


Synergistic era: practice and reflection on TACE combined with systemic therapy in hepatocellular carcinoma
 0
0 Abstract
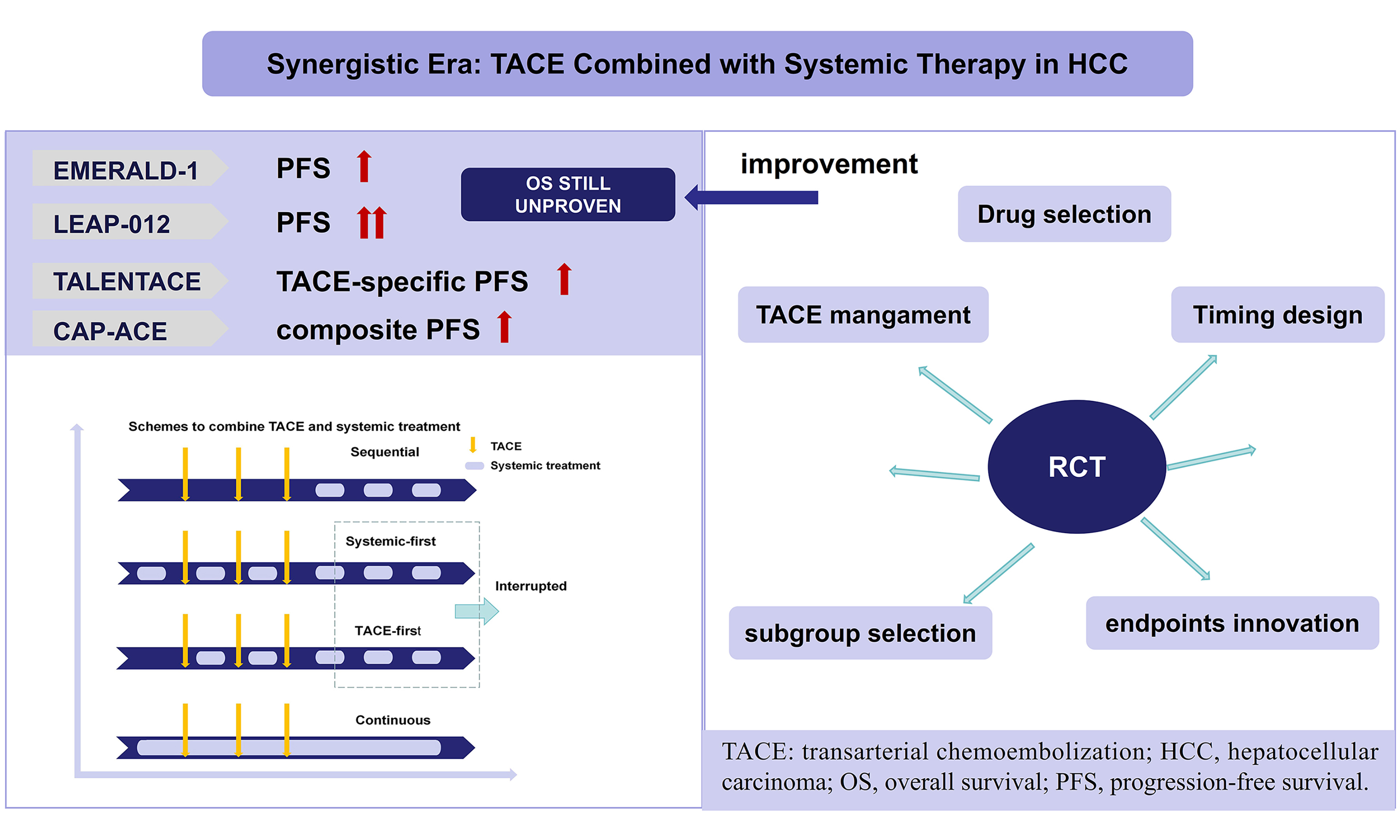
The treatment paradigm for hepatocellular carcinoma (HCC) is evolving from traditional stage-based monotherapy toward a synergistic era characterized by the proactive integration of locoregional and systemic therapies. Transarterial chemoembolization (TACE) plus systemic therapy has shown notable synergistic potential, but it also complicates clinical decision-making and trial interpretation. This review briefly outlines advances in TACE and systemic therapy, summarizes key randomized controlled trials of combination therapy, and analyzes core challenges in treatment timing, TACE implementation, endpoint design, and patient selection. Although combination regimens have not consistently demonstrated significant overall survival (OS) benefits in intermediate-to-advanced HCC, improvements in progression-free survival, objective response rate, and other disease-control endpoints have been increasingly reported. Future prospective trials should look beyond drug selection alone and refine combination timing, clinically meaningful endpoints, TACE standardization, and subgroup selection. These refinements may help determine how early disease-control gains can be translated more consistently into durable OS benefit, particularly for patients with intermediate-stage HCC.
Keywords
INTRODUCTION
Hepatocellular carcinoma (HCC) continues to pose a major global health burden and is among the most common malignancies and leading causes of cancer-related death worldwide[1]. Although vaccination and antiviral treatment have reduced the incidence of hepatitis B virus (HBV)-related HCC in some regions[2], the burden of HCC associated with metabolic dysfunction-associated steatotic liver disease (MASLD) is increasing[3]. Many patients are diagnosed after the window for curative resection, or ablation has passed[4], making locoregional and systemic therapies the therapeutic backbone for many patients.
For decades, transarterial chemoembolization (TACE) has been the standard option for intermediate-stage HCC[5-7]. However, clinical outcomes after TACE are heterogeneous, and recurrence remains common. Mechanistically, TACE induces tumor ischemia and immunogenic cell death[8], but may also trigger hypoxia-driven angiogenesis and reinforce an immunosuppressive tumor microenvironment[9,10], thereby limiting durable disease control. Systemic treatments, including immune checkpoint inhibitors (ICIs) and targeted agents including anti-vascular endothelial growth factor (VEGF) antibody and tyrosine kinase inhibitors (TKIs) - can potentially counteract these adaptive changes after TACE, providing a strong biological rationale for combination strategies[11]. Accordingly, multiple randomized trials have evaluated TACE in combination with systemic therapy across different sequencing paradigms and endpoint frameworks. Recent results from four key trials, including EMERALD-1[12], LEAP-012[13], TALENTACE[14], and CAP-ACE[15], have demonstrated significant benefits in progression-free survival (PFS) and objective response rate (ORR), accelerating guideline discussions and clinical adoption in selected settings[16]. Nevertheless, clinicians now face a more complex decision landscape: overall survival (OS) remains immature or not yet definitively improved in several trials, and substantial heterogeneity persists regarding when to initiate systemic therapy relative to TACE, how to operationalize on-demand TACE in trial protocols, and which endpoints best reflect clinically meaningful benefit in the modern era of effective post-progression therapy.
In contrast to previous reviews that mainly summarized efficacy outcomes, this narrative review interprets available evidence from a trial-design perspective. Relevant literature was identified through searches of PubMed, Web of Science, Embase, and ClinicalTrials.gov up to 2025 using core terms: “HCC”, “TACE”, “systemic therapy”, and “combination therapy”. Randomized trials, landmark studies, and major clinical guidelines were prioritized. Trials were highlighted if they impacted clinical practice or illustrated methodological issues critical to interpreting TACE-based combination therapy. A preliminary version of this study has been published as a preprint[17].
