

Recent advances in synergetic catalysis of single-atom catalysts in biomass conversion, CO2 reduction, and cascade reaction
 0
0  ,
, Abstract
Single-atom catalysts (SACs) have demonstrated immense potential in the fields of energy and biochemical conversions. Their unique properties make them particularly efficient and selective. Characterized by their unique isolated state on supports, SACs often showcase poor synergy in the multiple molecule conversion because of the infrequent communications with others. Recently, some interesting synergetic processes of SACs in some special reactions have been found; however, to our best knowledge, the synergetic catalytic effect of SACs has not been systematically summarized. This article comprehensively overviews the basic concepts, mainstream synthetic methods, and modification strategies of SACs and emphasizes the synergetic catalysis effect in biomass conversion, CO2 reduction, and cascade reaction by itself or other media. To maximize the advantage of SACs, various synthetic methods for them, including impregnation, co-impregnation, co-precipitation, atomic layer deposition, electrochemical methods, photochemical methods, pyrolysis synthesis, spray pyrolysis method, ball-milling and chemical vapor deposition method are discussed. This review provides a comprehensive discussion concerning the dynamic interplay between precisely engineered atomic architectures and their synergistic functionalities in modulating catalytic efficacy, with focused exploration of activity enhancement strategies and operational stability optimization. Distinctively, it pioneers the establishment of comprehensive theoretical frameworks that unify SAC-driven synergistic catalytic mechanisms across three critical domains: biomass valorization, electrochemical CO2 conversion, and multi-step cascade processes. Through systematic synthesis of cutting-edge developments in coordination microenvironment modulation, inter-site electronic communication, and fundamental mechanistic studies, this work affords some interesting insight into rational design of synergistic SAC systems, thereby addressing current challenges in translating atomic-scale precision to heterogeneous catalytic performance.
Keywords
INTRODUCTION
Single-atom catalysts (SACs) are defined as isolated atom active sites, which can be fabricated through precisely anchoring precious or non-precious metal atoms onto a carrier, wherein, each isolated single atom serves as the pivotal active center of the reaction[1-4]. In 2011, Zhang et al. made a remarkable breakthrough by preparing the single-atom Pt1/FeOx catalysts and pioneering the concept of single-atom catalysis[5]. Through an array of advanced characterization techniques such as X-ray absorption fine structure (XAFS), aberration-corrected (AC) electron microscopy, and infrared spectroscopy, the dispersed Pt single atomic states were successfully observed. This catalyst not only demonstrated robust activity and remarkable selectivity but also afforded outstanding stability in the CO oxidation reaction. This seminal study laid a solid foundation for the subsequent explosive development of SACs[6]. The isolated metal atoms within SACs are firmly anchored to the support, either via the strong interactions between the metal and the support or by forging stable chemical bonds with the support material. Depending on these distinct interaction mechanisms, SACs can be categorized into various types, prominently featuring metal single atoms supported on metal oxide surfaces, metals, carbon-based materials, metal-organic-framework materials (MOFs), covalent organic framework (COFs), composite materials, etc.
SACs are widely regarded as a crucial bridge connecting homogeneous and heterogeneous catalysts. Each of their active centers exhibits an identical geometric structure, mirroring the characteristics of homogeneous catalysts, while simultaneously retaining the inherent advantages of heterogeneous ones. The homogeneity of single atomic sites endows SACs with an exceptional capacity to confer high selectivity for specific products[7]. This unique combination effectively overcomes the long-standing limitations of both homogeneous and heterogeneous catalysts. Moreover, SACs possess several remarkable attributes, including the highest atomic utilization efficiency, a one-of-a-kind atomic structure and electronic properties, an unsaturated coordination environment, and the resulting extraordinary catalytic activity[4]. Diverging fundamentally from conventional nanoparticle (NP) catalysts constrained by limited surface atom accessibility (typically < 15% active site exposure), SACs achieve near-unity atomic utilization through site-isolated configurations while enabling unprecedented cooperative electronic interplay. For instance, Pt NPs typically exhibit low turnover frequencies (TOFs) in CO oxidation due to the dominance of inert bulk atoms, whereas SACs such as Pt1/FeOx achieve near-100% atomic efficiency with exceptional activity and selectivity[5,8]. Similarly, clusters (sub-nm aggregates of 2-10 atoms) often suffer from nonuniform active sites and dynamic structural instability under reaction conditions, whereas SACs possess uniform coordination environments, enabling precise structure-activity correlations[9]. These features have spotlighted SACs as a game-changer in the rational utilization of metal resources, attracting extensive attention from the scientific community.
However, it is imperative to note that SACs still suffer from some big challenges. Since the size of active species was tailored to the atomic level, the high surface free energy inevitably causes the later migration or aggregation of these isolated atoms. This high activity of SACs would cause the compromise of stability, which are two critical factors for its practical application. Secondly, the isolated atom active centers are generally powerless for the complex multiple molecule activation process, especially since the communication of adjacent atom sites is infrequent, which would showcase the poor synergy in the reaction process. The synergetic effect of SACs with other components has inspired the pioneering development of SACs in the complex reaction process especially for biomass conversion, CO2 reduction, and cascade reaction.
In light of these issues, the rational modulation of the synergistic catalytic effect between single atoms and other active centers emerges as a top priority in this field. However, to our best knowledge, there is no related review reported for the rational synergetic catalysis of SACs. To this end, this work comprehensively overviews recent experimental advancements in synergetic catalysis using SACs, with a focus on their synthesis, modification, and application in biomass conversion, CO2 reduction, and cascade reactions. Meanwhile, theoretical insights [e.g., density functional theory (DFT) calculations] are occasionally integrated to elucidate mechanistic principles. This review prioritizes experimental methodologies - from synthetic strategies and characterization techniques to performance evaluations - to highlight practical progress in optimizing the catalytic efficiency and stability of SACs. By emphasizing empirical studies, this work aims to bridge the gap between laboratory-scale innovations and scalable catalytic solutions for sustainable energy and environmental challenges.
SYNTHESIS STRATEGIES
Impregnation and co-impregnation method
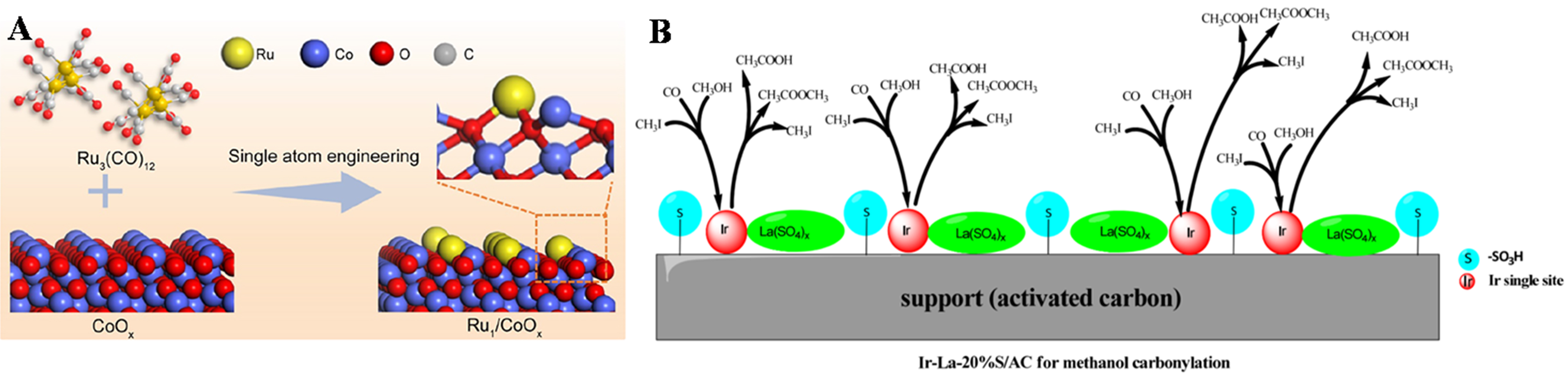
The impregnation method through introducing active components onto the surface of the carrier within the solution, plays a significant role in fabricating SACs. In this typical process, a solution containing metal salt solution mixed with the carrier easily triggers the anchoring of atomic active sites on the surface of the support. The surface adsorption and capillary condensation would maximize the contact of main metal ions, which allows for the controlled loading of metal ions by altering the concentration and the amount of dipping solution. For instance, Gu et al.[10] successfully prepared the Ru1/CoOx catalyst using impregnation, wherein Ru single atoms (Ru1) were anchored on the CoOx surface at an ultra-low loading of 0.5 wt% [Figure 1A]. The catalyst demonstrated exceptional efficiency in the selective electro-oxidation of
Co-precipitation method
The co-precipitation method is capable of obtaining uniformly dispersed supported metal catalysts with different active species by simultaneously precipitating insoluble compounds from two or more soluble salts in a mixed solution. The active metal atoms can be incorporated into the support when occurring a
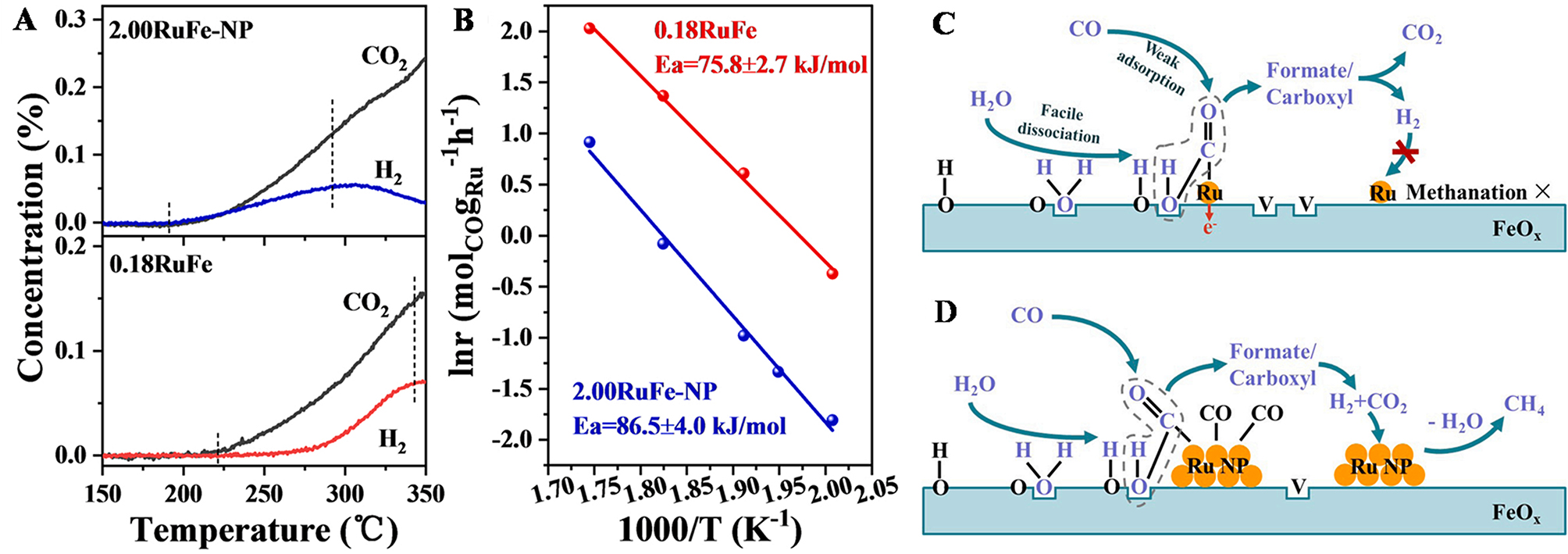
Figure 2. (A) MS analysis of introducing CO with temperatures on 0.18RuFe and 2.00RuFe-NP catalysts preadsorbed with 10% H2O at 250 °C, respectively; (B) Arrhenius plots of the reaction rate vs. 1/T for the WGS reaction on 0.18RuFe and 2.00RuFe-NP catalysts, with Ea indicated in the plot. Schematic illustration of the associative mechanism with the methanation side reaction during WGS reaction over 0.18RuFe (C) and 2.00RuFe-NP (D) catalysts. Adapted with permission[14]. Copyright 2022, Elsevier B.V. MS: Mass spectrometry; WGS: water-gas shift.
Atomic layer deposition
Atomic layer deposition (ALD) allows substances to be deposited layer by layer on the substrate surface in the form of a single atomic layer. It utilizes gas-phase precursor pulses that alternately enter the reactor, undergo chemical adsorption and reaction on the deposited substrate, and form a deposited layer. Based on the self-limitation of surface reactions in ALD, repeating this self-limiting reaction creates the desired thin film. Liu et al.[15] explored the synthesis of single-atom platinum (SA Pt) catalysts on vertical graphene (VG) using ALD [Figure 3]. By controlling the nominal thickness of Pt at the atomic level with the self-limiting effect, and leveraging the large number of graphene edges as energy-favorable nucleation sites for SA Pt, they achieved enhanced stability. Compared with nano Pt, Pt single atoms had higher stability near the VG edge state, and their sensitivity and response speed were more than an order of magnitude higher. Song et al.[16] selectively anchored isolated Ir single atoms onto Pt NPs through ALD, modifying the Pt NP surface. The formation of the SA-NP composite structure significantly improved the performance of the
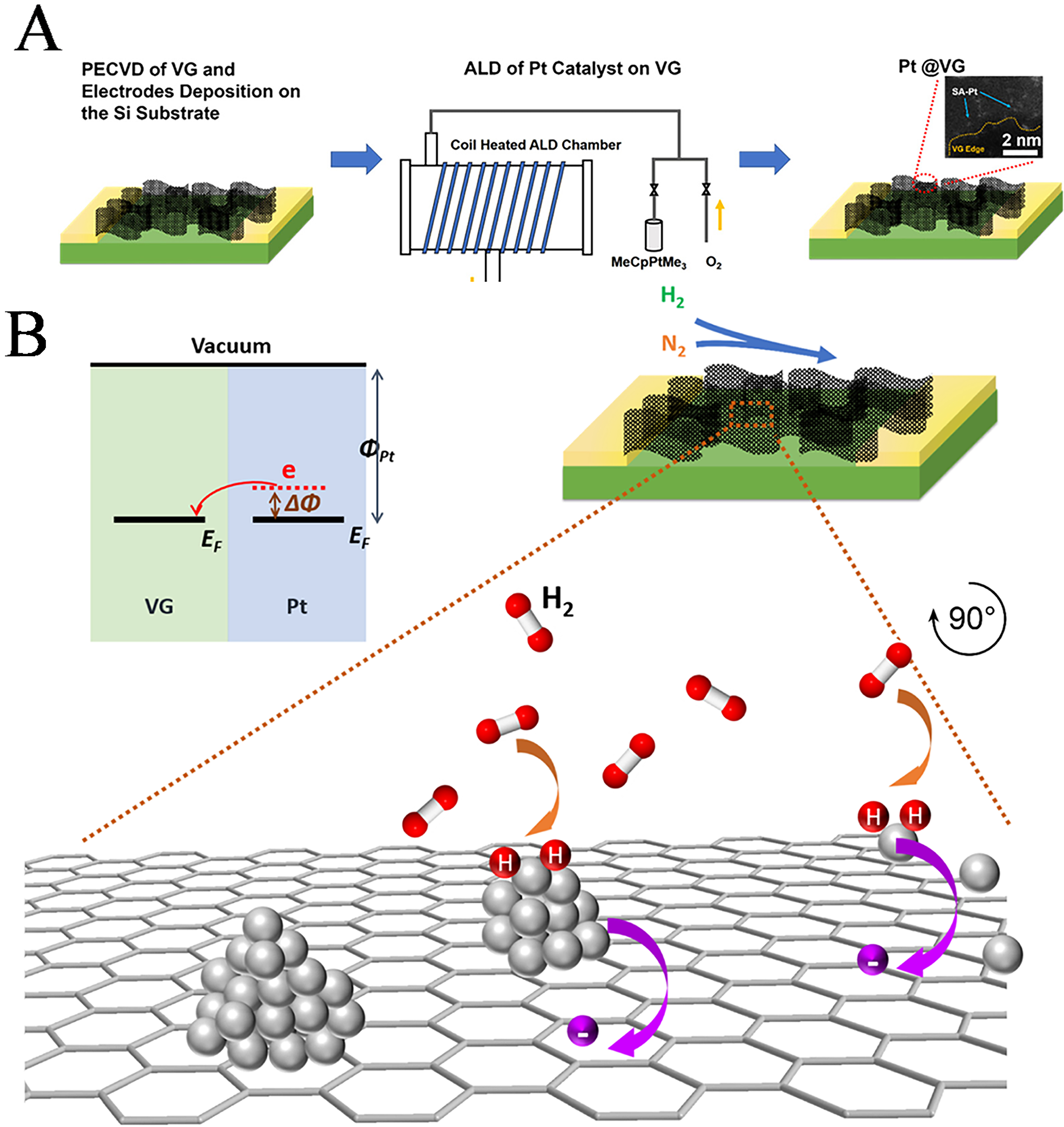
Figure 3. (A) Scheme of Fabrication of ALD Pt@VG; (B) Schematic illustration of the H2-sensing mechanism on the Pt@VG sensor with the role of the Pt catalyst in the adsorption and decomposition of H2 molecules, followed by a charge transfer process. Inset (left): schematic diagram showing charge transfer after H is adsorbed on Pt. EF represents the Fermi energy level and ΦPt represents the work function of Pt. Inset (right): schematic illustration of Pt-decorated VG grown on SiO2/Si substrates used for H2 sensing. Adapted with permission[15]. Copyright 2024, American Chemical Society. ALD: Atomic layer deposition; VG: vertical graphene.
Electrochemical methods and photochemical method
Electrochemical methods are generally allowed to operate in simple two- or three-electrode systems under ambient conditions. The synthesis parameters can be adjusted by altering input techniques such as current, voltage, scan rate, and reaction time to achieve desired products. Wu et al.[19] used electrochemical methods to embed 29 atomic transition metals into defective boron phosphide (BP) monolayers. Selecting Nb/BP and W/BP SAC as qualified electrocatalysts, they achieved high stability, low initial potential (as low as 0.25 and 0.19 V, respectively), and excellent selectivity (90.5% and 100%, respectively) for electrochemical N2 reduction reaction (NRR). Lv et al.[20] proposed a universal one-step electrochemical synthesis strategy to obtain various zeolite imidazolate frame-8 (ZIF-8) supported SACs by simply replacing different metal precursors [Figure 4]. Evaluating the platinum-based catalyst for electrosynthesis, it exhibited excellent activity and stability in the electrocatalytic hydrogen evolution reaction (HER). These electrochemical methods enable the in-situ generation and precise control of single-atom active sites, enhancing catalytic performance through effective synergy. Nitrogen-doped carbon-supported Pt single atom/cluster catalysts were prepared through electrochemical synthesis and subsequent pyrolysis processes Pt@NC. The active sites of Pt clusters/PtN3 (Pt clusters and nitrogen-doped carbon carriers) are mainly located on Pt clusters. However, the Pt clusters/PtN3 active species could further release the synergy, whose d-band center is closer to the Fermi level, which is obviously optimized compared to pure PtN3 and Pt clusters, helping to regulate the electronic structure of Pt clusters, optimize the adsorption strength of H, and thus improve HER activity.
