


Amide-functionalized copolymer electrolyte with high ionic conductivity and transference number for high-performance lithium metal batteries
 0
0 
Abstract
Solid polymer electrolytes are regarded as promising candidates for high-energy-density lithium metal batteries, yet they typically suffer from a trade-off between ionic conductivity and lithium-ion transference number, alongside interfacial instability. In this work, a molecular engineering strategy is proposed by copolymerizing an amide-functionalized acrylate monomer, bis[2-(acrylamido)ethyl] heptanedioate, into a polyacrylate backbone. Theoretical calculations and spectroscopic analyses reveal that the incorporated amide groups establish a hydrogen bonding network interacting preferentially with succinonitrile and bis(trifluoromethanesulphonyl)imide anions. This interaction effectively anchors the solvent molecules and anions to the polymer chain, thereby restricting anion mobility while facilitating Li+ dissociation. Therefore, the optimized copolymer electrolyte achieves a high room-temperature ionic conductivity of 0.92 mS·cm-1 and a remarkable Li+ transference number of 0.82. Moreover, the hydrogen-bond-modulated solvation structure promotes the formation of contact ion pairs and aggregates, which subsequently induces the formation of a robust inorganic-rich (LiF/Li3N) solid electrolyte interphase. As a result, the Li||Li symmetric cell demonstrates stable cycling for over 1,700 h at a current density of 0.5 mA·cm-2. The Li||LiNi0.8Co0.1Mn0.1O2 full cell delivers an initial discharge capacity of 220.8 mAh·g-1 at 0.1 C and exhibits superior long-term cyclability (85.3% capacity retention after 500 cycles at 1 C). This work highlights the critical role of intermolecular hydrogen bonding in designing advanced electrolytes for next-generation batteries.
Keywords
INTRODUCTION
The rapid electrification of the transportation sector and the proliferation of portable electronics have intensified the demand for energy storage systems with higher energy density and enhanced safety[1-5]. Lithium metal batteries are considered the ultimate solution due to the ultra-high theoretical capacity
Solid polymer electrolytes (SPEs) have emerged as a compelling alternative, offering intrinsic safety, leak-free operation, and excellent flexibility[9-11]. However, conventional SPEs, such as poly(ethylene oxide) and aliphatic polyesters, are intrinsically limited by their unsatisfactory room-temperature ionic conductivity and low Li+ transference number[12-14]. In these systems, Li+ transport is typically coupled with the segmental motion of the polymer chains, whereas anions often exhibit higher mobility than the solvated cations. A low Li+ transference number leads to the accumulation of anions near the anode, causing severe concentration polarization and increased internal resistance, which in turn aggravates lithium dendrite nucleation and cell failure[15,16]. Consequently, overcoming these intrinsic limitations to achieve high ionic conductivity and high cation selectivity remains a prerequisite for the practical deployment of high-performance solid-state batteries.
To address these issues, various strategies have been explored in recent years. For instance, the incorporation of inorganic fillers utilizes interfacial interactions to facilitate lithium salt dissociation; however, filler agglomeration and high interfacial resistance often impede further performance improvements[17]. Similarly, the design of single-ion conducting polymers maximizes the Li+ transference number via the covalent tethering of anions but typically compromises ionic conductivity due to the restricted segmental motion of the rigid backbone[18,19]. Furthermore, introducing plasticizers is an effective method to increase free volume and segmental mobility, thereby enhancing ionic conductivity[20]. However, this strategy typically accelerates both cations and anions indiscriminately, resulting in a reduced Li+ transference number. These existing approaches struggle to balance the trade-off between ion transport speed and selectivity, often improving one property at the expense of the other. Therefore, developing SPEs that simultaneously achieve high ionic conductivity and a high Li+ transference number is critical yet challenging.
In this work, a molecular engineering strategy is proposed to regulate intermolecular interactions by developing a novel copolymer electrolyte. This electrolyte is synthesized by copolymerizing an amide-functionalized acrylate monomer {bis[2-(acrylamido)ethyl] heptanedioate (Am-DAH)} with an acrylate monomer {bis[2-(acryloyloxy)ethyl] heptanedioate (DAH)}, denoted as p(Am-DAH-co-DAH). Specifically, the introduction of amide groups into the polyacrylate backbone allows them to act as hydrogen bond donors. By leveraging these groups to interact selectively with anions or plasticizers, it is theoretically possible to immobilize anions without hindering Li+ transport, thereby resolving the conductivity-selectivity trade-off. Theoretical calculations and spectroscopic evidence demonstrate that these amide groups preferentially interact with succinonitrile (SN) plasticizer and bis(trifluoromethanesulphonyl)imide anions (TFSI-). This interaction effectively anchors the anions and solvent molecules to the polymer backbone, restricting their mobility and significantly enhancing the Li+ transference number to 0.82. Simultaneously, this specific interaction weakens the coordination between Li+ and the polymer backbone, promoting fast Li+ conduction of 0.92 mS·cm-1. Furthermore, this unique solvation structure promotes anion-derived decomposition, facilitating the formation of a stable, inorganic-rich solid electrolyte interphase (SEI) that suppresses dendrite growth. As a result, the electrolyte enables the stable operation of high-voltage Li||LiNi0.8Co0.1Mn0.1O2 full cells with excellent long-term cycling stability.
EXPERIMENTAL
Chemicals
2-hydroxyethyl acrylate (> 98.0%), 2-hydroxyethyl acrylamide (> 98.0%), succinonitrile (SN, 98%), 2,2′-azobis(2-methylpropionitrile) (AIBN), dichloromethane (99.5%, water ≤ 50 ppm), anhydrous dichloromethane (97.0%), pyridine (99.5%), magnesium sulfate (MgSO4, 98%), hydrochloric acid (37.0%), petroleum ether (99.0%), ethyl acetate (99.0%) and deuterated chloroform (99.8%) were purchased from Energy Chemical Technology (Shanghai) Co., Ltd. Lithium bis(trifluoromethanesulphonyl)imide (LiTFSI) and lithium difluoro(oxalato)borate (LiDFOB) were acquired from Suzhou DodoChem Technology Co., Ltd. Malonylchloride and pimeloylchloride were sourced from Shanghai Macklin Biochemical Co., Ltd. Polyvinylidene difluoride (PVDF), Super P, N-methyl pyrrolidone (NMP) and LiNi0.8Co0.1Mn0.1O2 (NCM811) were provided by Canrd Technology Co., Ltd. All chemicals were used as received without further purification.
Material preparation
Am-DAH
2-hydroxyethyl acrylamide (10.0 g, 86.21 mmol) was dissolved in 40.0 mL of anhydrous dichloromethane in a 250 mL three-necked flask under a nitrogen atmosphere. The solution was chilled to 0 °C, followed by the slow, dropwise addition of pyridine (6.80 g, 86.12 mmol) and pimeloyl chloride (8.38 g, 42.56 mmol). Subsequently, the reaction mixture was stirred at 25 °C with a rotational speed of 300 rpm for 24 h. Upon completion, the resulting mixture underwent washing with 1.0 M aqueous HCl, filtration, and drying over anhydrous MgSO4. The solvent was then removed under reduced pressure to yield a white solution. Final purification by silica gel column chromatography with petroleum ether/ethyl acetate (2:1 v/v) yielded the target product as a solid powder after solvent evaporation.
DAH
The DAH was synthesized by the same procedure as Am-DAH, except that 2-hydroxyethyl acrylate (9.88 g, 85.12 mmol) was used instead of 2-hydroxyethyl acrylamide.
Polymer electrolyte membrane
The precursor blend containing 100.0 mg of DAH/Am-DAH monomers (weight ratios of 9:1, 8:2, 7:3, 6:4 and 5:5) and 200.0 mg SN was initially formulated. This mixture was dried under vacuum at 80 °C for 12 h to remove trace water. Following dehydration, 120.0 mg of lithium salts (LiTFSI:LiDFOB = 9:1 by weight) was added and stirred at 25 °C with a rotational speed of 300 rpm until complete dissolution. Next, 5.0 mg of AIBN (thermal initiator) was incorporated into the system. The obtained homogeneous solution was cast onto a glass fiber matrix and thermal polymerized at 80 °C for 8 h. For comparison, pristine poly-DAH and poly-Am-DAH membranes were fabricated via an identical protocol, simply replacing the binary monomer blend with 100.0 mg of pure DAH or pure Am-DAH.
Physiochemical characterizations
A Nicolet iS50 spectrometer (Thermo Scientific, USA) was utilized to acquire Fourier transform infrared (FTIR) spectra. For surface analysis, a Thermo K-Alpha XPS system (Thermo Scientific, USA) with a monochromatic Al-Kα X-ray source was employed to record X-ray photoelectron spectroscopy (XPS) data. Scanning electron microscopy (SEM, SU8220, Hitachi, Japan) was used to characterize the morphology. Thermal stability was evaluated using a Q50 thermogravimetric analyzer (TA Instruments, USA); TGA measurements were conducted at a heating rate of 5 °C·min-1 under an argon atmosphere. Bruker AVANCE III HD 400 instrumentation (Bruker, Germany) was used to analyze Nuclear magnetic resonance (NMR) spectra.
