


Evaluating normalization approaches for the better identification of aberrant microRNAs associated with hepatocellular carcinoma


Abstract
Aim: Dysregulated microRNAs (miRNAs) have been identified in hepatocellular carcinoma (HCC), but only a small proportion have been confirmed. An appropriate normalizer is crucial to determining the accuracy and reliability of data from miRNA studies.
Methods: Different normalization strategies were used to validate genome-wide miRNA profiles in HCC tumor and non-tumor tissues, and to determine the consistency and discrepancy of data on dysregulated miRNAs.
Results: Two sets of stable miRNAs (miR-30c/miR-30b and miR-30c/miR-126) were identified in HCC tissues by geNorm and NormFinder tools, respectively. The mean of global miRNAs also showed good stability for ranking the top 1-2 miRNAs, but the stabilities of the manufacturer-recommended ncRNAs controls were poor. Four panels of miRNAs were significantly associated with HCC by separately using various normalizers, and 14 miRNAs were consistently identified by three normalization strategies. Although fewer miRNAs (17-26) were dysregulated in HCC using the global mean or the 2 stable miRNAs as normalizers, perfect clustering of tissues was also obtained with only 1 to 2 misclassifications, suggesting the efficiency of the miRNA panels. Using global mean as the normalizer, the authors identified 7 miRNAs, including 2 novel (miR-324-5p and miR-550) significantly upregulated in HCC that were omitted when using 3 endogenous controls as the normalizer.
Conclusion: An optimal normalization strategy to identify biologically important miRNAs in HCC tissue studies of miRNA may be the combination of global mean and 2 stable miRNAs. Selection of appropriate normalization strategies to adjust miRNAs levels is particularly important for epidemiological studies dealing with large data sets and covering multiple experimental batches.
Keywords
Introduction
MicroRNAs (miRNAs) have important functions in negatively regulating coding genes’ expression and controlling multiple biological processes (DNA damage/repair, apoptosis, proliferation, differentiation, etc.) involved in tumorigenesis and progression.[1,2] Genome-wide and candidate gene approaches have been broadly used to identify miRNA biomarkers in order to better understand the effects of carcinogenic exposure, pathogenesis, and cancer risk, as well as for early diagnosis and prognostic prediction. Currently, over 100 mature miRNAs have been found to be dysregulated in hepatocellular carcinoma (HCC) tissues or blood.[3,4] Many are also associated with various HCC risk factors, such as hepatitis B/C virus infection,[5,6] aflatoxin B1 exposure,[7] alcohol drinking, non-alcoholic fatty liver disease or non-alcoholic steatohepatitis.[8-10] However, only a small proportion of miRNAs (miR-1, miR-9, miR-16, miR-18a, miR-21, miR-92a, miR-101, miR-122, miR-199a, miR-221, miR-222/223/224, miR-375, miR-483-5p)[11,12] were consistently confirmed by different studies for their role in hepatocarcinogenesis.[13,14] These discrepant results may be attributed to a variety of factors that potentially impact miRNA patterns but differ by studies. These factors include the difference in study design (cross-sectional, retrospective or prospective); heterogeneity of cancer patients (tumor types, stages, progression, treatment, hepatitis B, C or mixed viral etiologies); comparison groups (healthy or hepatitis infection controls or non-tumor tissues); types of biospecimens (fresh, frozen or formalin-fixed, paraffin-embedded tissue, serum, plasma, or exosome); and variations in sample collection, preservation and processing. Differences of RNA isolation assays, the input RNA quantity/quality and detection methods can also impact miRNA expression levels.
Even if a careful study design is used and consistent implementation is applied to pre-analytical and analytical procedures, different methods and “housekeeping” transcripts used to normalize miRNA expression levels may also bias the results and lead to misinterpretation of the biological role of miRNAs in tumorigenesis. The purpose of normalization is to remove as much non-biological variations as possible to ensure accurate miRNA results within or between experiments.[15] Therefore, how to select an appropriate normalizer to adjust miRNA expression profiles are crucial to obtaining comparable results. This is particularly important for epidemiological studies dealing with large data sets usually covering multiple experimental batches.
The most common methods to quantitate miRNA levels are quantitative real-time polymerase chain reaction (qPCR) and hybridization microarrays. The methods apply stem-loop reverse transcription and TaqMan probes (TaqMan low density arrays, TLDA, Life Technologies) or locked nucleic acid (LNA) primers (miRCURY LNA™ miRNA arrays, miRCURY, Exiqon) or poly (A)-tailed primers (miScript miRNA PCR arrays, miScript, QIAGEN) and SYBR Green detection. The normalization for miRNA levels usually uses “housekeeping” transcripts, i.e. a reference-gene-based method. Because no universal references have been accepted by all researches, a variety of endogenous or exogenous transcripts have been selected as references by different microarray and qPCR assays for data normalization. TLDA includes a total of 5 endogenous controls (U6 snRNA, RNU44, RNU48, RNU24 and MammU6) and miScript uses spike in cel-miR-39 and 6 references (SNORD61, SNORD68, SNORD72, SNORD95, SNORD96A and RNU6B/RNU6-2) as normalizers, while miRCURY recommends 5 most stable miRNAs (hsa-let-7i-5p, hsa-miR-222-3p, hsa-miR-425-5p, hsa-miR-93-5p, hsa-miR-152) as endogenous references, rather than small RNA species (snoRNA and snRNA). Even using the same microarray, different studies may artificially select various numbers of references to normalize their results. One study applied miScript array to profile expression of 84 miRNAs in hepatitis B virus-related HCC and controls, but only used 2 (SNORD61, RNU6-2) out of 6 snRNAs and spiked in cel-miR-39 as the normalizer to standardize miRNAs expression.[16] Another study examined miRNAs in HCC patients and matched controls by the miRCURY assay using the median of 50% quantile intensity to normalize data.[12] Seven published studies including ours have screened miRNA profiles by TLDA in either HCC tissue or serum/plasma. Two used four endogenous controls (U6 snRNA, RNU24, RNU44 and RNU48) to normalize target miRNA expression in HCC tissues;[17,18] four studies only used one reference (U6 snRNA[11,19,20] or RNU48);[21] and one study did not indicate the reference.[22] More importantly, whether those endogenous normalizers are stable among tested samples is unknown.[11,17-22]
Global normalization is another strategy which uses either the mean or median of detectable miRNAs in each sample as the calibrator to adjust miRNA expression profiles; this method is adopted from mRNA microarray data normalization protocols.[23] It is assumed that the mean or the median level of global or most miRNAs is constant across different tissues or conditions.[24] Although many studies have demonstrated the advantages of using global normalization,[23,25] the total number of detectable miRNAs are much less than mRNAs, which makes it susceptible to extreme values and may bias miRNAs expression patterns.[15,26] In addition, large epidemiological studies usually require independent validation for a limited number of miRNAs identified in a discovery set. Practically, it is also not feasible to use global miRNA profiles to normalize expression of candidate miRNAs.
