in n-octadecane (a phase-change material, PCM), designed for the photothermal reforming of glycerol. The microcapsules were prepared via a hydrothermal route, yielding rectangular particles (70 nm), where gold nanoparticles were deposited on TiO2 nanosheets and embedded in a the PCM matrix. These microcapsules acted as integrated solar energy harvesting systems, absorbing across a broad spectral range including the infrared region, serving simultaneously as thermal storage media and photocatalytic supports. Under simulated solar irradiation (Xe lamp, 300 W; 200-2,500 nm), the photothermal effect was quantified by monitoring a temperature increase in the glycerol suspension. The Au/TiO2@PCM system significantly outperformed conventional systems without PCM, where the internal thermal storage maintained elevated reaction temperatures even under fluctuating irradiance, sustaining a high hydrogen generation rate. Specifically, the microcapsules resulted in a H2 production rate of 5.4 mmol g-1·h-1, compared to
Interestingly, Oliveira et al. reported the design of photocatalytic plates containing immobilized TiO2/Pt, with a comparable performance to that of the powder photocatalyst[142]. The preparation involved the photodeposition of Pt onto TiO2 (P25), followed by homogenization with Nafion™ and ethanol and application onto acrylic substrates (5.0 × 1.0 cm) by solvent evaporation, with a final thickness of 150 µm. The optimal mass ratio per plate was fixed at 25 mg of TiO2-Pt, sufficient to prevent charge recombination without increasing Pt-related costs. The authors evaluated the stability and recyclability of the plates for up to 10 reaction cycles (3 h each), observing that at 0.3 wt.% Pt, the system maintained ~361 mmol H2·h-1·m-2 evolution from glycerol aqueous solutions up to the seventh cycle, while at 0.1 wt.% Pt, a noticeable performance decline occurred after the fourth cycle, likely due to catalyst loss from washing or leaching.
Several studies have explored replacing noble metals such as platinum and gold in TiO2-based photocatalytic systems for glycerol photoreforming, with copper emerging as a particularly promising and widely studied alternative due to its effectiveness and lower cost[141,162-164,166,167]. Pecoraro et al. presented a simple and cost-effective approach for synthesizing TiO2 (P25) photocatalysts modified with copper oxides using mechanical milling as the preparation method[72]. Among the tested samples, the catalyst containing 3 wt.% Cu2O-TiO2 exhibited a performance comparable to that of the benchmark TiO2-Pt system, achieving a glycerol conversion of approximately 33% and a hydrogen production rate of 0.17 mmol·h-1·g-1. Notably, the Cu2O-TiO2 catalyst showed good selectivity for the formation of high-value compounds such as 1,3- DHA and GA, the latter of which was not detected with the platinum-based catalyst. The enhanced efficiency of the system was attributed to the formation of a p-n heterojunction between Cu2O (p-type) and TiO2 (n-type), which enables a direct Z-scheme charge transfer mechanism. Additionally, a detailed investigation into the mechanisms of glycerol photoreforming using physical mixtures of CuO and TiO2 was reported by Martín-Gómez and coauthors[165]. Through EPR and cyclic voltammetry (CV), the origin of the improved photocatalytic activity was analyzed, with particular emphasis on the role of copper species. CuO nanoparticles with varying particle sizes were physically combined with TiO2 P25, and their performance was evaluated. While pure CuO and TiO2 showed no or limited activity for hydrogen evolution, the CuO:TiO2 mixtures demonstrated markedly superior performance, achieving up to
Accordingly, the proposed mechanism is based on a redox Cu(II)/Cu0 catalytic cycle, where photogenerated electrons migrate from TiO2 to CuO. The resulting Cu0 species act as active sites that facilitate proton reduction[165]. Subsequently, Cu0 is reoxidized back to Cu2+, completing the catalytic redox cycle. EPR analyses provided direct evidence for the presence and transformation of paramagnetic Cu2+ species during the photocatalytic process, confirming the dynamic operation of the Cu(II)/Cu0 cycle. Complementary CV measurements identified the characteristic redox potentials of copper species, consistent with the proposed mechanism.
In a similar study, Umair et al. investigated the pilot-scale photoreforming of glycerol for hydrogen production using Cu2O-TiO2 (P25) catalysts[30]. Conducted in a large-scale solar reactor, the work exemplifies translating laboratory-scale photocatalytic processes into pre-industrial applications. The catalyst was synthesized through a mechanical milling method, where structural analyses confirmed the formation of a p-n heterojunction between Cu2O and TiO2. In this configuration, Cu2O acts as a visible-light antenna due to its narrow band gap (1.8-2.2 eV), and both semiconductors are activated, promoting electron transfer from the Cu2O conduction band to that of TiO2, and holes from TiO2 migrate to the valence band of Cu2O. The pilot reactor, operated with suspended catalyst and fed with biomass-derived substrates (glycerol, glucose, and ethanol) under simulated solar exposure, demonstrated technical feasibility of scaling up such systems. EPR measurements confirmed the formation of hydroxyl radicals (•OH), which play a key role in substrate oxidation pathways.
During glycerol photoreforming, formic acid (formate) was identified as the main oxidation product, formed through intermediates such as GA and DHA, which were rapidly converted before full mineralization to CO2, as detected[30]. The optimal glycerol concentration was 0.075 M (6.9 g L-1), corresponding to a solar-to-hydrogen (STH) conversion efficiency of 1.71%. Lowering the concentration to 0.05 M led to a marked decrease in hydrogen generation (STH = 1.26%). Among the tested substrates, glycerol proved the most efficient, producing 57 mmol H2, followed by glucose (20.96 mmol) and ethanol (17.63 mmol). This superior performance was attributed to glycerol’s three easily oxidizable hydroxyl groups and low redox potential. Overall, the study demonstrated that Cu2O-TiO2 catalysts prepared by mechanochemical methods can be effectively scaled up, maintaining high performance, operational stability, and reproducibility under semi-industrial conditions.
Clarizia et al. investigated the effect of surface properties on copper-modified TiO2 photocatalysts using in situ photoinduced deposition of Cu onto three different TiO2 samples (100% anatase, 100% rutile, and P25)[161]. Characterization through XRD, UV-Vis, TEM, XPS, and XANES/EXAFS confirmed the oxidized nature of copper species and their interaction with the TiO2 support. However, under UV irradiation, copper was present as metallic Cu0 nanoparticles, formed via in situ photodeposition of Cu2+ ions. Interestingly, a big difference in photocatalytic performance was observed between methanol and glycerol as sacrificial agents. Superior activity for glycerol was observed and attributed to its molecular structure, which contains three hydroxyl groups, along with higher polarity and a greater number of hydrogen atoms in α-positions, favoring stronger adsorption on the photocatalyst surface. Glycerol also undergoes oxidation more readily than methanol, reacting more efficiently with photogenerated holes. Photocatalysts containing 1-10 wt.% Cu exhibited significant activity under UV irradiation, demonstrating that both the thermal pre-treatment and the TiO2 polymorphic form play crucial roles in determining photocatalytic performance. Among the tested supports, P25 consistently outperformed pure anatase and rutile. This was attributed to the intrinsic homojunction between the anatase and rutile phases in P25, and its surface displaying a higher density of hydroxyl groups, serving as preferential nucleation sites for copper deposition. This leads to the formation of smaller, more uniformly dispersed Cu0 nanoparticles, maximizing the interfacial area between metal and semiconductor. The incorporation of copper also modifies the electronic properties of the Cu-TiO2 heterostructure, primarily through the formation of a Schottky barrier at the metal-semiconductor interface. Additionally, plasmonic and optical effects may further enhance performance.
Interestingly, the study by Wang et al. reported the photoreforming of biopolyols and sugars to produce methanol and syngas (CO + H2) under UV irradiation at room temperature[160]. The photocatalyst was synthesized from anatase TiO2, which was first transformed into protonated titanate nanotubes (H-TNTs), subsequently exchanged with ammonium (NH4-TNTs), and finally subjected to ion exchange with Cu(NO3)2 followed by calcination to form defect-rich titanium oxide nanorods (TNRs). For comparison, a wide range of other semiconductor photocatalysts, including TiO2 P25 and other metal oxides, were also evaluated. Using Cu2+ species dispersed on defect-rich TiO2 nanorods (Cu/TNR), characterized by abundant Ov, the study reported the selective C-C bond cleavage of substrates such as glycerol, glucose, and even lignocellulosic biomass residues, leading to the formation of methanol and a mixture of CO and H2. The Cu loading was varied between 0.01 and 5 wt.%, with 1-2 wt.% found to be optimal for methanol generation and 0.01 wt.% for high CO selectivity. The authors proposed that glycerol photoreforming proceeds via an initial hydrolysis step, forming intermediates such as GA and 1,3-DHA, concurrently with the production of H2 and CO2. Water acts as the primary hydrogen source, while glycerol undergoes dehydrogenation, with the overall efficiency governed by selective C-C bond cleavage without overoxidation. Under optimized conditions, the 2 wt.% Cu/TNR catalyst using glycerol achieved a methanol yield of approximately 40% and H2 production of 22 mmol·g-1 after 12 h of irradiation, with an AQE of 3.4%. The same system also yielded 45% CO, an H2/CO ratio ≈ of 0.8, and 96% glycerol conversion. The method was further applied to various polyols (e.g., ethylene glycol, sorbitol, erythritol, xylitol) and sugars (e.g., fructose, glucose, sucrose), achieving methanol yields of 6%-28%, CO selectivity of 43%-52%, CO2 yields of 5%-6%, and H2 production ranging from 5 to 40 mmol·g-1, depending on the carbon chain length and water content of the substrate. Remarkably, the approach was successfully extended to biomass-derived materials such as cellulose and wood sawdust, after pre-treatment via hydrolysis or hydrogenolysis, enabling methanol production up to 14%, along with syngas mixtures rich in H2 and CO (up to 45% CO and 9.5 mmol·g-1 H2) using Cu/TNR catalysts with low or high Cu loadings, respectively. The copper loading was also shown to tune the selectivity between CO and CO2 through controlled degradation of formic acid intermediates; low Cu content favored CO formation, while higher Cu loadings promoted CO2 evolution.
Another possible modification involves using nickel-based co-catalysts. Eisapour et al. proposed the design of a p-n heterojunction between TiO2 and NiO to enable the simultaneous generation of H2 and selective production of DHA and GA from glycerol[168]. The system employed two-dimensional anatase TiO2 nanosheets combined with nickel oxide (NiO) nanoparticles. NiO (5 wt%) was deposited onto the TiO2 nanosheet surface via an impregnation-calcination method, leading to well-dispersed NiO nanoparticles. As observed in microscopy images, dark contrast points on the nanosheet surface confirmed the nanometric nature and uniform distribution of NiO particles. Under Xe-lamp irradiation (300 W), the 7.5 wt% NiO/TiO2 composite achieved an H2 production rate of approximately 8.0 mmol g-1·h. Recognizing that hydrogen production alone may be economically limited, the authors conducted a techno-economic analysis (TEA) to evaluate the integrated value of co-generated chemical products. From an initial 1.37 M glycerol solution, approximately 20% of glycerol was converted into high-value chemicals. GA accounted for ~89% of the projected annual revenue, followed by DHA (~11%) and H2 (0.03%), highlighting the economic advantage of coupling energy generation with chemical valorization. Mechanistically, the formation of the p-n heterojunction between TiO2 and NiO markedly enhanced charge carrier separation, steering the oxidation of glycerol toward selective formation of GA and DHA, rather than full mineralization to CO2 as commonly observed for bare TiO2.