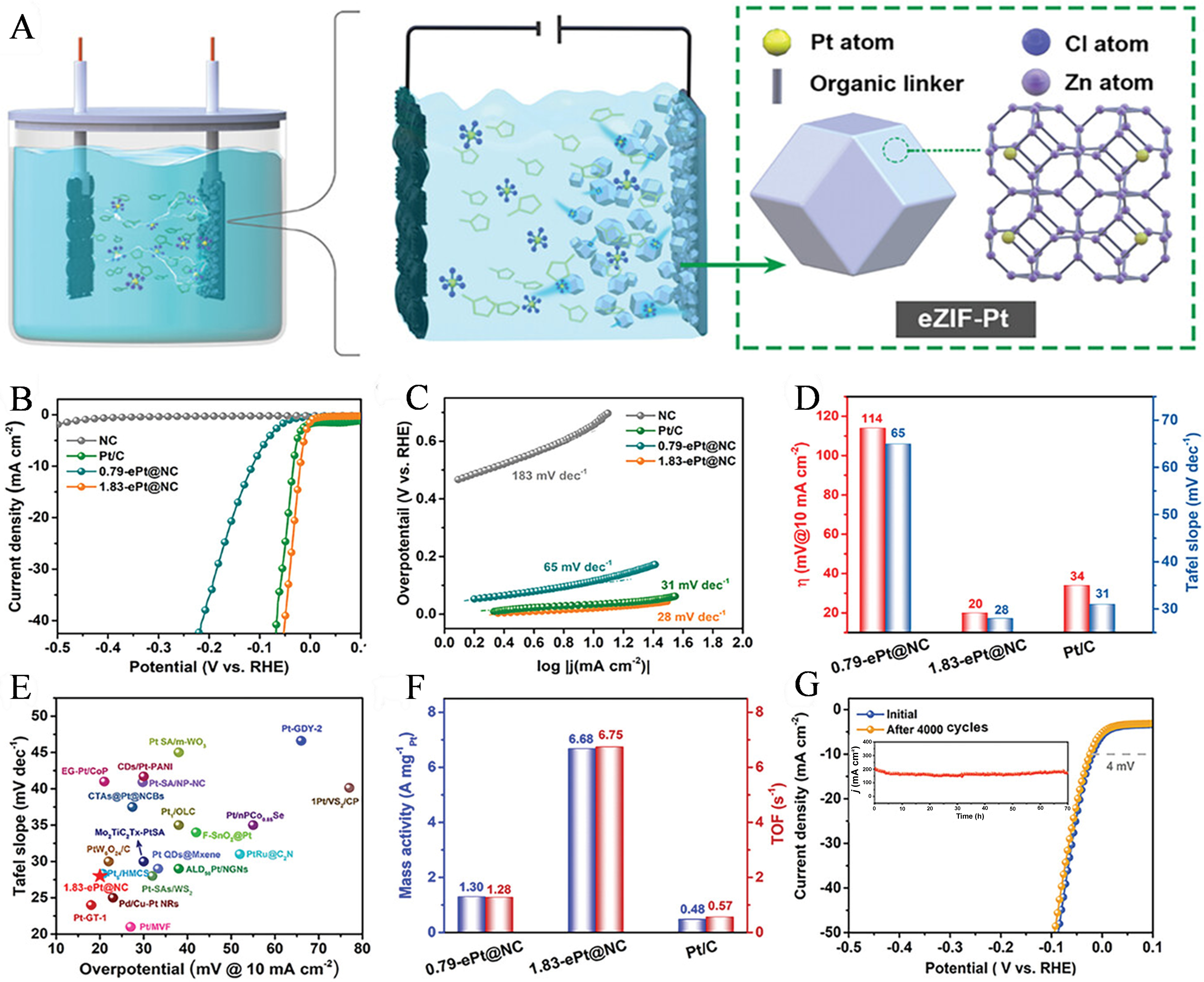
Figure 4. Preparation and characterizations of eZIF-Pt. (A) Schematic illustrating the synthesis of eZIF-Pt. Electrocatalytic HER performance and DFT calculations; (B) LSV curves of the 0.79-ePt@NC, 1.83-Pt@NC, commercial Pt/C, and NC; (C) The corresponding Tafel slope originated from LSV curves; (D) Comparison of overpotentials at 10 mA·cm-2 and Tafel slopes; (E) Comparison of the HER activity for 1.83-Pt@NC with reported catalysts; (F) Mass activity and TOF values at an overpotential of 100 mV; (G) Stability test of 1.83-Pt@NC through cyclic potential scanning (inset: chronoamperometry method). Adapted with permission[20]. Copyright 2023, Wiley-VCH GmbH. eZIF-Pt: Electrochemically synthesized zeolite imidazolate frame-8 with platinum single atoms; HER: hydrogen evolution reaction; LSV: linear sweep voltammetry; TOF: turnover frequency.
The photochemical method leverages ultraviolet (UV) light instead of electrochemical reduction, which requires the system to contain substances that can absorb UV light and release electrons. Liu et al.[21] employed a room-temperature photochemical strategy to prepare highly stable atomic level dispersed Pd catalysts (Pd1/TiO2) by irradiating ultra-thin TiO2 nanosheets stabilized with ethylene glycol (EG) through UV light. The Pd1/TiO2 catalyst exhibited extremely high activity in the hydrogenation reactions of C=C and C=O, nearly nine times enhancement compared to that of the Pd atoms on the surface of commercial Pd catalysts without any decay after 20 cycles. Ge et al.[22] used photochemical reduction to reduce PdCl42- on the surface of TiO2, forming the Pd single atom PdSA/TiO2 photocatalyst [Figure 5]. Due to the high dispersion of Pd single atoms on TiO2 and the high density of active sites, PdSA/TiO2 exhibited higher photocatalytic activity than Pd NPs PdNPs/TiO2, with a photocatalytic activity (10 mol·g-1·h-1) much higher than that of PdNPs/TiO2 (1.95 mol·g-1·h-1). This defines the synergetic promotion of Pd single atom with TiO2 support, through the improved photocurrent response and energy gap, which contributes to the higher photocatalytic properties.
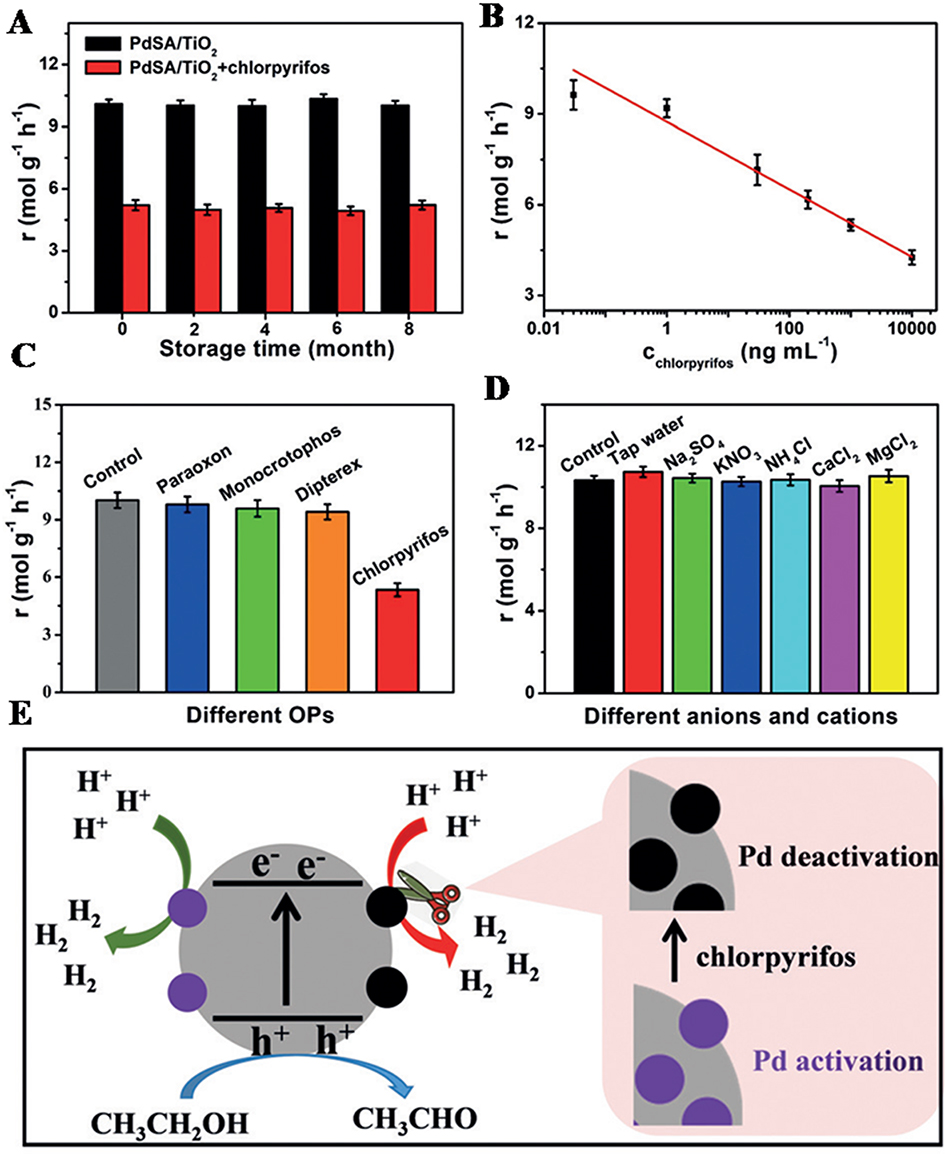
Figure 5. (A) The photocatalytic HER stability of PdSA/TiO2 before and after the introduction of chlorpyrifos; (B) The typically photocatalytic response of PdSA/TiO2 toward different concentrations of chlorpyrifos, 0.03, 1, 30, 200 ng·mL-1, 1, 10 μg·mL-1; (C) The interference assessment of other commonly used organophosphorus pesticides on chlorpyrifos determination; (D) The interference analysis of the anions and cations common in vegetable samples; (E) The principle of the PdSA/TiO2-based sensing platform utilizing photocatalytic H2 production. Adapted with permission[22]. Copyright 2020, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. HER: Hydrogen evolution reaction.
Pyrolysis synthesis method
The pyrolysis synthesis method requires heat treatment of metal precursors and carrier materials at high temperatures, usually in an inert atmosphere, to prevent oxidation of metal atoms. At high temperatures, the oxidation process easily triggers the aggregation of metal atoms, because of the loss of carbon framework, which can reduce the dispersion of active sites in the catalyst, thereby lowering catalytic efficiency and potentially leading to structural damage to the material. For example, in the pyrolysis process of certain MOFs, the oxidation of metals catalyzes the graphitization of carbon, leading to the collapse of MOF structures[23]. This is applicable for the abundant combination of various metals precursor and carrier materials, with a production scale of several kilograms per hour that can be used for large-scale production of SAC, which is of great significance for industrial applications[24].
Jiang et al.[25] developed high-density Fe single-atom assemblies through a carbothermal coordination-mediated synthesis strategy, utilizing graphitic carbon matrices to anchor metal complexes during pyrolysis [Figure 6A]. Through precise precursor dosage control on surface-engineered carbon matrices, the team achieved a record 7.5 wt% metal loading while maintaining atomic dispersion. Critical stabilization arises from atomic-scale chelation between Fe species and nitrogen dopants, where structural confinement effects through ordered M-N4 coordination motifs effectively mitigate metal clustering. Systematic evaluation revealed the ORR performance enhancement follows an active-site density dependency, with catalytic currents scaling proportionally to the concentration of intact Fe-N4 moieties. Ping et al.[26] developed a CO2RR catalyst (MgAl2O4/Ni-N-C) with abundant single Ni sites and MgAl2O4-NP (~14 nm) via a confinement pyrolysis strategy using NiMgAl LDHs as metal precursor [Figure 6B]. The MgAl2O4 NP with abundant oxygen vacancies stabilized Ni atoms and regulated the electronic structure of adjacent Ni atoms, promoting the conversion of CO2 to *COOH. MgAl2O4/Ni-N-C has a unique three-dimensional porous structure, which helps to expose active sites and disperse Ni atoms. The MgAl2O4/Ni-N4-C model exhibits a higher *H formation barrier than Ni-N4-C, indicating that the competitive HER process is significantly suppressed compared to MgAl2O4/Ni-N4-C. Note that the oxygen vacancy of support facilitates the trapping effect of CO2, coupling with the existing single atom Ni to reach the synergetic catalytic conversion of CO2[26,27]. The pyrolysis synthesis method helps to enhance the interaction between metal atoms and the support, facilitating the formation of stable single-atom sites that contribute to improved catalytic synergy.
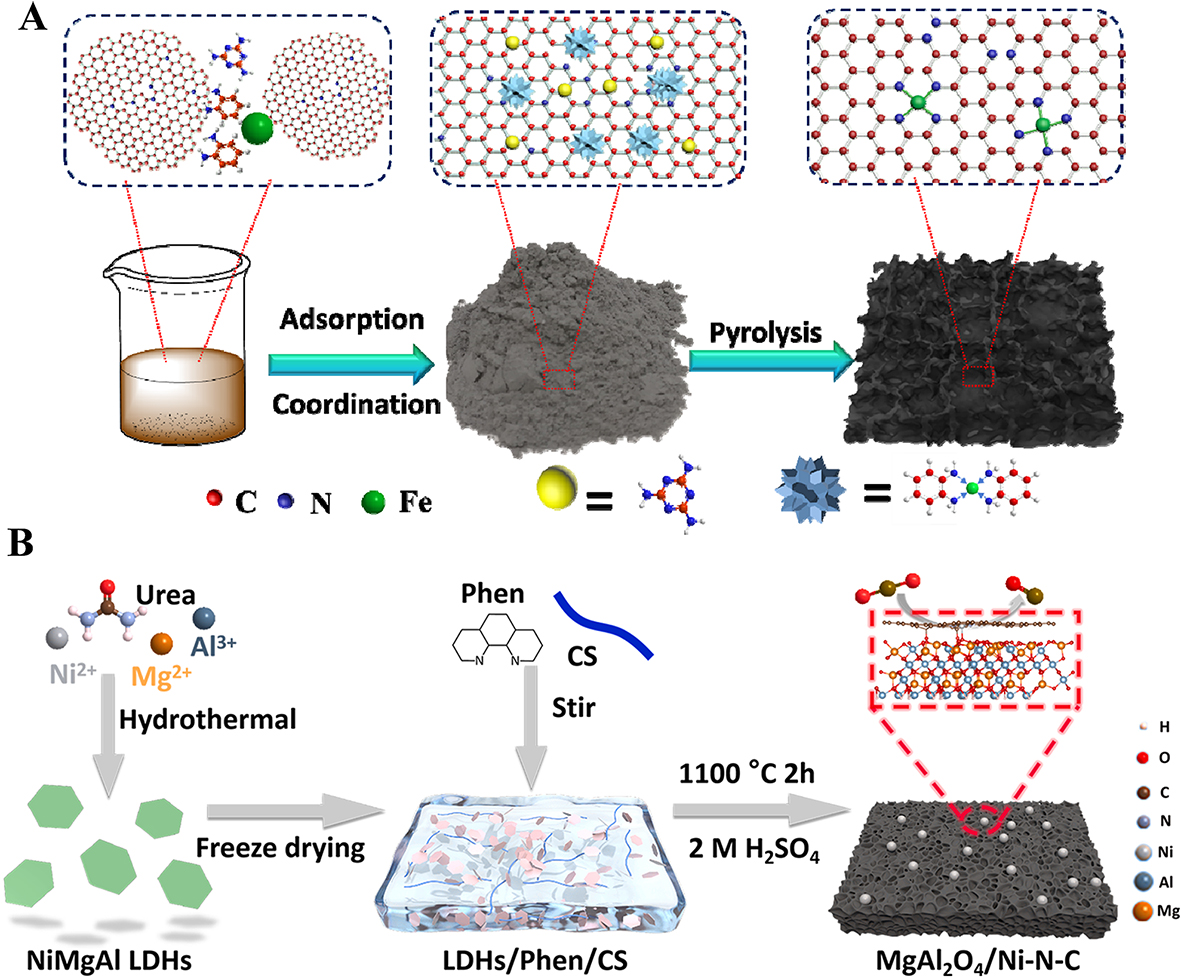
Figure 6. (A) Schematic illustration of the formation of xFe-SA/NPC-T samples; yellow and blue-grey balls represent melamine and the Fe-o-PD complex, respectively. Adapted with permission[23]. Copyright 2021, Elsevier; (B) The preparation scheme of the MgAl2O4/Ni-N-C. Adapted with permission[24]. Copyright 2024, Elsevier B.V. SA: Single-atom; NPC: N-doped porous carbon; T: pyrolysis temperature; o-PD: ortho-phenylenediamine.
Spray pyrolysis method
Spray pyrolysis is a scalable and versatile technique for synthesizing SACs, particularly suited for large-scale production. In this process, a precursor solution containing metal salts and support materials is atomized into fine droplets, which are rapidly dried and pyrolyzed at high temperatures. The confined environment of the droplets facilitates the uniform dispersion of metal precursors, while the fast thermal decomposition kinetics suppress metal aggregation, enabling the formation of isolated single-atom sites anchored on the support.
Recent studies highlight the efficacy of spray pyrolysis in SAC fabrication. For instance, Ding et al.[28] demonstrated the synthesis of Fe-N-C SACs via spray pyrolysis, achieving high Fe loading (3.2 wt%) with excellent ORR activity. The method’s rapid processing and compatibility with diverse precursors (e.g., metal-organic frameworks or carbon-based supports) make it advantageous for industrial applications. Similarly, Zheng et al.[29] utilized a modified spray pyrolysis approach to prepare Co SACs on N-doped carbon, where the synergistic interaction between Co single atoms and nitrogen defects significantly enhanced CO2 reduction performance. These studies underscore spray pyrolysis as a robust method for tailoring SACs with precise atomic coordination and high stability.