Measurement of miRNAs by qPCR is considered the gold standard for specific and sensitive detection of interesting miRNAs that may be present at very low levels. This approach is usually used to validate the findings from previous large-scale microarray profiling. Although commercial ready-to-use kits are available for almost all human mature miRNAs, how to select an appropriate endogenous control to normalize miRNA expression is still a challenge. A number of endogenous miRNAs, snRNA/snoRNA, and synthesized exogenous RNAs have been used as controls to normalize target miRNA expression in different studies because there is no widely accepted endogenous control. Most are based on previous literature or because a low standard deviation (SD) was observed in the microarray data. Several small RNAs (U6 snRNA,[16,19,27] RNU44,[18] cel-miR-39,[16,20] cel-miR-54)[22] have been frequently used as calibrators in previous HCC studies. One study also used a standard curve approach for absolute quantitation by spiking in an artificial reference (ath-miR-156a).[12] Therefore, it is not surprising that many previously identified miRNA panels are quite different between studies.
Here, we utilized genome-wide miRNA expression data derived from HCC tumor and non-tumor tissues to compare the miRNA panels identified as differentially expressed by using different normalization strategies. We sought to identify an optimal strategy to select stable references for miRNA normalization that can generate the most concordant miRNA panel deregulated in HCC. This strategy should also be feasible for large epidemiological studies, and more likely to reproducibly identify HCC-associated miRNAs in different studies.
Methods
Participants come from a previous HCC tissue study[28-30] conducted at Columbia University Medical Center (CUMC) and approved by the Institutional Review Board of CUMC. A waiver of consent was given in the study because the majority of patients died before the research was carried out.
A total of 16 paired frozen tumor and adjacent non-tumor tissues were screened for miRNA profiling. Tissue samples were collected and stored in the Molecular Pathology Shared Resource of the Herbert Irving Comprehensive Cancer Center. Tumor samples were microdissected to ensure > 80% purity of tumor. Tumor stage was determined according to the American Joint Committee on Cancer criteria.[31] To insure adjacent non-tumor tissue did not contain any tumor cells, tissue sections were cut from frozen tissues, and hematoxylin and eosin stained. The stained sections were carefully observed under a microscope by the study pathologist (HR) to ensure no tumor tissues or cells were present in the whole sections. Frozen tissue blocks of adjacent tissue were also evaluated with respect to the presence (Batts-Ludwig stage of 4) or absence of cirrhosis (Batts-Ludwig stage < 4).
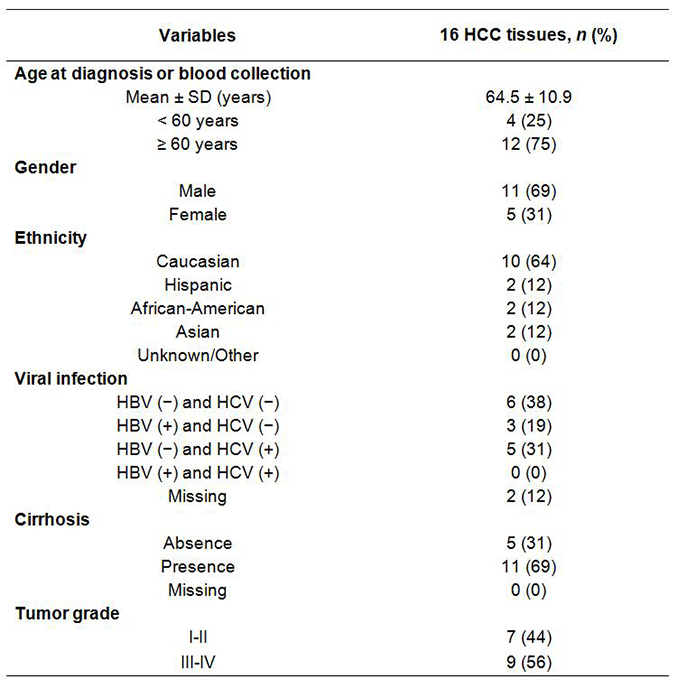
The demographic and clinic pathological data were collected from medical and pathological records including age, gender, ethnicity, viral infection (hepatitis B, hepatitis C), α-fetoprotein, tumor size and number, tumor grade, presence of vascular invasion, and capsular infiltration. Hepatitis B virus surface antigen and antibody against hepatitis C virus determined by immunoassay were also obtained [Supplementary Table 1].
Clinical and pathological characteristics of study subjects
 |
Total RNA, including miRNAs was isolated from 32 tissues by RNeasy Microarray Tissue Mini Kits (Qiagen, Frederick, MA) according to the manufacturer’s protocols. TaqMan Low Density Arrays (TLDA, Applied Biosystems, Foster City, CA), covering 733 miRNAs (670 unique human mature miRNAs), were used to quantify genome-wide miRNAs levels using a 7900HT Fast Real-Time PCR System. The same amount (750 ng) of total RNA was used for each array measurement. The means of RNA integrity number and A260/A280 ratio were respectively 5.9 and 2.1. The quantification cycle (Cq) defined as the cycle number when fluorescence passes the detectable threshold was obtained and raw Cq values ≥ 40 were excluded. These data have been deposited in NCBI’s Gene Expression Omnibus database (accession number GSE54751).[29,30]
The first strategy used the endogenous controls recommended by the TLDA array manufacturer as the normalizer. Three (U6 snRNA, RNU44 and RNU48), detected in all tissue samples, were selected as normalizers in order to obtain reliable results. The second strategy used the mean of global miRNAs obtained from miRNAs detectable in all tested samples as the normalizer. There were 157 miRNAs/ncRNAs detected in 100% of HCC tissues [Supplementary Table 2]; means of all miRNAs were separately calculated for each sample and then used as the normalizer. The third strategy evaluated the stabilities of those 100% detectable miRNAs/ncRNAs by statistical algorithms. The most stable 2 miRNAs were selected as normalizers.
The Cq values of 157 miRNAs/ncRNAs detectable in 100% of HCC tissues, which were used to determine the most stable 2 normalizers
 |
Two statistical algorithms (geNorm[32] and NormFinder[33]) were used to estimate the stabilities of the miRNAs profiles. The algorithm of geNorm is to calculate the average pairwise variation (V) for each miRNA with all others across the samples, and estimate a stability score (M) defined as the average V of a miRNA with all others. The less stable miRNA with the highest M is gradually removed until the 2 most stable normalizers are obtained. The algorithm of NormFinder is to calculate the inter- and intra-group variances of the log-transformed miRNAs expression data, and integrate it into a stability value to represent the systematic error of each miRNA. A lower value of systematic error indicates a more stable miRNA, and the combination of the most stable miRNAs is selected as the normalizer. Different types of normalizers (array recommended ncRNAs, mean of global miRNAs and the most stable miRNAs combination) were separately used to generate miRNA expression profiles from HCC tissue samples for future statistical data analysis.
Before performing any statistical analysis, the genome-wide miRNA profiles were checked to ensure the reliability and abundance of miRNAs. If the missing data (Cq ≥ 40) for any miRNA exceeded 50% of samples, this miRNA was excluded from further data analyses. Paired t-test was used to identify miRNAs that were significantly different by the univariate test (P < 0.001) with at least a 2-fold expression change between paired HCC tumor/non-tumor tissues or HCC cases and matched controls. Volcano plots were generated to describe the distribution of significant miRNAs with over 2-fold changes. Hierarchical clustering and heat maps were produced with average linkage and Pearson correlations to examine the classification of samples based on significant miRNAs. All statistical analyses were performed using BRB-ArrayTools (version 4.4) developed by Dr. Richard Simon and the BRB-ArrayTools Development Team (http://linus.nci.nih.gov/BRB-ArrayTools.html)[34] and Statistical Analysis System 9.0 (SAS Institute). The panels of significant miRNAs identified by different normalizers were compared using a web-based InteractiVenn tool (http://www.interactivenn.net/)[35] to determine the consistent and discrepant miRNAs.
