


Senescence-driven pathogenesis in chronic lung diseases: from mechanistic insights to translational approaches
 0
0 Abstract
Chronic lung diseases (CLDs) include a variety of disorders of the pulmonary vasculature and alveolar compartments, and they have become a considerable global health challenge due to their high morbidity and limited therapeutic options. Increasing evidence shows that cellular senescence, an irreversible cell-cycle arrest accompanied by altered secretory activity and resistance to apoptosis, plays a crucial role in the onset and progression of CLDs. One of the major consequences of senescence-associated secretory phenotype activity in senescent cells is the further exacerbation of CLD pathogenesis through the maintenance of chronic inflammation, tissue remodeling, and structural vascular changes. Although individual CLDs exhibit distinct pathological features, they share common mechanisms, including persistent inflammation and dysregulated tissue repair. Experimental models and new methods have advanced, demonstrating the promise of targeted approaches, such as senolytics and regulators of senescence-associated traits, for treating these diseases. This review evaluates how cell senescence is involved with pulmonary arterial hypertension, pulmonary fibrosis and chronic obstructive pulmonary disease, highlights both shared and different pathologic routes, and discusses using seno-therapeutic methods within precision medicine plans.
Keywords
INTRODUCTION
Chronic lung diseases (CLDs) consist of a group of disorders including pulmonary arterial hypertension (PAH), pulmonary fibrosis (PF) and chronic obstructive pulmonary disease (COPD)[1]. These conditions have the characteristics of structural and functional disturbances of the pulmonary vasculature and/or alveolar compartments in different subtypes. Characteristic pathology includes persistent inflammation, endothelial dysfunction and abnormal vascular remodeling that cause ongoing pulmonary injury in the majority of cases[2,3]. Despite the different clinical and histopathologic features of individual CLD subtypes, they can also share common pathogenic mechanisms, such as chronic inflammation, capillary rarefaction, progressive vascular remodeling, and cellular senescence[4-7]. All of these possible mechanisms are associated with worse clinical outcomes. However, the exact molecular and cellular mechanisms triggering CLD initiation and development are not completely known.
Cellular senescence has gradually become recognized as an important factor in the development of CLDs, and increasing evidence supports its involvement. This irreversible cell-cycle arrest state is characterized by the senescence-associated secretory phenotype (SASP), which includes cytokines, chemokines, proteases, and growth regulators. The SASP factors can also induce cellular metabolism, enhance resistance to apoptosis and drive vascular remodeling and functional impairment[8,9]. Accumulation of senescent cells in pulmonary vasculature and parenchymal compartments due to hypoxia, mechanical stress, oxidative injury, and inflammation has been observed in multiple types of CLDs. However, the extent to which senescence-related mechanisms are shared with disease-specific mechanisms remains unclear, nor does the therapeutic potential of selectively targeting senescent cells. This review integrates recent progress in understanding the contribution of cellular senescence to various CLDs, highlights common and distinct molecular pathways, and discusses emerging technologies and strategies for regulating senescence in clinical applications.
CHARACTERISTICS AND BIOLOGY OF CELLULAR SENESCENCE
Cellular senescence refers to a state of stable, generally irreversible cell-cycle arrest, typically triggered by various cellular stresses. This process is predominantly regulated by the p16INK4a/Rb and p53/p21CIP1 signaling axes, which suppress cyclin-dependent kinases and thereby block cell cycle progression[10,11]. The concept was first recognized in the early 1960s, when Paul Moorhead and Leonard Hayflick described the finite replicative lifespan of normal human cells, later termed the Hayflick limit, and identified these replication-exhausted cells as senescent[12]. Initially associated with replicative exhaustion due to progressive telomere shortening, senescence can be seen as a broad biological response occurring in vivo, contributing to organismal aging and a plethora of diseases. Telomere attrition is not the only way to trigger senescence; various stimuli, such as DNA damage, oxidative stress, oncogene overexpression, mitochondrial dysfunction, and epigenetic changes, also cause senescence[10,13]. Initiating triggers lead to different major subtypes: (i) replicative senescence (RS): telomere-dependent cell-cycle arrest[14], (ii) stress-induced premature senescence (SIPS): due to telomere-independent stressors such as genotoxic damage, oxidative stress, and oncogenic signaling[14], (iii) developmentally programmed senescence: induced during morphogenesis by physiological signals[15], (iv) mitochondrial dysfunction-associated senescence (MiDAS): triggered by bioenergetic collapse and activated through p53-p21 signaling[16,17]; (v) specialized variants include cisplatin- and doxorubicin-induced senescence, chemotherapy-induced senescence (CIS), and paracrine senescence mediated by SASP-driven cytokine/growth factor release[18]. The senescent cells can be identified by a few hallmark features. The dominant biomarker of senescence is senescence-associated β-galactosidase
Another significant feature of senescence is SASP, which involves the release of proinflammatory cytokines, chemokines, growth factors, proteases, and extracellular vesicles[20]. SASP components reshape the tissue microenvironment by maintaining chronic inflammation, remodeling the composition of extracellular matrix (ECM), and inducing secondary senescence of neighboring cells, thereby accelerating tissue dysfunction and disease progression. Cellular senescence can serve as an initial tumor-suppressive mechanism; however, accumulating evidence suggests that it is also detrimental to degeneration of tissue and progression of chronic inflammation and age-related disease. In the context of CLDs, senescent cells promote pathological vascular remodeling and functional decline by maintaining chronic inflammatory signaling and impairing tissue repair. These processes position cellular senescence at the core of the pathogenic driver in the progression and development of CLDs [Figure 1].
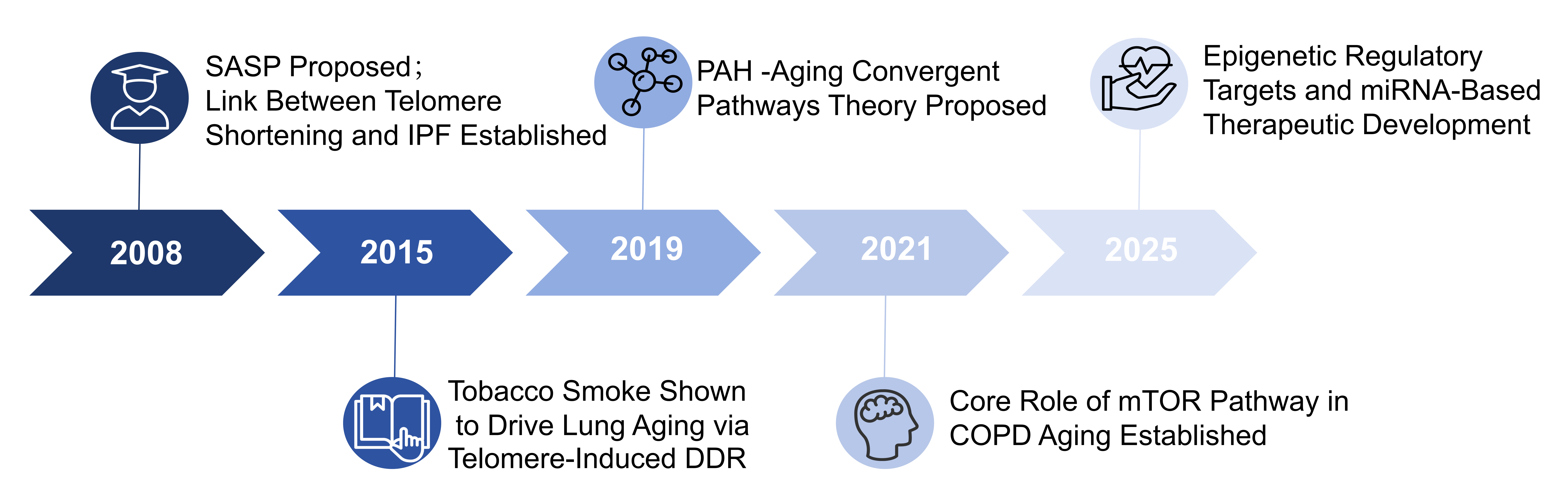
Figure 1. Timeline of key discoveries in aging and chronic lung diseases, with associated mechanisms. Major findings include the identification of telomere shortening and SASP in idiopathic pulmonary fibrosis (IPF) (2008); the role of tobacco smoke-induced DNA damage response (DDR) in lung aging (2019); the proposal of convergent pathways in pulmonary arterial hypertension (PAH) and aging (2015); the central role of mTOR in chronic obstructive pulmonary disease (COPD) (2021); and emerging directions in epigenetic- and miRNA-based therapeutics (2025). SASP: Senescence-associated secretory phenotype.
