


Phenotypic delineation of SET-related neurodevelopmental disorder: two case reports and literature review
 0
0 Abstract
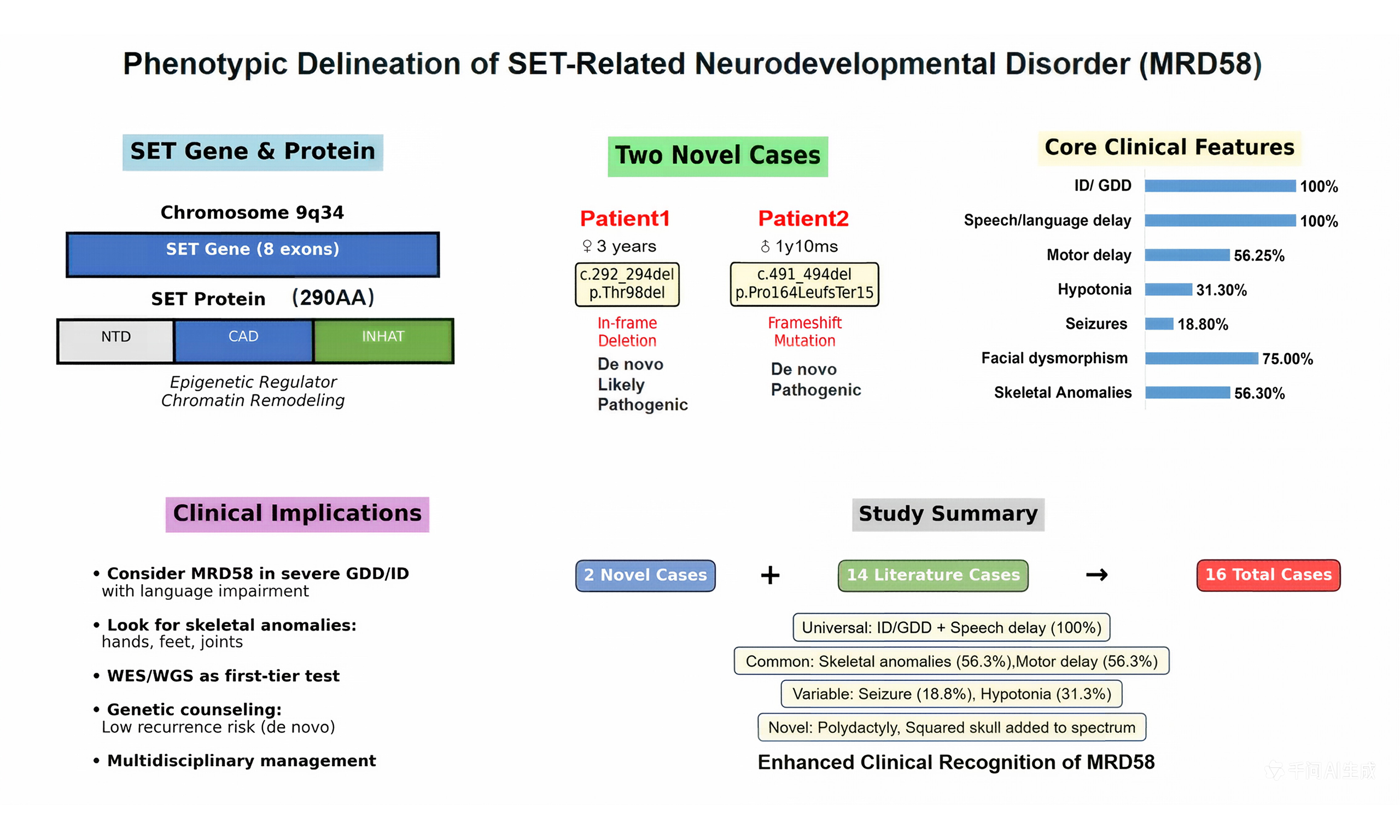
The SET nuclear proto-oncogene (SET; MIM# 600960) is a recently identified cause of autosomal dominant mental retardation-58 (MRD58), characterized by intellectual disability (ID), developmental delay, and variable additional clinical features. This report details two novel cases of MRD58 caused by de novo mutations in the SET gene (one in-frame deletion and one frameshift mutation), broadening the known phenotypic spectrum. Patient 1, a 3-year-old girl, presented with global developmental delay (GDD), seizures, right thumb polydactyly, pes planus and strabismus. Patient 2, a boy aged 1 year and 10 months, exhibited severe GDD, hypotonia, squared skull, and pes planus without seizures. Trio whole-exome sequencing identified novel likely pathogenic SET variants: c.292_294del (p.Thr98del) in Patient 1 and c.491_494del (p.Pro164LeufsTer15) in Patient 2. A review of 14 previously reported cases highlights ID and speech delay as universal features, with motor delay, facial dysmorphism, and skeletal anomalies being common; seizures appear less frequent. These cases underscore MRD58's variable expressivity and emphasize speech/language impairment as a core, persistent deficit. The report aims to enhance clinical recognition and understanding of this rare neurodevelopmental disorder.