Results
The stabilities of global miRNAs/ncRNAs
Using geNorm and NormFinder tools, we separately examined the stabilities of global miRNA profiles in HCC tissues. Among 157 miRNAs/ncRNAs tested in HCC tissues, the combination of miR-30c/miR-30b had the smallest M score of 0.024 by geNorm [Supplementary Figure 1], and miR-30c/miR-126 together had the lowest systematic error (stability value) of 0.133 by NormFinder [Supplementary Table 3], suggesting their good stabilities for normalization. If we included the means of global miRNAs in the stability analyses, the M score and stability value for miR-30c/mean of miRNAs combination were, respectively 0.022 and 0.088, lower than the combination of miR-30c with miR-30b or miR-126 [Supplementary Table 3]. This suggests that the mean of global miRNAs may be a good normalizer due to its high stability among samples. The stabilities of the manufacturer-recommended normalizer ncRNAs (U6 snRNA, RNU44 and RNU48) ranked much lower in the 17th to 140th range out of a total 157 candidates, indicating their poor stability in HCC tissue.
Stability tests (geNorm and NormFinder) for 157 miRNAs/ncRNAs that were detectable in 100% of HCC tissues
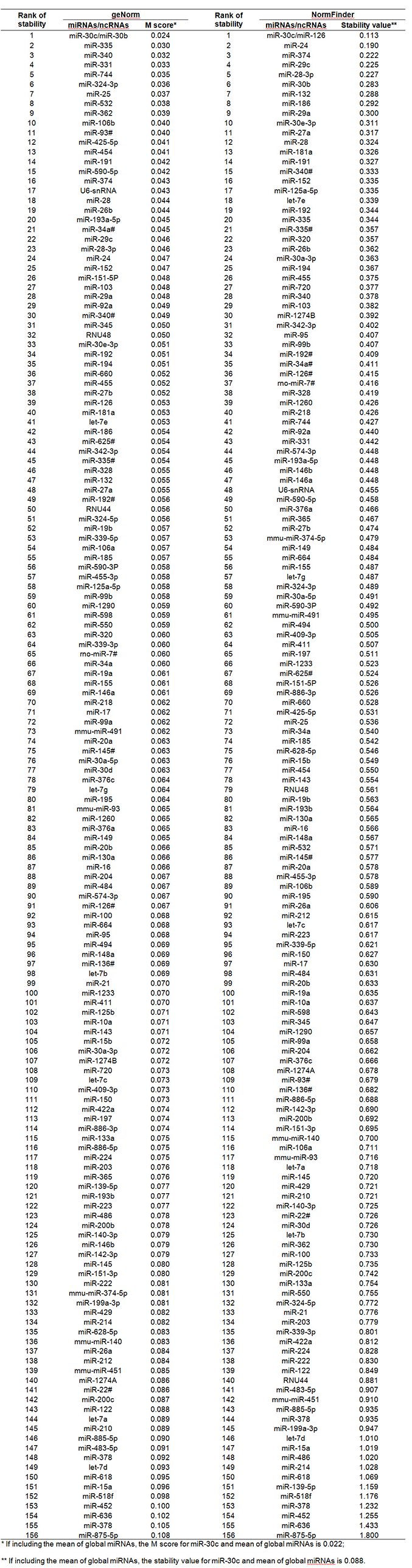 |

Supplementary Figure 1. The distribution of M scores for 157 miRNAs/ncRNAs tested by geNorm tool in HCC tissues. The combination of miR-30c/miR-30b had the smallest M score of 0.024. The stabilities of U6 snRNA, RNU48 and RNU44 ranked 17th, 32nd and 50th respectively, out of 157 candidates, indicating their unsuitability as normalizers
Aberrant miRNA panels identified by using varied normalizers
After excluding non-abundant miRNAs and those with missing data in over 50% of samples, a total of 361 miRNAs were finally analyzed. Using 3 endogenous controls (U6 snRNA, RNU44 and RNU48) as the normalizer, we found 46 miRNAs significantly dysregulated (P < 0.001) in HCC tumor tissues with at least 2-fold changes in expression [Supplementary Table 4, Figure 1A]. Most miRNAs (43) were significantly down-regulated in HCC tumor tissue (from 2 to 10-fold), and only 3 miRNAs were significantly up-regulated with fold changes of 5 to 9.
Using 3 endogenous controls to normalize miRNAs expression identified 46 miRNAs significantly aberrantly expressed in HCC tumor tissue
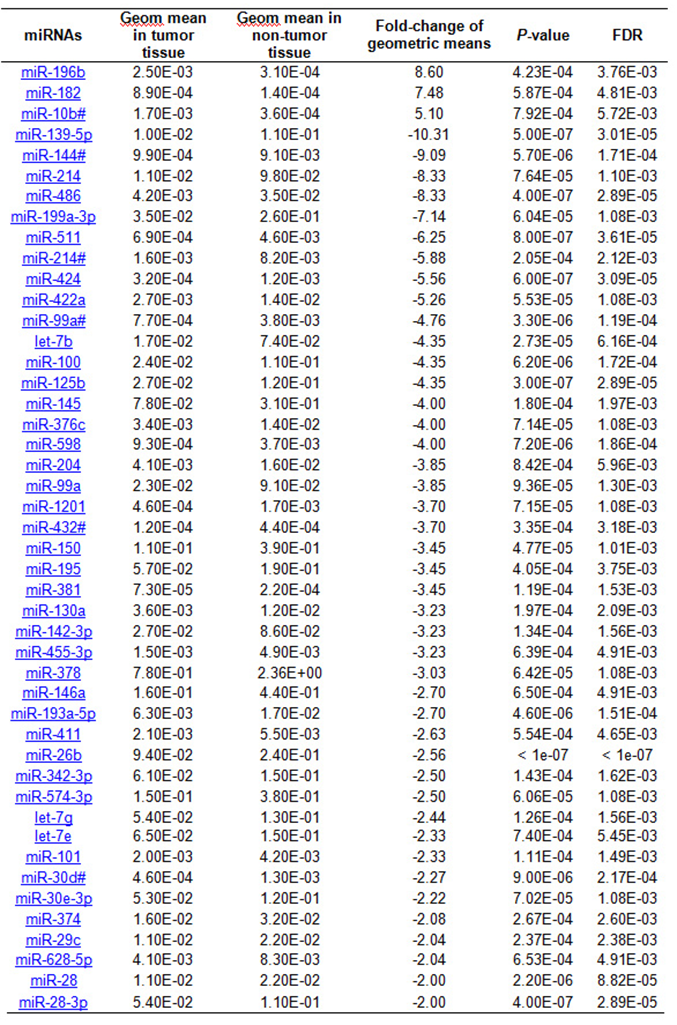 |

Figure 1. Volcano plots of the microRNAs (miRNAs) with significant (P < 0.001) over 2-fold expression changes in hepatocellular carcinoma (HCC) tumor compared to nontumor tissues. (A) There were more downregulated miRNAs aberrantly expressed in HCC tumor tissue with only 3 miRNAs significantly up-regulated if using 3 endogenous controls (U6 snRNA, RNU44 and RNU48) as the normalize; (B) Expression of 26 miRNAs (17 upregulated and 9 downregulated) significantly differed between tumor and non-tumor tissues; (C) Seventeen miRNAs were aberrantly expressed in HCC tumor tissue using 2 stable miRNAs (miR-30c and miR-30b) identified by geNorm tool as the normalize; (D) Twenty miRNAs were significantly deregulated in HCC tumor tissues using 2 stable miRNAs (miR-30c and miR-126) identified by NormFinder tool as the normalizer. The overall expression patterns of miRNAs in (B, C and D) were similar but different from (A) with many more down-regulated miRNAs
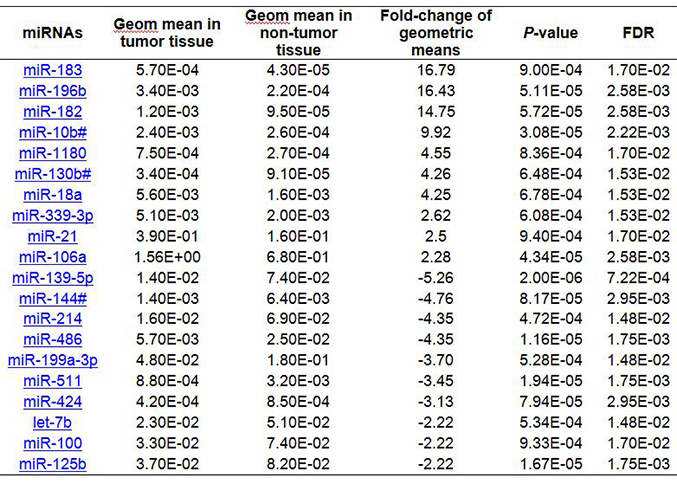
