
Parkinson’s disease and inflammatory bowel disease: exploring the connection between the two
 0
0 Abstract
Parkinson’s disease (PD) is a neurodegenerative disorder characterized by prominent inflammatory features, while inflammatory bowel disease (IBD) is a typical chronic inflammatory disorder.They share similarities in epidemiological patterns, genetic predispositions, immune mechanisms, and gut microbiota composition. Mounting data support a two-way relationship between gastrointestinal (GI) inflammation and neurodegeneration, aligning with the gut-brain axis framework. Certain therapies for IBD, such as fecal microbiota transplantation and probiotics, show potential protective effects for PD and hold promise for application in the treatment of both diseases. In the present review, we explore both supportive and contradictory data derived from epidemiological investigations, while emphasizing recent discoveries regarding the connection between these seemingly unrelated disorders that share genetic susceptibility. This study delves into the role of the gut-brain axis in the pathogenesis of PD and IBD, with a particular focus on the pivotal roles of gut microbiota and intestinal inflammation. Our analysis aims to offer a new perspective on understanding and exploring the relationship between these diseases.
Keywords
INTRODUCTION
Inflammatory bowel disease (IBD), comprising Crohn’s disease (CD) and ulcerative colitis (UC), are chronic gastrointestinal disorders defined by dysregulated mucosal immunity and localized inflammation. While intestinal symptoms (e.g., diarrhea, abdominal pain) dominate their clinical presentations, IBD exhibits connections to various extraintestinal manifestations while concurrently elevating the systemic inflammatory burden[1]. In addition, the peak incidence occurs in young adults[2,3]. Research using genome-wide association studies (GWAS) has uncovered over 200 genetic loci associated with IBD and a number of them have also been connected to other disease conditions[4,5]. In contrast, Parkinson’s disease (PD) is a progressive neurodegenerative disorder and predominantly affects the elderly[6]. Pathologically, the condition is characterized primarily by degenerative alterations in dopaminergic cells located within the substantia nigra pars compacta (SNpc), alongside the presence of Lewy bodies in both brainstem nuclei and cortical areas[7]. Clinically, manifestations of PD include a spectrum of signs[8]. Core motor signs are tremor, akinesia, rigidity, and postural instability, accompanied by non-motor manifestations, such as constipation, diminished olfactory function, and cognitive deficits[9]. Notably, these two seemingly disparate conditions, PD and IBD, share an unexpected epidemiological link: GI dysfunction, particularly constipation, frequently precedes motor symptoms in PD by decades[10], raising questions about the role of gut physiology in neurodegeneration.
A mounting body of evidence lends support to the hypothesis centered on the gut-brain axis (GBA), which points to bidirectional communication that connects the gut with the brain[11]. Chronic intestinal inflammation in IBD may act as a trigger for PD pathogenesis through multiple mechanisms. First, Lewy-related pathological changes might initially originate from the enteric nervous system (ENS) before spreading onward to the central nervous system (CNS) via the vagus nerve[12]. Furthermore, dysbiosis of the gut microbiota may disrupt the balance between protective and harmful metabolites, triggering a series of effects, such as increased intestinal permeability, amplification of local inflammatory responses, and widespread systemic inflammation. Once this inflammatory state is activated, it can further affect the CNS and promote the pathological progression of PD.
Despite these mechanistic insights, the exact connection linking IBD and PD has not been fully elucidated. Epidemiological studies[13-20] have reported conflicting results regarding whether IBD confers an increased risk of PD, while the genetic and molecular pathways linking gut inflammation to α-synucleinopathy require further clarification. This review consolidates epidemiological and genetic findings that link IBD with PD and examines potential biological mechanisms connecting the two disorders. By integrating data from population studies, genetic analyses, and preclinical models, our work is designed to unravel the complex connections between these diseases, ultimately to explore novel treatment approaches targeting the GBA.
EPIDEMIOLOGICAL STUDIES ON PD AND IBD
Risk of PD in patients with IBD
A number of studies[13-17] [Table 1] have collectively demonstrated that patients with IBD face a heightened risk of developing PD compared to control groups. Notably, a retrospective cohort study from Taiwan[13] revealed a 35% increase in PD risk among IBD patients compared to those without IBD. Similarly, a U.S.-based retrospective cohort study[17] found a 28% greater incidence of PD among IBD patients relative to the control group. In the nationwide Danish cohort study[14], a significant association was observed between IBD and late-onset PD. In addition, stratified analysis of this cohort study further demonstrated that the risk of PD was significantly higher in patients with UC, whereas no significant difference was observed in patients with CD. Nevertheless, earlier studies reporting a positive IBD-PD association, however, failed to consider surveillance bias. For instance, a Swedish study initially identified a strong link between IBD and elevated PD risk. However, after adjusting for the frequency of medical visits, this association was no longer evident[15], implying that surveillance or detection bias may have influenced the results. In contrast, the South Korean study found an elevated likelihood of PD among individuals with IBD, even following adjustment for healthcare utilization[16].
Summary of studies on the relationship between IBD and PD
| Study | Study population | Number of patients age | Study design | Study period | Age at inclusion | ICD code | aHR/aIR/aOR (95%CI) |
| Lin et al.[13] Taiwan (2016) | The National Health Insurance Research Database | IBD: 8373 Control: 33492 | Cohort study | 2000-2011 | ≥ 20 | ICD-9 | IBD: aHR = 1.4 (1.1 to 1.7) |
| Villumsen et al.[14] Denmark (2019) | The National Patient Register | IBD: 76477 Control: 7548259 | Cohort study | 1977-2014 | ≥ 15 | ICD-8, ICD-10 | IBD: aHR = 1.2 (1.1 to 1.4) |
| Weimers et al.[15] Sweden (2019) | The Swedish National Patient Register | IBD: 39652 Control: 396520 | Cohort study | 2002-2014 | ICD-7~ICD-10 | IBD: aHR = 0.9 (0.7 to 1.1) | |
| Peter et al.[17] USA (2018) | The Truven Health MarketScan Commercial Database and the Medicare Supplemental Database | IBD: 144018 Control: 720090 | Cohort study | 2000-2016 | ≥ 18 | ICD-9 | IBD: aIR = 1.3 (1.1 to 1.4) |
| Park et al.[16] South Korean (2019) | The Korean National Health care Insurance service | IBD: 38861 Control: 116583 | Cohort study | 2010-2013 | All age | ICD-10 | IBD: aHR = 1.9 (1.4 to 2.4) |
These epidemiological findings support a higher risk of PD in IBD patients, though some studies disagree. A re-analysis of the Truven Health Marketscan database showed that no statistically significant correlation between IBD and PD was observed after adjusting for a range of potential external confounders, with an adjusted HR of 1.01 (95%CI: 0.72-1.42)[18]. Prior observed associations between these disorders may have been influenced by environmental and socioeconomic confounding factors. A prospective analysis[19] of 468,556 individuals within the UK Biobank similarly revealed no material link of IBD with PD (HR: 1.356, 95%CI: 0.941-1.955, P = 0.103), though increased PD risk was observed in certain subgroups (e.g., females, those with high polygenic risk scores, and UC patients without hypertension). The variation in results may be due to baseline characteristics that might change during follow-up. An analysis[20] of German primary care records (N = 35,988) also revealed no markedly raised PD frequency in the overall IBD cohort. However, a rising but non-significant tendency was noted among men with CD (HR: 1.55, 95%CI: 0.98-2.45, P = 0.064).
The epidemiological link between IBD and PD is complex and appears to be influenced by population-specific factors[21]. While a cohort study have demonstrated a positive association[16], the other one has reported null findings or effect modification by sex and age[19]. These discrepancies may partly be attributed to detection bias, as patients with IBD might undergo more frequent medical examinations[15]. Moreover, the residual confounding effect by lifestyle factors - particularly smoking, which is known to be protective in PD, has been highlighted as a potential source of overestimation in studies that did not adequately adjust for these variables[22]. In the future, it is necessary to further validate the true association between the two and their underlying mechanisms through prospective designs, refined phenotypic stratification (such as age, disease subtypes, and treatment history), and integration of multi-omics data.