Keywords
INTRODUCTION
Autosomal dominant intellectual disability (AD-ID) disorders represent a genetically heterogeneous group. The human SET nuclear proto-oncogene (SET; MIM# 600960), located on chromosome 9q34, encodes a widely expressed multifunctional nuclear protein involved in chromatin remodeling, histone modification, and regulation of gene expression[1,2]. It plays crucial roles in neurogenesis, neuronal differentiation, and synaptic plasticity. SET interacts with several chromatin-modifying proteins encoded by known AD-ID genes [e.g., EP300 (E1A-binding protein p300), CREBBP (CREB binding protein), SETBP1 (SET binding protein 1), KMT2A (lysine-specific methyltransferase 2A)], placing it within a critical neurodevelopmental network[2,3].
In 2014, Hamdan et al. first implicated heterozygous loss-of-function mutations in the SET gene as a novel cause of AD-ID, designated Mental Retardation, Autosomal Dominant 58 (MRD58; MIM# 618106)[4]. The initial phenotype encompassed ID, developmental delay (DD), and variable dysmorphic features. Since then, a limited number of cases have been reported[1,2,4-7], which has slowly delineated the associated clinical spectrum.
To date, approximately 14 cases of MRD58 have been documented in the medical literature[1,2,4-7]. The phenotypic spectrum remains incompletely defined, with variable reports of motor delay, speech impairment, seizures, and skeletal anomalies. Herein, we report two novel pediatric cases with de novo mutations (one in-frame deletion and one frameshift mutation) in the SET gene, presenting with both overlapping and distinct features. We provide a detailed clinical description, genetic findings, and integrate these with a comprehensive review of the existing literature to expand and refine the phenotypic spectrum of SET-related MRD58.
CLINICAL REPORTS
Patient 1
Patient 1 is a 3-year-old girl with a de novo heterozygous in-frame deletion in the SET gene (NM_001122821.2:c.292_294del, p.Thr98del). She presented with severe global developmental delay (GDD), with a developmental age of 13 months at 2 years and 8 months (< 1st percentile on the Griffiths Scales). Speech/language impairment was the most severe domain, with only single words at age 3. Motor milestones were delayed: independent sitting at 10 months and unsteady walking at 26 months. Dysmorphic and skeletal features included right thumb polydactyly (surgically corrected) [Figure 1], pes planus, and congenital strabismus [Figure 2]. At 22 months, she developed recurrent afebrile focal seizures, which were well-controlled with levetiracetam. Electroencephalogram (EEG) showed occipital slowing during wakefulness and frontal-central epileptiform discharges during sleep. Brain magnetic resonance imaging (MRI) and metabolic screening were unremarkable. Her clinical presentation—combining severe ID, distinctive skeletal anomalies, and epilepsy—both aligns with and expands the recognized phenotypic spectrum of SET-related MRD58[2].