EVOLUTION OF TACE TECHNIQUES: FROM EMPIRICAL EMBOLIZATION TO PRECISION AND PERSONALIZED THERAPY
The development of TACE is rooted in the hemodynamic hallmark of HCC, in which tumor blood supply progressively shifts toward the hepatic artery, whereas the non-tumoral liver parenchyma remains predominantly portal venous[18]. This physiological contrast enables selective intra-arterial drug delivery and embolization to achieve a dual effect - tumor starvation and cytotoxic exposure - while limiting collateral injury to surrounding liver tissue. Its origins can be traced to the first hepatic artery embolization performed by Goldstein in 1976[19]. Subsequently, Nakakuma in the 1980s innovatively introduced gelatin sponge and iodized oil as a mitomycin carrier, establishing the classic model of conventional TACE (cTACE)[20]. Two randomized trials published in 2002 subsequently demonstrated a survival advantage of TACE over supportive care and established its role as standard therapy[21,22]. Since then, technical evolution has largely pursued three goals: higher precision, improved safety, and more consistent efficacy.
Precision and standardization of TACE
Contemporary TACE is best understood as a refined, standardized, and increasingly personalized procedure rather than a single uniform technique[23]. Superselective catheterization at the segmental or subsegmental level is now widely adopted to maximize tumor devascularization while preserving functional liver parenchyma and minimizing cumulative injury from non-target embolization[24]. The primary objectives are to maximize protection of normal liver parenchyma, avoiding cumulative liver damage from non-target embolization, and to achieve complete embolization by occluding the terminal tumor vasculature. Cone-beam computed tomography (CBCT) has become a central intra-procedural tool, providing three-dimensional guidance for superselective delivery and enabling immediate post-embolization assessment of coverage. On this basis, determining the embolization endpoint follows a strict individualized principle, requiring comprehensive consideration of tumor morphology, blood supply characteristics, and the patient’s hepatic functional reserve. The ideal endpoint is complete tumor devascularization, a hallmark of which in conventional cTACE is the visualization of small peritumoral portal vein branches[23,25]. Furthermore, the rational combination of differently sized embolic particles during the procedure, tailored to the vascular anatomy, helps achieve more extensive and uniform intratumoral embolization, thereby promoting complete necrosis.
Iterative optimization of embolic materials
Advances in embolic materials have progressed from mechanical occlusion toward engineered systems designed for controllable drug delivery and multifunctional integration. The evolution of embolic materials began in the 1980s with the advent of iodized oil. Its subsequent refined versions, represented by ultra-stable ethiodized oil, laid the foundation for angiography and drug delivery due to their excellent flow characteristics and selective tumor retention capability. Drug-eluting bead TACE (DEB-TACE) represents an innovative breakthrough in embolic materials, including drug-coated and drug-loaded microspheres (e.g., HepaSphere, LifePearl, TANDEM), which combine sustained embolization with controlled drug release[26-28]. The pivotal randomized controlled trial PRECISION V demonstrated that DEB-TACE was associated with significantly lower doxorubicin-related systemic and hepatic toxicity compared with cTACE[29]. A meta-analysis of patients with intermediate-stage HCC showed that DEB-TACE achieved significantly higher complete response and disease control rates than cTACE[30].
Current cutting-edge research further focuses on the integration of multiple functions, with the fundamental logic being the development of “smart” materials built upon embolic materials. The smart drug delivery system triggered by the tumor microenvironment is one of the representative strategies. Such materials[31-33] (e.g., temperature-and pH-sensitive hydrogels/microspheres) can passively sense unique signals in the lesion area within the body and accordingly alter their own structure or properties, thereby achieving targeted drug release or prodrug activation at the intended site. Meanwhile, external energy-responsive materials can be precisely activated via external energy sources (such as light, ultrasound, or magnetic fields) to achieve active thermal, mechanical, or chemical cytotoxic effects[34-36]. Collectively, these innovations reflect a broader shift from single-agent intra-arterial chemotherapy toward synergistic payload strategies that may include immunomodulation, anti-angiogenic mechanisms, and localized energy-based enhancement.
THE EVOLUTION OF SYSTEMIC THERAPY
Systemic treatment for HCC has progressed from a period of limited benefit to a rapidly expanding therapeutic field. Table 1 summarizes pivotal phase III first-line systemic therapy trials in advanced HCC. Historically, advanced HCC relied primarily on chemotherapy and targeted agents. A pivotal SHARP trial in 2008 demonstrated that the multi-target TKI sorafenib significantly improved OS (10.7 months vs. 7.9 months; HR = 0.69) compared to placebo, establishing sorafenib as the sole first-line standard for years[37]. Lenvatinib subsequently demonstrated non-inferiority in OS while improving progression-related endpoints and response rates, expanding first-line options[38]. The Chinese-developed TKI donafenib also showed superior OS to sorafenib in the ZGDH3 trial (12.1 months vs. 10.3 months; HR = 0.83), with lower drug-related grade ≥ 3 adverse events (AEs). Sorafenib, lenvatinib, and donafenib are all oral multi-target TKIs, yet their mechanisms and clinical profiles differ. Sorafenib primarily inhibits VEGFR-2/3, platelet-derived growth factor receptor (PDGFR)-β, and (rapidly accelerated fibrosarcoma) RAF kinases, establishing its historical standard role by blocking angiogenesis and tumor proliferation. Lenvatinib demonstrates broader and more potent inhibition of the VEGFR family (1-3) and, most distinctively, strongly targets fibroblast growth factor receptors (FGFR) 1-4[39]. This concurrent blockade of the FGFR pathway is the key mechanism behind its significantly higher ORR observed in the REFLECT trial. Donafenib is a deuterated derivative of sorafenib[40], sharing a similar core target profile but with optimized pharmacokinetics, which explains the better balance between efficacy and safety.
Efficacy summary of phase III first-line systemic therapy trials in advanced HCC
| Trial name | Year* | Regimen | Primary endpoint | Key efficacy results (months) |
| SHARP[37] | 2008 | Sorafenib vs. placebo | OS & TTP | OS: 10.7 vs. 7.9 (HR = 0.69) TTP: 5.5 vs. 2.8 (HR = 0.58) |
| REFLECT[38] | 2018 | Lenvatinib vs. sorafenib | OS non-inferiority | OS: 13.6 vs. 12.3 (HR = 0.92) PFS: 7.4 vs. 3.7 (HR = 0.66) |
| ZGDH3[40]# | 2021 | Donafenib vs. sorafenib | OS | OS: 12.1 vs. 10.3 (HR = 0.83) PFS: 3.7 vs. 3.6 (HR = 0.91) |
| ORIENT-32[45]# | 2021 | Sintilimab plus bevacizumab biosimilar vs. sorafenib | OS & PFS | OS: NE vs. 10.4 (HR = 0.57) PFS: 4.6 vs. 2.8 (HR = 0.56) |
| IMbrave150[42] | 2022 | Atezolizumab plus bevacizumab vs. sorafenib | OS & PFS | OS: 19.2 vs. 13.4 (HR = 0.66) PFS: 6.9 vs. 4.3 (HR = 0.65) |
| RATIONALE-301[46]# | 2023 | Tislelizumab vs. sorafenib | OS non-inferiority | OS: 15.9 vs. 14.1 (HR = 0.85) PFS: 2.1 vs. 3.4 (HR = 1.11) |
| CARES-310[47]# | 2025 | Camrelizumab plus apatinib vs. sorafenib | OS & PFS | OS: 23.8 vs. 15.2 (HR = 0.64) PFS: 5.6 vs. 3.7 (HR = 0.54) |
| HIMALAYA[43] | 2025 | Tremelimumab plus durvalumab vs. sorafenib | OS | OS: 16.4 vs. 13.8 (HR = 0.76) PFS: 3.8 vs. 4.1 (HR = 0.90) |
| CheckMate 9DW[44] | 2025 | Nivolumab plus ipilimumab vs. lenvatinib/sorafenib | OS | OS: 23.7 vs. 20.6 (HR = 0.79) PFS: 9.1 vs. 9.2 (HR = 0.87) |
| SCT-I10A-C301[48]# | 2025 | Finotonlimab plus bevacizumab biosimilar vs. sorafenib | OS & PFS | OS: 22.0 vs. 14.5 (HR = 0.76) PFS: 7.1 vs. 2.9 (HR = 0.50) |
| HEPATORCH[49]# | 2025 | Toripalimab plus bevacizumab vs. sorafenib | OS & PFS | OS: 20.0 vs. 14.5 (HR = 0.76) PFS: 5.8 vs. 4.0 (HR = 0.69) |
The advance of ICIs, mainly including programmed cell death-(ligand) protein 1 [PD-(L)1] inhibitors and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), have fundamentally reshaped the treatment paradigm. Although initial attempts with PD-(L)1 inhibitors in the first-line setting (e.g., nivolumab in CheckMate 459[41]) failed to meet their primary survival endpoints, combination strategies proved revolutionary. The updated landmark IMbrave150 trial established the combination of the PD-(L)1 inhibitor atezolizumab and the VEGF inhibitor bevacizumab as a new global standard, demonstrating significantly superior OS compared to sorafenib (19.2 months vs. 13.4 months; HR = 0.66)[42]. The STRIDE regimen [CTLA-4 inhibitor tremelimumab plus PD-(L)1 inhibitor durvalumab] in the HIMALAYA trial also outperformed sorafenib in OS (16.4 months vs. 13.8 months; HR = 0.76)[43]. The recently reported CheckMate 9DW trial[44] demonstrated a significant OS improvement for the combination of nivolumab and ipilimumab vs. lenvatinib/sorafenib (23.7 months vs. 20.6 months; HR = 0.79). Together with the results of the HIMALAYA trial, these findings confirm the efficacy of the combination protocol of CTLA-4 inhibitor plus PD-(L)1 inhibitor.