THE ROLE OF CELLULAR SENESCENCE IN PAH, PF AND COPD
As a basic biological reaction to chronic stress and injury, cellular senescence has been ascertained as a major pathological driver in various CLDs, such as PAH, PF and COPD. Under these conditions, cellular senescence serves as a common mechanism for promoting these pathological changes and maintaining the chronic proinflammatory environment during disease development[21,22].
Cellular senescence in PAH
PAH manifests through progressive vascular restructuring in distal pulmonary arteries, driven by endothelial dysfunction, smooth muscle hyperplasia, and persistent vascular inflammation. Senescence has been detected in various pulmonary vascular cell types, notably pulmonary artery endothelial cells (PAECs) and pulmonary artery smooth muscle cells (PASMCs)[7,23-25]. PAECs predominantly undergo MiDAS, whereas PASMCs primarily exhibit SIPS driven by oncogenic signaling. Additionally, right ventricular cardiomyocytes are susceptible to paracrine senescence propagated by SASP factors.
Senescent PAECs exhibit impaired proliferative capacity and barrier integrity, thereby promoting vascular remodeling and increased pulmonary vascular resistance. Mechanistically, this involves DDR activation [ataxia-telangiectasia mutated (ATM) kinase/ATM-Rad3-related kinase (ATR), γ-H2AX], p53/p21/p16 signaling, mitochondrial dysfunction, telomere erosion, and a pro-inflammatory SASP profile [e.g., interleukin (IL)-6, IL-1β, transforming growth factor-β (TGF-β)][24,26-29]. Specifically, van der Feen et al. used monocrotaline (MCT) plus shunt-induced PAH-associated congenital heart disease (PAH-CHD) rat models, together with tissues from patients with PAH-CHD, to show that endothelial cells are susceptible to shear stress-induced senescence. Notably, treatment with the senolytic agent ABT263 selectively eliminated senescent endothelial cells and reversed vascular lesions in rats with irreversible PAH[8]. In a related study, Wang et al. further confirmed, using MCT-induced PAH rat models and in vitro assays, that aldosterone promotes endothelial progenitor cell senescence by downregulating sirtuin 1 (SIRT1) and activating the p53/p21 pathway[27]. Notably, reduced expression of ATM has been specifically linked to aggravated endothelial senescence and disease progression in PAH[30]. In PASMCs, reactive oxygen species (ROS)-induced mitochondrial injury further drives both senescence and abnormal proliferation, exacerbating vascular remodeling[31]. Transcriptomic profiling of senescence-related gene (SRG) has identified distinct molecular subtypes of PAH, suggesting potential for precision-targeted therapies[32].
Cellular senescence and PF
Idiopathic pulmonary fibrosis (IPF) is a relentless and life-threatening interstitial lung disease characterized by dysregulated ECM deposition and irreversible distortion of alveolar structures. Accumulating evidence implicates cellular senescence in key cell populations, including alveolar type II (ATII) epithelial cells, PAECs, and fibroblasts/myofibroblasts[33-35]. Senescent ATII cells exhibit impaired regenerative capacity and adopt a pro-fibrotic SASP, activating fibroblasts and driving ECM remodeling[36]. Yao et al. demonstrated that p53-driven ATII cell senescence can initiate and sustain pulmonary fibrosis, as evidenced by single-cell RNA sequencing of IPF lung tissues and a conditional SIN3 transcription regulator homolog A (Sin3a) knockout mouse model targeting ATII cells. Importantly, this process was preventable through genetic or pharmacologic intervention[34]. Duckworth et al. employed Mendelian randomization analysis of large-scale biobank datasets and identified telomere shortening as a causal determinant of IPF[37]. Mechanistically, telomere dysfunction, oxidative stress, and chronic DNA damage contribute to this phenotype, with telomere shortening and pathogenic variants in telomerase pathway genes representing strong risk factors for IPF[37-39]. High levels of ROS can activate the NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome, which increases inflammation and promotes cell senescence[40,41]. Fibroblasts also exhibit mitochondrial dysfunction and impaired mitophagy, which boost ROS generation and prolong the senescent state. ATII epithelial cells and fibroblasts primarily undergo SIPS in pulmonary fibrosis, driven by oxidative stress, genotoxic stress, and inflammatory signaling. Interestingly, bone morphogenetic protein 4 can reverse fibroblast senescence, restore mitophagy, and reduce fibrosis progression[42].
Cellular senescence and COPD
The pathophysiology of COPD involves persistent airflow limitation, alveolar destruction, pulmonary vascular remodeling, and chronic inflammation[43]. Accelerated cellular senescence is a key feature of COPD pathophysiology, impacting airway and alveolar epithelial cells[44], microvascular endothelial cells (MVECs)[45], and airway smooth muscle cells. Cigarette smoke-induced oxidative stress is the main instigator for SIPS in airway epithelial cells that leads to DNA damage, mitochondrial dysfunction, telomere shortening and p16/p21 signaling[43,46,47]. Araya et al. demonstrated in both cellular and animal models that loss-of-function of parkin ring-between-ring (RBR) E3 ubiquitin protein ligase (PRKN) impairs mitophagy, resulting in mitochondrial dysfunction and accelerated senescence[46]. Similarly, Peng et al. found that the exposure to cadmium chloride promotes alveolar epithelial senescence via downregulation of sirtuin 3 (SIRT3) and mitochondrial dysfunction, effects that can be attenuated by overexpression of SIRT3 or nicotinamide adenine dinucleotide (NAD+) precursor[48]. Oxidative stress also compromises the nuclear factor erythroid 2-related factor 2 (Nrf2)/antioxidant response element (ARE) antioxidant defense system, thus increasing ROS accumulation and senescence[49]. Mitochondrial injury from cigarette smoke further compromises mitophagy, intensifying ROS generation and driving senescence in both epithelial and endothelial cells[46,48]. Senescent cells are abundant in COPD lungs - particularly within the airway epithelium, alveolar septa, and vascular walls - and their accumulation correlates with disease severity[9,50]. The secretion of pro-inflammatory cytokines and matrix metalloproteinases (MMPs) as key SASP factors underlies the role of these cells in perpetuating inflammation, tissue degradation, and vascular remodeling[51]. Regulators such as klotho and specific genetic variants have emerged as modulators of senescence and differentiation in COPD, offering promising therapeutic targets[52-54].
