


Maternal overnutrition impairs myocardial geometry and function in conjunction with proinflammatory activation in fetal sheep hearts
 0
0 Abstract
Aim: Maternal overnutrition predisposes offspring to an increased prevalence of cardiovascular disease, yet the specific underlying mechanisms remain elusive.
Methods: This study examined the roles of inflammation and ferroptosis in driving fetal cardiac structural and functional alterations following maternal overnutrition. Multiparous ewes were fed either a control diet [100% of National Research Council (NRC) recommended requirements] or an overnutrition diet (150% of NRC requirement) from 60 days preconception until gestational day 135. Cardiac geometry, histomorphology, immune cell infiltration, cardiomyocyte function, intracellular Ca2+ handling, and expression of inflammatory and ferroptotic markers were assessed in fetal hearts.
Results: Despite comparable fetal crown-rump lengths and organ weights (e.g., brain and liver), fetuses from overnourished ewes exhibited significantly lower body and heart weights. Maternal overnutrition induced marked cardiac atrophy, interstitial fibrosis, lipid deposition, and oxidative damage, coupled with increased neutrophil and monocyte infiltration. At the cellular level, cardiomyocytes from overnourished fetuses exhibited impaired contractile and intracellular Ca2+ properties. Molecular profiling revealed that maternal overnutrition significantly upregulated proinflammatory markers (CD14, CD68, IL1A, IL1B, IL6, TLR4, and iNOS), and increased IκB phosphorylation (indicating NFκB activation). Concurrently, overnutrition suppressed IL18 and M-CSF expression, alongside the ferroptosis-defense proteins GPX4 and SLC7A11.
Conclusion: These findings demonstrate that maternal overnutrition creates a proinflammatory and ferroptosis-prone myocardial environment, resulting in pathological structural remodeling and functional impairment of fetal hearts. Our data suggest that targeting the inflammation-ferroptosis signaling may help to mitigate the developmental programming of cardiovascular disease.
Keywords
INTRODUCTION
Obesity remains a critical global public health concern, with its prevalence continuing to rise at an alarming rate[1-6]. In the United States, over 30% of population is classified as obese, with data from the National Health and Nutrition Examination Survey (NHANES) highlighting an alarming obesity rate of 35.5% among females over 20 years of age[7]. This trend is mirrored globally, where the increasing incidence of obesity among women of reproductive age poses significant risks to both maternal and fetal health[4,8,9]. Gestational overnutrition often precipitates severe complications, including gestational diabetes mellitus, preeclampsia, and fetal macrosomia[8,10]. Beyond immediate perinatal outcomes, the “Developmental Origins of Health and Disease” (DOHaD) hypothesis posits that the intrauterine environment serves as a critical window for physiological programming. Consequently, offspring born to obese mothers face a 2-fold greater risk of developing insulin resistance, childhood obesity, and type 2 diabetes later in life[11].
Obesity, particularly when driven by high-fat, high-energy dietary intake, is inextricably linked to chronic low-grade inflammation. This systemic state is characterized by the elevation of proinflammatory cytokines across metabolic hubs, including the liver, adipose tissue, and skeletal muscle[1,4,12,13]. These inflammatory responses are pivotal in the pathogenesis of insulin resistance and cardiovascular disease in both mothers and their offspring[8,14]. A key mediator of this process is the elevation of circulating free fatty acids (FFAs), which function as endogenous ligands for proinflammatory toll-like receptors (TLRs), specifically TLR4[15]. Upon activation by saturated fatty acids, TLR4 recruits downstream adaptor proteins, such as MyD88, to trigger the nuclear factor κB (NFκB) signaling cascade. Activation of this pathway drives the transcription of diverse proinflammatory mediators that can traverse the placental barrier, potentially programming the fetal immune system toward a state of chronic inflammatory dysfunction[16,17].
Emerging evidence suggests that maternal overnutrition significantly impairs fetal cardiovascular development[18-22], though the precise molecular drivers remain poorly defined. A novel and potentially critical mechanism involves the induction of regulated cell death pathways, most notably ferroptosis, an iron-dependent form of cell death driven by lipid peroxidation. Ferroptosis has been increasingly implicated in metabolic and cardiovascular pathologies and may act in concert with inflammatory signaling to exacerbate maladaptive fetal cardiac remodeling[23,24]. In obese pregnancies, excessive lipid accumulation within fetal myocardial tissues not only amplifies inflammatory flux but also sensitizes cardiomyocytes to ferroptotic injury by overwhelming the glutathione-dependent antioxidant defense system[25-27]. This dual burden of inflammation and ferroptosis likely drives myocardial remodeling and impairs cardiomyocyte contractility, establishing a pathological substrate for long-term cardiovascular risk.
To elucidate these mechanisms, we utilized a well-established gestational overfeeding model in multiparous ewes[28,29]. Sheep represent an excellent translational model for studying developmental programming, as their gestational milestones and frequent singleton or twin pregnancies closely parallel to human physiology. This model enabled the precise characterization of inflammatory signaling and regulated cell death pathways in fetal left ventricular tissues during late gestation[30]. In this study, we evaluated cardiac geometry, histomorphology, lipid deposition, and immune cell infiltration (neutrophils and monocytes), alongside proinflammatory cytokine profiles and cardiomyocyte mechanical properties in fetal hearts. Furthermore, to determine the specific role of ferroptotic stress in mediating these morphometric and functional alterations, we analyzed key markers for ferroptosis resistance, including glutathione peroxidase 4 (GPX4) and SLC7A11, in fetal cardiac tissues.
MATERIALS AND METHODS
Experimental models
All animal procedures were performed in accordance with the protocols approved by the Institutional Animal Care and Use Committee (No: 20171009SF00291-02). In brief, multiparous ewes (Rambouillet/Columbia cross) were utilized to evaluate the effects of maternal overnutrition on fetal development. To establish a distinct pre-gestational metabolic state, weight-matched ewes were randomly assigned to one of two dietary groups beginning 60 days prior to conception (conception was designated as gestational day 0): a control group (100% of National Research Council [NRC] recommendations) or an overnutrition group (150% of NRC recommendations). Dietary rations were precisely adjusted based on metabolic body weight (BW0.75). The differential feeding regimen was maintained until gestational day 75 (D75), a critical window for placental and early organ development. After D75, all ewes were maintained under standard husbandry conditions until near-term (D135). At D135, ewes were anesthetized with ketamine and euthanized via intravenous sodium phenobarbital administration followed by exsanguination. The gravid uterus was rapidly excised, and comprehensive fetal biometric data were collected for both male and female fetuses. Parameters including total fetal body weight, crown-rump length, and individual weights of major organs (heart, liver, and brain) were recorded and normalized to total body weight for subsequent morphometric analyses[29,30].
RNA-sequencing analysis
RNA-sequencing data from fetal hematopoietic stem and progenitor cells (HSPCs) were retrieved from the Gene Expression Omnibus (GEO; GSE219095), comparing maternal Western diet (mWSD) and control diet (mCD) in a rhesus macaque model. Analyses were performed in R (version 4.0 or later), starting with a filtered FPKM matrix (excluding genes with FPKM < 1 in < 2 samples) followed by log2(x + 1) transformation. Principal component analysis (PCA) was utilized to assess global sample clustering and transcriptomic variance. Differential gene expression (DEG) was determined using the limma package, with P-values adjusted (adj.P.Val) for multiple testing via the Benjamini-Hochberg method to control False Discovery Rate (FDR). For pathway enrichment, gene symbols were mapped to Entrez IDs using clusterProfiler and the org.Hs.eg.db database, leveraging human-macaque gene homology. Gene Set Enrichment Analysis (GSEA) was conducted via the gseKEGG function to identify significantly modulated metabolic and signaling pathways. Data visualization was executed using pheatmap for hierarchical clustering (based on row-scaled log2-FPKM values), and ggplot2 or enrichplot for generating PCA and GSEA diagrams[31].
Histopathological analysis
Fetal left ventricular (LV) tissues were harvested and fixed in 10% neutral-buffered formalin for 24 h. Following fixation, samples underwent automated dehydration through a graded ethanol series. Samples were then washed in xylene, and embedded in paraffin. Paraffin-embedded tissues were sectioned at 5-μm thickness before staining using hematoxylin and eosin (H&E) for morphological evaluation. For interstitial fibrosis, Masson Trichrome staining was employed by normalizing the blue-colored area to the entire microscopic field. For lipid droplet detection, LV samples were processed in a solution containing optimal cutting temperature (OCT) compound. Cryosections (10-μm thick) were detected using oil red O staining to evaluate lipid droplet buildup. Assessments of cardiomyocyte cross-sectional area, blue-colored interstitial fibrosis (normalized to the entire field) and lipid accumulation were performed using the NIH ImageJ software[21,30].
Free radical injury
To evaluate intracellular reactive oxygen species (ROS) levels, myocardial tissues were loaded with 5 μM 2',7'-dichlorodihydrofluorescein diacetate (H2DCF-DA; Molecular Probes, Eugene, OR, USA). This cell-permeable non-fluorescent probe is de-esterified and turns into highly fluorescent DCF upon oxidation by free radicals. Tissues were washed with phosphate-buffered saline (PBS) to remove excess dye. Fluorescence images were captured using an Olympus BX51 microscope (Olympus, Tokyo, Japan). Fluorescence intensity, representing the intracellular oxidative burden, was quantified across multiple representative fields of view using Image-Pro Plus software (Media Cybernetics, Bethesda, MD, USA)[21].
Immunohistochemistry
Fetal LV tissues from control and overnourished ewes were processed in OCT (Sakura Finetek USA, Inc., Torrance, CA, USA) before storage at -80 °C. Cryosections (9-μm thick) were blocked with 10% goat serum at room temperature to minimize nonspecific binding. All samples were maintained at 4 °C with primary antibodies targeting neutrophil elastin or monocyte/macrophage CD68. After thorough washing, samples were exposed to an Alexa Fluor® 594-conjugated goat anti-rabbit IgG secondary antibody (Invitrogen). Fluorescent signals were displayed through fluorescence microscopy[32].