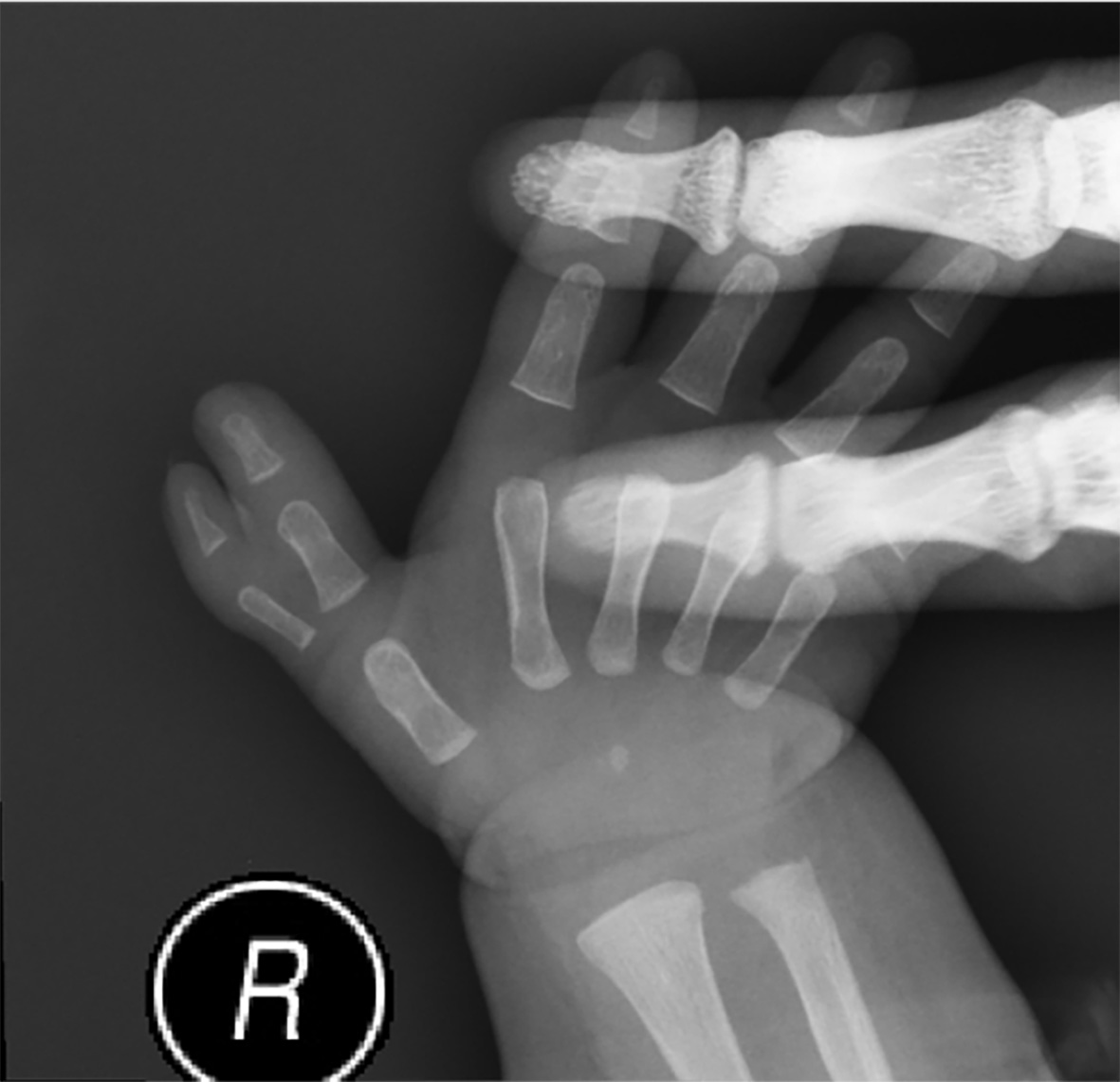
Figure 1. Clinical photograph of the right hand of Patient 1 showing postaxial polydactyly (surgically corrected). This figure is original. Written informed consent for publication was obtained from the patient’s parents.

Figure 2. Facial features of Patient 1 demonstrating broad nasal bridge, low-set ears and congenital strabismus. This is an original figure. Consent for publication was obtained.
Patient 2
Patient 2 is a boy aged 1 year and 10 months with a de novo heterozygous frameshift mutation in the SET gene (NM_001122821.2:c.491_494del, p.Pro164LeufsTer15). He presented with severe GDD, with an overall developmental age of approximately 11 months. Formal assessments confirmed profound motor delay (Gross Motor Quotient 55) and cognitive impairment (Bayley-III cognitive score < 50). Speech development was absent, with no meaningful words. Physical examination revealed distinctive craniofacial (squared skull) and skeletal (pes planus) features, along with generalized hypotonia and muscle weakness. Notably, he had no history of seizures. EEG showed a slow posterior dominant rhythm, while brain MRI was normal. Visual evoked potentials indicated prolonged P100 latency. This case further delineates the severe neurodevelopmental phenotype of MRD58, characterized by profound DD, hypotonia, and characteristic dysmorphism, while also illustrating the variable presence of seizures within this disorder.
DIAGNOSTIC ASSESSMENT AND GENETIC FINDINGS
This study was approved by the Ethics Committee of Shenzhen Children's Hospital (Approval Number: 202107402). Written informed consent was obtained from the parents or legal guardians of all participants for genetic testing, publication of clinical information, and use of clinical photographs. All procedures were performed in accordance with the Declaration of Helsinki and relevant guidelines and regulations.