Using the mean of all miRNAs as a normalizer, a total of 26 miRNAs were significantly different between HCC tumor and non-tumor tissues with over 2-fold changes [Table 1, Figure 1B]. The aberrant expression pattern was quite different from that identified by using 3 endogenous controls. More miRNAs (17) were upregulated 2- to 16-fold compared with 9 significantly downregulated miRNAs (2-5 fold) in HCC tumor tissues. The expression levels of endogenous controls (U6 snRNA and RNU44) were increased while RNU48 was reduced in tumor tissue when using the mean of all miRNAs as a normalizer, but no significant difference was obtained (data not shown). The fold-changes between tumor and non-tumor tissues were varied from -1.28 to 4.09 times.
The 26 miRNAs significantly aberrantly expressed in HCC tumor compared to nontumor tissues using the mean of all miRNAs as the normalizer
| miRNAs | Geometric mean in tumor tissue | Geometric mean in non-tumor tissue | Fold-change | P-value | FDR |
|---|---|---|---|---|---|
| miR-196b | 1.10E-01 | 7.30E-03 | 16.56 | 2.88E-05 | 1.41E-03 |
| miR-183 | 1.90E-02 | 1.40E-03 | 16.29 | 1.86E-04 | 4.47E-03 |
| miR-182 | 4.10E-02 | 3.20E-03 | 14.11 | 1.77E-05 | 1.41E-03 |
| miR-10b# | 8.10E-02 | 8.60E-03 | 10.13 | 1.41E-05 | 1.41E-03 |
| miR-1180 | 2.50E-02 | 9.10E-03 | 4.56 | 2.90E-04 | 6.55E-03 |
| miR-221 | 3.10E-01 | 7.40E-02 | 4.13 | 5.87E-04 | 1.00E-02 |
| miR-18a | 1.90E-01 | 5.70E-02 | 4.00 | 1.39E-04 | 3.58E-03 |
| miR-130b# | 1.10E-02 | 3.20E-03 | 3.97 | 7.69E-04 | 1.06E-02 |
| miR-222 | 1.81E+01 | 6.42E+00 | 2.81 | 6.48E-04 | 1.02E-02 |
| miR-339-3p | 1.70E-01 | 6.50E-02 | 2.62 | 1.08E-04 | 3.54E-03 |
| miR-21 | 1.30E+01 | 5.20E+00 | 2.49 | 9.27E-04 | 1.12E-02 |
| miR-324-5p | 8.30E-02 | 3.40E-02 | 2.47 | 6.11E-04 | 1.00E-02 |
| miR-550 | 3.30E-02 | 1.40E-02 | 2.38 | 3.87E-04 | 7.76E-03 |
| miR-362 | 1.10E-01 | 4.90E-02 | 2.30 | 7.96E-04 | 1.06E-02 |
| miR-148b | 6.60E-02 | 3.00E-02 | 2.28 | 8.78E-04 | 1.09E-02 |
| miR-106a | 5.19E+01 | 2.29E+01 | 2.27 | 8.20E-06 | 1.41E-03 |
| miR-93# | 1.30E+00 | 6.10E-01 | 2.14 | 8.28E-04 | 1.07E-02 |
| miR-139-5p | 4.70E-01 | 2.47E+00 | -5.26 | 3.13E-05 | 1.41E-03 |
| miR-144# | 4.60E-02 | 2.10E-01 | -5.00 | 5.78E-05 | 2.09E-03 |
| miR-214 | 5.20E-01 | 2.29E+00 | -4.35 | 7.09E-04 | 1.02E-02 |
| miR-486 | 1.90E-01 | 8.20E-01 | -4.35 | 5.10E-06 | 1.41E-03 |
| miR-199a-3p | 1.61E+00 | 5.95E+00 | -3.70 | 6.84E-04 | 1.02E-02 |
| miR-511 | 3.00E-02 | 1.10E-01 | -3.33 | 1.28E-04 | 3.54E-03 |
| miR-424 | 1.40E-02 | 2.70E-02 | -2.94 | 4.02E-05 | 1.61E-03 |
| miR-125b | 1.23E+00 | 2.74E+00 | -2.22 | 2.24E-05 | 1.41E-03 |
| miR-598 | 4.30E-02 | 8.50E-02 | -2.00 | 5.96E-04 | 1.00E-02 |
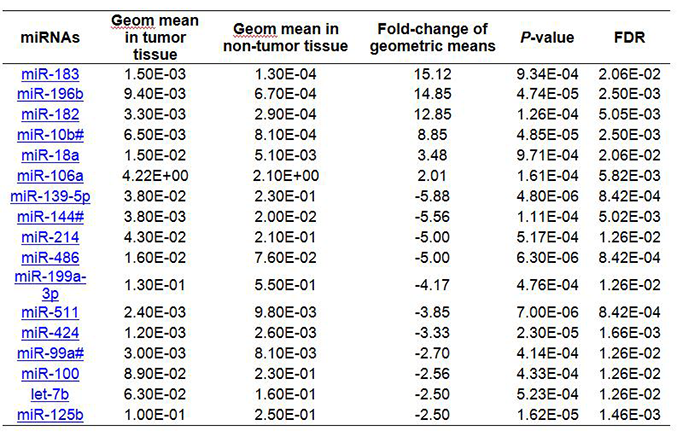
Supplementary Table 5 and Figure 1C display a panel of 17 miRNAs aberrantly expressed in HCC tumor tissue using 2 stable miRNAs (miR-30c and miR-30b) identified by the geNorm tool as the normalizer. Six were significantly upregulated 2- to 15-fold; 11 were downregulation 3- to 6-fold in HCC tumor tissue. Similarly, using 2 stable miRNAs (miR-30c and miR-126) identified by the NormFinder tool as the normalizer, we found 20 significantly deregulated miRNAs with over 2-fold changes between HCC tumor and non-tumor tissues [Supplementary Table 6, Figure 1D]. Half were upregulated 2- to 17-fold, and the others were downregulated 2- to 5-fold in HCC tumor tissue. Thus, the identified miRNA panels varied with the different normalization strategies.