SHARED AND DISEASE-SPECIFIC MECHANISMS OF CELLULAR SENESCENCE IN CLDS
Although the contribution of cellular senescence to the progression of CLDs varies across anatomical regions and dominant cell types, multiple aging-related pathways are conserved across disease subtypes. Despite differences in clinical manifestations and histopathology, CLDs share common molecular characteristics, including telomere shortening, genomic instability, mitochondrial dysfunction, epigenetic alterations, disrupted proteostasis, dysregulated nutrient-sensing pathways, and impaired intercellular communication[55]. Collectively, these features accelerate damage to the pulmonary vasculature and parenchyma. These processes drive senescence of PAECs, PASMCs, and fibroblasts, and thus promote pathological remodeling and structural degeneration.
A central unifying feature of CLDs is the accumulation of senescent cells and a pronounced SASP[56-58]. With aging, the lung undergoes structural, immunological, and regenerative decline, making it more susceptible to chronic pulmonary disease[56]. Under the continuous stimulation of factors such as hypoxia, oxidative damage, mechanical load and environmental toxins, senescent cells can accumulate in the pulmonary vasculature and parenchyma. Through SASP-mediated secretion of proinflammatory cytokines, chemokines, MMPs, and growth factors, these cells exacerbate inflammation, promote ECM degradation and induce paracrine senescence of adjacent cells[57,58]. This inflammatory and proteolytic environment promotes continuous tissue damage and remodeling in PF, PAH, and COPD. Oxidative stress is also one of the common pathogenic drivers, especially in PF and COPD. Excessive ROS leads to continuous DNA damage, mitochondrial dysfunction, and accelerated aging. Telomere shortening, common across CLD subtypes, further destabilizes the genome and promotes RS in lung epithelial cells, endothelial cells (ECs), and mesenchymal cells[37,39]. Meanwhile, mitochondrial impairment aggravates ROS accumulation and disrupts cellular energy homeostasis, further reinforcing cell-cycle arrest and stabilizing the senescent phenotype. Together, these interconnected mechanisms establish a self-amplifying loop that further progressively undermines the structure and function of the lungs.
Although these mechanisms are common, CLDs exhibit disease-specific patterns of senescence under the influence of the dominant cell types, the nature of pathological changes, and initiating stimuli. In PF, senescence is dominant in ATII epithelial cells and pulmonary microvascular ECs[33,37,59,60]. This senescence drives fibroblast activation and excessive ECM deposition, ultimately causing irreversible fibrosis and architectural distortion. In PAH, senescent PAECs and PASMCs promote abnormal proliferation, vascular obstruction, and maladaptive remodeling of distal pulmonary arteries[24,26]. In COPD, the most affected are airway epithelial cells, vascular ECs, and alveolar epithelial cells, which cause chronic airway inflammation, emphysematous destruction, and secondary vascular rarefaction[61-63]. The initiating triggers also differ across diseases. In IPF, telomere maintenance defects and replicative exhaustion represent the dominant risks[37-39]. In COPD, cigarette smoke-induced oxidative stress and mitochondrial dysfunction trigger premature senescence[46,48]. In PAH, chronic hypoxia, altered shear stress, and mechanical strain promote endothelial dysfunction and senescence. These context-dependent trajectories indicate that, although the biology of senescence is conserved, its pathogenic manifestations vary depending on disease-specific microenvironments, cellular susceptibility, and upstream insults [Figure 2]. Understanding these differences will be essential for developing tailored anti-senescence strategies for each type of CLD.
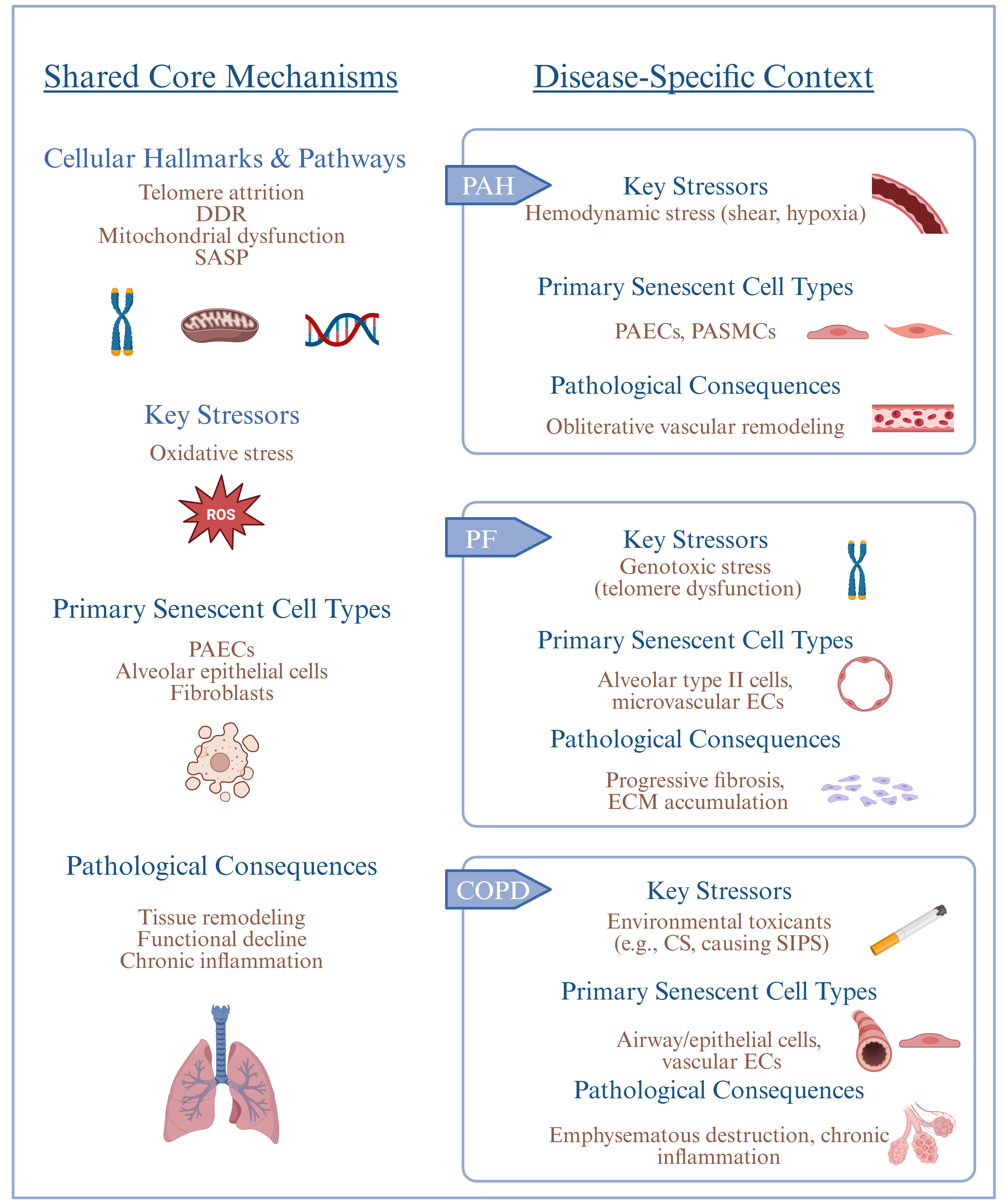
Figure 2. Shared and specific mechanisms of cellular senescence in chronic lung disease. Created in BioRender. Fan S (2025) https://BioRender.com/axpdt6p. PAH: Pulmonary arterial hypertension; IPF: idiopathic pulmonary fibrosis; COPD: chronic obstructive pulmonary disease; DDR: DNA damage response; SASP: senescence-associated secretory phenotype; PAECs: pulmonary arterial endothelial cells; PASMCs: pulmonary arterial smooth muscle cells; ECM: extracellular matrix; SIPS: stress-induced premature senescence; ECs: endothelial cells; CS: cigarette smoke.
