

fig3
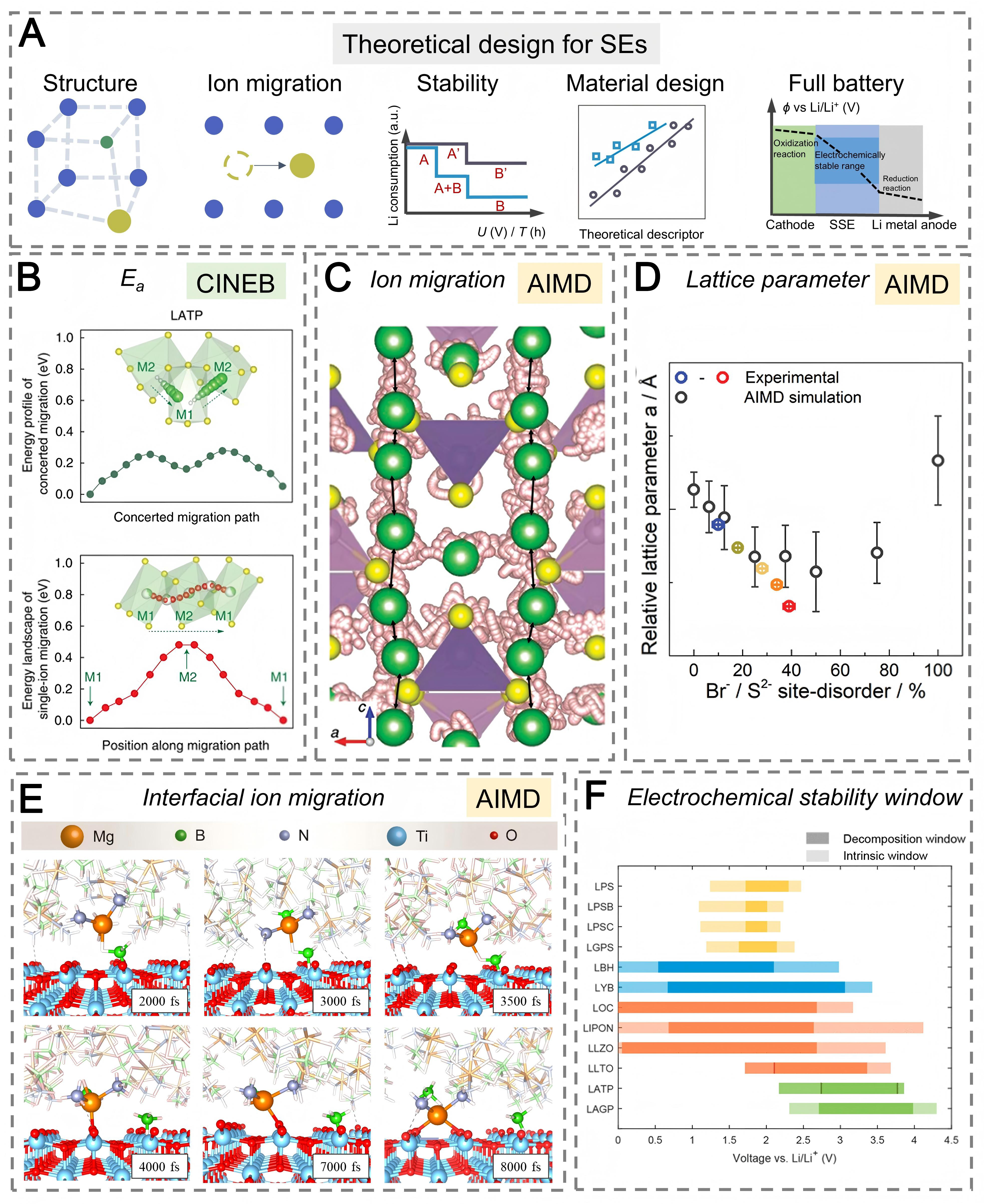
Figure 3. Computational simulation approaches for SEs. (A) Overview of theoretical techniques and representative predictive performance; (B) CI-NEB energy profiles showing Li+ migration pathway in LATP. Reproduced with permission from ref.[64]. Copyright 2017, Springer Nature; (C) AIMD trajectories of Li atoms (white) in LGPS, highlighting the one-dimensional c-axis diffusion. Reproduced with permission from ref.[26]. Copyright 2012, American Chemical Society; (D) Comparison between AIMD-predicted and experimentally measured lattice parameters of Li6PS5Br. Reproduced with permission from ref.[65]. Copyright 2021, Wiley-VCH; (E) Snapshots from AIMD simulations showing Mg2+ migration at the TiO2 (001) and Mg(BH4)2·1.5NH3 interface. Reproduced with permission from ref.[58]. Copyright 2022, Elsevier; (F) Calculated intrinsic and decomposition ESW of representative SEs. Reproduced with permission from ref.[66]. Copyright 2021, American Chemical Society. SE: Solid electrolyte; AIMD: ab initio molecular dynamics; CI-NEB: climbing-image nudged elastic band; LATP: Li1.3Al0.3Ti1.7(PO4)3; LGPS: Li10GeP2S12; ESW: electrochemical stability window.