Using geNorm tool identified 2 miRNAs (miR-30c and miR-30b) as a normalizer resulting in17 miRNAs being significantly aberrantly expressed in HCC tumor tissue
 |
Using NormFinder tool identified 2 miRNAs (miR-30c and miR-126) as a normalizer resulting in 20 miRNAs being significantly aberrantly expressed in HCC tumor tissue
 |
Comparisons of different miRNA panels in classification of HCC status
The hierarchical clustering and heat map showed that a panel of 46 significant miRNAs could well distinguish HCC tumor from non-tumor tissues using 3 endogenous controls as the normalizer [Figure 2]. Only 1 tumor and 1 non-tumor tissue were misclassified. Similarly, the other 3 panels of miRNAs identified using as normalizers the mean of all miRNAs or the 2 most stable miRNAs also well classified HCC tumor from non-tumor tissues with only 1 or 2 misclassifications. The later three panels consisting of 17 to 26 aberrant miRNAs contained many fewer miRNAs than that using 3 endogenous controls as the normalizer, suggesting a more efficient panel of HCC classification.

Figure 2. Comparisons of different microRNAs (miRNAs) panels identified with varied normalization strategies in distinguishing hepatocellular carcinoma (HCC) tumor from non-tumor tissues. (A) A panel of 46 miRNAs identified using 3 endogenous controls as the normalizer can distinguish HCC tumor from non-tumor tissues with 2 misclassifications. The other 3 panels of miRNAs identified by using as normalizers of the mean of all miRNAs (B), miR-30c/miR-30b (C) and miR-30c/miR-126 (D) also classified HCC tumor from non-tumor tissues with only 1 or 2 misclassifications
Consistence of identified miRNAs by different normalizers
Using InteractiVenn to compare different panels of miRNAs dysregulated in HCC, we found that most miRNAs identified using the mean of miRNAs overlapped with those identified when using 2 stable miRNAs as the normalizer [Supplementary Figure 2A]. There were, respectively 14 (54%) and 18 (69%) miRNAs consistent with the 26 significant miRNAs identified from the global mean analysis. A total of 14 miRNAs (miR-196b, miR-183, miR-182, miR-10b#, miR-18a, miR-106a, miR-139-5p, miR-144#, miR-214, miR-486, miR-199a-3p, miR-511, miR-424 and miR-125b) were consistently dysregulated by using the mean of miRNAs or 2 stable miRNAs as normalizers [Figure 3A], suggesting a high consistency for these normalization strategies. Using the mean of miRNAs as the normalizer, we found 8 additional dysregulated miRNAs (miR-221, miR-222, miR-324-5p, miR-550, miR-362, miR-148b, miR-93# and miR-598) in HCC tissue that were not significant when using 2 stable miRNAs as the normalizer [Supplementary Table 7]. Seven dysregulated miRNAs (miR-183, miR-1180, miR-18a, miR-130b#, miR-339-3p, miR-21 and miR-106a) were identified in HCC tumor tissues using either the mean of global miRNAs or miR-30c/miR-126 (selected as the most stable miRNAs by using NormFinder tool) as normalizers, including functionally important oncogenic miR-21, miR-18a, miR-106a and miR-183 [Supplementary Figure 3A]. Using either the mean of global miRNAs or miR-30c/miR-30b (selected as the most stable miRNAs by using geNorm tool) as normalizers, the same oncogenic miR-183, miR-18a and miR-106a were identified [Supplementary Figure 3B]. These miRNAs would not have been identified if using 3 endogenous ncRNAs as the normalizer. These data suggest that the combination of using the mean of miRNAs and 2 stable miRNAs identified by NormFinder as normalizers may be a good option to pinpoint biologically important miRNA.
Dysregulated miRNAs identified by using the mean of miRNAs and the 2 most stable miRNAs as normalizers
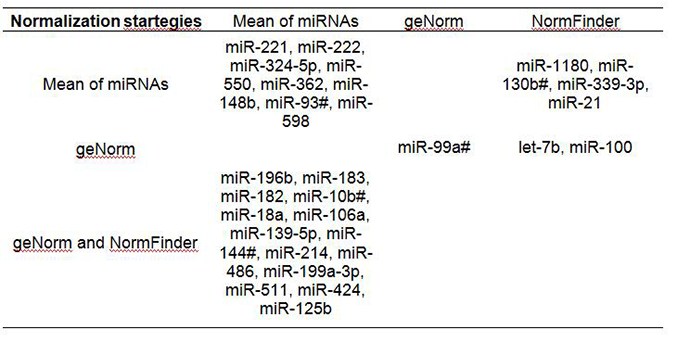 |

Figure 3. InteractiVenn determination of the consistent and discrepant microRNA (miRNA) panels identified using different normalization strategies. Using the mean of miRNAs and 2 stable miRNAs as normalizers (A), 14 miRNAs (miR-196b, miR-183, miR-182, miR-10b#, miR-18a, miR-106a, miR-139-5p, miR-144#, miR-214, miR-486, miR-199a-3p, miR-511, miR-424 and miR-125b) were consistently identified as dysregulated in hepatocellular carcinoma tumor tissue. (B) Compared to the panel identified using endogenous controls as the normalizer, a total of 11 miRNAs (miR-196b, miR-182, miR-10b#, miR-139-5p, miR-144#, miR-214, miR-486, miR-199a-3p, miR-511, miR-424 and miR-125b) were consistently identified by all normalization strategies

Supplementary Figure 2. Comparisons of the consistency and discrepancy of the identified miRNA panels using different normalization strategies. (A) Most miRNAs (14-18) identified by using the mean of miRNAs overlapped with those identified using 2 stable miRNAs as the normalizer. (B) 12-14 miRNAs overlapped with the 46 identified using 3 endogenous controls as the normalizer

Supplementary Figure 3. Comparisons of the consistency and discrepancy of miRNA panels identified using global mean or top stable miRNAs as normalizers. (A) Using the mean of global miRNAs or miR-30c/miR-126 as normalizers, 7 dysregulated miRNAs (miR-183, miR-1180, miR-18a, miR-130b#, miR-339-3p, miR-21 and miR-106a) were identified in HCC tumor tissues. (B) Using the mean of global miRNAs or miR-30c/miR-30b as normalizers, 3 dysregulated oncogenic miRNAs (miR-183, miR-18a and miR-106a) were identified. These miRNAs were not identified using 3 endogenous ncRNAs as normalizer
Comparing these miRNA panels with that identified by using 3 endogenous controls as the normalizer, less than one third (12-14) out of a total 46 miRNAs overlapped [Supplementary Figure 2B]. A total of 11 miRNAs (miR-196b, miR-182, miR-10b#, miR-139-5p, miR-144#, miR-214, miR-486, miR-199a-3p, miR-511, miR-424 and miR-125b) were consistently identified by all normalization strategies [Figure 3B, Supplementary Table 8], suggesting the importance of these miRNAs. Thirty-one miRNAs were only downregulated in HCC when using 3 ncRNAs (U6 snRNA, RNU44 and RNU48) as the normalizer, which may be due to the fact that the expression levels of the 3 ncRNAs were significantly enhanced in HCC tumor (data not shown). The mean Cq of ncRNAs in HCC tumor was significantly higher than in non-tumor tissues (22.2 vs. 23.0, P = 4.77E-07). Therefore, using the unstable and upregulated ncRNAs as the normalizer, we may falsely identify miRNAs downregulated in target tissue. In contrast, using the more stable global mean of miRNAs as the normalizer, an additional 7 miRNAs (miR-221, miR-222, miR-324-5p, miR-550, miR-362, miR-148b, miR-93#), including 2 novel (miR-324-5p, miR-550) were identified as significantly upregulated in HCC tumor tissues, which would not have been identified when using 3 endogenous controls as the normalizer [Supplementary Table 8].
