
Influence of type 2 diabetes on Parkinson’s disease: emerging insights into pathogenesis and progression
 0
0 
Abstract
Parkinson’s disease (PD) is the second most common neurodegenerative disorder worldwide and is characterized by progressive motor and non-motor manifestations. Its rising prevalence poses significant challenges for clinical management and public health. Type 2 diabetes mellitus (T2DM), the most frequent metabolic disorder, shares several pathogenic mechanisms with PD, including mitochondrial dysfunction, insulin resistance, and oxidative stress. Epidemiological studies suggest a higher incidence of PD among individuals with T2DM, with growing evidence that diabetes may accelerate the onset and progression of motor and cognitive symptoms. This narrative review summarizes current knowledge on the relationship between T2DM and PD, with emphasis on epidemiological associations, shared biological pathways, and the therapeutic implications of antidiabetic agents. Available data indicate that patients with both PD and T2DM tend to experience a faster progression of motor and cognitive decline. Clinical trials assessing hypoglycemic agents have reported heterogeneous findings: pioglitazone failed to demonstrate neuroprotective effects, while exenatide showed temporary improvements in motor function that were not sustained after treatment discontinuation. Experimental studies also suggest possible neuroprotective actions of metformin and dipeptidyl peptidase 4 inhibitors, although well-designed clinical trials are still lacking. The interaction between T2DM and PD highlights the importance of metabolic factors in neurodegeneration. Further studies are required to determine the impact of diabetes and its treatments on the PD course and to explore antidiabetic drugs as potential disease-modifying therapies.
Keywords
INTRODUCTION
Parkinson’s disease (PD) is recognized as the second-most prevalent neurodegenerative disorder, manifesting through an array of motor and non-motor symptoms[1]. According to the Global Burden of Disease (GBD) 2021 study, an estimated 11.8 million people worldwide were living with PD in 2021[2]. Concurrently, type 2 diabetes mellitus (T2DM), the most frequent chronic metabolic disorder, has emerged as a significant global health concern. In 2021, the GBD study estimated that 529 million people [95% uncertainty interval (UI) 500-564 million) were living with diabetes worldwide[3].
An increasing body of population-based studies suggests a heightened risk of PD development in individuals with T2DM[4-7]. The two conditions are posited to share pathophysiological pathways, including mitochondrial dysfunction, hyperglycemia, as well as inflammatory and oxidative stress[8]. Metabolic diseases are increasing worldwide, driven in part by lifestyle and environmental factors. Pesticides, heavy metals, and industrial pollutants can impair mitochondrial function and induce genetic or epigenetic alterations, linking environmental stressors to systemic metabolic dysfunction[9,10].
Neurons, which rely heavily on glucose oxidation to generate adenosine triphosphate (ATP), are especially susceptible when both glucose transport and its intracellular processing are compromised[11,12]. This energy shortfall undermines synaptic activity and fosters oxidative damage and neurodegeneration, patterns that are evident in conditions such as Alzheimer’s and Parkinson’s disease[12].
Mitochondrial dysfunction is a central feature of metabolic disturbances. Mitochondria are the primary organelles responsible for producing cellular energy in the form of ATP[13]. ATP synthesis occurs through oxidative phosphorylation (OXPHOS), in which electrons are transferred along the mitochondrial electron transport chain (ETC), ultimately driving ATP production. This process is highly efficient, generating ~36 ATP molecules per glucose molecule, and is especially critical in energy-demanding cells such as neurons, which rely almost exclusively on OXPHOS to sustain synaptic activity, ionic gradients, and long-term survival[14].
Cells can also generate energy through glycolysis, a cytoplasmic, mitochondria-independent pathway that breaks down glucose into pyruvate. Although glycolysis contributes to glucose metabolism and supports cellular signaling, including activation of glucose transporters, it produces far less ATP than OXPHOS, yielding only two ATP molecules per glucose molecule[15,16]. Consequently, when mitochondrial function is impaired, energy production is drastically reduced, making neurons particularly vulnerable. This energy deficit is increasingly recognized as a mechanistic link between systemic metabolic dysfunction, neuronal vulnerability, and the pathogenesis of neurodegenerative diseases such as PD[17].
Against this backdrop, ketone bodies, particularly β-hydroxybutyrate (BHB), offer a metabolic alternative that can bypass faulty glycolytic pathways and feed directly into the Krebs cycle within neurons[18-20]. Delivered to the brain via monocarboxylate transporters (MCTs), these ketones are converted into acetyl-CoA, thereby enhancing mitochondrial ATP synthesis and curbing the production of reactive oxygen species (ROS)[21,22]. This metabolic flexibility may provide a protective advantage in the context of mitochondrial impairment.
Beyond energy metabolism, T2DM is characterized by a chronic, low-grade inflammatory state, underscored by the sustained presence of predominantly proinflammatory cytokines such as tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β), and interleukin-6 (IL-6). This systemic inflammation can undermine the structural integrity of the blood–brain barrier (BBB), making it more permeable by disrupting tight junction proteins such as ZO-1 and occludin[23]. As a result, inflammatory molecules are more readily able to enter the central nervous system (CNS), where they can trigger the activation of glial cells, most notably microglia and astrocytes. Under these inflammatory conditions, microglia often shift from their typically neuroprotective, anti-inflammatory M2 state to a proinflammatory M1 phenotype[24]. This change is characterized by the release of neurotoxic cytokines and ROS, amplifying inflammatory responses in the brain. In individuals with PD, this diabetes-related inflammatory shift may worsen pre-existing neuroinflammation, potentially accelerating the degeneration of neurons and intensifying both motor and non-motor symptoms associated with the disease[25].
Insulin resistance adds an additional layer of vulnerability. In the brain, impaired insulin signaling alters phosphorylation of insulin receptor substrate (IRS) proteins in the substantia nigra, disrupting mitochondrial proteins, calcium homeostasis, and mitochondrial biogenesis[26-30]. These changes lead to mitochondrial depolarization, ROS overproduction, and oxidative stress, contributing to neuronal death. One proposed mechanism involves the Parkin–PARIS–PGC1α pathway, where loss of Parkin function inhibits PGC1α activity, impairing mitochondrial biogenesis and accelerating dopaminergic neuron loss in the substantia nigra[31-35]. Notably, abnormalities in glucose metabolism have been frequently observed in PD patients.