Cardiomyocyte isolation, mechanical and intracellular Ca2+ assessment
LV tissues from fetal sheep were enzymatically digested by perfusion with Krebs-Henseleit bicarbonate (KHB) buffer supplemented with Liberase Blendzyme 4 (Hoffmann-La Roche Inc., Indianapolis, IN, USA) for 20 min. Isolated cardiomyocytes exhibiting a rod-like shape with sharp, intact borders were selected for subsequent functional assessments. Cells were subjected to electrical pacing at 0.5 Hz, and were monitored through an IonOptix™ soft-edge detection system (Milton, MA, USA). The following parameters were evaluated including maximum rate of shortening/relengthening (± dL/dt), peak shortening (PS), time-to-PS (TPS), and time-to-90% relengthening (TR90). To examine intracellular Ca2+ handling, cardiomyocytes were incubated with Fura-2/AM (0.5 µM) for 15 min. Fluorescent signal was captured using alternating excitation wavelengths at 360 and 380 nm, and emission signals were collected between
Quantitative real-time PCR
Quantitative real-time PCR was conducted in total RNA derived from fetal LV tissues to assess mRNA levels of CD14, CD68, IL1A, IL1B, IL6, IL18, TLR4, macrophage colony-stimulating factor (M-CSF), inducible nitric oxide synthase (iNOS), glutathione peroxidase 4 (GPX4) and SLC7A11, with 18S rRNA serving as the internal control. Primer sequences used for amplification are provided in Table 1. Reactions were carried out using the QuantiTect SYBR Green Real-Time PCR Kit (Bio-Rad) following standard protocols as reported[30].
Primer sequence information
| Primer | Forward sequence | Reverse sequence |
| CD14 | 5′-CTC AGC GTG CTT GAT CTC AG-3′ | 5′-AAG GGA TTT CCG TCC AGA GT-3′ |
| CD68 | 5′-CAG GGG ACA GGG AAT GAC T-3′ | 5′-CCA AGT GGT GGT TCT GTG G-3′ |
| GPX4 | 5′-TGTGGTTTACGGATCCTGGC-3′ | 5′-GAGGGACGACTTTTCCCGAG-3′ |
| IL1A | 5′-GTG CTC AAA ATG AAG ACG AAC C-3′ | 5′- CCC AGA AGA AGA GGA GAT TGG T-3 |
| IL1B | 5′-CGT CTT CCT GGG ACG TTT TAG-3′ | 5′- CTG CGT ATG GCT TCT TTA GGG-3′ |
| IL6 | 5′-TCA TCC TGA GAA GCC TTG AGA-3′ | 5′- TTT CTG ACC AGA GGA GGG AAT-3′ |
| IL18 | 5′-ATG GCG AAG ACC TGG AAT C-3′ | 5′- CAG GTT GAT TTC CCT GGC TA-3′ |
| iNOS | 5′-CAT TTT ATT TCC CCA TCT CCT TC-3′ | 5′-CTC AAG AGA GAC AAA AGG TTC CA-3′ |
| M-CSF | 5′-ATG GAC TAT GAT GAG CAG GAC AA-3′ | 5′-TTC TTG ATC TTC TCC AGC AAC TG-3′ |
| SLC7A11 | 5′-GAACAGCAGTTCTCGCCACT-3′ | 5′-ACGATGCATATCTGGGCGTT-3′ |
| TLR4 | 5′-TGC TGG CTG CAA AAA GTA TG-3′ | 5′-CCC TGT AGT GAA GGC AGA GC-3′ |
| 18S | 5′-AGC CTG CGG CTT AAT TTG AC-3′ | 5′-CAA CTA AGA ACG GCC ATG CA-3′ |
Western blot analysis
Fetal LV tissues were homogenized and lysed. Total protein levels were evaluated using a Bio-Rad device (Bio-Rad Laboratories, Hercules, CA, USA). Equal amounts of protein (30 µg) were loaded onto 7%, 10%, or 12% SDS-PAGE gels, based on molecular sizes of target proteins. Samples were transferred onto nitrocellulose membranes prior to blocking in TBS-T containing 5% non-fat dry milk. Samples were then incubated overnight with primary antibodies against CD14, CD68, IL1A, IL1B, IL6, IL18, TLR4, iNOS, M-CSF, GPX4, SLC7A11, IκB (pan and phosphorylated), NFκB, α-tubulin and GAPDH (loading controls), all at 1:1000 dilution. All antibodies used were obtained from Santa Cruz Biotechnology (Dallas, TX, USA) or Cell Signaling Technology (Beverly, MA, USA) and demonstrated strong cross-reactivity with sheep myocardial tissues. After immunoblotting, membranes were visualized, and band intensities were determined using a calibrated densitometer (Bio-Rad)[30].
Statistical analysis
All data are presented as mean ± SEM. Statistical analyses were performed using GraphPad Prism (ver9.0, GraphPad Software, San Diego, CA, USA). Data normality was assessed using the Shapiro-Wilk test. For comparisons between control and overnourished groups, an unpaired Student’s t-test was employed for normally distributed data. For multiple comparisons across the diverse range of physiological and molecular variables measured (e.g., organ weights, protein expression, and cardiomyocyte mechanics), the Benjamini-Hochberg FDR correction was applied to adjust P-values and minimize risk of false positives. A corrected P-value < 0.05 was considered statistically significant.
RESULTS
Transcriptomic profile in fetal HSPC following maternal Western diet intake
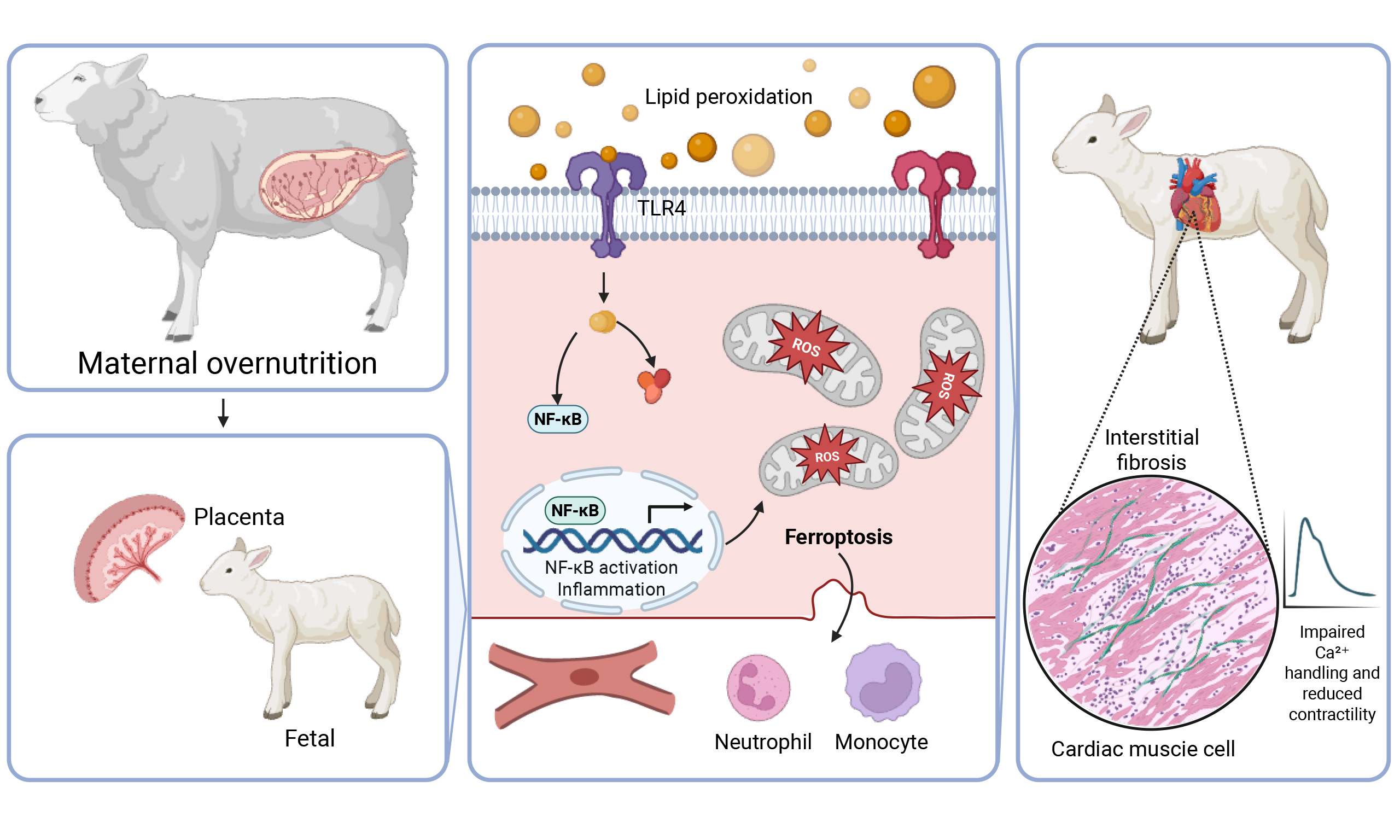
To evaluate the impact of in utero maternal Western diet exposure on fetal hematopoietic system, we performed a comprehensive transcriptomic analysis of rhesus macaque fetal bone marrow CD34+ HSPCs. PCA of global gene expression revealed distinct clustering and a robust separation between mWSD and control groups [Figure 1A]. This divergence was primarily driven by the first principal component (PC1), which accounted for 30% of the total variance, indicating a profound diet-induced shift in the cellular state. Hierarchical clustering and heatmap analysis of the top 30 differentially expressed genes further illustrated a highly consistent pattern of transcriptomic reprogramming across the mWSD cohort [Figure 1B]. To delineate the functional consequences of these molecular shifts, we employed GSEA. Notably, GSEA identified a significant downregulation of the 'Ribosome' pathway in mWSD-exposed fetuses [Figure 1C], suggesting an impairment in protein synthesis machinery or translational capacity within the progenitor pool. Collectively, these data demonstrate that mWSD exposure induces a consistent shift in the fetal HSPC transcriptome, impacting fundamental cellular processes. These systemic alterations provide a broader context for the immune cell infiltration observed in our fetal sheep myocardial model.
