


Diagnosis and therapy of Niemann-Pick disease type C1- what have we learned that can help veterinary patients?
 0
0 Abstract
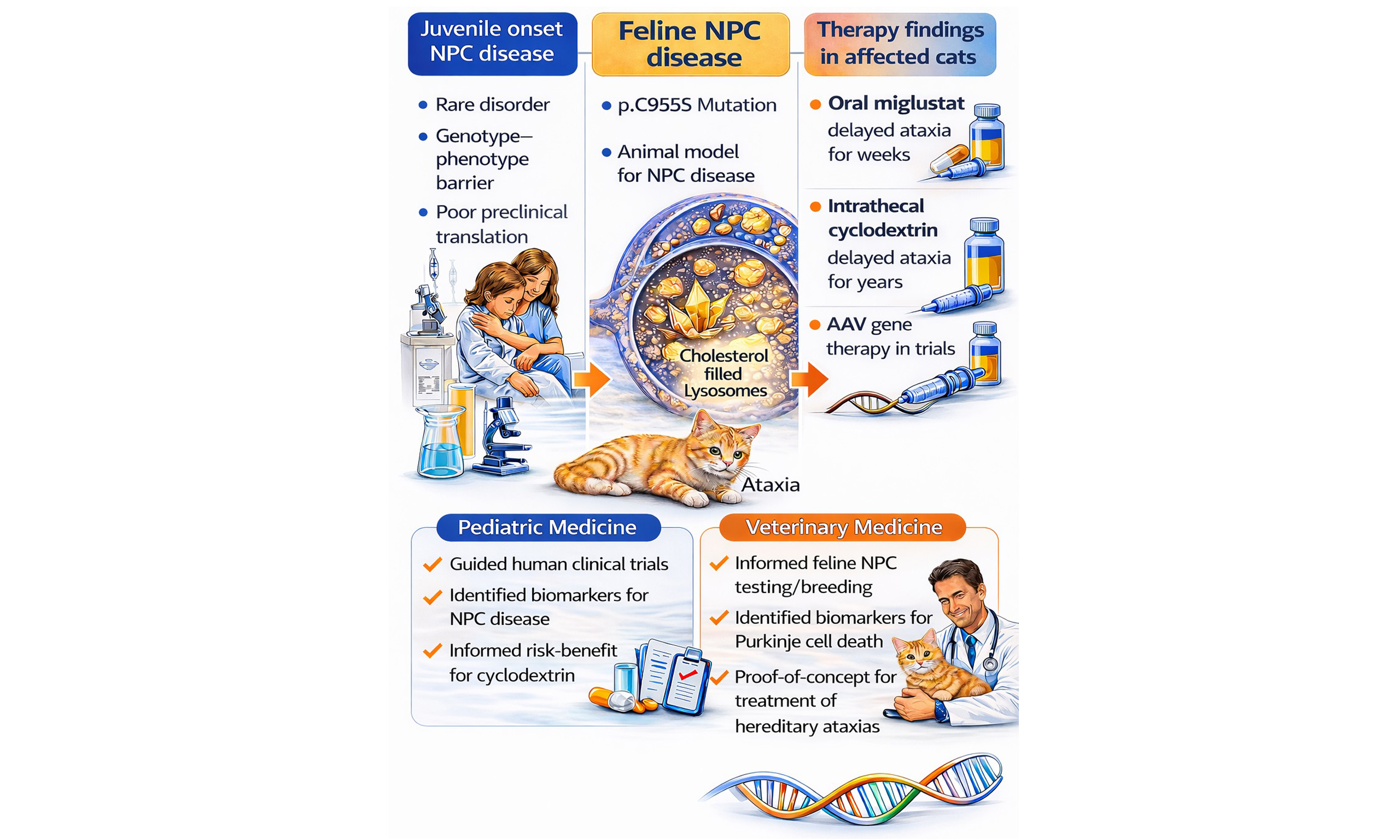
Naturally occurring hereditary diseases in dogs and cats can overcome major barriers in developing therapies for rare human genetic disorders, including limited natural history data, poor genotype-phenotype resolution, inadequate biomarkers, and weak translation from rodent models. By focusing on Niemann-Pick disease type C1 (NPC disease) in cats as a representative lysosomal disorder, the relevance of translating findings from large animal models to both pediatric medicine and veterinary neurology is discussed. The feline model of NPC disease closely mirrors the human disease genetically, biochemically, pathologically, and clinically, showing progressive cerebellar ataxia, Purkinje neuron loss, neuroaxonal dystrophy, hepatic disease, and abnormal cholesterol and sphingolipid storage. Studies in affected cats established the disease’s natural history, identified plasma 24(S)-hydroxycholesterol and spinal fluid calbindin D as biomarkers of disease progression, and enabled rigorous testing of therapies. Oral miglustat modestly delayed neurological decline, while intrathecal 2-hydroxypropyl-beta-cyclodextrin produced marked preservation of neurological function, prolonged survival, and reduced neuropathology, although ototoxicity emerged as an important adverse effect. These feline preclinical studies directly informed human clinical trial design, dosing, safety monitoring, and biomarker development, thereby accelerating therapeutic translation for children with NPC disease. Experimental Adeno-associated viral (AAV)-mediated gene therapy in cats is ongoing although current technical limitations of gene delivery for diffuse neurodegenerative disorders may limit efficacy. For veterinary clinical practice, the work supports improved and early diagnosis, breeding management, and the development of future treatment of feline NPC disease and other hereditary ataxias.