The same group proposed the development of a “sandwich” NiO-Ni-TiO2 heterojunction designed for the simultaneous coproduction of green hydrogen and value-added chemicals via glycerol photoreforming[169]. The structure consists of consecutive layers in which TiO2 and NiO are intercalated with an active metallic Ni layer, forming a p-n system with internal barriers that enhance electron-hole separation. The metallic nickel functions as a conductive layer, optimizing the internal electric field and consequently improving charge transport within the photocatalyst. For the synthesis, anatase TiO2 was employed, and after comparative photocatalytic tests using different morphologies, the nanosheet type (sample S3) was identified as the most effective for hydrogen production. NiO-TiO2 composite was obtained by mixing the TiO2 nanosheets with a 15 wt.% nickel acetate solution in ethanol, followed by calcination at 450 °C. In the second step, the sandwich-type NiO-Ni-TiO2 heterojunction (SPN) was synthesized by thermally treating the NiO-TiO2 composite under a hydrogen flow at 500 °C, generating the metallic Ni interlayer. Finally, the Ni-TiO2 sample was partially reoxidized in air at 350 °C. The optimized sample (10 wt.% Ni) achieved the highest activity (300 W Xenon lamp), producing 24.5 mmol g-1 h-1 of H2 with AQE of 6.9 % at 365 nm. Glycerol conversion reached approximately 58 % after 24 h, generating primarily DHA and GA, with DHA being the most selective product (~21 % selectivity, ~12 % yield). The system demonstrated operational stability, maintaining high photocatalytic activity over repeated cycles.
Liu et al. also demonstrated a p-n heterojunction photocatalyst composed of TiO2 and NiO, but featuring oxygen-bridged diatomic Ni-O-Ni sites instead of single-atom Ni sites, to optimize glycerol photoreforming through local electronic structure engineering[170]. The strategy involved anchoring Ni-O-Ni diatomic sites onto a highly porous TiO2 support derived from a MOF, employing oxalic acid chelation. The study demonstrated that atomic-level Ni deposition reduces the contact angle between TiO2 and the glycerol-water mixture, thereby enhancing substrate adsorption. Multiple characterization techniques were employed to elucidate the reaction mechanism and the structure-activity relationship. Femtosecond TAS revealed that O-Ni2/TiO2 exhibits the longest average carrier relaxation lifetime (54.3 ± 2.6 ps), confirming effective suppression of charge recombination. XAFS (XANES and EXAFS) analyses verified that Ni atoms are coordinated to five oxygen atoms, forming Ni-O-Ni dimers. Contact angle measurements and DFT calculations demonstrated a stronger glycerol affinity (adsorption energy of -0.82 eV) and a reduced energy barrier for the rate-determining step (0.88 eV). Finally, in situ DRIFTS spectroscopy detected key intermediates (*CHO, *COOH, *CO), validating the proposed reaction pathway. Beyond syngas (H2 and CO), the optimized catalyst also generated other valuable oxidized intermediates in both liquid and gas phases. 1H NMR and HPLC analyses identified formaldehyde (HCHO), formic acid (HCOOH), and ethylene glycol as major liquid products, with production rates of 581.2, 866.7, and 658.4 µmol g-1 h-1, respectively. These compounds are key intermediates in the oxidation pathway leading to CO and CO2, as confirmed by in situ DRIFTS analyses. Additionally, CO2 was detected in the gas phase (113.5 µmol g-1 h-1) but was maintained at levels consistent with high syngas selectivity (96.1%). The catalyst achieved remarkably high H2 and CO production rates of 2,542.6 and 361.7 µmol g-1 h-1, respectively. The study further demonstrated the versatility of O-Ni2/TiO2 by testing a variety of substrates, including C1 molecules (methanol, formaldehyde, formic acid) and polyols C4-C6 (erythritol, xylitol, sorbitol), all of which yielded H2 and CO. The proposed mechanism involves rapid migration of photogenerated electrons and holes to the catalyst surface, where Ni-O-Ni sites adsorb and oxidize glycerol into intermediates such as formaldehyde and formic acid, which subsequently form CO and CO2. In parallel, protons are reduced by electrons to form H2.
A pilot-scale hydrogen production via solar-driven photocatalysis using NiO-TiO2 mixtures and glycerol as a sacrificial agent was reported by Villachica- Villachica-Llamosas et al.[176] The aim was to evaluate the photocatalytic efficiency of this system under real solar irradiation and natural water conditions. Experiments were conducted in a 25 L pilot-scale compound parabolic collector (CPC) photoreactor with a 2.10 m2 sun-exposed surface area, using both demineralized and natural water. The catalysts, commercial P25-TiO2 and NiO, were mixed mechanically in different ratios (1:10, 2:1, and 5:10) and concentrations (25-200 mg L-1). The best performance was achieved with NiO:TiO2 = 1:10 at 50 mg L-1, producing the highest hydrogen yield and a STH efficiency of 1.44 % in demineralized water, considering only the UV fraction of sunlight. The study also found that in natural water, hydrogen production was significantly lower (STH 0.4%) due to photocatalyst sedimentation promoted by ionic species. During the glycerol photoreforming process, H2 and CO2 evolution were monitored, and intermediate carboxylic acids were identified, confirming partial oxidation before complete mineralization. The H2/CO2 ratio initially exceeded the theoretical value (2.3) due to the formation of these intermediates but tended toward stoichiometry over time. Overall, the study demonstrates that commercial NiO and TiO2 nanoparticles can achieve hydrogen production efficiencies comparable to those of more complex, synthesized photocatalysts. However, ionic strength, catalyst sedimentation, and water quality are key limiting factors under real solar conditions. The authors conclude that for large-scale solar hydrogen generation, flow photoreactors with higher catalyst loadings may be required, especially when using saline wastewater as feedstock. Similarly, Toledo-Camacho et al. also demonstrated hydrogen generation from water-glycerol mixtures using Pd/TiO2 photocatalysts in a 25 L solar CPC pilot plant[177]. The photocatalyst containing 0.25 wt% Pd exhibited a satisfactory activity, achieving a quantum yield of approximately 2.4%.
Li and Zhang reported the use of an anatase TiO2 modified with Ba2+ ions to optimize the photocatalytic conversion of glycerol and nitrate into glycine[178]. The incorporation of Ba2+ was found to be crucial, as it enhances charge separation, prolongs carrier lifetimes, and increases the surface density of hydroxyl groups on the catalyst. The reactions were conducted under UV irradiation (365 nm) at 50 °C, in a sealed argon atmosphere. The proposed mechanism involved a cascade of coupled reactions: glycerol undergoes photoreforming to produce glycol, while nitrate is reduced to ammonia (NH3). The key step is the coupling of glycol with NH3 to generate glycine. Simultaneously, nitrate acts as a nitrogen source and moderates overoxidation by scavenging hydroxyl radicals (•OH) to form •NO3, thereby enhancing reaction selectivity. Radical species such as •CHOHCH2OH and intermediates like glycol and methanol were detected using EPR experiments. The glycine production rate reached 765 µmol·g-1·h-1, with a 5.3 % yield and nearly 100 % glycerol conversion. The Ba2+-TiO2 catalyst outperformed other tested materials, including pure TiO2 and modified with Ag, Cu, Mg, Ca, and Sr. The system also demonstrated substrate versatility, successfully converting various polyols (glycerol, glycol, erythritol, xylitol, sorbitol), sugars (glucose, fructose, xylose), and even raw biomass (wood sawdust) hydrolysis, showing potential for glycine production from renewable feedstocks. Comparisons highlighted the essential roles of nitrate and Ba2+-TiO2, since glycerol alone with pure TiO2 showed poor selectivity, and replacing nitrate with NH4+ or NH3 was ineffective.
Kobayashi developed a cascade photoredox system designed to integrate photocatalytic and redox mechanisms in a coordinated manner, combining homogeneous and heterogeneous catalysts to improve substrate contact and enhance charge separation efficiency[173]. This approach primarily aims to overcome limitations related to the restricted interaction between solid catalysts and complex organic substrates in biomass, particularly those with low solubility, such as cellulose. The system, termed the Photoredox Cascade Catalyst (PRCC), consists of TiO2 nanoparticles loaded with platinum and functionalized with two polypyridine-type ruthenium dyes, forming a dual-dye-sensitized photocatalyst (DDSP). The DDSP absorbs blue light (λ ≈ 460 nm), generating electrons that reduce protons to hydrogen, while simultaneously oxidizing the homogeneous catalyst 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO). TEMPO acts as a redox mediator, being oxidized to TEMPO+ by DDSP and subsequently regenerated through oxidation of organic substrates such as glycerol or cellulose. Additionally, N-methylimidazole is incorporated to facilitate proton transfer, stabilizing the photocatalytic process and promoting efficient charge separation. The functional integration of DDSP, TEMPO, and the acid-base mediator creates a closed catalytic circuit that partially mimics the electron transport chain of natural photosynthesis. Experimental results showed high hydrogen production rates, reaching 2.7 mmol g-1·h-1 for glycerol photoreforming and 1.6 mmol g-1·h-1 for cellulose, the latter particularly notable given the substrate’s low solubility. The system also exhibited stability and reproducibility over multiple reaction cycles. Other substrates were evaluated, including 2-propanol and glucose. For 2-propanol, H2 production was significantly lower, about 0.7 mmol g-1·h-1, consistent with TEMPO’s selectivity for primary alcohols. In contrast, glucose outperformed glycerol, producing 1.3 times more H2 (3,430 μmol g-1·h-1), attributed to its greater reducing capability and the formation of carboxylate groups via primary alcohol oxidation. This study highlights the potential of hybrid photocatalytic systems for highly efficient and selective solar-driven hydrogen production, with promising applications in biomass-based fuel generation and sustainable chemical platforms.
PHOTOREFORMING OF OTHER BIOMASS DERIVATIVES
As it becomes clear from some of the above-cited examples, the photoreforming studies on glycerol can be readily extended to other polyols typically found on biomass residues. They contain predominantly lignocellulosic materials, whose main structural constituents are carbohydrates, cellulose and hemicellulose, and the aromatic polymer lignin[179]. Moreover, such residues are carbon-neutral sources of electrons for photocatalytic reactions, acting simultaneously as a scavenger for photogenerated holes to support hydrogen evolution, while converting renewable feedstocks into value-added chemicals[180,181]. Nevertheless, despite these advantages, the efficiency of current systems remains far from practical application. Major challenges stem from the structural complexity, poor solubility, and the recalcitrant nature of lignocellulosic biomass, which hinder its interaction and electron transfer with heterogeneous photocatalysts. Moreover, the intrinsic limitations of conventional photocatalysts, such as narrow light absorption and rapid charge recombination, call for advanced material design[179]. Therefore, the pursuit of more efficient and sustainable photocatalytic routes for biomass valorization has intensified in recent years, with particular attention to innovations involving TiO2-based composite materials.