Ball-milling method
The Ball Milling method is regarded as a mechanochemical synthesis technique, which could leverage the shearing force to tailor the size and distribution of active species and support, thereby improving the mixing, refinement, and activation of catalysts. It should be worth mentioning that the size of the support also can be further tailored to optimize the surface property. Wang et al.[30] enabled a large-scale synthesis by ball-milling method, receiving beyond 4.7 g of Co-SNC once [Figure 7A]. The introduction of S atoms changes the charge density of the central Co atom, making the charge of Co in Co-N4-S more negative than in Co-N4-C. This different surface charge state endows the catalyst with different catalytic properties through the synergetic effect of S and Co. This strategy can be leveraged to construct novel dual-sites SACs without the interference of the wet chemistry method, which easily reaches great synergy in the heterogeneous catalytic reaction. Tang et al.[31] combined mechanochemistry with SACs to prepare single atom Ni on graphitized carbon nitride (g-C3N4) catalyst using high-energy ball milling for photocatalytic CO2 reduction reaction (CO2RR) [Figure 7B] without sacrificial agents. The combined effects of impact, shear, and friction during ball milling allow Ni atoms to be highly dispersed, avoiding aggregation into clusters or NPs. The presence of single atom Ni alters the electronic structure of g-C3N4 and improves the separation efficiency of photogenerated charges, thus promoting the activation and reduction of CO2. When the loading amount of Ni is 0.5 atomic percent, the photocatalyst exhibits the best photocatalytic CO2 reduction performance. Such atomic abundance helps to stimulate more electrons to participate in photocatalytic reactions, thereby improving the efficiency of photocatalytic reactions.
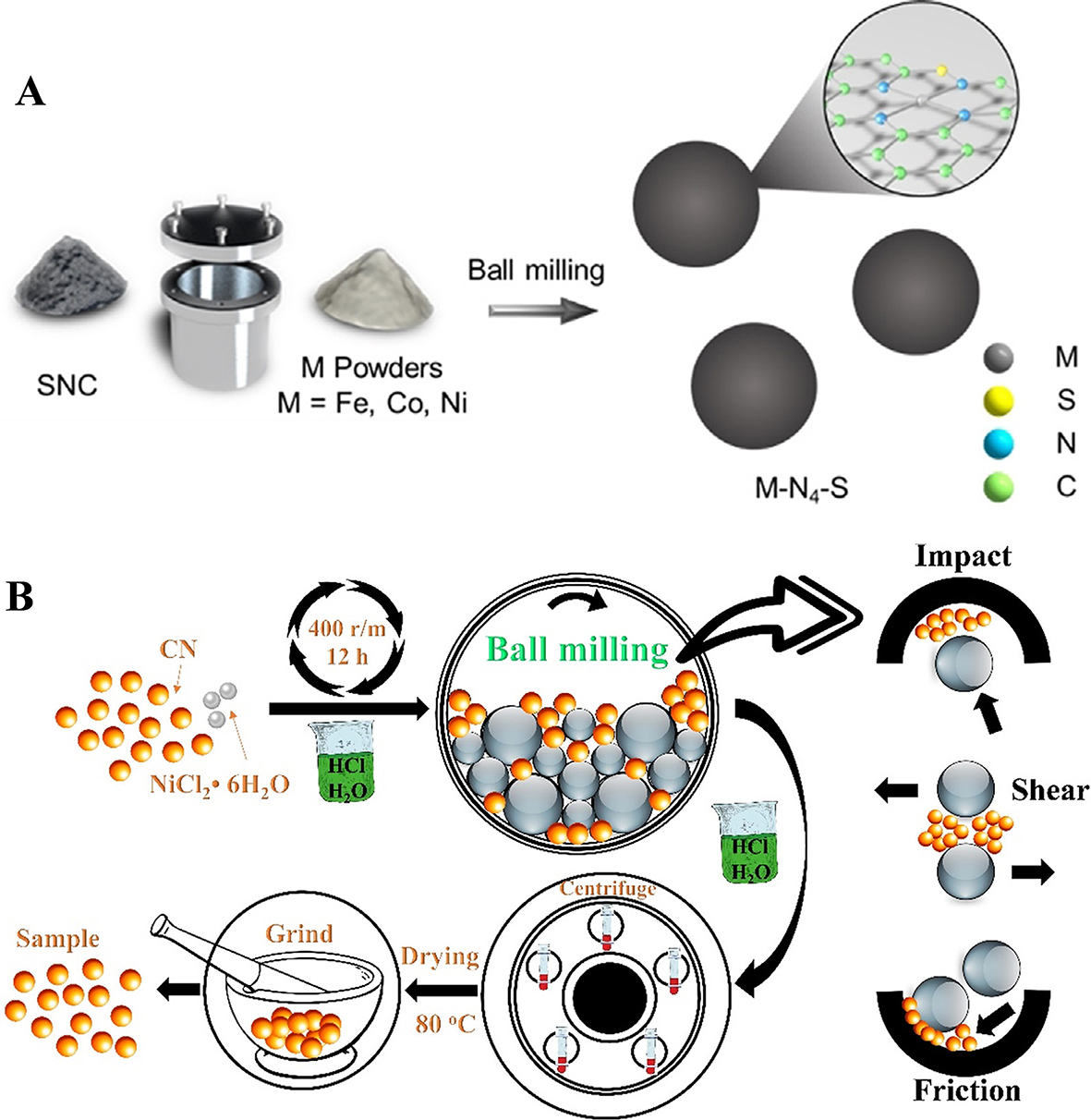
Figure 7. (A) Schematic illustration of the developed ball-milling approach. Adapted with permission[30]. Copyright 2021, Wiley-VCH GmbH; (B) Schematic diagram of Ni/CN-X synthesized via high-energy ball milling method. Enlarge diagram: a combination of impact, shear, and friction forces generated in a planetary-type ball mill. Adapted with permission[31]. Copyright 2023, Elsevier B.V.
Chemical vapor deposition method
Chemical vapor deposition (CVD) would be available for not only obtaining the fabrication of thin films and nanomaterials but also affording the promise in the synthesis of SACs[32]. In principle, gaseous or vaporous precursor substances would undergo chemical reactions at the gas or gas-solid interface, generating solid deposits, i.e., single-atom species onto the support. These precursors are typically composed of metal elements and ligands, which can be directly deposited onto the support with the formation of an isolated atom state by precisely altering the reaction conditions and precursor types. Shen et al.[33] constructed Fe-NxC moieties onto nanocellulose carbon aerogels through the synergistic application of directional freeze-casting and CVD methods. It not only substantially augmented the proportion of Fe-Nx active sites but also deftly steered the selectivity towards specific reaction pathways in four-electron ORR. Based on this approach, Han et al.[34] developed the catalyst VSACs@1T-WS2 for the HER, affording high catalytic activity and suitability, which is a great choice for developing efficient dual functional electrocatalysts. Single-atom vanadium (V) doped in 1T-WS2 (tungsten disulfide) monolayer as a catalyst was synthesized through a one-step CVD strategy. After the V atom replaces the W atom, a charge depletion region will be generated around the V atom. This local charge redistribution may alter the electronic properties of 1T-WS2 and enhance its adsorption capacity for hydrogen. V SACs have a significant impact on the hydrogen adsorption-free energy (ΔGH) at the edge sites of 1T-WS2; the synergistic effect between V atoms and 1T-WS2 carriers is crucial for HER activity. Even so, some challenges such as difficult precursor selection, complex control of reaction conditions, and relatively high costs still limit its widespread application[34]. Given that, the pioneering of novel precursors and supports is believed to obtain well-defined synthesis of SACs through CVD for broader application prospects [Table 1].
Advantages and disadvantages of different synthesis methods
| Entry | SACs synthetic method | Advantages | Disadvantages | Ref.Ref. |
| 1 | Co-impregnation method | Capable of achieving atomic level dispersion of metal precursors | Sensitive to synthesis conditions such as pH and temperature l | [10,11] |
| 2 | Co-precipitation method | Be suitable for preparing SACS on a carrier with a high specific surface area | Metal loadings are often low, limiting their usefulness in some applications | [13,14] |
| 3 | Atomic layer deposition | Accurately control the deposition of metal atoms on the carrier | The equipment and operation costs are high | [15,16] |
| 4 | Electrochemical method | The deposition position and quantity of the metal atoms can be accurately controlled | Specific electrochemical equipment and conditions are required | [19,20] |
| 5 | Photochemical method | The process can be carried out at room temperature, and the energy consumption is low | Photochemical reduction efficiency may be affected by light intensity and wavelength | [21,22] |
| 6 | Pyrolysis synthesis method | Directly used for the preparation of SACs; suitable for the synthesis of a series of metal-nitrogen-carbon SACs | Requires higher pyrolysis temperature and high energy consumption | [23,24] |
| 7 | Spray pyrolysis method | Single atoms exhibit high dispersion and uniformity, and the synthesis method is straightforward | The stringent process control requirements and high-temperature limitations pose challenges for applications in low-temperature-sensitive systems | [28,29] |
| 8 | Ball-milling method | Simple, green, can be produced on a large scale | Catalyst is easy to aggregate | [30,31] |
| 9 | Chemical vapor deposition method | CVD is suitable for complex-shaped substrates and can operate at lower temperatures, which is particularly important for thermally sensitive materials | The equipment is typically expensive, requires precise control of process conditions, and has a limited selection of precursors | [32,33] |
MODIFICATION STRATEGIES FOR SACS
The size tailoring of active components within the catalyst to the atomic scale generally causes a significant surge in surface free energy. This, in turn, sets off the process of atomic agglomeration, leading to a reduction in both the density and dispersion of single atoms. Hence, the quest for the development of an appropriate synthesis approach that can not only fortify the interaction between single atoms and the support but also proficiently suppress their agglomeration is challenged.
The uniform active site architecture of SACs paves the way for a more profound comprehension of the correlation between structure and resulting performance, which, in essence, unmasks the underlying catalytic mechanisms[35]. However, it is worth noting that the electronic structure of single-atom catalytic active sites is highly sensitive to alterations in the local coordination environment, such as defects, heteroatoms, the nature of supports, and the pore microstructure. These changes can potentially give rise to divergent reaction pathways, thereby impeding the elucidation of the structure-performance relationship[36]. Fortunately, carbon materials, metal oxides, MOFs, and COFs have proven to be apt candidates for implementing diverse regulatory strategies[37]. For instance, thermal treatment can be employed to introduce heteroatoms and carbon defects into carbon materials, while metal oxides are better suited for engineering oxygen defects to stabilize atomic metal sites. Similarly, subjecting MOFs and COFs to heat treatment can engender defective structures, and the inherent flexibility of their metal centers and organic ligands eases the incorporation of heteroatoms. Through these meticulously designed strategies, a certain level of precision in adjusting the coordination environment of SACs has been achieved, which plays a pivotal role in enhancing single atom synergistic catalytic effect. By optimizing the local coordination environment, the interaction between single atoms and supports is fine-tuned, allowing for more efficient electron transfer and reactant adsorption. This, in turn, facilitates the synchronous participation of multiple single atoms in the catalytic process, maximizing the synergistic effect and ultimately leading to improved catalytic activity and selectivity. In other words, these regulatory strategies act as the linchpin in harnessing the full potential of single atom synergistic catalytic effect, propelling the performance of SACs to new heights.
Regulation of structural composition with different supports
The cornerstone in the synthesis of SACs lies in the stabilization of densely populated individual metal atoms on a befitting support. Unsaturated chemical sites present on the support surface play a dual and indispensable role during both the synthesis and catalysis process[38,39]. Not only do they function as the physical anchors for single atoms, providing a firm footing, but they also wield the power to reshape the electronic structure of the metal centers. This electronic modulation, in turn, casts a profound catalytic or chemical influence on the overall performance of SACs[40].
When it comes to the typical supports for SACs, they can be broadly categorized into microporous matrices, two-dimensional material, and metal-containing supports. Microporous matrices, exemplified by zeolites, MOFs, and COFs, utilize their intricate micropore networks to lock metal single atoms[41]. Zeolites, in particular, flaunt well-developed pores replete with numerous surface hydroxyl groups. These hydroxyl-rich pores are akin to tiny cradles, effectively stabilizing a diverse array of species bearing single active sites[42]. A case in point is the synthesis of thermally stable M-SACs[43]. By impregnating a blend of metal salts and aqueous ethylenediamine onto modified commercial S-1 zeolites, followed by one-step pyrolysis under an argon blanket, the atomic site catalyst can be generated. XAFS and integrated differential phase contrast scanning transmission electron microscopy (iDPC-STEM) have unequivocally validated the presence of atomically dispersed transition metal atoms on the zeolite scaffold. Deeper investigations have unearthed that the dispersion of metal atoms owes its origin to the trapping prowess of silane nests. When assessing the proficiency of SACs supported on MFI-type Silicalite-1 zeolites in the conversion of CO2, these catalysts, courtesy of their potent interactions with the zeolite walls, exhibit diminished energy barriers and tenacious resistance to aggregation and sintering. This not only safeguards the integrity of the single-atom dispersion but also promotes efficient reactant diffusion and interaction, thereby supercharging the single-atom synergistic catalytic ability.
Two-dimensional materials, such as graphene, g-C3N4, boron nitride (BN), etc., offer a different yet equally effective avenue for enhancing catalytic activity. In these cases, individual metal atoms form covalent bonds with their coordinating atoms (such as C, O, N, and S)[44]. The key to unlocking their potential lies in the judicious selection of the appropriate support. When the right match is made, it triggers a cascade of changes in the electronic structure of the single-atom metal active sites [Figure 8A and B][45]. Taking graphene as a sample, its edges, provided they are not H-terminated, harbor C atoms with unpaired electrons. These reactive hotspots are the epicenters of activity in defect-free graphene[46]. Graphene’s repertoire of unique traits is nothing short of impressive. Its ultra-large theoretical surface area offers an expansive playground for catalytic reactions, while its excellent conductivity ensures rapid electron shuttling. Coupled with its remarkable chemical and electrochemical stability, two-dimensional structural elegances, and highly crystalline lattice, graphene emerges as a veritable powerhouse. Moreover, its ability to finesse active sites through defect engineering and chemical doping further cements its status as an ideal support[47]. In the context of graphene-supported single-atom transition metal catalysts for methane activation, researchers have harnessed the power of theoretical calculations, particularly those underpinned by DFT methods. By probing the activation dynamics of five magnetic transition metal atoms chromium (Cr), manganese (Mn), iron (Fe), cobalt (Co), and copper (Cu) in both adsorbed and intercalated states on graphene, a game-changing discovery has been made. Compared to their free metal methane counterparts, graphene-supported transition metal systems slash the activation energy barriers and streamline the reaction pathways, confining them to a single spin state[48]. This eradicates the erratic polymorphic reactivity witnessed in free systems, facilitating a more coordinated and efficient catalytic process. The enhanced electron transfer and reactant adsorption capabilities, courtesy of the graphene support, galvanize the single-atom synergistic catalytic effects, driving the reaction forward with greater gusto.
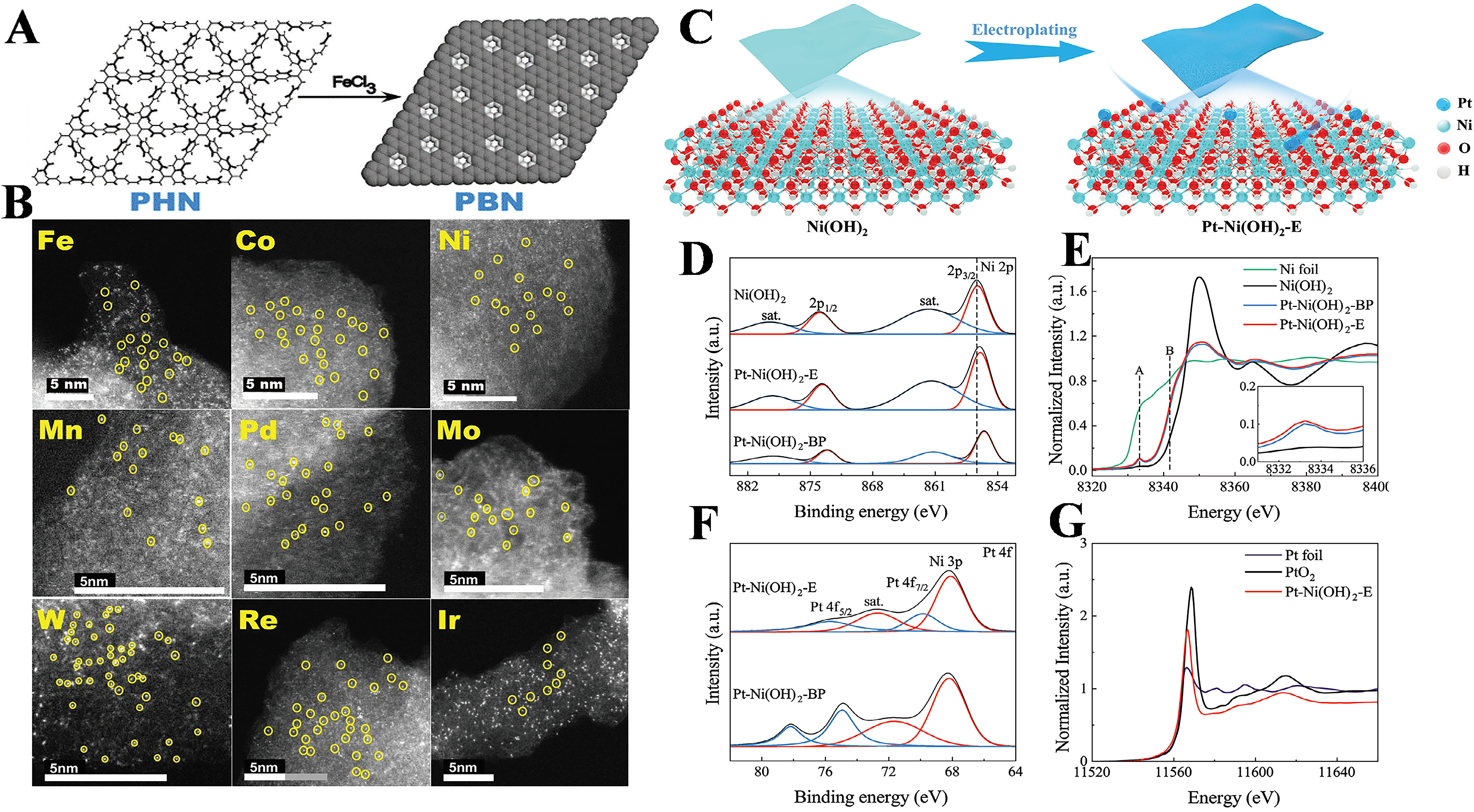
Figure 8. (A) The reaction scheme to prepare PBN; (B) ACHAADF-STEM imaging of PBN-300-M (M = Fe, Co, Ni, Mn, Pd, Mo, W, Re, Ir) samples. The atomic distribution of various metal elements was evident. Adapted with permission[45]. Copyright 2022, Wiley-VCH GmbH; (C) Schematic illustration of the preparation for the Pt-Ni(OH)2-E; (D) Ni 2p XPS spectra; (E) XANES spectra of Ni foil, Ni(OH)2, Pt-Ni(OH)2-BP, and Pt-Ni(OH)2-E at the Ni K-edge; (F) Pt 4f XPS spectra; (G) XANES spectra of Pt foil, PtO2, Pt-Ni(OH)2-E. Adapted with permission[49]. Copyright 2023, Wiley-VCH GmbH. ACHAADF-STEM: Aberration-corrected high-angle annular dark-field scanning transmission electron microscopy; PBM: pyrolytic boron nitride; XPS: X-ray photoelectron spectroscopy; XANES: X-ray absorption near-edge structure.