Electrochemical measurements
Battery assembly and test
The CR2032 coin-type cells were assembled in an argon-filled glovebox. The cells comprised the prepared NCM811 cathode and a lithium metal anode (500 μm thickness, 10 mm diameter). The cathodes were fabricated using the conventional doctor-blade technique. A slurry was prepared by dispersing NCM811 powder, Super P and PVDF in NMP solvent at a mass ratio of 8:1:1. The resulting mixture was cast onto aluminum foil and dried under vacuum at 80 °C for 12 h. Subsequently, the cathode sheets were punched into discs with a diameter of 10 mm (average active material mass loading was 3.5 mg·cm-2). To evaluate the compatibility with lithium metal anodes, galvanostatic cycling tests were performed on Li||Li symmetric cells at a current density of 0.5 mA·cm-2 with fixed deposition capacities of 0.5 mAh·cm-2. The electrochemical performance of the SPEs in full cells was systematically analyzed using an NCM811 cathode within specific voltage windows of 3.0-4.3 V and 2.8-4.6 V.
Electrochemical stability
The electrochemical stability window of the solid polymer electrolyte (SPE) was evaluated via linear sweep voltammetry (LSV). Measurements were conducted on a Li|SPE|SS (stainless steel) coin cell configuration across a potential range of 2.0 to 6.0 V at a scan rate of 1.0 mV·s-1.
Ionic conductivity
The ionic conductivity was determined using electrochemical impedance spectroscopy (EIS) on symmetric SS|SPE|SS cells using a Zahner Zennium workstation. The frequency range was set from 10-1 to 105 Hz with an AC amplitude of 10 mV. The intrinsic ionic conductivity (σ) was calculated according to standard electrochemical principles[21] as follows:
where L denotes the thickness of the SPE, Rb is the bulk resistance and S represents the effective contact area between the electrolyte and the electrode.
Activation energy
The activation energy (Ea) was derived from the temperature-dependent conductivity (30-80 °C) following the empirical Arrhenius equation[22]:
where A is the conductivity pre-exponential factor, T is the absolute temperature, and R is the universal gas constant.
Lithium ion transference number
The lithium ion transference number (
where I0 and Is denote the initial and steady-state currents, respectively, and R0 and Rs are the interfacial resistance before and after polarization.
Computational details
All Density Functional Theory (DFT) calculations were performed using the CP2K package[25]. A hybrid Gaussian and plane-wave (GPW) approach was employed, wherein the Kohn-Sham orbitals were expanded using Gaussian-type basis sets, while the electron density was represented by an auxiliary plane-wave basis. Specifically, molecularly optimized triple-zeta valence doubly polarized (TZV2P-MOLOPT) basis sets[26] were utilized for all elements (S, N, O, F, Li, C, and H), coupled with a plane-wave energy cutoff of 500 Ry. The exchange-correlation interactions were described by the Perdew-Burke-Ernzerhof (PBE) functional[27]. To account for weak interactions, Grimme’s D3 dispersion correction was applied throughout[28]. Wave function optimization was achieved via matrix diagonalization, and Fermi smearing at an electronic temperature of 300 K was used to aid self-consistent field (SCF) convergence. Geometry optimizations were conducted using the Broyden-Fletcher-Goldfarb-Shanno (BFGS) algorithm. Furthermore, the basis-set superposition error (BSSE) was evaluated for all adsorbate systems and found to be negligible (< 0.05 eV)[27].
RESULTS AND DISCUSSION
Synthesis and characterization
The DAH and Am-DAH monomers are synthesized via esterification and amidation reactions, respectively [Supplementary Figure 1]. In terms of molecular structure, DAH contains ester linkages connecting the polymerizable vinyl groups, whereas Am-DAH features amide functionalities. This substitution of oxygen with nitrogen (-CONH- vs. -COO-) endows Am-DAH with the capability to form hydrogen bonding networks. The molecular structures of the synthesized DAH and Am-DAH monomers are first verified via 1H nuclear magnetic resonance [Supplementary Figure 2]. Subsequently, the p(DAH-co-Am-DAH) solid polymer electrolyte (SPE) is fabricated by solution casting and thermal curing in the presence of SN plasticizers, lithium bis(trifluoromethanesulphonyl)imide (LiTFSI) and lithium difluoro(oxalato)borate[29]. The optimal Am-DAH dosage is determined by measuring the ionic conductivity of the copolymers with varying Am-DAH contents [Supplementary Figure 3]. Despite the relatively high conductivity of pristine p(DAH), the copolymer exhibits a non-monotonic trend, where conductivity initially increases but subsequently decreases with higher Am-DAH ratios. This drastic reduction is attributed to the dense hydrogen-bonding network introduced by the amide groups, which severely constrains polymer segmental motion and impedes Li+ transport[30,31]. By precisely regulating this composition, the copolymerization approach achieves a critical synergy. The optimized p(DAH-co-Am-DAH) effectively utilizes the hydrogen-bonding network to anchor molecules and enhance stability, while simultaneously avoiding excessive chain restriction to ensure unimpeded Li+ conduction. Therefore, the subsequent discussion primarily focuses on the comparative analysis of p(DAH) and the optimized p(DAH-co-Am-DAH) (DAH:Am-DAH = 4:1, by weight) to elucidate the effect of the amide groups.
With the optimal composition established, the polymerization process and structural evolution are examined. As illustrated in Figure 1A, the precursor solution transforms from a flowable liquid into a transparent, gel-like solid following free radical thermal polymerization. Figure 1B presents an optical photograph of the composite electrolyte supported on a glass fiber membrane, exhibiting a milky-white and semi-transparent appearance. The surface morphology is analyzed via scanning electron microscopy (SEM). The p(DAH-co-Am-DAH) surface appears smooth and uniform, devoid of significant protrusions [Figure 1C]. This high degree of surface flatness ensures intimate contact between the electrolyte and the electrode materials, thereby effectively reducing interfacial impedance and maintaining low internal resistance within the battery. FTIR spectroscopy is employed to monitor the evolution of functional groups [Figure 1D]. Notably, the characteristic absorption peak at 1,670 cm-1, attributed to the terminal C=C bonds of the precursors, completely disappears in the cured electrolyte, indicating a high degree of free radical polymerization. Furthermore, any trace unreacted monomers potentially remaining below the FTIR detection limit are effectively eliminated during the subsequent rigorous purification and vacuum drying steps, thereby ensuring the high purity of the final copolymer.
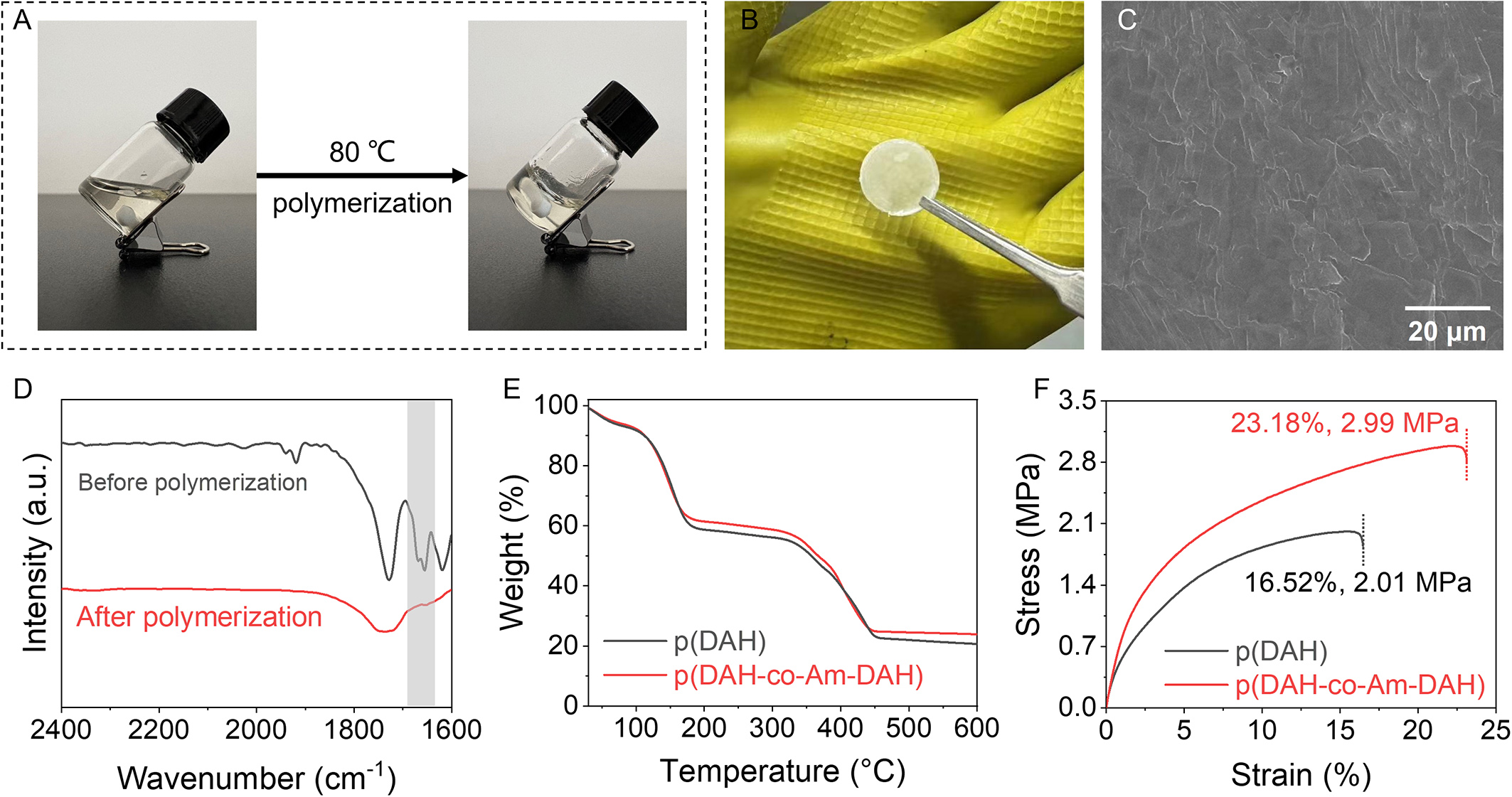
Figure 1. Optical photographs of (A) the electrolyte before and after polymerization and (B) the p(DAH-co-Am-DAH) electrolyte membrane [all photographs in (A) and (B) are taken by the authors]; (C) SEM image of the p(DAH-co-Am-DAH) electrolyte membrane; (D) FTIR spectra of the electrolyte before and after polymerization; (E) Thermogravimetric analysis and (F) Tensile testing of p(DAH-co-Am-DAH) and p(DAH) without glass-fiber support. DAH: Bis[2-(acryloyloxy)ethyl] heptanedioate; Am-DAH: bis[2-(acrylamido)ethyl] heptanedioate; SEM: scanning electron microscopy; FTIR: Fourier transform infrared.