Liu et al. report a composite of anatase-phase titanium dioxide (TiO2) with carbon nanosheets (a-TiO2@MC) applied for the photoreforming of xylose[182]. This approach addresses classic limitations of pure TiO2, such as its wide bandgap and slow electron-transfer dynamics. The developed method involves hybridizing TiO2 with carbon nanosheets derived from chitosan biochar and sodium carboxymethylcellulose, resulting in a reduced bandgap (2.55 eV) and notable improvements in charge separation and transfer, evidenced by techniques such as ultraviolet photoelectron spectroscopy (UPS) and Mott-Schottky plots. Xylose, a sugar derived from lignocellulosic biomass, was used as the substrate for selective conversion into xylonic acid and lactic acid, both high-value products. The proposed mechanism was based on a selective oxidation of xylose, with holes (h+) acting as the primary oxidizing agents alongside hydroxyl radicals (•OH) and singlet oxygen
Enhancing the efficiency of photocatalysts for the conversion of solid biomass, such as cellulose, has relied on both support engineering and the optimization of active metal dispersion. Therefore, Lan et al. aimed to elucidate the impact of TiO2 composition (pure anatase vs. anatase/rutile mixture) and platinum loading on the performance of Pt/TiO2 catalysts for hydrogen production via cellulose photoreforming[33]. The photocatalysts were prepared by the impregnation method and tested in aqueous photoreforming under UV-A irradiation at 40 °C, with Pt loadings ranging from 0.16% to 1.00%. The authors reported that the TiO2 support significantly influences the structure of Pt species, especially at low loadings (0.16%), promoting the formation of sub-nanometric particles and enhancing charge separation. This results in higher hydrogen production rates, particularly for mixed-phase TiO2 (80% anatase, 20% rutile). Conversely, increasing the Pt loading leads to the growth of metallic particles (> 1-2 nm), promoting charge recombination and a marked decrease in catalytic activity for both supports. Mechanistic studies revealed that H2 is generated via a formic acid-driven pathway, which was primarily formed during cellulose degradation. More recently, the group also conducted an in-depth mechanistic study of glucose photoreforming for hydrogen production, employing a combined approach of product analysis (HPLC, GC) and in situ spectroscopic techniques, such as attenuated total reflectance infrared spectroscopy (ATR-IR) and isotopic labeling, to compare the activity of TiO2 and Pt/m-TiO2 photocatalysts[183]. The results confirmed that glucose oxidation occurs predominantly via an α-cleavage mechanism, leading sequentially to the formation of arabinose (a C5 sugar) and formic acid. The platinum-modified catalyst (Pt/m-TiO2) exhibited an activity 13.4 times higher than that of pure TiO2, attributed to its superior efficiency in separating photogenerated electron-hole pairs. Another contribution of the work was the identification of the central role of formic acid, elucidated by ATR-IR analysis. This intermediate preferentially adsorbs on the catalyst surface, and its subsequent oxidation at Pt-TiO2 interfaces is the rate-determining step. Additionally, isotopic studies revealed that at low glucose concentrations, the protons for H2 generation are predominantly derived from water molecules. Therefore, this study highlights the system’s efficiency dependency, not only on the degradation of the initial substrate, but critically on the catalyst’s ability to oxidize key intermediates adsorbed on its surface.
Another strategy to enhance the efficiency of sugar photoreforming is the morphological engineering of TiO2-based catalysts, particularly by improving mass diffusion and catalytic accessibility[181]. Shi et al. report the development of three-dimensional hierarchical TiO2 microspheres (THM), obtained via a template-free hydrothermal route, aimed at optimizing the photocatalytic conversion of glucose into hydrogen and high-value compounds such as arabinose and formic acid[61]. The study explores the influence of synthesis parameters, such as TiCl4 concentration, reaction time, and temperature, on the growth mechanism and microsphere structure, elucidated as a “nucleation-dissolution-recrystallization-assembly” process. The resulting THMs exhibit a “sea urchin” morphology, an average diameter of approximately 5 μm, a high specific surface area, and the presence of Ov, which significantly reduce the optical bandgap (2.92 eV). The presence and concentration of Ov were quantified by EPR, showing a direct correlation between defect density, enhanced electron-hole separation, and improved photocatalytic efficiency. Photocatalytic tests were performed with Pt as a cocatalyst, in a H2O/MeCN solvent mixture, and demonstrated an H2 production rate of 9.44 mmol g cat-1 h-1, glucose conversion of 86%, arabinose selectivity of 11%, and formic acid selectivity of 47%. Modulation of reaction conditions, particularly the use of the H2O/MeCN solvent and pH adjustment with Na2CO3, was crucial to favor glucose adsorption and stabilize reactive intermediates, directly contributing to the observed selectivity. These results reflected in substantially superior photocatalytic performance compared to commercial TiO2, showing up to a 135-fold increase in H2 production. The reaction mechanism involves glucose oxidation mainly via ROS (•O2- and •OH), promoting α C1-C2 cleavage and Ruff degradation, which favors the formation of highly selective products. The study emphasizes that the 3D hierarchical architecture, combined with structural defects (Ov, Ti3+), not only enhances light absorption and charge separation but also facilitates glucose accessibility to active sites, making the photocatalytic process more efficient, sustainable, and applicable under mild operating conditions.
Interestingly, the introduction of Ov combined with thermal and photonic energy was studied by Wu et al., who performed the thermo-photocatalytic reforming of lignin using Pt/TiO2 nanosheets with varying concentrations of Ov[67]. Through controlled synthesis and advanced characterizations, including DFT calculations, the authors investigated how Ov modulate the material’s electronic structure to simultaneously produce H2 and value-added chemicals, such as aromatic aldehydes. It was demonstrated that Ov promote electron delocalization, shifting the Pt d-band center to lower energy levels. This modulation results in charge redistribution, with TiO2 becoming electron-deficient and Pt electron-enriched, which facilitates charge separation and the desorption of reactive intermediates. Consequently, the combination of thermo-photocatalytic catalysis (at 473 K) with defect engineering resulted in a remarkable H2 production rate close to 1 mmol g-1 h-1 from lignin, an 18.2-fold increase compared to the purely photocatalytic process, along with high selectivity toward value-added aromatic aldehydes. The synergy between energy sources was crucial for enhancing performance. Thermal energy accelerates the kinetics of radical formation (•OH, h+, e-) and mass transfer, promoting the selective cleavage of C-O bonds in lignin, while photonic energy initiates the process. The study concludes that oxygen vacancy engineering creates a bifunctional catalyst, optimizing oxidation on TiO2 and reduction on Pt, whereas the thermo-photocatalytic approach overcomes kinetic barriers, establishing an effective strategy for lignin valorization into H2 and sustainable chemicals.
The combination of hierarchical architectures with the optical properties of metallic nanoparticles was also explored by Zhao et al. Three-dimensional ordered macroporous titanium dioxide (3DOM TiO2) was functionalized with gold nanoparticles (Au) and applied for glucose photoreforming, with coproduction of arabinose and gaseous fuels (H2, CH4, CO) in pure aqueous medium[69]. The 3DOM TiO2-Au composite exhibited significantly superior photocatalytic performance, achieving 37% glucose conversion and 0.2 g/L arabinose production, corresponding to increases of 2.8 and 10 times, respectively, compared to pure TiO2. The enhancement in activity was attributed to the synergy between the 3DOM structure, which facilitates mass diffusion and optimizes light harvesting, and the Au nanoparticles, which promote more efficient charge separation and intensify light absorption via the localized surface plasmon resonance (LSPR) effect. The proposed reaction mechanism involves α-cleavage of the C1-C2 bond in the glucose molecule, resulting in the selective formation of arabinose and formic acid. The latter acts as an intermediate that is subsequently decomposed to generate the gaseous fuels (H2, CH4, CO). Zhong et al. showed the performance of a ternary TiO2-Au-CdS photocatalyst featuring a hierarchical 3DOM architecture[184]. It achieves a high hydrogen production rate from glucose (645.1 μmol h-1 g-1), which decreases with increasing substrate complexity, from cellobiose (273.9 μmol h-1 g-1) to dissolving pulp (79.7 μmol h-1 g-1). The generation of CO and CH4 further confirmed the partial conversion of biomass during photoreforming.
Moreover, the complex relationship between photocatalyst’s properties and its activity in biomass conversion remains a field of intense investigation. For instance, Umair et al. present a comprehensive comparative study on the activity of modified TiO2 photocatalysts (with Pt, Cu2O, and Nb) for the photoreforming of glucose and fructose under simulated sunlight and ambient temperature[34]. The main goal of the study was to correlate photoactivity with the structural and surface characteristics of the materials, aiming to elucidate the factors governing efficiency in H2 production and selective substrate oxidation. To achieve this, the authors investigated different TiO2 polymorphs (anatase, rutile, and brookite) modified via Pt deposition, Nb doping, and coupling with Cu2O. Photocatalytic tests revealed that activity depends on a complex interplay of factors. While pure TiO2 was inactive for H2 production, the modified photocatalysts exhibited significantly higher rates compared to commercial benchmarks. Cu2O modification emerged as a more economical alternative to platinum for this purpose. Nb doping, although improving charge separation, resulted in lower H2 production than Pt and Cu2O. Additionally, the study illustrated platinum’s multifaceted role. Beyond its well-established function as an electron sink that minimizes charge recombination (confirmed by EPR analyses), Diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) and TPD analyses revealed that Pt also significantly modifies the acid-base properties of the TiO2 surface. Although no direct or monotonic correlation was established, this surface modulation affects substrate interactions and, consequently, reaction selectivity, with home-prepared Pt (Pt-HP) standing out for partial sugar oxidation. Therefore, this work shows that optimizing photocatalysts for biomass valorization relies on a delicate balance between crystal phase, surface properties, and charge separation efficiency, highlighting the unusual and significant role of platinum in modulating surface acidity. Interestingly, Umair et al. reported that the combined presence of Pt and Nb2O5 on TiO2 generated strong basic sites[34]. The photoreforming of glucose and fructose was examined using TiO2 combined with 0.5 wt% Pt and 4 wt.% of Nb2O5, in which Pt improved charge separation and introduced additional basic sites, while niobium oxide reduced electron-hole recombination and modified surface acidity. Together, Pt and Nb2O5 created an optimal balance of acid-base properties and electronic structure, facilitating efficient partial oxidation of sugars and hydrogen evolution.