Risk of IBD in patients with PD
The role of IBD incidence among patients with PD remains a contentious topic, with a current lack of sufficient epidemiological studies. Research from a Taiwan-based cohort found that the observed variations in IBD incidence among subjects with PD was statistically insignificant[23]. An analysis of U.S. insurance records indicated a 15% reduction in IBD incidence among individuals with PD relative to the comparison group[24]. This inconsistency may arise from the distinct ages at which each condition typically emerges, with IBD presenting most frequently in early adult life and PD rarely developing before 50. Additionally, the presence of delayed gastric emptying and constipation symptoms in PD may mask the diarrhea symptoms associated with IBD, potentially leading to underdiagnosis of IBD in PD patients. These factors could contribute to a lower prevalence of IBD in PD patients compared to non-PD controls. A Swedish case-control study further reported an association between IBD and PD at the time of diagnosis (OR = 1.4, 95%CI: 1.2-1.8). This increased risk was consistent in subgroup analyses for both CD (OR = 1.6, 95%CI: 1.1-2.3) and UC (OR = 1.4, 95%CI: 1.1-1.9), suggesting a higher probability of pre-existing PD in these patients compared to non-PD controls[15]. This suggests that the probability of PD existing before the diagnosis of IBD is higher, indicating that PD may be a risk factor for the future development of IBD.
Causal relationship between PD and IBD
All the aforementioned studies are observational in nature, with the majority of them initially collecting administrative data for non-statistical purposes. This data may be influenced by confounding factors (such as history of poor lifestyle habits and medication use), making it challenging to distinguish true causal relationships. Mendelian Randomization (MR) is a causal inference method that leverages genetic variations (particularly single nucleotide polymorphisms, SNPs) highly correlated with the target exposure, while ensuring these variations being independent of confounding factors[25,26].
However, the validity of MR depends on three core assumptions: (1) the genetic variants are robustly associated with the exposure (IBD); (2) they are independent of confounders; and (3) they influence the outcome (PD) only through the exposure, with no horizontal pleiotropy (i.e., no alternative pathways bypassing the exposure). Several recent MR studies have applied this framework to investigate the IBD-PD relationship. For instance, Freuer et al. conducted an MR study analyzing data related to IBD (7,045 cases and 456,327 controls) and PD (56,306 cases and 1,417,791 controls) with a total of 463,372 IBD-related data points. The results indicated no association between IBD and PD risk [Odds Ratio-Inverse Variance Weighted (OR_IVW) = 0.98; 95%CI: (0.93; 1.04); P = 0.48][27]. Another MR analysis yielded similar results [IVW (95%CI): 1.01 (0.98-1.05), P = 0.50][28] although their analysis was one-directional and could not determine whether PD increases the risk of IBD.
The null findings from these MR studies warrant careful interpretation. First, while sensitivity analyses (e.g., MR-Egger, weighted median) can detect pleiotropy, the possibility of weak instrument bias or residual horizontal pleiotropy cannot be fully excluded, especially if the genetic variants explain only a modest proportion of IBD variance. Second, these studies predominantly involved populations of European ancestry, limiting generalizability to other ethnic groups. One study using multiple MR methods reported that while the IVW and Robust Adjusted Profile Score (RAPS) methods suggested an increased risk of IBD in patients with PD (OR_IVW = 1.062, P < 0.05), other methods did not, highlighting the sensitivity of findings to methodological choices[29]. A bidirectional MR analysis conducted in 2024 revealed that the IVW analysis showed no significant correlation between genetically predicted PD risk and IBD risk (OR = 1.04, 95%CI: 0.98-1.11; P = 0.182), similar results were obtained in the reverse analysis (OR = 0.98, 95%CI: 0.94-1.03; P = 0.530)[30].
Taken together, the current body of MR evidence does not support a strong causal relationship between IBD and PD. While observational studies have reported positive associations, these may be attributed to confounding factors, detection bias, or reverse causation rather than a direct biological link. Future MR studies with larger sample sizes, more diverse populations, and more refined genetic instruments for specific IBD subtypes may help clarify whether any subtle or subgroup-specific causal effects exist.
RESEARCH ON THE SHARED PATHOPHYSIOLOGICAL MECHANISMS OF PD AND IBD
Genetic overlap
Genetic susceptibility offers another perspective on the heightened risk of PD, and related studies further substantiate this notion. A 2017 analysis employing GWAS and pathway analyses identified 17 novel pleiotropic loci from cohorts of patients with PD and seven autoimmune diseases, such as CD and UC[31]. The most significant genome-wide pleiotropic enrichment was identified between PD and CD. Based on the National Genome Research Institute GWAS Catalog[32] and the evaluation by Lee et al.[33], six loci exhibit strong associations with both PD and IBD. This section summarizes several of these typical genes whose encoded proteins may functionally connect the two diseases.
leucine-rich repeat kinase 2
Among the most notable genetic links between IBD and PD, leucine-rich repeat kinase 2 (LRRK2) stands out. It contains multiple domains, including a catalytic core that exhibits both kinase and GTPase activities, flanked by structural scaffolding regions[34]. LRRK2 is implicated in the pathogenesis of sporadic as well as familial PD[35]. LRRK2 has been established through GWAS as a shared genetic predisposition for CD and UC, with a notably stronger association to CD[36]. Enhanced LRRK2 activity might increase vulnerability to intestinal inflammation, which can escalate into systemic inflammatory processes and act as a precursor to PD. Additionally, LRRK2 could facilitate the pathological accumulation of alpha-synuclein in the gut by influencing immune mechanisms, thereby accelerating neurodegeneration[37].
A number of pathogenic mutations in PD are located within the functional domains of LRRK2, such as N1437H, R1441C/G/H, Y1699C, I2012T, G2019S, and I2020T. Of these, the G2019S variant is the most prevalent one in PD patients[38]. This variant of LRRK2 is significantly more effective at phosphorylating α-synuclein compared to the wild-type. In addition to known pathogenic mutations in PD, the M2397T variant has been linked to sporadic CD and demonstrated to influence LRRK2 protein expression. Meanwhile, the N2081D variant is found to confer elevated risk for both CD and PD. In contrast, the N551K and R1398H variants demonstrate protective associations, correlating with a reduced risk for these disorders[39]. Notably, the LRRK2 N2081D CD risk allele and the G2019S pathogenic variant are situated within a shared kinase domain. The latter represents a major cause of familial as well as sporadic PD. Similar to G2019S, the N2081D variant correlates with elevated kinase activity. In contrast, neither N551K nor R1398H, located on a protective haplotype, affect its kinase function. Additionally, R1398H, not N551K, enhances GTPase activities, resulting in LRRK2 deactivation[40]. Although N551K is not located within any specific LRRK2 domain, studies suggest it is in linkage disequilibrium with R1398H[41]. These findings imply that the pathogenesis of both PD and CD may be closely associated with specific functional alterations in LRRK2, particularly involving its GTPase and kinase domains. Further studies[42] have shown that rare, high-impact genetic variations, such as the missense mutations G2019S and N2081D, are significantly associated with the comorbidity of IBD and PD. Additionally, phenotype-wide association studies (PheWAS) have identified 14 prioritized genes with strong genetic associations between IBD and PD, highlighting their possible involvement in the pathology of both conditions.
A key consideration is whether the common pathogenic effect of LRRK2 mutations in both IBD and PD indicates that LRRK2 kinase inhibitors could hold therapeutic potential for these two disorders. A review introduced that LRRK2 inhibitors as promising novel therapeutic strategies for managing the overlapping PD and IBD[43]. Preclinical evidence supports LRRK2 kinase inhibitors as a shared therapeutic strategy for both diseases. Using mouse models, Sun et al. revealed that the compound DN-9713, an LRRK2-targeting inhibitor, reversed Paneth cell abnormalities in G2019S LRRK2 transgenic mice and T300A Atg16l1 mutant mice following cigarette smoke exposure, which reduced crypt base apoptosis and reactivated autophagy through Beclin1[44]. These findings align with the shared mechanism of LRRK2 hyperactivity-induced autophagy impairment in both PD and CD, suggesting that targeting LRRK2 kinase activity may simultaneously address neurodegeneration and intestinal inflammation.