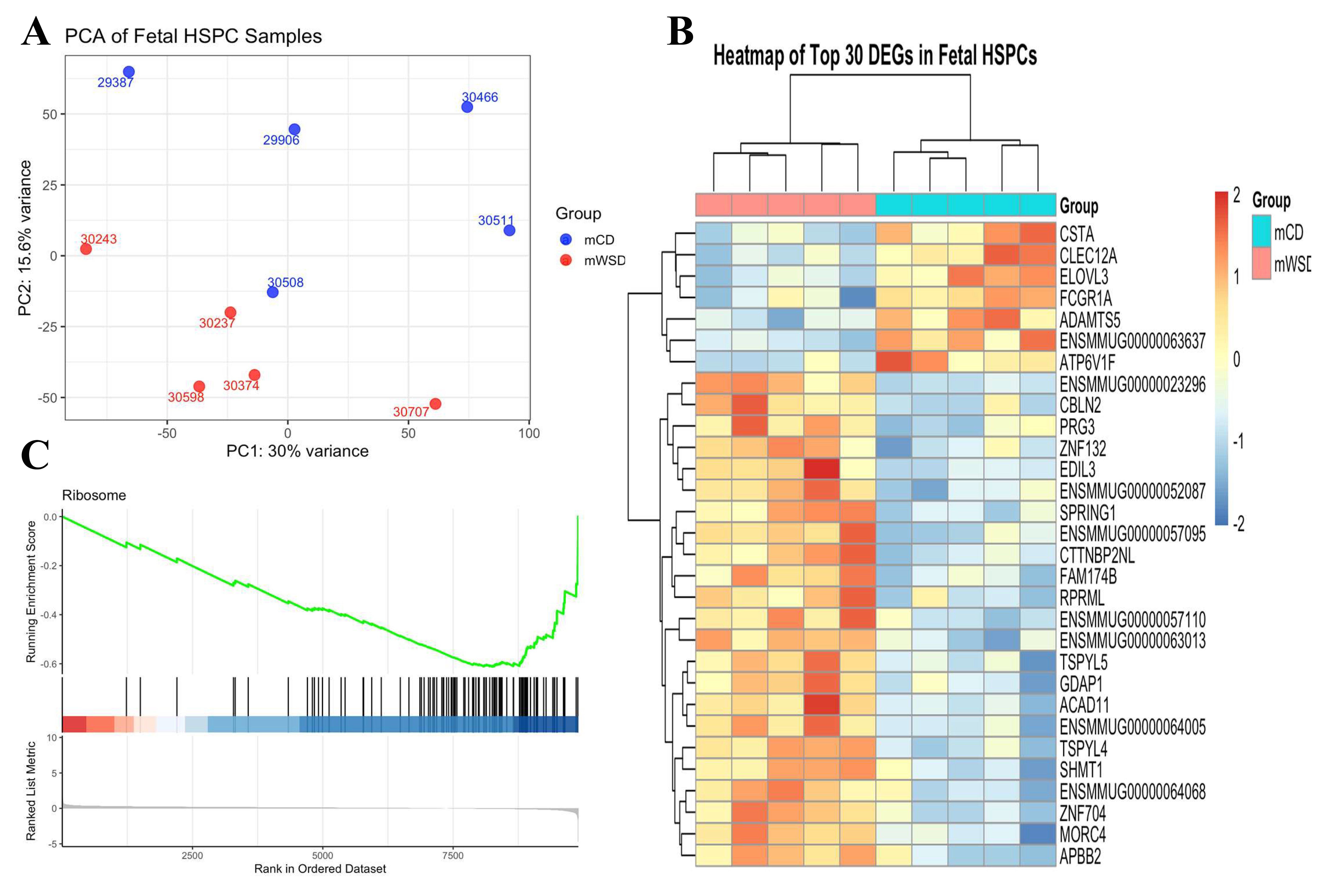
Figure 1. Principal component analysis (PCA), heatmap, and GSEA of fetal HSPC transcriptome. (A) PCA plot of log2-transformed FPKM gene expression from fetal HSPCs. Samples from maternal control diet (mCD, blue) and maternal Western-style diet (mWSD, red) groups form distinct clusters, primarily separated along PC1 (30% variance); (B) Heatmap of top 30 DEGs in fetal HSPCs. The heatmap displays the row-scaled log2-transformed expression values of the top 30 differentially expressed genes. The consistent pattern of gene expression correctly clusters the samples by group and concludes that maternal diet induces a targeted and robust reprogramming of fetal HSPC transcriptome; (C) GSEA enrichment plot for ribosome pathway. The plot shows significant downregulation of the KEGG "Ribosome" pathway in mWSD-exposed fetal HSPCs, indicating that maternal Western-style diet suppresses fundamental protein synthesis machinery in fetal progenitor cells, representing a key mechanism of adverse developmental programming.
Influence of maternal overnutrition on maternal and fetal biometric and morphometric profile
Maternal overnutrition from 60 days preconception through gestational D135 led to a significant gain in maternal liver weight without affecting the heart, left ventricle (LV), right ventricle (RV), myocardial septum, kidney and gravid uterus weights [Figure 2A-G]. At near-term (D135), the impact on the offspring was characterized by sex-specific vulnerabilities and selective organ atrophy. Specifically, female fetuses from overnourished ewes exhibited significantly decreased heart and thymus weights, whereas these differences were not statistically significant in males. Conversely, fetal subcutaneous fat deposition was markedly elevated in both sexes in the overnutrition group compared to controls. Other biometric parameters, including fetal crown-rump length, body weight, heart-to-body weight ratio, as well as weights of the liver, kidneys, brain, spleen, adrenal glands, pancreas, gonads, and visceral fat (pericardial and perirenal), were comparable between maternal overnutrition and control groups, regardless of sex [Figure 2H-V]. However, when data from both sexes were pooled, the larger sample size revealed a more pervasive phenotype. Combined analysis showed that maternal overnutrition significantly reduced fetal heart, pancreas, kidney, and thymus weights, while concurrently increasing subcutaneous fat accumulation in fetuses. Despite these changes, fetal crown-rump length, fetal heart-to-body weight ratio, fetal liver, brain, spleen, adrenal gland, gonad, and visceral fat (pericardial and perirenal) weights remained stable [Supplementary Figure 1A-O]. These results suggest that maternal overnutrition drives a pattern of asymmetric fetal development, prioritizing adipose deposition at the expense of vital organ mass, particularly the heart and lymphoid tissues.
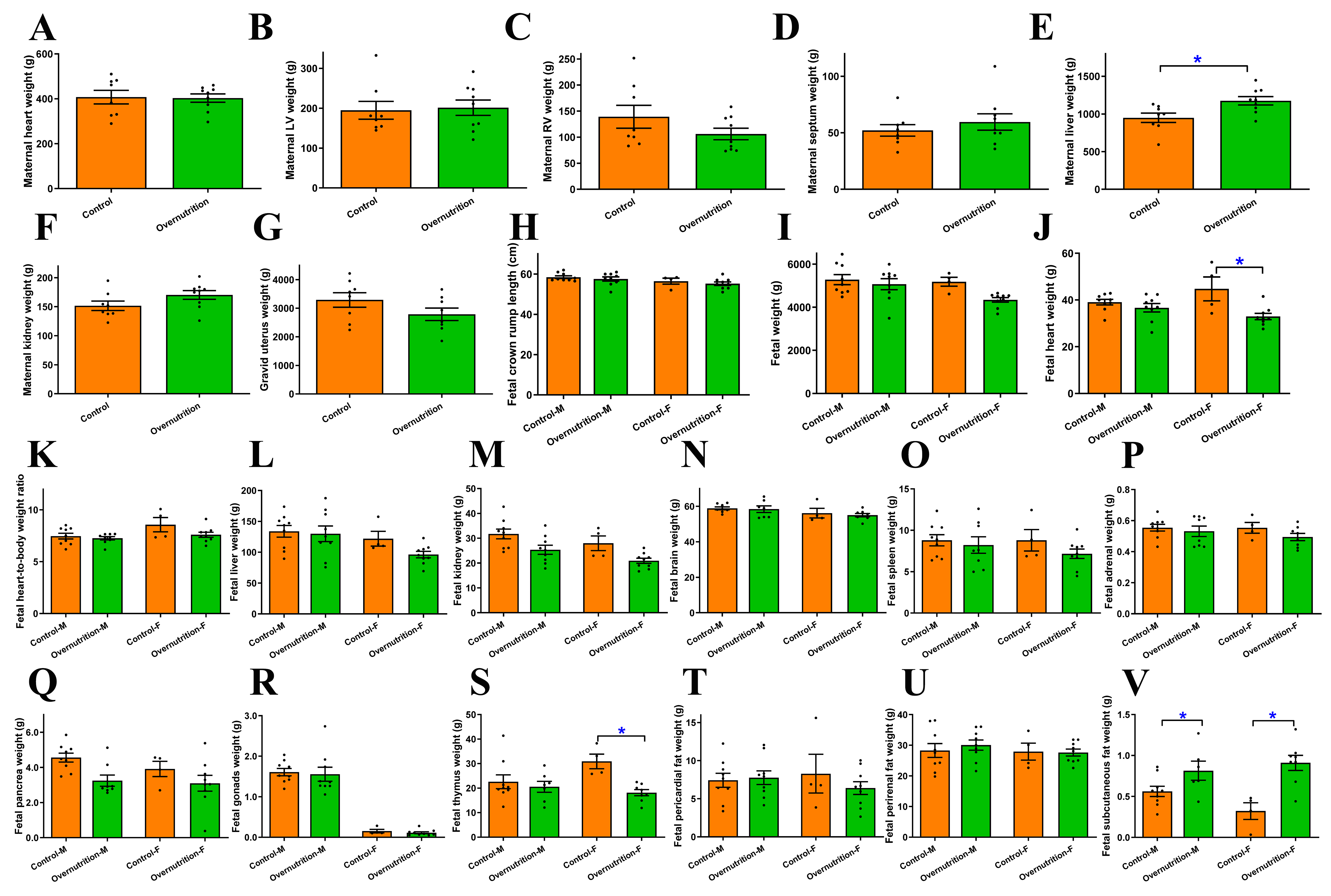
Figure 2. Morphometric and biometric profiles in dams receiving dietary overnutrition for 135 days and their offspring at D135 of gestation. Multiparous ewes received either 100% of NRC-recommended (control) or 150% of NRC-recommended nutrient (overnutrition) diets from 60 days before mating to D75. (A-G) Maternal morphometric profile: (A) Maternal heart weight; (B) Maternal LV weight; (C) Maternal RV weight; (D) Maternal septal weight; (E) Maternal liver weight; (F) Maternal kidney weight; (G) gravid uterus weight; (H-V) Fetal (D135) morphometric profile in males or females: (H) fetal crown rump length; (I) fetal weight; (J) fetal heart weight; (K) fetal heart-to-body weight; (L) fetal liver weight; (M) fetal kidney weight; (N) fetal brain weight; (O) fetal spleen weight; (P) fetal adrenal gland weight; (Q) fetal pancreas gland weight; (R) fetal gonad gland weight; (S) fetal thymus weight; (T) fetal pericardial fat weight; (U) fetal perirenal fat weight; (V) fetal subcutaneous fat weight. Mean ± SEM, *P < 0.05 between groups; n = 7 and 9 ewes for control and overnutrition groups (A-G), or 5 male/4 female for control group, and 4 male/5 female fetuses for overnutrition group (H-V).