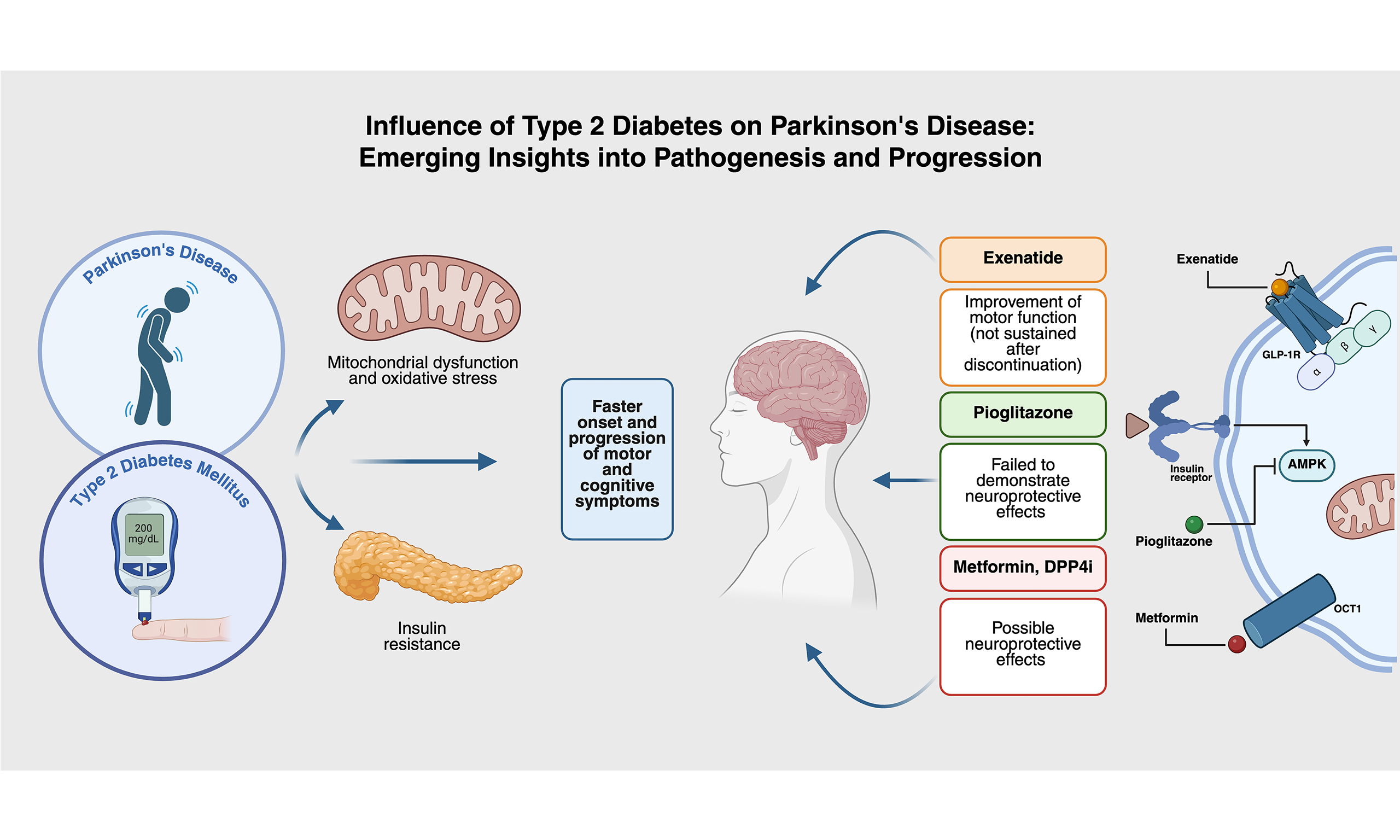
The prevalence of insulin resistance in patients solely with PD is approximately 50% to 80%, and the prevalence of T2DM in patients with PD is 8% to 30%[36-40]. The convergence of epidemiological and experimental data increasingly supports a linkage between these two prevalent chronic diseases. Figure 1 offers a comprehensive visual representation of the complex pathophysiological interplay between PD and T2DM. This narrative review aims to synthesize the existing literature on the potential role of DM in the pathogenesis of PD.
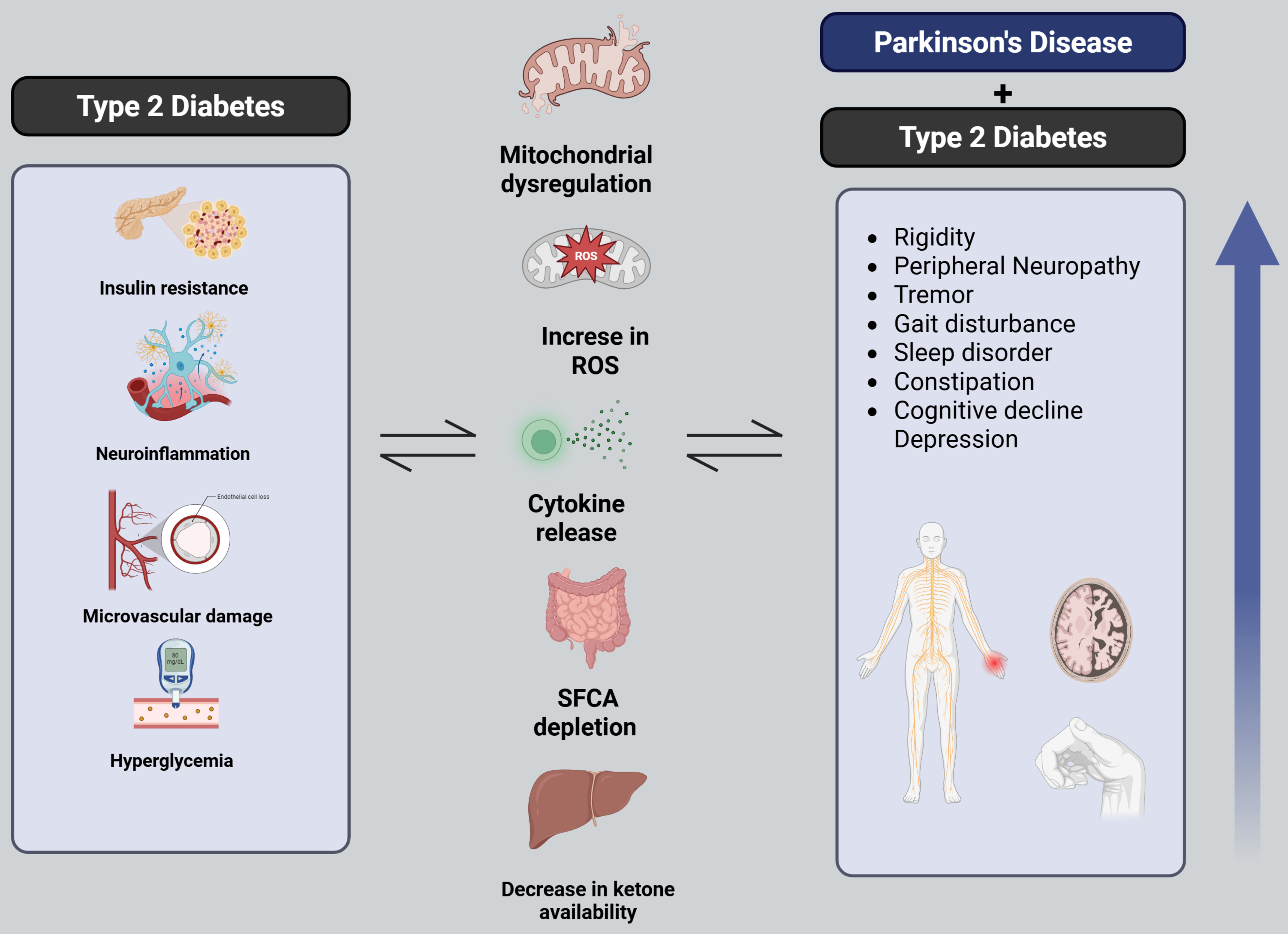
Figure 1. Pathophysiological interplay between PD and T2DM. Schematic representation summarizing the overlapping mechanisms between PD and T2DM, including mitochondrial dysfunction, insulin resistance, oxidative stress, and neuroinflammation. The figure also highlights the compounded clinical manifestations observed when both conditions coexist [Created in BioRender. Robls, E. (2025)]. PD: Parkinson’s disease; T2DM: type 2 diabetes mellitus.
EPIDEMIOLOGICAL INTERPLAY BETWEEN PD AND T2DM
Synthesis of epidemiological findings
This narrative review was conducted to synthesize current evidence on the pathophysiological and clinical interplay between T2DM and PD. A non-systematic search of the literature was performed using PubMed and Scopus databases, covering publications from January 2000 to September 2025. Keywords included “Parkinson’s disease”, “type 2 diabetes mellitus”, “insulin resistance”, “mitochondrial dysfunction”, “neuroinflammation”, “hypoglycemic agents”, and “neuroprotection”. Articles selected for inclusion comprised epidemiological studies, experimental research, and clinical trials published in English or Spanish. Reference lists from key publications were also reviewed to identify additional relevant studies. Case reports, editorials, and narrative summaries without primary data were excluded. This approach allowed the identification of major trends and hypotheses regarding the mechanistic links and potential therapeutic overlaps between both conditions.
As of 2021, the GBD study estimated that 529 million people (95%UI 500-564 million) were living with diabetes worldwide, with T2DM accounting for 96% of these cases. The global age-standardized prevalence of diabetes was 6.1% (5.8%-6.5%)[3]. Concurrently, PD was identified as the second most common neurodegenerative disorder and the fastest-growing neurological condition in prevalence and disability. Projections indicate that by 2050, 25.2 million people (95%UI 21.7-30.1 million) will be living with PD worldwide, representing a 112% increase from 2021, primarily driven by population aging[41].