LRRK2 also plays a role in the pathological progression and propagation of α-synuclein, exacerbating the pathological process of PD. Research by Kondo et al. indicates that in human neuroblastoma SH-SY5Y cells, co-transfection of LRRK2 with α-synuclein promotes α-synuclein aggregation, phosphorylation, release into the extracellular medium, and enhances its cell-to-cell transmission to neighboring cells. Moreover, the LRRK2 G2019S mutation has a direct link to the secretion pathway of α-synuclein[45]. Subsequent research has found that the LRRK2 G2019S mutation can trigger α-synuclein aggregation in neurons derived from PD patients and in primary neuronal cultures, leading to the development of pathological inclusions[46]. These studies suggest that the LRRK2 G2019S mutation not only affects the intracellular processing of α-synuclein but may also promote its intercellular spread. This enhanced propagation is thought to be driven by the hyperactive kinase activity of the mutant LRRK2, which phosphorylates a subset of Rab GTPases (e.g., Rab35), key regulators of intracellular vesicle trafficking and endocytosis. Such phosphorylation events are believed to disrupt the endolysosomal pathway and alter the balance between the clearance and exocytosis of pathogenic α-synuclein. Specifically, the aberrant vesicle sorting and exocytosis may lead to an increased release of α-synuclein into the extracellular space, thereby facilitating its uptake by neighboring neurons and propagating pathology.This propagation could be a key factor in the pathology of PD. Further supporting this, work from Bae et al. implicates that LRRK2 facilitates the pathological propagation of α-synuclein within the brain. LRRK2 regulates the spread of α-synuclein between neurons, including its long-distance transmission[47]. Interestingly, in another study where α-synuclein PFFs were intracranially injected into wildtype and transgenic mice carrying the LRRK2 G2019S mutation, it was revealed that this specific genetic alteration enhances retrograde spread of α-synuclein (from postsynaptic to presynaptic neurons) rather than anterograde propagation (from presynaptic to postsynaptic neurons), although the exact mechanism remains unclear[48]. Overall, these findings are pivotal for understanding PD pathology and developing therapeutic strategies targeting LRRK2.
CARD15/NOD2
The NOD2/CARD15 gene, along with LRRK2, is considered a common genetic factor influencing the susceptibility to both PD and IBD. NOD2, an intracellular bacterial sensor encoded by the CARD15 gene, demonstrates low expression in the healthy colon but is markedly overproduced in the macrophages and mucosal epithelial cells of individuals with CD[49]. Three specific polymorphisms in the CARD15/NOD2 gene (R702W, G908R, P268S, and 1007fs) were found to be independently linked to an increased risk of CD[50]. Furthermore, variations within the NOD2/CARD15 gene have been suggested as potential contributors to the development of sporadic PD[51]. The protein encoded by the CARD15/NOD2 gene plays a key role in microbial pattern recognition and the initiation of host immune responses against inflammation. Mutations in CARD15/NOD2 have been implicated in the pathogenesis of chronic IBD[52]. Additionally, monocytes and macrophages isolated from humans and murine models carrying these mutations exhibit dysregulated NF-κB activation and aberrant secretion of inflammatory cytokines[53]. The NOD2 receptor, an intracellular sensor for recognizing bacterial pathogens, is widely expressed in astrocytes and microglia. Its activation can trigger an inflammatory cascade within the CNS, which may have destructive effects on neuronal cells under infectious conditions[54].
The evidence discussed above points toward a convergent, multi-step pathogenic model linking IBD and PD, rather than a simple linear causality. A unifying mechanistic framework can be proposed as follows: genetic susceptibility, particularly involving shared risk loci like LRRK2 and NOD2/CARD15, establishes a baseline vulnerability by dysregulating immune responses and intracellular trafficking pathways in both the gut and the brain. This genetic priming creates an environment where environmental triggers (e.g., gut dysbiosis, infections, or mucosal barrier injury) can more easily precipitate chronic intestinal inflammation and loss of epithelial integrity. The ensuing “leaky gut” facilitates the translocation of microbial metabolites and pro-inflammatory mediators, which not only fuel local inflammation but also activate systemic immune pathways. Within this inflamed milieu, the expression and aggregation of α-synuclein in the ENS are promoted. Leveraging the anatomical and biochemical connections of the GBA - particularly the vagus nerve - this pathological α-synuclein can propagate in a prion-like manner to the CNS, ultimately triggering neuroinflammation and dopaminergic neuron loss in the substantia nigra. Conversely, CNS-driven stress responses can reciprocally exacerbate gut inflammation via the brain-to-gut axis, creating a self-perpetuating cycle. This integrated framework helps to explain why therapeutic strategies targeting intestinal inflammation or modulating the gut microbiome might hold promise not only for IBD, but also for mitigating the onset and progression of PD.
GBA
Growing evidence of gut-CNS interaction has inspired the “GBA” theory, involving two-way communication between the central and ENS. It influences diverse physiological functions within both the central nervous and gastrointestinal systems, and is involved in the pathogenesis of PD[55] [Figure 1]. A rodent study demonstrated that mouse fetuses exposed to small amounts of penicillin in the uterus or shortly after birth exhibited enduring consequences later in life. These effects included an altered gut microbiome, elevated frontal cortex cytokine expression, impaired blood brain barrier (BBB) integrity, and manifestations of anxiety-like behavioral abnormalities[56]. Utilizing metagenomic approaches, Yang and colleagues uncovered disrupted gut viral and bacterial ecology in patients with major depression, where notably decreased levels of Blautia and Eubacterium were clinically associated with symptom severity[57]. Collectively, these findings highlight a bidirectional interplay: the gut microenvironment influences behavior via microbial metabolites and inflammatory signals (gut-to-brain axis), while psychological stress reciprocally disrupts microbiota composition (brain-to-gut axis).