Metal-containing supports, including metal carbides, metal oxides, metal sulfides, and the like, bring their own set of advantages to the table. The diverse array of surface unsaturated sites on these supports is the linchpin in preventing metal agglomeration. They achieve this by forging strong chemical bonds between the metal atoms and the support surface [Figure 8C-G][49]. Metal compounds, in general, have emerged as the first choice for stabilizing individual metal atoms, especially in scenarios where carbon-based materials falter under high-temperature catalytic demands due to their inherent stability limitations[35]. In SACs, the interactions between metal atoms and oxide supports are a complex web of mechanisms, including defect-mediated interactions, oxygen-stabilized couplings, spatial confinement effects, strong metal-support interactions (SMSI), and covalent metal support linkages[50]. Consider the synthesis of SA Pt catalysts supported on nanoceria (Pt1@CeO2). These catalysts are fabricated through high-temperature calcination of Pt-impregnated porous ceria NPs, with a specific focus on the direct conversion of methane. A battery of advanced characterization techniques has been deployed to validate the atomic scale dispersion of Pt. The results speak for themselves, the Pt1@CeO2 catalyst achieves a methane conversion of 14.4% and a selectivity of 74.6% for C2 products (ethylene, ethane, and acetylene). This performance eclipses that of its NP analog by a wide margin. The secret sauce lies in the synergistic interplay between the Pt single atoms and the CeO2 support[51]. The support not only provides a stable anchoring point for the Pt atoms but also modulates their electronic properties, optimizing the adsorption and activation of methane. The enhanced interaction and coordination between the metal and support components turbocharge the single-atom synergistic catalytic effects, making it a force to be reckoned with in catalytic conversions.
The structural composition of supports plays a pivotal role in stabilizing SACs and modulating their electronic properties. Microporous matrices (e.g., zeolites, MOFs) confine metal atoms within well-defined pores, enabling precise spatial control and enhancing reactant diffusion. Two-dimensional materials (e.g., graphene, g-C3N4) leverage their large surface areas and conductive frameworks to optimize charge transfer, while metal-containing supports (e.g., CeO2, TiO2) utilize SMSI to prevent aggregation and tailor adsorption behavior[42,50,52]. For instance, Pt1@CeO2 achieves high methane conversion (14.4%) and C2 selectivity (74.6%) in methane activation[51], demonstrating the synergy between isolated Pt atoms and oxygen-rich CeO2 surfaces. The single-atom-support interaction steers the catalysis mechanism through two principal avenues: Firstly, the robust metal-support interaction, which exists between the isolated atom and its support, acts as a master tuner. It finely tunes parameters such as adsorption strength, electronic configuration, coordination environment, and binding energy. These adjustments have a direct bearing on the binding affinity of reaction intermediates at the active sites, dictating the reaction pathway and rate. Secondly, the co-catalytic interaction comes into play, where both the single atom and the support atoms actively engage and directly partake in binding the reactant species during the reaction progression[53-59]. This synchronized participation amplifies the catalytic efficiency, unlocking the true potential of single-atom synergistic catalytic effects. The carrier enhances the synergistic catalytic effect of single atoms by intensifying the adsorption, diffusion, and activation of substrates, thereby improving the overall activity of the catalyst. These strategies are particularly valuable in industrial catalysis, where stability and selectivity under harsh conditions (e.g., high temperatures, reactive atmospheres) are critical. By selecting supports that complement the target reaction’s requirements, SACs can be engineered for scalable applications such as biomass refining and CO2 conversion.
Regulation of defect sites
Mastering the manipulation of defect configurations on the support emerges as a highly efficacious strategy to curtail the migration of metal atoms thereon. These defects function as powerful modifiers, not only reconfiguring the coordination environment but also inducing significant alterations in the surrounding electronic structure. This, in turn, gives rise to the generation of vacancies and unsaturated coordination sites, setting the stage for enhanced catalytic performance[59]. Concurrently, during the post-treatment phase, these very defects on the support morph into crucial anchoring points for metal precursors and subsequently, the metal atoms themselves[60]. When embarking on the application of defect engineering strategies for the synthesis of SACs, the initial and pivotal step lies in meticulously optimizing the processing conditions [Figure 9A-C]. This optimization is geared towards guaranteeing the formation of uniformly distributed defect centers that can serve as reliable anchoring sites. By doing so, a uniform and stable dispersion of individual atoms is achieved, laying the foundation for efficient single-atom synergistic catalytic effects.
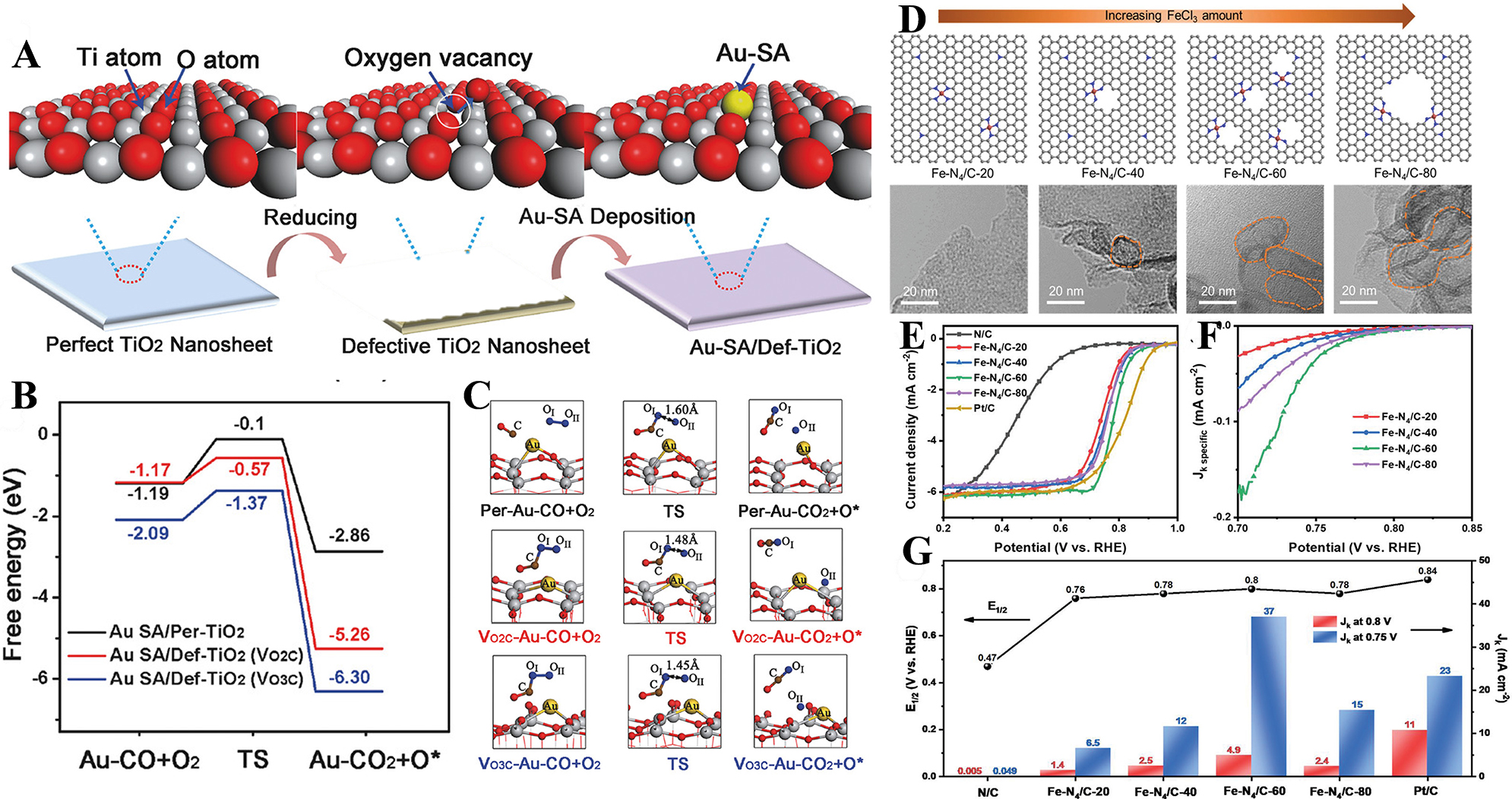
Figure 9. (A) Schematic illustration of the formation; (B) Free energy paths for Au-SA/Per-TiO2 and Au-SA/Def-TiO2; (C) Schematic models for the CO oxidation process. Adapted with permission[60]. Copyright 2018, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim; (D) The schematic illustration and TEM images of morphological evolution for Fe-N4-Cx samples when varying the amount of reactant FeCl3; (E) LSV curves for different catalysts in 0.1 M HClO4 electrolyte; (F) Potential dependence of Jk-specific for as-prepared various atomic Fe catalysts; (G) Comparison of E1/2 and Jk for different testing catalysts. Adapted with permission[63]. Copyright 2020, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. TEM: Transmission electron microscopy; LSV: linear sweep voltammetry.
Moreover, a clear identification of the defect types is of paramount importance. This knowledge empowers researchers to make informed decisions regarding the selection of appropriate metal species as precursors. Additionally, it enables the implementation of targeted measures to safeguard against the disappearance or unwanted transformation of defects during post-treatment[61]. The size and content of defects hold the key to fine-tuning the active sites of catalysts, a crucial factor in enhancing ORR performance and paving the way for the long-term stability and practical applications of the catalysts [Figure 9D-G][62,63]. Through precise control of these parameters, the design of more efficient and durable catalysts becomes a tangible reality.
A prime illustration of this concept is the successful fabrication of the single-atom gold catalyst (Au-SA/Def-TiO2). This was accomplished by deliberately introducing surface oxygen vacancy defects onto TiO2 nanosheets. On an ideal, defect-free TiO2 surface, gold atoms tend to form a four-center structure (Ti-Au-O-Ti) in conjunction with four Ti atoms. However, in the presence of a defective TiO2 surface, a remarkable transformation occurs. Coordination experiments and Density Functional Theory (DFT) results converge to reveal that the oxygen vacancies trigger a profound change in the coordination environment of gold atoms. Specifically, a Ti-Au-Ti structure takes shape[60]. This novel structure is the result of a direct interaction between the gold atom and two adjacent Ti atoms. It serves a dual purpose: firstly, it acts as a robust anchor, firmly securing individual gold atoms in place and effectively thwarting their migration and aggregation tendencies. Secondly, the presence of these defect sites works wonders for enhancing the dispersion of metal atoms on the support. It renders the gold atoms more isolated and uniformly distributed across the TiO2 nanosheets, a prerequisite for improved catalytic activity. Notably, the d-band center of Ti atoms close to the defect sites experiences a shift, drawing nearer to the Fermi level. This shift has far-reaching implications as it can modulate the electronic state of the gold atoms. In turn, this altered electronic state exerts a direct influence on the adsorption behavior of the gold atoms towards reactants such as CO and O2. By precisely tailoring the defect-induced changes in the electronic and coordination structures, the reaction kinetics are optimized, and the synergistic catalytic effect between single atoms and defect sites was enhanced, providing ideas for the design of highly efficient SACs.
Defect engineering is a powerful strategy to stabilize SACs and enhance their catalytic performance. Oxygen vacancies, lattice distortions, or edge defects act as anchoring sites for single atoms, reducing their surface free energy and suppressing migration. For example, Au-SA/Def-TiO2, with Ti-Au-Ti coordination at oxygen vacancies, exhibits superior CO oxidation activity compared to defect-free counterparts[60]. Defects also modify the electronic environment of active sites, lowering energy barriers for key intermediates (e.g.,
Regulation of heteroatoms
Heteroatom doping represents a strategic maneuver wherein diverse non-metal elements, such as N, S, P, etc., are deliberately incorporated into the support or coordination environment of a catalyst[64]. This seemingly simple addition of heteroatoms wields a profound impact on the catalyst’s performance. Firstly, they act as electronic architects, restructuring the catalyst’s electronic landscape. Through p-d coupling or long-range electron delocalization, heteroatoms can tweak the metal charge at the active center. This modulation of the electronic structure is not a passive change; it is the catalyst for enhanced single-atom synergistic catalytic effects. For example, in reactions demanding acid-base catalysis, heteroatom doping, especially N doping, can transform the acid-base properties of the catalyst. N-doping has been shown to augment the alkalinity of the support, endowing it with a heightened adsorption capacity for oxygen-containing reactants. This enhanced adsorption is a linchpin in facilitating more efficient reactions, as it allows reactants to interact more favorably with the catalyst’s active sites. Moreover, heteroatom doping serves as a stabilizing force, preventing the unruly aggregation or migration of metal atoms during the reaction process. This stability is the bedrock upon which long-term catalytic efficiency is built, ensuring that the catalyst remains active and effective over extended periods.
N modification
SACs with M-Nx coordination motifs anchored on nitrogen-enriched carbon frameworks have emerged as dominant architectures in heterogeneous catalysis, demonstrating superior catalytic metrics (activity/selectivity/stability triad) that position them as viable substitutes for noble metal systems[35]. The strategic incorporation of secondary heteroatoms (B/S/P) into M-Nx configurations enables the construction of dual-site electronic configurations, where synergistic heteroatomic pairs activate distinct reaction pathways[9,65]. Notably, nitrogen functionalities in the carbon matrix serve dual roles as electron-donating sites that modulate reactant adsorption/desorption energetics while establishing charge-transfer networks with anchored metal centers, collectively optimizing intermediate stabilization dynamics.
Fe-N4 monoatomic configuration catalyst system has become a strategic material system to replace platinum-based ORR catalysts because of its unique electronic regulatory properties[66,67] [Figure 10A and B]. By regulating the electronic coordination microenvironment of pyrrole type nitrogen (PN) and graphite type nitrogen (GN) in nitrogen-doped carbon carrier, the charge distribution state of iron active center can be accurately regulated. Theoretical simulations have confirmed that PN coordination with strong electron absorption properties induces the charge polarization effect at Fe-N4 site, induces the local electron hole state in the metal center, and then significantly improves the intrinsic activity of ORR by enhancing the interfacial electric field intensity. In typical M-N-C catalytic systems, metal-nitrogen electronegativity differences drive the formation of directional charge transfer channels, making the number of coordinated nitrogen atoms a key parameter in regulating the electronic microenvironment at the active site[68]. Systematic DFT analysis has shown that the asymmetric tricoordination transition metal site (TM-N3-C) exhibits more optimized adsorption free energy for oxygen-containing intermediates than the conventional tetarate configuration (TM-N4-C)[69]. Among them, Ni-N3-C system is outstanding in bifunctional catalytic performance, and its ORR/OER overpotential drops to 0.29 and 0.28 V, respectively, showing a unique collaborative catalytic mechanism.
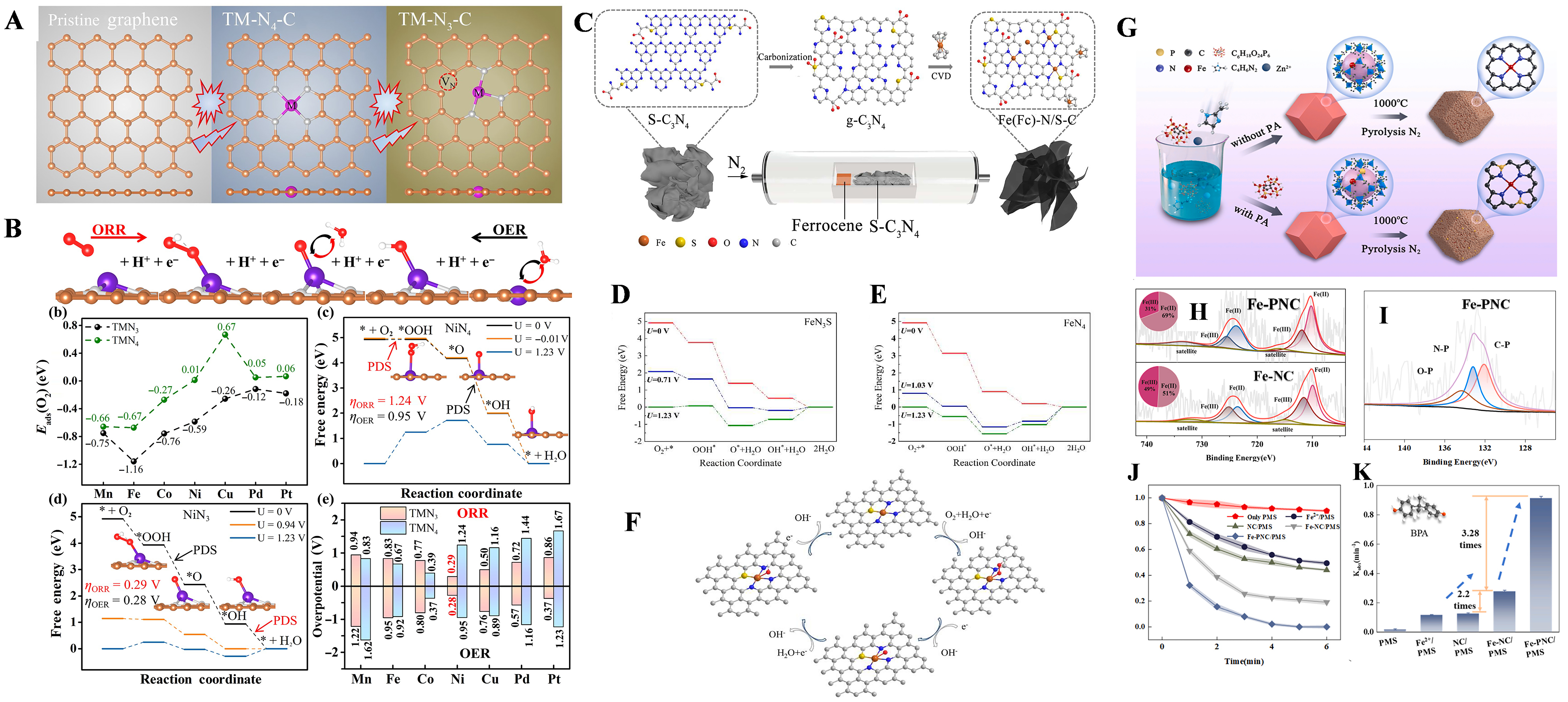
Figure 10. (A) Structure model of FeN4-PN; (B)XPS N 1s spectra of FeN4, FeN4-PN, and FeN4-GN. Adapted with permission[66]. Copyright 2021, American Chemical Society; (C) Typical CVD procedures to synthesize the Fe(Fc)-N/S-C catalyst. Relative energy profiles for the ORR processes on (D) FeN3S and (E) FeN4; (F) Reaction scheme with the intermediates in the ORR process on FeN3S (O: red, C: gray, N: blue, Fe: orange, S: yellow, H: white). Adapted with permission[75]. Copyright 2021, American Chemical Society; (G) Steps for the synthesis of the samples; SEM images of Fe-PNC at different scales; (H) Fe 2p XPS spectrum of Fe-PNC and Fe-NC; (I) P2p XPS spectrum of Fe-PNC; (J) Degradation performance of BPA in different catalytic systems; (K) The kobs values of BPA in different catalytic systems. Adapted with permission[81]. Copyright 2024, Elsevier B.V. PN: Pyrrole type nitrogen; XPS: X-ray photoelectron spectroscopy; CVD: chemical vapor deposition; ORR: oxygen reduction reaction; SEM: scanning electron microscopy; PNC: phosphorus and nitrogen co-doped carbon; NC: nitrogen-doped carbon; BPA: bisphenol A.