Similarly to other photocatalytic processes, the use of non-noble metals for biomass photoreforming is a highly explored approach aiming at reduced implementation costs[185-187]. Belda-Marco et al. present an investigation on hydrogen and organic acid production from cellulose applying commercial TiO2 (P25) modified with bimetallic cocatalysts composed of copper and nickel[188]. The study focused on optimizing photocatalyst preparation by evaluating the impact of two main variables: the total metal loading (1 and 5wt.%) and the sequence of metal deposition (simultaneous or sequential). Under UV light irradiation
Eqi et al. reported the synergistic effect of bimetallic Ni-Au nanoparticles for glucose photoreforming over TiO2[68]. Under light irradiation, electrons are transferred from TiO2 to the Au and Ni nanoparticles, which have high work functions, facilitating charge separation for photocatalysis. The presence of Au nanoparticles also enhances visible-light absorption through the LSPR effect. During the oxidation process, glucose is primarily converted into gluconic acid via the action of superoxide radicals (•O2-). Subsequently, gluconic acid undergoes decarboxylation through C1-C2 bond cleavage. This step is specifically promoted by the Ni nanoparticles, which are effective in breaking carbon-carbon bonds, resulting in the selective formation of arabinose. Simultaneously, the electrons accumulated on the metallic nanoparticles reduce protons in the aqueous solution to produce gaseous hydrogen. The results demonstrated superior catalytic performance of the bimetallic material compared to monometallic catalysts and pure TiO2. The optimized catalyst, Ni0.05Au0.45/TiO2, exhibited the highest hydrogen production rate, reaching 64 mmol h-1 g-1. This value is almost 120 times higher than pure TiO2, 31 times higher than Ni/TiO2, and almost two times higher than Au/TiO2. In addition, glucose conversion reached 95%, with an arabinose selectivity of 36%. The success of the process was attributed to the synergistic effect between Ni and Au, as well as the three-dimensional hierarchical structure of TiO2, which facilitates mass transport and accessibility of glucose to the active sites of the catalyst.
Moreover, the search for efficient and low-cost photocatalysts, free of noble metals, has driven the development of semiconductor heterojunctions. Zhao et al. developed an ultrathin n-p core-shell heterojunction nanostructure composed of TiO2 and NiO, synthesized via a one-pot hydrothermal method[70]. The main objective was to enhance the separation of photogenerated charges and, consequently, improve the efficiency of H2 production from the photocatalytic reforming of lignin. The optimized material, containing 3.25% NiO, exhibited a H2 production rate of 23.5 mmol h-1 g-1 in a water-methanol solution, significantly outperforming pure TiO2. When applied to lignin photocatalytic reforming in an alkaline medium, the catalyst was able to coproduce H2 (0.45 mmol h-1 g-1) and methane (CH4), in addition to converting the biomass into long-chain fatty acids, such as palmitic and stearic acids. This superior performance was attributed to the synergy between high crystallinity, the core-shell architecture, and, critically, the formation of an internal electric field at the n-p heterojunction interface, which efficiently suppresses charge carrier recombination. The study concludes that the ultrathin TiO2-NiO n-p heterojunction represents an effective strategy for solar-to-chemical energy conversion without the need for noble-metal cocatalysts.
CONCLUSIONS AND PERSPECTIVES
This review has shown that significant progress has been achieved in the field of TiO2-based photoreforming of alcohols and biomass-derived substrates as a viable pathway for low-carbon hydrogen production. Deep understanding of fundamental aspects concerning the selectivity of the oxidation reactions and the overall mechanism of the consecutive reactions following initial light-induced charge transfer steps has been described for several alcohols based on advanced spectroscopic techniques. Photoreforming of model alcohols such as methanol and ethanol has provided valuable insights into reaction mechanisms, charge carrier dynamics, and the identification of intermediate species. Such studies have guided the development of new photocatalyst systems with improved efficiency compared with pristine TiO2. This includes doping, heterojunction formation, and the use of co-catalysts, which enable enhanced charge separation, visible-light harvesting, and improved hydrogen evolution rates, although few reports deal with the challenge of tuning the photocatalytic oxidation selectivity.
The extension of photoreforming studies from methanol to more complex substrates, such as C2+ alcohols, glycerol, and other biomass derivatives, reveals the increasing challenges associated with molecular complexity. These include more intricate reaction pathways, formation of a wider range of intermediates, and a greater dependence on catalyst surface properties. Future research should focus on surface engineering to enhance the formation/stabilization of key radical intermediates to drive selectivity, as well as a deeper understanding of the role of physicochemical conditions (pH, ionic strength, etc.) on the overall efficiency and selectivity of the process.
Such advances can facilitate the design of systems capable of operating on real and complex biomass-derived streams. A deeper understanding of reaction mechanisms, particularly for more complex substrates, will be crucial for guiding photocatalyst design and improving selectivity toward desirable products. Moreover, integrating photoreforming processes with waste valorization strategies and renewable energy inputs could further enhance their sustainability and practical relevance. Overall, TiO2-based photoreforming of alcohols represents a promising and versatile approach for sustainable hydrogen production. Continued efforts in photocatalyst development, mechanistic understanding, and process integration will be key to advancing this technology toward practical and scalable applications in a future low-carbon energy landscape.
DECLARATIONS
Authors’ contributions
Writing original draft: Ribeiro, R. N.; Amaral, K. R. M.
Writing review & editing: Desordi, J. C.; Augusto, K. K. L.
Writing review & editing, validation: Gonçalves, P. J.; Alonso, C. G.; Pan, J. H.; Ribeiro, C.; Lopes, O. F.
Writing review & editing, Validation, supervision: Bahnemann, D. W.; Nunes, B. N.
Writing review & editing, validation, supervision, conceptualization and funding acquisition: Patrocinio, A. O. T.
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
AI assistance was used for language review and polishing of the graphical abstract (Gemini version 3.1). The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship
This work was supported by Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG, APQ-01044-21, APQ-02473-23, APQ-00366-24 and APQ-06609-24), Conselho Nacional de Desenvolvimento Científico e Tecnológico (BRICS CNPq 440177/2022-4, Universal CNPq 405627/2023-8), Fundação de Amparo à Pesquisa do Estado de Goias (FAPEG, 4/2023 - FAPEG/UFG/FUNAPE) and MCTI/FINEP/FNDCT (Grant No. 0966/24 #01.25.0086.00).
Conflicts of interest
Pan, J. H. and Ribeiro, R. N. are Guest Editors of the Special Topic "Solar Energy Conversion and Storage” of the journal Energy Materials. but were not involved in any steps of editorial processing, notably including reviewers’ selection, manuscript handling, or decision-making, while the other authors have declared that they have no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. International Energy Agency. Global Hydrogen Review 2024. Available from: https://www.iea.org/reports/global-hydrogen-review-2024 [Last accessed on 24 Jun 2026].
4. Bhattacharjee, S.; Linley, S.; Reisner, E. Solar reforming as an emerging technology for circular chemical industries. Nat. Rev. Chem. 2024, 8, 87-105.
5. Yue, S.; Zhao, Z.; Zhang, T.; Li, F.; Wang, P.; Zhan, S. Photoreforming of plastic waste to sustainable fuels and chemicals: waste to energy. Environ. Sci. Technol. 2024, 58, 22865-79.
6. Yao, Y.; Zhang, J.; Shi, L.; Wang, S.; Duan, X. Solar fuels production from plastics and biomass photoreforming. Energy. Fuels. 2025, 39, 14455-82.
7. Toe, C. Y.; Tsounis, C.; Zhang, J.; et al. Advancing photoreforming of organics: highlights on photocatalyst and system designs for selective oxidation reactions. Energy. Environ. Sci. 2021, 14, 1140-75.
8. Ashraf, M.; Ullah, N.; Khan, I.; Tremel, W.; Ahmad, S.; Tahir, M. N. Photoreforming of waste polymers for sustainable hydrogen fuel and chemicals feedstock: waste to energy. Chem. Rev. 2023, 123, 4443-509.
9. Atilano-Camino, M. M.; García-González, A.; Olivo-Alanís, D. S.; García-Reyes, R. B. Photoreforming of fermentation byproducts by TiO2 and Pt/TiO2 to enhance hydrogen production: insight into a real perspective. J. Environ. Chem. Eng. 2024, 12, 112017.
10. Karimi Estahbanati, M.; Babin, A.; Feilizadeh, M.; Nayernia, Z.; Mahinpey, N.; Iliuta, M. C. Photocatalytic conversion of alcohols to hydrogen and carbon-containing products: a cleaner alcohol valorization approach. J. Clean. Prod. 2021, 318, 128546.
11. Liu, Y.; Miao, J.; Zhang, W.; Wei, A.; Wang, J. In-situ photodeposition of co-catalyst Ni2P on CdS for photocatalytic conversion of ethanol for synergistic hydrogen production. J. Fuel. Chem. Technol. 2024, 52, 1629-40.
12. Sanwald, K. E.; Berto, T. F.; Eisenreich, W.; Gutiérrez, O. Y.; Lercher, J. A. Catalytic routes and oxidation mechanisms in photoreforming of polyols. J. Catal. 2016, 344, 806-16.
13. Pecoraro, C. M.; Di Franco, F.; Bellardita, M.; Loddo, V.; Santamaria, M. Enhancing H2 production rate in PGM-free photoelectrochemical cells by glycerol photo-oxidation. Int. J. Hydrogen. Energy. 2024, 49, 322-36.
14. Wahab, A.; Idriss, H. Study of the photocatalytic reforming and oxidation of Glycerol over Ag-Pd/TiO2. Int. J. Hydrogen. Energy. 2024, 52, 159-71.
15. Hai, H. T. N.; Nguyen, T. T.; Nishibori, M.; Ishihara, T.; Edalati, K. Photoreforming of plastic waste into valuable products and hydrogen using a high-entropy oxynitride with distorted atomic-scale structure. Appl. Catal. B. Environ. Energy. 2025, 365, 124968.
16. Tam Nguyen, T.; Edalati, K. Efficient photoreforming of plastic waste using a high-entropy oxide catalyst. J. Catal. 2024, 440, 115808.
17. Zhao, H.; Yu, X.; Hu, G.; et al. Confined synthesis of BiVO4 nanodot and ZnO cluster co-decorated 3DOM TiO2 for formic acid production from the xylan-based hemicellulose photorefinery. Green. Chem. 2021, 23, 8124-30.
18. Gaya, U. I. Heterogeneous photocatalysis using inorganic semiconductor solids. Dordrecht: Springer Netherlands; 2014.
19. Shelake, S. P.; Sutar, D. N.; Abraham, B. M.; Banerjee, T.; Sainath, A. V. S.; Pal, U. Emerging photoreforming process to hydrogen production: a future energy. Adv. Funct. Mater. 2024, 34, 2403795.
20. Alsalka, Y.; Al-Madanat, O.; Hakki, A. TiO2-based photocatalytic hydrogen production: how to transfer it to an applicable approach? Appl. Catal. A. Gen. 2023, 662, 119287.
21. Du, M.; Zhang, Y.; Kang, S.; et al. Trash to treasure: photoreforming of plastic waste into commodity chemicals and hydrogen over MoS2-tipped CdS nanorods. ACS. Catal. 2022, 12, 12823-32.