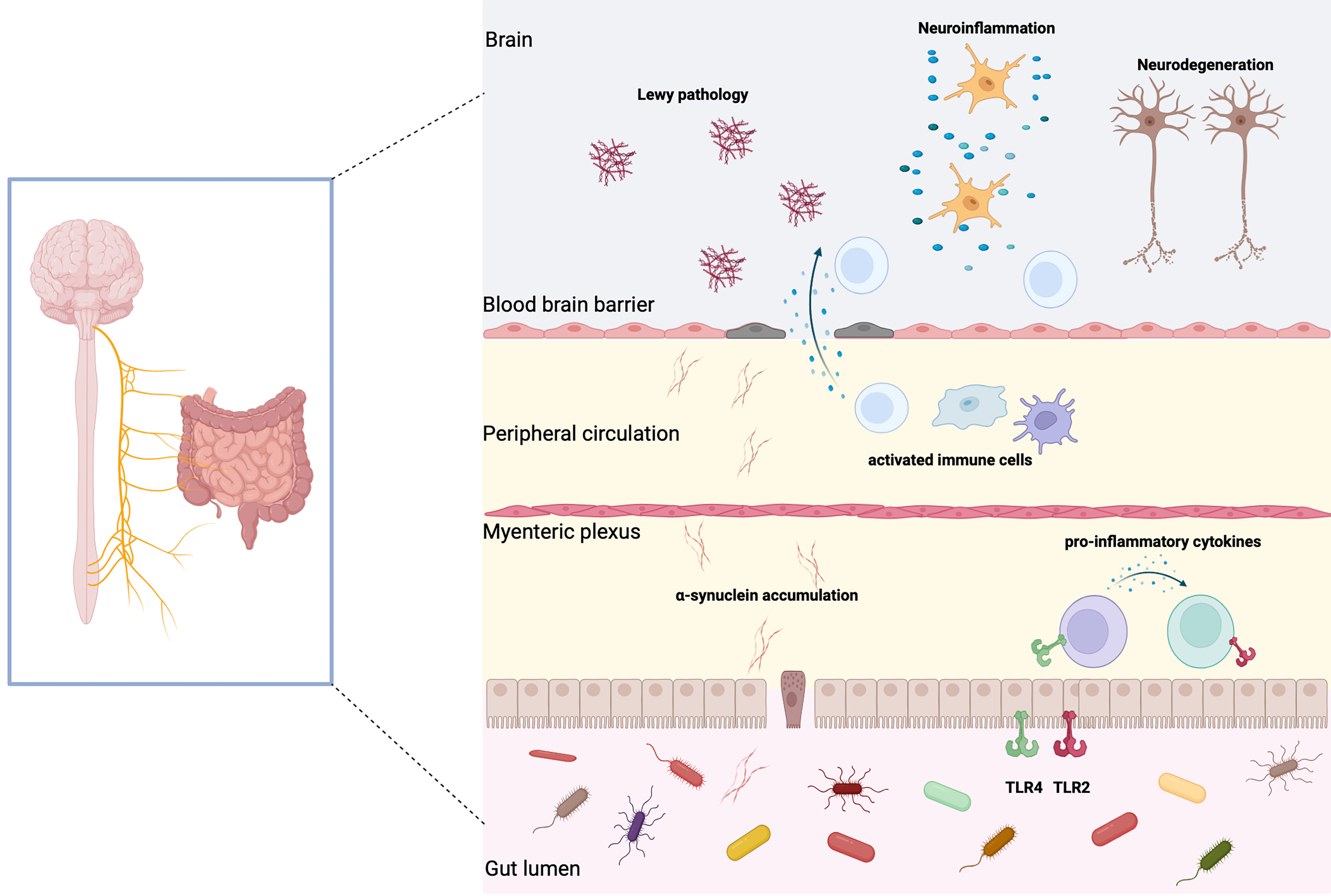
Figure 1. Schematic diagram illustrating changes in the intestinal microenvironment of PD and their impact on the pathophysiology. In PD, observable manifestations include intestinal α synaptic nuclear protein deposits, gastrointestinal inflammation, and impaired intestinal epithelial barrier function. Pathological α synaptic nuclear proteins are found in intestinal EECs and neurons, which transmit to the brainstem via the vagus nerve, leading to Lewy body pathology in the central nervous system. These proteins also trigger inflammatory responses that compromise intestinal barrier integrity. Concurrently, alterations in the gut microbiota composition may accelerate α synaptic nuclear protein aggregation. Dysbiosis of gut microbiota can induce intestinal inflammation, increase gut permeability, and allow systemic infiltration of microbial toxins and metabolites. This process activates immune responses while enhancing blood-brain barrier permeability, ultimately contributing to neuroinflammation and dopaminergic neuron degeneration. Created in BioRender. xin, Z. (2026) https://biorender.com/i7lju4w. PD: Parkinson’s disease; α-synuclein: alpha-synuclein; EECs: endocrine cells; CNS: central nervous system; BBB: blood-brain barrier; TLR2: Toll-like receptor 2; TLR4: Toll-like receptor 4.
More evidence indicates that PD extends beyond a movement disorder to include significant gastrointestinal involvement. PD may be associated with gut dysbiosis or intestinal barrier dysfunction, or both. According to research conducted by Cersosimo and Benarroch, the gastrointestinal profile of PD is characterized by sialopenia, dysphagia, gastroparesis, constipation, and dysfunctional defecation. Notably, these problems often precede motor symptoms by decades and affect a significant majority of patients, over 80%. Their research further indicates that a strong link between these gastrointestinal manifestations and neurodegenerative changes involving α-synuclein within the ENS[58]. Evidence indicates that α-synuclein accumulation occurs not only in the gastrointestinal tract of PD patients exhibiting gastrointestinal symptoms[59], but also in individuals prior to the manifestation of PD motor symptoms[60]. These findings support the idea that PD may originate from the ENS, where α-synuclein misfolding first occurs. However, it is important to note that this “bottom-up” model is not universally accepted, as some studies have failed to observe a consistent caudal-to-rostral spread of pathology in all PD patients, suggesting that alternative or parallel initiation sites (e.g., the olfactory bulb or CNS itself) may also exist[61].
Serving as a major conduit between the gut and the brain, the vagus nerve enables bidirectional signaling. Kim et al. demonstrated that duodenal and pyloric administration of α-synuclein in a murine model led to its translocation via the vagus nerve to multiple areas of the brain, such as the cerebellum, olfactory bulb, brainstem, and substantia nigra[62]. This process was linked to the emergence of PD symptoms, encompassing motor and non-motor dysfunctions. Additionally, the research highlighted vagotomy can decrease the risk of PD[63]. Several reports have demonstrated a preferential influence of the right vagus nerve on the SNpc, which displays a characteristic pattern of asymmetric degeneration. For example, a study[64] on 28-day and 63-day DSS (dextran sulfate sodium) treatment models found that in the 28-day DSS model, a 40% loss of dopaminergic neurons was observed in the right SNpc, while the left SN showed no significant loss, reflecting asymmetry. However, in the 63-day DSS model, both sides of the SN showed significant dopaminergic neuronal loss, suggesting that neurodegeneration starts unilaterally and later spreads bilaterally.
Gut microbiota and their metabolites
The gut microbiota is a critical component of GBA and has been widely studied for its role in regulating brain activity. It facilitates GBA communication by producing various bioactive compounds and metabolites[65]. Compounds, such as short-chain fatty acids (SCFAs), neurotransmitters, and other biochemical messengers generated by the microbiota, can regulate host physiological systems, including nervous system activity, immune regulation, and overall bodily responses. The brain and intestinal microbiota are interconnected, with communication occurring either through specific chemical signals or via pathways, such as the GBA. Similarly, neural activities can reshape the microbial composition by altering gut conditions through direct interventions or secondary effects[66].
The gut ecosystem comprises diverse inhabitants. These intestinal organisms and their metabolic products are strongly linked to the development of PD and contribute significantly to its pathological process. Newly identified pathways involve shaping the gastrointestinal environment, preserving the stability of microbial communities, and influencing gut-derived neurotransmitters, inflammatory mediators, and metabolic products[67]. Possible routes of influence include: (i) the movement of α-synuclein through neural pathways, particularly via the vagus nerve; (ii) pathways governed by the neuroendocrine system, particularly the hypothalamic-pituitary-adrenal (HPA) axis, along with the activity of specific neurochemicals including 5-hydroxytryptamine (5-HT) and γ-aminobutyric acid (GABA); (iii) immune modulation involving immune cell activity and the secretion of inflammatory mediators; and (iv) the circulatory system transporting molecules like bile acids (BAS), SCFAs, lipopolysaccharides (LPS), and hydrogen.
Changes in the gut microbial community have been observed in patients with both PD and IBD. This imbalance can lead to increased gut barrier leakage, inflammation, disturbances in SCFA balance, and oxidative damage. These factors may further encourage the clumping of α-synuclein. When compared to samples from healthy controls, the introduction of stool samples from PD patients into a transgenic murine system expressing human α-synuclein resulted in a more pronounced exacerbation of motor dysfunction[68]. Collectively, these results imply that microbial alterations correlate with elevated PD risk, lending direct support to the concept that gut dysbiosis participates in the pathogenesis of this disorder. Both IBD and PD patients show reduced bacterial diversity[69]. Additionally, oral antibiotic use is linked to a markedly lower likelihood of developing PD[70]. The intestinal environment in PD patients shows increased prevalence of proinflammatory bacteria like Proteobacteria and a reduction in microbes that produce SCFAs[71]. Additionally, the intestinal microbiome can stimulate immune cells in the CNS, including resident microglia. Following activation, microglia secrete various inflammatory mediators, including reactive oxygen/nitrogen compounds and pro-inflammatory signaling molecules, which collectively contribute to neuroinflammatory processes[72]. Emerging data demonstrate a potential role of the intestinal microbiome in modulating the pharmacokinetics of PD drugs. The microbiota may increase the rate of drug inactivation or reduce drug absorption. For example, tyrosine decarboxylase produced by gut bacteria in rats can lower the levels of levodopa in plasma[73].