Dysregulated miRNAs identified by using 3 endogenous controls, the mean of miRNAs and the 2 most stable miRNAs as normalizers
 |
Discussion
Our study for the first time, demonstrated that using different normalizers identifies diverse aberrant miRNA patterns in HCC tumors [Figures 1 and 2], and a combination of global mean and the top stable miRNAs as normalizer might be an optimal strategy to identify biologically meaningful miRNAs [Table 1 and Figure 3]. We derive this conclusion based on the assumption that overall miRNA expression levels are invariable because all up- and down-regulated miRNAs are similarly distributed[15,26] and only a small proportion of specific miRNAs significantly vary across samples due to different biological conditions,[23,36] such as hepatocarcinogenesis. Therefore, selection of the most stable candidate as the normalizer is the key principle to adjust for variations from sample and technical differences during miRNA measurements. We found that both global mean and 2 sets of miRNAs as normalizers in the current study ranked high in terms of stability, while the endogenous controls recommended by the manufacturer were not stable and usually up-regulated in HCC tumors [Supplementary Figure 1]. If using the endogenous controls as normalizer, we would obtain more miRNAs that were significantly down-regulated in HCC tumor tissue, but many of them might be false positive findings due to using an inappropriate normalizer to adjust miRNA expression [Figures 1-3]. In contrast, using the global mean of miRNAs and miR-30c/miR-126 as normalizers, several functionally important oncogenic miRNAs (miR-21,[37-40] miR-18a,[41] miR-106a[41,42] and miR-183[37,43-45]) were identified as dysregulated in HCC tumor tissues [Supplementary Figure 3]. Several well-known oncogenic miRNAs (miR-221[46-49], miR-222[46,48,49], miR-362[50,51]) and two miRNAs (miR-324-5p and miR-550) first identified in HCC tumor tissue were significantly over-expressed by using miRNA global mean as the normalizer [Supplementary Table 7]. These miRNAs would not have been discovered using the 3 endogenous controls as normalizer. Our results were strongly supported by the evidence obtained from previous studies that using global expression mean as normalizer significantly reduces technical variation (standard deviations) across samples and faithfully represents the input amount of total RNA.[23,52] More importantly, this approach also showed maximum separation for biologically different samples and significantly reduces false positive findings of down-regulated miRNAs.[23,52] It suggests that the combination of global mean and the top stable miRNAs as the normalizer may be a good option to identify biologically important miRNA in hepatocarcinogenesis.
Although using this strategy may raise concern that measuring global miRNA profiles for all participants in a large epidemiological study is not feasible, we strongly recommended running at least a subset of representative samples or samples mixed from all subjects to select the most stable candidates among detectable miRNAs for normalization. This additional step is necessary to ensure a proper normalization strategy for miRNA quantification and comparison. Mestdagh et al.[23] proposed a similar strategy that first obtained miRNA expression levels by using global mean of miRNAs as normalizer, and then identified the most stable miRNAs by comparing the normalized miRNAs, which may be influenced by the extreme values of specific miRNAs that were used to estimate the global mean.[26] We simplified the procedure by directly evaluating the stabilities of fully detectable miRNAs using unadjusted raw Cq value to exclude this potential impact of extreme data. Most aberrant miRNAs (54-69%) identified by using global mean and 2 stable miRNAs as normalizers overlapped [Supplementary Figure 2A] in the current study, which is consistent with a previous study that showed over 65% of miRNAs displaying significant correlation coefficients of above 0.9 using global mean and stable miRNAs as normalizers.[23] Therefore, this normalization strategy outperforms other available approaches and is also straightforward to be performed in future large epidemiological studies.
Three miRNAs (miR-30c, miR-30b and miR-126) were identified as normalizers in the current study, indicating their expression levels remained stable in liver tissue regardless of HCC status. In contrast, several previous studies found significant dysregulation of miR-30c,[21,53] miR-30b[21] and miR-126[54,55] in HCC tumor tissue and blood samples, suggesting their potential etiologic or diagnostic roles. However, none of those previous studies used global mean of miRNAs as the normalizer and did not evaluate miRNAs expression stabilities that might lead to false positive findings. Several studies also identified these 3 miRNAs as dysregulated in other types of human cancers.[56-58] Because miRNAs have a characteristic of tissue type specificity,[24] the expression stabilities of miR-30c, miR-30b and miR-126 should be separately validated in relevant tissues before drawing conclusions on their role in carcinogenesis.
Overall, the optimal normalization strategy described here can help identify the most concordant miRNA panel differentiating HCC tumor from non-tumor tissues that are feasible to be used for future validation in large epidemiological studies. This strategy can also prevent potential false positive findings, largely down-regulated miRNAs. Of course, this strategy may improve the identification of novel miRNAs. Two novel miRNAs (miR-324-5p and miR-550) were identified as significantly over-expressed in HCC tumor tissue using the optimal normalization strategy. However, the weaknesses of current study need to be recognized, such as a small sample size, no data for different arrays and lack of validation for identified miRNAs in larger and independent patients.
In summary, normalization methods impact on miRNAs that are differentially expressed in tumors; often many studies do not consider how normalization methods impact their findings. We first ascertained two sets of stable miRNAs in liver tissue that are independent of HCC status, and emphasize the importance of using a proper normalization strategy to identify aberrant miRNAs associated with HCC. In combination with the global mean of miRNA profiles as the normalizer, we finally identified a panel of miRNAs dysregulated in HCC, and were able to exclude potential false positive findings in hepatocarcinogenesis. Our results need to be further validated in other independent studies to ensure distinguishing of biologically meaningful miRNAs in HCC. Studies using different approaches (TLDA, miScript, miRCURY, etc.) but the same set of stable miRNAs to validate our findings are also warranted to strengthen the significance.