Impact of maternal overnutrition on fetal heart morphological, free radical damage, lipid deposition and proinflammatory infiltration profiles
Morphological evaluations using H&E and Masson Trichrome staining demonstrated that maternal overnutrition induced significant pathological remodeling in the fetal heart. This was characterized by a marked decrease in cardiomyocyte cross-sectional area and elevated interstitial fibrosis in overnourished fetal group compared to the control group. Similarly, free radical damage assessed using DCF staining indicated significantly elevated free radical accumulation in fetal hearts following maternal overnutrition [Figure 3A-D]. These observations are reminiscent of our earlier finding[30].
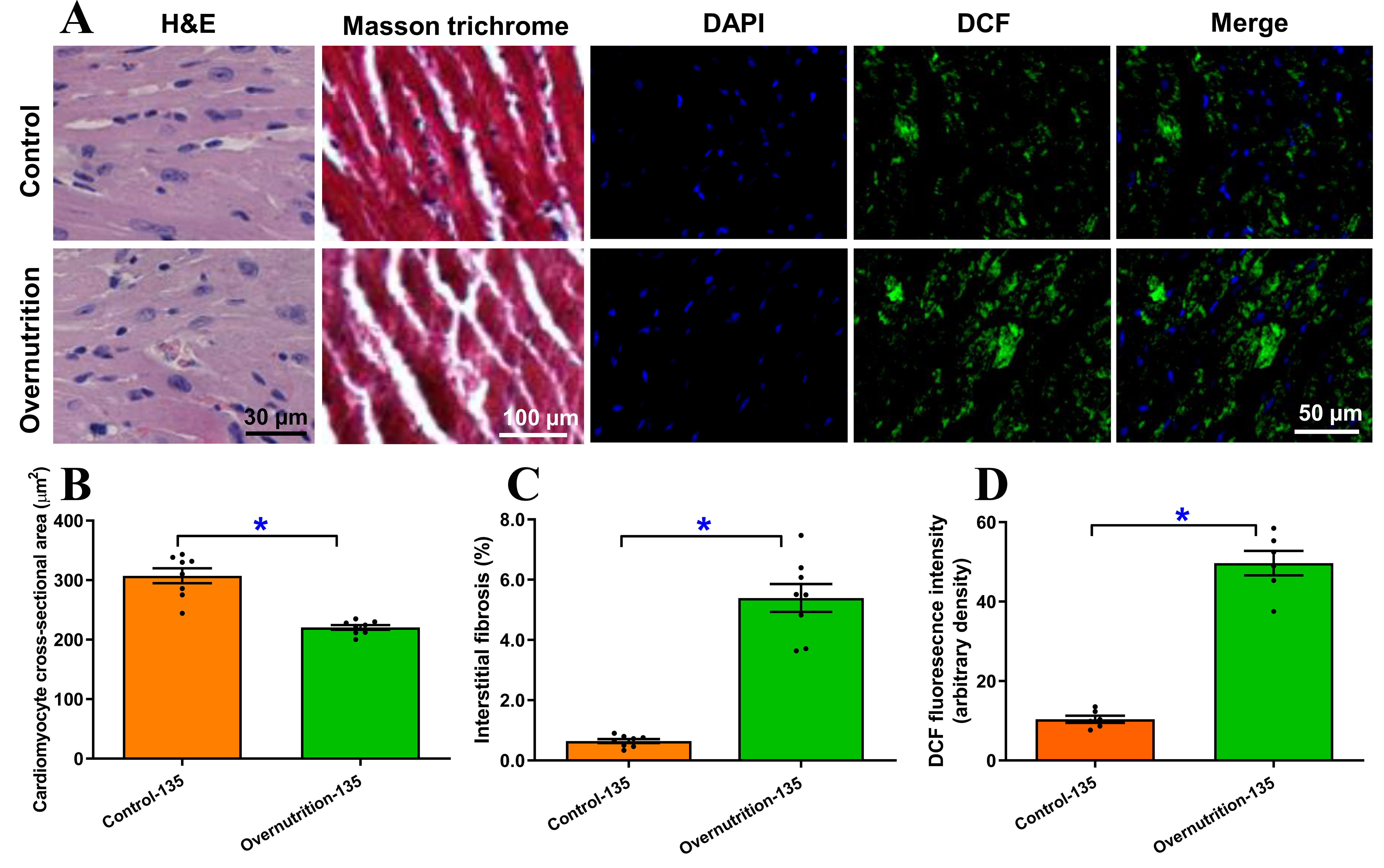
Figure 3. Morphological and free radical accumulation of fetal (D135) hearts from ewes receiving either control or overnutrition diet (150% NRC recommendation) for 135 days. (A) Representative micrographs depicting H&E staining, Masson Trichrome staining and DCF staining from fetal hearts from ewes receiving control or overnutrition diet. DCF staining includes DAPI (nucleus), DCF and merged images in 3 respective columns; (B) Pooled cardiomyocyte cross-sectional area from H&E staining; (C) Pooled myocardial interstitial fibrosis from Masson Trichrome staining; and (D) Pooled myocardial free radical accumulation from DCF staining. Mean ± SEM, *P < 0.05 between groups; n = 8 (B and C) or 6 (D) fetuses per group.
To assess the metabolic impact of overnutrition, we evaluated neutral lipid accumulation via oil red O staining. Fetal myocardial tissues from overnourished ewes displayed an evident rise in lipid deposition compared to controls [Figure 4A and B]. This metabolic shift was accompanied by a robust inflammatory response. To delineate the infiltration of proinflammatory immune cells (neutrophils and monocytes), levels of elastin and CD68, markers for neutrophils and monocytes, respectively, were evaluated using fluorescent immunostaining. As depicted in Figure 4C-F, maternal overnutrition overtly promoted neutrophil and monocyte infiltration compared with controls.