22. Kumai, Y.; Nagata, M. Hydrogen production by photoreforming of organic waste using Cd-ZnS solid solution photocatalyst via MOF precursor. Meet. Abstr. 2024, MA2024-02, 4006.
23. Liang, E.; Cheng, K.; Liu, X.; et al. Zinc cadmium sulphide-based photoreforming of biomass-based monosaccharides to lactic acid and efficient hydrogen production. J. Colloid. Interface. Sci. 2025, 683, 432-45.
24. Song, J.; Rao, C.; Zhang, Z.; Yang, X.; Zhang, Y. CdS quantum dots with sulfur defects for photoreforming plastics into valuable chemicals coupled with hydrogen production. Mol. Catal. 2025, 579, 115049.
25. Yan, J.; Sun, D.; Huang, J. Synergistic poly(lactic acid) photoreforming and H2 generation over ternary NixCo1-xP/reduced graphene oxide/g-C3N4 composite. Chemosphere 2022, 286, 131905.
26. Mohanty, C.; Samal, A.; Kumar, J.; Behera, A. K.; Das, R.; Das, N. Design and first-principles investigation of step-scheme (S-scheme) g-C3N4/α-MnO2 nanojunction for polystyrene photoreforming into value-added chemicals and hydrogen. Int. J. Hydrogen. Energy. 2025, 120, 628-41.
27. Mohanty, C.; Samal, A.; Das, N. Concurrent photoreforming of polyethylene into commercial chemicals and hydrogen generation utilizing g-C3N4/Co3O4 Z-scheme heterostructure: a waste-to-wealth concept. Int. J. Hydrogen. Energy. 2024, 61, 84-93.
28. Munusamy, T. D.; Chin, S. Y.; Tarek, M.; Khan, M. R. Sustainable hydrogen production by CdO/exfoliated g-C3N4 via photoreforming of formaldehyde containing wastewater. Int. J. Hydrogen. Energy. 2021, 46, 30988-99.
29. Sulaiman, N. H. M.; Wang, S.; Yue, H.; Wei, J.; Schmuki, P.; Zhou, X. Hydrogen evolution using alloyed AuPd/TiO2 hollow spheres by photoreforming of polyethylene terephthalate waste. J. Mater. Chem. A. 2025, 13, 12545-52.
30. Umair, M.; Ruiz-Aguirre, A.; Berruti, I.; et al. Biomass derivatives photoreforming in pilot plant scale to obtain H2 under green conditions by using ball milling Cu2O-TiO2 P25 photocatalysts. Chem. Eng. J. 2025, 504, 158585.
31. Hippargi, G.; Anjankar, S.; Krupadam, R. J.; Rayalu, S. S. Simultaneous wastewater treatment and generation of blended fuel methane and hydrogen using Au-Pt/TiO2 photo-reforming catalytic material. Fuel 2021, 291, 120113.
32. Alshehri, A.; Narasimharao, K. PtOx-TiO2 anatase nanomaterials for photocatalytic reformation of methanol to hydrogen: effect of TiO2 morphology. J. Mater. Res. Technol. 2020, 9, 14907-21.
33. Lan, L.; Daly, H.; Jiao, Y.; Yan, Y.; Hardacre, C.; Fan, X. Comparative study of the effect of TiO2 support composition and Pt loading on the performance of Pt/TiO2 photocatalysts for catalytic photoreforming of cellulose. Int. J. Hydrogen. Energy. 2021, 46, 31054-66.
34. Umair, M.; Loddo, V.; Palmisano, L.; et al. Investigating the activity of modified TiO2 photocatalysts used for the photoreforming of biomass derivatives. J. Photochem. Photobiol. A. Chem. 2024, 453, 115654.
35. Iervolino, G.; Vaiano, V.; Sannino, D.; Rizzo, L.; Palma, V. Enhanced photocatalytic hydrogen production from glucose aqueous matrices on Ru-doped LaFeO3. Appl. Catal. B. Environ. 2017, 207, 182-94.
36. Wei, Z.; Liu, J.; Fang, W.; Qin, Z.; Jiang, Z.; Shangguan, W. A visible-light driven novel layered perovskite oxyhalide Bi4MO8X (M = Nb, Ta; X = Cl, Br) constructed using BiOX (X = Cl, Br) for enhanced photocatalytic hydrogen evolution. Catal. Sci. Technol. 2018, 8, 3774-84.
37. López-Vásquez, A.; Delgado-Niño, P.; Salas-Siado, D. Photocatalytic hydrogen production by strontium titanate-based perovskite doped europium (Sr0.97Eu0.02Zr0.1Ti0.9O3). Environ. Sci. Pollut. Res. 2018, 26, 4202-14.
38. Umair, M.; Palmisano, L.; Bellardita, M. Comparison between the efficiency of bare and Pt-loaded TiO2 and ZnIn2S4 for H2 production in the presence of triethanolamine, methanol, furfuryl alcohol used as sacrificial agents. J. Mol. Struct. 2025, 1340, 142567.
39. Eddy, D. R.; Permana, M. D.; Sakti, L. K.; et al. Heterophase Polymorph of TiO2 (Anatase, Rutile, Brookite, TiO2(B)) for efficient photocatalyst: fabrication and activity. Nanomaterials 2023, 13, 704.
40. Chang, L.; Yong, S.; Chai, S.; Putri, L.; Tan, L.; Mohamed, A. A review of methanol photoreforming: elucidating the mechanisms, photocatalysts and recent advancement strategies. Mater. Today. Chem. 2023, 27, 101334.
41. Barba-Nieto, I.; Caudillo-Flores, U.; Gómez-Cerezo, M. N.; Kubacka, A.; Fernández-García, M. Boosting Pt/TiO2 hydrogen photoproduction through Zr doping of the anatase structure: a spectroscopic and mechanistic study. Chem. Eng. J. 2020, 398, 125665.
42. Musso, M.; Veiga, S.; De León, M. A.; Quevedo, A.; Bussi, J. Hydrogen production from pure and crude glycerol photoreforming using platinum supported on TiO2 and N-TiO2. Mater. Lett. 2024, 357, 135714.
43. Yang, G.; Yan, Z.; Xiao, T. Preparation and characterization of SnO2/ZnO/TiO2 composite semiconductor with enhanced photocatalytic activity. Appl. Surf. Sci. 2012, 258, 8704-12.
44. Wang, W.; Ye, Y.; Li, G.; et al. High-efficiency photocathodic protection performance of novel MnIn2S4/TiO2 n-n heterojunction films for Q235 carbon steel in chloride-containing simulated concrete pore solution. J. Alloys. Compd. 2023, 941, 168957.
45. Kumari, M. A.; Devi, L. G.; Maia, G.; Chen, T.; Al-zaqri, N.; Ali, M. A. Mechanochemical synthesis of ternary heterojunctions TiO2(A)/TiO2(R)/ZnO and TiO2(A)/TiO2(R)/SnO2 for effective charge separation in semiconductor photocatalysis: a comparative study. Environ. Res. 2022, 203, 111841.
46. Ding, M.; Jia, M.; Wang, J. Insights into performance and mechanism of improving methanol photoreforming on the ternary catalysts with p-n junction. Int. J. Hydrogen. Energy. 2025, 98, 67-77.
47. Drobná, H.; Meinhardová, V.; Dubnová, L.; et al. Partially reduced Ni-NiO-TiO2 photocatalysts for hydrogen production from methanol-water solution. Catalysts 2023, 13, 293.
48. Caudillo-Flores, U.; Barba-Nieto, I.; Muñoz-Batista, M. J.; Motta, Meira. D.; Fernández-García, M.; Kubacka, A. Thermo-photo production of hydrogen using ternary Pt-CeO2-TiO2 catalysts: a spectroscopic and mechanistic study. Chem. Eng. J. 2021, 425, 130641.
49. Li, W. L.; Zhang, Q. Q.; Dong, L.; Zhang, W. J.; Lin, L. L.; Chen, Z. P. Selective photosynthesis of imines from biomass-derived aldehydes over Ni/TiO2. Rare. Metals. 2023, 43, 1116-24.
50. Afrin, M. F.; Furukawa, M.; Tateishi, I.; Katsumata, H.; Uzzaman, M.; Kaneco, S. Enhanced photocatalytic hydrogen generation from methanol solutions via in situ Ni/Pt Co-deposition on TiO2. J. Compos. Sci. 2025, 9, 68.
51. Jakimińska, A.; Spilarewicz, K.; Macyk, W. Phototransformations of TiO2/Ag2O composites and their influence on photocatalytic water splitting accompanied by methanol photoreforming. Nanoscale. Adv. 2023, 5, 1926-35.
52. Platero, F.; López-Martín, A.; Caballero, A.; Rojas, T. C.; Nolan, M.; Colón, G. Overcoming Pd-TiO2 deactivation during H2 production from photoreforming using Cu@Pd nanoparticles supported on TiO2. ACS. Appl. Nano. Mater. 2021, 4, 3204-19.
53. Silvaino, P. F.; Ferreira, J. C.; Carminati, S. A.; Vaz, J. M.; Spinacé, E. V. Vinasse photoreforming for hydrogen production using Pt/TiO2 as catalyst under UV irradiation. RSC. Adv. 2025, 15, 5795-800.
54. Piątkowska, A.; Janus, M.; Szymański, K.; Mozia, S. C-,N- and S-doped TiO2 Photocatalysts: a review. Catalysts 2021, 11, 144.
55. Rani, S.; Singh, S.; Pal, B. Core-shell structure of reduced graphene oxide@Ag-TiO2 for photocatalytic H2O splitting and CH3OH dehydrogenation under UV light irradiation. Int. J. Hydrogen. Energy. 2024, 49, 910-24.
56. Bu, E.; Chen, X.; López-Cartes, C.; et al. Effect of the TiO2-carbon interface on charge transfer and ethanol photo-reforming. Catal. Today. 2023, 422, 114220.
57. De Souza Niero, A. L.; Mozzaquatro Pasini, S.; Daguer, P. H.; et al. Structured macroporous Brown TiO2-SiC catalysts for ethanol-water photoreforming under visible light for hydrogen production. Int. J. Hydrogen. Energy. 2025, 140, 26-35.
58. Šalipur, H.; Fronczak, M.; Prašnikar, A.; et al. Metal doped TiO2 decorated carbon nanostructured materials as an emerging photocatalysts for solar fuels production. Catal. Today. 2024, 436, 114724.
59. Mansoor, S.; Hu, Z.; Zhang, Y.; et al. Simultaneous hydrogen production with photo reforming of lactic acid over MXene derived MoS2/TiO2/Ti3C2 nanowires. Chin. J. Catal. 2025, 71, 234-45.
60. Lang, M.; Klahn, M.; Strunk, J. Supported titanium oxide species as photocatalysts in 2-propanol oxidation: linking selectivity to structural and electronic properties. Appl. Surf. Sci. 2023, 611, 155623.