To further evaluate the electrolyte’s suitability for practical applications, thermogravimetric analysis (TGA) and tensile testing are employed to characterize the fundamental physicochemical properties. Figure 1E displays the TGA profiles, where both electrolytes exhibit two distinct thermal decomposition stages. The first stage at 119.30 °C corresponds approximately to the flash point of SN, while the second at 325.40 °C represents the decomposition of the polymer backbone. The thermal degradation curves of p(DAH-co-Am-DAH) and p(DAH) are highly coincident, indicating that the introduction of the amide modifying group does not significantly alter the thermal decomposition behavior. With backbone decomposition temperatures exceeding 300 °C, both electrolytes demonstrate good flame resistance. To evaluate the intrinsic mechanical properties of the polymer networks, tensile testing is performed on free-standing pure polymer films prepared without glass-fiber support [Figure 1F]. The p(DAH) exhibits an elongation at break of 16.52% and a tensile strength of 2.01 MPa. In comparison, p(DAH-co-Am-DAH) shows superior mechanical performance, with an elongation at break of 21.18% and a tensile strength of
Solvation structure analysis
Density functional theory (DFT) calculations and FTIR spectroscopy are employed to characterize and verify the hydrogen bonding interactions between the polymer backbone and components such as SN, Li+ and TFSI- in the p(DAH-co-Am-DAH) electrolyte. As shown in Figure 2A, DFT results show that Am-DAH exhibits a relatively lower binding energy with Li+ (-0.59 eV) compared to DAH (-0.95 eV). This suggests that the introduction of Am-DAH effectively reduces the overall affinity of the polymer backbone for Li+, thereby facilitating Li+ dissociation[34]. In contrast to this weak Li+ affinity, Am-DAH demonstrates significantly stronger interactions with TFSI- and SN compared to DAH, owing to the presence of the amide groups. Specifically, the binding energies of Am-DAH with TFSI- and SN increase to -1.82 and -1.70 eV, respectively, markedly surpassing the values of -1.41 and -1.21 eV observed for DAH. This significant enhancement confirms the formation of a robust hydrogen bond network.
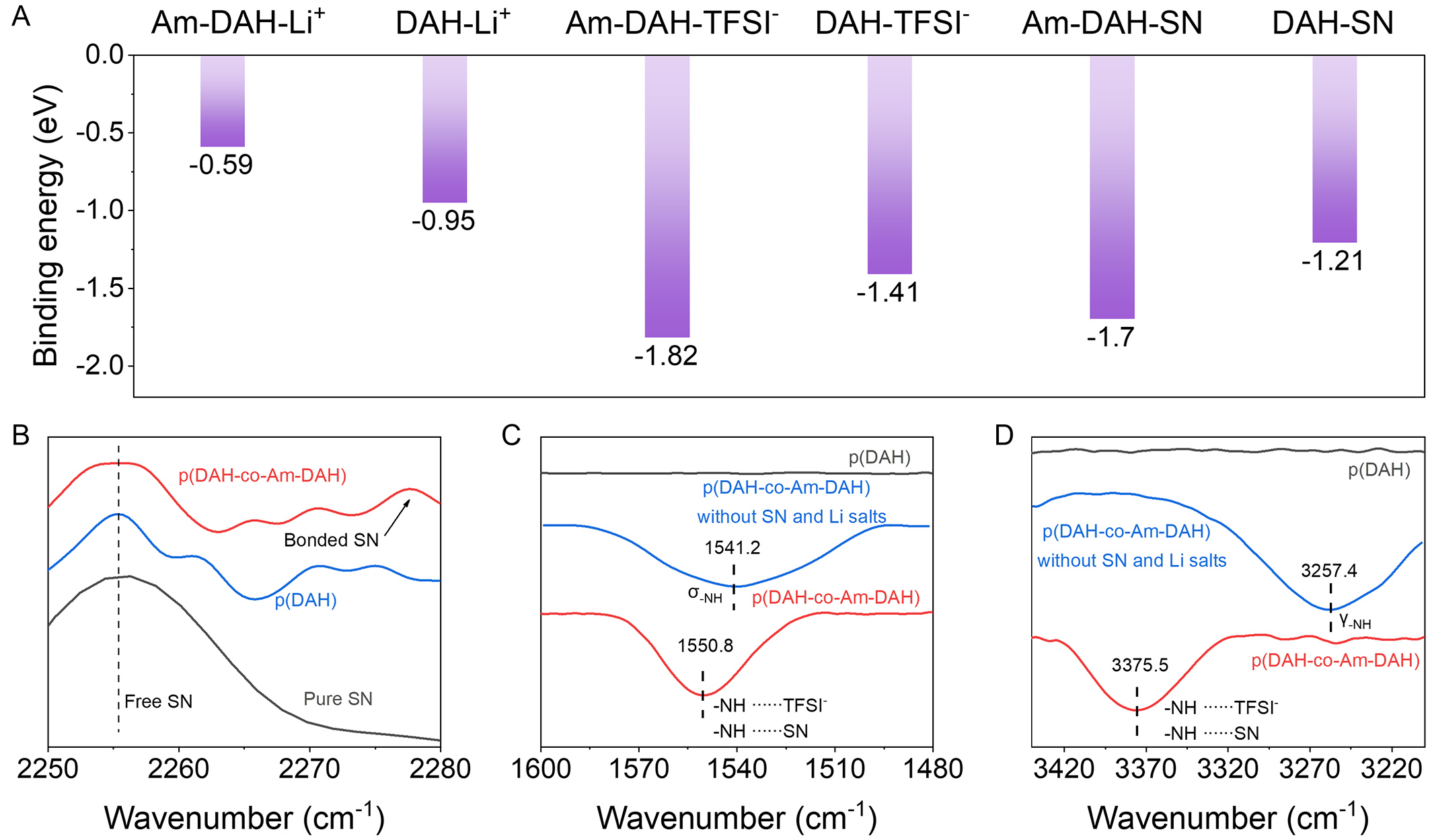
Figure 2. (A) Binding energies of DAH/Am-DAH monomers with Li+, SN, and TFSI-; (B-D) FTIR spectra of p(DAH-co-Am-DAH), p(DAH) and SN. DAH: Bis[2-(acryloyloxy)ethyl] heptanedioate; Am-DAH: bis[2-(acrylamido)ethyl] heptanedioate; SN: succinonitrile; TFSI-: bis(trifluoromethanesulphonyl)imide anions; FTIR: Fourier transform infrared.
Subsequently, FTIR spectroscopy is employed to investigate the interactions of the terminal nitrile group (-C≡N) of SN with the p(DAH) and p(DAH-co-Am-DAH) electrolytes. As shown in Figure 2B, in the p(DAH) electrolyte, a peak appears at 2,275.1 cm-1, corresponding to the Lewis acid-base interaction between the nitrile group and Li+. However, in the p(DAH-co-Am-DAH) system, the introduction of the amide hydrogen bond donor induces a blue shift of this peak to 2,277.6 cm-1 This shift indicates that, in addition to interacting with Li+, the SN molecules also form hydrogen bonds with the amide groups on the polymer backbone. To corroborate these hydrogen bonding interactions, the response of the secondary amide (-NH-) groups on the polymer backbone is further analyzed. Upon the incorporation of SN and lithium salts into the p(DAH-co-Am-DAH) matrix, the bending peak shifts from 1,541.2 to 1,550.8 cm-1 [Figure 2C], and the stretching peak shifts from 3,257.4 to 3,375.5 cm-1 [Figure 2D]. These significant blue shifts provide compelling evidence of the strong interaction network formed between the polymer backbone, SN, and TFSI-. In contrast, the p(DAH) spectrum naturally lacks these amide-specific features. Consequently, these spectral results confirm that the strong intermolecular forces in p(DAH-co-Am-DAH) effectively anchors the SN and TFSI- components, thereby ensuring the stable localization of these species within the polymer matrix.