THERAPEUTIC STRATEGIES TARGETING CELLULAR SENESCENCE
Given that cellular senescence is a key driver of chronic inflammation, pathological remodeling and vascular dysfunction in CLDs, targeting cellular senescence has become an attractive approach. Currently, there are three major types of senescence targeted therapies: (i) senolytics, which selectively kill senescent cells[64-66], (ii) senomorphics, which target and reduce the harmful effects of the SASP but do not kill senescent cells[67], and (iii) senopreventative approaches, which decrease oxidative stress, DNA damage, or mitochondrial dysfunction[68-70]. Preclinical studies show that these modalities can dampen pathological remodeling and restore organ function in age-related lung disorders. Mechanistically, therapeutic efforts have focused on modulating pathways that are key to the initiation and persistence of senescence, such as p53/p21 and p16/Rb signaling governing cell-cycle arrest, as well as nuclear factor kappa B (NF-κB) and Nrf2 pathways regulating inflammation and oxidative stress[71-73]. Targeting particular factors of the SASP such as IL-6, IL-1β, and TGF-β may offer an avenue to dampen signaling cascades that fuel inflammation and fibrosis.
Despite many advances, significant barriers remain to the clinical application of anti-senescence therapies. Senescent cell populations are heterogeneous and context-dependent, making it challenging to develop broadly effective treatments. The specificity of senolytics is also a concern, as off-target effects may harm non-senescent cells. Optimal timing, dosage, and treatment duration remain unclear, and effective in vivo biomarkers for detecting senescence and monitoring therapy are lacking. Consequently, there is an urgent need to develop noninvasive, organ-level technologies to assess senescence. Currently, highly specific probes are being developed, such as the fluorogenic probe PGal-FA, which operates in stages by sequentially activating in response to senescence markers β-galactosidase (β-Gal) and formaldehyde (FA). This probe shows good selectivity for detecting senescent cells in bleomycin-induced PF model tissue[74], and it is expected to be an extensively applicable tool for diagnosis and treatment monitoring. At the same time, targeting specific SASP signaling pathways offers an alternative therapeutic approach. For example, in radiation-induced lung injury, inhibition of the epithelial-derived C-C motif chemokine ligand 2 (CCL2), an SASP mediator, preserves the function of pulmonary endothelial cells, reduces the acute inflammatory response, and slows the progression of fibrosis[75]. These results suggest that pathway-specific SASP modulation could protect the pulmonary vasculature and improve disease outcomes. Overcoming these challenges will be essential to fully realize the potential of senescence-focused therapies and integrate them into the clinical management of CLDs.
TRANSLATIONAL ADVANCES LINKING SENESCENCE MECHANISMS AND THERAPIES IN CLDS AND CURRENT CHALLENGES
Advances in new technologies, including single-cell RNA sequencing (scRNA-seq) and precision-cut lung slices techniques, have deepened the research on cellular senescence in CLDs, yet they also face preclinical challenges.
Novel insights from single-cell transcriptomics in PF and COPD
Emerging scRNA-seq studies are providing unprecedented insight into cellular heterogeneity and intercellular communication networks underlying senescence in PF and COPD, revealing previously unrecognized therapeutic targets.
In IPF, scRNA-seq has refined the traditional view that senescence of ATII epithelial cells is a central pathological event, revealing a high degree of cell type specificity. Increasing evidence indicates that aging disrupts communication between cells in both the vascular and alveolar compartments, leading to impaired repair and prolonged fibrosis. At the vascular interface, analyses of young and aged mouse lungs show that atypical chemokine receptor 1 (Ackr1+) venous ECs and tropomyosin receptor kinase B (TrkB+) capillary ECs in aged lungs fail to return to a quiescent state after injury. Instead, they remain in a persistently active state due to stimulation of Yes-associated protein (YAP)/transcriptional coactivator with PDZ-binding motif (TAZ) signaling, thereby disrupting vascular homeostasis and potentially influencing alveolar repair through altered secretory programs[76]. Meanwhile, aging enhances pathological epithelial-mesenchymal interactions in the alveolar niche. Genetic models of PF demonstrate that dysfunctional ATII epithelial cells drive fibroblasts toward a pro-inflammatory and pro-fibrotic state, accompanied by an age-related increase in human serum amyloid A3 (SAA3+) inflammatory fibroblasts.
Notably, these fibroblasts also express high levels of IL-1R1, making them more sensitive to IL-1 and creating a self-reinforcing loop between abnormal epithelial cells and activated fibroblasts[77]. Aging not only impairs the normal function of endothelial and epithelial cells but also disrupts their coordinated communication. Failure to resolve repair signals after injury shifts the tissue response toward persistent inflammation, abnormal matrix deposition, and progressive fibrosis.
Single-cell transcriptomics have also characterized the multicellular senescence networks driving COPD. In the airway epithelium, scRNA-seq reveals elevated senescence markers in basal and club cells and identifies upstream regulatory mechanisms. Loss of the anti-aging protein klotho promotes senescence and abnormal differentiation of club cells via the neddylation-heterogeneous nuclear ribonucleoprotein A2/B1 (hnRNPA2/B1) axis[52]. In parallel, aberrant activation of type I and II interferon signaling has emerged as a major driver of epithelial senescence. This activation increases the SASP and locks cells into a senescent state via the Janus kinase (JAK) /signal transducers and activators of transcription (STAT) and cyclic guanosine monophosphate - adenosine monophosphate (GMP-AMP) synthase (cGAS) - stimulator of interferon genes (STING) pathways. Importantly, this process can be effectively suppressed by pharmacologic intervention with baricitinib[78]. At the level of pulmonary vasculature, deficiency of protein arginine methyltransferase 1 (PRMT1) causes hyperactivation of NF-κB signaling in ECs, promoting endothelial dysfunction and senescence, and aggravating vascular pathology in COPD[79]. Together, these findings reveal a multicellular and multi-pathway senescence network in COPD: senescence of epithelial cells caused by both intrinsic klotho deficiency and extrinsic interferon signaling reduces airway barrier integrity and repair; EC senescence due to PRMT1-NF-κB dysregulation impairs vascular homeostasis. Through cell-cell communication, SASP production, and interaction with the immune microenvironment, these senescence programs converge to generate chronic, self-sustaining inflammation and tissue destruction in COPD. Targeting these specific cell types and their key pathways (hnRNPA2/B1, interferon signaling, PRMT1, etc.) offers new ideas for the development of precise anti-senescence therapies.
Recent preclinical advances in targeting senescence and remodeling in PAH
In addition to classical senolytics, recent preclinical studies have revealed new strategies that directly regulate the senescence program or target the downstream effects of senescence-induced vascular remodeling in PAH.
Cellular senescence and aberrant proliferative phenotypes in PAH pathophysiology have prompted investigation into therapeutic strategies that simultaneously target both processes. Inhibition of Aurora kinase B (AURKB) can stop the cell cycle and reverse the disease-specific transcriptional signature in PASMCs of PAH. Importantly, AURKB inhibition induces SASP in apoptosis-resistant PAH-PASMCs. In animal models, pharmacologic inhibition of AURKB by barasertib improves hemodynamics and attenuates pulmonary vascular remodeling. Notably, the combination of barasertib with a p21 attenuator has a synergistic effect on remodeling attenuation, which is better than either drug alone, suggesting a new combination strategy of manipulating senescence pathways for the treatment of PAH[80].