The gut microbiota may contribute to neurodegeneration of PD by gradually accumulating microbial metabolites throughout the entire disease course, encompassing the pre-symptomatic phase. These microbial metabolites, derived from dietary fiber fermentation, are known for their neuroprotective properties[74]. SCFAs are involved in PD pathogenesis through their influence on BBB function, microglial activity, neuronal autophagy and apoptosis, as well as intestinal barrier integrity and inflammation. Moreover, SCFAs mediate immune regulation in IBD through influencing the differentiation of both innate and adaptive immune cell lineages[75]. Studies have shown that levels of SCFAs in both serum and fecal specimens are markedly different when comparing PD patients to healthy subjects, with lower concentrations observed in the PD group[76]. Furthermore, research by Tan et al. indicated that butyrate levels were inversely related to both the severity of postural instability-gait disorder and constipation. Thus, SCFAs may serve as diagnostic biomarkers for both PD and IBD[77]. Future treatment approaches may aim to modulate gut microbiota composition and enhance the yield of beneficial SCFAs.
In addition to SCFAs, trimethylamine N-oxide (TMAO) is also a gut microbial metabolite though its role in PD appears complex. Some studies suggest that TMAO may have protective effects. For instance, Jamal et al. observed that TMAO can induce conformational changes in α-synuclein, potentially inhibiting its aggregation and the formation of insoluble fibrils associated with PD[78]. Uversky et al. found that at higher TMAO concentrations, α-synuclein forms oligomers that may represent its physiological folding form of the protein, possibly beneficial for avoiding pathological aggregation[79]. However, studies by Chen et al. and Sankowski et al. indicate that high TMAO levels may be associated with disease progression[80,81]. It is important to note that these studies do not directly prove whether the increased TMAO levels are the cause or the result of disease progression. Additionally, research by Chung et al. demonstrated diminished plasma TMAO concentrations among untreated patients in the early stages of PD when compared with control subjects, proposing an association of TMAO with PD onset[82]. Conversely, Sankowski et al. reported elevated circulating TMAO in PD patients (mean disease duration: 8.1 years) relative to controls, with even greater levels observed in those experiencing motor fluctuations[81]. It is proposed that TMAO concentrations are linked to advanced stages of PD and the emergence of motor complications. Taken together, these seemingly contradictory findings suggest that TMAO may exert stage-dependent dual effects in PD pathophysiology. It is plausible that physiological levels of TMAO contribute to maintaining α-synuclein in a non-pathological conformation, potentially offering neuroprotection in the early, pre-symptomatic phases. However, in established disease, elevated TMAO, whether as a consequence of gut dysbiosis, altered host metabolism, or renal dysfunction, might serve as a biomarker of disease progression or even actively contribute to neuroinflammation and motor complications. This interpretation remains speculative, as the current evidence is largely derived from cross-sectional studies that cannot establish temporality or causality. Longitudinal studies tracking TMAO levels from the prodromal phase through the advanced stages of PD are urgently needed to clarify whether TMAO fluctuations are a cause, a consequence, or an epiphenomenon of disease progression.
The above studies suggest the possibility that PD may originate from the gut. The gut microbiota influences the disease process of PD through various mechanisms, including affecting neural pathways, altering host metabolism, regulating immunity, and impacting drug efficacy. Therefore, the influence of gut microbiota on the onset of PD and IBD may occur through the microbiota’s direct involvement in inflammatory responses, or through its metabolites, which in turn affect the inflammatory process.
Intestinal inflammation
Intestinal inflammation is critically involved in the pathogenic mechanisms underlying PD and IBD. A growing body of research demonstrates that chronic gastrointestinal inflammation may promote neuroinflammatory responses through the GBA[83]. Persistent gut inflammation may promote the aggregation of α-synuclein in the intestinal tract. This pathology could subsequently propagate toward the CNS through vagal pathways[60]. αSyn, TLR activation, and changes in intestinal epithelial cells (IECs) all play crucial roles in this process. In the context of inflammation, αSyn and its aggregated forms may promote TLR-dependent inflammatory signaling. This indicates that TLR activation could trigger pro-inflammatory cytokine production in the intestinal environment, possibly via targeted activation of IECs. IECs are regarded as pivotal contributors to inflammatory processes owing to their capacity to generate barrier functions that shield the host from luminal microbes and immunogens, which is essential for maintaining gastrointestinal equilibrium[84]. Moreover, inflammation in the intestine is associated with a rise in gut barrier permeability and a disruption in blood-brain barrier integrity[85]. Elevated gut barrier permeability enables intestinal microbes and their metabolic byproducts to trigger pro-inflammatory cytokine production in the ENS and promote pathological α-synuclein aggregation. This process enables the spread of abnormal α-synuclein to the midbrain via vagal nerve routes, while inflammatory cytokines may either cross into the brain through the bloodstream or activate the vagus nerve to promote neuroinflammation in the CNS. Pro-inflammatory cytokines, along with brain α-synuclein pathology, can induce neuronal damage and eventually lead to cell death, further exacerbating acute inflammatory responses. In addition to the “bottom-up” hypothesis, the “top-down” hypothesis, where gastrointestinal inflammation is caused by vagus nerve pathway impairment, is also plausible[86].
Chronic gut inflammation and elevated permeability contribute to systemic inflammation, which can, in turn, compromise BBB integrity. Together, intestinal inflammation, systemic immune activation, and cerebral synuclein pathology collectively drive neuroinflammation, whereas neuroinflammation is a major driver of neurodegeneration in PD. Therefore, intestinal inflammation serves as a critical link between gut and brain pathologies, and further elucidating the mechanisms by which gut inflammation influences CNS dysfunction, and vice versa. These will be essential for advancing our understanding of disease pathogenesis. Future studies ought to investigate intervention strategies targeting intestinal inflammation, with the goal of mitigating PD risk or slowing its progression.
THE IMPACT OF IBD TREATMENT METHODS ON PD
Drugs
The previous discussion explored the connection between PD and IBD, with IBD increasing the incidence of PD. Therefore, could medications used to treat IBD reduce the risk of PD? Common medications used to treat IBD include aminosalicylates, corticosteroids, immunosuppressants, and biologic agents. According to a retrospective cohort study, early treatment with anti-TNF (tumor necrosis factor) was associated with a significantly lower risk of developing PD among IBD subjects. The treated cohort showed a 78% reduction in incidence compared to that without a history of anti-TNF exposure[17]. More investigations are needed to ascertain whether TNF inhibition therapy ought to be implemented in individuals with IBD to reduce PD risk. A recent MR study assessed how long-term inhibition of TNF affects the risk of PD onset and the age at which it develops, and the findings failed to verify the idea that inhibiting the TNF-TNFR1 signaling pathway can prevent or delay PD[87]. Unlike the study by Peter et al.[17], this research primarily focused on selective inhibition of TNF-TNFR1 signaling, a pathway believed to be central to TNF-related neurotoxicity in PD. Additionally, a cross-sectional study in Spain revealed that individuals under 65 using 5-aminosalicylic acid (5-ASA) had a significantly lower likelihood of receiving PD medication (OR = 0.28, P = 0.01)[88]. Moreover, administration of these medications (azathioprine and corticosteroids) demonstrated a significant correlation with decreased PD incidence[89]. The above studies suggest that some common medications used to treat IBD may have a protective effect on the onset of PD, providing a reference for future PD treatments and the potential for “repurposing” existing drugs. MR analyses of PD and IBD indicate that the Cxcr4 gene represents a promising drug target. Encoding the chemokine receptor CXCR4, this gene represents a target whereby flavonoids might offer therapeutic benefits in both PD and IBD via CXCR4 protein inhibition[90]. Consequently, the identification of drug-related genetic targets may advance the development of novel therapeutic strategies targeting PD and IBD.
Fecal microbiota transplantation
Fecal microbiota transplantation (FMT) is an intervention to modulate the host microbiome by restoring gut microbial balance, aiming to treat dysbiosis-related diseases. The first recorded medical application of FMT dates back to 1958, for treating pseudomembranous colitis[91]. The involvement of gut dysbiosis in the progression of PD indicates that the restoration of gut microbiota may serve as a new therapeutic option for PD, thereby attracting considerable interest in FMT as a potential treatment strategy. A case report indicates that FMT may alleviate gastrointestinal and motor dysfunctions in PD patients[92]. Additionally, animal studies have proposed several mechanisms by which FMT could influence PD, including altering gut microbiota composition, enhancing gut barrier function, inhibiting pathogens, and modulating immune responses. Research by Sun et al. indicated that FMT was shown to mitigate manifestations in a murine PD model induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), through the amelioration of both microbial imbalance and central inflammation[93]. Zhao et al also showed that FMT improved rotenone-induced PD mouse models by correcting dysbiosis and inhibiting the lipopolysaccharide-TLR4 signaling pathway[94].