Notably, China-developed combination regimens have also demonstrated equally remarkable efficacy. In the ORIENT-32 trial, sintilimab plus a bevacizumab biosimilar significantly improved OS and PFS compared with sorafenib[45]. Subsequently, the RATIONALE-301 trial[46], which compared PD-(L)1 inhibitor tislelizumab with sorafenib, reported a non-inferior OS outcome (15.9 months vs. 14.1 months; HR = 0.85), providing crucial evidence for global research on single-agent immunotherapy. Furthermore, in the CARES-310 trial, camrelizumab plus apatinib achieved a median OS of 23.8 months (vs. 15.2 months for sorafenib, HR = 0.64)[47]. Recently, the SCT-I10A-C301 trial showed that finotonlimab plus a bevacizumab biosimilar significantly prolonged OS compared with sorafenib (22.1 months vs. 14.2 months; HR = 0.60)[48]. The HEPATORCH trial demonstrated that toripalimab plus bevacizumab improved OS compared with sorafenib (20.0 months vs. 14.5 months; HR = 0.76)[49]. Together, these studies have expanded the range of first-line systemic treatment options for HCC, particularly in China, and underscore the growing contribution of regionally developed regimens to the global evidence base.
EXPLORATION OF TACE COMBINED WITH SYSTEMIC THERAPY
Since the introduction of sorafenib into clinical practice, continuous efforts have been made to enhance the efficacy of TACE by combining with systemic therapy. The three combination strategies-sequential, interrupted, and continuous- were systematically proposed by Strebel in 2008[50], established a crucial theoretical framework for subsequent clinical trial designs, as shown in Figure 1. Early randomized trials combining TACE with TKIs or antiangiogenic agents in intermediate-to-advanced HCC yielded largely negative results, whereas later studies demonstrated that trial success is highly sensitive to (i) the timing of systemic exposure around TACE; (ii) progression/response assessment criteria that govern TACE continuation; and (iii) whether TACE is delivered in a protocol-mandated “scheduled” manner or as response-guided “on-demand” treatment.
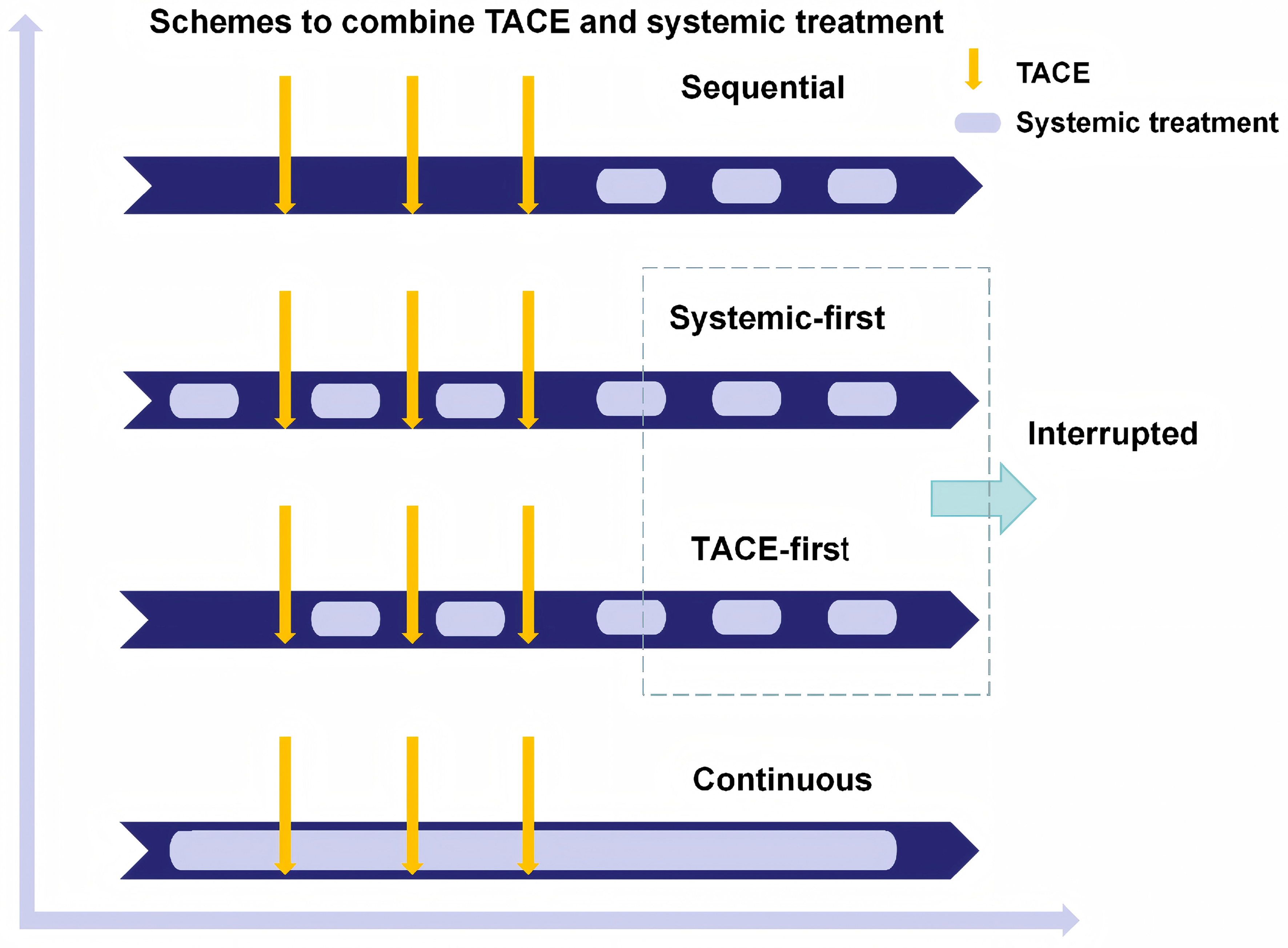
Figure 1. Schemes to combine TACE and systemic treatment. This schematic was independently illustrated based on the original combination framework proposed by Strebel[50], with modern timing strategies (TACE-first, Systemic-first) newly integrated into clinical practice. The three core therapeutic schedules derived from the original framework are: Sequential: TACE and systemic therapy are administered in succession with clear intervals. Interrupted: TACE and systemic therapy are alternated with treatment breaks. Continuous: TACE and systemic therapy are co-administered without interruption to maintain sustained exposure. TACE: Transarterial chemoembolization.