Raman spectroscopy is employed to investigate the variations in ion solvation structure induced by the Am-DAH hydrogen bond donor. First, the coordination states of SN are examined [Figure 3A and B]. SN exists primarily in two forms, identified as bound SN (interacting with the backbone or ions) at 2,254.0 cm-1 and free SN at 2,278.0 cm-1[35,36]. In the p(DAH) electrolyte, SN exists predominantly as free solvent molecules (90.1%), with only 9.9% in the bound state. In comparison, the p(DAH-co-Am-DAH) system exhibits a significantly higher proportion of bound SN, which reaches 30.4%. This increase occurs because the introduction of Am-DAH facilitates hydrogen bonding between the polymer backbone and SN molecules. Simultaneously, the weakened interaction between the backbone and Li+ releases more Li+, which further promotes SN coordination. Subsequently, the solvation states of the TFSI- are analyzed [Figure 3C and D]. The anion exists in three forms, identified as free TFSI- at 738.0 cm-1, contact ion pairs (CIPs) at 741.6 cm-1, and aggregates (AGGs) at 751.2 cm-1[37,38]. In the p(DAH) electrolyte, TFSI- exists primarily as free anions (44.5%), with CIPs and AGGs accounting for 31.2% and 24.3%, respectively. In comparison, the p(DAH-co-Am-DAH) system exhibits an aggregate-rich solvation structure, characterized by a lower fraction of free TFSI- (40.9%) and a higher proportion of AGGs (27.3%). Similarly, the Am-DAH backbone anchors TFSI- while releasing Li+, which collectively drives the formation of ion aggregates. These distinct solvation structures have profound implications for interfacial chemistry. The abundance of free SN in p(DAH) tends to undergo parasitic decomposition, leading to the formation of an unstable, organic-rich solid electrolyte interphase (SEI). Conversely, the enrichment of CIPs and AGGs in p(DAH-co-Am-DAH) promotes anion-derived reduction, fostering a robust, inorganic-rich SEI that enhances electrochemical stability[39].
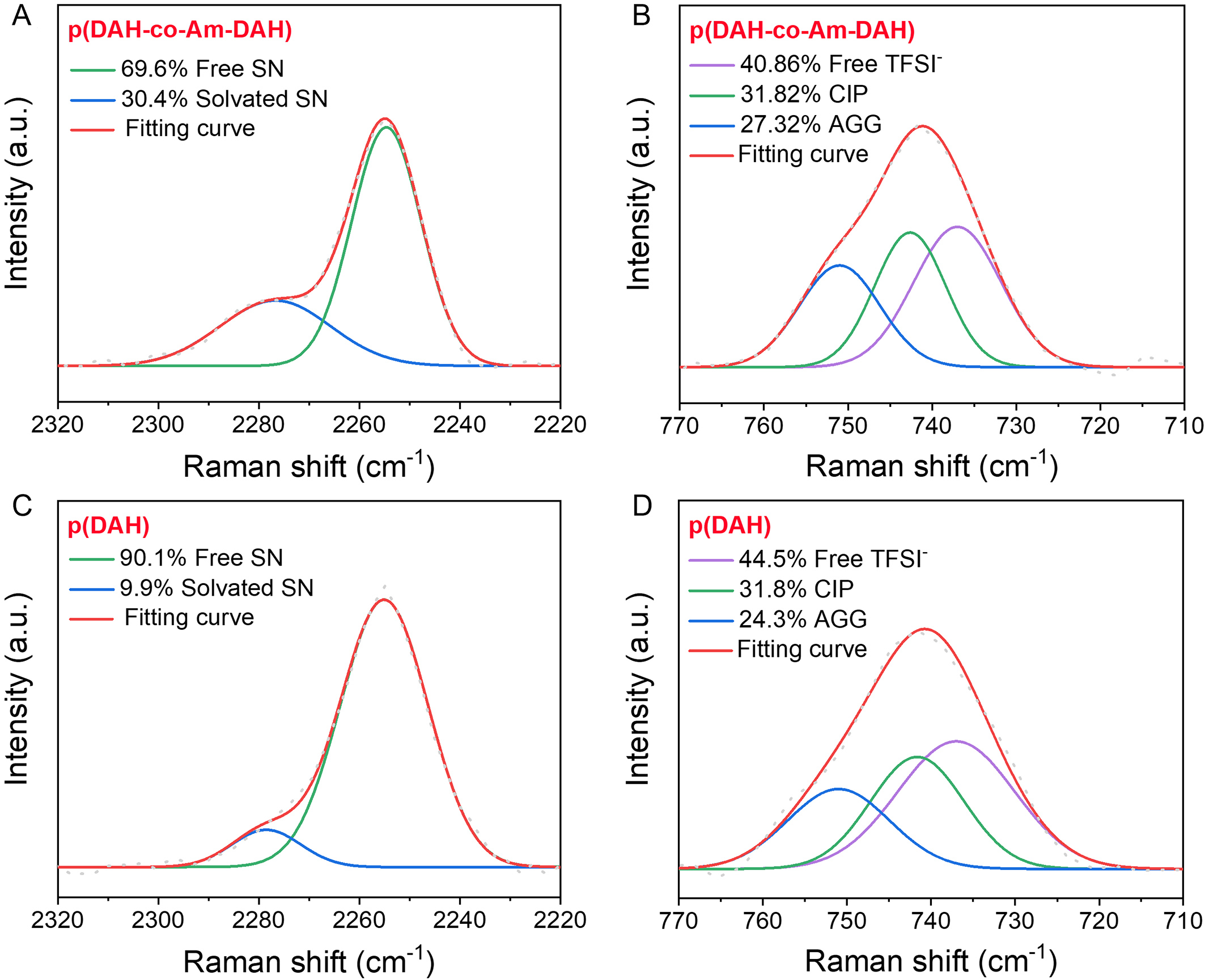
Figure 3. Raman spectra of SN in (A) p(DAH) and (B) p(DAH-co-Am-DAH), and of TFSI- anions in (C) p(DAH) and (D) p(DAH-co-Am-DAH). SN: Succinonitrile; DAH: bis[2-(acryloyloxy)ethyl] heptanedioate; Am-DAH: bis[2-(acrylamido)ethyl] heptanedioate; TFSI-: bis(trifluoromethanesulphonyl)imide anions; CIP: contact ion pair; AGG: aggregate.
Electrochemical properties
To evaluate the impact of the Am-DAH donor on electrochemical performance, room-temperature ionic conductivity and exchange current density are measured. As shown in Figure 4A, the p(DAH-co-Am-DAH) electrolyte exhibits a higher ionic conductivity (0.92 mS·cm-1) than the p(DAH) (0.79 mS·cm-1). This enhancement is initially attributed to the weakened interaction between the polymer backbone and Li+, which facilitates Li+ dissociation. To validate this mechanism, the ion migration activation energy is determined by fitting the temperature-dependent ionic conductivity [Supplementary Figure 4] to the Arrhenius equation [Figure 4B]. The p(DAH-co-Am-DAH) electrolyte exhibits a lower ion transport activation energy of 0.258 eV compared to 0.278 eV for p(DAH). This reduction confirms that the Am-DAH modification weakens the interaction between the polymer backbone and Li+, thereby lowering the energy barrier for Li+ dissociation into the SN/TFSI- mobile phase. Beyond bulk ion transport, the interfacial Li+ plating/stripping kinetics are also assessed using Tafel plots [Figure 4C]. The p(DAH-co-Am-DAH) electrolyte exhibits a higher exchange current density of 0.162 mA·cm-2 compared to 0.147 mA·cm-2 for p(DAH), indicating that the optimized solvation structure facilitates faster interfacial charge transfer[33]. These superior kinetic parameters are crucial for reducing internal resistance, improving rate capability and mitigating heat-induced degradation.