Therefore, FMT holds significant potential as a future treatment for PD. Nevertheless, it is essential to consider the potential risks linked to FMT, including infections, sepsis, the spread of harmful intestinal organisms, bleeding, reactivation of dormant viruses like cytomegalovirus, and respiratory complications, such as pneumonia[95].
Despite promising preclinical evidence, several critical limitations must be acknowledged when translating these findings to human PD. First, current animal models of PD, whether neurotoxin-induced (e.g., MPTP, rotenone) or transgenic, fail to fully recapitulate the chronic, progressive nature of human sporadic PD, particularly the age-dependent accumulation of α-synuclein pathology and the complex gut-brain interactions over decades[96]. Second, most FMT studies on PD models have been short-term, whereas PD is a lifelong condition, making it difficult to assess long-term efficacy and safety. Third, the optimal donor selection, route of administration, dosing frequency, and timing of intervention (prodromal vs. established disease) remain undefined.
To advance the broader clinical application of FMT in PD, larger-scale, high-quality, randomized, double-blind, placebo-controlled clinical trials with long-term follow-up are urgently needed to gather sufficient clinical evidence and to determine whether the benefits observed in animals can be replicated in PD patients.
Probiotics
Most existing studies report that the administration of probiotics yields neuroprotective effects and alleviates cognitive impairment[97]. Probiotics are esteemed for possessing antioxidant, anti-inflammatory, and neuroprotective efficacies. Their impact on neurological processes is mediated by multifaceted biological interactions, encompassing the mitigation of oxidative stress, immunomodulation, and the governance of apoptotic pathways[98]. According to Liu et al., combined probiotic-prebiotic treatment yielded significantly greater efficacy than either monotherapy in murine models of PD[99]. A study led by Sun et al. revealed that supplementing standard PD treatments with Probio-M8, a probiotic formulation, significantly enhanced therapeutic effectiveness. This intervention further modified the intestinal microbiota, shifted microbial metabolic capabilities, and altered serum biochemical markers in the host[100]. An in vitro study revealed that probiotics (lactobacilli and bifidobacteria) inhibit the production of inflammatory cytokines and reactive oxygen species (ROS) in PD patients[101].
Psychobiotics refer to a specific class of probiotics known to confer benefits to psychological health[102]. They are under investigation as stand-alone or adjunctive treatments for psychiatric, neurodevelopmental, and neurodegenerative disorders, showing promise as future clinical interventions for brain-related conditions. Recent studies have identified Lactobacillus plantarum DP189 as an effective psychobiotic capable of mitigating α-synuclein degradation in MPTP-induced PD mice through modulation of oxidative stress, inflammatory response, and gut microbiota dysfunction[103]. The above studies suggest that gut microbiota modulation holds potential in the treatment of both PD and IBD. However, it is crucial to interpret these findings with caution due to substantial translational barriers. The majority of evidence for probiotics in PD is derived from in vitro assays or animal models, which do not account for the complexity of the human gut ecosystem, including host-microbe interactions, inter-individual variability in microbiota composition, and the confounding effects of diet and co-medications. Furthermore, the viability, colonization capacity, and metabolic activity of probiotic strains in the human gastrointestinal tract are highly variable and often poorly characterized.
In conclusion, although PD and IBD appear to be distinctly different diseases at first glance, existing research suggests a connection between them in terms of epidemiology and pathophysiological mechanisms. The dual issues of chronic gut inflammation and dysbiosis are pivotal drivers in the pathogenesis of these conditions. The abnormal aggregation of α-synuclein is hypothesized to connect the pathological processes of IBD and PD. Additionally, treatment for IBD has shown some protective effects against PD, and current IBD therapies might be cautiously repurposed for PD treatment. Despite recent progress, our understanding of the complex interactions between these two diseases remains limited. Future research needs to further explore the molecular basis of these connections and assess the efficacy of existing IBD treatments in the management of PD. Furthermore, these results carry substantial clinical relevance, indicating that neurological symptom surveillance warrants greater emphasis during extended care for individuals with IBD. Collectively, these insights could lay the foundation for advanced management strategies, ultimately improving quality of life of individuals with IBD and PD.
DECLARATIONS
Authors’ contributions
Made substantial contributions to conception and design of the study and performed data analysis and interpretation as well as writing the manuscript: Zhou X, Bi Y
Performed data acquisition, as well as provided administrative, technical, and material support: Zhang X, Liang H
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This work was supported by grants from National Natural Science Foundation of China (No.82174158), Shanghai Municipal Commission of Health and Family Planning (No. 202340293), Shanghai Pudong New Area Leading Health System Personnel Training Program (PWR12023-06 to Bi Y).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Argollo M, Gilardi D, Peyrin-Biroulet C, Chabot JF, Peyrin-Biroulet L, Danese S. Comorbidities in inflammatory bowel disease: a call for action. Lancet Gastroenterol Hepatol. 2019;4:643-54.
2. Agrawal M, Jess T. Implications of the changing epidemiology of inflammatory bowel disease in a changing world. United European Gastroenterol J. 2022;10:1113-20.
3. Kaplan GG, Windsor JW. The four epidemiological stages in the global evolution of inflammatory bowel disease. Nat Rev Gastroenterol Hepatol. 2021;18:56-66.
4. Cho JH. The genetics and immunopathogenesis of inflammatory bowel disease. Nat Rev Immunol. 2008;8:458-66.
5. Liu JZ, van Sommeren S, Huang H, et al. ; International Multiple Sclerosis Genetics Consortium, International IBD Genetics Consortium. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. 2015;47:979-86.
7. Wong YC, Krainc D. α-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat Med. 2017;23:1-13.
8. Emamzadeh FN, Surguchov A. Parkinson’s disease: biomarkers, treatment, and risk factors. Front Neurosci. 2018;12:612.
9. Opara J, Małecki A, Małecka E, Socha T. Motor assessment in Parkinson’s disease. Ann Agric Environ Med. 2017;24:411-5.
10. Abbott RD, Petrovitch H, White LR, et al. Frequency of bowel movements and the future risk of Parkinson’s disease. Neurology. 2001;57:456-62.
11. Yoo BB, Mazmanian SK. The enteric network: interactions between the immune and nervous systems of the gut. Immunity. 2017;46:910-26.
12. Braak H, Rüb U, Gai WP, Del Tredici K. Idiopathic Parkinson’s disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm. 2003;110:517-36.
13. Lin JC, Lin CS, Hsu CW, Lin CL, Kao CH. Association between Parkinson’s disease and inflammatory bowel disease: a nationwide taiwanese retrospective cohort study. Inflamm Bowel Dis. 2016;22:1049-55.
14. Villumsen M, Aznar S, Pakkenberg B, Jess T, Brudek T. Inflammatory bowel disease increases the risk of Parkinson’s disease: a Danish nationwide cohort study 1977-2014. Gut. 2019;68:18-24.
15. Weimers P, Halfvarson J, Sachs MC, et al. Inflammatory bowel disease and Parkinson’s disease: a nationwide Swedish cohort study. Inflamm Bowel Dis. 2019;25:111-23.
16. Park S, Kim J, Chun J, et al. Patients with inflammatory bowel disease are at an increased risk of Parkinson’s disease: a South Korean nationwide population-based study. J Clin Med. 2019;8:1191.
17. Peter I, Dubinsky M, Bressman S, et al. Anti-tumor necrosis factor therapy and incidence of Parkinson disease among patients with inflammatory bowel disease. JAMA Neurol. 2018;75:939-46.
18. Coates MD, Ba DM, Liu G, Dalessio S, Leslie DL, Huang X. Revisiting the association between inflammatory bowel disease and Parkinson’s disease. Inflamm Bowel Dis. 2022;28:850-4.
19. Wang HL, Wang ZY, Tian J, Ma DR, Shi CH. Association between inflammatory bowel disease and Parkinson’s disease: a prospective cohort study of 468,556 UK biobank participants. Front Aging Neurosci. 2023;15:1294879.