61. Shi, C.; Eqi, M.; Shi, J.; Huang, Z.; Qi, H. Constructing 3D hierarchical TiO2 microspheres with enhanced mass diffusion for efficient glucose photoreforming under modulated reaction conditions. J. Colloid. Interface. Sci. 2023, 650, 1736-48.
62. Al-madanat, O.; Curti, M.; Günnemann, C.; Alsalka, Y.; Dillert, R.; Bahnemann, D. W. TiO2 photocatalysis: impact of the platinum loading method on reductive and oxidative half-reactions. Catal. Today. 2021, 380, 3-15.
63. Chen, W.; Dong, Y.; Yadav, P.; Aughterson, R. D.; Sun-Waterhouse, D.; Waterhouse, G. I. Effect of alcohol sacrificial agent on the performance of Cu/TiO2 photocatalysts for UV-driven hydrogen production. Appl. Catal. A. Gen. 2020, 602, 117703.
64. Cwieka, K.; Bojarska, Z.; Czelej, K.; et al. Zero carbon footprint hydrogen generation by photoreforming of methanol over Cu/TiO2 nanocatalyst. Chem. Eng. J. 2023, 474, 145687.
65. Piedra-López, J.; Calzada, L. A.; Guerra-Blanco, P.; et al. Enhancing photocatalytic H2 production and dye degradation: comparative analysis of gold reduction techniques on Au/TiO2 nanocomposites. Catal. Today. 2024, 432, 114610.
66. Courtois, C.; Walenta, C. A.; Tschurl, M.; Heiz, U.; Friend, C. M. Regulating photochemical selectivity with temperature: isobutanol on TiO2(110). J. Am. Chem. Soc. 2020, 142, 13072-80.
67. Wu, W.; Liu, B.; Qiu, X.; et al. Modulating oxygen vacancy in Pt/TiO2 for thermo-photo reforming lignin and its derivatives to H2 and value-added product. ACS. Sustain. Chem. Eng. 2024, 12, 9027-36.
68. Eqi, M.; Shi, C.; Xie, J.; et al. Synergetic effect of Ni-Au bimetal nanoparticles on urchin-like TiO2 for hydrogen and arabinose co-production by glucose photoreforming. Adv. Compos. Hybrid. Mater. 2022, 6, 5.
69. Zhao, H.; Liu, P.; Wu, X.; et al. Plasmon enhanced glucose photoreforming for arabinose and gas fuel co-production over 3DOM TiO2-Au. App. Catal. B. Environ. 2021, 291, 120055.
70. Zhao, H.; Li, C.; Liu, L.; et al. n-p Heterojunction of TiO2-NiO core-shell structure for efficient hydrogen generation and lignin photoreforming. J. Colloid. Interface. Sci. 2021, 585, 694-704.
71. Ding, M.; Wang, J. Photocatalytic reforming of ethanol to produce hydrogen: effect of anatase doped with NiO on products and reaction pathways. Int. J. Hydrogen. Energy. 2024, 56, 92-101.
72. Pecoraro, C. M.; Bellardita, M.; Loddo, V.; Di Franco, F.; Palmisano, L.; Santamaria, M. A facile way to synthesize noble metal free TiO2 based catalysts for glycerol photoreforming. J. Ind. Eng. Chem. 2023, 118, 247-58.
73. Liu, R.; Yin, H.; Guo, P.; Liu, X.; Yin, Z. Photoreforming light alcohols for value-added resources: a mini review. Energy. Technol. 2024, 13, 2301708.
74. Walenta, C. A.; Tschurl, M.; Heiz, U. Introducing catalysis in photocatalysis: what can be understood from surface science studies of alcohol photoreforming on TiO2. J. Phys. Condens. Matter. 2019, 31, 473002.
75. Garcia-Muñoz, P.; Fresno, F. Oxidation of alcohols in photocatalytic hydrogen production: from sacrifice to valorization. Curr. Opin. Chem. Eng. 2025, 49, 101146.
76. Yao, Y.; Gao, X.; Li, Z.; Meng, X. Photocatalytic reforming for hydrogen evolution: a review. Catalysts 2020, 10, 335.
77. Zhu, B.; Dong, B.; Wang, F.; et al. Unraveling a bifunctional mechanism for methanol-to-formate electro-oxidation on nickel-based hydroxides. Nat. Commun. 2023, 14, 1686.
78. Miyake, M. An electrochemical study of the photocatalytic oxidation of methanol on rutile. J. Catal. 1979, 58, 22-7.
79. Kawai, T.; Sakata, T. Photocatalytic hydrogen production from liquid methanol and water. J. Chem. Soc. Chem. Commun. 1980, 694-5.
80. Kruczynski, L.; Gesser, H. D.; Turner, C. W.; Speers, E. A. Porous titania glass as a photocatalyst for hydrogen production from water. Nature 1981, 291, 399-401.
81. Kawai, M.; Naito, S.; Tamaru, K.; Kawai, T. The mechanism of photocatalytic hydrogen production from gaseous methanol and water: IR spectroscopic approach. Chem. Phys. Lett. 1983, 98, 377-80.
82. Lindan, P. J. D.; Harrison, N. M.; Gillan, M. J.; White, J. A. First-principles spin-polarized calculations on the reduced and reconstructed TiO2(110) surface. Phys. Rev. B. 1997, 55, 15919-27.
83. Chrétien, S.; Metiu, H. Electronic structure of partially reduced rutile TiO2(110) surface: where are the unpaired electrons located? J. Phys. Chem. C. 2011, 115, 4696-705.
84. Ichihara, F.; Pang, H.; Kako, T.; Bahnemann, D. W.; Ye, J. Photogenerated charge carrier dynamics on Pt-loaded SrTiO3 nanoparticles studied via transient-absorption spectroscopy. Nanoscale 2025, 17, 2567-76.
85. Ma, J.; Miao, T. J.; Tang, J. Charge carrier dynamics and reaction intermediates in heterogeneous photocatalysis by time-resolved spectroscopies. Chem. Soc. Rev. 2022, 51, 5777-94.
86. Qian, R.; Zong, H.; Schneider, J.; et al. Charge carrier trapping, recombination and transfer during TiO2 photocatalysis: an overview. Catal. Today. 2019, 335, 78-90.
87. Park, C.; Cho, K. R.; Fujitsuka, M.; et al. Time-resolved spectroelectrochemical observation of overlayer-induced charge carrier dynamics in water photooxidation. Angew. Chem. Int. Ed. 2025, 64, e202502805.
88. Miao, T. J.; Tang, J. Characterization of charge carrier behavior in photocatalysis using transient absorption spectroscopy. J. Chem. Phys. 2020, 152, 194201.
89. Lai, T. H.; Katsumata, K. I.; Hsu, Y. J. In situ charge carrier dynamics of semiconductor nanostructures for advanced photoelectrochemical and photocatalytic applications. Nanophotonics 2020, 10, 777-95.
90. Tamaki, Y.; Furube, A.; Murai, M.; Hara, K.; Katoh, R.; Tachiya, M. Dynamics of efficient electron-hole separation in TiO2 nanoparticles revealed by femtosecond transient absorption spectroscopy under the weak-excitation condition. Phys. Chem. Chem. Phys. 2007, 9, 1453-60.
91. Hoffmann, M. R.; Martin, S. T.; Choi, W.; Bahnemann, D. W. Environmental applications of semiconductor photocatalysis. Chem. Rev. 2002, 95, 69-96.
92. Kohtani, S.; Kawashima, A.; Miyabe, H. Reactivity of trapped and accumulated electrons in titanium dioxide photocatalysis. Catalysts 2017, 7, 303.
93. Schneider, J.; Matsuoka, M.; Takeuchi, M.; et al. Understanding TiO2 photocatalysis: mechanisms and materials. Chem. Rev. 2014, 114, 9919-86.
94. Tamaki, Y.; Furube, A.; Murai, M.; Hara, K.; Katoh, R.; Tachiya, M. Direct observation of reactive trapped holes in TiO2 undergoing photocatalytic oxidation of adsorbed alcohols: evaluation of the reaction rates and yields. J. Am. Chem. Soc. 2005, 128, 416-7.
95. Walle, L. E.; Borg, A.; Uvdal, P.; Sandell, A. Experimental evidence for mixed dissociative and molecular adsorption of water on a rutile TiO2(110) surface without oxygen vacancies. Phys. Rev. B. 2009, 80, 235436.
96. Muir, J. N.; Choi, Y.; Idriss, H. Computational study of ethanol adsorption and reaction over rutile TiO2(110) surfaces. Phys. Chem. Chem. Phys. 2012, 14, 11910.
97. Panayotov, D. A.; Burrows, S. P.; Morris, J. R. Photooxidation mechanism of methanol on rutile TiO2 nanoparticles. J. Phys. Chem. C. 2012, 116, 6623-35.
98. Hykaway, N.; Sears, W. M.; Morisaki, H.; Morrison, S. R. Current-doubling reactions on titanium dioxide photoanodes. J. Phys. Chem. 2002, 90, 6663-7.
99. Shen, M.; Henderson, M. A. Role of water in methanol photochemistry on rutile TiO2(110). J. Phys. Chem. C. 2012, 116, 18788-95.
100. Asmus, K. D.; Moeckel, H.; Henglein, A. Pulse radiolytic study of the site of hydroxyl radical attack on aliphatic alcohols in aqueous solution. J. Phys. Chem. 2002, 77, 1218-21.
101. Lilie, J.; Beck, G.; Henglein, A. Pulsradiolyse und Polarographie: Halbstufenpotentiale für die Oxydation und Reduktion von kurzlebigen organischen Radikalen an der Hg-Elektrode. Ber. Bunsenges. Phys. Chem. 2014, 75, 458-65.
102. Xia, S.; Wang, T.; Ren, Z.; Yang, X.; Guo, Q.; Zhou, C. Adsorption structure-activity correlation in the photocatalytic chemistry of methanol and water on TiO2(110). Acc. Chem. Res. 2024, 57, 3407-18.
103. Dong, S.; Hu, J.; Xia, S.; et al. Origin of the adsorption-state-dependent photoactivity of methanol on TiO2(110). ACS. Catal. 2021, 11, 2620-30.
104. Setvin, M.; Shi, X.; Hulva, J.; et al. Methanol on anatase TiO2 (101): mechanistic insights into photocatalysis. ACS. Catal. 2017, 7, 7081-91.
105. Liu, H.; Liu, M.; Nakamura, R.; Tachibana, Y. Primary photocatalytic water reduction and oxidation at an anatase TiO2 and Pt-TiO2 nanocrystalline electrode revealed by quantitative transient absorption studies. Appl. Catal. B. Environ. 2021, 296, 120226.
106. Patrocinio, A. O. T.; Schneider, J.; França, M. D.; et al. Charge carrier dynamics and photocatalytic behavior of TiO2 nanopowders submitted to hydrothermal or conventional heat treatment. RSC. Adv. 2015, 5, 70536-45.