After obtaining informed consent, peripheral blood samples were collected from the probands and their parents. Whole-exome sequencing was performed on the proband-parent trios. Genomic DNA was captured using the IDT xGen Exome Research Panel v2.0, followed by high-throughput sequencing on an Illumina platform. Sequencing data quality control metrics included a target region coverage ≥ 99.6%, an average depth > 30× over ≥ 96.6% of the target, and a Q20 score (percentage of bases with a Phred quality score ≥ 20) ≥ 0.95. Bioinformatic analysis focused on variants in genes associated with neurodevelopmental disorders (Online Mendelian Inheritance in Man (OMIM), MedGen). Candidate variants were confirmed and segregation analyzed via Sanger sequencing.
In Patient 1, the novel in-frame deletion c.292_294del (p.Thr98del) was absent from population databases (gnomAD) (PM2), occurred de novo (PS2), and is located within the critical central acidic domain (CAD) of the SET protein. As an in-frame deletion within a non-repeat functional domain, it is predicted to alter protein length and potentially disrupt structure/function (PM4). According to the ACMG (American College of Medical Genetics and Genomics)/AMP (the Association for Molecular Pathology) guidelines, this variant was classified as “Likely Pathogenic” based on the combined evidence of PS2 and PM4, supported by PM2.
In Patient 2, the novel frameshift variant c.491_494del (p.Pro164LeufsTer15) introduces a premature termination codon (PTC), consistent with a loss-of-function mechanism (PVS1). It was absent from population databases (gnomAD) (PM2), occurred de novo (PS2), and is located within the inhibitor of histone acetyltransferases (INHAT) domain. According to the ACMG/AMP guidelines, this variant was classified as “Pathogenic” based on the combined evidence of PVS1 and PS2, supported by PM2.
Both novel variants have been submitted to ClinVar databases: Patient 1 (c.292_294del, p.Thr98del): ClinVar Submission ID: SUB16051245; Patient 2 (c.491_494del, p.Pro164LeufsTer15): ClinVar Submission ID: SUB16051893.
LITERATURE REVIEW
We conducted a systematic PubMed search (inception to November 2025) using keywords "SET" AND ("intellectual disability" OR "mental retardation" OR "MRD58") with database link https://pubmed.ncbi.nlm.nih.gov/. We identified six primary studies reporting a total of 14 patients with MRD58 due to SET mutations [Table 1]. Including our two cases, the total number of genetically confirmed cases analyzed here is 16 (9 males, 6 females, and 1 not specified), aged 2 months to 49 years. The core phenotype is highly consistent: all 16 patients (100%) presented with ID/GDD and speech/language delay [Table 2]. Motor DD was reported in 9/16 (56.25%). Hypotonia or motor incoordination was noted in several cases. Dysmorphic features were common but variable. Facial features reported across the literature include a prominent forehead, hypertelorism, downslanting palpebral fissures, a broad nasal bridge, and a thin upper lip[1]. Skeletal/limb anomalies were frequent, including pes planus (3/11, including our cases), scoliosis, joint laxity, and digit anomalies (e.g., clinodactyly, syndactyly). Our Patient 1 adds polydactyly to this spectrum. Seizures were reported in only 2/16 patients prior to our report; our Patient 1 represents the third case, suggesting it is an infrequent but part of the spectrum. Genetically, all reported pathogenic variants are heterozygous, consistent with autosomal dominant inheritance. The mutational spectrum includes frameshift (most common), nonsense, splice-site, missense, and in-frame deletions [Figure 3], all predicted to cause haploinsufficiency or disrupt critical protein function.