Diabetes mellitus as a risk factor for PD
Numerous factors, both genetic and environmental, such as advancing age, exposure to pesticides, and traumatic brain injuries, have been implicated in elevating the risk of PD onset[9,40,41]. The earliest inquiries into oral glucose tolerance in PD patients can be traced to the 1960s[42,43]. Subsequent research revealed that 83% of PD patients experienced glucose metabolism disorders, with T2DM diagnosed in 52% of these cases[44-46]. These seminal findings have catalyzed a growing scholarly interest in the PD-T2DM nexus. Table 1 consolidates recent studies that have identified T2DM as a potential risk factor for PD.
Epidemiological evidence linking DM with increased risk of PD
| Authors | Associated risk | Outcome |
| Aune et al.[45] | RR 1.27 (95%CI 1.20-1.35) | Patients with DM have a 27% increased relative risk of developing PD |
| De Pablo-Fernandez et al.[46] | HR 1.32 (95%CI 1.29-1.35) | Increased risk with complicated DM |
| De Pablo-Fernandez et al.[47] | OR 3.27 (95%CI 1.21-8.85) | Duration of > 10 years with T2DM was associated with an increased risk of PD |
| Yang et al.[48] | HR adjusted of 1.19 (95%CI 1.08-1.32) | Incidence density rate was higher in the DM vs. non-DM group, 1.53 vs. 2.08 per 100,000 person-years |
| Sun et al.[49] | The adjusted HR of 1.61 (95%CI 1.56-1.66) | The incidence of PD was 3.59 and 2.15 per 10,000 person-years for the DM and non-DM groups, respectively |
| Schernhammer et al.[50] | OR 1.36 (95%CI 1.08-1.71) | Having DM associated with increased risk of developing PD |
| Xu et al.[51] | OR 1.41 (95%CI 1.2-1.66) | Baseline DM associated with higher risk of PD |
| Hu et al.[52] | HR 1.85 (95%CI 1.23-2.8) | T2DM associated with increased risk of PD |
| Chohan et al.[53] | SMD of 0.55 (95%CI 0.39-0.72) | T2DM associated with faster progression in the severity of motor symptoms |
A recent meta-analysis reported a combined relative risk for developing PD in individuals with T2DM compared to those without T2DM of 1.27, indicating a 27% higher risk in the diabetic population[45]. In addition, when comparing populations with and without T2DM, the incidence rate of PD in the T2DM cohort was 1.32 times higher than in the non-DM cohort[46]. Investigations into the impact of antidiabetic medications on PD risk have yielded mixed outcomes. A case-control study examining the use of various antidiabetic drugs including insulins, biguanides, sulfonylureas, dipeptidyl peptidase 4 (DPP4) inhibitors, glucagon-like peptide 1 (GLP-1) analogs, and glinides, found no significant link to PD, except for thiazolidinediones (TZDs), which were associated with a marginally reduced risk[54].
SHARED PATHOPHYSIOLOGICAL MECHANISMS
Insulin resistance and dopaminergic dysfunction
Research on the influence of diabetes mellitus (DM) on levodopa-induced complications in PD remains limited. Mohamed Ibrahim et al. examined this association in a cohort of 72 PD patients, of whom 21 (29.2%) had T2DM. Among these, 15 patients developed T2DM prior to PD diagnosis (71.4%) with a median of 4 years [interquartile range (IQR), 5], while the remaining six developed T2DM after PD onset (28.6%) with a median of 3.5 years (IQR, 4). The study found that motor complications appeared significantly earlier in the diabetic group, with a median of 12 months (IQR, 6), compared to 24 months (IQR, 6) in non-diabetic patients (P < 0.001). This difference was observed despite comparable levodopa equivalent daily doses, overall levodopa use, disease duration, and stage. The authors acknowledged limitations, including the small sample size and potential recall bias when determining the onset of motor complications, and no significant differences were reported in the incidence of dyskinesia[55].
Complementing these clinical findings, Zagare et al. investigated insulin resistance as a potential disease-modifying factor in PD through a translational study entitled “Insulin Resistance Is a Modifying Factor for Parkinson’s Disease”[56]. Using midbrain organoids derived from carriers of glucocerebrosidase gene (GBA1) mutations (GBA-PD) and healthy controls, they identified dysregulation of insulin signaling pathways in GBA-PD organoids. Statistical analyses were performed with the non-parametric Kruskal–Wallis test followed by Dunn’s post hoc test, with p values adjusted using the Benjamini–Hochberg method (P < 0.05; *P < 0.01; **P < 0.001). Under insulin-resistant conditions, GBA-PD organoids showed a significant reduction in TH-positive dopaminergic neurons and dopamine levels compared with wild-type organoids. Furthermore, probabilistic Boolean modeling predicted forkhead box protein O1 (FOXO1)-driven phenotypes under insulin resistance, including α-synuclein accumulation and neuronal death. Together, these results support the concept that insulin resistance contributes to dopaminergic vulnerability in PD, consistent with clinical observations that diabetic patients often present with a more aggressive disease course[56].
Gut dysbiosis, type 2 diabetes, and the enteric hypothesis of PD
An increasing body of evidence points to a strong mechanistic connection between changes in gut microbiota and the development of both T2DM and PD. In T2DM, microbial imbalance typically involves a decline in overall diversity, enrichment of proinflammatory strains, and reduced levels of beneficial short-chain fatty acid (SCFA)-producing genera such as Faecalibacterium and Rose[23,57-59]. These microbial disruptions weaken the intestinal barrier, allowing endotoxins, particularly lipopolysaccharide (LPS), to leak into the blood, fueling systemic inflammation that may, in turn, impair the BBB and drive neuroinflammatory responses[57,59,60].
Reduced SCFA levels, notably butyrate, further exacerbate gut vulnerability by impairing mucosal repair, altering immune responses, and diminishing vagal nerve signaling, an essential communication route in the gut–brain axis. Such alterations are particularly relevant to the Braak hypothesis, which suggests that PD may originate in the enteric nervous system and ascend to the brainstem via the vagus[57,58,61,62].
Emerging findings have also identified microbial amyloids such as curli produced by Escherichia coli and other gut bacteria as potential contributors to α-synuclein aggregation via cross-seeding mechanisms[57,63]. Animal studies have demonstrated that colonization with curli-producing bacteria triggers α-synuclein buildup in both the enteric and CNSs, accompanied by motor symptoms and gastrointestinal disturbances[62,63]. Moreover, PD patients have shown heightened immune responses to curli, underscoring its possible role in gut-driven neuroinflammation[59,64-66].
Additional markers such as elevated zonulin and fecal calprotectin further point to compromised intestinal barrier integrity and inflammation in PD, reinforcing the idea that gastrointestinal dysfunction may not only precede but also promote neurodegeneration[60]. This shared pathological thread between T2DM and PD, marked by leaky gut, chronic inflammation, metabolic imbalances, and α-synuclein misfolding, offers a unified model for understanding disease progression and identifying potential intervention points.
In light of this, restoring microbial balance has become a focus of therapeutic research. Fecal microbiota transplantation (FMT), for instance, has shown neuroprotective potential in animal models of PD. However, clinical outcomes remain inconclusive[67,68]. A recent Finnish double-blind, placebo-controlled trial assessed the effects of a one-time anaerobically prepared FMT via colonoscopy in PD patients with confirmed dysbiosis. Although the intervention proved safe, it did not yield significant motor improvements after six months. Notably, dysbiosis resolved more frequently in the placebo group, suggesting that factors such as donor selection, host compatibility, and ecological dynamics may critically influence outcomes[67]. Together, these insights reinforce the role of gut dysbiosis in PD, particularly among individuals with T2DM. Through intersecting pathways involving immune activation, metabolic disruption, and pathological protein aggregation, microbiome alterations may lower the threshold for neurodegeneration. The microbiota–gut–brain axis thus emerges as both a compelling pathogenic framework and a promising therapeutic frontier.