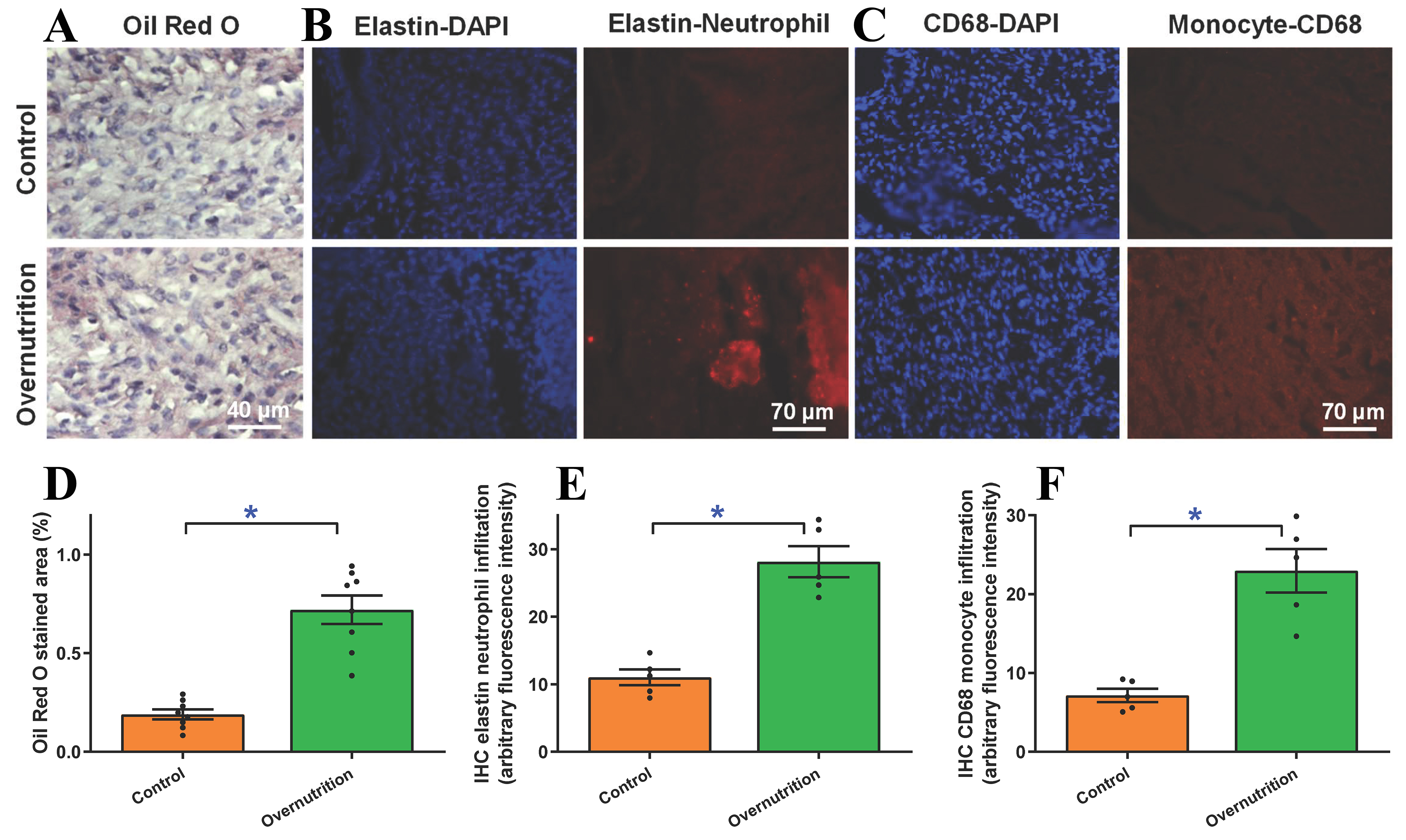
Figure 4. Myocardial lipid deposition and inflammatory immunohistochemistry (IHC) infiltration in fetal (D135) hearts from ewes receiving either control or overnutrition diet (150% NRC recommendation) for 135 days. (A) Representative oil red O staining depicting lipid deposition; (B) Pooled lipid deposition from oil red O staining; (C) Representative elastin staining depicting neutrophil infiltration (blue staining = DAPI staining for nucleus); (D) Pooled neutrophil infiltration status from elastin staining; (E) Representative CD68 staining depicting monocyte infiltration (blue staining = DAPI staining for nucleus); and (F) Pooled monocyte infiltration status from CD68 IHC staining. Mean ± SEM, *P < 0.05 between groups; n = 8 (D) or 5 (E and F) fetuses per group.
Impact of maternal overnutrition on fetal cardiomyocyte mechanical features
Further scrutiny of cardiomyocyte contractile function revealed that maternal overfeeding significantly reduced peak shortening and maximal velocity of shortening/relengthening (± dL/dt). Additionally, these cells exhibited elongated relengthening (TR90), although cardiomyocyte length and shortening period (TPS) remained unchanged [Figure 5A-F]. To discern potential mechanisms underlying overnutrition-prompted changes in cardiomyocyte mechanics, intracellular Ca2+ homeostasis was evaluated using Fura-2 fluorescence. Results displayed in Figure 5G-I indicated significantly declined electrically evoked Fura-2 fluorescence intensity (ΔFFI) and intracellular Ca2+ clearance alongside unaltered baseline intracellular Ca2+ level in fetal cardiomyocytes from maternal overnutrition group compared to the controls.
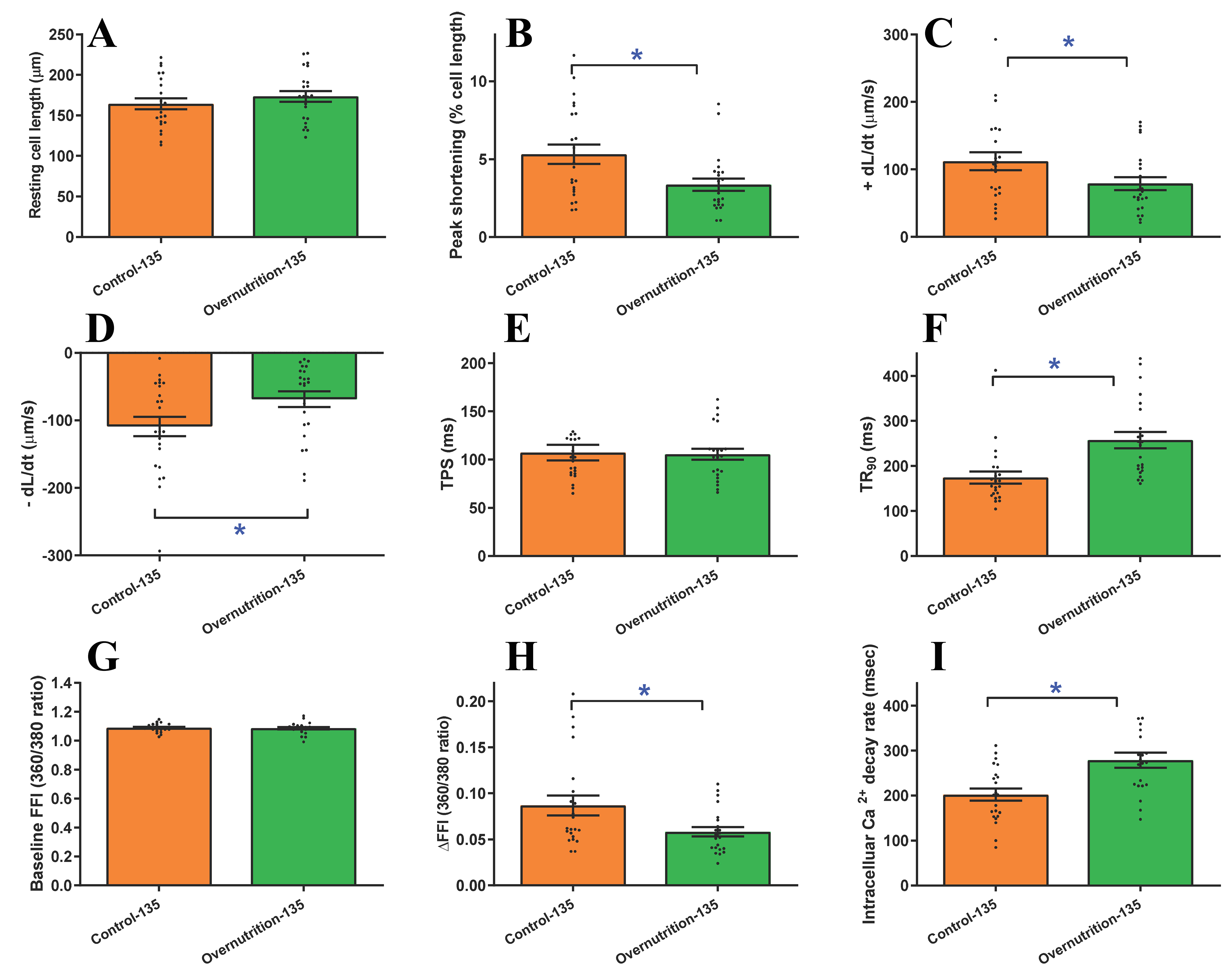
Figure 5. Cardiomyocyte contractile and intracellular Ca2+ properties in fetal (D135) hearts from ewes receiving either control or overnutrition diet (150% NRC recommendation) for 135 days. (A) Resting cell length; (B) PS; (C) +dL/dt; (D) -dL/dt; (E) TPS; (F) TR90; (G) Baseline intracellular Ca2+ FFI; (H) ΔFFI; and (I) Intracellular Ca2+ decay constant. Mean ± SEM, *P < 0.05 between indicated groups, n = 23 (A-F) or 22 (G-I) cells (technical replicates) from 3 fetuses (biological replicates) per group.