S modification
The strategic incorporation of sulfur heteroatoms into M-N-C architectures leverages their distinct atomic characteristics-larger ionic radius and reduced electronegativity compared to nitrogen[70]-to engineer enhanced oxygen reduction performance through coordination environment modulation[71,72]. While sulfur atoms primarily function as secondary coordination elements, their long-range electronic coupling with metal centers via M-N-C-S charge transfer networks enables effective electronic structure tailoring[73]. This approach proves particularly valuable in addressing the critical challenge of developing efficient electrocatalysts for the nitrogen reduction reaction (e-NRR), where conventional systems struggle with nitrogen activation kinetics[74].
Li et al.[75,76] demonstrate that sulfur-mediated spin-state modulation in Fe-N-S-C configurations induces metastable intermediate spin states (Feδ+), creating electron-deficient metal centers that synergistically interact with N2 molecules through optimized adsorption geometries. This electronic restructuring facilitates nitrogen protonation via stabilized NNH* intermediates while suppressing competing carbide formation pathways. Comparative mechanistic studies reveal Fe-N3S moieties significantly lower the activation barrier of potential-determining steps relative to conventional Fe-N4 configurations [Figure 10C-F]. The synergistic effects of N/S co-doping have been successfully implemented in bifunctional catalyst design, where atomic Fe-Nx sites anchored on sulfur-modified carbon matrices demonstrate enhanced oxygen electrocatalysis (OER/ORR) through improved charge transfer kinetics and optimized intermediate binding energetics[77]. When integrated into zinc-air battery systems, these catalysts surpass commercial Pt/C benchmarks in both cycling stability and power density.
P modification
P atoms, with their strong electronegativity, have been proven to help regulate the electronic structure of carbon and enhance the ORR activity of catalysts[78]. They also contribute to improving the stability and methanol resistance of catalysts, which are crucial for fuel cells and metal-air batteries in practical applications. The doping of P atoms aids in modulating the electron density of the active center. Through DFT calculations, researchers found that P atoms, together with N atoms, can break the symmetry of the Co-N4 structure, thereby modulating the electron density distribution[79]. A P-doped single Sn atom catalyst was developed by gently pyrolyzing tin phthalocyanine (SnPc), a typical SnN4 molecule, onto carbon nanotubes[80]. P atoms were successfully introduced into the second coordination shell of the Sn center to regulate the electronic structure of the metallic Sn center. The results showed that the doping of P atoms favored the activation and hydrogenation steps of CO2, significantly outperforming the control sample without P doping. It also facilitated the reduction of Sn4+ to Sn2+ during the reaction, with Sn2+ identified as the true active site for CO generation. The SAC (Fe-NC) was modified through an electronic structure modulation strategy by introducing P into the second shell, which reduced the ligand splitting field energy and the energy gap between pollutants and the catalyst [Figure 10G-K][81]. The enhanced interfacial electron flux between surface-bound contaminants and Fe active sites sustains the metastable high-spin configuration of iron centers, thereby reinforcing cooperative catalytic durability through dynamic charge equilibration mechanisms.
Heteroatom doping (N, S, P) fine-tunes the coordination environment and electronic structure of SACs, significantly impacting their activity and selectivity. Nitrogen-doped carbon supports (M-Nx) enhance Lewis basicity and metal-support interactions, as seen in Fe-N4 catalysts for ORRs. Sulfur doping induces charge redistribution (e.g., FeN3S sites), accelerating N2 activation in ammonia synthesis, while phosphorus modulates d-band centers (e.g., Sn-N4-P) to optimize CO2 adsorption and reduction. These modifications are pivotal for sustainable energy applications: N,S-coordinated FeSA catalysts achieve bifunctional OER/ORR activity in zinc-air batteries, and P-doped Sn sites improve CO selectivity in CO2 electroreduction. By tailoring heteroatom types and coordination numbers, SACs can be adapted for industrial-scale processes such as green chemical synthesis and renewable energy storage, bridging the gap between atomic-level design and macroscopic catalytic performance. Taken together, the cooperative regulatory mechanism of SACs including dynamic structural evolution, coordination-electronic synergistic optimization, interface charge transfer mechanism, and dynamic intermediate-driven effect[82].
Strategies for enhancing stability
Ligand modification and local coordination engineering
Functional ligands (e.g., N-, S-, or P-containing molecules) can stabilize metal atoms by modulating their coordination environment. For example, Wu et al.[83] synthesized a stable single-atom Fe catalyst by pyrolyzing an urchin-like FeOOH-polyaniline precursor, where pyridinic and graphitic nitrogen derived from polyaniline acted as ligands to form a stable Fe-N4 coordination structure. During pyrolysis, the formation of a 3D porous carbon nanorod framework facilitated uniform Fe dispersion, while acid washing removed uncoordinated Fe particles, ensuring the purity of single-atom sites. X-ray photoelectron spectroscopy (XPS) and XAFS analyses confirmed the atomic-level dispersion of Fe within the carbon matrix and its tetra-nitrogen coordination environment. This design, integrating ligand modification and local coordination engineering, achieved 95% CO Faradaic efficiency (at 530 mV overpotential) in CO2 electroreduction, highlighting structural stability and efficient utilization of active sites.
Design of innovative support materials
Porous frameworks (MOFs/COFs) are also used to stabilize metals. Confinement effects in MOF pores stabilize single atoms. Ma et al.[84] successfully anchored Fe single atoms within ZIF-8. After pyrolysis, the single-atom iron catalysts (SA-Fe/CNs) retained the morphology of ZIF-8 and its surface unsaturated single-atom sites. Among them, SA-Fe-c/CN derived from ZIF-8-c with exposed facets exhibited the best ORR performance and cycling stability. Two-dimensional materials (e.g., g-C3N4, MXenes) are also star materials with potential. Edge defects and high surface areas of two-dimensional supports provide abundant anchoring sites. Giannakakia et al.[85] stabilized Pd single atoms on N-vacancy-rich g-C3N4. Pd1@C3N4 showed excellent activity and selectivity in the Buchwald-Hartwig amination reaction. It was able to efficiently pair a variety of aryl halides (e.g., chlorides, brominates) to amine substrates and showed high chemical selectivity towards amines (superior to amides).
Advanced anchoring techniques
Defect engineering plays a critical role in stabilizing SACs by creating anchoring sites for metal atoms. Tan et al.[86] demonstrated the synthesis of NHMs@Fe catalyst through pyrolysis of a Fe-N-coordinated polyphenylenediamine hollow microsphere precursor (PHMs@Fe-N), in which single Fe atoms are anchored within carbon vacancies. The predefined Fe-N coordination structure ensures atomic dispersion of Fe during thermal treatment, effectively suppressing agglomeration. While co-anchoring strategies synergistically enhance stability through dual-site coordination, effectively inhibiting metal atom migration and agglomeration. Chen et al.[87] developed a tungsten SAC (W-SAC) anchored in MOF-derived nitrogen-doped carbon, which demonstrates remarkable HER activity and exceptional stability. The stability is attributed to the multiple anchoring sites provided by the N-doped carbon framework inherited from the MOF precursor.
CHARACTERIZATION OF SINGLE-ATOM CATALYSTS
The characterization techniques for SACs encompass a variety of high-precision analytical methods, providing vital insights into their structure and performance[36,88]. Scanning tunneling microscopy (STM) is a type of scanning probe microscopy that leverages the quantum tunneling effect. When the tip of a sharp conducting probe is brought extremely close (approximately 0.5-1 nanometer) to a conductive sample surface, an applied voltage generates a tunneling current between the tip and the sample. By systematically scanning the surface and precisely recording minute variations in this tunneling current, STM reconstructs atomic-scale surface topography with subnanometer resolution. This technique directly visualizes surface features, defects, and adsorbed species, making it a cornerstone tool for studying electronic structures and chemical processes at the atomic level. STM has been effectively used to reveal the chemical reactivity of SACs[89]. Notably, high-angle annular dark-field (HAADF)-STEM technology, by correcting for imaging aberrations, achieves Angstrom-level resolution, enabling direct observation of the distribution of isolated metal atoms on supports. In combination with the HAADF detector, this technology has been widely employed to distinguish individual metal atoms, offering fundamental guidance for research into the structure-property relationships of SACs. Furthermore, electron energy loss spectroscopy (EELS), used in conjunction with AC-HAADF-STEM, can reveal the composition of individual active sites[49,90,91]. In situ transmission electron microscopy (in situ ETEM) technology enables the monitoring of dynamic processes involving single metal atoms, aiding in the understanding of catalytic mechanisms and the design of more effective SACs. Additional spectroscopic characterization methods, such as Fourier transform infrared spectroscopy (FTIR) and X-ray absorption spectroscopy (XAS), serve as auxiliary tools for identifying isolated metal atoms. In particular, extended XAFS (EXAFS) technology is often used to determine the presence or absence of metal-metal bonds in SACs. XPS is employed to obtain surface structural information of SACs, such as the oxidation state of individual metal atoms[92-94]. Meanwhile, X-ray absorption near-edge structure (XANES) characterization is highly sensitive to the oxidation state of the atoms being probed, and its fitting results exhibit subtle differences between different structural models[95-97]. Together, these techniques constitute a powerful toolkit for the characterization of SACs, providing robust support for their research and development.
APPLICATION OF SINGLE-ATOM CATALYSTS
SACs have demonstrated tremendous potential in biomass conversion, CO2 reduction, and cascade reaction due to their high activity and selectivity[98,99].
Biomass conversion
As the global energy crisis and environmental pollution issues intensify, biomass, as a renewable carbon resource, has received extensive attention in research and development of conversion technologies. Biomass can be converted into various energy sources and chemicals through chemical catalytic conversion technologies, which is of great significance for reducing dependence on fossil fuels, mitigating the energy crisis, and decreasing greenhouse gas emissions[100-102]. In biomass conversion, the isolated single atoms act as highly efficient active sites. Their unique electronic structures enable them to interact synergistically with the functional groups present in biomass-derived molecules. However, generally, the general reaction conditions of biomass conversion are harsh, and the carrier or other active center can achieve high-activity and high-stability conversion by co-catalysis with single atoms.
Thermocatalytic conversion of biomass
Tian et al.[33] demonstrated the superior catalytic performance of Ir1/def-TiO2(B) in the selective hydrogenation of furfural to furfuryl alcohol, achieving a conversion rate of 99%, high selectivity of 99%, and good stability, which outperformed Ir samples supported on mesoporous graphitic carbon nitride and Ir NPs. By adjusting the geometric and electronic structure of the single atom, the carrier can produce synergistic interaction with the single atom, affecting the interaction intensity with the reactant molecule, and then regulating the catalytic performance. Wang et al.[103] utilized Pt/TiO2 SACs to catalyze the conversion of 4-propyl guaiacol, achieving a conversion rate of 96.9% and a demethoxylation selectivity of 93.3%. In the catalytic refining of lignin-derived monomers, the oxygen vacancies enable the adsorption and dispersion of Pt atoms. There is an electron transfer between Pt and the support. The methoxy group of the reactant is preferentially adsorbed on the oxygen vacancy. Pt dissociates, and the single-atom Pt has less spillover hydrogen to inhibit side reactions, realizing the synergistic catalysis between the support and the single atom.
Wang et al.[104] utilized Pd/ZnO/C catalysts for the efficient depolymerization of lignocellulose to produce high-value-added products, yielding 40.7 wt% phenolic monomers, close to its theoretical monomer yield. Each mole of Pd metal atoms could catalyze the production of up to 375 moles of phenolic monomer products in a single reaction (TON = 375 mol phenolic monomers/mol palladium). These research advancements have promoted the industrialization process of SACs. In this study, the synergistic catalytic effect of Pd/ZnO/C catalyst is significant. Firstly, atomically dispersed Pd is closely bound to ZnO in its structure. XAFS analysis shows that Pd is in a cationic environment and there is Pd-O coordination with the carrier, which lays the foundation for the catalytic reaction. When catalyzing the reaction of birch sawdust, on the one hand, the lignin can be effectively depolymerized, the molecular weight of the liquid product is greatly reduced, the key bonding bond is broken and specific allyl related signal peaks are generated, and highly selective allyl functional aromatic monomers are generated. This is due to the synergistic effect of ZnO promoting C–O bond hydrolysis and atomically dispersed Pd weakening hydrogenation ability. On the other hand, a large amount of cellulose and hemicellulose can be retained in the solid phase, and the remaining cellulose can be effectively hydrolyzed to glucose, reflecting the collaborative regulation of the transformation and retention of different biomass components. In the process of recycling, the components of the catalyst remain stable and can be regenerated by calcination after deactivation, maintaining a good catalytic performance, which further proves the effectiveness of its synergistic catalysis in multiple reactions. Liu et al.[105] engineered a boron-modulated palladium single-atom architecture (Pd1/BNC) via a supramolecular confinement-assisted pyrolysis protocol, creating unique Pd-N-B coordination environments with tailored electronic characteristics. This precisely engineered atomic coordination architecture demonstrates exceptional catalytic proficiency in valorizing diverse biomass-derived carbonyl compounds (aldehydes/ketones). Multimodal spectroscopic analysis coupled with DFT simulations elucidates the critical role of polarized M-N-B coordination motifs in governing substrate activation thermodynamics. The system achieves unprecedented atomic efficiency in HMF reductive amination, exhibiting 5.5 times enhancement in Pd utilization metrics compared to conventional PdNP/N-C counterparts, attributed to the elimination of metallic ensemble effects through atomic dispersion engineering.
Photoelectrocatalytic conversion of biomass
Zhou et al.[106] designed and synthesized a Pt single atom supported TiO2 (PtSA-TiO2) photocatalyst for photocatalytic methyl activation and C–C coupling reactions [Figure 11A-E]. They predicted and experimentally verified that the PtSA-TiO2 photocatalyst can achieve methyl activation, formation of CH3COCH2• radicals, and production of 2,5-hexanedione (HDN) and hydrogen with a selectivity of up to 93%. Compared to other noble metal single-atom (such as Ru, Rh, Ir) or Pt NP-supported photocatalysts, PtSA-TiO2 exhibited at least 13 times higher activity[12,14,106]. Through in-situ attenuated total reflection infrared spectroscopy and in-situ electron spin resonance spectroscopy, it was confirmed that Pt single atoms can promote effective methyl activation and further facilitate the formation of CH3COCH2• radicals, which is a critical step in C–C coupling reactions. The PtSA-TiO2 photocatalyst maintained good stability and activity during prolonged photocatalytic reactions. Xiong et al.[107] synthesized a highly selective catalyst composed of nickel single atoms confined on the surface of titania was proposed for the conversion of glycerol into the high-value product glycolaldehyde. Driven by light, the catalyst utilizes air as a green oxidant for the reaction under ambient conditions. The optimized catalyst exhibited selectivity for glycolaldehyde exceeding 60% with a yield of 1,058 μmol·gCat-1·h-1, and the turnover number was nearly three times higher than that of NiOx NP-modified TiO2 photocatalysts.
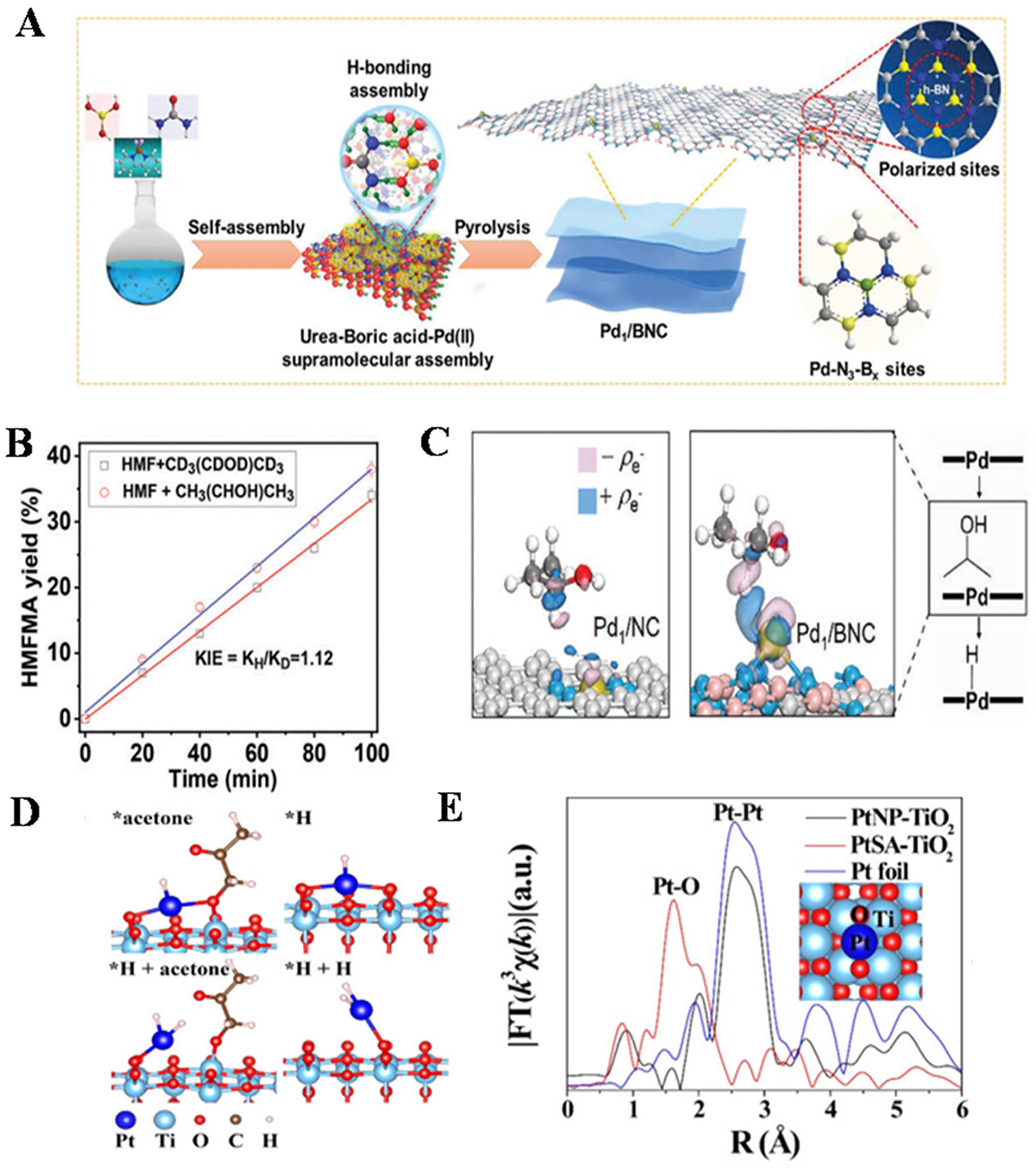
Figure 11. (A) Schematic illustration of the synthesis of the Pd1/BNC single-atom catalyst; (B) Kinetic isotope effect for reductive amination with the catalysis of Pd1/BNC; (C) Electron density difference plots of the adsorbed isopropanol molecule at
The integration of SACs into biomass conversion processes holds transformative potential for sustainable biorefineries. For instance, the Pd/ZnO/C catalyst achieves a high TON in lignocellulose depolymerization, rivaling traditional NP catalysts while minimizing metal usage and waste generation[104]. This efficiency is critical for industrial-scale biofuel production, where cost and scalability are paramount. Such advancements align with global efforts to replace fossil-derived chemicals with renewable alternatives, particularly in sectors such as polymer manufacturing and pharmaceuticals. Future industrial adoption will depend on optimizing catalyst stability and developing continuous-flow systems to handle complex biomass feedstocks.