CLINICAL IMPACT OF DM ON PD
Influence on motor symptomatology
Recent studies have clarified the aggravating effect of DM on the motor symptoms of PD. Kotagal et al. conducted a case-control study comparing motor symptom severity between PD patients with and without T2DM. The study involved 39 PD subjects, 13 with T2DM and 26 without. The following variables were controlled: gender, age, duration of disease, striatal dihydrotetrabenazine distribution volume ratio, body mass index (BMI), levodopa equivalent dose, history of hypertension, and history of statin use. Findings revealed a significant exacerbation of motor symptoms in the PD-T2DM cohort, notably in bradykinesia (scores: 13.7 vs. 12.3, P = 0.002), rigidity (scores: 5.0 vs. 4.6, P = 0.048), and postural instability with gait difficulty (scores: 5.0 vs. 3.6, P < 0.001), while tremor scores did not show significant differences (scores: 5.2 vs. 3.9, P = 0.62)[69]. Additionally, it should be noted that the study from which this information was obtained does not explicitly mention that corrections for multiple comparisons were conducted.
Complementing these results, Cereda et al. conducted a population-based retrospective case-control study involving 178 PD patients to investigate whether prior onset of diabetes contributes to the severity of motor symptoms and disease progression in PD, after systematically excluding secondary causes of parkinsonism. They analyzed 89 PD cases with diabetes and 89 PD controls without diabetes, matched (1:1) by gender, BMI, and disease duration (> 1 year). Using the Unified Parkinson’s Disease Rating Scale (UPDRS) scale, PD patients with diabetes (PD-DM) scored a mean of 33.7 (SD 15.0) on Part III, significantly higher than PD patients without diabetes, who scored a mean of 18.3 (SD 7.9)[70]. These findings underscore that motor symptoms tend to be more severe in PD patients with diabetes compared to those without, highlighting significant statistical differences[69,70].
A comprehensive systematic review and meta-analysis by Chohan et al. included 28 articles meeting inclusion criteria as observational studies that specifically investigated preceding T2DM and its effect on PD risk, as well as studies analyzing how diabetes was associated with PD progression. They used Part III of the UPDRS to measure the progression of motor symptoms in PD. They found that T2DM correlates with accelerated progression of motor symptoms in PD measured by UPDRS Part III [IVW odds ratio (OR) 1.10, 95% confidence interval (CI) 1.01-1.20; P = 0.032][53].
Cognitive decline
Research has begun to elucidate the adverse impact of T2DM on cognitive function in PD. Bohnen et al. observed that PD patients with concurrent T2DM exhibited more pronounced cognitive deficits than non-diabetic PD patients, with significantly lower global Z scores for cognition (-0.91 vs. -0.36, P = 0.006)[71]. Similarly, Petrou et al. included an assessment of total gray matter volume and found that the PD-T2DM group had reduced gray matter volume, which correlated with lower Z scores across visuospatial, executive, and composite cognitive domains[72]. Longitudinal analysis by Ong et al. involving biannual cognitive assessments over three years using the Mini-Mental State Examination (MMSE) and the Montreal Cognitive Assessment (MoCA), demonstrated a greater decline in cognitive scores in the PD-T2DM group compared to the non-T2DM group, with differences in the MMSE and MoCA being -2.29 vs. -0.5 (P = 0.024) and -3.29 vs. -0.55 (P = 0.016), respectively[73].
Conversely, a systematic review and meta-analysis by Chohan et al. did not find significant evidence of accelerated cognitive decline in PD patients with T2DM as measured by the MoCA (OR 0.81, 95%CI 0.49-1.33; P = 0.399) or MMSE (OR 0.99, 95%CI 0.85-1.14; P = 0.848)[53]. These inconsistencies may be attributed to methodological differences such as variations in cognitive assessment tools, particularly the sensitivity of longitudinal applications of MoCA and MMSE vs. single-time-point assessments, as well as differences in study design, including follow-up duration and sample size. Additionally, heterogeneity among patient populations, including variation in glycemic control, duration of diabetes, and presence of vascular comorbidities, may contribute to the conflicting findings across studies.
THERAPEUTIC IMPLICATIONS
Hypoglycemic agents in clinical trials for PD
A variety of antidiabetic medications have demonstrated potential neuroprotective effects in preclinical models[74-77]. This discussion focuses on those agents that have progressed to clinical trials, specifically TZDs and glucagon-like peptide (GLP-1) analogs. These drugs have been subject to further scrutiny to assess their therapeutic potential in PD management. Figure 2 provides a visual representation of the impact of GLP-1 analog agents on dopaminergic neurons.
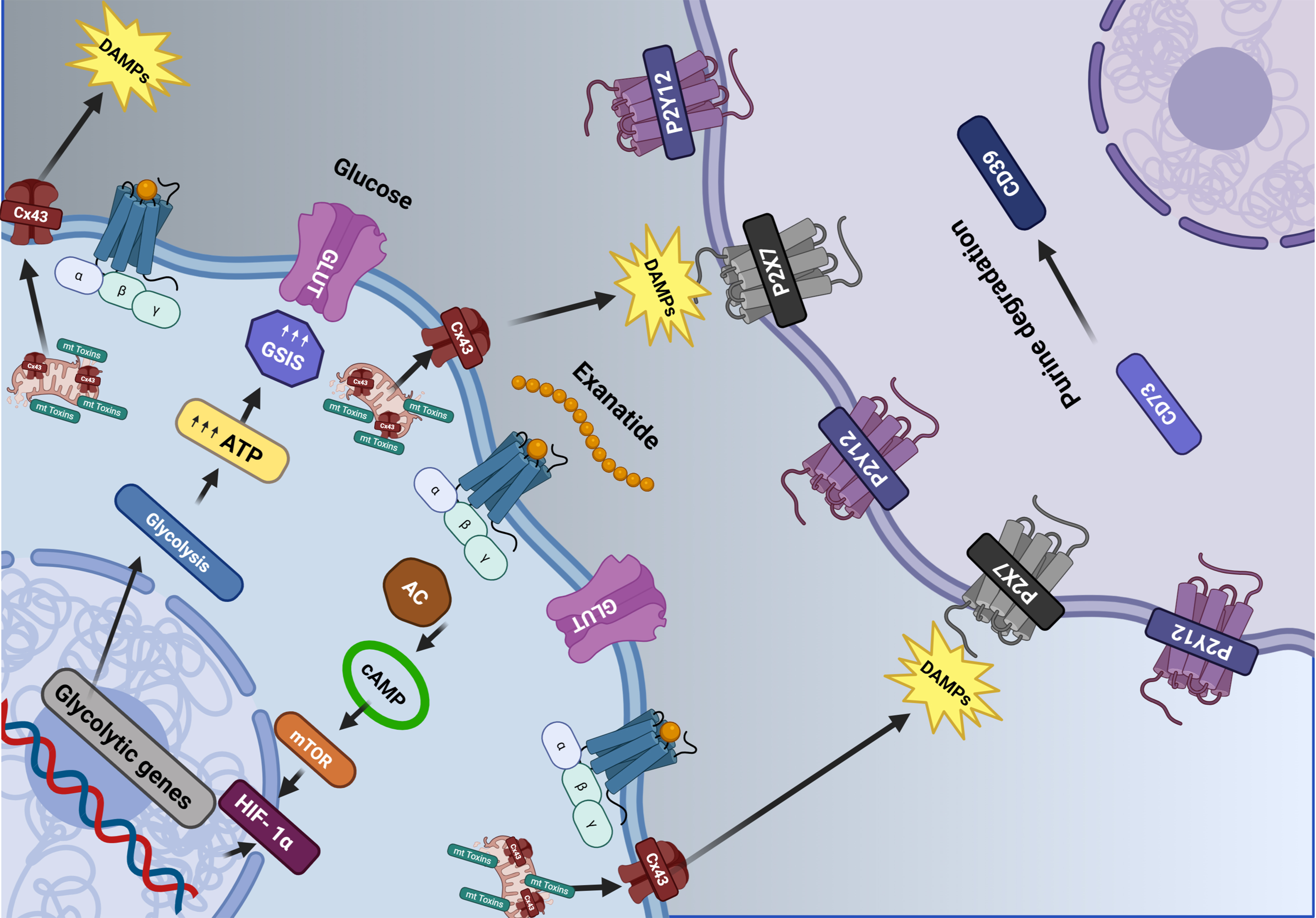
Figure 2. Modulatory impacts of antidiabetic medications on dopaminergic neuron function. The image illustrates how Exenatide (a GLP-1 agonist) enhances glucose metabolism within the cell, increasing energy production and reducing the release of DAMPs. If left uncontrolled, these signals activate purinergic receptors in neighboring cells and promote inflammation. Exenatide promotes metabolic resilience by enhancing glucose utilization and energy production, thereby limiting mitochondrial dysfunction and the release of proinflammatory danger signals. This dual action links energy metabolism with purinergic signaling and may explain therapeutic benefits in the context of diabetes-related metabolic stress and neurodegeneration in PD [Created in BioRender. Robls, E. (2025)]. GLP-1: Glucagon-like peptide 1; DAMPs: damage-associated molecular patterns; PD: Parkinson’s disease.