This work is supported by NIH grants R01 ES005116 (RMS), P30 ES009089 (RMS), R03 CA156629 (JS).
Conflicts of interestThere are no conflicts of interest.
Patient consentA waiver of consent was given in the study because the majority of patients died before the research was carried out.
Ethics approvalThe study was approved by the Institutional Review Board of Columbia University Medical Center.
REFERENCES
1. Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, Downing JR, Jacks T, Horvitz HR, Golub TR. MicroRNA expression profiles classify human cancers. Nature 2005;435:834-8.
2. Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet 2009;10:704-14.
3. Sidhu K, Kapoor NR, Pandey V, Kumar V. The "macro" world of microRNAs in hepatocellular carcinoma. Front Oncol 2015;5:68.
4. Borel F, Konstantinova P, Jansen PL. Diagnostic and therapeutic potential of miRNA signatures in patients with hepatocellular carcinoma. J Hepatol 2012;56:1371-83.
5. Van Keuren-Jensen KR, Malenica I, Courtright AL, Ghaffari LT, Starr AP, Metpally RP, Beecroft TA, Carlson EW, Kiefer JA, Pockros PJ, Rakela J. microRNA changes in liver tissue associated with fibrosis progression in patients with hepatitis C. Liver Int 2016;36:334-43.
7. Valencia-Quintana R, Sánchez-Alarcón J, Tenorio-Arvide MG, Deng Y, Montiel-González JM, Gómez-Arroyo S, Villalobos-Pietrini R, Cortés-Eslava J, Flores-Márquez AR, Arenas-Huertero F. The microRNAs as potential biomarkers for predicting the onset of aflatoxin exposure in human beings: a review. Front Microbiol 2014;5:102.
8. Yang Z, Cappello T, Wang L. Emerging role of microRNAs in lipid metabolism. Acta Pharm Sin B 2015;5:145-50.
9. Zarfeshani A, Ngo S, Sheppard AM. MicroRNA expression relating to dietary-induced liver steatosis and NASH. J Clin Med 2015;4:1938-50.
10. Zhang S, Ouyang X, Jiang X, Gu D, Lin Y, Kong SK, Xie W. Dysregulated serum microRNA expression profile and potential biomarkers in hepatitis C virus-infected patients. Int J Med Sci 2015;12:590-8.
11. Shen J, Wang A, Wang Q, Gurvich I, Siegel AB, Remotti H, Santella RM. Exploration of genome-wide circulating microRNA in hepatocellular carcinoma: MiR-483-5p as a potential biomarker. Cancer Epidemiol Biomarkers Prev 2013;22:2364-73.
12. Zhang Z, Ge S, Wang X, Yuan Q, Yan Q, Ye H, Che Y, Lin Y, Zhang J, Liu P. Serum miR-483-5p as a potential biomarker to detect hepatocellular carcinoma. Hepatol Int 2013;7:199-207.
13. Bronte F, Bronte G, Fanale D, Caruso S, Bronte E, Bavetta MG, Fiorentino E, Rolfo C, Bazan V, Di Marco V, Russo A. HepatomiRNoma: the proposal of a new network of targets for diagnosis, prognosis and therapy in hepatocellular carcinoma. Crit Rev Oncol Hematol 2016;97:312-21.
14. Mizuguchi Y, Takizawa T, Yoshida H, Uchida E. Dysregulated microRNAs in progression of hepatocellular carcinoma: a systematic review. Hepatol Res 2016;46:391-406.
15. Boisen MK, Dehlendorff C, Linnemann D, Schultz NA, Jensen BV, Høgdall EV, Johansen JS. MicroRNA expression in formalin-fixed paraffin-embedded cancer tissue: identifying reference microRNAs and variability. BMC Cancer 2015;15:1024.
16. Wang Y, Gao Y, Shi W, Zhai D, Rao Q, Jia X, Liu J, Jiao X, Du Z. Profiles of differential expression of circulating microRNAs in hepatitis B virus-positive small hepatocellular carcinoma. Cancer Biomark 2015;15:171-80.
17. Wong CM, Wong CC, Lee JM, Fan DN, Au SL, Ng IO. Sequential alterations of microRNA expression in hepatocellular carcinoma development and venous metastasis. Hepatology 2012;55:1453-61.
18. Augello C, Vaira V, Caruso L, Destro A, Maggioni M, Park YN, Montorsi M, Santambrogio R, Roncalli M, Bosari S. MicroRNA profiling of hepatocarcinogenesis identifies C19MC cluster as a novel prognostic biomarker in hepatocellular carcinoma. Liver Int 2012;32:772-82.
19. Wang W, Zhao LJ, Tan YX, Ren H, Qi ZT. Identification of deregulated miRNAs and their targets in hepatitis B virus-associated hepatocellular carcinoma. World J Gastroenterol 2012;18:5442-53.
20. Wen Y, Han J, Chen J, Dong J, Xia Y, Liu J, Jiang Y, Dai J, Lu J, Jin G, Han J, Wei Q, Shen H, Sun B, Hu Z. Plasma miRNAs as early biomarkers for detecting hepatocellular carcinoma. Int J Cancer 2015;137:1679-90.
21. Oksuz Z, Serin MS, Kaplan E, Dogen A, Tezcan S, Aslan G, Emekdas G, Sezgin O, Altintas E, Tiftik EN. Serum microRNAs; miR-30c-5p, miR-223-3p, miR-302c-3p and miR-17-5p could be used as novel non-invasive biomarkers for HCV-positive cirrhosis and hepatocellular carcinoma. Mol Biol Rep 2015;42:713-20.
22. Zhuang L, Xu L, Wang P, Meng Z. Serum miR-128-2 serves as a prognostic marker for patients with hepatocellular carcinoma. PLoS One 2015;10:e0117274.
23. Mestdagh P, Van Vlierberghe P, De Weer A, Muth D, Westermann F, Speleman F, Vandesompele J. A novel and universal method for microRNA RT-qPCR data normalization. Genome Biol 2009;10:R64.
24. Liang Y, Ridzon D, Wong L, Chen C. Characterization of microRNA expression profiles in normal human tissues. BMC Genomics 2007;8:166.
25. Chugh P, Dittmer DP. Potential pitfalls in microRNA profiling. Wiley Interdiscip Rev RNA 2012;3:601-16.
26. Wang X, Gardiner EJ, Cairns MJ. Optimal consistency in microRNA expression analysis using reference-gene-based normalization. Mol Biosyst 2015;11:1235-40.
27. Lin L, Lu B, Yu J, Liu W, Zhou A. Serum miR-224 as a biomarker for detection of hepatocellular carcinoma at early stage. Clin Res Hepatol Gastroenterol 2016;40:397-404.
28. Shen J, Wang S, Zhang YJ, Wu HC, Kibriya MG, Jasmine F, Ahsan H, Wu DP, Siegel AB, Remotti H, Santella RM. Exploring genome-wide DNA methylation profiles altered in hepatocellular carcinoma using Infinium HumanMethylation 450 BeadChips. Epigenetics 2013;8:34-43.
29. Shen J, LeFave C, Sirosh I, Siegel AB, Tycko B, Santella RM. Integrative epigenomic and genomic filtering for methylation markers in hepatocellular carcinomas. BMC Med Genomics 2015;8:28.