CO2 reduction reaction
The system of single-atom catalysis for CO2 reduction is frequently encountered in photocatalysis, electrocatalysis, or photoelectrocatalysis[108]. Visible light-driven carbon dioxide (CO2) reduction reaction (CO2RR) into valuable chemicals and fuels is a promising strategy to utilize CO2 as an abundant and nontoxic C1 resource while storing clean and renewable solar energy[109]. The single atoms, often anchored on suitable supports, work in concert with the support materials. The support not only stabilizes the single atoms but also participates in the catalytic process. In the electrochemical reduction of CO2, metal single atoms can catalyze the initial electron transfer to CO2, while the support can provide additional binding sites and modify the local electronic density. This cooperation between the single atom and the support enhances the adsorption and activation of CO2, steering the reaction toward the formation of valuable products such as carbon monoxide or hydrocarbons. The well-defined single-atom active sites also minimize side reactions, such as the formation of methane or carbon deposition, leading to high selectivity in CO2 reduction.
Photocatalysis provides a sustainable avenue toward syngas with tunable CO/H2 ratios under mild conditions[4]. However, the photogeneration of charges occurs rapidly with an electron lifetime of several picoseconds. At the same time, the CO2 reduction rate is relatively slow with the reaction time within milliseconds or longer, attributable to the inertness of CO2 molecules[110]. SACs exhibit robust catalytic active sites that can accelerate charge transfer in CO2 reduction systems, making them highly suitable for application in CO2RR. These catalysts are gradually enhancing their stability and activity in such systems[111]. Many scientists have conducted explorations to address the ongoing issues in the field of CO2RR. Various catalysts, including metal oxides, metal-organic complexes, carbon nanotubes, carbon-based materials, and COFs, have been reported in multiple literature sources to support metal single-atom active centers for use in CO2RR[29,108,112,113].
Addressing the challenges of low catalytic efficiency and poor selectivity in CO2RR in pure water systems, Ji et al.[114] reported a novel atomic confinement and coordination (ACC) strategy for synthesizing rare earth erbium (Er) SACs supported on carbon nitride nanotubes (denoted as Er1/CN-NT) [Figure 12A and B]. The synergistic catalysis between single-atom Er and the carbon nitride nanotube support is highly remarkable. During the preparation of the catalyst through the unique atom-confinement and coordination strategy, the structural characteristics of the sponge cooperate with the nitrogen atoms, urea, and the low-temperature environment of liquid nitrogen therein, successfully realizing the high dispersion of Er atoms on the support and constructing a stable and unique catalytic structure foundation. The oxidation state of Er atoms exhibits a specific variation pattern, and with the increase of the loading amount, charge redistribution is induced. Such a change in electronic properties is closely related to the catalytic activity, providing a suitable electronic environment for the catalytic reaction. The single Er atom and the support act synergistically to significantly enhance the absorption intensity of visible light. Meanwhile, in the process of charge separation, the high-density single Er atoms promote the efficient separation of charges, effectively prolong the decay lifetime of photogenerated carriers, reduce the interfacial resistance, increase the photocurrent density and carrier density, and comprehensively enhance the photoelectrochemical performance. The catalyst containing single-atom Er shows higher efficiency in catalyzing the conversion of CO2 into CH4 and CO compared with the support without Er. Among them, a catalyst with a high density of rare-earth single Er atoms supported on a carbon nitride nanotube (HD-Er1/CN-NT) performs particularly outstandingly due to the higher density of single Er atoms. Moreover, through means such as CO2-temperature-programmed desorption (TPD) measurement and electrochemical reduction measurement, the close synergistic relationship between the single atom and the support in enhancing the catalytic activity is further fully verified, providing an efficient catalytic system for the photocatalytic CO2RR.
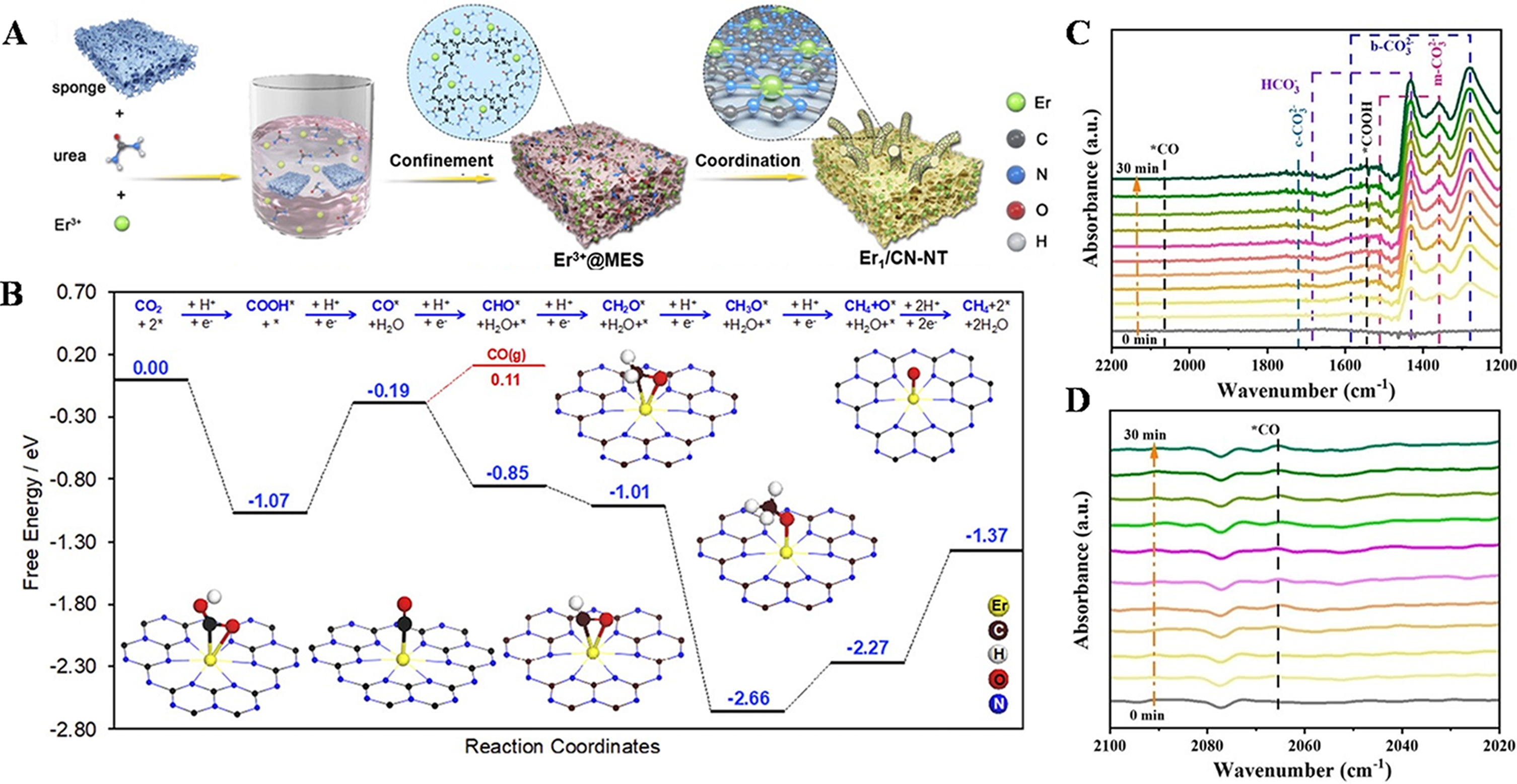
Figure 12. (A) Schematic process of the synthesis of single-atom Er1/CN-NT catalysts; (B) Calculated free-energy diagram for CO2 reduction to CO and CH4 on the Er1/CN-NT catalyst, as well as the adsorption configurations of key intermediates. The Er, C, H, O, and N atoms are given in yellow, brown, white, red, and blue, respectively. Adapted with permission[114]. Copyright 2020, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim; (C and D) In situ ATR-SEIRAS spectra for the adsorption and photocatalytic activation of CO2 on TPy-COF-Co. Adapted with permission[111]. Copyright 2024, Wiley-VCH GmbH.
Addressing the primary issues in carbon dioxide reduction, namely the rapid recombination of photogenerated charges and the sluggish surface reduction reaction of carbon dioxide, Fu et al.[111] constructed a unique CoN4Cl2 single atom site by loading Co species within a triazine-based COF
Regarding the issue of catalytic activity and selectivity in the CO2RR, Cheng et al.[112] prepared Co-Nx (x = 2, 3, 4) edge site model SACs by anchoring Co complexes with a specific number of pyridine-type ligands at the edges of carbon black (CB) via conjugated pyrazine linkers. They employed electrochemical measurements and DFT simulations to assess the catalytic performance of different Co-Nx sites. It was demonstrated that the Co-N4 edge site is the most active site for producing CO in the CO2RR, exhibiting a higher TOF. Furthermore, DFT simulations confirmed that the Co-N4 site possesses a lower energy barrier for the CO2RR and a higher energy barrier for the HER. The findings of this study provide strategies for designing efficient and selective SACs for the CO2RR.
To explore the synergistic mechanism of SAC in CO2RR, Jeong et al.[115] used in situ electrochemical STM to observe the restructuring of Cu-phthalocyanine (CuPC) into CuxO nanoclusters during CO2 reduction. This restructuring creates active sites where Cu single atoms interact synergistically with oxygen vacancies on the TiO2 support, enhancing CO2 activation and selectivity for hydrocarbons. This example illustrates how local geometric distortions and charge redistribution between single atoms and the support drive synergy.
SACs offer a promising pathway for scalable CO2 valorization, addressing both climate change and energy storage challenges. The Er1/CN-NT catalyst achieves a CO2-to-CH4 conversion rate of 105 μmol·g-1·h-1 under visible light, demonstrating the feasibility of solar-driven CO2 utilization. This technology could integrate with existing carbon capture infrastructure to produce syngas (CO/H2) or methane for industrial fuel synthesis. However, transitioning SACs from lab-scale to industrial reactors requires addressing challenges such as long-term stability under fluctuating CO2 concentrations and scalability of synthesis methods. Partnerships with energy sectors could accelerate pilot projects, particularly in regions with abundant renewable energy for powering CO2 electrolysis or photocatalysis.
Cascade catalysis
In cascade reactions, the strength of SACs truly comes to the fore. These reactions involve multiple sequential steps, and SACs can seamlessly guide the reaction from one stage to the next. The ability of SACs to maintain high activity and selectivity throughout the complex cascade reaction sequence is a result of their atomic-level precision. Each single atom can interact with different reactants and intermediates in a coordinated manner, ensuring that the overall reaction proceeds smoothly and efficiently toward the desired final products. Overall, SACs are revolutionizing these areas by unlocking new reaction pathways and enhancing catalytic efficiencies. SACs, endowed with unique catalytic properties due to the distinctive adsorption characteristics of their isolated active sites, exhibit exceptional activity and selectivity in cascade catalysis. This enables them to efficiently promote key reaction steps and enhance overall catalytic efficiency. Furthermore, by altering the support, ligands, or reaction conditions, the activity, selectivity, and stability of SACs can be conveniently tuned to meet the requirements of different steps in cascade catalysis. Notably, SACs typically possess high structural stability, maintaining their catalytic activity over extended reaction times, which is particularly crucial for reactions in cascade catalysis that require multiple consecutive steps.
Metal-enzyme cascade catalysis
The recent integration of chemical catalysts with biological catalysts for one-pot linear cascade reactions has garnered significant attention, particularly highlighting the advantages of noble metal-enzyme cascade catalysts in the dynamic kinetic resolution of chiral compounds[116]. This approach not only conserves solvent and time costs but also enhances reaction efficiency. However, the incompatibility of reaction conditions between the two active centers poses challenges for achieving efficient and stable synergistic catalysis[117]. Consequently, improving the catalytic activity of transition metals while reducing the severity of their reaction conditions can elevate the synergistic catalytic efficiency of the dual active centers[118]. Additionally, exploring the impact of the microenvironment of immobilization carriers on enzyme structure, rigidity, and selectivity, along with conducting reaction kinetic studies, is crucial for optimizing the synergistic catalytic efficiency.
SACs and enzymes each exhibit unique advantages in their respective catalytic domains. SACs boast high catalytic activity, selectivity, and stability, while enzymes are renowned for their high biological specificity and mild reaction conditions[119]. By combining these two, the complementary advantages of chemical and biological catalysis can be realized, offering new pathways for the synthesis of complex compounds[120]. By designing and synthesizing combinations of SACs and enzymes with specific catalytic activities, an efficient cascade catalytic system can be constructed to achieve the selective synthesis of complex compounds. For instance, in fields such as pharmaceutical synthesis and fine chemical production, research findings on single atom and enzyme cascade catalysis hold the potential to propel the development and advancement of related industries. Professor Ge and Li et al.[121] have conducted in-depthstudies in this regard. They synthesized a lipase-palladium single-atom composite catalyst Pd1/CALB-Pluronic through a photochemical method and investigated its catalytic performance in the synthesis of biphenyl chiral alcohols via a bio-chemical one-pot cascade reaction [Figure 13A-D]. This composite catalyst exhibited excellent activity and good stability in
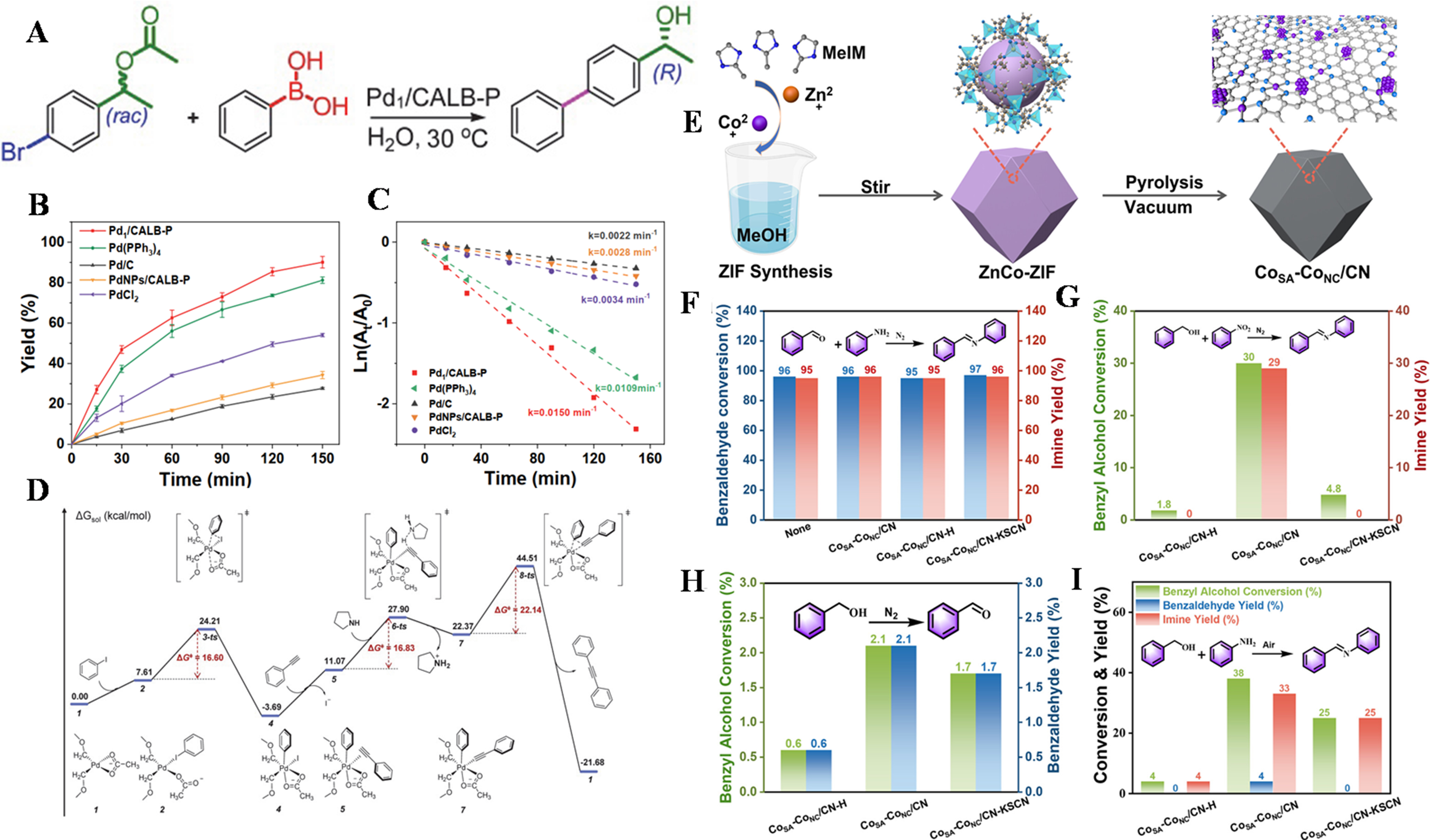
Figure 13. (A) Synthesis of (R)‐1‐(4‐biphenyl)ethanol by the one‐pot chemoenzymatic reaction; (B) Conversion versus reaction time; (C) ln(At/A0) versus reaction time, At and A0 stand for the concentration of iodobenzene at the intervals and at the initial stage; (D) Calculated free energy profile of the Sonogashira reaction pathway catalyzed by the model catalyst for Pd1/CALB‐P. Adapted with permission[121]. Copyright 2022, Elsevier B.V.; (D) Schematic illustration of the CoSA-CoNC/CN synthesis; (E) Condensation of aldehydes and amines; (F) CHT cascade reaction; (G) Oxidation of benzyl alcohol under N2; (H) Alcohol-amine coupling under air. Adapted with permission[124]. Copyright 2024, American Chemical Society. CHT: Catalytic hydrogen transfer.