TZDs
The Neuroprotection Exploratory Trials in Parkinson’s Disease (NET-PD) program conducted a comprehensive systematic review to identify neuroprotective agents for potential use in early-stage PD[78]. From this review, pioglitazone emerged as a candidate for clinical trials due to its mechanism of action involving the activation of peroxisome proliferator-activated receptor gamma, a nuclear receptor integral to lipid and carbohydrate metabolism regulation[78]. In a multicenter, randomized, double-blind, placebo-controlled phase 2 trial by NET-PD researchers, pioglitazone was evaluated for its effects on PD progression. Unfortunately, the trial results did not support pioglitazone as an agent capable of slowing disease progression or improving clinical outcomes in PD[79].
GLP-1 analogs
Exenatide, a synthetic form of exendin-4, functions by activating GLP-1 receptors. Stimulation of the GLP-1 receptor leads to an increase in intracellular cAMP, which subsequently activates protein kinase A (PKA) and phosphoinositide 3-kinase (PI3K), responsible for phosphorylating and activating several downstream signaling pathways that can be simplified into two pathways. The first is the MAPK/ERK pathway and the PI3K/AKT pathways, which can modulate events such as activation of calcium channels, increased protein synthesis, mitochondrial biogenesis, inhibition of apoptosis, inflammation, and protein aggregation, leading to improved cell survival[74,80]. Following promising results, Athauda et al. conducted a randomized, placebo-controlled study to determine exenatide’s effects on motor severity in PD[81]. Patients were treated with 2 mg of exenatide weekly for 48 weeks, followed by a 12-week withdrawal period. The study found a significant adjusted difference of -3.5 points (-6.7 to -0.3, P = 0.0318) as measured by the MDS-UPDRS part 3, favoring exenatide treatment. There were no notable differences in non-motor symptoms, dyskinesia, or quality of life measures[81]. A subsequent post hoc analysis explored the drug’s effects on individual non-motor symptoms, revealing improvements in mood/depression and emotional well-being that were not maintained after drug withdrawal[82]. To corroborate these findings, the Exenatide-PD3 phase 3 trial is currently underway, aiming to enroll 200 participants over a 96-week treatment period[83].
In a Phase 2, double-blind, randomized, placebo-controlled trial, Meissner et al. evaluated the effect of lixisenatide (20 μg/d) vs. placebo, administered as add-on therapy with the usual antiparkinsonian treatment, on the progression of motor disability in patients with early PD to assess its potential disease-modifying effect[84]. Participants, diagnosed within the past three years, maintaining stable doses of symptomatic medications (dopamine agonist and/or L-dopa and/or MAO-B inhibitor) without motor complications, were randomly allocated in a 1:1 ratio to receive daily subcutaneous injections of either Lixisenatide or placebo over a period of 12 months. The primary endpoint measured was the change in MDS-UPDRS part III scores from baseline. A total of 156 subjects were enrolled, evenly distributed with 78 in each group. Baseline MDS-UPDRS part III scores averaged around 15 in both groups. By the end of the 12-month period, scores on MDS-UPDRS part III had improved by -0.04 points in the Lixisenatide group and worsened by 3.04 points in the placebo group (difference of 3.08; 95%CI 0.86-5.30; P = 0.007). It is important to mention that during this trial, nausea occurred in 46% of participants receiving lixisenatide, and vomiting occurred in 13%. Subsequent evaluations will be necessary to establish a statistically significant link between GLP-1 and its effects in PD[84].
Exploration of additional hypoglycemic agents in preclinical PD models
GLP-1 analogs, such as liraglutide, along with dual GLP-1 and glucose-dependent insulinotropic peptide (GIP) analogs, have demonstrated neuroprotective effects in animal models, suggesting potential applicability for PD treatment strategies[85-87].
Metformin operates via dual pathways, one of which involves the activation of the cAMP protein kinase pathway, culminating in reduced hepatic glucose production, decreased intestinal glucose absorption, and enhanced insulin sensitivity[75,88,89]. Preclinical in vitro and in vivo investigations have revealed metformin’s neuroprotective capabilities in PD[90-92]. In murine models, metformin has been shown to safeguard dopaminergic neurons by modulating mitochondrial complex I and attenuating α-synuclein phosphorylation via AMP-activated protein kinase (AMPK) pathway activation, mTOR pathway inhibition, and mitigation of MPTP-induced cellular damage[93-95]. This results in decreased aggregation of α-synuclein proteins and reduced oxidative stress generated by mitochondrial dysfunction. Nonetheless, clinical trials to assess metformin’s symptomatic impacts on PD are yet to be conducted.
DPP4 inhibitors, which enhance the bioavailability of active GLP-1 by inhibiting the degradation of incretins, have also been associated with anti-inflammatory and anti-apoptotic effects in murine models exhibiting parkinsonian characteristics[76,77,96,97]. Recent observational studies have indicated that DPP4 inhibitors may exert a positive influence on baseline nigrostriatal dopamine degeneration and may improve long-term motor outcomes in PD patients with concurrent diabetes[97-101].
Dietary interventions and metabolic modulators
Both experimental and clinical findings support the neuroprotective potential of ketogenic interventions, whether through diet or exogenous ketones, by improving mitochondrial efficiency, reducing oxidative stress, and promoting neuronal viability[18,101]. A pilot randomized controlled trial by Phillips et al. assessed the impact of a ketogenic diet compared to a low-fat diet in 47 individuals with PD over an 8-week period[102]. Both groups showed meaningful improvements in motor and non-motor symptoms as measured by the MDS-UPDRS, but the ketogenic group experienced a significantly greater reduction in non-motor symptoms (Part I), with an average change of -4.58 ± 2.17 points vs. -0.99 ± 3.63 in the low-fat group (P < 0.001), amounting to a 41% improvement from baseline. This difference held up across various sensitivity checks. Elevated BHB levels (~1.15 mmol/L) confirmed sustained physiological ketosis, lending support to the theory that ketone bodies may boost neuronal energy metabolism and reduce oxidative stress. Methodological strengths, such as evaluator blinding, stratified randomization, and consistent levodopa administration, add weight to the study’s reliability and underscore the potential role of ketones in Parkinson’s treatment. However, in type 2 diabetes, hepatic insulin resistance and diminished fatty acid oxidation may impair ketone production, thereby limiting access to this alternative fuel source[102-105]. This scenario introduces a possible double hit for individuals with both T2DM and PD: impaired glucose metabolism compounded by reduced ketogenesis, jointly intensifying neuronal susceptibility to dopaminergic degeneration in PD[103,105]. These findings suggest that while ketogenic interventions show promise in improving symptoms in PD, their metabolic efficacy may be significantly limited in patients with type 2 diabetes. Recent studies demonstrate that impaired ketogenesis is a frequent finding in T2DM and may hinder the generation and utilization of ketone bodies as an energy source[102,104,106]. This dual metabolic vulnerability highlights the need for tailored strategies to overcome the impaired ketogenesis and fully realize the neuroprotective potential of ketogenic therapies in the PD–T2DM population. Ultimately, the ketogenic diet remains a promising therapeutic tool in PD, even within the context of metabolic comorbidities, provided its application is carefully adapted to the patient’s metabolic profile.