mRNA and protein expression of proinflammatory and ferroptotic markers
To decipher the mechanisms underlying maternal overnutrition-induced changes in fetal myocardial morphology and function, we examined markers of inflammation and ferroptosis, including CD14, CD68, IL1A, IL1B, IL6, IL18, TLR4, iNOS, M-CSF, GPX4 and SLC7A11. An assessment of mRNA levels using RT-PCR revealed that gestational overnutrition significantly upregulated CD14, CD68, IL1A, IL1B, IL6, TLR4, and iNOS mRNA levels while downregulating that of IL18, M-CSF, GPX4 and SLC7A11
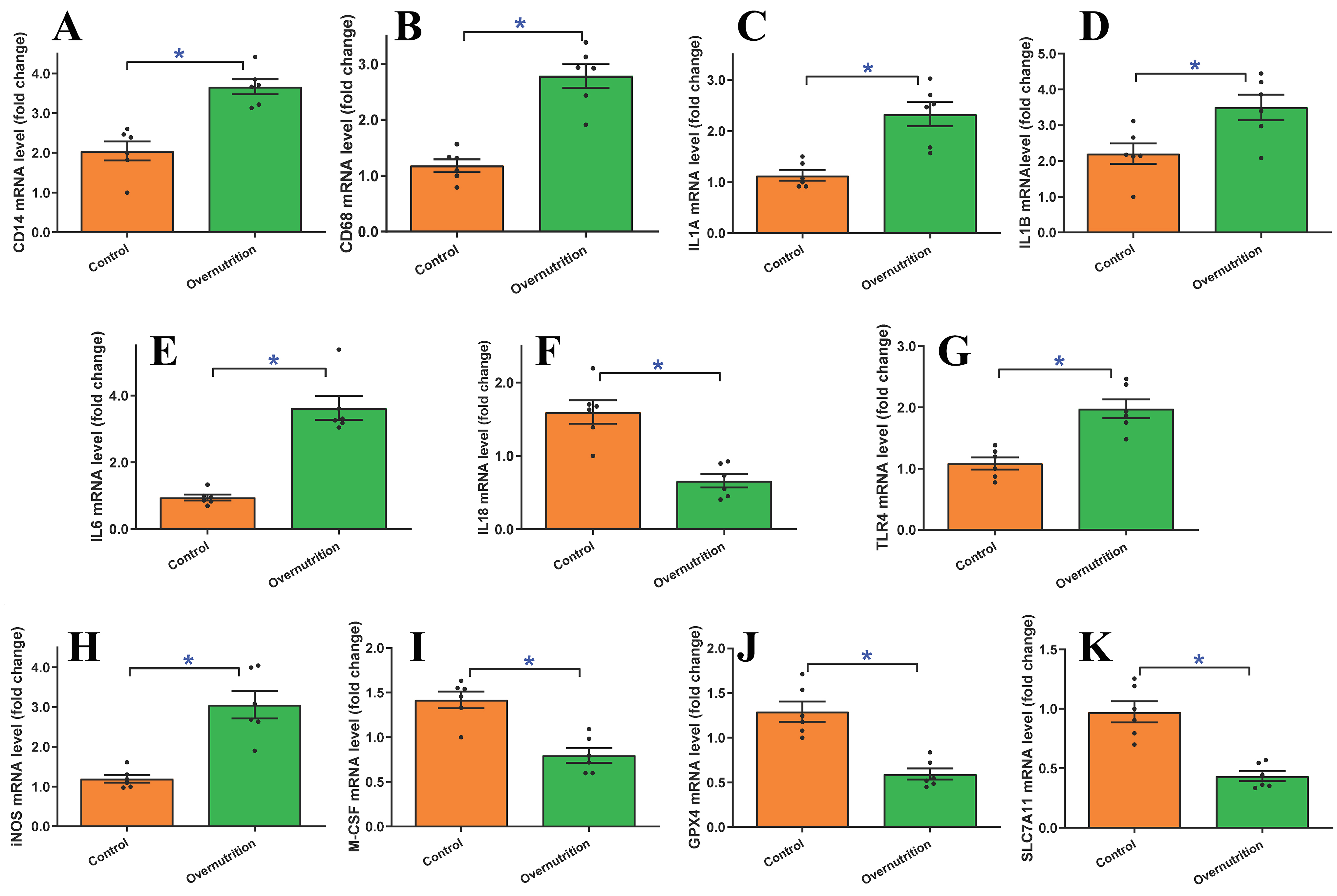
Figure 6. mRNA expression of proinflammatory and ferroptotic markers in fetal (D135) hearts from ewes receiving either control or overnutrition (150% NRC recommendation) for 135 days assessed using quantitative real-time PCR (qPCR). (A) CD14 mRNA; (B) CD68 mRNA; (C) IL1A mRNA; (D) IL1B mRNA; (E) IL6 mRNA; (F) IL18 mRNA; (G) TLR4 mRNA; (H) iNOS mRNA; (I) M-CSF mRNA; (J) GPX4 mRNA; and (K) SLC7A11 mRNA. All gene expressions were normalized to that of 18S (as house-keeping gene). Mean ± SEM, *P < 0.05 between groups, n = 6 fetuses per group.
Further evaluation of the protein-level expressions of proinflammatory and ferroptotic markers corroborated these transcriptomic shifts. By D135 of gestation, maternal overnutrition resulted in marked upregulation of CD14, CD68, IL1A, IL1B, IL6, TLR4, iNOS protein expression. In parallel, a significant reduction was observed in the protein levels of IL18, M-CSF, GPX4 and SLC7A11 [Figure 7A-L]. To further delineate the underlying proinflammatory signaling mechanisms, we assessed the NFκB pathway. Maternal overnutrition was found to downregulate pan NFκB level while facilitating phosphorylation of the NFκB inhibitory molecule IκB, denoting elevated NFκB activity [Figure 7M and N]. Collectively, these data suggest that maternal overfeeding triggers systemic inflammatory activation and suppresses antioxidant defenses in fetus, creating a myocardial environment highly susceptible to ferroptotic injury.
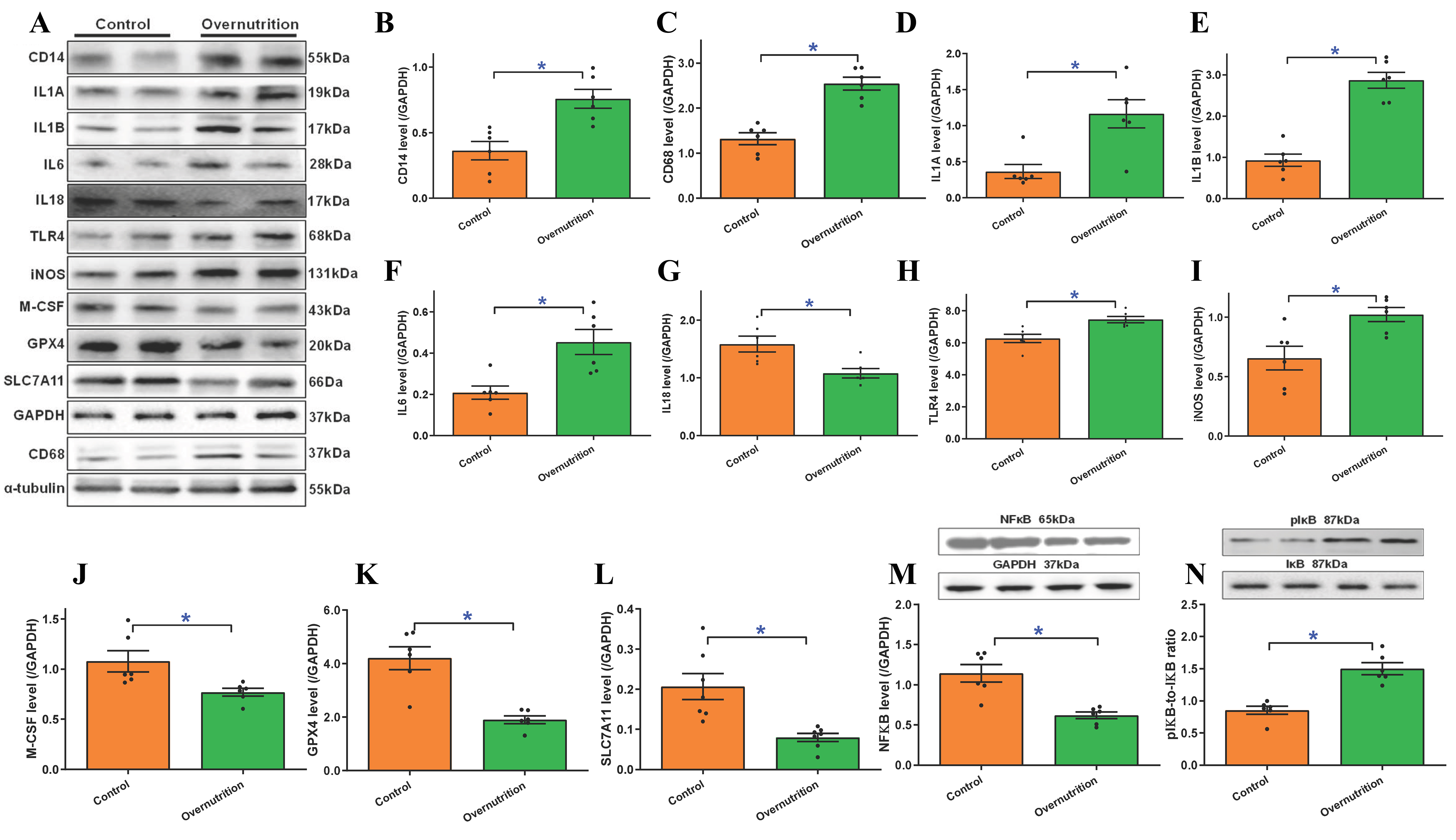
Figure 7. Protein levels of proinflammatory and ferroptotic markers in fetal (D135) hearts from ewes receiving either control or overnutrition (150% NRC recommendation) for 135 days. (A) Representative immunoblots of CD14, CD68, IL1A (IL1α), IL1B (IL1β), IL6, IL18, TLR4, iNOS, M-CSF, GPX4, and SLC7A11 using specific antibodies (using GAPDH or α-tubulin as loading controls);(B) CD14 protein level; (C) CD68 protein level; (D) IL1A (IL1α) protein level; (E) IL1B (IL1β) protein level; (F) IL6 protein level; (G) IL18 protein level; (H) TLR4 protein level; (I) iNOS protein level; (J) M-CSF protein level; (K) GPX4 protein level; (L) SLC7A11 protein level; (M) NFκB level; and (N) phosphorylated IκB (pIκB)-to-IκB ratio. Insets: Representative immunoblots of NFκB, pIκB, and IκB using specific antibodies (GAPDH as the loading control). Mean ± SEM, *P < 0.05 between groups, n = 6-7 fetuses per group.
DISCUSSION
This study was designed to elucidate the mechanistic pathways underlying fetal cardiac alterations in the setting of maternal overnutrition, utilizing a well-established pregnant sheep model. Previous investigations have identified various adverse sequelae of maternal obesity affecting the fetal heart[33], placenta[34], skeletal muscle[35,36], and gut[37], as well as broader metabolic programming[35,38,39]. Our findings extend these observations by offering novel insights into how maternal overnutrition specifically drives inflammatory signaling and ferroptotic stress within the fetal myocardium. These results indicate that maternal overnutrition, both prior to and during gestation, triggers systemic inflammation, immune cell infiltration, and ferroptosis, which collectively compromise fetal heart homeostasis. These data highlight a critical pathophysiological link between maternal obesity, inflammation, and ferroptosis during fetal cardiac development.