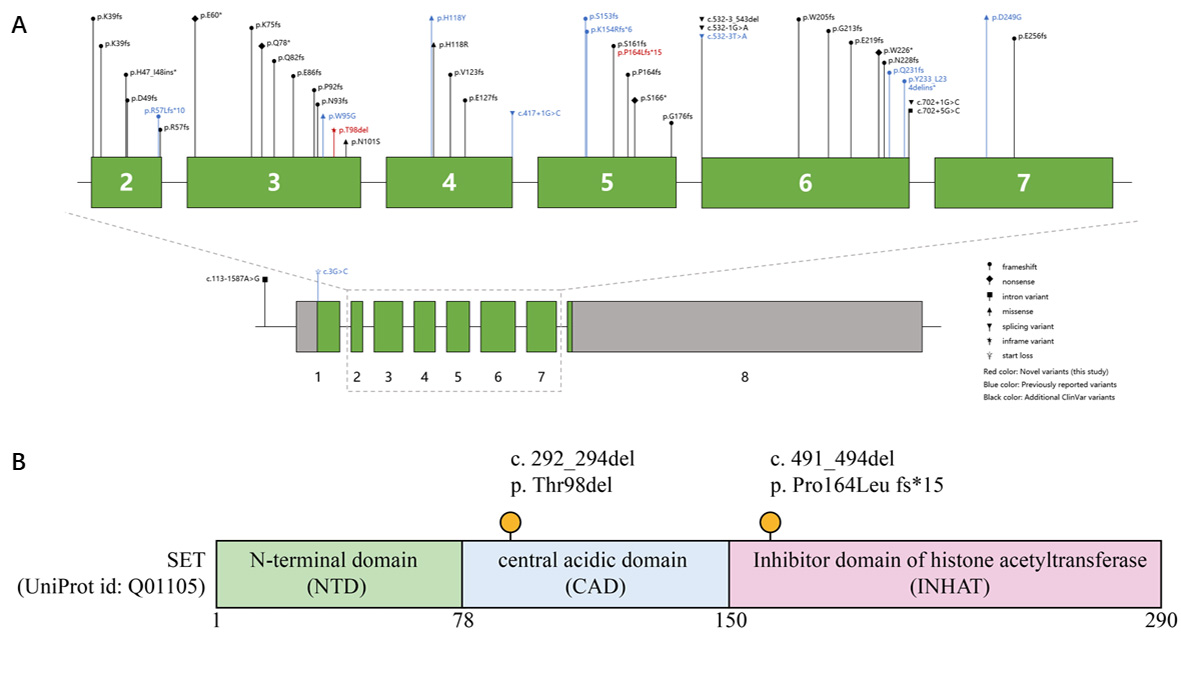
Figure 3. SET gene structure, protein domains, and mutation locations. (A) Schematic of the SET gene (exons 1-8 shown; exon 2-7 detailed above). Pathogenic variants: novel (this study, red), previously reported (blue), ClinVar - additional (black). Variant types (frameshift, nonsense, etc.) are indicated. (B) SET protein domains (UniProt ID: Q01105): N - terminal (NTD), central acidic (CAD), histone acetyltransferase inhibitor (INHAT). Novel mutations (c.292_294del, p.Thr98del; c.491_494del, p.Pro164LeufsTer15) are marked with orange dots.
Summary of published clinical cases with SET gene variants (Transcript: NM_001122821.2)
| Case ID | Ref. | Age at report | Sex | cDNA change | Protein change | Variant type | Inheritance pattern | Key clinical phenotypes | ClinVar ID |
| 1 | Hamdan et al., 2014[4] | 12 years | M | c.699_701delCTT | p.Tyr233* | Frameshift (nonsense) | De novo | Congenital microcephaly, moderate ID, normal brain MRI, no seizures | VCV000560209.3 |
| 2 (Patient 1a) | Stevens et al., 2018[2] | 16 years | M | c.167_170del | p.Arg57Leufs*10 | Frameshift | Inherited from affected mother (Patient 1b) | Moderate ID, speech delay, developmental delay, motor delay | SUB16055850 |
| 3 (Patient 1b) | Stevens et al., 2018[2] | 49 years | F | c.167_170del | p.Arg57Leufs*10 | Frameshift | De novo (transmitted to son) | Moderate ID, speech delay, developmental delay, motor delay | SUB16055850 |
| 4 | Stevens et al., 2018[2] | 5.5 years | M | c.283T>G | p.Trp95Gly | Missense | De novo | ID, speech delay, developmental delay, motor delay, mild facial asymmetry | VCV000560206.3 |
| 5 | Stevens et al., 2018[2] | 4.25 years | M | c.352C>T | p.His118Tyr | Missense | De novo | ID, speech delay, developmental delay, motor delay | VCV000560207.33 |
| 6 | Stevens et al., 2018[2] | 4.25 years | M | c.417+1G>C | r.spl? | Canonical splice site | De novo | ID, speech delay, developmental delay, motor delay | SUB16055890 |
| 7 | Stevens et al., 2018[2] | 5.7 years | M | c.689_690dup | p.Gln231Tyrfs*29 | Frameshift | De novo | ID, speech delay, developmental delay, motor delay; Epilepsy | VCV000560208.2 |
| 8 | Richardson et al., 2018[7] | 10 years | F | c.167_170delACAG | p.Arg57Leufs*10 | Frameshift | De novo | Moderate ID, attends special school | SUB16055905 |
| 9 | Richardson et al., 2018[7] | 17 years | M | c.167_170delACAG | p.Arg57Leufs*10 | Frameshift | De novo | Moderate ID, attends special school | SUB16055905 |
| 10 | Richardson et al., 2018[7] | 15 years | M | c.459_460delCA | p.Lys154ArgfsTer6 | Frameshift | De novo | Moderate ID, attends mainstream and special schools | SUB16055913 |
| 11 | Brunet et al., 2020[5] | Not specified | Not specified | c.457_458del, | p.Ser153Glnfs*7 | Frameshift | Not specified | ID / Neurodevelopmental disorder | VCV000807681.3 |
| 12 | Zhang et al., 2020[6] | 2 months | F | c.746A>G | p.Asp249Gly | Missense | De novo | Epileptic encephalopathy (co-occurring with STXBP1 variant) | SUB16055921 |