Significant progress has also been made in therapies that do not directly target senescence but effectively reverse remodeling. Sotatercept acts by targeting and restoring the imbalance between TGF-β and bone morphogenetic protein pathway signaling[81]. Recently, integrin α5β1 has been identified as a key pathogenic driver. Its expression is elevated in vascular cells of patients with PAH, and α5β1 blockade suppresses forkhead box protein M1 (FOXM1)-regulated gene networks, reversing the proliferative and anti-apoptotic phenotype. In various preclinical models and ex vivo precision-cut human lung slices, targeted inhibition of α5β1 safely mitigates pulmonary vascular remodeling, enhances hemodynamics and right ventricular performance, and shows efficacy comparable to - or even exceeding - that of standard therapies and Sotatercept[82]. These findings highlight a diversification of PAH therapeutic strategies, including direct targeting of the cellular senescence cycle, rebalancing signaling pathways, and inhibiting cell - matrix interactions.
Established senotherapeutic strategies and translational challenges
Among senotherapeutic approaches for CLDs, dasatinib and/or quercetin remain the most extensively studied senolytic regimen, showing efficacy in models of PAH, PF, and COPD[64,65,83]. However, some studies have suggested that quercetin may paradoxically induce PAH under certain conditions[84]. In PF, Bcl-2-associated X protein (BAX) trigger site activator 1 (BTSA1) has emerged as a novel agent that selectively activates BAX to inhibit fibroblast senescence[85]. Shen et al. used senescent myofibroblast cultures and bleomycin-induced PF mouse models to characterize BTSA1 as a novel senolytic that induces BAX-dependent apoptosis specifically in senescent cells[85]. Zhuo et al. employed primary lung fibroblasts of bleomycin-induced PF model and A549 epithelial cells to show that indole-3-acetic acid alleviates fibrosis by regulating lung microbiota and inhibiting cellular senescence[86].
Building on cellular senescence theories, multiple small-molecular drugs and bioactive natural compounds are under clinical investigation. In PF, agents such as indole-3-acetic acid, YAP activators, honokiol, and pentoxifylline have been shown in preclinical studies to mitigate epithelial cellular senescence and suppress fibrotic remodeling[86-89]. Senolytics such as valproic acid (VPA) and mitochondria-targeted antioxidants (MTAs) provide enhanced specificity by directly alleviating mitochondrial oxidative stress, a key upstream driver of senescence[90,91]. In COPD, therapeutic strategies targeting SIRT1 and PRMT1 are being evaluated for their ability to modulate oxidative injury, inflammation, and senescence-related signaling[79,92-96]. Tran et al. used endothelial-specific PRMT1 knockout mice, COPD models, and in vitro EC assays to demonstrate that loss of PRMT1 aggravates COPD pathology through NF-κB-mediated senescence and endothelial dysfunction[79]. In PAH, interventions aimed at suppressing PASMC senescence and limiting SASP-driven vascular remodeling are under active development[80,97,98]. Wang et al. showed in hypoxia-induced PH models and cultured PASMCs that senescent PASMCs promote the proliferation of neighboring cells via IL-6-dependent paracrine signaling[97]. Similarly, Lemay et al. combined transcriptomic analysis of patient-derived PASMCs, preclinical PAH models and human precision-cut lung slices to validate AURKB inhibition as a therapeutic strategy[80]. Recent findings also highlight lipid metabolism pathways[99,100] and specific microRNAs[101], including miR-93-5p[102], as potential regulators of senescence, representing promising molecular targets for future therapy.
Although preclinical studies show promise, significant barriers remain for clinical translation. First, there is a lack of CLD-specific senolytics that are highly selective and exhibit low off-target toxicity. Second, the heterogeneity and plasticity of senescent cell populations across different lung regions make it difficult to identify common drug targets. Third, systemic clearance of senescent cells may disrupt their context-dependent protective functions, such as tissue repair, raising safety concerns. Finally, few reliable, noninvasive biomarkers exist to detect senescence in lung tissue or circulation, complicating patient stratification and real-time monitoring of treatment efficacy.
To overcome these limitations, future senotherapeutic strategies should be combined with existing antifibrotic, anti-inflammatory, or vasodilatory treatments to achieve synergistic effects. Concurrent advances in gene therapy and targeted drug delivery systems may enhance precision, reduce side effects, and broaden the clinical applicability of these approaches in CLDs management[103-105]. For example, Sun et al. demonstrated in a progeria mouse model (LmnaG609G) that endothelial-targeted sirtuin-7 (Sirt7) gene therapy improves vascular dysfunction and extends lifespan[103]. Likewise, Lu et al. developed an inhalable gene-editable nanoplatform that has been validated in lung-on-chip models and IPF mice. This platform comprises a clustered regularly interspaced short palindromic repeat (CRISPR) - CRISPR-associated protein 9 (Cas9) system linked to a core FePt diatomic catalyst and coated with a biocompatible hyaluronic acid (HA) layer, combining ROS scavenging with lysine acetyltransferase 7 (KAT7) gene editing to reverse ATII cell senescence[105]. Moving forward, the design of clinical trials should therefore focus on addressing the challenges of specificity, long-term safety and durable response to fully unleash the potential of senotherapies.
TOWARD PRECISION SENOTHERAPY IN CLDS
The concept of precision senotherapy in CLDs is gaining popularity as the mechanism of cellular senescence becomes gradually clearer. New evidence shows that different molecular profiles can be used to define different disease subtypes and to guide treatment. For instance, miR-125a-5p has been found to be one of the important factors influencing the senescence of lung epithelial cells, and its expression level may be a biomarker to identify a subgroup of COPD patients for whom miR-125a-5p inhibitors would be particularly effective[106]. In addition to molecular signatures, organelle-specific dysfunction is also an important part of senescence biology that can be targeted for intervention. Disruption of mitochondria-endoplasmic reticulum crosstalk is now recognized as a major driver of oxidative stress and cellular senescence, providing a rationale for interventions targeting specific pathways, such as mitochondria-targeted antioxidants[107]. Senescence has significant effects on the pulmonary vasculature. Recent studies suggest that inhaled corticosteroids (ICS) protect endothelial progenitor cells from senescence, increasing the likelihood that COPD patients with severe endothelial dysfunction represent a treatment-responsive subgroup[108].
Future precision medicine strategies have the potential to refine molecular endotyping of CLD patients by combining multiple biomarkers, including miRNA profiles, indicators of organellar function, and markers of endothelial senescence. This approach could enable patient subgroups to be matched with the most effective targeted therapies, such as miRNA inhibitors, organelle-stabilizing agents, or vascular-protective anti-inflammatory treatments. The overarching goal of precision senotherapy is to protect the pulmonary vasculature and improve clinical outcomes.
CONCLUSION
Cellular senescence has emerged as a fundamental pathological mechanism in the initiation and progression of CLDs. Through the release of SASP components, senescent cells perpetuate chronic inflammation, disrupt tissue homeostasis, and drive maladaptive remodeling of pulmonary structures. Although CLDs share core senescence-associated pathways, the dominant senescent cell populations, upstream triggers, and downstream effects vary across disease contexts. Therapeutically, targeting senescence - using senolytics, senomorphics, or agents that modulate oxidative stress, DDR, or mitochondrial function - offers a strategy to address the root causes of disease rather than merely treating symptoms. Clinical translation, however, requires: (i) accurate identification of senescent cell subsets in each CLD; (ii) development of selective, potent compounds with minimal off-target effects; and (iii) identification of robust, noninvasive biomarkers for diagnosis, prognosis, and treatment monitoring. Looking forward, well-designed clinical trials are essential to establish the safety and efficacy of senescence-targeted therapies across different CLD subtypes. Moreover, preventive strategies targeting senescence may hold substantial promise for disease prevention. Collectively, these efforts could shift the management paradigm from downstream damage control to upstream disease modification, with the potential to halt - or even reverse - disease progression by addressing its root causes.