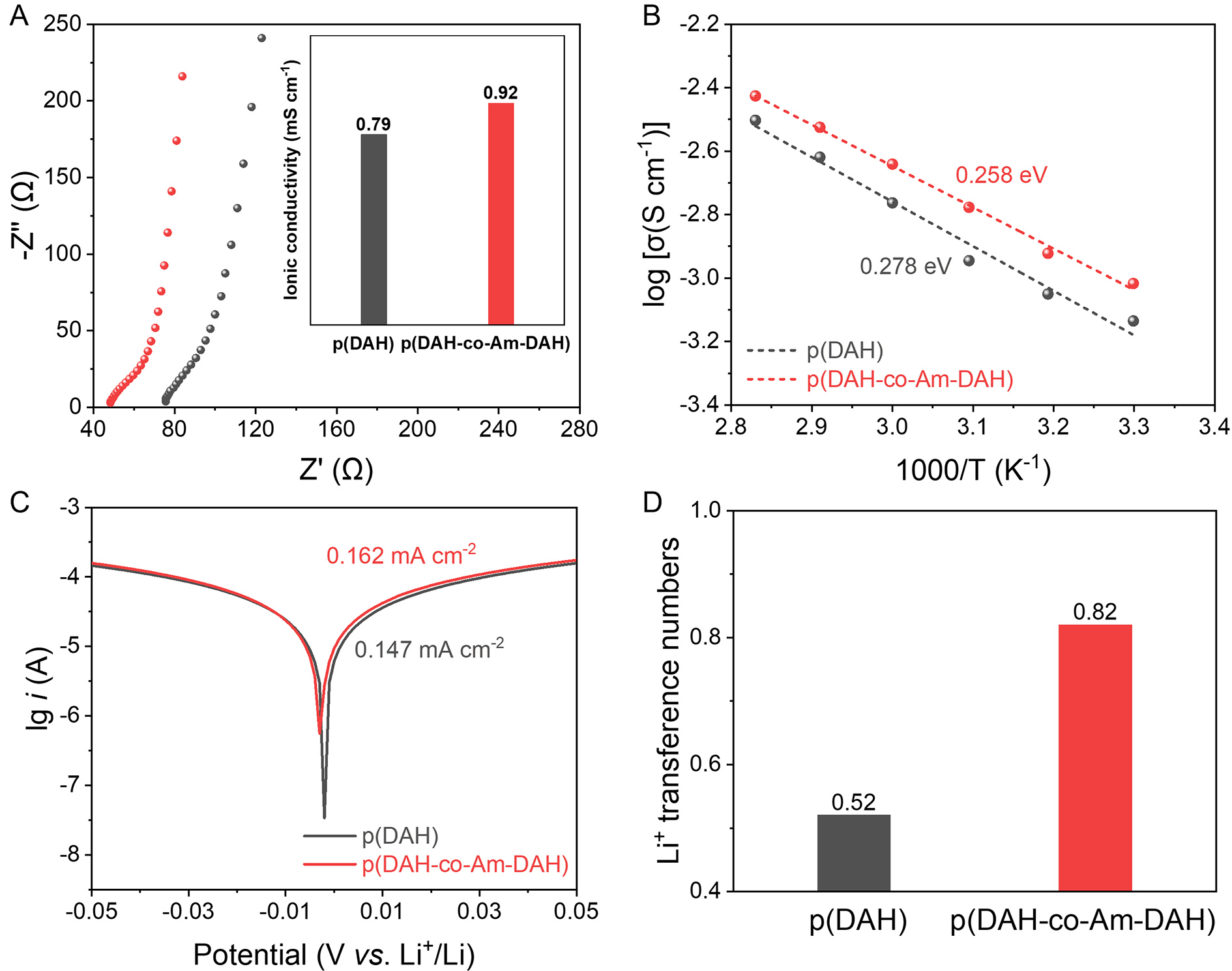
Figure 4. (A) Nyquist plots, (B) Arrhenius plots of the ionic conductivities from 30 to 80 °C, (C) Tafel plots, and (D) Li+ transference number of p(DAH) and p(DAH-co-Am-DAH). DAH: Bis[2-(acryloyloxy)ethyl] heptanedioate; Am-DAH: bis[2-(acrylamido)ethyl] heptanedioate.
While high conductivity and fast kinetics are essential, a high Li+ transference number is equally critical to ensure efficient Li+ transport. The Li+ transference number is determined using the Evans method [Supplementary Figure 5, combining AC impedance and DC polarization]. The p(DAH-co-Am-DAH) electrolyte achieves a remarkably high Li+ transference number of 0.82, significantly surpassing the 0.52 of p(DAH) [Figure 4D]. This improvement stems from a unique structural configuration where SN and TFSI- are effectively anchored to the polymer backbone via hydrogen bonds, thereby restricting their mobility. Consequently, the ionic current is predominantly carried by highly mobile Li+. A high Li+ transference number enhances energy efficiency, reduces concentration polarization, and promotes uniform Li deposition, effectively suppressing dendrite growth and extending cycle life[40]. Furthermore, the intrinsic electrochemical stability windows of p(DAH) and p(DAH-co-Am-DAH) are evaluated via linear sweep voltammetry. Both electrolytes demonstrate impressive oxidation resistance, with onset potentials of 5.53 and 4.81 V, respectively [Supplementary Figure 6]. Such wide electrochemical windows ensure full compatibility with commercial cathodes operating at elevated voltages, confirming their suitability for high-energy-density battery systems[41].
Interface chemistry
To evaluate the long-term compatibility of the electrolytes with lithium metal anodes, Li||Li symmetric cells are assembled using p(DAH) and p(DAH-co-Am-DAH) electrolytes and subjected to galvanostatic cycling tests [Figure 5A]. The p(DAH-co-Am-DAH) cell exhibits superior stability, operating for 1,700 h at a current density of 0.5 mA·cm-2 with a stable overpotential. In comparison, the p(DAH) succumbs to an internal short circuit after approximately 1,600 h, accompanied by a noticeably higher overpotential. This electrochemical durability is further corroborated by post-cycling morphological analysis via SEM [Figure 5B and C]. The lithium surface cycled with p(DAH-co-Am-DAH) retains a smooth and dense texture free of obvious dendrites, whereas the p(DAH)-cycled surface appears significantly rougher, confirming that the modified electrolyte promotes more uniform lithium deposition.
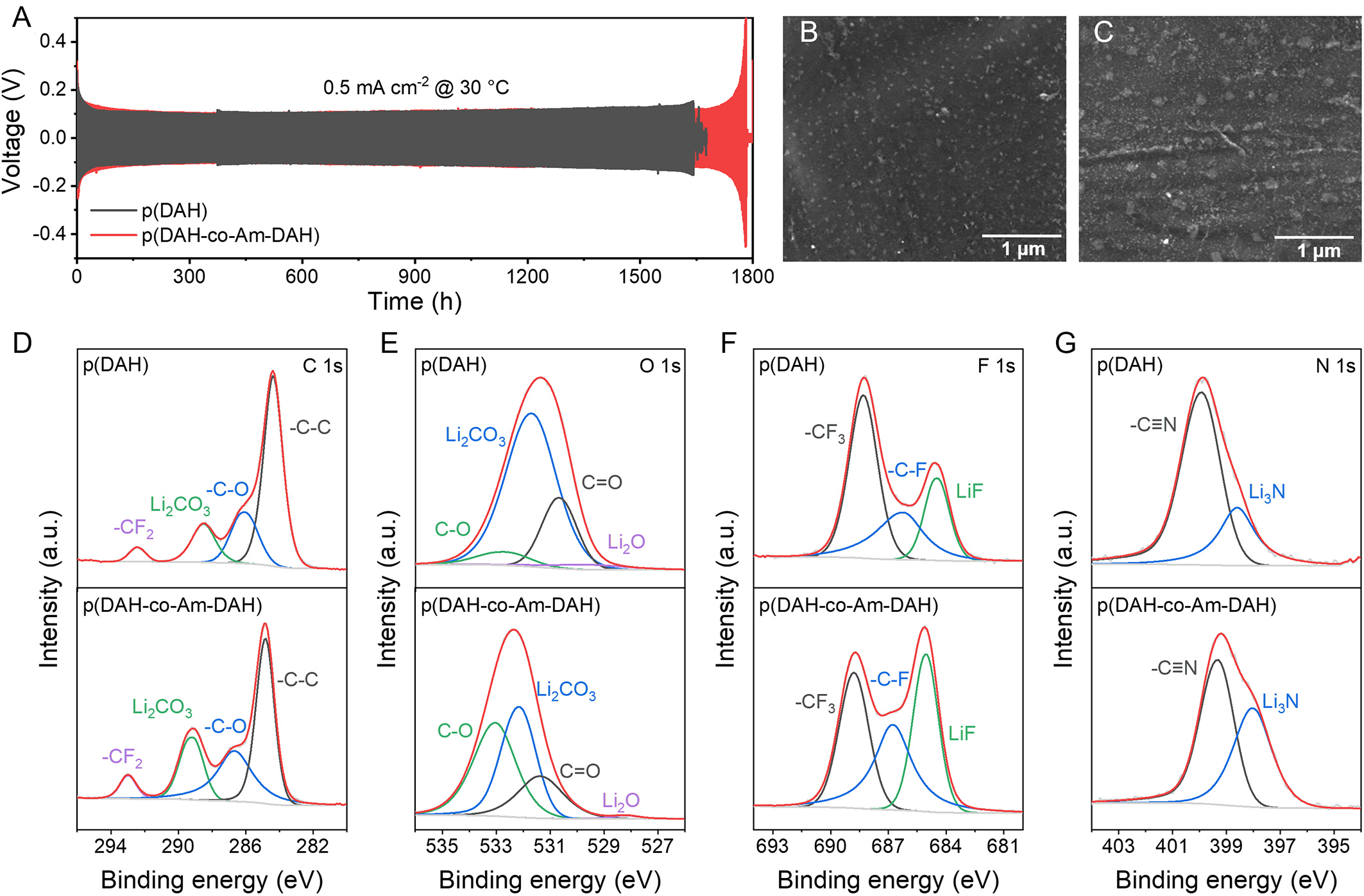
Figure 5. (A) Cycling performances of Li||Li symmetric cells. SEM images of Li metal surface after 100 h of cycling at 0.5 mA·cm-2 with (B) p(DAH-co-Am-DAH) and (C) p(DAH). XPS spectra and fitting results of (D) C 1s, (E) O 1s, (F) F 1s, and (G) N 1s for p(DAH) and p(DAH-co-Am-DAH) after cycling. SEM: Scanning electron microscopy; DAH: bis[2-(acryloyloxy)ethyl] heptanedioate; Am-DAH: bis[2-(acrylamido)ethyl] heptanedioate; XPS: X-ray photoelectron spectroscopy.