DUAL THERAPY: TACE PLUS TARGETED OR ANTIANGIOGENIC THERAPY
Evolution and heterogeneity of TACE-TKI timing strategies
Early TACE-TKI studies showed substantial heterogeneity in systemic-treatment timing and TACE scheduling. The Post-TACE trial[51] adopted a delayed sequential strategy, initiating sorafenib only after planned TACE had been completed and objective response achieved; more than half of patients started sorafenib beyond 9 weeks after TACE, and time to progression (TTP) was not significantly improved (5.4 months vs. 3.7 months; HR = 0.87). Subsequent trials moved systemic therapy closer to TACE but used different models. SPACE[52] started sorafenib 3-7 days before scheduled DEB-TACE, yet TTP remained statistically negative (169 days vs. 166 days; HR = 0.80). TACE-2[53] initiated sorafenib/placebo and followed by DEB-TACE 2-5 weeks later and response-guided retreatment, but failed to improve PFS (7.8 months vs. 7.5 months; HR = 0.99) or OS (19.5 months vs. 18.4 months; HR = 0.91). In contrast, TACTICS[54] moved toward a systemic-first and on-demand strategy, administering sorafenib 2-3 weeks before cTACE and continuing treatment until unTACEable progression, improving TACE-specific PFS (22.8 months vs. 13.5 months; HR = 0.66) but not OS (36.2 months vs. 30.8 months; HR = 0.86). The DEB-TACE-apatinib trial[55] adopted a TACE-first strategy, with apatinib started 3-5 days after DEB-TACE and held before subsequent sessions, improving PFS (7.1 months vs. 5.2 months) and OS (23.3 months vs. 18.9 months). LAUNCH[56] represented a different paradigm in advanced HCC, where lenvatinib served as the systemic backbone, and TACE was added for intrahepatic control, improving PFS (10.6 months vs. 6.4 months; HR = 0.43) and OS (17.8 months vs. 11.5 months; HR = 0.45) with a systemic-first strategy. Taken together, these trials suggest an evolution from delayed sequential therapy toward earlier, more sustained, and more operationally integrated systemic exposure, but the optimal sequence remains dependent on TACE technique, retreatment rules, drug interruption, endpoint definition, and disease stage.
Treatment discontinuation criteria
Assessment criteria are central to TACE-TKI trial design because they determine when treatment is judged ineffective and whether TACE or systemic therapy should be discontinued. RECIST 1.1 is widely used in systemic therapy trials but may underestimate post-TACE necrosis, whereas mRECIST and RECICL better capture viable enhancing tumor and HCC-specific post-embolization response. TACE-specific endpoints, such as time to untreatable progression (TTUP) or TACE-specific PFS, further address whether patients can still safely benefit from additional TACE. In the TACE-apatinib trial, adequate treatment exposure may partly explain the positive results: ≥ 3 DEB-TACE sessions were delivered in 74.6% vs. 65.3% of patients, and apatinib was continued for > 3 months in 68.9%. This suggests that sustained antiangiogenic coverage and sufficient repeat TACE may be critical for translating biological synergy into PFS, OS, and ORR benefits. Earlier sorafenib-based studies suggest why these design details matter. In SPACE, 35.9% of patients in the sorafenib arm received only one DEB-TACE session, and sorafenib exposure was lower than planned (566 mg/day; 21.0 weeks). TACE-2 used response-guided DEB-TACE, but sorafenib exposure remained limited (660 mg/day; 120 days), with fewer TACE procedures delivered in the sorafenib arm. In contrast, TACTICS adopted RECICL-based TACE-specific PFS and did not regard new intrahepatic lesions alone as treatment failure, allowing on-demand TACE until unTACEable progression, TACE refractoriness, toxicity, or withdrawal. Although OS remained nonsignificant, post-trial active treatment was more frequent in the TACE-alone arm (76.3% vs. 58.8%). Thus, the available evidence suggests that effective TACE exposure and adequate systemic treatment duration are crucial; future trials should optimize stopping rules by integrating radiographic progression, technical TACE feasibility, liver reserve, drug tolerability, and post-progression therapy.
Refining the population most likely to benefit
Across these trials, the target population has gradually shifted from broadly defined TACE-eligible patients to clinically enriched subgroups with higher intrahepatic tumor burden but preserved Child-Pugh/ALBI function[57]. Earlier sorafenib-based trials, such as SPACE and TACE-2, mainly enrolled intermediate-stage or liver-confined HCC without clear enrichment for patients at high risk of TACE failure, which may have diluted potential benefit. Although the overall OS benefit of TACTICS was not statistically significant, post hoc analysis showed that the PFS and OS advantages of TACE-sorafenib were more evident in patients beyond the up-to-seven criteria, but only one-third of patients were beyond the up-to-seven criteria at baseline. This suggested that combination therapy may be more relevant for patients whose tumor burden is too high for durable control with repeated TACE alone, but who still have sufficient liver reserve to tolerate both TACE and systemic therapy. Recent positive trials further support this selection trend. LAUNCH tested TACE-lenvatinib in advanced HCC, a population including large intrahepatic tumor burden, portal vein thrombosis, and extrahepatic spread, and demonstrated significant OS and PFS benefits over lenvatinib alone. Therefore, future trials should not simply ask whether all TACE-eligible patients benefit from combination therapy, but should prospectively define risk-enriched populations, particularly high tumor burden, preserved Child-Pugh/ALBI function, technically feasible selective TACE, and limited systemic spread.
Therefore, the lessons from TACE-targeted therapy are not merely historical. They define the key design requirements for modern triple-therapy trials: early and sustained systemic exposure, clinically meaningful TACE continuation rules, and enrichment of patients likely to fail TACE alone but still retain adequate hepatic reserve.
TRIPLE THERAPY (TACE PLUS ICI AND ANTI-VEGF/TKI): PERSISTENT OS CHALLENGES
With immunotherapy plus anti-angiogenic therapy emerging as a dominant systemic backbone, triple therapy integrating TACE with ICI and anti-VEGF/TKI has become a logical next step. Real-world evidence and target-trial emulation studies have reported encouraging disease control[58,59]. Meanwhile, several randomized trials have provided more rigorous signals of benefit, most consistently in PFS and ORR. However, OS remains immature or difficult to improve definitively in intermediate-stage populations where subsequent effective therapies and crossover can dilute survival differences. Below, we summarized and reflected pivotal randomized trials using a standardized evidence-focused template.
THE EMERALD-1 TRIAL
EMERALD-1[12] was the first reported phase III trial evaluating TACE combined with immunotherapy and anti-angiogenic therapy. This multiregional, randomized, double-blind, placebo-controlled study compared TACE plus durvalumab and bevacizumab, TACE plus durvalumab, and TACE plus placebo in patients with unresectable HCC eligible for embolization [Table 2]. The enrolled population was broad and clinically heterogeneous, including patients with Barcelona Clinic Liver Cancer (BCLC) stage A-C disease; approximately 26% had stage A disease, about 98% had Child-Pugh class A liver function, and nearly half were beyond the up-to-seven criteria. This broad eligibility enhanced external validity but also complicated the interpretation of OS by introducing heterogeneity in tumor burden, disease stage, TACE suitability, and subsequent treatment pathways.