While our study did not include longitudinal maternal metabolic profiling, such as blood glucose or insulin levels, several key physiological indicators confirm the successful establishment of the maternal overnutrition model. Specifically, ewes receiving the 150% NRC diet exhibited a significant increase in maternal liver weight, a hallmark of metabolic redirection and hepatic fat accumulation in overfed ruminants. Correspondingly, fetuses from overnourished ewes displayed a significant increase in subcutaneous fat and evident myocardial lipid deposition. These systemic and tissue-specific changes, combined with the significant upregulation of fetal myocardial TLR4, a receptor known to be activated by saturated fatty acids during nutritional stress, provide compelling evidence that the overnutrition regimen induced a robust state of maternal and fetal metabolic excess, thereby establishing a pathological substrate for developmental programming.
Our findings of localized inflammation in the fetal heart are further corroborated by evidence of systemic fetal programming at the progenitor cell level. Indeed, transcriptomic analysis from a nonhuman primate model suggests that maternal dietary stress fundamentally reprograms fetal hematopoietic stem cells - the precursors to infiltrating immune cells - by suppressing essential pathways, including ribosomal biogenesis [Figure 1B and C]. This cross-species evidence suggests that the pro-inflammatory environment within myocardium may be amplified by a pre-existing, systemic priming of the fetal immune system, originating at the earliest stages of hematopoietic development.
Building on our previous report of impaired myocardial contractility in the late-gestation offspring of obese ewes[29], our current study identifies a comprehensive profile of maladaptive remodeling. While biometric data revealed restricted fetal growth and cardiac atrophy (although with unchanged heart size), many organ weights and crown-rump lengths remained unchanged, reflecting complex compensatory transplacental adjustments to maternal metabolic stress. At the cellular level, we observed a “latent” structural remodeling phase, where histological and molecular damage precedes overt changes in total cardiac mass. Specifically, fetal hearts exhibited cardiomyocyte atrophy, interstitial fibrosis, myofibrillar disarray, and myocardial steatosis, alongside robust neutrophil and monocyte infiltration. These structural abnormalities were mechanistically linked to compromised contractile capacity, intracellular Ca2+ derangement, and the potent activation of the TLR4/NFκB inflammatory axis - evidenced by the significant upregulation of iNOS, IL1α (IL1A), IL1β (IL1B), and IL6. It is noteworthy that the fibrotic remodeling in obese offspring may represent a proportional substitution of collagen for atrophying contractile units, a compensatory mechanism to maintain structural integrity amidst intrauterine growth restriction (IUGR). This pathological adaptation, characterized by reduced fetal organ weights despite stable body length, underscores a critical window of subclinical remodeling that may dictate long-term cardiovascular vulnerability.
Crucially, the discrepancy between stable heart-to-body weight ratio and profound internal changes observed here suggests that morphological indices, such as cardiomyocyte cross-sectional area, are far more sensitive markers of programming-induced injury than gross biometry. This lag identifies a critical window of subclinical pathological transition driven by oxidative stress and impaired nutrient exchange - often rooted in diminished placental proliferation and angiogenic factor expression during mid-gestation[40]. In this context, maternal overnutrition disrupts transplacental nutrient exchange by impairing both placental growth and vascular development, exerting a profound negative impact on fetal growth. Ultimately, the morphological integrity of the sheep myocardium is compromised far earlier than gross heart size suggests, highlighting the subtle yet profound impact of maternal nutritional status on fetal cardiac development.
Prolonged maternal overnutrition and metabolic excess induce inflammatory responses across multiple tissues, including adipose tissue, skeletal muscle, and the gastrointestinal tract, in both mothers and their offspring[41,42]. Evidence from human and animal studies has revealed pronounced elevations in proinflammatory cytokines and mediators - such as C-reactive protein, TLR4, IL-6, CD14 and CD68 - across various organs including gut, liver, adipose tissues, skeletal muscles, and placenta[15,35]. Notably, our present study revealed elevated mRNA and protein expression of CD14 and CD68 in overnourished fetuses, corresponding with our IHC results. TLR4, a key pattern-recognition receptor involved in initiation of innate immune responses, was also significantly upregulated. TLR4 is uniquely positioned at the intersection of metabolism and immunity, as it is activated not only by bacterial lipopolysaccharides but also by saturated fatty acids, which are likely elevated under conditions of nutritional stress[43,44]. Upon ligand binding, TLR4, in conjunction with its co-receptor CD14 (cluster of differentiation 14), recruits the adaptor protein MyD88 to activate the NFκB signaling cascade[16]. This cascade prompts the transcriptional activation of genes implicated in inflammation, immunity, cell proliferation, and regulated cell death. Proinflammatory cytokines, including IL1α, IL1β, IL6, and IL18, further amplify this response by acting as activators of the NFκB pathway, which is initiated by the phosphorylation of the NFκB inhibitory protein IκB. This mechanism is strongly supported by our experimental findings of increased NFκB activity (demonstrated by elevated IκB phosphorylation) despite downregulated pan NFκB protein level in overnourished fetus
A novel and significant contribution of our study is the identification of ferroptosis, a recently characterized regulated cell death modality, as a critical mediator of cardiac maladaptation following maternal overnutrition[27]. While ferroptosis has not been classically considered in the framework of the DOHaD fetal programming hypothesis, emerging evidence has implicated this iron-dependent pathway in myocardial injury, particularly within metabolically stressed or lipid-rich environments[24]. Excessive lipid accumulation within fetal cardiomyocytes, as observed in our overnourished group, creates a biochemical substrate highly susceptible to ferroptotic damage, particularly when antioxidant defenses (e.g., GPX4) are overwhelmed. Furthermore, the chronic inflammatory milieu and sustained TLR4 activation observed in these fetuses may enhance iron uptake and lipid peroxidation, further facilitating the ferroptotic cascade. These findings suggest that maternal obesity does not merely drive structural and inflammatory remodeling, but actively modulates programmed cell death pathways in the fetal heart. This molecular switch toward ferroptosis likely contributes to the impaired contractile function observed in late gestation and provides a novel therapeutic target for mitigating long-term myocardial dysfunction in offspring.
Several experimental limitations warrant consideration. First, while the ovine model offers significant developmental parallels to human pregnancy (particularly when compared to rodents), species differences in placental architecture and gestational timeline must be acknowledged when translating these findings into clinical contexts. Second, the integration of transcriptomic data from a nonhuman primate (rhesus macaque) model with the physiological findings from fetal sheep may introduce a layer of interspecies variability. We leveraged the macaque dataset (GSE219095) specifically to delineate how maternal dietary stress reprograms fetal hematopoietic stem and progenitor cells, precursors to the immune cells infiltrating sheep myocardium. This cross-species approach facilitated the identification of pathways such as "Ribosomal Biogenesis" by utilizing the superior gene annotation of the macaque genome. However, potential divergence in inflammatory responses and developmental kinetics between species may limit the generalizability of this integrated analysis. Third, this observational approach lacks direct animal rescue or cellular experiments to confirm specific causal mechanisms. While the correlation between maternal diet and fetal proinflammatory activation is robust, mechanistic causality among inflammation, ferroptosis, and long-term cardiac function in offspring remains to be established. Fourth, despite evidence of immune activation, we observed suppressed IL-18 expression during late gestation. Given IL-18’s role in macrophage recruitment and systemic inflammation, its reduction may reflect a stage-dependent shift in immune dynamics, exhaustion of compensatory signaling, or feedback suppression by ferroptosis-related oxidative stress[45,46]. Regarding our electrophysiological methodology, cells were loaded with Fura-2 AM and excited at 360/380 nm. Although the 340/380 nm ratio is the conventional standard, 360 nm was utilized as it sits near the Fura-2 isosbestic point. In this configuration, the 360 nm signal remains relatively insensitive to [Ca2+] fluctuations, effectively normalizing for dye loading and cell thickness, while the 380 nm signal captures calcium binding. While this approach may provide a different dynamic range than the 340/380 nm ratio, results remained consistent across all experimental groups analyzed under identical optical conditions. Finally, we acknowledge that our cardiomyocyte functional analysis utilized a cell-based sample size derived from a smaller biological cohort of three fetuses per group. While this captures cellular heterogeneity and contractile distribution, treating individual cells as independent observations may introduce pseudoreplication. We recognize the fetus as the primary biological unit; therefore, statistical power may be overestimated in cell-based analysis compared to animal-level analyses. To mitigate this, results were cross-referenced with tissue-level molecular data. Future studies should employ nested or mixed-effect models to more rigorously account for animal-to-animal variability.
In conclusion, our findings demonstrate that maternal overnutrition imposes multifaceted stress on the developing fetal heart by triggering inflammatory signaling, promoting oxidative damage, and disrupting cardiac architecture. These alterations culminate in a heightened myocardial vulnerability to ferroptosis. These pathophysiological alterations likely carry enduring consequences for cardiovascular health from the postnatal period into adulthood. Given that maternal obesity remains a critical global health concern, our data suggest that excessive gestational nutrient intake amplifies redox-immune crosstalk within the fetal myocardium, potentially driving pathological remodeling and intrinsic contractile dysfunction. These insights underscore the necessity of further research into the molecular mechanisms of developmental programming and highlight ferroptosis-targeted interventions as a promising therapeutic strategy to mitigate the cardiovascular risks associated with maternal obesity. Ultimately, extending these observations into neonatal and adult offspring stages is essential to confirm the persistence of these defects and solidify the clinical relevance of these findings within the framework of the developmental origins of adult heart disease.
DECLARATIONS
Acknowledgments
The authors wish to pay tribute to the memory of the late Professor Steven P. Ford of the University of Wyoming Fetal Programming Center, who initiated this research and animal protocol. This work traced its origins to the period between 2003 and 2020, during which Drs. Jun Ren, Wei Guo and Qiurong Wang were members of the Fetal Programming Center, where they were trained by and collaborated closely with Professor Ford. His pioneering vision and foundational contributions to the field of fetal programming remain a guiding influence on this research. The graphical abstract was created with BioRender.com (https://biorender.com/446inzq).