20. Loosen SH, Yaqubi K, May P, et al. Association between inflammatory bowel disease and subsequent development of restless legs syndrome and Parkinson’s disease: a retrospective cohort study of 35,988 primary care patients in Germany. Life. 2023;13:897.
21. Choi K, Lee HJ, Han K, Koh SJ, Im JP, Kim JS. Depression in patients with inflammatory bowel disease is associated with increased risk of dementia and Parkinson’s disease: a nationwide, population-based study. Front Med. 2022;9:1014290.
23. Hsu YT, Liao CC, Chang SN, et al. Increased risk of depression in patients with Parkinson disease: a nationwide cohort study. Am J Geriatr Psychiatry. 2015;23:934-40.
24. Camacho-Soto A, Gross A, Searles Nielsen S, Dey N, Racette BA. Inflammatory bowel disease and risk of Parkinson’s disease in Medicare beneficiaries. Parkinsonism Relat Disord. 2018;50:23-8.
25. Smith GD, Ebrahim S. ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol 2003;32:1-22.
26. Lawlor DA, Harbord RM, Sterne JA, Timpson N, Davey Smith G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med. 2008;27:1133-63.
27. Freuer D, Meisinger C. Association between inflammatory bowel disease and Parkinson’s disease: a Mendelian randomization study. NPJ Parkinsons Dis. 2022;8:55.
28. Zeng R, Wang J, Jiang R, et al. Investigating causality and shared genetic architecture between neurodegenerative disorders and inflammatory bowel disease. Aging Dis. 2023;14:1349-59.
29. Cui G, Li S, Ye H, et al. Are neurodegenerative diseases associated with an increased risk of inflammatory bowel disease? A two-sample Mendelian randomization study. Front Immunol. 2022;13:956005.
30. Yang J, Lin W, Ma Y, et al. Investigation of the causal association between Parkinson’s disease and autoimmune disorders: a bidirectional Mendelian randomization study. Front Immunol. 2024;15:1370831.
31. Witoelar A, Jansen IE, Wang Y, et al. ; International Parkinson’s Disease Genomics Consortium (IPDGC), North American Brain Expression Consortium (NABEC), and United Kingdom Brain Expression Consortium (UKBEC) Investigators. Genome-wide pleiotropy between Parkinson disease and autoimmune diseases. JAMA Neurol. 2017;74:780-92.
32. Buniello A, MacArthur JAL, Cerezo M, et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019;47:D1005-12.
33. Lee HS, Lobbestael E, Vermeire S, Sabino J, Cleynen I. Inflammatory bowel disease and Parkinson’s disease: common pathophysiological links. Gut. 2021;70:408-17.
35. Zimprich A, Biskup S, Leitner P, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601-7.
36. Umeno J, Asano K, Matsushita T, et al. Meta-analysis of published studies identified eight additional common susceptibility loci for Crohn’s disease and ulcerative colitis. Inflamm Bowel Dis. 2011;17:2407-15.
37. Li Y, Chen Y, Jiang L, et al. Intestinal inflammation and Parkinson’s disease. Aging Dis. 2021;12:2052-68.
39. Herrick MK, Tansey MG. Is LRRK2 the missing link between inflammatory bowel disease and Parkinson’s disease? NPJ Parkinsons Dis 2021;7:26.
40. Hui KY, Fernandez-Hernandez H, Hu J, et al. Functional variants in the LRRK2 gene confer shared effects on risk for Crohn’s disease and Parkinson’s disease. Sci Transl Med. 2018;10:eaai7795.
41. Gopalai AA, Lim JL, Li HH, et al. LRRK2 N551K and R1398H variants are protective in Malays and Chinese in Malaysia: a case-control association study for Parkinson’s disease. Mol Genet Genomic Med. 2019;7:e604.
42. Kars ME, Wu Y, Stenson PD, et al. The landscape of rare genetic variation associated with inflammatory bowel disease and Parkinson’s disease comorbidity. Genome Med. 2024;16:66.
43. Lashgari NA, Roudsari NM, Niknejad A, et al. LRRK2; communicative role in the treatment of Parkinson’s disease and ulcerative colitis overlapping. CNS Neurol Disord Drug Targets. 2024;23:1177-88.
44. Sun S, Hodel M, Wang X, et al. Macrophage LRRK2 hyperactivity impairs autophagy and induces Paneth cell dysfunction. Sci Immunol. 2024;9:eadi7907.
45. Kondo K, Obitsu S, Teshima R. α-Synuclein aggregation and transmission are enhanced by leucine-rich repeat kinase 2 in human neuroblastoma SH-SY5Y cells. Biol Pharm Bull. 2011;34:1078-83.
46. Bieri G, Brahic M, Bousset L, et al. LRRK2 modifies α-syn pathology and spread in mouse models and human neurons. Acta Neuropathol. 2019;137:961-80.
47. Bae EJ, Kim DK, Kim C, et al. LRRK2 kinase regulates α-synuclein propagation via RAB35 phosphorylation. Nat Commun. 2018;9:3465.
48. Henderson MX, Cornblath EJ, Darwich A, et al. Spread of α-synuclein pathology through the brain connectome is modulated by selective vulnerability and predicted by network analysis. Nat Neurosci. 2019;22:1248-57.
49. Berrebi D, Maudinas R, Hugot JP, et al. Card15 gene overexpression in mononuclear and epithelial cells of the inflamed Crohn’s disease colon. Gut. 2003;52:840-6.
50. Lesage S, Zouali H, Cézard JP, et al. ; EPWG-IBD Group, EPIMAD Group, GETAID Group. CARD15/NOD2 mutational analysis and genotype-phenotype correlation in 612 patients with inflammatory bowel disease. Am J Hum Genet. 2002;70:845-57.
51. Bialecka M, Kurzawski M, Klodowska-Duda G, et al. CARD15 variants in patients with sporadic Parkinson’s disease. Neurosci Res. 2007;57:473-6.
52. Sterka D Jr, Rati DM, Marriott I. Functional expression of NOD2, a novel pattern recognition receptor for bacterial motifs, in primary murine astrocytes. Glia. 2006;53:322-30.
53. Beynon V, Cotofana S, Brand S, et al. NOD2/CARD15 genotype influences MDP-induced cytokine release and basal IL-12p40 levels in primary isolated peripheral blood monocytes. Inflamm Bowel Dis. 2008;14:1033-40.
54. Chauhan VS, Sterka DG Jr, Furr SR, Young AB, Marriott I. NOD2 plays an important role in the inflammatory responses of microglia and astrocytes to bacterial CNS pathogens. Glia. 2009;57:414-23.
55. Carabotti M, Scirocco A, Maselli MA, Severi C. The gut-brain axis: interactions between enteric microbiota, central and enteric nervous systems. Ann Gastroenterol. 2015;28:203-9.
56. Leclercq S, Mian FM, Stanisz AM, et al. Low-dose penicillin in early life induces long-term changes in murine gut microbiota, brain cytokines and behavior. Nat Commun. 2017;8:15062.
57. Yang J, Zheng P, Li Y, et al. Landscapes of bacterial and metabolic signatures and their interaction in major depressive disorders. Sci Adv. 2020;6:eaba8555.
58. Cersosimo MG, Benarroch EE. Pathological correlates of gastrointestinal dysfunction in Parkinson’s disease. Neurobiol Dis. 2012;46:559-64.
59. Lebouvier T, Chaumette T, Damier P, et al. Pathological lesions in colonic biopsies during Parkinson’s disease. Gut. 2008;57:1741-3.
60. Stokholm MG, Danielsen EH, Hamilton-Dutoit SJ, Borghammer P. Pathological α-synuclein in gastrointestinal tissues from prodromal Parkinson disease patients. Ann Neurol. 2016;79:940-9.
61. Del Tredici K, Rüb U, De Vos RA, Bohl JR, Braak H. Where does parkinson disease pathology begin in the brain? J Neuropathol Exp Neurol 2002;61:413-26.
62. Kim S, Kwon SH, Kam TI, et al. Transneuronal propagation of pathologic α-synuclein from the gut to the brain models Parkinson’s disease. Neuron. 2019;103:627-41.e7.