Key design features and outcomes of phase II/III trials evaluating TACE-based combination therapies in HCC
| Trial name (Phase) | Key population | Treatment arms | Sequence & interval | Primary endpoint & outcome |
| SPACE (Phase II)[52] | BCLC B; Child-Pugh A | DEB-TACE plus sorafenib vs. DEB-TACE plus placebo | Systemic-first: sorafenib initiated 3-7 day before the first TACE procedure | TTP: 169 vs. 166 days (HR = 0.80, P = 0.07) |
| TACE-2 (Phase III)[53] | Unresectable HCC; Child-Pugh A; ECOG PS ≤ 1; Patent main portal vein; No extrahepatic spread | DEB-TACE plus sorafenib vs. DEB-TACE plus placebo | Systemic-first: sorafenib initiated 2-5 weeks before the first TACE procedure | PFS: 7.8 vs. 7.5 months (HR = 0.99, P = 0.94); OS: 19.5 vs. 18.4 months (HR = 0.91, P = 0.57) |
| TACTICS (Phase II)[54] | Unresectable HCC; Child-Pugh A/B7; ECOG PS 0-1 | cTACE plus sorafenib vs. cTACE alone | Systemic-first: sorafenib initiated 2-3 weeks before the first TACE procedure | PFS: 22.8 vs. 13.5 months (HR = 0.66, P = 0.020); OS: 36.2 vs. 30.8 months (HR = 0.86, P = 0.400) |
| DEB-DEB-TACE-apatinib (Phase III)[55] | BCLC B/C; Child-Pugh A/B; ECOG PS 0-1; Vascular invasion allowed | DEB-TACE plus apatinib vs. DEB-TACE alone | TACE-first: apatinib initiated 3-5 days after DEB-TACE | PFS: 7.1 vs. 5.2 months (HR = 0.564, P < 0.001); OS: 23.3 vs. 18.9 months (HR = 0.526, P < 0.001) |
| LAUNCH (Phase III)[56] | Advanced HCC; Child-Pugh A | TACE plus lenvatinib vs. lenvatinib | Systemic-first: lenvatinib initiated 1 day before the first TACE procedure | PFS: 10.6 vs. 6.4 months (HR = 0.43, P < 0.001); OS: 17.8 vs. 11.5 months (HR = 0.45, P < 0.001) |
| EMERALD-1 (Phase III)[12] | BCLC A-C; Child-Pugh A-B7; ECOG PS 0-1; No extrahepatic spread | TACE plus durvalumab plus bevacizumab vs. TACE plus durvalumab vs. TACE plus placebo | TACE-first: durvalumab initiated 1 week after the first TACE procedure; bevacizumab initiated ≥ 2 weeks after completion of all planned TACE procedures | PFS: 15.0 vs. 8.2 months (HR = 0.77, P = 0.032); OS: immature |
| LEAP-012 (Phase III)[13] | Non-metastatic HCC; Child-Pugh A; ECOG PS 0-1; Extreme tumor burden excluded | TACE plus lenvatinib plus pembrolizumab vs. TACE plus dual placebo | Systemic-first: lenvatinib plus pembrolizumab initiated 2-4 weeks before the first TACE procedure | PFS: 14.6 vs. 10.0 months (HR = 0.66, P = 0.0002); 24-month OS: 75% vs. 69% (HR = 0.80, P = 0.087) |
| TALENTACE (Phase III; preliminary conference data)[14] | Intermediate-to-high tumor burden; Child-Pugh A; Mainly BCLC B, selected BCLC C/Vp1-2 | TACE plus atezolizumab and bevacizumab vs. TACE | TACE-first: atezolizumab plus bevacizumab initiated 2 to 8 weeks after the first TACE procedure | TACE-PFS: 11.3 vs. 7.0 months (HR = 0.71, P = 0.009); OS: 34.5 vs. 35.4 months (HR = 0.96, P > 0.05) |
| CAP-ACE (Phase II)[15] | Unresectable HCC; Child-Pugh A; ECOG PS 0-1; No extrahepatic metastasis | TACE plus camrelizumab and rivoceranib/apatinib vs. TACE alone | TACE-first: systemic therapy initiated within 2 weeks after the first TACE procedure | Composite PFS: 10.8 months vs. 3.2 months (HR = 0.34, P < 0.001); OS: 24.0 months vs. 21.5 months (HR = 0.87, P > 0.05) |
The primary endpoint was PFS assessed according to RECIST 1.1. TACE was performed according to local practice, with one to four procedures permitted and all planned sessions required to be completed within 16 weeks after the first procedure. Durvalumab was initiated at least 7 days after the first TACE session, whereas bevacizumab was delayed until at least 14 days after the final session. This design clearly prioritized peri-procedural safety, particularly by avoiding concurrent exposure to bevacizumab during embolization. Mechanistically, however, the potentially long interval between the first embolization and bevacizumab initiation may have reduced coverage of the early post-embolization window characterized by hypoxia and VEGF upregulation[60,61].
The trial showed that triple therapy significantly prolonged PFS compared with TACE plus placebo (15.0 months vs. 8.2 months, HR = 0.77). In contrast, the TACE plus durvalumab group did not show PFS benefit (10.0 months vs. 8.2 months, HR = 0.94). The finding suggests that bevacizumab is not merely an additional component, but may be essential for effective synergy. Notably, curve separation appeared closer to the period after bevacizumab introduction rather than immediately after embolization, further supporting the contribution of anti-angiogenic therapy. Nevertheless, OS remains immature. Given its exploratory role in triple therapy development, the broad eligibility criteria of EMERALD-1 are understandable and provide useful subgroup signals for future trial design. Current PFS-based subgroup analyses suggest potentially greater benefit in patients with no vascular invasion, multifocal intrahepatic disease, and ECOG PS 0, however, these findings remain hypothesis-generating and require further validation. In addition, RECIST 1.1 may trigger early reassessment when new intrahepatic lesions appear, and treatment discontinuation due to adverse events occurred in approximately 28% of patients in the triple therapy group. Therefore, EMERALD-1 provides proof of concept that adding both durvalumab and bevacizumab can improve early disease control, but future trials must better optimize anti-angiogenic timing, response criteria, toxicity management, and post-progression treatment pathways.
THE LEAP-012 TRIAL
LEAP-012[13] was a global, randomized, double-blind phase III trial evaluating TACE plus lenvatinib and pembrolizumab vs. TACE plus dual placebo in unresectable, non-metastatic HCC. Unlike EMERALD-1, which enrolled a broader embolization-eligible population, LEAP-012 used stricter selection criteria: patients had Child-Pugh class A liver function, Eastern Cooperative Oncology Group performance status 0-1, liver-confined disease, and tumors technically suitable for embolization. Extreme tumor burden was excluded, including tumors ≥ 10 cm, more than 10 tumors, or tumor occupation of ≥ 50% of the liver. Therefore, LEAP-012 tested triple therapy in a more standardized, TACE-suitable population rather than in all embolization-eligible patients.
The design was also more synchronized with the biological window of TACE. Lenvatinib and pembrolizumab were started before embolization, with the first TACE performed 2-4 weeks after randomization. Lenvatinib was administered at 8 or 12 mg once daily according to body weight, and pembrolizumab was administered at 400 mg every 6 weeks. To improve safety, lenvatinib was interrupted around the time of TACE, and each tumor could receive no more than 2 TACE sessions. This approach reduced procedural heterogeneity and the risk of excessive embolization; however, the fixed per-tumor TACE cap may also have limited effective locoregional exposure in selected patients who could still have benefited from additional selective embolization.
LEAP-012 met its PFS endpoint, with a median of 14.6 months vs. 10.0 months and a hazard ratio of 0.66. The primary endpoint was RECIST 1.1 by blinded central review, modified to allow up to five intrahepatic target lesions and to require new intrahepatic tumors to meet LI-RADS 5 criteria before being considered progression. This design attempted to balance regulatory rigor with the clinical complexity of post-TACE intrahepatic changes. OS remained immature at the published interim analysis, with 24-month survival rates of 75% vs. 69% (HR = 0.80). Grade 3 or worse treatment-related adverse events were frequent, occurring in 71% vs. 32%, indicating that systemic-first triple therapy requires careful toxicity management.