DECLARATIONS
Acknowledgment
The Graphical Abstract was created with BioRender.com (Created in BioRender. Fan S. (2025) https://BioRender.com/f70vcef).
Authors’ contributions
Writing - original draft and figure preparation: Liu SF, Fan SC, Wang RQ, Wang XJ, Lu TY, Song JW, Zhu YJ
Supervision, writing - review and editing: Guo Z, Zhu N, Yuan P
Availability of data and materials
Not applicable.
Financial support and sponsorship
This work was supported by the Natural Science Foundation of Shanghai (S.L., 25ZR1401394; P.Y., 22ZR1452400), the Key Research and Development Projects in Hainan Province (P.Y., ZDYF2024SHFZ057), the Joint Research and Development Project between Shenkang and United Imaging on Clinical Research and Translation (P.Y., SKLY2022CRT202), and the National Natural Science Foundation of China (P.Y., 82370057; N.Z., 82570414).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
Statement of the use of AI tools
Generative AI tools (e.g., ChatGPT and DeepL) were used in a limited manner during manuscript preparation to improve readability and clarity. All conceptual development and final content decisions were made independently by the authors. The final manuscript was thoroughly reviewed and edited by the authors, who assume full responsibility for its content.
REFERENCES
1. Elia D, Caminati A, Zompatori M, et al. Pulmonary hypertension and chronic lung disease: where are we headed? Eur Respir Rev. 2019;28:190065.
2. Ackermann M, Werlein C, Plucinski E, et al. The role of vasculature and angiogenesis in respiratory diseases. Angiogenesis. 2024;27:293-310.
3. Kolb TM, Hassoun PM. Right ventricular dysfunction in chronic lung disease. Cardiol Clin. 2012;30:243-56.
5. Moss BJ, Ryter SW, Rosas IO. Pathogenic mechanisms underlying idiopathic pulmonary fibrosis. Annu Rev Pathol. 2022;17:515-46.
6. Borek I, Birnhuber A, Voelkel NF, Marsh LM, Kwapiszewska G. The vascular perspective on acute and chronic lung disease. J Clin Invest. 2023:133.
7. van der Feen DE, Berger RMF, Bartelds B. Converging paths of pulmonary arterial hypertension and cellular senescence. Am J Respir Cell Mol Biol. 2019;61:11-20.
8. van der Feen DE, Bossers GPL, Hagdorn QAJ, et al. Cellular senescence impairs the reversibility of pulmonary arterial hypertension. Sci Transl Med. 2020;12:eaaw4974.
9. Adnot S, Amsellem V, Boyer L, et al. Telomere dysfunction and cell senescence in chronic lung diseases: therapeutic potential. Pharmacol Ther. 2015;153:125-34.
10. Sikora E, Arendt T, Bennett M, Narita M. Impact of cellular senescence signature on ageing research. Ageing Res Rev. 2011;10:146-52.
11. Uyar B, Palmer D, Kowald A, et al. Single-cell analyses of aging, inflammation and senescence. Ageing Res Rev. 2020;64:101156.
12. Zhu Y, Anastasiadis ZP, Espindola Netto JM, Evans T, Tchkonia T, Kirkland JL. Past and future directions for research on cellular senescence. Cold Spring Harb Perspect Med. 2024;14:a041205.
13. Zhu Y, Liu X, Ding X, Wang F, Geng X. Telomere and its role in the aging pathways: telomere shortening, cell senescence and mitochondria dysfunction. Biogerontology. 2019;20:1-16.
14. Mohamad Kamal NS, Safuan S, Shamsuddin S, Foroozandeh P. Aging of the cells: insight into cellular senescence and detection Methods. Eur J Cell Biol. 2020;99:151108.
15. Muñoz-Espín D, Cañamero M, Maraver A, et al. Programmed cell senescence during mammalian embryonic development. Cell. 2013;155:1104-18.
16. Wiley CD, Velarde MC, Lecot P, et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 2016;23:303-14.
17. Sizek H, Deritei D, Fleig K, et al. Unlocking mitochondrial dysfunction-associated senescence (MiDAS) with NAD+ - A Boolean model of mitochondrial dynamics and cell cycle control. Transl Oncol. 2024;49:102084.
18. Acosta JC, Banito A, Wuestefeld T, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. 2013;15:978-90.
19. Corpet A, Stucki M. Chromatin maintenance and dynamics in senescence: a spotlight on SAHF formation and the epigenome of senescent cells. Chromosoma. 2014;123:423-36.
20. Zhang L, Pitcher LE, Yousefzadeh MJ, Niedernhofer LJ, Robbins PD, Zhu Y. Cellular senescence: a key therapeutic target in aging and diseases. J Clin Invest. 2022:132.
21. Barnes PJ, Baker J, Donnelly LE. Cellular Senescence as a Mechanism and Target in Chronic Lung Diseases. Am J Respir Crit Care Med. 2019;200:556-64.
22. Schneider JL, Rowe JH, Garcia-de-Alba C, Kim CF, Sharpe AH, Haigis MC. The aging lung: physiology, disease, and immunity. Cell. 2021;184:1990-2019.
23. Lawrie A, Francis SE. Frataxin and endothelial cell senescence in pulmonary hypertension. J Clin Invest. 2021;131::e149721.
24. Liu L, Wei Y, Giunta S, He Q, Xia S. Potential role of cellular senescence in pulmonary arterial hypertension. Clin Exp Pharmacol Physiol. 2022;49:1042-9.
25. Stenmark KR, Frid MG, Graham BB, Tuder RM. Dynamic and diverse changes in the functional properties of vascular smooth muscle cells in pulmonary hypertension. Cardiovasc Res. 2018;114:551-64.
26. Qamsari E, Stewart DJ. Cellular senescence in the pathogenesis of pulmonary arterial hypertension: the good, the bad and the uncertain. Front Immunol. 2024;15:1403669.
27. Wang Y, Zhong B, Wu Q, Tong J, Zhu T, Zhang M. Effect of aldosterone on senescence and proliferation inhibition of endothelial progenitor cells induced by Sirtuin 1 (SIRT1) in pulmonary arterial hypertension. Med Sci Monit. 2020;26:e920678.
28. Noureddine H, Gary-Bobo G, Alifano M, et al. Pulmonary artery smooth muscle cell senescence is a pathogenic mechanism for pulmonary hypertension in chronic lung disease. Circ Res. 2011;109:543-53.
29. Miwa S, Kashyap S, Chini E, von Zglinicki T. Mitochondrial dysfunction in cell senescence and aging. J Clin Invest. 2022;132:e158447.
30. Zhan H, Suzuki T, Aizawa K, Miyagawa K, Nagai R. Ataxia telangiectasia mutated (ATM)-mediated DNA damage response in oxidative stress-induced vascular endothelial cell senescence. J Biol Chem. 2010;285:29662-70.
31. Zhang L, Ma C, Zhang C, et al. Reactive oxygen species effect PASMCs apoptosis via regulation of dynamin-related protein 1 in hypoxic pulmonary hypertension. Histochem Cell Biol. 2016;146:71-84.
32. Meng ZY, Lu CH, Li J, et al. Identification and experimental verification of senescence-related gene signatures and molecular subtypes in idiopathic pulmonary arterial hypertension. Sci Rep. 2024;14:22157.
33. Venosa A. Senescence in pulmonary fibrosis: between aging and exposure. Front Med. 2020;7:606462.
34. Yao C, Guan X, Carraro G, et al. Senescence of alveolar type 2 cells drives progressive pulmonary fibrosis. Am J Respir Crit Care Med. 2021;203:707-17.