30. Shen J, Wang S, Siegel AB, Remotti H, Wang Q, Sirosh I, Santella RM. Genome-wide expression of microRNAs is regulated by DNA methylation in hepatocarcinogenesis. Gastroenterol Res Pract 2015;2015:230642.
31. Edge SB, Compton CC. The American Joint Committee on Cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol 2010;17:1471-4.
32. Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 2002;3:1-11.
33. Andersen CL, Jensen JL, Orntoft TF. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res 2004;64:5245-50.
34. Simon R, Lam A, Li MC, Ngan M, Menenzes S, Zhao Y. Analysis of gene expression data using BRB-ArrayTools. Cancer Inform 2007;3:11-7.
35. Heberle H, Meirelles GV, da Silva FR, Telles GP, Minghim R. InteractiVenn: a web-based tool for the analysis of sets through Venn diagrams. BMC Bioinformatics 2015;16:169.
36. Pritchard CC, Cheng HH, Tewari M. MicroRNA profiling: approaches and considerations. Nat Rev Genet 2012;13:358-69.
37. Li ZB, Li ZZ, Li L, Chu HT, Jia M. MiR-21 and miR-183 can simultaneously target SOCS6 and modulate growth and invasion of hepatocellular carcinoma (HCC) cells. Eur Rev Med Pharmacol Sci 2015;19:3208-17.
38. Thurnherr T, Mah WC, Lei Z, Jin Y, Rozen SG, Lee CG. Differentially expressed miRNAs in hepatocellular carcinoma target genes in the genetic information processing and metabolism pathways. Sci Rep 2016;6:20065.
39. Huang CS, Yu W, Cui H, Wang YJ, Zhang L, Han F, Huang T. Increased expression of miR-21 predicts poor prognosis in patients with hepatocellular carcinoma. Int J Clin Exp Pathol 2015;8:7234-8.
40. Shi KQ, Lin Z, Chen XJ, Song M, Wang YQ, Cai YJ, Yang NB, Zheng MH, Dong JZ, Zhang L, Chen YP. Hepatocellular carcinoma associated microRNA expression signature: integrated bioinformatics analysis, experimental validation and clinical significance. Oncotarget 2015;6:25093-108.
41. Zhang ZZ, Liu X, Wang DQ, Teng MK, Niu LW, Huang AL, Liang Z. Hepatitis B virus and hepatocellular carcinoma at the miRNA level. World J Gastroenterol 2011;17:3353-8.
42. Yuan R, Zhi Q, Zhao H, Han Y, Gao L, Wang B, Kou Z, Guo Z, He S, Xue X, Hu H. Upregulated expression of miR-106a by DNA hypomethylation plays an oncogenic role in hepatocellular carcinoma. Tumour Biol 2015;36:3093-100.
43. Zhang QH, Sun HM, Zheng RZ, Li YC, Zhang Q, Cheng P, Tang ZH, Huang F. Meta-analysis of microRNA-183 family expression in human cancer studies comparing cancer tissues with noncancerous tissues. Gene 2013;527:26-32.
44. Wojcicka A, Swierniak M, Kornasiewicz O, Gierlikowski W, Maciag M, Kolanowska M, Kotlarek M, Gornicka B, Koperski L, Niewinski G, Krawczyk M, Jazdzewski K. Next generation sequencing reveals microRNA isoforms in liver cirrhosis and hepatocellular carcinoma. Int J Biochem Cell Biol 2014;53:208-17.
45. Li J, Fu H, Xu C, Tie Y, Xing R, Zhu J, Qin Y, Sun Z, Zheng X. miR-183 inhibits TGF-beta1-induced apoptosis by downregulation of PDCD4 expression in human hepatocellular carcinoma cells. BMC Cancer 2010;10:354.
46. Li J, Wang Y, Yu W, Chen J, Luo J. Expression of serum miR-221 in human hepatocellular carcinoma and its prognostic significance. Biochem Biophys Res Commun 2011;406:70-3.
47. He XX, Guo AY, Xu CR, Chang Y, Xiang GY, Gong J, Dan ZL, Tian DA, Liao JZ, Lin JS. Bioinformatics analysis identifies miR-221 as a core regulator in hepatocellular carcinoma and its silencing suppresses tumor properties. Oncol Rep 2014;32:1200-10.
48. Sohn W, Kim J, Kang SH, Yang SR, Cho JY, Cho HC, Shim SG, Paik YH. Serum exosomal microRNAs as novel biomarkers for hepatocellular carcinoma. Exp Mol Med 2015;47:e184.
49. Pineau P, Volinia S, McJunkin K, Marchio A, Battiston C, Terris B, Mazzaferro V, Lowe SW, Croce CM, Dejean A. miR-221 overexpression contributes to liver tumorigenesis. Proc Natl Acad Sci U S A 2010;107:264-9.
50. Shen H, Li W, Tian Y, Xu P, Wang H, Zhang J, Li Y. Upregulation of miR-362-3p modulates proliferation and anchorage-independent growth by directly targeting Tob2 in hepatocellular carcinoma. J Cell Biochem 2015;116:1563-73.
51. Ni F, Zhao H, Cui H, Wu Z, Chen L, Hu Z, Guo C, Liu Y, Chen Z, Wang X, Chen D, Wei H, Wang S. MicroRNA-362-5p promotes tumor growth and metastasis by targeting CYLD in hepatocellular carcinoma. Cancer Lett 2015;356:809-18.
52. Wylie D, Shelton J, Choudhary A, Adai AT. A novel mean-centering method for normalizing microRNA expression from high-throughput RT-qPCR data. BMC Res Notes 2011;4:555.
53. El-Halawany MS, Ismail HM, Zeeneldin AA, Elfiky A, Tantawy M, Kobaisi MH, Hamed I, Abdel Wahab AH. Investigating the pretreatment miRNA expression patterns of advanced hepatocellular carcinoma patients in association with response to TACE treatment. Biomed Res Int 2015;2015:649750.
54. Du C, Lv Z, Cao L, Ding C, Gyabaah OA, Xie H, Zhou L, Wu J, Zheng S. MiR-126-3p suppresses tumor metastasis and angiogenesis of hepatocellular carcinoma by targeting LRP6 and PIK3R2. J Transl Med 2014;12:259.
55. Ghosh A, Ghosh A, Datta S, Dasgupta D, Das S, Ray S, Gupta S, Datta S, Chowdhury A, Chatterjee R, Mohapatra SK, Banerjee S. Hepatic miR-126 is a potential plasma biomarker for detection of hepatitis B virus infected hepatocellular carcinoma. Int J Cancer 2016;138:2732-44.
56. Ebrahimi F, Gopalan V, Smith RA, Lam AK. miR-126 in human cancers: clinical roles and current perspectives. Exp Mol Pathol 2014;96:98-107.
57. Yanokura M, Banno K, Iida M, Irie H, Umene K, Masuda K, Kobayashi Y, Tominaga E, Aoki D. MicroRNAS in endometrial cancer: recent advances and potential clinical applications. EXCLI J 2015;14:190-8.
Cite This Article
 , ... Regina M. Santella
, ... Regina M. SantellaHow to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].