More importantly, LEAP-012 provides useful clues for future population refinement. Subgroup results suggested stronger PFS benefit in patients with BCLC stage B disease, ECOG PS 0, ALBI grade 1, Child-Pugh score 5, alpha-fetoprotein > 400 ng/mL, and medium-to-high tumor burden. However, OS data remain immature, and these subgroup findings should be interpreted as exploratory. Thus, the major lesson of LEAP-012 is not simply that systemic-first triple therapy improves early disease control, but that future trials should prospectively enrich for patients with sufficient risk of TACE failure to justify systemic intensification, while still preserving enough liver function and performance status to tolerate sustained combination therapy.
THE TALENTACE TRIAL
TALENTACE was a phase III, open-label, randomized trial comparing on-demand TACE plus atezolizumab and bevacizumab with on-demand TACE alone in systemically untreated, unresectable HCC. Its key design feature was population enrichment. Eligible patients had intermediate-to-high tumor burden, defined as maximum tumor diameter plus tumor number ≥ 6; most had BCLC stage B disease, while selected BCLC stage C patients with limited portal vein invasion, mainly Vp1-2, or ECOG PS 1 were also allowed. Thus, TALENTACE focused on a clinically important boundary population: patients who are still technically suitable for TACE, but at sufficient risk of TACE failure to justify systemic intensification.
In the experimental arm, atezolizumab 1,200 mg plus bevacizumab 15 mg/kg every 3 weeks was initiated at least 14 days and within 8 weeks after the first TACE. This TACE-first strategy differs from the systemic-first design of LEAP-012 and preserves real-world retreatment flexibility by avoiding a fixed per-tumor TACE cap. The co-primary endpoints were TACE-PFS and OS. TACE-PFS was defined as time to untreatable progression, TACE failure/refractoriness, or death, making it closer to a treatment-strategy failure endpoint than conventional RECIST progression alone.
At the first interim analysis, TALENTACE met its TACE-PFS endpoint, with a median of 11.30 months vs. 7.03 months (HR = 0.71). RECIST 1.1 PFS also favored the combination arm, 10.32 months vs. 6.37 months (HR = 0.64). However, OS remained immature, with only 38.6% of events reported; median OS was 34.53 months vs. 35.38 months (HR = 0.96). The apparently neutral early survival result may reflect several factors: immature follow-up, unexpectedly long survival in the control arm, subsequent treatment after progression, and the possibility that improved TACE-PFS mainly delays treatment-strategy failure rather than immediately translating into death reduction.
The current subgroup data should be interpreted cautiously. Publicly available reports suggest generally consistent TACE-PFS benefit across predefined subgroups, but detailed subgroup hazard ratios have not yet been fully published. Therefore, TALENTACE should not yet be used to define a definitive biomarker-like beneficiary subgroup. Its deeper implication is conceptual: future trials may need to enrich for patients with both high risk of inadequate TACE control and sufficient liver and performance reserve to tolerate sustained systemic therapy. Toxicity remains a key constraint, as grade 3-4 treatment-related adverse events occurred in 60.8% vs. 40.5%, and treatment withdrawal due to adverse events occurred in 21.1% vs. 2.3%. Thus, TALENTACE represents a shift from asking whether triple therapy works in all embolization-eligible patients to asking whether it works in a risk-enriched population using an endpoint that captures TACE-strategy failure rather than radiographic progression alone.
THE CAP-ACE TRIAL
The CAP-ACE, also reported as CARES-005, was a multicenter, open-label, randomized phase II trial conducted in China that compared TACE plus camrelizumab and rivoceranib/apatinib with TACE alone in patients with unresectable HCC eligible for embolization. Eligible patients had no extrahepatic metastasis, Child-Pugh class A liver function, and ECOG PS 0-1. Most patients had BCLC stage B or C disease.
A key feature of CAP-ACE was its crossover design. Patients initially assigned to TACE alone were allowed to cross over to receive camrelizumab plus rivoceranib/apatinib after progression. This design was clinically and ethically reasonable and also resembles real-world practice, in which patients are often switched to combined systemic therapy after TACE failure. However, this inevitably reduced the ability of the trial to detect an OS difference because the control group could later receive the active systemic regimen. The primary endpoint was RECICL-assessed PFS, which is particularly relevant in TACE-based trials because it better captures viable tumor response after embolization than size-based criteria alone. CAP-ACE showed a strong PFS advantage: median PFS was 10.8 months vs. 3.2 months (HR = 0.34), and time to unTACEable progression was also prolonged, 13.7 months vs. 3.9 months, suggesting that early systemic intensification can delay the point at which a TACE-based strategy becomes ineffective. Subgroup analyses showed RECICL-PFS benefit in both BCLC A/B and BCLC C disease, with hazard ratios of 0.34 and 0.39, respectively; however, OS benefit was not confirmed. Safety is another important limitation. Grade 3 or higher treatment-related adverse events were more frequent with the combination, including aspartate aminotransferase elevation, alanine aminotransferase elevation, hypertension, and platelet count decrease. These toxicities were described as manageable, but they highlight the recurring challenge in TACE-based triple therapy: stronger early disease control may come at the cost of greater hepatic and systemic toxicity.
Overall, CAP-ACE reinforces two important concepts. First, RECICL-based PFS and time to unTACEable progression may better capture the benefit of delaying TACE-strategy failure than conventional radiographic endpoints alone. Second, when crossover is allowed, OS becomes strongly influenced by post-progression treatment pathways and may underestimate the value of early combination therapy. Thus, CAP-ACE provides strong randomized evidence that early TACE plus camrelizumab and rivoceranib/apatinib improves disease control and delays unTACEable progression, but it should not be overinterpreted as definitive proof of OS benefit.
FUTURE CONSIDERATIONS
The central challenge for future TACE-based combination trials is no longer whether systemic therapy can enhance early tumor control, but how to design treatment strategies that preserve enough effective TACE and systemic exposure to convert early disease control into durable survival benefit. Biologically, this PFS-OS dissociation may reflect the fact that triple therapy primarily enhances early intrahepatic tumor control rather than fully modifying the later determinants of survival in HCC. TACE can reduce viable tumor burden and promote antigen release, while anti-VEGF/TKI therapy may suppress post-embolization angiogenic rebound and render the tumor microenvironment more permissive to ICI activity[8-10]. These mechanisms are well suited to improve radiographic response and delay progression. However, OS is additionally influenced by hepatic functional reserve, cirrhosis-related competing mortality, cumulative liver injury after repeated TACE, treatment-related toxicities and interruptions, and effective post-progression therapies or crossover in control arms. Therefore, PFS may serve as a more sensitive signal of biological synergy, whereas OS requires sustained tumor control together with preservation of liver function. This discrepancy underscores several critical areas that require attention in future research.
First, the optimal timing and sequencing of TACE combined with systemic therapies must be directly compared. Current studies utilize heterogeneous strategies, including systemic-first and TACE-first, without definitive comparative evidence. Prospective trials specifically designed to test the impact of different sequencing approaches - while controlling for TACE technique, retreatment protocols, and continuity of systemic therapies - are essential to determine whether sequencing significantly influences synergy and long-term outcomes.
Second, the endpoints used in these trials need to be modernized to better capture clinically meaningful benefits. In settings where active subsequent therapies and crossover are common, OS may be diluted, making it statistically challenging to measure. Therefore, composite endpoints incorporating time to untreatable progression, liver function preservation, conversion to curative therapy rates, and patient-reported outcomes should be considered, as they more accurately reflect the true clinical benefit. When using TACE-specific endpoints, simplicity and clinical interpretability should be prioritized to facilitate broad adoption and reproducibility.
Third, TACE operational standardization should align more closely with real-world practice. A persistent gap exists between the on-demand TACE approach used in clinical practice and the protocol-driven session limits seen in trials. Future studies should incorporate adaptive, response-guided TACE algorithms, considering imaging response, liver reserve, and performance status, rather than relying on fixed session caps that may under-treat or over-treat certain patient subgroups.