Keywords
INTRODUCTION
Many rare genetic disorders of human patients lack effective therapies due to limited natural history data, an incomplete understanding of genotype-phenotype relationships, a lack of validated surrogate disease markers, and the inability to translate experimental therapies from mouse models into other species. These challenges may be addressed, in part, through the study of naturally occurring hereditary diseases of dogs and cats. This concept is not a new one. Indeed, the Referral Center for Animal Models of Human Genetic Disease (RCAM) was established in 1974 at the School of Veterinary Medicine of the University of Pennsylvania to serve as a national referral site and resource for the discovery and characterization of dogs and cats with hereditary diseases homologous to those found in human patients. These naturally occurring large animal models of human disease represented true orthologs of their respective human diseases, involving defects in homologous genes resulting in similar molecular, biochemical, pathological, and clinical phenotypes as in human patients[1]. In collaboration with the School of Veterinary Medicine’s Section of Medical Genetics and various hospital clinical services, naturally occurring canine and feline models of human genetic diseases found by veterinarians and breeders internationally were identified using accurate phenotypic description including clinical evaluation, diagnostic imaging, clinical laboratory testing, and histological studies[1-3]. Laboratory testing included biochemical screening of blood, urine, cerebrospinal fluid, and tissue samples for abnormal metabolites indicative of defective metabolic pathways. Mode of inheritance was determined along with genomic and candidate gene analyses. Over its nearly 50-year existence, the Center discovered, characterized, and/or maintained breeding colonies or germplasm from over 40 different hereditary diseases resulting in hundreds of papers on the natural histories, pathogeneses, or experimental therapies for these diseases. Data from these large-animal models contributed to the development of small molecules and biologics for human trials[1-3].
LYSOSOMAL DISEASES
Of the diseases characterized by the Center, 11 were lysosomal diseases. Lysosomal diseases are a group of ~70 disorders defined by the lysosomal accumulation of a substrate due to deficiency of a soluble lysosomal acid hydrolase or an activator protein, or due to defective lysosomal membrane transport preventing the contents from reaching the cytoplasm[4,5]. As a group, lysosomal diseases affect ~1 in 5000 live births[6]; however, each individual lysosomal storage disorder is rare making therapies difficult to develop due to the low incidence as well as to disease heterogeneity in the patient population.
Neuronopathic lysosomal diseases are those with central nervous system (CNS) manifestations including neurodegeneration, impaired axonal transport, synaptic dysfunction, and neuroinflammation[5,7,8]. This group of diseases include alpha- and beta-mannosidosis, fucosidosis, globoid cell leukodystrophy, glucocerebrosidosis, GM1- and GM2-gangliosidoses, mucolipidosis II, the mucopolysaccharidoses (I, II, III, VII), neuronal ceroid lipofuscinosis, acid sphingomyelinase deficiency, and Niemann-Pick disease type C[3,5]. A review of how large animal models contribute to the development of therapies for these diseases has been published[3].