35. Parimon T, Hohmann MS, Yao C. Cellular senescence: pathogenic mechanisms in lung fibrosis. Int J Mol Sci. 2021;22:6214.
36. Wan R, Wang L, Zhu M, Li W, Duan Y, Yu G. Cellular senescence: a troy horse in pulmonary fibrosis. Int J Mol Sci. 2023;24:16410.
37. Duckworth A, Gibbons MA, Allen RJ, et al. Telomere length and risk of idiopathic pulmonary fibrosis and chronic obstructive pulmonary disease: a mendelian randomisation study. Lancet Respir Med. 2021;9:285-94.
39. Snetselaar R, van Batenburg AA, van Oosterhout MFM, et al. Short telomere length in IPF lung associates with fibrotic lesions and predicts survival. PLoS One. 2017;12:e0189467.
40. Feng J, Liu H, Jiang K, et al. Enhanced oxidative stress aggravates BLM-induced pulmonary fibrosis by promoting cellular senescence through enhancing NLRP3 activation. Life Sci. 2024;358:123128.
41. Yin Y, Zhou Z, Liu W, Chang Q, Sun G, Dai Y. Vascular endothelial cells senescence is associated with NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome activation via reactive oxygen species (ROS)/thioredoxin-interacting protein (TXNIP) pathway. Int J Biochem Cell Biol. 2017;84:22-34.
42. Guan R, Yuan L, Li J, et al. Bone morphogenetic protein 4 inhibits pulmonary fibrosis by modulating cellular senescence and mitophagy in lung fibroblasts. Eur Respir J. 2022;60:2102307.
43. Easter M, Bollenbecker S, Barnes JW, Krick S. Targeting aging pathways in chronic obstructive pulmonary disease. Int J Mol Sci. 2020;21:6924.
44. Zhang Y, Huang W, Zheng Z, et al. Cigarette smoke-inactivated SIRT1 promotes autophagy-dependent senescence of alveolar epithelial type 2 cells to induce pulmonary fibrosis. Free Radic Biol Med. 2021;166:116-27.
45. Cheng PP, Yu F, Chen SJ, et al. PM2.5 exposure-induced senescence-associated secretory phenotype in airway smooth muscle cells contributes to airway remodeling. Environ Pollut. 2024;347:123674.
46. Araya J, Tsubouchi K, Sato N, et al. PRKN-regulated mitophagy and cellular senescence during COPD pathogenesis. Autophagy. 2019;15:510-26.
47. Savale L, Chaouat A, Bastuji-Garin S, et al. Shortened telomeres in circulating leukocytes of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2009;179:566-71.
48. Peng K, Yao YX, Lu X, et al. Mitochondrial dysfunction-associated alveolar epithelial senescence is involved in CdCl2-induced COPD-like lung injury. J Hazard Mater. 2024;476:135103.
49. Kominkova E, Petrek M, Navratilova Z. Protective factors against oxidative stress in COPD: focus on Nrf2-dependent antioxidant gene expression. Front Med. 2025;12:1492256.
50. Barnes PJ. Targeting cellular senescence as a new approach to chronic obstructive pulmonary disease therapy. Curr Opin Pharmacol. 2021;56:68-73.
51. Tao L, Lu X, Fu Z, et al. Tong Sai granule improves AECOPD via regulation of MAPK-SIRT1-NF-κB pathway and cellular senescence alleviation. J Ethnopharmacol. 2023;314:116622.
52. Li M, Chen B, Sun S, Wang K, Wang Y, Wu J. Klotho regulates club cell senescence and differentiation in chronic obstructive pulmonary disease. Cell Prolif. 2025;58:e70000.
53. da Silva CO, de Souza Nogueira J, do Nascimento AP, et al. COPD patients exhibit distinct gene expression, accelerated cellular aging, and bias to M2 macrophages. Int J Mol Sci. 2023;24:9913.
54. Luo X, Zeng W, Tang J, et al. Multi-modal transcriptomic analysis reveals metabolic dysregulation and immune responses in chronic obstructive pulmonary disease. Sci Rep. 2024;14:22699.
55. Budde J, Skloot G. Aging and susceptibility to pulmonary disease. Compr Physiol. 2022;12:3509-22.
57. Aghali A, Koloko Ngassie ML, Pabelick CM, Prakash YS. Cellular senescence in aging lungs and diseases. Cells. 2022;11:1781.
58. Birch J, Barnes PJ, Passos JF. Mitochondria, telomeres and cell senescence: Implications for lung ageing and disease. Pharmacol Ther. 2018;183:34-49.
59. Zhou S, Zhu J, Zhou PK, Gu Y. Alveolar type 2 epithelial cell senescence and radiation-induced pulmonary fibrosis. Front Cell Dev Biol. 2022;10:999600.
60. Schuliga M, Kanwal A, Read J, et al. A cGAS-dependent response links DNA damage and senescence in alveolar epithelial cells: a potential drug target in IPF. Am J Physiol Lung Cell Mol Physiol. 2021;321:L859-71.
61. Bateman G, Guo-Parke H, Rodgers AM, et al. Airway epithelium senescence as a driving mechanism in COPD pathogenesis. Biomedicines. 2023;11:2072.
62. Chilosi M, Carloni A, Rossi A, Poletti V. Premature lung aging and cellular senescence in the pathogenesis of idiopathic pulmonary fibrosis and COPD/emphysema. Transl Res. 2013;162:156-73.
63. Tuder RM, Yoshida T, Arap W, Pasqualini R, Petrache I. State of the art. Cellular and molecular mechanisms of alveolar destruction in emphysema: an evolutionary perspective. Proc Am Thorac Soc. 2006;3:503-10.
64. Nambiar A, Kellogg D 3rd, Justice J, et al. Senolytics dasatinib and quercetin in idiopathic pulmonary fibrosis: results of a phase I, single-blind, single-center, randomized, placebo-controlled pilot trial on feasibility and tolerability. EBioMedicine. 2023;90:104481.
65. Bloom SI, Tuday E, Islam T, Gogulamudi VR, Lesniewski LA, Donato AJ. Senolytics reduce endothelial cell DNA damage and telomere dysfunction despite reductions in telomere length. Aging Biol. 2023;1:20230007.
66. Baker JR, Daly L, Hassibi S, et al. Senolytic therapy reduces inflammation in epithelial cells from COPD patients and in smoke-exposure mice. Front Med. 2025;12:1451056.
67. Schafer MJ, Haak AJ, Tschumperlin DJ, LeBrasseur NK. Targeting senescent cells in fibrosis: pathology, paradox, and practical considerations. Curr Rheumatol Rep. 2018;20:3.
68. Roth-Walter F, Adcock IM, Benito-Villalvilla C, et al. Metabolic pathways in immune senescence and inflammaging: Novel therapeutic strategy for chronic inflammatory lung diseases. An EAACI position paper from the Task Force for Immunopharmacology. Allergy. 2024;79:1089-122.
69. Barnes PJ. Senotherapy for lung diseases. In: Fred Wong WS, Editor. Advances in Pharmacology. Elsevier; 2023. pp. 249-71.
70. Baker JR, Donnelly LE, Barnes PJ. Senotherapy: a new horizon for COPD therapy. Chest. 2020;158:562-70.
71. Mouraret N, Marcos E, Abid S, et al. Activation of lung p53 by Nutlin-3a prevents and reverses experimental pulmonary hypertension. Circulation. 2013;127:1664-76.