107. Kandiel, T. A.; Ivanova, I.; Bahnemann, D. W. Long-term investigation of the photocatalytic hydrogen production on platinized TiO2: an isotopic study. Energy. Environ. Sci. 2014, 7, 1420.
108. Mandelbaum, P. A.; Regazzoni, A. E.; Blesa, M. A.; Bilmes, S. A. Photo-electro-oxidation of alcohols on titanium dioxide thin film electrodes. J. Phys. Chem. B. 1999, 103, 5505-11.
109. Ahmed, A. Y.; Kandiel, T. A.; Ivanova, I.; Bahnemann, D. Photocatalytic and photoelectrochemical oxidation mechanisms of methanol on TiO2 in aqueous solution. Appl. Surf. Sci. 2014, 319, 44-9.
110. Hao, Q.; Wang, Z.; Wang, T.; Ren, Z.; Zhou, C.; Yang, X. Role of Pt loading in the photocatalytic chemistry of methanol on rutile TiO2(110). ACS. Catal. 2018, 9, 286-94.
111. Walenta, C. A.; Courtois, C.; Kollmannsberger, S. L.; Eder, M.; Tschurl, M.; Heiz, U. Surface species in photocatalytic methanol reforming on Pt/TiO2(110): learning from surface science experiments for catalytically relevant conditions. ACS. Catal. 2020, 10, 4080-91.
112. Kollmannsberger, S. L.; Walenta, C. A.; Courtois, C.; Tschurl, M.; Heiz, U. Thermal control of selectivity in photocatalytic, water-free alcohol photoreforming. ACS. Catal. 2018, 8, 11076-84.
113. Kandiel, T. A.; Dillert, R.; Robben, L.; Bahnemann, D. W. Photonic efficiency and mechanism of photocatalytic molecular hydrogen production over platinized titanium dioxide from aqueous methanol solutions. Catal. Today. 2011, 161, 196-201.
114. Eder, M.; Tschurl, M.; Heiz, U. Toward a comprehensive understanding of photocatalysis: what systematic studies and alcohol surface chemistry on TiO2(110) have to offer for future developments. J. Phys. Chem. Lett. 2023, 14, 6193-201.
115. Courtois, C.; Eder, M.; Kollmannsberger, S. L.; Tschurl, M.; Walenta, C. A.; Heiz, U. Origin of poisoning in methanol photoreforming on TiO2(110): the importance of thermal back-reaction steps in photocatalysis. ACS. Catal. 2020, 10, 7747-52.
116. Wang, Z.; Henderson, M. A.; Lyubinetsky, I. Origin of coverage dependence in photoreactivity of carboxylate on TiO2(110): hindering by charged coadsorbed hydroxyls. ACS. Catal. 2015, 5, 6463-7.
117. Yamazaki, Y.; Toyonaga, T.; Doshita, N.; et al. Crystal facet engineering and hydrogen spillover-assisted synthesis of defective Pt/TiOx nanorods with enhanced visible light-driven photocatalytic activity. ACS. Appl. Mater. Interfaces. 2021, 14, 2291-300.
118. Hejazi, S. M. H.; Shahrezaei, M.; Błoński, P.; et al. Defect engineering over anisotropic brookite toward substrate-specific photo-oxidation of alcohols. Chem. Catal. 2022, 2, 1177-90.
119. Eder, M.; Courtois, C.; Petzoldt, P.; Mackewicz, S.; Tschurl, M.; Heiz, U. Size and coverage effects of Ni and Pt Co-catalysts in the photocatalytic hydrogen evolution from methanol on TiO2(110). ACS. Catal. 2022, 12, 9579-88.
120. Kennedy, J.; Hayward, J.; Davies, P. R.; Bowker, M. Hydrogen production by the photoreforming of methanol and the photocatalytic water-gas shift reaction. J. Phys. Energy. 2021, 3, 024007.
121. Chiarello, G. L.; Bernareggi, M.; Selli, E. Redox dynamics of Pt and Cu nanoparticles on TiO2 during the photocatalytic oxidation of methanol under aerobic and anaerobic conditions studied by in situ modulated excitation X-ray absorption spectroscopy. ACS. Catal. 2022, 12, 12879-89.
122. Muscetta, M.; Clarizia, L.; Race, M.; Andreozzi, R.; Marotta, R.; Di Somma, I. Visible-light driven systems: effect of the parameters affecting hydrogen production through photoreforming of organics in presence of Cu2O/TiO2 nanocomposite photocatalyst. Appl. Sci. 2023, 13, 2337.
123. Clarizia, L.; Spasiano, D.; Di Somma, I.; Marotta, R.; Andreozzi, R.; Dionysiou, D. D. Copper modified-TiO2 catalysts for hydrogen generation through photoreforming of organics. A short review. Int. J. Hydrogen. Energy. 2014, 39, 16812-31.
124. Christoforidis, K. C.; Fornasiero, P. Photocatalysis for hydrogen production and CO2 reduction: the case of copper-catalysts. ChemCatChem 2018, 11, 368-82.
125. Shams Ghamsari, Z.; Bashiri, H. Hydrogen production through photoreforming of methanol by Cus/TiO2 nanocatalyst: optimization and simulation. Surf. Interfaces. 2020, 21, 100709.
126. Plascencia-Hernández, F.; Albiter, E.; Mohamed Nawfal, G.; et al. Unraveling the effect of low Cu2O loading on P25 TiO2 and its self-reduction during methanol photoreforming. Inorg. Chem. Commun. 2023, 158, 111541.
127. Khan, A.; Le Pivert, M.; Ranjbari, A.; et al. Cu-based MOF/TiO2 composite nanomaterials for photocatalytic hydrogen generation and the role of copper. Adv. Funct. Mater. 2025, 36, 2501736.
128. Nomikos, G. N.; Panagiotopoulou, P.; Kondarides, D. I.; Verykios, X. E. Kinetic and mechanistic study of the photocatalytic reforming of methanol over Pt/TiO2 catalyst. Appl. Catal. B. Environ. 2014, 146, 249-57.
129. Phillips, K. R.; Jensen, S. C.; Baron, M.; Li, S.; Friend, C. M. Sequential photo-oxidation of methanol to methyl formate on TiO2(110). J. Am. Chem. Soc. 2013, 135, 574-7.
130. Coronado, J. M.; Kataoka, S.; Tejedor-Tejedor, I.; Anderson, M. A. Dynamic phenomena during the photocatalytic oxidation of ethanol and acetone over nanocrystalline TiO2: simultaneous FTIR analysis of gas and surface species. J. Catal. 2003, 219, 219-30.
131. Zhang, J.; Toe, C. Y.; Kumar, P.; Scott, J.; Amal, R. Engineering defects in TiO2 for the simultaneous production of hydrogen and organic products. Appl. Catal. B. Environ. Energy. 2023, 333, 122765.
132. Deas, R.; Pearce, S.; Goss, K.; Wang, Q.; Chen, W.; Waterhouse, G. I. Hierarchical Au/TiO2 nanoflower photocatalysts with outstanding performance for alcohol photoreforming under UV irradiation. Appl. Catal. A. Gen. 2020, 602, 117706.
133. Luo, L.; Zhang, T.; Zhang, X.; et al. Enhanced hydrogen production from ethanol photoreforming by site-specific deposition of Au on Cu2O/TiO2 p-n junction. Catalysts 2020, 10, 539.
134. Esteves, M. A.; Fresno, F.; Fernandes, V. R.; Oropeza, F. E.; De La Peña O’shea, V. A.; Rangel, C. TiO2-reduced graphene oxide-Pt nanocomposites for the photogeneration of hydrogen from ethanol liquid and gas phases. Catal. Today. 2021, 380, 41-52.
135. Shahpal, A.; Strunk, J.; Khare, R.; Lercher, J. A. Enhancing photocatalytic hydrogen production: a systematic approach to improving g-C3N4/TiO2 nano-composites. J. Environ. Chem. Eng. 2024, 12, 114803.
136. Šalipur, H.; ManojloviC, D.; MiloševiC, K.; et al. Unraveling the solar and visible light-induced deactivation mechanism of Pt-decorated carbon/TiO2 nanocomposite in photocatalytic hydrogen production. J. Environ. Chem. Eng. 2024, 12, 112862.
137. Roebuck, L.; Daly, H.; Lan, L.; et al. H2 production from the photocatalytic reforming of ethylene glycol: effect of TiO2 crystalline phase on photo-oxidation mechanism. J. Catal. 2025, 442, 115876.
138. El Hakim, S.; Bathias, M.; Chave, T.; Nikitenko, S. I. Influence of butanol isomerization on photothermal hydrogen production over Ti@TiO2 core-shell nanoparticles. Catalysts 2022, 12, 1662.
139. Courtois, C.; Eder, M.; Schnabl, K.; Walenta, C. A.; Tschurl, M.; Heiz, U. Reactions in the photocatalytic conversion of tertiary alcohols on rutile TiO2(110). Angew. Chem. Int. Ed. 2019, 58, 14255-9.
140. Özdemir, P.; Yıldırım, R. Photocatalytic glycerol reforming on Pt, Au and Cu supported by reduced TiO2 under visible light irradiation. Int. J. Hydrogen. Energy. 2024, 52, 283-94.
141. Herrera-Beurnio, M. C.; López-Tenllado, F. J.; Hidalgo-Carrillo, J.; et al. Glycerol photoreforming for photocatalytic hydrogen production on binary and ternary Pt-g-C3N4-TiO2 systems: a comparative study. Catal. Today. 2024, 430, 114548.
142. Oliveira, M. M. R. D.; Sousa, E. J. R.; Silva, A. M. P. D.; Araújo, R. D. S.; Salgado, B. C. B. Construction of photocatalytic plates for hydrogen production from photoreforming of glycerol. Int. J. Hydrogen. Energy. 2023, 48, 28792-802.
143. Tran, N. H.; Kannangara, G. S. K. Conversion of glycerol to hydrogen rich gas. Chem. Soc. Rev. 2013, 42, 9454.
144. Nascimento, L. L.; Carvalho Souza, R. A.; Zacour Marinho, J.; Wang, C.; Patrocinio, A. O. T. Light-driven conversion of biomass-derived compounds into green fuels and chemicals. J. Clean. Prod. 2024, 449, 141709.
145. Nascimento, L. L.; Souza, R. A. C.; Horta, Nunes. P. H.; et al. Photoreforming of real biomass-derived waste streams using Nb-doped BiVO4 photoanodes for sustainable hydrogen production. RSC. Sustain. 2025, 3, 4504-13.
146. Marinho, J. Z.; Nascimento, L. L.; Santos, A. L. R.; Faria, A. M.; Machado, A. E. H.; Patrocinio, A. O. T. On the influence of hydrothermal treatment pH on the performance of Bi2WO6 as photocatalyst in the glycerol photoreforming. Photochem. Photobiol. Sci. 2022, 21, 1659-75.