Here we have chosen to focus on how a single lysosomal disease affecting cats, Niemann-Pick disease, type C1 (NPC disease), has contributed to therapy in human patients and how studies in the NPC1 cat have impacted veterinary care. NPC disease in human patients is characterized by progressive ataxia, dementia, gaze palsy, dysphagia, hepatopathy, and early death[9-11]. Dysfunction of the NPC1 protein, which is present in the lysosomal membrane, prevents lysosomal fusion with endosomes and autophagosomes[12] resulting in lysosomal storage of cholesterol and sphingolipids[13-17] as well as impaired export of lipoprotein-derived cholesterol. The disease manifests in several forms associated with age of onset[18-20]: perinatal presentation characterized primarily by hepatosplenomegaly and cholestatic icterus; early infantile presentation characterized by hepatosplenomegaly, delayed motor development, spasticity, and intention tremor; late infantile/juvenile presentation characterized by ataxia, intellectual impairment, dysarthria, dysphagia, seizures, and vertical supranuclear gaze palsy; and adult presentation characterized by dementia, psychiatric symptoms, ataxia, and dystonia[10]. Histologic abnormalities include neuronal vacuolization, Purkinje neuron death, neuroaxonal dystrophy, gliosis, and CNS inflammation[11,13,16,21,22].
STUDIES IN NIEMANN-PICK DISEASE, TYPE C1 CATS
A 9-week-old domestic short-haired kitten presented to the Cornell University College of Veterinary Medicine with signs of progressive cerebellar ataxia beginning at 6 weeks of age[23]. At histopathological review, the CNS showed diffuse vacuolization of many neuronal populations, neuroaxonal dystrophy, and gliosis. Lysosomal storage was extensive in lung, liver, and lymphoid tissues. Liver and brain showed accumulation of cholesterol, sphingomyelin, and glycolipids (glucosylceramide, lactosylceramide, GM2 and GM3 gangliosides). Cultured fibroblasts had a decreased ability to esterify exogenous cholesterol. Clinical, morphological, and biochemical findings suggested a diagnosis of NPC disease most similar to the juvenile-onset form[23]. A breeding colony was established at Colorado State University where natural history studies and further histological studies identified progressive Purkinje neuron death and meganeurite formation with ectopic dendritogenesis as characteristics of the disease[13,24-27]. Dietary cholesterol restriction was ineffective at ameliorating disease[28], while oral administration of miglustat, an inhibitor of glucosylceramide synthase resulting in the reduction in the biosynthesis of glucosylceramide and complex glycosphingolipids, to three post-symptomatic cats delayed the onset and progression of cerebellar dysfunction[15]. In 2003, NPC disease in these cats was identified as being due to a spontaneously-occurring missense mutation in NPC1 (p.C955S; c.2864G>C substitution)[29].
The breeding colony moved to the RCAM in 2005 and subsequently moved to the University of Florida in 2023. In the last 20 years, a more extensive natural history study was performed describing onset of specific neurological deficits, patterned Purkinje neuron loss, peripheral and central demyelination, pulmonary abnormalities, as well as progressive hepatic disease in affected cats[30-33]. Biomarkers of disease presence and progression were identified[34-39]. Therapy trials followed. In 2012, a larger study showed the pharmacokinetics and activity of orally-administered miglustat in slowing feline NPC disease progression, increasing lifespan, decreasing GM2 ganglioside accumulation, improving Purkinje neuron survival, and modulating microglial immunophenotype and function[40]. In 2015, a study showed biweekly intrathecal administration of 2-hydroxypropyl-beta-cyclodextrin (HPßCD), a small molecule capable of binding to and exporting accumulated lysosomal cholesterol, to presymptomatic cats prevented the onset of cerebellar dysfunction for greater than a year (untreated cats do not live beyond 30 weeks), reduced Purkinje neuron loss, and resulted in near normal concentrations of brain cholesterol and sphingolipids. An elevation of hearing threshold was a notable adverse event[41-43].
Gene therapy studies in the NPC1 cat are ongoing in hopes of providing consistent expression of a functional protein to Purkinje neurons as was done in rodents[44-47]. In these studies, affected NPC1 kittens are being dosed intrathecally at the cisterna magna at a pre-symptomatic age of three weeks old with an Adeno-associated virus serotype-9 (AAV9) vector carrying a copy of the feline NPC1 gene. Preliminary findings are encouraging and the hope is Purkinje neurons may express functional protein and neuronal loss may be avoided.