Single atoms-metal cascade catalysis
Zhao et al.[123] have reported a biomimetic composite material, namely Pd1@Fe1 with a yolk-shell structure, which features two kinds of single atom sites (CSAS) for the simultaneous execution of oxidation and reduction reactions. This catalyst is capable of concurrently catalyzing the hydrogenation reduction of nitroaromatics and the epoxidation of olefins, followed by a cascade reaction to synthesize amino alcohols. During the reaction process, O2 and H2 are activated at the Fe1 and Pd1 sites, respectively, further facilitating the oxidation of styrene and the reduction of nitrobenzene. The resultant styrene oxide and aniline spontaneously react within the voids of the CSAS, ultimately yielding 1-phenyl-2-(phenylamino)ethanol. This catalyst exhibits exceptional conversion rates (≥ 99%) and selectivity (85%). It offers a novel pathway for the continuous and rapid synthesis of various complex compounds, holding promising applications across a wide range of fields. Chen et al.[124] designed a tandem catalyst, CoSA-CoNC/CN, which couples cobalt single atoms with adjacent cobalt clusters for catalyzing hydrogen transfer cascade reactions
SACs excel in cascade reactions by enabling sequential transformations in a single reactor, simplifying processes and reducing waste. Such systems are ideal for fine chemical industries, where multi-step syntheses dominate production costs. For instance, the chemoenzymatic cascade using Pd1/CALB-Pluronic combines enzymatic specificity with SAC efficiency, streamlining chiral alcohol synthesis for pharmaceuticals[121]. Industrial adoption would benefit from modular reactor designs that accommodate SAC-enzyme hybrids and automated separation systems. Furthermore, SAC-driven cascade processes could revolutionize green ammonia synthesis or polymer upcycling, aligning with circular economy goals by minimizing solvent use and energy input.
Other areas
In various other applications, single atoms have demonstrated remarkable potential, particularly in biomedical fields such as cancer treatment[126]. Cai et al.[127] reported that SACs can efficiently convert tumor-overexpressed H2O2 into •OH, initiating ferroptosis through the Fenton reaction and thereby achieving efficient oxidative therapy for tumors. Researchers have developed MitoCAT-g nanoamplifiers, which amplify oxidative stress in mitochondria by generating reactive oxygen species (ROS) and consuming the antioxidant glutathione (GSH), ultimately triggering cell apoptosis for cancer treatment. Moreover, single atoms find use in wound disinfection. M-N-C SACs (SAzymes), which possess structures similar to natural metalloenzymes, mimic the catalytic activity of natural enzymes, inhibiting bacterial proliferation and promoting wound regeneration. By loading hemoglobin-like single Fe atoms onto amorphous carbon structures, peroxidase-mimetic catalysis is achieved, generating •OH to inhibit the growth of both Gram-positive and Gram-negative bacteria.
Single atoms also exhibit exceptional performance in biosensing and antioxidant enzyme mimicry. In the field of biosensing, SACs have demonstrated their prowess in detecting biomolecules such as ascorbic acid (AA), H2O2, and glucose with high sensitivity and selectivity. By optimizing the structure and performance of SACs, researchers can achieve precise detection of these biomolecules. Furthermore, SACs can mimic the catalytic activity of antioxidant enzymes, protecting cells from oxidative stress damage. Given that natural antioxidant enzymes are prone to denaturation under harsh environmental conditions, exploring alternative catalysts to mimic their activity is of significant importance. Beyond these applications, SACs have found additional uses in the biomedical field, including drug metabolism, biomolecule recognition, and separation. By further researching and optimizing the performance and stability of SACs, we can expand their applications within the biomedical domain.
Moreover, SACs have potential applications in energy conversion and storage[96,128]. They can enhance the efficiency of reactions such as fuel cells and water electrolysis. Additionally, they can be utilized in organic synthesis and environmental catalysis, which could trigger the oriented generation of reactive active species for contaminants degradation, offering new solutions to various challenges in these fields[129,130]. The versatility and potential of single atoms in diverse applications make them a promising area of research for future advancements.
CONCLUSION AND OUTLOOK
In conclusion, significant progress has been made in the applications of SACs in energy conversion and chemoenzymatic cascade catalysis. However, the preparation process for SACs is often complex and costly. During prolonged reactions, these catalysts may aggregate or deactivate, leading to a decline in catalytic performance. Therefore, there still exist some challenges in the field of SACs preparation and application. For example: (1) the synthesis of SACs with high loadings; (2) enhancing their high-temperature stability; (3) achieving precise control over the active sites. Among them, the synthesis of high-load SACs in large quantities is particularly challenging due to the aggregation and sintering of metal atoms at high temperatures. Besides, despite some strategies such as ligand modification, support design, and dynamic anchoring have improved SAC stability, challenges remain in achieving precise control during large-scale synthesis. Future work should integrate machine learning and high-throughput screening to develop universal stabilization strategies and explore long-term stability mechanisms under extreme conditions (e.g., high temperature, acidic/alkaline environments).
The design of highly efficient synergistic SAC also requires several considerations including precisely regulating the monatomic coordination environment, selecting suitable carrier materials to enhance stability, designing bifunctional active sites to facilitate multi-step reactions, using advanced characterization techniques and theoretical calculations to guide design, and exploring new synthetic methods to achieve more accurate monatomic dispersion. These principles aim to bridge the gap between fundamental mechanisms and practical applications, inspiring innovations in energy conversion, environmental remediation, and fine chemical synthesis.
Therefore, future research needs to focus on simplifying the preparation process, improving the stability and reusability of the catalysts, and further exploring the applications of SAC catalysts in more cascade catalytic reactions, especially in the context of sustainable energy conversion and environmental remediation. It is also necessary to concentrate on developing new synthesis methods to produce SACs with high loadings, improved stability, and precise control over active sites. Theoretical calculations and simulations will play a crucial role in guiding the design of more effective catalysts by providing in-depth insights into their structural and electronic properties. In addition, feasibility studies are essential for evaluating the potential of SACs for industrial applications, including their scalability, cost-effectiveness, and environmental impact. By addressing these challenges and focusing on the development of novel catalysts and optimized conversion processes, we can pave the way for more efficient, cost-effective, and sustainable biomass conversion technologies and contribute to a greener future.
DECLARATIONS
Authors’ contributions
Conceived the project: Wang, Q.; Yang, F.
Wrote and edited the manuscript: Zheng, L.; Ni, R. T.; Ma, F. P.; Pan, J. M.
All authors reviewed and approved the final version of the manuscript.
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
The authors greatly thank the National Natural Science Foundation of China (No. 22408309). They also extend sincere gratitude to the China Postdoctoral Science Foundation (Nos. 2020M671621, 2020Z291), Jiangsu Provincial Natural Science Foundation of Universities (Nos. 20KJB530003, BK20241950), and Yangzhou University ‘Qinglan Project’.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Cao, L.; Liu, W.; Luo, Q.; et al. Atomically dispersed iron hydroxide anchored on Pt for preferential oxidation of CO in H2. Nature 2019, 565, 631-5.
2. Bing, H.; Rui, L.; Botao, Q.; Aiqin, W.; Tao, Z. Highlights of the major progress in single-atom catalysis in 2015 and 2016. Chin. J. Catal. 2017, 38, 1498-1507.
3. Zhang, L.; Banis, M. N.; Sun, X. Single-atom catalysts by the atomic layer deposition technique. Natl. Sci. Rev. 2018, 5, 628-30.
4. Ji, S.; Chen, Y.; Wang, X.; Zhang, Z.; Wang, D.; Li, Y. Chemical synthesis of single atomic site catalysts. Chem. Rev. 2020, 120, 11900-55.
5. Qiao, B.; Wang, A.; Yang, X.; et al. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 2011, 3, 634-41.
6. He, H.; Wang, H. H.; Liu, J.; Liu, X.; Li, W.; Wang, Y. Research progress and application of single-atom catalysts: a review. Molecules 2021, 26, 6501.
7. Fonseca, J.; Lu, J. Single-atom catalysts designed and prepared by the atomic layer deposition technique. ACS. Catal. 2021, 11, 7018-59.
8. Gu, W.; Pei, A.; Zhang, S.; et al. Atomic-interface effect of single-atom Ru/CoOx for selective electrooxidation of 5-hydroxymethylfurfural. ACS. Appl. Mater. Interfaces. 2023, 15, 28036-43.
9. Cheng, N.; Stambula, S.; Wang, D.; et al. Platinum single-atom and cluster catalysis of the hydrogen evolution reaction. Nat. Commun. 2016, 7, 13638.
10. Yang, L.; Zhang, X.; Yu, L.; Hou, J.; Zhou, Z.; Lv, R. Atomic Fe-N4/C in flexible carbon fiber membrane as binder-free air cathode for Zn-air batteries with stable cycling over 1,000 h. Adv. Mater. 2022, 34, e2105410.
11. Baskaran, S.; Xu, C. Q.; Wang, Y. G.; Garzón, I. L.; Li, J. Catalytic mechanism and bonding analyses of Au-Pd single atom alloy (SAA): CO oxidation reaction. Sci. China. Mater. 2020, 63, 993-1002.
12. Ren, Z.; Lyu, Y.; Feng, S.; Song, X.; Ding, Y. Acid-promoted Ir-La-S/AC-catalyzed methanol carbonylation on single atomic active sites. Chinese. J. Catal. 2018, 39, 1060-9.
13. Yang, X.; Chen, L.; Yu, X.; et al. Exploration of atomic interfaces with inherent oxygen vacancies in zirconia for toluene oxidation. J. Mater. Chem. A. 2022, 11, 287-96.
14. Sun, L.; Cao, L.; Su, Y.; Wang, C.; Lin, J.; Wang, X. Ru1/FeOx single-atom catalyst with dual active sites for water gas shift reaction without methanation. Appl. Catal. B-Environ. 2022, 318, 121841.
15. Liu, B.; Han, Z.; Bendavid, A.; et al. Atomic-layer deposition of the single-atom Pt catalyst on vertical graphene for H2 sensing. ACS. Appl. Nano. Mater. 2024, 7, 22605-16.
16. Song, Z.; Wang, Q.; Li, J.; et al. Single-atom surface anchoring strategy via atomic layer deposition to achieve dual catalysts with remarkable electrochemical performance. EcoMat 2023, 5, e12351.
17. Ye, X.; Wang, H.; Lin, Y.; et al. Insight of the stability and activity of platinum single atoms on ceria. Nano. Res. 2019, 12, 1401-9.
18. Wang, C.; Gu, X.; Yan, H.; et al. Water-mediated Mars-van Krevelen mechanism for CO oxidation on ceria-supported single-atom Pt1 catalyst. ACS. Catal. 2017, 7, 887-91.
19. Wu, J.; Li, J. H.; Yu, Y. X. Single Nb or W atom-embedded BP monolayers as highly selective and stable electrocatalysts for nitrogen fixation with low-onset potentials. ACS. Appl. Mater. Interfaces. 2021, 13, 10026-36.
20. Lv, Y. K.; Wang, K.; Sun, W. Y.; Peng, P.; Zang, S. Q. A universal electrochemical synthetic strategy for the direct assembly of single-atom catalysts. Adv. Sci. 2023, 10, e2304656.
21. Liu, P.; Zhao, Y.; Qin, R.; et al. Photochemical route for synthesizing atomically dispersed palladium catalysts. Science 2016, 352, 797-801.
22. Ge, X.; Zhou, P.; Zhang, Q.; et al. Palladium single atoms on TiO2 as a photocatalytic sensing platform for analyzing the organophosphorus pesticide chlorpyrifos. Angew. Chem. Int. Ed. Engl. 2020, 59, 232-6.
23. Huang, Y.; Chen, Y.; Xu, M.; et al. Catalysts by pyrolysis: transforming metal-organic frameworks (MOFs) precursors into metal-nitrogen-carbon (M-N-C) materials. Mater. Today. 2023, 69, 66-78.
24. Tepluchin, M.; Pham, D. K.; Casapu, M.; Mädler, L.; Kureti, S.; Grunwaldt, J. D. Influence of single- and double-flame spray pyrolysis on the structure of MnOx/γ-Al2O3 and FeOx/γ-Al2O3 catalysts and their behaviour in CO removal under lean exhaust gas conditions. Catal. Sci. Technol. 2015, 5, 455-64.
25. Jiang, J. S.; Wei, H. L.; Tan, A. D.; Si, R.; Zhang, W. D.; Yu, Y. X. Fabricating high-loading Fe-N4 single-atom catalysts for oxygen reduction reaction by carbon-assisted pyrolysis of metal complexes. Chin. J. Catal. 2021, 42, 753-61.
26. Ping, D.; Feng, Y.; Wu, S.; et al. Engineering single-atomic Ni sites stabilized with adjacent spinel nanoparticles to boost CO2 electroreduction. Sep. Purif. Technol. 2025, 357, 130193.
27. Seo, B.; Ko, E. H.; Boo, J.; Kim, M.; Kang, D.; Park, N. CO2 Hydrogenation on NixMg1-xAl2O4: a comparative study of MgAl2O4 and NiAl2O4. Catalysts 2021, 11, 1026.
28. Ding, S.; Chen, H. A.; Mekasuwandumrong, O.; et al. High-temperature flame spray pyrolysis induced stabilization of Pt single-atom catalysts. Appl. Catal. B-Environ. 2021, 281, 119471.
29. Zheng, Y.; Zhang, L.; Jiang, H.; Li, C.; Hu, Y. Pd single-atom loaded Ce-Zr solid solution catalysts prepared by flame spray pyrolysis for efficient CO catalytic oxidation. Small 2024, 20, e2311346.
30. Wang, H.; Wang, X.; Pan, J.; et al. Ball-milling induced debonding of surface atoms from metal bulk for construing high-performance dual-site single-atom catalysts. Angew. Chem. Int. Ed. Engl. 2021, 60, 23154-8.
31. Tang, R.; Wang, H.; Dong, X.; Zhang, S.; Zhang, L.; Dong, F. A ball milling method for highly dispersed Ni atoms on g-C3N4 to boost CO2 photoreduction. J. Colloid. Interface. Sci. 2023, 630, 290-300.
32. Li, J.; Yang, Z.; Li, Y.; Zhang, G. Advances in single-atom catalysts: design, synthesis and environmental applications. J. Hazard. Mater. 2022, 429, 128285.
33. Shen, M.; Qi, J.; Gao, K.; et al. Chemical vapor deposition strategy for inserting atomic FeN4 sites into 3D porous honeycomb carbon aerogels as oxygen reduction reaction catalysts in high-performance Zn-air batteries. Chem. Eng. J. 2023, 464, 142719.
34. Han, A.; Zhou, X.; Wang, X.; et al. One-step synthesis of single-site vanadium substitution in 1T-WS2 monolayers for enhanced hydrogen evolution catalysis. Nat. Commun. 2021, 12, 709.
35. Liu, D.; He, Q.; Ding, S.; Song, L. Structural regulation and support coupling effect of single-atom catalysts for heterogeneous catalysis. Adv. Energy. Mater. 2020, 10, 2001482.
36. Zhang, X.; Sun, Z.; Wang, B.; et al. C–C coupling on single-atom-based heterogeneous catalyst. J. Am. Chem. Soc. 2018, 140, 954-62.
37. Zhang, W.; Zhao, Y.; Huang, W.; Huang, T.; Wu, B. Coordination environment manipulation of single atom catalysts: regulation strategies, characterization techniques and applications. Coordin. Chem. Rev. 2024, 515, 215952.
39. Lai, W. H.; Miao, Z.; Wang, Y. X.; Wang, J. Z.; Chou, S. L. Atomic-local environments of single-atom catalysts: synthesis, electronic structure, and activity. Adv. Energy. Mater. 2019, 9, 1900722.
40. Pan, Y.; Zhang, C.; Liu, Z.; Chen, C.; Li, Y. Structural regulation with atomic-level precision: from single-atomic site to diatomic and atomic interface catalysis. Matter 2020, 2, 78-110.
41. Li, H.; Wang, M.; Luo, L.; Zeng, J. Static regulation and dynamic evolution of single-atom catalysts in thermal catalytic reactions. Adv. Sci. 2019, 6, 1801471.
42. Zheng, Y.; Jiao, Y.; Zhu, Y.; et al. Molecule-level g-C3N4 coordinated transition metals as a new class of electrocatalysts for oxygen electrode reactions. J. Am. Chem. Soc. 2017, 139, 3336-9.
43. Liu, Q.; Wang, J.; Zhao, K.; Yang, Y.; Wang, X. General synthesis for supported single-atom catalysts using hydroxyl nests in zeolites. J. Mater. Chem. C. 2024, 12, 14858-64.
44. Farid, J.; Kadhimi, H.; Al-jubouri, A. The role of two-dimensional materials (graphene, g-C3N4, and MXene) in enhancing photocatalytic performance: a review. Inorg. Chem. Commun. 2026, 184, 115981.
45. Liu, C.; Pan, G.; Liang, N.; Hong, S.; Ma, J.; Liu, Y. Ir single atom catalyst loaded on amorphous carbon materials with high HER activity. Adv. Sci. 2022, 9, e2105392.
46. Rivera-Cárcamo, C.; Serp, P. Single atom catalysts on carbon-based materials. ChemCatChem 2018, 10, 5058-91.
47. Fei, H.; Dong, J.; Chen, D.; et al. Single atom electrocatalysts supported on graphene or graphene-like carbons. Chem. Soc. Rev. 2019, 48, 5207-41.