63. Liu B, Fang F, Pedersen NL, et al. Vagotomy and Parkinson disease: a Swedish register-based matched-cohort study. Neurology. 2017;88:1996-2002.
64. Espinosa-Oliva AM, Ruiz R, Soto MS, et al. Inflammatory bowel disease induces pathological α-synuclein aggregation in the human gut and brain. Neuropathol Appl Neurobiol. 2024;50:e12962.
65. Socała K, Doboszewska U, Szopa A, et al. The role of microbiota-gut-brain axis in neuropsychiatric and neurological disorders. Pharmacol Res. 2021;172:105840.
67. Jia X, Chen Q, Zhang Y, Asakawa T. Multidirectional associations between the gut microbiota and Parkinson’s disease, updated information from the perspectives of humoral pathway, cellular immune pathway and neuronal pathway. Front Cell Infect Microbiol. 2023;13:1296713.
68. Sampson TR, Debelius JW, Thron T, et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell. 2016;167:1469-80.e12.
69. Cirstea MS, Yu AC, Golz E, et al. Microbiota composition and metabolism are associated with gut function in Parkinson’s disease. Mov Disord. 2020;35:1208-17.
70. Mertsalmi TH, Pekkonen E, Scheperjans F. Antibiotic exposure and risk of Parkinson’s disease in Finland: a nationwide case-control study. Mov Disord. 2020;35:431-42.
71. Sun MF, Shen YQ. Dysbiosis of gut microbiota and microbial metabolites in Parkinson’s disease. Ageing Res Rev. 2018;45:53-61.
72. Phani S, Loike JD, Przedborski S. Neurodegeneration and inflammation in Parkinson’s disease. Parkinsonism Relat Disord. 2012;18:S207-9.
73. van Kessel SP, Frye AK, El-Gendy AO, et al. Gut bacterial tyrosine decarboxylases restrict levels of levodopa in the treatment of Parkinson’s disease. Nat Commun. 2019;10:310.
74. Kim CS. Roles of diet-associated gut microbial metabolites on brain health: cell-to-cell interactions between gut bacteria and the central nervous system. Adv Nutr. 2024;15:100136.
75. Gonçalves P, Araújo JR, Di Santo JP. A cross-talk between microbiota-derived short-chain fatty acids and the host mucosal immune system regulates intestinal homeostasis and inflammatory bowel disease. Inflamm Bowel Dis. 2018;24:558-72.
76. Unger MM, Spiegel J, Dillmann KU, et al. Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Parkinsonism Relat Disord. 2016;32:66-72.
77. Tan AH, Chong CW, Lim SY, et al. Gut microbial ecosystem in Parkinson disease: new clinicobiological insights from multi-omics. Ann Neurol. 2021;89:546-59.
78. Jamal S, Kumari A, Singh A, Goyal S, Grover A. Conformational ensembles of α-synuclein derived peptide with different osmolytes from temperature replica exchange sampling. Front Neurosci. 2017;11:684.
79. Uversky VN, Li J, Fink AL. Trimethylamine-N-oxide-induced folding of alpha-synuclein. FEBS Lett. 2001;509:31-5.
80. Chen SJ, Kuo CH, Kuo HC, et al. The gut metabolite trimethylamine N-oxide is associated with Parkinson’s disease severity and progression. Mov Disord. 2020;35:2115-6.
81. Sankowski B, Księżarczyk K, Raćkowska E, Szlufik S, Koziorowski D, Giebułtowicz J. Higher cerebrospinal fluid to plasma ratio of p-cresol sulfate and indoxyl sulfate in patients with Parkinson’s disease. Clin Chim Acta. 2020;501:165-73.
82. Chung SJ, Rim JH, Ji D, et al. Gut microbiota-derived metabolite trimethylamine N-oxide as a biomarker in early Parkinson’s disease. Nutrition. 2021;83:111090.
83. Chen QQ, Haikal C, Li W, Li JY. Gut inflammation in association with pathogenesis of Parkinson’s disease. Front Mol Neurosci. 2019;12:218.
84. Drobny A, Ngo PA, Neurath MF, Zunke F, López-Posadas R. Molecular communication between neuronal networks and intestinal epithelial cells in gut inflammation and Parkinson’s disease. Front Med. 2021;8:655123.
85. Dogra N, Mani RJ, Katare DP. The gut-brain axis: two ways signaling in Parkinson’s disease. Cell Mol Neurobiol. 2022;42:315-32.
86. Rolli-Derkinderen M, Leclair-Visonneau L, Bourreille A, Coron E, Neunlist M, Derkinderen P. Is Parkinson’s disease a chronic low-grade inflammatory bowel disease? J Neurol 2020;267:2207-13.
87. Kang X, Ploner A, Pedersen NL, et al. Tumor necrosis factor inhibition and Parkinson disease: a Mendelian randomization study. Neurology. 2021;96:e1672-9.
88. Pinel Ríos J, Madrid Navarro CJ, Pérez Navarro MJ, et al. Association of Parkinson’s disease and treatment with aminosalicylates in inflammatory bowel disease: a cross-sectional study in a Spain drug dispensation records. BMJ Open. 2019;9:e025574.
89. Racette BA, Gross A, Vouri SM, Camacho-Soto A, Willis AW, Searles Nielsen S. Immunosuppressants and risk of Parkinson disease. Ann Clin Transl Neurol. 2018;5:870-5.
90. Dogra N, Jakhmola-Mani R, Potshangbam AM, Buch S, Pande Katare D. CXCR4 as possible druggable target linking inflammatory bowel disease and Parkinson’s disease. Metab Brain Dis. 2023;38:1079-96.
91. Eiseman B, Silen W, Bascom GS, Kauvar AJ. Fecal enema as an adjunct in the treatment of pseudomembranous enterocolitis. Surgery. 1958;44:854-9.
92. Huang H, Xu H, Luo Q, et al. Fecal microbiota transplantation to treat Parkinson’s disease with constipation: a case report. Medicine. 2019;98:e16163.
93. Sun MF, Zhu YL, Zhou ZL, et al. Neuroprotective effects of fecal microbiota transplantation on MPTP-induced Parkinson’s disease mice: gut microbiota, glial reaction and TLR4/TNF-α signaling pathway. Brain Behav Immun. 2018;70:48-60.
94. Zhao Z, Ning J, Bao XQ, et al. Fecal microbiota transplantation protects rotenone-induced Parkinson’s disease mice via suppressing inflammation mediated by the lipopolysaccharide-TLR4 signaling pathway through the microbiota-gut-brain axis. Microbiome. 2021;9:226.
95. Dailey FE, Turse EP, Daglilar E, Tahan V. The dirty aspects of fecal microbiota transplantation: a review of its adverse effects and complications. Curr Opin Pharmacol. 2019;49:29-33.
96. Reinares-Sebastián A, Esteban-García N, Takada M, Trigo-Damas I. Drug discovery and development for Parkinson’s disease: are preclinical models good enough? Front Aging Neurosci 2025;17:1692592.
97. Ji HF, Shen L. Probiotics as potential therapeutic options for Alzheimer’s disease. Appl Microbiol Biotechnol. 2021;105:7721-30.
98. Gao J, Zhao L, Cheng Y, et al. Probiotics for the treatment of depression and its comorbidities: a systemic review. Front Cell Infect Microbiol. 2023;13:1167116.
99. Liu X, Du ZR, Wang X, et al. Polymannuronic acid prebiotic plus Lacticaseibacillus rhamnosus GG probiotic as a novel synbiotic promoted their separate neuroprotection against Parkinson’s disease. Food Res Int. 2022;155:111067.
100. Sun H, Zhao F, Liu Y, et al. Probiotics synergized with conventional regimen in managing Parkinson’s disease. NPJ Parkinsons Dis. 2022;8:62.
101. Magistrelli L, Amoruso A, Mogna L, et al. Probiotics may have beneficial effects in Parkinson’s disease: in vitro evidence. Front Immunol. 2019;10:969.
102. Dinan TG, Stanton C, Cryan JF. Psychobiotics: a novel class of psychotropic. Biol Psychiatry. 2013;74:720-6.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].