| 13 | Pan et al., 2022[1] | Not specified | F | c.532-3T>A | r.? (Exon 6 skipping) | Splice site | Unknown (parental testing not done) | Moderate ID, mild facial dysmorphism (broad nasal bridge, strabismus) | SUB16055925 |
| 14 | Pan et al., 2022[1] | 11 years | F | c.3G>C | p.0? (Start loss) | Start loss | Unknown (parental testing not done) | Moderate ID, mild facial dysmorphism, height <3rd percentile | SUB16055930 |
| 15 | Our Study | 3 years | F | c.292_294del | p.Thr98del | In-frame deletion | De novo | ID, speech delay, developmental delay, motor delay; Epilepsy; high palate; polydactyly; Pes planus; left eye strabismus; Patent Foramen Ovale | SUB16051245 |
| 16 | Our Study | 1 years and 10 months | M | c.491_494del | p.Pro164LeufsTer15 | Frameshift | De novo | ID, speech delay, developmental delay, motor delay; Pes planus; hypotonia | SUB16051893 |
Summary of clinical features in all reported MRD58 cases (n = 16)
| Clinical feature | Number affected | Percentage |
| Core neurological features | ||
| Intellectual disability/Global developmental delay | 16/16 | 100% |
| Speech/language delay | 16/16 | 100% |
| Motor developmental delay | 9/16 | 56.25% |
| Hypotonia | 5/16 | 31.25% |
| Seizures/Epilepsy | 3/16 | 18.75% |
| Dysmorphic features | ||
| Facial dysmorphism | 12/16 | 75.00% |
| Skeletal Anomalies | 9/16 | 56.30% |
DISCUSSION
This report describes two new pediatric cases of MRD58 (SET-related neurodevelopmental disorder) caused by de novo mutations (one in-frame deletion and one frameshift mutation) in the SET gene. By combining our findings with the existing literature[2,4-9], we aim to summarize the key clinical features and genetic insights for clinicians.
Core clinical picture: severe delay with prominent language impairment
The most consistent finding in our patients and in the literature is severe GDD, with language skills being disproportionately affected[2,4,5,7]. Both our patients had minimal speech and severe comprehension deficits. In our patients and some reported cases, language impairment appears disproportionately severe relative to motor delay. However, further data are needed to confirm if this is a consistent hallmark of the disorder[2,5,7]. For clinicians, this profile in a child with unexplained ID or GDD should prompt consideration of SET gene testing. Biologically, the SET protein helps regulate gene activity, and its deficiency appears to particularly disrupt the development of brain circuits involved in language.
Skeletal anomalies: a common and useful diagnostic clue
A key feature extending beyond the nervous system is the presence of skeletal abnormalities. Our Patient 1 had thumb polydactyly, and Patient 2 had pes planus. Other reported features include brachydactyly, syndactyly, and joint laxity[2,5,7]. This consistent involvement of the limbs and digits is an important clinical clue, suggesting SET haploinsufficiency disrupts early limb development. Therefore, a thorough physical examination of the hands, feet, and joints is crucial in evaluating any child with ID/GDD, as these findings can help narrow the differential diagnosis.