FUTURE DIRECTIONS AND RESEARCH GAPS
Despite growing evidence suggesting a relationship between PD and T2DM, and the increased risk of T2DM accelerating the clinical course of PD, a significant knowledge gap remains. To better understand the mechanisms underlying this association, more focused and targeted research is warranted.
Most studies exploring this relationship are either cross-sectional or rely on retrospective analyses, which limits the ability to draw causal inferences. To accurately track metabolic changes, neurodegenerative markers, and clinical outcomes over time, large-scale, prospective cohort studies are needed. These studies should involve diverse populations to capture the genetic, environmental, and lifestyle variations that may influence both conditions.
Currently, there are no validated biomarkers that serve as prognostic or diagnostic indicators specific to the PD-T2DM interplay. Identifying molecular or imaging biomarkers in this patient population could facilitate earlier diagnosis, enable targeted interventions, and support the monitoring of disease progression.
There is also an urgent need for standardized clinical outcome measures that encompass both the motor and non-motor symptoms of PD, as well as the metabolic parameters relevant to T2DM. Such measures would support a more unified assessment of the bidirectional impact of these diseases and enhance the design and interpretation of clinical trials.
Addressing these gaps is essential for advancing our understanding of the shared pathophysiology between T2DM and PD. This, in turn, could pave the way for the development of new treatment strategies capable of modifying the course of both diseases and ultimately improving the quality of life for patients and their families.
CONCLUSIONS
PD is a neurodegenerative disorder whose incidence is escalating globally. This emergent health concern underscores the necessity of identifying risk factors that contribute to the onset, exacerbation, and progression of the disease. Enhanced understanding of these factors is vital for the development of early intervention strategies. There exists a growing body of basic and clinical research suggesting an elevated risk of PD in individuals with T2DM. Nonetheless, there is a need for more comprehensive research to elucidate the extent of DM’s influence on the manifestation and severity of PD symptoms, especially non-motor symptoms. This represents a significant opportunity for further inquiry and may have profound implications for the management and treatment of PD.
DECLARATIONS
Acknowledgments
The graphical abstract was created with BioRender.com [Created in BioRender. https://BioRender.com/6gduzrc. Robls, E. (2025)].
Authors’ contributions
Conceptualization, methodology, writing - original draft: Rodríguez-González IC
Conceptualization, project administration, writing - original draft: Botello-Villagrana F
Investigation, writing - review and editing: Gomez-Villalobos SR, González-Cantú A, González-González M
Investigation, visualization, writing - original draft: Torres-Mancilla XM, Robles E
Methodology, visualization, writing - original draft: Luna-Range FA, Gonzalez-Bedolla B
Conceptualization, supervision, writing - review and editing: Martinez-Ramirez D
All authors of this manuscript meet the authorship criteria established by the International Committee of Medical Journal Editors (ICMJE). Specifically, each author has: (1) made substantial contributions to the conception or design of the work, or to the acquisition, analysis, or interpretation of data; (2) participated in drafting the work or critically revising it for important intellectual content; (3) provided final approval of the version to be published; and (4) agreed to be accountable for all aspects of the work, ensuring that any questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
2. Luo Y, Qiao L, Li M, Wen X, Zhang W, Li X. Global, regional, national epidemiology and trends of Parkinson’s disease from 1990 to 2021: findings from the Global Burden of Disease Study 2021. Front Aging Neurosci. 2024;16:1498756.
3. GBD 2021 Diabetes Collaborators. Global, regional, and national burden of diabetes from 1990 to 2021, with projections of prevalence to 2050: a systematic analysis for the Global Burden of Disease Study 2021. Lancet. 2023;402:203-34.
4. Deischinger C, Dervic E, Kaleta M, Klimek P, Kautzky-Willer A. Diabetes mellitus is associated with a higher relative risk for Parkinson’s disease in women than in men. J Parkinsons Dis. 2021;11:793-800.
5. Labandeira CM, Fraga-Bau A, Arias Ron D, et al. Parkinson’s disease and diabetes mellitus: common mechanisms and treatment repurposing. Neural Regen Res. 2022;17:1652-8.
6. Sánchez-Gómez A, Díaz Y, Duarte-Salles T, Compta Y, Martí MJ. Prediabetes, type 2 diabetes mellitus and risk of Parkinson’s disease: a population-based cohort study. Parkinsonism Relat Disord. 2021;89:22-7.
7. Rhee SY, Han KD, Kwon H, et al. Association between glycemic status and the risk of Parkinson disease: a nationwide population-based study. Diabetes Care. 2020;43:2169-75.
8. Hassan A, Sharma Kandel R, Mishra R, Gautam J, Alaref A, Jahan N. Diabetes mellitus and Parkinson’s disease: shared pathophysiological links and possible therapeutic implications. Cureus. 2020;12:e9853.
9. Elbaz A, Clavel J, Rathouz PJ, et al. Professional exposure to pesticides and Parkinson disease. Ann Neurol. 2009;66:494-504.
10. Reddam A, McLarnan S, Kupsco A. Environmental chemical exposures and mitochondrial dysfunction: a review of recent literature. Curr Environ Health Rep. 2022;9:631-49.
12. Han R, Liang J, Zhou B. Glucose metabolic dysfunction in neurodegenerative diseases-new mechanistic insights and the potential of hypoxia as a prospective therapy targeting metabolic reprogramming. Int J Mol Sci. 2021;22:5887.
13. Xiao Liang K. Interplay of mitochondria and diabetes: unveiling novel therapeutic strategies. Mitochondrion. 2024;75:101850.
14. Nesci S, Trombetti F, Pagliarani A, et al. Molecular and supramolecular structure of the mitochondrial oxidative phosphorylation system: implications for pathology. Life. 2021;11:242.
15. Kierans SJ, Taylor CT. Glycolysis: a multifaceted metabolic pathway and signaling hub. J Biol Chem. 2024;300:107906.
16. Zhao Y, Wieman HL, Jacobs SR, Rathmell JC. Mechanisms and methods in glucose metabolism and cell death. Methods Enzymol. 2008;442:439-57.
17. Sian-Hulsmann J, Riederer P, Michel TM. Metabolic dysfunction in Parkinson’s disease: unraveling the glucose-lipid connection. Biomedicines. 2024;12:2841.
18. Jensen NJ, Wodschow HZ, Nilsson M, Rungby J. Effects of ketone bodies on brain metabolism and function in neurodegenerative diseases. Int J Mol Sci. 2020;21:8767.
20. García-Rodríguez D, Giménez-Cassina A. Ketone bodies in the brain beyond fuel metabolism: from excitability to gene expression and cell signaling. Front Mol Neurosci. 2021;14:732120.
21. Blanco A, Blanco G. Chapter 15 - Lipid metabolism. In: Medical biochemistry (second edition). Elsevier; 2022. pp. 359-400.
22. Pierre K, Pellerin L. Monocarboxylate transporters in the central nervous system: distribution, regulation and function. J Neurochem. 2005;94:1-14.
23. Rohm TV, Meier DT, Olefsky JM, Donath MY. Inflammation in obesity, diabetes, and related disorders. Immunity. 2022;55:31-55.
24. Tian Y, Jing G, Ma M, Yin R, Zhang M. Microglial activation and polarization in type 2 diabetes-related cognitive impairment: a focused review of pathogenesis. Neurosci Biobehav Rev. 2024;165:105848.
25. Yu H, Sun T, He X, et al. Association between Parkinson’s disease and diabetes mellitus: from epidemiology, pathophysiology and prevention to treatment. Aging Dis. 2022;13:1591-605.
26. Pomytkin I, Pinelis V. Brain insulin resistance: focus on insulin receptor-mitochondria interactions. Life. 2021;11:262.
27. Ruiz-Pozo VA, Tamayo-Trujillo R, Cadena-Ullauri S, et al. The molecular mechanisms of the relationship between insulin resistance and Parkinson’s disease pathogenesis. Nutrients. 2023;15:3585.
28. Bassil F, Delamarre A, Canron MH, et al. Impaired brain insulin signalling in Parkinson’s disease. Neuropathol Appl Neurobiol. 2022;48:e12760.