CONCLUSION
Future progress hinges on large-scale, intelligently designed trials that simultaneously address these intertwined questions, including timing, endpoint selection, and TACE standardization. The goal is to move beyond demonstrating PFS superiority and to reliably deliver the OS benefit that defines meaningful therapeutic advancement for patients with intermediate-to-advanced HCC.
DECLARATIONS
Authors’ contributions
Responsible for the review design, manuscript revisions, and funding acquisition: Zhu G, Guo J, Chen L, Li C
The main authors responsible for writing the manuscript: Li C, Chen L
Contributed to sections of the manuscript and the creation of tables: Bao H, Wang Y
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
During the preparation of this manuscript, the AI tool ChatGPT (version 5.1, released 2025-08-07) was used solely for language editing and grammatical polishing. The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship
This work was supported by Noncommunicable Chronic Diseases-National Science and Technology Major Project (2024ZD0520400, 2024ZD0520403), National Natural Science Foundation of China (82372066,82572339), Interventional Medicine Research Special Fund Project of Jiangsu Medical Association [SYH-3201140-0045 (2022002)], Jiangsu Provincial Natural Science Foundation Youth Fund (BK20251691), The Natural Science Foundation of Jiangsu Province (BG2024007). The funding sources had no role in the writing of the report or the decision to submit the paper for publication.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Bray F, Laversanne M, Sung H, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74:229-63.
2. Liu CJ, Chen PJ. Changing epidemiology of liver disease in Asia: dual infection of HBV and HCV. Liver Int. 2022;42:1945-54.
3. Ioannou GN. Epidemiology and risk-stratification of NAFLD-associated HCC. J Hepatol. 2021;75:1476-84.
4. Vogel A, Meyer T, Sapisochin G, Salem R, Saborowski A. Hepatocellular carcinoma. Lancet. 2022;400:1345-62.
5. European Association for the Study of the Liver. EASL Clinical Practice Guidelines on the management of hepatocellular carcinoma. J Hepatol. 2025;82:315-74.
6. Taddei TH, Brown DB, Yarchoan M, Mendiratta-Lala M, Llovet JM. Critical update: AASLD Practice Guidance on prevention, diagnosis, and treatment of hepatocellular carcinoma. Hepatology. 2025;82:272-4.
7. Vogel A, Chan SL, Dawson LA, et al. ; ESMO Guidelines Committee. Electronic address: [email protected]. Hepatocellular carcinoma: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann Oncol. 2025;36:491-506.
8. Pinato DJ, Murray SM, Forner A, et al. Trans-arterial chemoembolization as a loco-regional inducer of immunogenic cell death in hepatocellular carcinoma: implications for immunotherapy. J Immunother Cancer. 2021;9:e003311.
9. Tan J, Fan W, Liu T, et al. TREM2+ macrophages suppress CD8+ T-cell infiltration after transarterial chemoembolisation in hepatocellular carcinoma. J Hepatol. 2023;79:126-40.
10. Wang Z, Li Q, Liang B. Hypoxia as a target for combination with transarterial chemoembolization in hepatocellular carcinoma. Pharmaceuticals. 2024;17:1057.
11. Romero D. Early promising results with addition of an ICI and an anti-angiogenic to TACE. Nat Rev Clin Oncol. 2025;22:157.
12. Sangro B, Kudo M, Erinjeri JP, et al. ; EMERALD-1 Investigators. Durvalumab with or without bevacizumab with transarterial chemoembolisation in hepatocellular carcinoma (EMERALD-1): a multiregional, randomised, double-blind, placebo-controlled, phase 3 study. Lancet. 2025;405:216-32.
13. Kudo M, Ren Z, Guo Y, et al. ; LEAP-012 investigators. Transarterial chemoembolisation combined with lenvatinib plus pembrolizumab versus dual placebo for unresectable, non-metastatic hepatocellular carcinoma (LEAP-012): a multicentre, randomised, double-blind, phase 3 study. Lancet. 2025;405:203-15.
14. Dong J, Han G, Ogasawara S, et al. LBA2 TALENTACE: a phase III, open-label, randomized study of on-demand transarterial chemoembolization (TACE) combined with atezolizumab + bevacizumab (Atezo+Bev) or on-demand TACE alone in patients with systemically untreated, intermediate-to-high burden unresectable hepatocellular carcinoma (uHCC). Ann Oncol. 2025;36:S62.
15. Zhu H, Teng G, Fan W, et al. Transarterial chemoembolization (TACE) combined with camrelizumab and apatinib versus TACE alone in the treatment of unresectable hepatocellular carcinoma eligible for embolization: a multicenter, open-label, randomized, phase 2 study (CAP-ACE). JCO. 2025;43:LBA522.
16. Zhou J, Sun H, Wang Z, et al. China liver cancer guidelines for the diagnosis and treatment of hepatocellular carcinoma (2024 Edition). Liver Cancer. 2025;14:779-835.
17. Li C, Chen L, Bao H, Wang Y, Zhu G. Synergistic era: Practice and reflection on TACE combined with systemic therapy in hepatocellular carcinoma. SciEriXiv. 2025.
18. Matsui O, Kobayashi S, Sanada J, et al. Hepatocelluar nodules in liver cirrhosis: hemodynamic evaluation (angiography-assisted CT) with special reference to multi-step hepatocarcinogenesis. Abdom Imaging. 2011;36:264-72.
19. Goldstein HM, Wallace S, Anderson JH, Bree RL, Gianturco C. Transcatheter occlusion of abdominal tumors. Radiology. 1976;120:539-45.
20. Nakamura H, Hashimoto T, Oi H, Sawada S. Transcatheter oily chemoembolization of hepatocellular carcinoma. Radiology. 1989;170:783-6.
21. Llovet JM, Real MI, Montaña X, et al. ; Barcelona Liver Cancer Group. Arterial embolisation or chemoembolisation versus symptomatic treatment in patients with unresectable hepatocellular carcinoma: a randomised controlled trial. Lancet. 2002;359:1734-9.
22. Lo CM, Ngan H, Tso WK, et al. Randomized controlled trial of transarterial lipiodol chemoembolization for unresectable hepatocellular carcinoma. Hepatology. 2002;35:1164-71.
23. Zhu HD, Liu R, Jia ZZ, et al. Transarterial chemoembolization for hepatocellular carcinoma: treatment algorithm proposed by Chinese College of Interventionalists(CCI). EngMedicine. 2024;1:1-9.
24. Miyayama S, Matsui O. Superselective conventional transarterial chemoembolization for hepatocellular carcinoma: rationale, technique, and outcome. J Vasc Interv Radiol. 2016;27:1269-78.
25. de Baere T, Ronot M, Chung JW, et al. Initiative on superselective conventional transarterial chemoembolization results (INSPIRE). Cardiovasc Intervent Radiol. 2022;45:1430-40.
26. Liu B, Gao S, Guo J, et al. Efficacy and safety of hepasphere drug-eluting bead transarterial chemoembolization combined with hepatic arterial infusion chemotherapy as the second-line treatment in advanced hepatocellular carcinoma. J Hepatocell Carcinoma. 2024;11:477-88.
27. Gjoreski A, Popova-Jovanovska R, Eftimovska-Rogac I, Vejseli J. Safety profile and efficacy of chemoembolization with doxorubicin - loaded polyethylene glycol microspheres in patients with hepatocellular carcinoma. Open Access Maced J Med Sci. 2019;7:742-6.
28. Lin CY, Liu YS, Pan KT, Chen CB, Hung CF, Chou CT. The short-term safety and efficacy of TANDEM microspheres of various sizes and doxorubicin loading concentrations for hepatocellular carcinoma treatment. Sci Rep. 2021;11:12277.
29. Vogl TJ, Lammer J, Lencioni R, et al. Liver, gastrointestinal, and cardiac toxicity in intermediate hepatocellular carcinoma treated with PRECISION TACE with drug-eluting beads: results from the PRECISION V randomized trial. AJR Am J Roentgenol. 2011;197:W562-70.