Epilepsy: a variable, not core, feature
The link between SET mutations and epilepsy needs careful interpretation. While our Patient 1 had seizures, Patient 2 did not, supporting the observation from the literature that epilepsy is an inconsistent or variable feature of MRD58[5,7,9]. An important lesson from the literature is that one early case linking SET to severe epilepsy[8] was later found to have a co-occurring mutation in the STXBP1 (syntaxin-binding protein 1) gene, a well-known cause of epileptic encephalopathy. This highlights that seizures in a patient with a SET mutation may be due to other genetic or environmental factors, rather than being a direct, core feature of MRD58 itself. For clinicians, this means that while epilepsy can occur, its absence does not rule out the diagnosis, and its presence should not automatically be attributed solely to the SET variant without considering other causes.
Genotype and inheritance: insights for genetic counseling
All pathogenic SET variants reported to date, including ours, are predicted to cause a loss of function (haploinsufficiency) and are predominantly de novo[2,5-7]. This consistent genetic mechanism explains the overlapping core phenotype. While the total number of known cases is still small, there is a suggestion from the literature that protein-truncating variants (e.g., frameshifts and nonsense mutations) may lead to a more consistent and severe phenotype than some missense changes[4,7]. As more cases are gathered, we will better understand if specific mutations correlate with certain features. For now, identifying a pathogenic de novo SET mutation provides a definitive diagnosis and allows for accurate genetic counseling regarding the very low recurrence risk for future pregnancies.
SET's role: an epigenetic regulator affecting multiple systems
The co-occurrence of neurodevelopmental and skeletal anomalies in MRD58 can be explained by the core epigenetic regulatory functions of the SET protein[2-4,6,7,10]. As a multifunctional chromatin modulator, SET primarily inhibits histone acetyltransferases (e.g., p300 (adenoviral E1A binding protein of 300 kDa)/CBP (CREB binding protein)) via its INHAT domain and suppresses protein phosphatase 2A (PP2A) through its CAD, thereby broadly regulating gene transcription and cellular signaling[11]. Haploinsufficiency resulting from heterozygous SET mutations disrupts the precise spatiotemporal dynamics of gene expression during critical windows of early embryonic development. Tissues with exceptionally high demands for genetic precision, such as the developing central nervous system and limb buds, are particularly vulnerable to this epigenetic dysregulation, explaining the multi-system involvement observed in this disorder.
Functional domain analysis of novel mutations
Both novel mutations identified in this study are located within critical functional domains of the SET protein [Figure 3]. The p.Thr98del in-frame deletion in Patient 1 resides within the CAD, which is essential for PP2A inhibition[11] . Although an in-frame deletion, the loss of this conserved threonine residue could perturb local protein structure or a potential modification site within this critical functional region, thereby affecting its interaction with PP2A and related regulatory functions. The p.Pro164LeufsTer15 frameshift mutation in Patient 2 is located within the C-terminal INHAT domain, which is central to SET's chromatin-silencing activity by masking histone lysine residues to block acetylation[11]. This mutation introduces a PTC, and the resulting truncated protein is expected to be eliminated via nonsense-mediated mRNA (messenger RNA) decay (NMD), leading to a complete loss of function of the mutant allele—a canonical loss-of-function mechanism[12]. This directly disrupts the core chromatin-regulatory function of the INHAT domain.
Ultimately, both mutations—whether through structural perturbation of the CAD or direct truncation of the INHAT domain—compromise SET-mediated chromatin regulation. This leads to dysregulation of the chromatin landscape and gene expression programs during critical neurodevelopmental periods, as recently demonstrated in cellular models of SET haploinsufficiency[1].
Clinical recommendations for genetic testing
For clinicians, the combination of severe ID/GDD with disproportionately severe language impairment, along with skeletal anomalies (particularly involving hands and feet), should raise suspicion for MRD58. However, given the nonspecific nature of these features and the genetic heterogeneity of neurodevelopmental disorders, we do not recommend targeted single-gene testing for SET. Instead, when resources permit, first-tier whole-exome or whole-genome sequencing strategies are the most efficient approach for early diagnosis in patients with neurodevelopmental disorders[13-15]. Identification of a pathogenic SET variant should prompt careful evaluation for associated features, particularly skeletal anomalies, and appropriate genetic counseling.
CONCLUSION
We report two novel cases of MRD58, reinforcing its core characteristics of severe DD with profound language impairment and frequent skeletal anomalies. For the child neurologist or clinical geneticist, this syndrome should be considered in a child with unexplained severe ID/GDD, a profile of significantly worse language delay, and the presence of hand/foot anomalies or joint laxity. A diagnosis of MRD58, achieved through genetic testing such as trio whole-exome sequencing, enables targeted management, provides families with a clear cause, and offers accurate recurrence risk counseling.