To elucidate the chemical origin of this enhanced stability, the SEI composition is probed using XPS. As a highly surface-sensitive technique (typically probing the top 5-10 nm), XPS specifically reveals the chemical composition and robust nature of the SEI formed on the electrode surface, rather than the bulk electrolyte. Accordingly, Figure 5D-G displays the high-resolution spectra of C 1s, O 1s, F 1s, and N 1s for p(DAH) and p(DAH-co-Am-DAH), respectively. The C 1s spectra are deconvoluted into four peaks at 284.8 eV (C-C), 286.8 eV (C-O), 289.4 eV (-C≡N) and 293.1 eV (-CF3)[42,43]. The C-C and C-O signals originate from the decomposition of the polymer backbone, while the -C≡N and -CF3 are derived from the decomposition of SN and TFSI-, respectively[44]. The O 1s spectra further confirm the partial decomposition of the polymer skeleton with peaks at 528.1 eV (Li2O), 530.8 eV (C=O), 531.7 eV (Li2CO3) and 532.5 eV (C-O)[44]. Crucially, the F 1s and N 1s spectra reveal peaks at 684.8 eV (LiF), 687.1/689.0 eV (-C-F/-CF3), 398.4 eV (Li3N), and 399.5 eV (-C≡N)[44-46]. Notably, compared to p(DAH), the p(DAH-co-Am-DAH) exhibits higher relative intensities for LiF and Li3N peaks, indicating that a greater proportion of TFSI- anions participated in the SEI formation[47]. This high-quality interface effectively suppresses dendrite growth and minimizes side reactions, thereby ensuring the superior long-term cycling stability and compatibility of the p(DAH-co-Am-DAH) electrolyte.
Battery performance
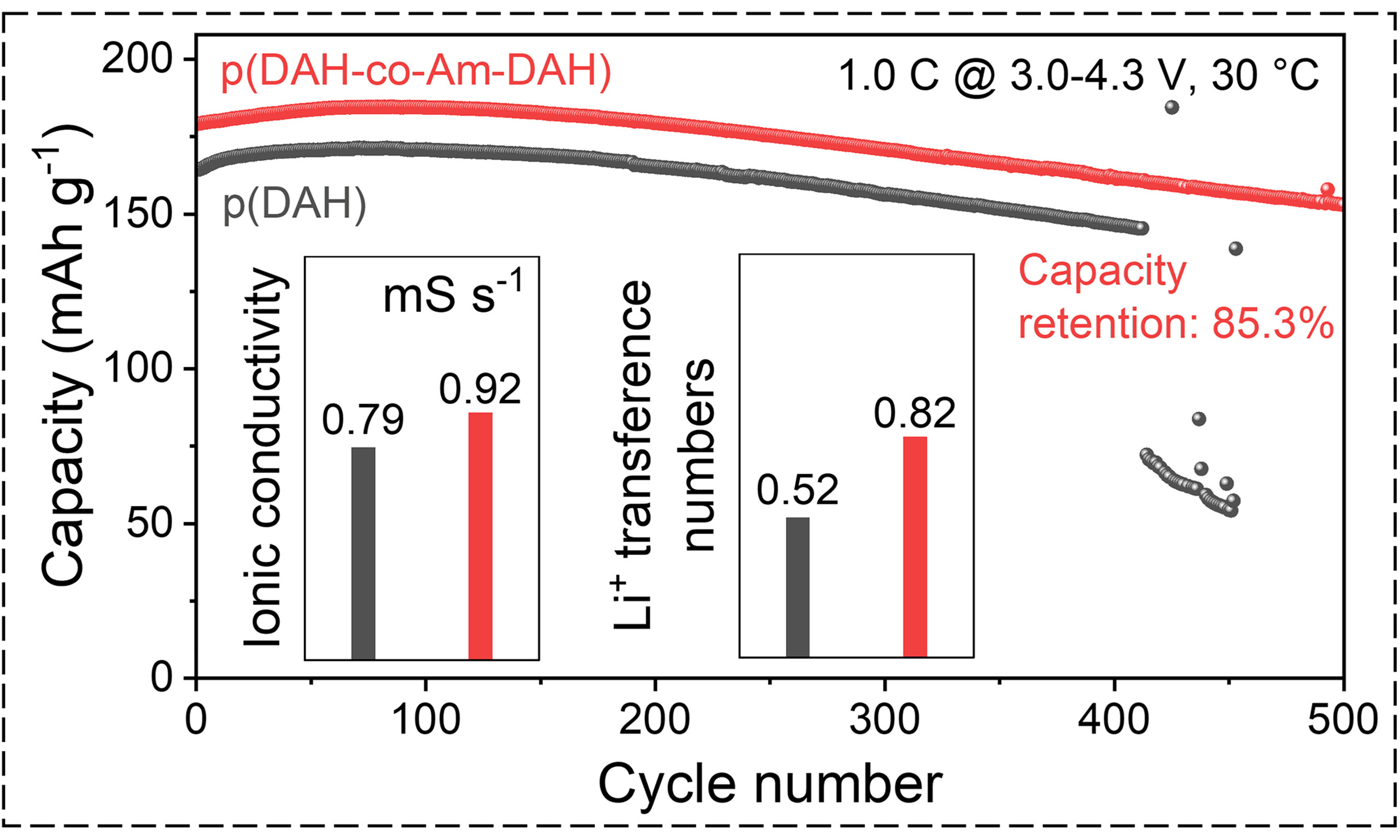
To further validate the practical applicability of the electrolytes beyond symmetric cells, full cells are assembled using a nickel-rich LiNi0.8Co0.1Mn0.1O2 (NCM811) cathode. NCM811 is regarded as one of the most promising cathode materials for high-energy-density lithium-ion batteries due to its high specific capacity and high operating voltage. The Li|p(DAH-co-Am-DAH)|NCM811 and Li|p(DAH)|NCM811 coin cells are assembled and subjected to rate capability and long-term cycling tests within a voltage range of 3.0-4.3 V. Figure 6A and B displays the rate capability of the cells at currents of 0.1, 0.2, 0.5, 1, and 2 C (1 C = 200.0 mAh·g-1). The Li|p(DAH-co-Am-DAH)|NCM811 cell delivers discharge capacities of 220.8, 212.8, 200.5, 187.3, and 152.6 mAh·g-1, respectively. In comparison, the Li|p(DAH)|NCM811 cell yields lower capacities of 200.6, 193.4, 181.1, 167.4, and 137.1 mAh·g-1, respectively. This superior rate performance is attributed to the higher ionic conductivity and higher Li+ transference number of p(DAH-co-Am-DAH) compared to p(DAH), which enables faster charge/discharge kinetics. The robustness of the system is further confirmed by the rate recovery test [Figure 6C]. After undergoing the variable-rate testing protocol, the Li|p(DAH-co-Am-DAH)|NCM811 cell recovers a discharge capacity of 214.4 mAh·g-1 at 0.1 C, retaining 97.1% of its initial capacity.