29. Moroo I, Yamada T, Makino H, et al. Loss of insulin receptor immunoreactivity from the substantia nigra pars compacta neurons in Parkinson’s disease. Acta Neuropathol. 1994;87:343-8.
30. Arnold SE, Arvanitakis Z, Macauley-Rambach SL, et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat Rev Neurol. 2018;14:168-81.
31. Pereira SL, Grossmann D, Delcambre S, Hermann A, Grünewald A. Novel insights into Parkin-mediated mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Curr Opin Neurobiol. 2023;80:102720.
32. Pirooznia SK, Yuan C, Khan MR, et al. PARIS induced defects in mitochondrial biogenesis drive dopamine neuron loss under conditions of parkin or PINK1 deficiency. Mol Neurodegener. 2020;15:17.
33. Gao XY, Yang T, Gu Y, Sun XH. Mitochondrial dysfunction in Parkinson’s disease: from mechanistic insights to therapy. Front Aging Neurosci. 2022;14:885500.
34. Alzahrani MA, Almutairi MA, Alsiary KA, et al. Type-2 diabetes prevalence and severity of Parkinson’s disease at a tertiary hospital. Int J Med Dev Ctries. 2024;8:1433-8.
35. Sandyk R, Awerbuch GI. The association of diabetes mellitus with dementia in Parkinson’s disease. Int J Neurosci. 1992;64:209-12.
36. Van Woert MH, Mueller PS. Glucose, insulin, and free fatty acid metabolism in Parkinson’s disease treated with levodopa. Clin Pharmacol Ther. 1971;12:360-7.
37. Hogg E, Athreya K, Basile C, Tan EE, Kaminski J, Tagliati M. High prevalence of undiagnosed insulin resistance in non-diabetic subjects with Parkinson’s disease. J Parkinsons Dis. 2018;8:259-65.
38. Pezzoli G, Cereda E, Amami P, et al. Onset and mortality of Parkinson’s disease in relation to type II diabetes. J Neurol. 2023;270:1564-72.
39. Su D, Cui Y, He C, et al. Projections for prevalence of Parkinson’s disease and its driving factors in 195 countries and territories to 2050: modelling study of Global Burden of Disease Study 2021. BMJ. 2025;388:e080952.
40. Jafari S, Etminan M, Aminzadeh F, Samii A. Head injury and risk of Parkinson disease: a systematic review and meta-analysis. Mov Disord. 2013;28:1222-9.
41. Klein C, Westenberger A. Genetics of Parkinson’s disease. Cold Spring Harb Perspect Med. 2012;2:a008888.
42. Lipman IJ, Boykin ME, Flora RE. Glucose intolerance in Parkinson’s disease. J Chronic Dis. 1974;27:573-9.
43. Elner AM, Kandel EI. Studies on the carbohydrate metabolism in Parkinsonism. (Relation of carbohydrate metabolism disorders to the clinical picture of the disease). Zh Nevropatol Psikhiatr Im S S Korsakova. 1965;65:46-50.
44. Sandyk R. The relationship between diabetes mellitus and Parkinson’s disease. Int J Neurosci. 1993;69:125-30.
45. Aune D, Schlesinger S, Mahamat-Saleh Y, Zheng B, Udeh-Momoh CT, Middleton LT. Diabetes mellitus, prediabetes and the risk of Parkinson’s disease: a systematic review and meta-analysis of 15 cohort studies with 29.9 million participants and 86,345 cases. Eur J Epidemiol. 2023;38:591-604.
46. De Pablo-Fernandez E, Goldacre R, Pakpoor J, Noyce AJ, Warner TT. Association between diabetes and subsequent Parkinson disease: a record-linkage cohort study. Neurology. 2018;91:e139-42.
47. De Pablo-Fernandez E, Sierra-Hidalgo F, Benito-León J, Bermejo-Pareja F. Association between Parkinson’s disease and diabetes: data from NEDICES study. Acta Neurol Scand. 2017;136:732-6.
48. Yang YW, Hsieh TF, Li CI, et al. Increased risk of Parkinson disease with diabetes mellitus in a population-based study. Medicine. 2017;96:e5921.
49. Sun Y, Chang YH, Chen HF, Su YH, Su HF, Li CY. Risk of Parkinson disease onset in patients with diabetes: a 9-year population-based cohort study with age and sex stratifications. Diabetes Care. 2012;35:1047-9.
50. Schernhammer E, Hansen J, Rugbjerg K, Wermuth L, Ritz B. Diabetes and the risk of developing Parkinson’s disease in Denmark. Diabetes Care. 2011;34:1102-8.
51. Xu Q, Park Y, Huang X, et al. Diabetes and risk of Parkinson’s disease. Diabetes Care. 2011;34:910-5.
52. Hu G, Jousilahti P, Bidel S, Antikainen R, Tuomilehto J. Type 2 diabetes and the risk of Parkinson’s disease. Diabetes Care. 2007;30:842-7.
53. Chohan H, Senkevich K, Patel RK, et al. Type 2 diabetes as a determinant of Parkinson’s disease risk and progression. Mov Disord. 2021;36:1420-9.
54. Sunnarborg K, Tiihonen M, Huovinen M, Koponen M, Hartikainen S, Tolppanen AM. Association between different diabetes medication classes and risk of Parkinson’s disease in people with diabetes. Pharmacoepidemiol Drug Saf. 2022;31:875-82.
55. Mohamed Ibrahim N, Ramli R, Koya Kutty S, Shah SA. Earlier onset of motor complications in Parkinson’s patients with comorbid diabetes mellitus. Mov Disord. 2018;33:1967-8.
56. Zagare A, Hemedan A, Almeida C, et al. Insulin resistance is a modifying factor for Parkinson’s disease. Mov Disord. 2025;40:67-76.
57. Kaur G, Behl T, Bungau S, et al. Dysregulation of the gut-brain axis, dysbiosis and influence of numerous factors on gut microbiota associated Parkinson’s disease. Curr Neuropharmacol. 2021;19:233-47.
58. Sobral J, Empadinhas N, Esteves AR, Cardoso SM. Impact of nutrition on the gut microbiota: implications for Parkinson’s disease. Nutr Rev. 2025;83:713-27.
59. Campos-Acuña J, Elgueta D, Pacheco R. T-cell-driven inflammation as a mediator of the gut-brain axis involved in Parkinson’s disease. Front Immunol. 2019;10:239.
60. Dumitrescu L, Marta D, Dănău A, et al. Serum and fecal markers of intestinal inflammation and intestinal barrier permeability are elevated in Parkinson’s disease. Front Neurosci. 2021;15:689723.
61. Rietdijk CD, Perez-Pardo P, Garssen J, van Wezel RJ, Kraneveld AD. Exploring Braak’s hypothesis of Parkinson’s disease. Front Neurol. 2017;8:37.
62. Montanari M, Imbriani P, Bonsi P, Martella G, Peppe A. Beyond the microbiota: understanding the role of the enteric nervous system in Parkinson’s disease from mice to human. Biomedicines. 2023;11:1560.
63. Sampson TR, Challis C, Jain N, et al. A gut bacterial amyloid promotes α-synuclein aggregation and motor impairment in mice. Elife. 2020;9:e53111.
64. Oliver PJ, Civitelli L, Hu MT. The gut-brain axis in early Parkinson’s disease: from prodrome to prevention. J Neurol. 2025;272:413.
65. Guo M, Gao H, Wang Y, Xiang Y. Exploring the role of gut microbiota in Parkinson’s disease: insights from fecal microbiota transplantation. Front Neurosci. 2025;19:1574512.
66. Kustrimovic N, Balkhi S, Bilato G, Mortara L. Gut microbiota and immune system dynamics in Parkinson’s and Alzheimer’s diseases. Int J Mol Sci. 2024;25:12164.
67. Scheperjans F, Levo R, Bosch B, et al. Fecal microbiota transplantation for treatment of Parkinson disease: a randomized clinical trial. JAMA Neurol. 2024;81:925-38.
68. Menozzi E, Schapira AHV, Borghammer P. The gut-brain axis in Parkinson disease: emerging concepts and therapeutic implications. Mov Disord Clin Pract. 2025;12:904-16.