DECLARATIONS
Acknowledgments
We thank all patients who participated in this study.
Authors’ contributions
Conceived and designed the study: Mai J, Chen L, Wen F
Collected clinical data and performed the clinical evaluations: Mai J, Ao L, Zhang Q, Liu L, Zeng Q, Luo X, Cao D
Drafted the manuscript: Mai J, Ao L
Revised the manuscript for important intellectual content: Chen L, Wen F
All authors read and approved the final manuscript.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request. The novel variants have been submitted to ClinVar (Submission IDs: SUB16051245, SUB16051893). This case report was prepared following the CARE (CAse REport) guidelines. A completed CARE checklist is available as Supplementary Material.
AI and AI-assisted tools statement
During the preparation of this manuscript, the AI tool Ima.copilot was used solely for language editing and polishing. The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship
This work was supported by grants from the Guangdong High-Level Hospital Construction Fund (No. ynkt2022-zz04), Shenzhen Key Medical Discipline Construction Fund (No. SZXK033), Shenzhen Science and Technology Program (No. SGDX20211123142200001), Sanming Project of Medicine in Shenzhen (No. SZSM202311028), and Guangdong High-level Hospital Construction Fund Clinical Research Project of Shenzhen Children's Hospital (LCYJ2022062).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
This study was approved by the Ethics Committee of Shenzhen Children's Hospital (Approval Number: 202107402). All procedures were performed in accordance with the Declaration of Helsinki and relevant guidelines and regulations.
Consent for publication
Written informed consent was obtained from the parents or legal guardians for publication of identifying information and images of the patients included in this study.
Copyright
© The Author(s) 2026
Supplementary Materials
REFERENCES
1. Pan X, Liu S, Feng X, et al. Whole exome sequencing and transcriptome analysis in two unrelated patients with novel SET mutations. J Hum Genet. 2023;68:867-74.
2. Stevens SJC, van der Schoot V, Leduc MS, et al. De novo mutations in the SET nuclear proto-oncogene, encoding a component of the inhibitor of histone acetyltransferases (INHAT) complex in patients with nonsyndromic intellectual disability. Hum Mutat. 2018;39:1014-23.
3. Weerts MJA, Lanko K, Guzmán-Vega FJ, et al. Delineating the molecular and phenotypic spectrum of the SETD1B-related syndrome. Genet Med. 2021;23:2122-37.
4. Hamdan FF, Srour M, Capo-Chichi JM, et al. De novo mutations in moderate or severe intellectual disability. PLoS Genet. 2014;10:e1004772.
5. Brunet T, Jech R, Brugger M, et al. De novo variants in neurodevelopmental disorders-experiences from a tertiary care center. Clin Genet. 2021;100:14-28.
6. Zhang L, Gao J, Liu H, et al. Pathogenic variants identified by whole-exome sequencing in 43 patients with epilepsy. Hum Genomics. 2020;14:44.
7. Richardson R, Splitt M, Newbury-Ecob R, Hulbert A, Kennedy J, Weber A, DDD Study. SET de novo frameshift variants associated with developmental delay and intellectual disabilities. Eur J Hum Genet. 2018;26:1306-11.
8. Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature. 2017;542:433-8.
9. Trinh J, Kandaswamy KK, Werber M, et al. Novel pathogenic variants and multiple molecular diagnoses in neurodevelopmental disorders. J Neurodev Disord. 2019;11:11.
10. Roston A, Evans D, Gill H, et al. SETD1B-associated neurodevelopmental disorder. J Med Genet. 2021;58:196-204.
11. Seo SB, McNamara P, Heo S, Turner A, Lane WS, Chakravarti D. Regulation of histone acetylation and transcription by INHAT, a human cellular complex containing the set oncoprotein. Cell. 2001;104:119-30.
12. Kurosaki T, Popp MW, Maquat LE. Quality and quantity control of gene expression by nonsense-mediated mRNA decay. Nat Rev Mol Cell Biol. 2019;20:406-20.
13. Stark Z, Schofield D, Alam K, et al. Prospective comparison of the cost-effectiveness of clinical whole-exome sequencing with that of usual care overwhelmingly supports early use and reimbursement. Genet Med. 2017;19:867-74.
14. Qin Y, Cao H, Liu L, et al. Genetic characterization of 128 chinese individuals with neurodevelopmental disorders via whole-exome sequencing. Dev Neurosci. ;2025:1-17.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].