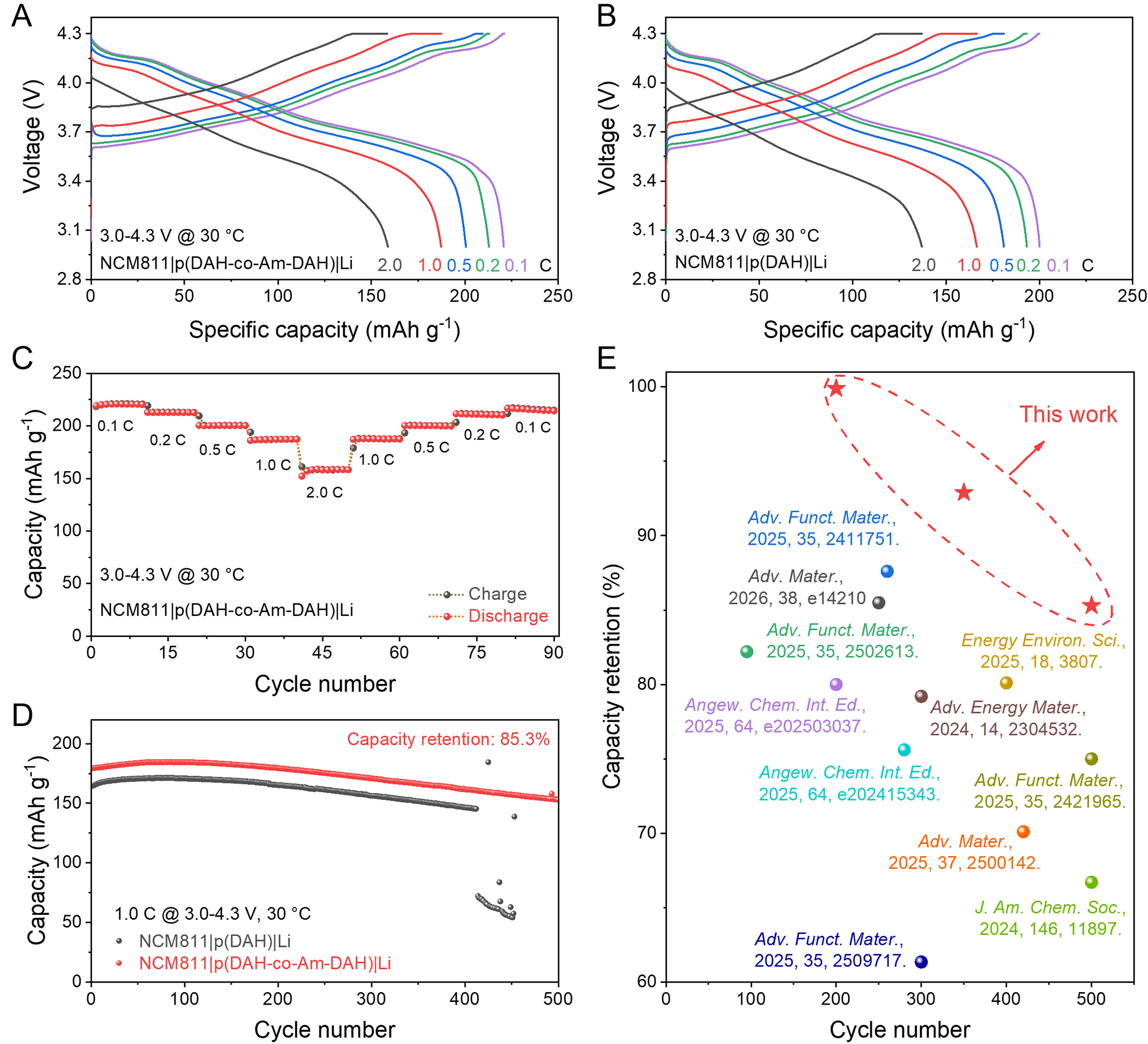
Figure 6. Voltage-capacity profile of Li||NCM811 with (A) p(DAH-co-Am-DAH) and (B) p(DAH); (C) Rate performance of Li|p(DAH-co-Am-DAH)|NCM811; (D) Cycling performance of Li|p(DAH-co-Am-DAH)|NCM811 and Li|p(DAH)|NCM811; (E) Performance comparison of Li|p(DAH-co-Am-DAH)|NCM811 with other representative SPEs in the literature[33,48-57]. DAH: Bis[2-(acryloyloxy)ethyl] heptanedioate; Am-DAH: bis[2-(acrylamido)ethyl] heptanedioate; SPEs: solid polymer electrolytes.
Finally, the long-term cycling stability at 1 C is evaluated, as shown in Figure 6D. The p(DAH-co-Am-DAH) system demonstrates exceptional durability, with capacity retentions of 99.9%, 92.9% and 85.3% at the 200th, 350th, and 500th cycles, respectively. In comparison, the p(DAH) system suffers a sudden capacity decay after 413 cycles and fails to recover, accompanied by severe fluctuations in Coulombic efficiency. To confirm that the enhanced performance originates from intrinsic material properties rather than experimental variations, a stringent high-voltage cycling test (2.8-4.6 V) is conducted [Supplementary Figure 7]. Under these harsh conditions, the performance gap becomes highly pronounced. The p(DAH-co-Am-DAH) SPE maintains stable operation for 200 cycles with 92.2% capacity retention. In contrast, the p(DAH) cell experiences a sudden capacity drop at the 114th cycle, followed by rapid degradation. This contrast demonstrates that the p(DAH-co-Am-DAH) exhibits superior interfacial compatibility and structural stability with the NCM811 cathode, ensuring a prolonged lifespan under severe electrochemical stress. Post-cycling SEM analysis on the NCM811 cathodes with p(DAH-co-Am-DAH) and p(DAH) [Supplementary Figure 8] provides direct morphological evidence to support this conclusion. This long-term stability significantly outperforms that of representative electrolytes reported in recent literature [Figure 6E and Supplementary Table 1], further demonstrating the immense potential of the p(DAH-co-Am-DAH) polymer electrolyte for high-voltage cathode applications.
CONCLUSIONS
In summary, to address the challenges of lithium metal batteries, this work presents the design and fabrication of a high-performance polymer electrolyte, p(DAH-co-Am-DAH), engineered with amide-based hydrogen bond donors. The core of this research highlights that regulating intermolecular interactions within the electrolyte effectively optimizes the solvation structure, facilitates lithium-ion transport, and stabilizes the solid electrolyte interphase, thereby significantly enhancing the overall cycling stability and electrochemical performance of the batteries. This study demonstrates that regulating intermolecular interactions is a potent strategy for developing advanced high-energy-density lithium metal batteries.
DECLARATIONS
Authors’ contributions
Methodology, investigation, data curation, formal analysis, writing-original draft: Ren, T.
Formal analysis, methodology: Zhang, J.; Wan, K.
Conceptualization, writing-review and editing, supervision, funding acquisition: Fu, Z.; Liang, Z.; Wan, K.
Availability of data and materials
The original contributions presented in this study are included in the article/Supplementary Materials. Further inquiries can be directed to the corresponding authors.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This work was supported by the Guangdong Major Project of Basic and Applied Basic Research (No. 2023B0303000002), the National Natural Science Foundation of China (Nos. 22325802, U22A20417, 92372106, 22578135, 22538003), the Commanding Heights of Science and Technology of Chinese Academy of Sciences (No. LDES150000), the Guangdong Basic and Applied Basic Research Foundation (No. 2023B1515120005), the Guangdong Natural Science Foundation (No. 2024A1515011963), the State Key Laboratory of Catalysis (No. 2024SKL-A-009), the Energy Revolution S&T Program of Yulin Innovation Institute of Clean Energy (No. E411120705).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
Supplementary Materials
REFERENCES
1. Alsaç, E. P.; Nelson, D. L.; Yoon, S. G.; et al. Characterizing electrode materials and interfaces in solid-state batteries. Chem. Rev. 2025, 125, 2009-119.
2. He, M.; Hector, L. G.; Dai, F.; et al. Industry needs for practical lithium-metal battery designs in electric vehicles. Nat. Energy. 2024, 9, 1199-205.
3. Tang, C.; Yu, L.; Jiang, Q.; et al. Polymer-based electrolyte for lithium-based high-energy-density and safe energy storages devices: strategy and mechanisms. Renewables 2024, 2, 297-340.
4. Wu, F. Jointly Create a brilliant future of superior development for the global lithium battery industry. Energy. Mater. Adv. 2025, 6, 0186.
5. Zhang, H.; Zeng, Z.; Cheng, S.; Xie, J. Recent progress and perspective on lithium metal battery with nickel-rich layered oxide cathode. eScience 2024, 4, 100265.
6. Wang, Z.; Xia, J.; Ji, X.; et al. Lithium anode interlayer design for all-solid-state lithium-metal batteries. Nat. Energy. 2024, 9, 251-62.
7. Geng, N.; Lu, C.; Li, W.; et al. Fluoro-functionalized polyether electrolytes enabling high-energy-density solid-state lithium-metal batteries. Energy. Environ. Mater. 2026, 9, e70188.
8. Song, W.; Sun, R.; Chen, K.; Wang, Y.; Jian, X.; Hu, F. Molecular engineering of polymer electrolytes for lithium and sodium metal batteries. Small 2026, 22, e14635.
9. Luo, C.; Ning, C.; Huang, X.; et al. Molecular engineering of weakly solvating dinitrile electrolytes for long-lasting room-temperature lithium metal batteries. Angew. Chem. Int. Ed. 2025, 64, e202507051.
10. Kou, W.; Zhang, Y.; Wu, W.; Guo, Z.; Hua, Q.; Wang, J. Thin polymer electrolyte with MXene functional layer for uniform Li+ deposition in all-solid-state lithium battery. Green. Energy. Environ. 2024, 9, 71-80.
11. Tang, W.; Zhou, T.; Duan, Y.; Zhou, M.; Li, Z.; Liu, R. Nonflammable in situ PDOL-based gel polymer electrolyte for high-energy-density and high safety lithium metal batteries. Carbon. Neutraliz. 2024, 3, 386-95.
12. Huang, Z.; Lyu, H.; Greenburg, L. C.; Cui, Y.; Bao, Z. Stabilizing lithium-metal electrodes with polymer coatings. Nat. Energy. 2025, 10, 811-23.
13. Wang, Y.; Wu, Z.; Azad, F. M.; et al. Fluorination in advanced battery design. Nat. Rev. Mater. 2023, 9, 119-33.
14. Zhang, C.; Li, Z.; Wang, S.; et al. Boosting high-rate lithium metal batteries by using ether-based gel polymer electrolyte. Energy. Mater. Adv. 2025, 6, 0188.
15. Jiang, D.; Wang, X.; Yin, S.; et al. Solid-state electrolytes with vertically aligned Li+ transport channels for lithium batteries: a comprehensive review. Energy. Storage. Mater. 2025, 74, 103986.
16. Wang, Z.; Chen, J.; Fu, J.; Li, Z.; Guo, X. Polymer-based electrolytes for high-voltage solid-state lithium batteries. Energy. Mater. 2024, 4, 400050.