48. Li, W.; Zhang, J.; Duan, X.; Yi, D. Regulating spin states of single transition metal atoms on N-doped graphene for efficient ammonia synthesis. J. Mater. Chem. A. 2025, 13, 11760-6.
49. Jin, C.; Huo, L.; Tang, J.; et al. Precise atomic structure regulation of single-atom platinum catalysts toward highly efficient hydrogen evolution reaction. Small 2024, 20, e2309509.
50. Lang, R.; Du, X.; Huang, Y.; et al. Single-atom catalysts based on the metal-oxide interaction. Chem. Rev. 2020, 120, 11986-2043.
51. Xie, P.; Pu, T.; Nie, A.; et al. Nanoceria-supported single-atom platinum catalysts for direct methane conversion. ACS. Catal. 2018, 8, 4044-8.
52. Wang, Y.; Zhang, W.; Deng, D.; Bao, X. Two-dimensional materials confining single atoms for catalysis. Chinese. J. Catal. 2017, 38, 1443-53.
53. Gloag, L.; Somerville, S. V.; Gooding, J. J.; Tilley, R. D. Co-catalytic metal-support interactions in single-atom electrocatalysts. Nat. Rev. Mater. 2024, 9, 173-89.
54. Li, Z.; Wu, R.; Zhao, L.; et al. Metal-support interactions in designing noble metal-based catalysts for electrochemical CO2 reduction: recent advances and future perspectives. Nano. Res. 2021, 14, 3795-809.
55. Yang, J.; Li, W.; Wang, D.; Li, Y. Electronic metal-support interaction of single-atom catalysts and applications in electrocatalysis. Adv. Mater. 2020, 32, e2003300.
56. Zheng, W.; Zhu, R.; Wu, H.; et al. Tailoring bond microenvironments and reaction pathways of single-atom catalysts for efficient water electrolysis. Angew. Chem. Int. Ed. Engl. 2022, 61, e202208667.
57. Chen, Y.; Lin, J.; Jia, B.; Wang, X.; Jiang, S.; Ma, T. Isolating single and few atoms for enhanced catalysis. Adv. Mater. 2022, 34, e2201796.
58. Luo, Z.; Zhao, G.; Pan, H.; Sun, W. Strong metal-support interaction in heterogeneous catalysts. Adv. Energy. Mater. 2022, 12, 2201395.
59. Zhang, B. W.; Wang, Y. X.; Chou, S. L.; Liu, H. K.; Dou, S. X. Fabrication of superior single-atom catalysts toward diverse electrochemical reactions. Small. Methods. 2019, 3, 1800497.
60. Wan, J.; Chen, W.; Jia, C.; et al. Defect effects on TiO2 nanosheets: stabilizing single atomic site Au and promoting catalytic properties. Adv. Mater. 2018, 30, 1705369.
61. Fu, X.; Li, N.; Ren, B.; et al. Tailoring FeN4 sites with edge enrichment for boosted oxygen reduction performance in proton exchange membrane fuel cell. Adv. Energy. Mater. 2019, 9, 1803737.
62. Xiao, M.; Xing, Z.; Jin, Z.; et al. Preferentially engineering FeN4 edge sites onto graphitic nanosheets for highly active and durable oxygen electrocatalysis in rechargeable Zn-air batteries. Adv. Mater. 2020, 32, e2004900.
63. Wang, X.; Jia, Y.; Mao, X.; et al. Edge-rich Fe-N4 active sites in defective carbon for oxygen reduction catalysis. Adv. Mater. 2020, 32, e2000966.
64. Yin, H.; Yuan, P.; Lu, B. A.; et al. Phosphorus-driven electron delocalization on edge-type FeN4 active sites for oxygen reduction in acid medium. ACS. Catal. 2021, 11, 12754-62.
65. Fan, M.; Cui, J.; Wu, J.; Vajtai, R.; Sun, D.; Ajayan, P. M. Improving the catalytic activity of carbon-supported single atom catalysts by polynary metal or heteroatom doping. Small 2020, 16, e1906782.
66. Lin, Y.; Liu, K.; Chen, K.; et al. Tuning charge distribution of FeN4 via external N for enhanced oxygen reduction reaction. ACS. Catal. 2021, 11, 6304-15.
67. Yang, L.; Cheng, D.; Xu, H.; et al. Unveiling the high-activity origin of single-atom iron catalysts for oxygen reduction reaction. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 6626-31.
68. Wang, J.; Li, Z.; Wu, Y.; Li, Y. Fabrication of single-atom catalysts with precise structure and high metal loading. Adv. Mater. 2018, 30, e1801649.
69. Xiao, G.; Lu, R.; Liu, J.; Liao, X.; Wang, Z.; Zhao, Y. Coordination environments tune the activity of oxygen catalysis on single atom catalysts: a computational study. Nano. Res. 2022, 15, 3073-81.
70. Fu, S.; Zhu, C.; Song, J.; et al. Template-directed synthesis of nitrogen- and sulfur-codoped carbon nanowire aerogels with enhanced electrocatalytic performance for oxygen reduction. Nano. Res. 2017, 10, 1888-95.
71. Haile, A. S.; Hansen, H. A.; Yohannes, W.; Mekonnen, Y. S. The role of nitrogen and sulfur dual coordination of cobalt in Co-N4-xSx/C single atom catalysts in the oxygen reduction reaction. Sustain. Energy. Fuels. 2021, 6, 179-87.
72. Li, L.; Huang, S.; Cao, R.; et al. Optimizing microenvironment of asymmetric N,S-coordinated single-atom Fe via axial fifth coordination toward efficient oxygen electroreduction. Small 2022, 18, e2105387.
73. Hu, B.; Huang, A.; Zhang, X.; et al. Atomic Co/Ni dual sites with N/P-coordination as bifunctional oxygen electrocatalyst for rechargeable zinc-air batteries. Nano. Res. 2021, 14, 3482-8.
74. Li, Y.; Ji, Y.; Zhao, Y.; et al. Local spin-state tuning of iron single-atom electrocatalyst by S-coordinated doping for kinetics-boosted ammonia synthesis. Adv. Mater. 2022, 34, e2202240.
75. Li, X.; Yang, X.; Liu, L.; et al. Chemical vapor deposition for N/S-doped single Fe site catalysts for the oxygen reduction in direct methanol fuel cells. ACS. Catal. 2021, 11, 7450-9.
76. Chen, P.; Zhou, T.; Xing, L.; et al. Atomically dispersed iron-nitrogen species as electrocatalysts for bifunctional oxygen evolution and reduction reactions. Angew. Chem. Int. Ed. Engl. 2017, 56, 610-4.
77. Kwak, D. H.; Han, S. B.; Lee, Y. W.; et al. Fe/N/S-doped mesoporous carbon nanostructures as electrocatalysts for oxygen reduction reaction in acid medium. Appl. Catal. B-Environ. 2017, 203, 889-98.
78. Jin, H.; Zhou, H.; Ji, P.; et al. ZIF-8/LiFePO4 derived Fe-N-P Co-doped carbon nanotube encapsulated Fe2P nanoparticles for efficient oxygen reduction and Zn-air batteries. Nano. Res. 2020, 13, 818-23.
79. Chen, Y.; Gao, R.; Ji, S.; et al. Atomic-level modulation of electronic density at cobalt single-atom sites derived from metal-organic frameworks: enhanced oxygen reduction performance. Angew. Chem. Int. Ed. Engl. 2021, 60, 3212-21.
80. Yue, C.; Yang, X.; Zhang, X.; et al. Secondary coordination sphere engineering of single-Sn-atom catalyst via P doping for efficient CO2 electroreduction. Adv. Energy. Mater. 2024, 14, 2401448.
81. Chai, Y.; Dai, H.; Duan, X.; et al. Elucidation of the mechanistic origin of spin-state-dependent P-doped Fe single-atom catalysts for the oxidation of organic pollutants through peroxymonosulfate activation. Appl. Catal. B-Environ. 2024, 341, 123289.
82. Sun, H.; Sun, L.; Liao, Y.; et al. Atomically imaging single atom catalysts and their behaviors by scanning tunneling microscopy. EES. Catal. 2023, 1, 794-809.
83. Wu, S.; Lv, X.; Ping, D.; et al. Highly exposed atomic Fe-N active sites within carbon nanorods towards electrocatalytic reduction of CO2 to CO. Electrochim. Acta. 2020, 340, 135930.
84. Ma, R.; Li, Q.; Yan, J.; et al. Thermodynamically controllable synthesis of ZIF-8 exposing different facets and their applications in single atom catalytic oxygen reduction reactions. Nano. Res. 2023, 16, 9618-24.
85. Giannakakis, G.; Usteri, M. E.; Bugaev, A.; et al. Reactivity and mechanism of recoverable Pd1@C3N4 single-atom catalyst in Buchwald-Hartwig aminations. ACS. Catal. 2025, 15, 284-95.
86. Tan, F.; Li, W.; Wang, J.; et al. Clarifying the critical roles of iron in boosting oxygen reduction: single Fe atoms anchored on carbon vacancies as efficient active sites. Appl. Catal. B-Environ. 2022, 305, 121035.
87. Chen, W.; Pei, J.; He, C. T.; et al. Single tungsten atoms supported on MOF-derived N-doped carbon for robust electrochemical hydrogen evolution. Adv. Mater. 2018, 30, e1800396.
88. Peng, J. X.; Yang, W.; Jia, Z.; Jiao, L.; Jiang, H. L. Axial coordination regulation of MOF-based single-atom Ni catalysts by halogen atoms for enhanced CO2 electroreduction. Nano. Res. 2022, 15, 10063-9.
89. Hulva, J.; Meier, M.; Bliem, R.; et al. Unraveling CO adsorption on model single-atom catalysts. Science 2021, 371, 375-9.
90. Zhang, L.; Meng, Q.; Zheng, R.; et al. Microenvironment regulation of M-N-C single-atom catalysts towards oxygen reduction reaction. Nano. Res. 2023, 16, 4468-87.
91. Huang, Y.; Xiong, J.; Zou, Z.; Chen, Z. Emerging strategies for the synthesis of correlated single atom catalysts. Adv. Mater. 2025, 37, e2312182.
92. Zhang, H.; Li, Y.; Zeng, L.; Pan, Y. Atomic-level regulation of Cu-based electrocatalyst for enhancing oxygen reduction reaction: from single atoms to polymetallic active sites. Small 2024, 20, e2307384.
93. Ma, T.; Li, H.; Yu, Y.; et al. Lattice-confined single-atom catalyst: preparation, application and electron regulation mechanism. Small. Methods. 2024, 8, e2400530.
94. Guan, W.; Cheng, W.; Pei, S.; Chen, X.; Yuan, Z.; Lu, C. Probing coordination number of single-atom catalysts by d-band center-regulated luminescence. Angew. Chem. Int. Ed. Engl. 2024, 63, e202401214.
95. Wang, L.; Li, J.; Ji, S.; Xiong, Y.; Wang, D. Microenvironment engineering of covalent organic framework based single/dual-atom catalysts toward sustainable energy conversion and storage. Energy. Environ. Sci. 2024, 17, 8482-528.
96. Liu, H.; Tian, L.; Zhang, Z.; et al. Atomic-level asymmetric tuning of the Co1-N3P1 catalyst for highly efficient N-alkylation of amines with alcohols. J. Am. Chem. Soc. 2024, 146, 20518-29.
97. Wu, J.; Zhu, X.; Li, Q.; et al. Enhancing radiation-resistance and peroxidase-like activity of single-atom copper nanozyme via local coordination manipulation. Nat. Commun. 2024, 15, 6174.
98. Ma, Z.; Kuloor, C.; Kreyenschulte, C.; et al. Development of iron-based single atom materials for general and efficient synthesis of amines. Angew. Chem. Int. Ed. Engl. 2024, 63, e202407859.
99. Chen, J. Y.; Xiao, Y.; Guo, F. S.; Li, K. M.; Huang, Y. B.; Lu, Q. Single-atom metal catalysts for catalytic chemical conversion of biomass to chemicals and fuels. ACS. Catal. 2024, 14, 5198-226.
100. Modak, A. Recent progress and opportunity of metal single-atom catalysts for biomass conversion reactions. Chem. Asian. J. 2023, 18, e202300671.
101. Liu, X.; Chen, Z.; Lu, S.; et al. Heterogeneous photocatalytic conversion of biomass to biofuels: a review. Chem. Eng. J. 2023, 476, 146794.
102. Wang, D.; Shan, H.; Yin, W.; Li, H. Defect engineering of single-atom catalysts in biomass conversion. Fuel 2024, 355, 129439.
103. Wang, W.; Li, S.; Qiang, Q.; et al. Catalytic refining lignin-derived monomers: seesaw effect between nanoparticle and single-atom Pt. Angew. Chem. Int. Ed. Engl. 2024, 63, e202404683.
104. Wang, S.; Li, X.; Fu, C.; Li, H.; Song, G. Atomically dispersed palladium driving reductive catalytic fractionation of lignocellulose into alkene-functionalized phenols. ACS. Catal. 2024, 14, 3565-74.
105. Liu, W. J.; Zhou, X.; Min, Y.; et al. Engineering of local coordination microenvironment in single-atom catalysts enabling sustainable conversion of biomass into a broad range of amines. Adv. Mater. 2024, 36, e2305924.
106. Zhou, P.; Chao, Y.; Lv, F.; et al. Metal single atom strategy greatly boosts photocatalytic methyl activation and C–C coupling for the coproduction of high-value-added multicarbon compounds and hydrogen. ACS. Catal. 2020, 10, 9109-14.
107. Xiong, L.; Qi, H.; Zhang, S.; et al. Highly selective transformation of biomass derivatives to valuable chemicals by single-atom photocatalyst Ni/TiO2. Adv. Mater. 2023, 35, e2209646.
108. Wang, M.; Li, M.; Liu, Y.; Zhang, C.; Pan, Y. Structural regulation of single-atomic site catalysts for enhanced electrocatalytic CO2 reduction. Nano. Res. 2022, 15, 4925-41.
109. Yin, H. Q.; Zhang, Z. M.; Lu, T. B. Ordered integration and heterogenization of catalysts and photosensitizers in metal-/covalent-organic frameworks for boosting CO2 photoreduction. Acc. Chem. Res. 2023, 56, 2676-87.
110. Liu, H.; Cheng, M.; Liu, Y.; et al. Single atoms meet metal-organic frameworks: collaborative efforts for efficient photocatalysis. Energy. Environ. Sci. 2022, 15, 3722-49.
111. Fu, P.; Chen, C.; Wu, C.; et al. Covalent organic framework stabilized single CoN4Cl2 site boosts photocatalytic CO2 reduction into tunable syngas. Angew. Chem. Int. Ed. Engl. 2025, 64, e202415202.
112. Cheng, Y. T.; Peng, J. Z.; Lai, G. T.; et al. Edge-site Co-Nx model single-atom catalysts for CO2 electroreduction. ACS. Catal. 2024, 14, 8446-55.
113. Chen, H.; Liu, W.; Laemont, A.; et al. A visible-light-harvesting covalent organic framework bearing single nickel sites as a highly efficient sulfur-carbon cross-coupling dual catalyst. Angew. Chem. Int. Ed. Engl. 2021, 60, 10820-7.
114. Ji, S.; Qu, Y.; Wang, T.; et al. Rare-earth single erbium atoms for enhanced photocatalytic CO2 reduction. Angew. Chem. Int. Ed. Engl. 2020, 59, 10651-7.
115. Jeong, Y.; Kim, Y.; Kim, Y. J.; Park, J. Y. In situ probing of CO2 reduction on Cu-phthalocyanine-derived CuxO complex. Adv. Sci. 2024, 11, e2304735.
116. Chang, F.; Wang, C.; Chen, Q.; Zhang, Y.; Liu, G. A chemoenzymatic cascade combining a hydration catalyst with an amine dehydrogenase: synthesis of chiral amines. Angew. Chem. Int. Ed. Engl. 2022, 61, e202114809.
117. Gao, D.; Song, W.; Wu, J.; et al. Efficient production of l-homophenylalanine by enzymatic-chemical cascade catalysis. Angew. Chem. Int. Ed. Engl. 2022, 61, e202207077.
118. González-Granda, S.; Escot, L.; Lavandera, I.; Gotor-Fernández, V. Chemoenzymatic cascades combining biocatalysis and transition metal catalysis for asymmetric synthesis. Angew. Chem. Int. Ed. Engl. 2023, 62, e202217713.
119. Cabré, A.; Verdaguer, X.; Riera, A. Recent advances in the enantioselective synthesis of chiral amines via transition metal-catalyzed asymmetric hydrogenation. Chem. Rev. 2022, 122, 269-339.
120. Yang, L. C.; Deng, H.; Renata, H. Recent progress and developments in chemoenzymatic and biocatalytic dynamic kinetic resolution. Org. Process. Res. Dev. 2022, 26, 1925-43.
121. Li, X.; Cao, Y.; Xiong, J.; et al. Enzyme-metal-single-atom hybrid catalysts for one-pot chemoenzymatic reactions. Chinese. J. Catal. 2023, 44, 139-45.
122. Debecker, D. P.; Smeets, V.; Van, V. M.; Meersseman, A. H.; Kinnaer, M.; Devred, F. Hybrid chemoenzymatic heterogeneous catalysts. Curr. Opin. Green. Sustain. Chem. 2021, 28, 100437.
123. Zhao, Y.; Zhou, H.; Zhu, X.; et al. Simultaneous oxidative and reductive reactions in one system by atomic design. Nat. Catal. 2021, 4, 134-43.
124. Chen, Z.; Yang, S.; Yang, J.; et al. Boosting catalytic hydrogen transfer cascade reactions via tandem catalyst design by coupling Co single atoms with adjacent Co clusters. ACS. Catal. 2024, 14, 18256-67.
125. Wu, R.; Meng, Q.; Yan, J.; et al. Intermetallic synergy in platinum-cobalt electrocatalysts for selective C–O bond cleavage. Nat. Catal. 2024, 7, 702-18.
126. Xiang, H.; Feng, W.; Chen, Y. Single-atom catalysts in catalytic biomedicine. Adv. Mater. 2020, 32, e1905994.
127. Cai, S.; Liu, J.; Ding, J.; et al. Tumor-microenvironment-responsive cascade reactions by a cobalt-single-atom nanozyme for synergistic nanocatalytic chemotherapy. Angew. Chem. Int. Ed. Engl. 2022, 61, e202204502.
128. Lu, Y.; Ke, Z. Strategies for the preparation of single-atom catalysts using low-dimensional metal-organic frameworks. Small 2024, 20, e2403767.
129. Saptal, V. B.; Ruta, V.; Bajada, M. A.; Vilé, G. Single-atom catalysis in organic synthesis. Angew. Chem. Int. Ed. Engl. 2023, 62, e202219306.
Cite This Article
 , ... Fu Yang
, ... Fu YangHow to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].