72. Gao S, Chen L, Lin Z, et al. 8-Oxoguanine DNA glycosylase protects cells from senescence via the p53-p21 pathway. Acta Biochim Biophys Sin. 2024;56:184-98.
73. You B, Liu Y, Chen J, et al. Vascular peroxidase 1 mediates hypoxia-induced pulmonary artery smooth muscle cell proliferation, apoptosis resistance and migration. Cardiovasc Res. 2018;114:188-99.
74. Zhou L, Zhang X, Dong Y, et al. A Tandemly activated fluorescence probe for detecting senescent cells with improved selectivity by targeting a biomarker combination. ACS Sens. 2022;7:1958-66.
75. Wiesemann A, Ketteler J, Slama A, et al. Inhibition of radiation-induced Ccl2 signaling protects lungs from vascular dysfunction and endothelial cell loss. Antioxid Redox Signal. 2019;30:213-31.
76. Raslan AA, Pham TX, Lee J, et al. Lung injury-induced activated endothelial cell states persist in aging-associated progressive fibrosis. Nat Commun. 2024;15:5449.
77. Banaschewski BJH, Michki SN, Sitaraman S, et al. Emergence of inflammatory fibroblasts with aging in Hermansky-Pudlak syndrome associated pulmonary fibrosis. Commun Biol. 2025;8:284.
78. Guo-Parke H, Cappa O, Linden DA, et al. IFN-mediated bronchial epithelium cellular senescence in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2025;73:871-83.
79. Tran TTV, Jeong Y, Kim S, et al. PRMT1 ablation in endothelial cells causes endothelial dysfunction and aggravates COPD attributable to dysregulated NF-κB signaling. Adv Sci. 2025;12:e2411514.
80. Lemay SE, Mougin M, Sauvaget M, et al. Unraveling AURKB as a potential therapeutic target in pulmonary hypertension using integrated transcriptomic analysis and pre-clinical studies. Cell Rep Med. 2025;6:101964.
81. Fujiwara T, Ishii S, Minatsuki S, Hatano M, Takeda N. Exploring novel therapeutics for pulmonary arterial hypertension: from the bench to the bedside. Int Heart J. 2025;66:3-12.
82. Lemay SE, Montesinos MS, Grobs Y, et al. Exploring integrin α5β1 as a potential therapeutic target for pulmonary arterial hypertension: insights from comprehensive multicenter preclinical studies. Circulation. 2025;151:1162-83.
83. He Y, Cao X, Liu X, et al. Quercetin reverses experimental pulmonary arterial hypertension by modulating the TrkA pathway. Exp Cell Res. 2015;339:122-34.
84. Guignabert C, Phan C, Seferian A, et al. Dasatinib induces lung vascular toxicity and predisposes to pulmonary hypertension. J Clin Invest. 2016;126:3207-18.
85. Shen M, Fu J, Zhang Y, et al. A novel senolytic drug for pulmonary fibrosis: BTSA1 targets apoptosis of senescent myofibroblasts by activating BAX. Aging Cell. 2024;23:e14229.
86. Zhuo J, Liu D, Yu Q, et al. Indole-3-acetic acid attenuates pulmonary fibrosis by modulating lung microbiota, inhibiting fibroblast activation, and alleviating alveolar epithelial cell senescence. Life Sci. 2024;359:123191.
87. Su W, Guo Y, Wang Q, et al. YAP1 inhibits the senescence of alveolar epithelial cells by targeting Prdx3 to alleviate pulmonary fibrosis. Exp Mol Med. 2024;56:1643-54.
88. Zhou Q, Yi G, Chang M, et al. Activation of Sirtuin3 by honokiol ameliorates alveolar epithelial cell senescence in experimental silicosis via the cGAS-STING pathway. Redox Biol. 2024;74:103224.
89. Lin Y, Xu Z, Zhou B, Ma K, Jiang M. Pentoxifylline inhibits pulmonary fibrosis by regulating cellular senescence in mice. Front Pharmacol. 2022;13:848263.
90. Sun W, Gao Y, Wu Y, et al. Targeted apoptosis of senescent cells by valproic acid alleviates therapy-induced cellular senescence and lung aging. Phytomedicine. 2024;135:156131.
92. Sun Q, Liu L, Mandal J, et al. PDGF-BB induces PRMT1 expression through ERK1/2 dependent STAT1 activation and regulates remodeling in primary human lung fibroblasts. Cell Signal. 2016;28:307-15.
93. Li S, Huang Q, He B. SIRT1 as a potential therapeutic target for chronic obstructive pulmonary disease. Lung. 2023;201:201-15.
94. Wang Y, Chen J, Chen W, et al. LINC00987 ameliorates COPD by regulating LPS-induced cell apoptosis, oxidative stress, inflammation and autophagy through let-7b-5p/SIRT1 Axis. Int J Chron Obstruct Pulmon Dis. 2020;15:3213-25.
95. Yao H, Rahman I. Perspectives on translational and therapeutic aspects of SIRT1 in inflammaging and senescence. Biochem Pharmacol. 2012;84:1332-9.
96. Yang F, Qin H, Qin C, et al. SIRT1 regulates cigarette smoke extractinduced alveolar macrophage polarization and inflammation by inhibiting the TRAF6/NLRP3 signaling pathway. Mol Med Rep. 2025;31:43.
97. Wang AP, Yang F, Tian Y, et al. Pulmonary artery smooth muscle cell senescence promotes the proliferation of PASMCs by paracrine IL-6 in hypoxia-induced pulmonary hypertension. Front Physiol. 2021;12:656139.
98. Boucherat O, Bonnet S, Provencher S, Potus F. Anti-remodeling therapies in pulmonary arterial hypertension. Trends Pharmacol Sci. 2025;46:674-91.
99. Zeng Q, Gong Y, Zhu N, Shi Y, Zhang C, Qin L. Lipids and lipid metabolism in cellular senescence: emerging targets for age-related diseases. Ageing Res Rev. 2024;97:102294.
100. Liang J, Huang G, Liu X, et al. Lipid deficiency contributes to impaired alveolar progenitor cell function in aging and idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol. 2024;71:242-53.
101. Zhu C, Zhang L, Heidari M, et al. Small RNA deep sequencing revealed microRNAs’ involvement in modulating cellular senescence and immortalization state. Poult Sci. 2023;102:102474.
102. Choi JY, Shin HJ, Bae IH. miR-93-5p suppresses cellular senescence by directly targeting Bcl-w and p21. Biochem Biophys Res Commun. 2018;505:1134-40.
103. Sun S, Qin W, Tang X, et al. Vascular endothelium-targeted Sirt7 gene therapy rejuvenates blood vessels and extends life span in a Hutchinson-Gilford progeria model. Sci Adv. 2020;6:eaay5556.
104. Katz MG, Hadas Y, Vincek A, et al. Acid ceramidase gene therapy ameliorates pulmonary arterial hypertension with right heart dysfunction. Respir Res. 2023;24:197.
105. Lu Q, Ye C, Mao W, et al. Targeting senescent alveolar type 2 cells with a gene-editable FePt dual-atom catalyst for mitigating idiopathic pulmonary fibrosis. ACS Nano. 2025;19:23162-76.
106. Wu H, Ma H, Wang L, et al. Regulation of lung epithelial cell senescence in smoking-induced COPD/emphysema by microR-125a-5p via Sp1 mediation of SIRT1/HIF-1a. Int J Biol Sci. 2022;18:661-74.
107. Manevski M, Muthumalage T, Devadoss D, et al. Cellular stress responses and dysfunctional Mitochondrial-cellular senescence, and therapeutics in chronic respiratory diseases. Redox Biol. 2020;33:101443.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0











Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].