17. Shen, Z.; Cheng, Y.; Sun, S.; Ke, X.; Liu, L.; Shi, Z. The critical role of inorganic nanofillers in solid polymer composite electrolyte for Li+ transportation. Carbon. Energy. 2021, 3, 482-508.
18. Shan, C.; Wang, Y.; Liang, M.; et al. A comprehensive review of single ion-conducting polymer electrolytes as a key component of lithium metal batteries: from structural design to applications. Energy. Storage. Mater. 2023, 63, 102955.
19. Shi, J.; Nguyen, H.; Chen, Z.; et al. Nanostructured block copolymer single-ion conductors for low-temperature, high-voltage and fast charging lithium-metal batteries. Energy. Mater. 2023, 3, 300036.
20. Han, J.; Lee, M. J.; Lee, K.; et al. Role of bicontinuous structure in elastomeric electrolytes for high-energy solid-state lithium-metal batteries. Adv. Mater. 2022, 35, 2205194.
21. Bard, A.J.; Faulkner, L.R.; White, H.S. Electrochemical methods: fundamentals and applications, 3rd ed.; John Wiley & Sons, 2022. https://www.wiley.com/en-us/Electrochemical+Methods%3A+Fundamentals+and+Applications%2C+3rd+Edition-p-9781119334057 (accessed 2026-04-28).
22. Atkins, P.W.; De Paula, J.; Keeler, J. Atkins’ physical chemistry, 12th ed.; Oxford University Press, 2023. https://global.oup.com/ukhe/product/atkins-physical-chemistry-9780198847816?cc=us&lang=en& (accessed 2026-04-28).
23. Evans, J.; Vincent, C. A.; Bruce, P. G. Electrochemical measurement of transference numbers in polymer electrolytes. Polymer 1987, 28, 2324-8.
24. Bruce, P. G.; Vincent, C. A. Steady state current flow in solid binary electrolyte cells. J. Electroanal. Chem. Interfacial. Electrochem. 1987, 225, 1-17.
25. Hutter, J.; Iannuzzi, M.; Schiffmann, F.; Vandevondele, J. cp2k: atomistic simulations of condensed matter systems. WIREs. Comput. Mol. Sci. 2013, 4, 15-25.
26. Vandevondele, J.; Hutter, J. Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases. J. Chem. Phys. 2007, 127, 114105.
27. Buin, A.; Pietsch, P.; Xu, J.; et al. Materials processing routes to trap-free halide perovskites. Nano. Lett. 2014, 14, 6281-6.
28. Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104.
29. Cui, J.; Liu, F.; Lu, Z.; et al. Repeatedly recyclable 3D printing catalyst-free dynamic thermosetting photopolymers. Adv. Mater. 2023, 35, 2211417.
30. Han, D.; Kim, S.; Kim, Y.; et al. Regulating segmental dynamics for ion clusters in polymer binders to realize high-areal-capacity electrodes in lithium batteries. Energy. Environ. Sci. 2025, 18, 7514-26.
31. Gerdroodbar, A. E.; Alihemmati, H.; Safavi-mirmahaleh, S.; et al. A review on ion transport pathways and coordination chemistry between ions and electrolytes in energy storage devices. J. Energy. Storage. 2023, 74, 109311.
32. He, Y.; Wang, C.; Zou, P.; Lin, R.; Hu, E.; Xin, H. L. Anion-tethered single lithium-ion conducting polyelectrolytes through UV-induced free radical polymerization for improved morphological stability of lithium metal anodes. Angew. Chem. Int. Ed. 2023, 62, e202308309.
33. Tao, F.; Yan, K.; Dong, C.; et al. Electric-dipole coupling ion-dipole engineering induced rational solvation-desolvation behavior for constructing stable solid-state lithium metal batteries. Angew. Chem. Int. Ed. 2025, 64, e202503037.
34. Liu, Y.; Jin, Z.; Liu, Z.; et al. Regulating the solvation structure in polymer electrolytes for high-voltage lithium metal batteries. Angew. Chem. Int. Ed. 2024, 63, e202405802.
35. Hong, H.; Zhu, J.; Wang, Y.; et al. Metal-free eutectic electrolyte with weak hydrogen bonds for high-rate and ultra-stable ammonium-ion batteries. Adv. Mater. 2023, 36, 2308210.
36. Xia, M.; Fu, H.; Lin, K.; et al. Hydrogen-bond regulation in organic/aqueous hybrid electrolyte for safe and high-voltage K-ion batteries. Energy. Environ. Sci. 2024, 17, 1255-65.
37. Xue, S.; Zhou, Y.; Zhang, Z.; Hou, K.; He, M.; Liu, X. “Dragging” effect induced formation of AGGs-rich solvation structures in lithium metal batteries. J. Energy. Storage. 2024, 95, 112558.
38. Shi, T.; Hou, R.; Zheng, L.; et al. Modulating double-layer solvation structure via dual-weak-interaction for stable sodium-metal batteries. Adv. Energy. Mater. 2025, 15, 2405803.
39. Yi, T.; Zhao, E.; He, Y.; Liang, T.; Wang, H. Quantification and visualization of spatial distribution of dendrites in solid polymer electrolytes. eScience 2024, 4, 100182.
40. Zhou, P.; Zhang, X.; Xiang, Y.; Liu, K. Strategies to enhance Li+ transference number in liquid electrolytes for better lithium batteries. Nano. Res. 2022, 16, 8055-71.
41. Han, X.; Jiang, H.; Mu, P.; et al. Helmholtz plane regulation empowers PF6- permselectivity towards high coulombic efficiency dual ion battery of 5.5 V. Angew. Chem. Int. Ed. 2024, 64, e202412753.
42. Li, Y.; Ma, S.; Zhao, Y.; et al. Synergetic control of Li+ transport ability and solid electrolyte interphase by boron-rich hexagonal skeleton structured all-solid-state polymer electrolyte. Energy. Environ. Mater. 2023, 7, e12648.
43. Pan, Q.; Zhang, S.; Gui, X.; et al. Carbonyl-engineered acrylate-based solid polymer electrolyte for high-performance lithium metal batteries. Adv. Energy. Mater. 2025, 15, e02815.
44. Zhang, H.; Deng, J.; Xu, H.; et al. Molecule crowding strategy in polymer electrolytes inducing stable interfaces for all-solid-state lithium batteries. Adv. Mater. 2024, 36, 2403848.
45. Qi, S.; Li, M.; Gao, Y.; et al. Enabling scalable polymer electrolyte with dual-reinforced stable interface for 4.5 V lithium-metal batteries. Adv. Mater. 2023, 35, 2304951.
46. Park, J.; Lee, D.; Kim, S.; et al. Fluorine-, nitrogen-, and boron-functionalized polymer electrolytes for advanced lithium metal batteries. ACS. Nano. 2025, 19, 15220-55.
47. Yao, N.; Sun, S. Y.; Chen, X.; et al. The anionic chemistry in regulating the reductive stability of electrolytes for lithium metal batteries. Angew. Chem. Int. Ed. 2022, 61, e202210859.
48. Zhao, C.; Lu, Y.; Yan, K.; et al. Tailoring the chemical/electrochemical response in a quasi-solid polymer electrolyte enables the simultaneous in situ construction of superior cathodic and anodic interfaces. Adv. Energy. Mater. 2024, 14, 2304532.
49. Chen, X.; Chu, F.; Li, D.; Si, M.; Liu, M.; Wu, F. In situ polymerized fluorine-free ether gel polymer electrolyte with stable interface for high-voltage lithium metal batteries. Adv. Funct. Mater. 2024, 35, 2421965.
50. Jiang, Y.; Liu, L.; Liu, Y.; et al. A novel orientation aliphatic ketone-based liquid crystal polymer electrolyte for high-voltage solid-state lithium metal batteries. Adv. Funct. Mater. 2025, 35, 2502613.
51. Li, C.; Zhong, Y.; Liao, R.; et al. Robust and antioxidative quasi-solid-state polymer electrolytes for long-cycling 4.6 V lithium metal batteries. Adv. Mater. 2025, 37, 2500142.
52. Miao, X.; Hong, J.; Huang, S.; et al. In situ gel polymer electrolyte with rapid Li+ transport channels and anchored anion sites for high-current-density lithium-ion batteries. Adv. Funct. Mater. 2024, 35, 2411751.
53. Peng, J.; Lu, D.; Wu, S.; et al. Lithium superionic conductive nanofiber-reinforcing high-performance polymer electrolytes for solid-state batteries. J. Am. Chem. Soc. 2024, 146, 11897-905.
54. Zhang, Q.; Bian, T.; Wang, X.; Shi, R.; Zhao, Y. Unlocking mechanism of anion and cation interaction on ion conduction of polymer based electrolyte in metal batteries. Angew. Chem. Int. Ed. 2024, 64, e202415343.
55. Zhang, S.; Li, Z.; Zhang, Y.; et al. Moderate Li+-solvent binding for gel polymer electrolytes with stable cycling toward lithium metal batteries. Energy. Environ. Sci. 2025, 18, 3807-16.
56. Wang, Y.; Zhang, S.; Chen, Z.; et al. Long-life lithium metal batteries enabled by in situ solidified polyphosphoester-based electrolyte. Adv. Mater. 2025, 38, e14210.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].