147. Nascimento, L. L.; Marinho, J. Z.; Dos Santos, A. L. R.; et al. Photoelectrochemical reforming of glycerol by Bi2WO6 photoanodes: role of the electrolyte pH on the H2 evolution efficiency and product selectivity. Appl. Catal. A. Gen. 2022, 646, 118867.
148. Karimi Estahbanati, M. R.; Feilizadeh, M.; Attar, F.; Iliuta, M. C. Current developments and future trends in photocatalytic glycerol valorization: process analysis. React. Chem. Eng. 2021, 6, 197-219.
149. Kozlova, E. A.; Kurenkova, A. Y.; Gerasimov, E. Y.; et al. Comparative study of photoreforming of glycerol on Pt/TiO2 and CuOx/TiO2 photocatalysts under UV light. Mater. Lett. 2021, 283, 128901.
150. Maslova, V.; Quadrelli, E. A.; Gaval, P.; Fasolini, A.; Albonetti, S.; Basile, F. Highly-dispersed ultrafine Pt nanoparticles on microemulsion-mediated TiO2 for production of hydrogen and valuable chemicals via oxidative photo-dehydrogenation of glycerol. J. Environ. Chem. Eng. 2021, 9, 105070.
151. Pecoraro, C. M.; Mino, L.; Kozyr, E.; et al. Pt-TiO2 catalysts for glycerol photoreforming: comparison of anatase, brookite and rutile polymorphs. Chem. Commun. 2024, 60, 3782-5.
152. Escamilla-Mejía, J. C.; Hidalgo-Carrillo, J.; Martín-Gómez, J.; et al. Biochars from olive stones as carbonaceous support in Pt/TiO2-carbon photocatalysts and application in hydrogen production from aqueous glycerol photoreforming. Nanomaterials 2023, 13, 1511.
153. López-Tenllado, F. J.; Estévez, R.; Hidalgo-Carrillo, J.; López-Fernández, S.; Urbano, F. J.; Marinas, A. Hydrogen photo-production from glycerol on platinum, gold and silver-modified TiO2-USY62 catalysts. Catal. Today. 2022, 390-1, 92-8.
154. Escamilla-Mejía, J. C.; Hidalgo-Carrillo, J.; Martín-Gómez, J.; et al. Pt preferential incorporation onto TiO2 in TiO2-carbon composites for hydrogen production from glycerol photoreforming. Catal. Today. 2023, 413-5, 113943.
155. Chen, W.; Chan, A.; Sun-Waterhouse, D.; Llorca, J.; Idriss, H.; Waterhouse, G. I. Performance comparison of Ni/TiO2 and Au/TiO2 photocatalysts for H2 production in different alcohol-water mixtures. J. Catal. 2018, 367, 27-42.
156. Zhong, W.; Wang, C.; Jiang, B.; et al. Investigation on multifunctional Au/TiO2@n-octadecane microcapsules towards catalytic photoreforming hydrogen production and photothermal conversion. Int. J. Hydrogen. Energy. 2022, 47, 41540-52.
157. Balsamo, S. A.; Fiorenza, R.; Iapichino, M. T. A.; Lopez-Tenllado, F. J.; Urbano, F. J.; Sciré, S. H2 production through glycerol photoreforming using one-pot prepared TiO2-rGO-Au photocatalysts. Mol. Catal. 2023, 547, 113346.
158. Majeed, I.; Arif, A.; Idrees, A.; et al. Synergistic effect of Pd co-catalyst and rGO-TiO2 hybrid support for enhanced photoreforming of oxygenates. Hydrogen 2023, 4, 192-209.
159. Martín-Gómez, J.; Reca-Expósito, S.; López-Tenllado, F. J.; Hidalgo-Carrillo, J.; Marinas, A.; Urbano, F. J. Synthesis of Fe-TiO2 and Cu-TiO2 based materials by olive leaves biotemplating—application to hydrogen production from glycerol photoreforming. Nanomaterials 2023, 13, 664.
160. Wang, M.; Liu, M.; Lu, J.; Wang, F. Photo splitting of bio-polyols and sugars to methanol and syngas. Nat. Commun. 2020, 11, 1083.
161. Clarizia, L.; Vitiello, G.; Pallotti, D. K.; et al. Effect of surface properties of copper-modified commercial titanium dioxide photocatalysts on hydrogen production through photoreforming of alcohols. Int. J. Hydrogen. Energy. 2017, 42, 28349-62.
162. Martín-Gómez, J.; Escamilla, J. C.; Hidalgo-Carrillo, J.; et al. Influence of sacrificial agent on Cu photodeposition over TiO2/MCH composites for photocatalytic hydrogen production. Catal. Today. 2023, 413-5, 113928.
163. Martín-Gómez, J.; Hidalgo-Carrillo, J.; Estévez, R. C.; Urbano, F. J.; Marinas, A. Hydrogen photoproduction on TiO2-CuO artificial olive leaves. Appl. Catal. A. Gen. 2021, 620, 118178.
164. Villachica-Llamosas, J.; Ruiz-Aguirre, A.; Colón, G.; Peral, J.; Malato, S. CuO-TiO2 pilot-plant system performance for solar photocatalytic hydrogen production. Int. J. Hydrogen. Energy. 2024, 51, 1069-77.
165. Martín-Gómez, J.; Hidalgo-Carrillo, J.; Montes, V.; et al. EPR and CV studies cast further light on the origin of the enhanced hydrogen production through glycerol photoreforming on CuO:TiO2 physical mixtures. J. Environ. Chem. Eng. 2021, 9, 105336.
166. Segovia-Guzmán, M. O.; Román-Aguirre, M.; Verde-Gomez, J. Y.; Collins-Martínez, V. H.; Zaragoza-Galán, G.; Ramos-Sánchez, V. H. Green Cu2O/TiO2 heterojunction for glycerol photoreforming. Catal. Today. 2020, 349, 88-97.
167. Villachica-Llamosas, J.; Ruiz-Aguirre, A.; Colón, G.; Peral, J.; Malato, S. H2 production based on a ternary mixture of commercial CuO-NiO-TiO2 in a solar pilot plant. Catal. Today. 2024, 431, 114608.
168. Eisapour, M.; Zhao, H.; Zhao, J.; et al. p-n heterojunction of nickel oxide on titanium dioxide nanosheets for hydrogen and value-added chemicals coproduction from glycerol photoreforming. J. Colloid. Interface. Sci. 2023, 647, 255-63.
169. Eisapour, M.; Huang, R.; Roostaei, T.; Zhao, H.; Hu, J.; Chen, Z. Sandwich-like heterojunction of NiO-Ni-TiO2 for simultaneous production of hydrogen and value-added products from glycerol photoreforming. Environ. Surf. Interfaces. 2025, 3, 46-54.
170. Liu, J.; Zhou, Y.; Fan, Y.; et al. Regulating the local electronic structure by constructing Ni-O-Ni sites confined in TiO2 for selective photocatalytic glycerol reforming. J. Energy. Chem. 2025, 108, 129-37.
171. Falara, P. P.; Antoniadou, M.; Zourou, A.; Sakellis, E.; Kordatos, K. V. Carbon dot-titanium dioxide (CD/TiO2) nanocomposites: reusable photocatalyst for sustainable H2 production via photoreforming of green organic compounds. Coatings 2024, 14, 131.
172. Escamilla, J. C.; Hidalgo-Carrillo, J.; Martín-Gómez, J.; et al. Hydrogen production through glycerol photoreforming on TiO2/mesoporous carbon: influence of the synthetic method. Materials 2020, 13, 3800.
173. Kobayashi, A. Photoredox cascade catalyst for efficient hydrogen production with biomass photoreforming. Angew. Chem. Int. Ed. 2023, 62, e202313014.
174. Panagiotopoulou, P.; Karamerou, E. E.; Kondarides, D. I. Kinetics and mechanism of glycerol photo-oxidation and photo-reforming reactions in aqueous TiO2 and Pt/TiO2 suspensions. Catal. Today. 2013, 209, 91-8.
175. Hu, Y.; Qian, Q.; Wang, J.; Cao, F.; Shen, Y. TiO2-mediated photoreforming of glycerol to formic acid in acetonitrile-free aqueous solutions: mechanism of crystal phase-regulated oxygen radicals influencing photoreforming. Chem. Eng. J. 2025, 524, 169428.
176. Villachica-Llamosas, J.; Sowik, J.; Ruiz-Aguirre, A.; Colón, G.; Peral, J.; Malato, S. Photoreforming of glycerol to produce hydrogen from natural water in a compound parabolic collector solar photoreactor. J. Environ. . Chem. Eng. 2023, 11, 111216.
177. Toledo-Camacho, S.; Rey, A.; Maldonado, M.; Llorca, J.; Contreras, S.; Medina, F. Photocatalytic hydrogen production from water-methanol and -glycerol mixtures using Pd/TiO2(-WO3) catalysts and validation in a solar pilot plant. Int. J. Hydrogen. Energy. 2021, 46, 36152-66.
178. Li, P.; Zhang, B. Photocatalytic conversion of biomass and nitrate into glycine. ACS. Catal. 2024, 14, 18345-53.
179. Granone, L. I.; Sieland, F.; Zheng, N.; Dillert, R.; Bahnemann, D. W. Photocatalytic conversion of biomass into valuable products: a meaningful approach? Green. Chem. 2018, 20, 1169-92.
180. Nwosu, U.; Wang, A.; Palma, B.; et al. Selective biomass photoreforming for valuable chemicals and fuels: a critical review. Renew. Sustain. Energy. Rev. 2021, 148, 111266.
181. Zhao, H.; Liu, J.; Zhong, N.; et al. Biomass photoreforming for hydrogen and value-added chemicals co-production on hierarchically porous photocatalysts. Adv. Energy. Mater. 2023, 13, 2300257.
182. Liu, K.; Ma, J.; Yang, X.; et al. Boosting electron kinetics of anatase TiO2 with carbon nanosheet for efficient photo-reforming of xylose into biomass-derived organic acids. J. Alloys. Compd. 2022, 906, 164276.
183. Lan, L.; Daly, H.; Sung, R.; et al. Mechanistic study of glucose photoreforming over TiO2-based catalysts for H2 production. ACS. Catal. 2023, 13, 8574-87.
184. Zhong, N.; Yu, X.; Zhao, H.; Hu, J.; Gates, I. D. Biomass photoreforming for hydrogen production over hierarchical 3DOM TiO2-Au-CdS. Catalysts 2022, 12, 819.
185. Bahadori, E.; Ramis, G.; Zanardo, D.; et al. Photoreforming of glucose over CuO/TiO2. Catalysts 2020, 10, 477.
186. Belda-Marco, S.; Lillo-Ródenas, M. A.; Román-Martínez, M. C. H2 production by cellulose photoreforming with TiO2-Cu photocatalysts bearing different Cu species. Catal. Today. 2023, 413-5, 113945.
187. Belda-Marco, S.; Lillo-Ródenas, MÁ.; Román-Martínez, M. C. Optimization of active sites in TiO2-Cu photocatalysts for H2 generation via cellulose photo-reforming. ChemCatChem 2025, 17, e202500250.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].