Authors’ contributions
Conducted the study: Min J, Xu Z, Wang Q, Yang L, Zhang M, Turdi S, Guo W, Ren J
Drafted the initial manuscript: Ren J
Analyzed data and edited the manuscript: Wang M, Li F, Tao J, Pei Z
Revised manuscript: Reiter RJ
Availability of data and materials
All original data will be provided upon reasonable request to the corresponding authors.
AI and AI-assisted tools statement
During the preparation of this manuscript, the AI tool Google Gemini (version 3, released 2025-11-18) was used solely for language editing. The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship
None.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
This work was performed under the approved animal protocol (20171009SF00291-02). No human subject was involved.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
Supplementary Materials
REFERENCES
1. Ng M, Gakidou E, Lo J, et al. Global, regional, and national prevalence of adult overweight and obesity, 1990-2021, with forecasts to 2050: a forecasting study for the Global Burden of Disease Study 2021. Lancet. 2025;405:813-38.
2. Kerr JA, Patton GC, Cini KI, et al. Global, regional, and national prevalence of child and adolescent overweight and obesity, 1990-2021, with forecasts to 2050: a forecasting study for the Global Burden of Disease Study 2021. Lancet. 2025;405:785-812.
3. Dörr M. Big data and cardiovascular risk—insights into obesity, diabetes, and coronary heart disease. Herz. 2025;50:260-7.
5. Zhou X, Targher G, Byrne CD, Shapiro MD, Chen L, Zheng M. Metabolic dysfunction-associated fatty liver disease: bridging cardiology and hepatology. Cardiol Plus. 2024;9:275-82.
6. Zisis K, Athanasakis K. Obesity in childhood and adolescence: epidemiology and financial implications. Horm Res Paediatr. 2025;99:169-74.
8. Kent L, Mcgirr M, Eastwood K. Global trends in prevalence of maternal overweight and obesity: A systematic review and meta-analysis of routinely collected data retrospective cohorts. Int J Popul Data Sci. 2024;9:2401.
9. Wierzchowska-Opoka M, Domańska A, Masiarz A, Olbrot N, Oleszczuk A. Metabolic roles of fatty acid binding protein 4 (FABP4) in fetal and maternal health and maintenance of pregnancy in women with obesity: a review. Med Sci Monit. 2025;31:947679.
10. Wang L, O'kane AM, Zhang Y, Ren J. Maternal obesity and offspring health: adapting metabolic changes through autophagy and mitophagy. Obes Rev. 2023;24:e13567.
11. Armitage JA, Poston L, Taylor PD. Developmental origins of obesity and the metabolic syndrome: the role of maternal obesity. Front Horm Res. 2008;36:73-84.
12. Rubino F, Cummings DE, Eckel RH, et al. Definition and diagnostic criteria of clinical obesity. Lancet Diabetes Endocrinol. 2025;13:221-62.
13. De Oliveira MP, Da Silva LE, Fernandes BB, et al. The impact of obesity on mitochondrial dysfunction during pregnancy. Mol Cell Endocrinol. 2025;598:112463.
14. Zatterale F, Longo M, Naderi J, et al. Chronic adipose tissue inflammation linking obesity to insulin resistance and type 2 diabetes. Front Physiol. 2020;10:1607.
15. Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Investig. 2006;116:3015-25.
16. Reyna SM, Ghosh S, Tantiwong P, et al. Elevated toll-like receptor 4 expression and signaling in muscle from insulin-resistant subjects. Diabetes. 2008;57:2595-602.
17. Mhatre UA, Jindal I, Mojavery A, et al. The role of polyunsaturated fatty acids in neurodevelopmental pediatric outcomes in the settings of gestational diabetes and obesity: a comprehensive review. Neuroscience. 2025;579:227-38.
18. Guillaumin MCC, Peleg-Raibstein D. Maternal over- and malnutrition and increased risk for addictive and eating disorders in the offspring. Nutrients. 2023;15:51095.
19. Seneviratne SN, Rajindrajith S. Fetal programming of obesity and type 2 diabetes. World J Diabetes. 2022;13:482-97.
20. Peleg-Raibstein D. Understanding the link between maternal overnutrition, cardio-metabolic dysfunction and cognitive aging. Front Neurosci. 2021;15:645569.
21. Turdi S, Ge W, Hu N, Bradley KM, Wang X, Ren J. Interaction between maternal and postnatal high fat diet leads to a greater risk of myocardial dysfunction in offspring via enhanced lipotoxicity, IRS-1 serine phosphorylation and mitochondrial defects. J Mol Cell Cardiol. 2013;55:117-29.
22. Dong M, Zheng Q, Ford SP, Nathanielsz PW, Ren J. Maternal obesity, lipotoxicity and cardiovascular diseases in offspring. J Mol Cell Cardiol. 2013;55:111-6.
23. Wu L, Zhang Y, Wang G, Ren J. Molecular mechanisms and therapeutic targeting of ferroptosis in doxorubicin-induced cardiotoxicity. JACC Basic Transl Sci. 2024;9:811-26.
24. Ajoolabady A, Aslkhodapasandhokmabad H, Libby P, et al. Ferritinophagy and ferroptosis in the management of metabolic diseases. Trends Endocrinol Metab. 2021;32:444-62.
25. Larsen TD, Sabey KH, Knutson AJ, et al. Diabetic pregnancy and maternal high-fat diet impair mitochondrial dynamism in the developing fetal rat heart by sex-specific mechanisms. Int J Mol Sci. 2019;20:3090.
26. Yujiao C, Meng Z, Shanshan L, Wei W, Yipeng W, Chenghong Y. Exposure to bisphenol a induces abnormal fetal heart development by promoting ferroptosis. Ecotoxicol Environ Saf. 2023;255:114753.
27. Fisher AL, Sangkhae V, Balušíková K, Palaskas NJ, Ganz T, Nemeth E. Iron-dependent apoptosis causes embryotoxicity in inflamed and obese pregnancy. Nat Commun. 2021;12:4026.
28. Huang Y, Yan X, Zhao JX, et al. Maternal obesity induces fibrosis in fetal myocardium of sheep. Am J Physiol Endocrinol Metab. 2010;299:E968-75.
29. Wang Q, Zhu C, Sun M, et al. Maternal obesity impairs fetal cardiomyocyte contractile function in sheep. FASEB J. 2018;33:2587-98.
30. Fan X, Turdi S, Ford SP, et al. Influence of gestational overfeeding on cardiac morphometry and hypertrophic protein markers in fetal sheep. J Nutr Biochem. 2011;22:30-7.
31. Zhuang Z, Zhu Y, Tao J, et al. UCF101 rescues against diabetes-evoked cardiac remodeling and contractile anomalies through AMP-activated protein kinase-mediated induction of mitophagy. Pharmacology. 2024;110:127-40.
32. Yang L, Ma J, Tan Y, et al. Cardiac‐specific overexpression of metallothionein attenuates L‐NAME‐induced myocardial contractile anomalies and apoptosis. J Cell Mol Med. 2019;23:4640-52.
33. Wang J, Ma H, Tong C, et al. Overnutrition and maternal obesity in sheep pregnancy alter the JNK‐IRS‐1 signaling cascades and cardiac function in the fetal heart. FASEB J. 2010;24:2066-76.
34. Zhu M, Du M, Nathanielsz P, Ford S. Maternal obesity up-regulates inflammatory signaling pathways and enhances cytokine expression in the mid-gestation sheep placenta. Placenta. 2010;31:387-91.
35. Yan X, Zhu MJ, Xu W, et al. Up-regulation of toll-like receptor 4/nuclear factor-κB signaling is associated with enhanced adipogenesis and insulin resistance in fetal skeletal muscle of obese sheep at late gestation. Endocrinology. 2010;151:380-7.
36. Huang Y, Yan X, Zhu MJ, et al. Enhanced transforming growth factor-β signaling and fibrogenesis in ovine fetal skeletal muscle of obese dams at late gestation. Am J Physiol Endocrinol Metab. 2010;298:E1254-60.
37. Yan X, Huang Y, Wang H, et al. Maternal obesity induces sustained inflammation in both fetal and offspring large intestine of sheep. Inflamm Bowel Dis. 2011;17:1513-22.
38. Yan X, Huang Y, Zhao J, et al. Maternal obesity-impaired insulin signaling in sheep and induced lipid accumulation and fibrosis in skeletal muscle of offspring1. Biol Reprod. 2011;85:172-8.
39. Long NM, George LA, Uthlaut AB, et al. Maternal obesity and increased nutrient intake before and during gestation in the ewe results in altered growth, adiposity, and glucose tolerance in adult offspring1. J Anim Sci. 2010;88:3546-53.
40. Wallace J, Luther J, Milne J, et al. Nutritional modulation of adolescent pregnancy outcome - a review. Placenta. 2006;27:61-8.
41. Tallarek AC, Zeller T, Goßling A, et al. High‐sensitivity CRP is elevated in pregnant women with overweight and obesity and modulated by gestational weight gain. Acta Obstet Gynecol Scand. 2025;104:1339-46.
42. Loperfido F, Sottotetti F, Bianco I, et al. Folic acid supplementation in European women of reproductive age and during pregnancy with excessive weight: a systematic review. Reprod Health. 2025;22:13.
43. Poltorak A, He X, Smirnova I, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr Mice: mutations in Tlr4 gene. Science. 1998;282:2085-8.
44. Lee JY, Ye J, Gao Z, et al. Reciprocal modulation of toll-like receptor-4 signaling pathways involving MyD88 and phosphatidylinositol 3-kinase/AKT by saturated and polyunsaturated fatty acids. J Biol Chem. 2003;278:37041-51.
45. Esposito K, Marfella R, Giugliano D. Plasma interleukin-18 concentrations are elevated in type 2 diabetes. Diabetes Care. 2004;27:272.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0











Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].