30. Ayyub J, Dabhi KN, Gohil NV, et al. Evaluation of the safety and efficacy of conventional transarterial chemoembolization (cTACE) and drug-eluting bead (DEB)-TACE in the management of unresectable hepatocellular carcinoma: a systematic review. Cureus. 2023;15:e41943.
31. Shi D, Ren Y, Liu Y, et al. Temperature-sensitive nanogels combined with polyphosphate and cisplatin for the enhancement of tumor artery embolization by coagulation activation. Acta Biomater. 2024;185:240-53.
32. Li Q, Shi J, Ruan X, et al. Hypoxia‐ and pH‐responsive metal‐phenolic network‐engineered ferroptosis‐immuno microspheres for transarterial chemoembolization in hepatocellular carcinoma. Adv Funct Mater. 2025;35:e10798.
33. Wen K, Ren Y, Huang J, et al. Dual-stage drug delivery microspheres amplify tumor starvation through coagulation-enhanced vascular occlusion. Small. 2025;21:e08070.
34. Zhang M, Zhou J, Jiang X, et al. MoS2 nanozyme-chlorella hydrogels: pioneering a hepatocellular carcinoma integrative therapy. Adv Funct Mater. 2025;35:2417125.
35. Xu J, Pei Z, Wang Y, et al. Bioactive microspheres to enhance sonodynamic-embolization-metalloimmune therapy for orthotopic liver cancer. Biomaterials. 2025;317:123063.
36. Yang N, Sun X, Zhou Y, et al. Liquid metal microspheres with an eddy-thermal effect for magnetic hyperthermia-enhanced cancer embolization-immunotherapy. Sci Bull. 2023;68:1772-83.
37. Llovet JM, Ricci S, Mazzaferro V, et al. ; SHARP Investigators Study Group. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378-90.
38. Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018;391:1163-73.
39. Matsui J, Funahashi Y, Uenaka T, Watanabe T, Tsuruoka A, Asada M. Multi-kinase inhibitor E7080 suppresses lymph node and lung metastases of human mammary breast tumor MDA-MB-231 via inhibition of vascular endothelial growth factor-receptor (VEGF-R) 2 and VEGF-R3 kinase. Clin Cancer Res. 2008;14:5459-65.
40. Li X, Qiu M, Wang S, Zhu H, Feng B, Zheng L. A phase I dose-escalation, pharmacokinetics and food-effect study of oral donafenib in patients with advanced solid tumours. Cancer Chemother Pharmacol. 2020;85:593-604.
41. Yau T, Park JW, Finn RS, et al. Nivolumab versus sorafenib in advanced hepatocellular carcinoma (CheckMate 459): a randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 2022;23:77-90.
42. Cheng AL, Qin S, Ikeda M, et al. Updated efficacy and safety data from IMbrave150: Atezolizumab plus bevacizumab vs. sorafenib for unresectable hepatocellular carcinoma. J Hepatol. 2022;76:862-73.
43. Rimassa L, Chan SL, Sangro B, et al. Five-year overall survival update from the HIMALAYA study of tremelimumab plus durvalumab in unresectable HCC. J Hepatol. 2025;83:899-908.
44. Yau T, Galle PR, Decaens T, et al. ; CheckMate 9DW investigators. Nivolumab plus ipilimumab versus lenvatinib or sorafenib as first-line treatment for unresectable hepatocellular carcinoma (CheckMate 9DW): an open-label, randomised, phase 3 trial. Lancet. 2025;405:1851-64.
45. Ren Z, Xu J, Bai Y, et al. ; ORIENT-32 study group. Sintilimab plus a bevacizumab biosimilar (IBI305) versus sorafenib in unresectable hepatocellular carcinoma (ORIENT-32): a randomised, open-label, phase 2-3 study. Lancet Oncol. 2021;22:977-90.
46. Qin S, Kudo M, Meyer T, et al. Tislelizumab vs sorafenib as first-line treatment for unresectable hepatocellular carcinoma: a phase 3 randomized clinical trial. JAMA Oncol. 2023;9:1651-9.
47. Qin S, Gu S, Chan SL, et al. ; CARES-310 Study Group. Camrelizumab plus rivoceranib versus sorafenib as first-line therapy for unresectable hepatocellular carcinoma (CARES-310): final analysis of a randomised, open-label, international, phase 3 study. Lancet Oncol. 2025;26:1598-611.
48. Zhao C, Zhang Y, Wang G, et al. Finotonlimab (PD-1 inhibitor) plus bevacizumab (bevacizumab biosimilar) as first-tier therapy for late-stage hepatocellular carcinoma: a randomized phase 2/3 trial. Signal Transduct Target Ther. 2025;10:249.
49. Shi Y, Han G, Zhou J, et al. Toripalimab plus bevacizumab versus sorafenib as first-line treatment for advanced hepatocellular carcinoma (HEPATORCH): a randomised, open-label, phase 3 trial. Lancet Gastroenterol Hepatol. 2025;10:658-70.
50. Strebel BM, Dufour JF. Combined approach to hepatocellular carcinoma: a new treatment concept for nonresectable disease. Expert Rev Anticancer Ther. 2008;8:1743-9.
51. Kudo M, Imanaka K, Chida N, et al. Phase III study of sorafenib after transarterial chemoembolisation in Japanese and Korean patients with unresectable hepatocellular carcinoma. Eur J Cancer. 2011;47:2117-27.
52. Lencioni R, Llovet JM, Han G, et al. Sorafenib or placebo plus TACE with doxorubicin-eluting beads for intermediate stage HCC: The SPACE trial. J Hepatol. 2016;64:1090-8.
53. Meyer T, Fox R, Ma YT, et al. Sorafenib in combination with transarterial chemoembolisation in patients with unresectable hepatocellular carcinoma (TACE 2): a randomised placebo-controlled, double-blind, phase 3 trial. Lancet Gastroenterol Hepatol. 2017;2:565-75.
54. Kudo M, Ueshima K, Ikeda M, et al. Final results of TACTICS: a randomized, prospective trial comparing transarterial chemoembolization plus sorafenib to transarterial chemoembolization alone in patients with unresectable hepatocellular carcinoma. Liver Cancer. 2022;11:354-67.
55. Duan X, Li H, Kuang D, et al. Comparison of drug-eluting bead transarterial chemoembolization combined with apatinib versus drug-eluting bead transarterial chemoembolization for the treatment of unresectable hepatocellular carcinoma: a randomized, prospective, multicenter phase III trial. Signal Transduct Target Ther. 2024;9:304.
56. Peng Z, Fan W, Zhu B, et al. Lenvatinib combined with transarterial chemoembolization as first-line treatment for advanced hepatocellular carcinoma: a phase III, randomized clinical trial (LAUNCH). J Clin Oncol. 2023;41:117-27.
57. Koroki K, Ogasawara S, Ooka Y, et al. Analyses of intermediate-stage hepatocellular carcinoma patients receiving transarterial chemoembolization prior to designing clinical trials. Liver Cancer. 2020;9:596-612.
58. Zhu HD, Li HL, Huang MS, et al. ; CHANCE001 Investigators. Transarterial chemoembolization with PD-(L)1 inhibitors plus molecular targeted therapies for hepatocellular carcinoma (CHANCE001). Signal Transduct Target Ther. 2023;8:58.
59. Jin ZC, Chen JJ, Zhu XL, et al. ; CHANCE2201 Investigators. Immune checkpoint inhibitors and anti-vascular endothelial growth factor antibody/tyrosine kinase inhibitors with or without transarterial chemoembolization as first-line treatment for advanced hepatocellular carcinoma (CHANCE2201): a target trial emulation study. EClinicalMedicine. 2024;72:102622.
60. Liu K, Yang L, Zhang X-M, et al. HIF-1α and VEGF levels for monitoring hepatocellular carcinoma treatment response to transcatheter arterial chemoembolization. Transl Cancer Res. 2017;6:1043-9.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].