69. Kotagal V, Albin RL, Müller ML, Koeppe RA, Frey KA, Bohnen NI. Diabetes is associated with postural instability and gait difficulty in Parkinson disease. Parkinsonism Relat Disord. 2013;19:522-6.
70. Cereda E, Barichella M, Cassani E, Caccialanza R, Pezzoli G. Clinical features of Parkinson disease when onset of diabetes came first: a case-control study. Neurology. 2012;78:1507-11.
71. Bohnen NI, Kotagal V, Müller ML, et al. Diabetes mellitus is independently associated with more severe cognitive impairment in Parkinson disease. Parkinsonism Relat Disord. 2014;20:1394-8.
72. Petrou M, Davatzikos C, Hsieh M, et al. Diabetes, gray matter loss, and cognition in the setting of Parkinson disease. Acad Radiol. 2016;23:577-81.
73. Ong M, Foo H, Chander RJ, et al. Influence of diabetes mellitus on longitudinal atrophy and cognition in Parkinson’s disease. J Neurol Sci. 2017;377:122-6.
74. Foltynie T, Athauda D. Chapter 13 - Repurposing anti-diabetic drugs for the treatment of Parkinson’s disease: rationale and clinical experience. Prog Brain Res. 2020;252:493-523.
75. Rotermund C, Machetanz G, Fitzgerald JC. The therapeutic potential of metformin in neurodegenerative diseases. Front Endocrinol. 2018;9:400.
76. Pariyar R, Bastola T, Lee DH, Seo J. Neuroprotective effects of the DPP4 inhibitor vildagliptin in in vivo and in vitro models of Parkinson’s disease. Int J Mol Sci. 2022;23:2388.
77. Abdelsalam RM, Safar MM. Neuroprotective effects of vildagliptin in rat rotenone Parkinson’s disease model: role of RAGE-NFκB and Nrf2-antioxidant signaling pathways. J Neurochem. 2015;133:700-7.
78. Ravina BM, Fagan SC, Hart RG, et al. Neuroprotective agents for clinical trials in Parkinson’s disease: a systematic assessment. Neurology. 2003;60:1234-40.
79. NINDS Exploratory Trials in Parkinson Disease (NET-PD) FS-ZONE Investigators. Pioglitazone in early Parkinson’s disease: a phase 2, multicentre, double-blind, randomised trial. Lancet Neurol. 2015;14:795-803.
80. Athauda D, Foltynie T. The glucagon-like peptide 1 (GLP) receptor as a therapeutic target in Parkinson’s disease: mechanisms of action. Drug Discov Today. 2016;21:802-18.
81. Athauda D, Maclagan K, Skene SS, et al. Exenatide once weekly versus placebo in Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet. 2017;390:1664-75.
82. Athauda D, Maclagan K, Budnik N, et al. What effects might exenatide have on non-motor symptoms in Parkinson’s disease: a post hoc analysis. J Parkinsons Dis. 2018;8:247-58.
83. Vijiaratnam N, Girges C, Auld G, et al. Exenatide once weekly over 2 years as a potential disease-modifying treatment for Parkinson’s disease: protocol for a multicentre, randomised, double blind, parallel group, placebo controlled, phase 3 trial: the ‘Exenatide-PD3’ study. BMJ Open. 2021;11:e047993.
84. Meissner WG, Remy P, Giordana C, et al; LIXIPARK Study Group. Trial of lixisenatide in early Parkinson’s disease. N Engl J Med. 2024;390:1176-85.
85. Roy A, Dawson VL, Dawson TM. From metabolism to mind: the expanding role of the GLP-1 receptor in neurotherapeutics. Neurotherapeutics. 2025;22:e00712.
86. Hölscher C. Protective properties of GLP-1 and associated peptide hormones in neurodegenerative disorders. Br J Pharmacol. 2022;179:695-714.
87. Yang X, Feng P, Ji R, Ren Y, Wei W, Hölscher C. Therapeutic application of GLP-1 and GIP receptor agonists in Parkinson’s disease. Expert Opin Ther Targets. 2022;26:445-60.
88. Lu M, Chen H, Nie F, Wei X, Tao Z, Ma J. The potential role of metformin in the treatment of Parkinson’s disease. J Bio X Res. 2020;03:27-35.
89. Cao G, Gong T, Du Y, Wang Y, Ge T, Liu J. Mechanism of metformin regulation in central nervous system: progression and future perspectives. Biomed Pharmacother. 2022;156:113686.
90. Patil SP, Jain PD, Ghumatkar PJ, Tambe R, Sathaye S. Neuroprotective effect of metformin in MPTP-induced Parkinson’s disease in mice. Neuroscience. 2014;277:747-54.
91. Reed S, Taka E, Darling-Reed S, Soliman KFA. Neuroprotective effects of metformin through the modulation of neuroinflammation and oxidative stress. Cells. 2025;14:1064.
92. Paudel YN, Angelopoulou E, Piperi C, Shaikh MF, Othman I. Emerging neuroprotective effect of metformin in Parkinson’ disease: a molecular crosstalk. Pharmacol Res. 2020;152:104593.
93. Lu M, Su C, Qiao C, Bian Y, Ding J, Hu G. Metformin prevents dopaminergic neuron death in MPTP/P-induced mouse model of Parkinson’s disease via autophagy and mitochondrial ROS clearance. Int J Neuropsychopharmacol. 2016;19:pyw047.
94. Bayliss JA, Lemus MB, Santos VV, et al. Metformin prevents nigrostriatal dopamine degeneration independent of AMPK activation in dopamine neurons. PLoS One. 2016;11:e0159381.
95. Ay M, Charli A, Langley M, et al. Mito-metformin protects against mitochondrial dysfunction and dopaminergic neuronal degeneration by activating upstream PKD1 signaling in cell culture and MitoPark animal models of Parkinson’s disease. Front Neurosci. 2024;18:1356703.
96. Jeong SH, Chung SJ, Yoo HS, et al. Beneficial effects of dipeptidyl peptidase-4 inhibitors in diabetic Parkinson’s disease. Brain. 2021;144:1127-37.
97. Singh AK. Dipeptidyl peptidase-4 inhibitors: novel mechanism of actions. Indian J Endocrinol Metab. 2014;18:753-9.
98. Kumari S, Deshmukh R. Dipeptidyl peptidase 4 (DPP4) inhibitors stride up the management of Parkinson’s disease. Eur J Pharmacol. 2023;939:175426.
99. Bang Y, Moon SH, Lee S, Choi HJ. Anti-inflammatory effects of dipeptidyl peptidase-4 inhibitors and their therapeutic application for Parkinson’s disease. Drug Targets Ther. 2024;3:83-93.
100. Yu SJ, Wang Y, Shen H, et al. DPP-4 inhibitors sitagliptin and PF-00734,200 mitigate dopaminergic neurodegeneration, neuroinflammation and behavioral impairment in the rat 6-OHDA model of Parkinson’s disease. Geroscience. 2024;46:4349-71.
101. Jang J, Kim SR, Lee JE, et al. Molecular mechanisms of neuroprotection by ketone bodies and ketogenic diet in cerebral ischemia and neurodegenerative diseases. Int J Mol Sci. 2023;25:124.
102. Phillips MCL, Murtagh DKJ, Gilbertson LJ, Asztely FJS, Lynch CDP. Low-fat versus ketogenic diet in Parkinson’s disease: a pilot randomized controlled trial. Mov Disord. 2018;33:1306-14.
103. Lee S, Bae J, Kim SU, et al. Intact ketogenesis predicted reduced risk of moderate-severe metabolic-associated fatty liver disease assessed by liver transient elastography in newly diagnosed type 2 diabetes. Front Endocrinol. 2023;14:1306134.
104. Wallenius K, Kroon T, Hagstedt T, et al. The SGLT2 inhibitor dapagliflozin promotes systemic FFA mobilization, enhances hepatic β-oxidation, and induces ketosis. J Lipid Res. 2022;63:100176.
105. Chen L, Wang C, Qin L, Zhang H. Parkinson’s disease and glucose metabolism impairment. Transl Neurodegener. 2025;14:10.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].