HOW STUDIES IN THE NPC1 CAT BENEFIT PEDIATRIC PATIENTS
Preclinical testing of HPßCD in the NPC1 cat model had a major positive impact on the development of this drug for the treatment of individuals with NPC disease. Clinical, laboratory and pathological safety data from NPC1 cats treated with HPßCD was a key component of the initial Investigational Drug Application to the Food and Drug Administration (FDA). These preclinical studies identified ototoxicity as a major toxic effect and not only allowed for integration of appropriate audiological testing into the phase I/II clinical trial[41,42,48,49], but also allowed the FDA, Institutional Review Boards and parents to make an informed decision with respect to the risks and potential benefit of this investigational drug. The marked efficacy of this drug that was demonstrated by videos of treated NPC1 cats, clearly demonstrated the potential clinical efficacy. The NPC1 cat model also provided invaluable data with respect to determining drug dosing and proof of concept that 24(S)-hydroxycholesterol, an oxysterol reflective of neuronal cholesterol homeostasis, could serve as a pharmacodynamic biomarker to guide dose escalation in the phase I/II trial[35,48]. In addition, biomarker studies in the HPßCD treated cats demonstrated that calbindin-D, a biomarker reflective of Purkinje neuron damage, could be used to monitor biochemical efficacy[37,48]. Knowledge of ototoxicity, availability of biomarkers, and demonstration of clinical efficacy was invaluable in translating laboratory studies to a clinical trial.
HOW STUDIES IN THE NPC1 CAT BENEFIT CARE OF VETERINARY PATIENTS
In most cases, the signalment, history, clinical signs, and basic laboratory findings suggest the presence of a lysosomal disorder and the breeder or veterinary practitioner will contact a genetic testing lab. Typically, at the metabolic screening lab, urine and serum samples are examined for abnormal metabolites and the presence or absence of specific lysosomal enzymes and together with the clinicopathological findings, a diagnosis, or at least a list of differential diagnoses, is made. Whole genome sequencing has allowed for quicker and accurate discovery of disease-causing variants in specific genes. Once a diagnosis is made, DNA-based testing is then provided by the laboratory that developed the test or passed on to larger commercial laboratories. This provides the tools to test related animals to determine their genotypes. Testing then allows the breeder or veterinarian to make informed breeding decisions, not only to prevent the production of affected animals but also to allow carrier animals to be bred to homozygous normal animals to retain otherwise excellent genetic potential in the carrier animal.
For feline NPC disease, genetic screening is only performed once clinical signs have developed and testing is limited to the c.2864G>C substitution[29] or for the only other identified mutation in a single cat where clinical signs were not described[50]. The prevalence of the disease in cat has not been determined but evidence suggests it is rare.
Treating feline NPC disease
Once a definitive diagnosis is made, therapeutic intervention is discussed but is unlikely to be pursued. Why? Orally-administered miglustat is an option, however, miglustat therapy in children costs over $300,000/year, making cost a significant limiting factor[51]. Cyclodextrin is a less expensive option, however, the need for biweekly intrathecal administration makes therapy highly inconvenient; also, disease amelioration is considerably less when administered post-symptomatically[42]. What about gene therapy - a one-time, albeit expensive therapy that has proven safe and effective in many studies in cats[52-58]? Affected animals could be tested for the absence of neutralizing antibodies to specific AAV serotypes. Then, species-specific, codon-optimized NPC1 cDNA could be delivered intrathecally in a suitable AAV vector potentially under the control of a constitutive promoter to express functioning wild type protein as had been done in other lysosomal disease[59]. Voila - animal is cured. Well, not exactly… gene therapy, as well as other treatments for neurodegenerative diseases, are most effective prior to the onset of significant clinical signs[56]; however, most client-owned animals would be substantially affected at the time of diagnosis and treatment institution. In human patients, newborn screening is being developed in order to begin therapy prior to the onset of clinical signs; newborn screening could potentially be done in veterinary patients assuming that previous litters indicated the presence of NPC disease[60]. However, even if pre- or post-symptomatic therapy were effective in slowing disease progression, pre-existing and/or acquired immune responses to either the vector or the transgene product may limit the efficacy of therapy[61]. Finally, unlike many lysosomal diseases in which a transduced/corrected cell can exocytose proteins to correct nearby untransduced cells (a process known as “cross-correction”[62]), NPC1 protein is membrane-associated and is not exocytosed. Therefore, in order to correct the entire NPC1 brain, each neuron must be transduced - a process not possible using the methods of administration and the vectors that are currently available. To continue the argument, let’s assume that the majority of Purkinje neurons are corrected in the treated cat brain and that ataxia does not develop. However, the remainder of the brain’s neurons are uncorrected which can result in the future development of non-cerebellar neurological dysfunction. In a canine model of globoid cell leukodystrophy, the authors were disheartened that insufficient dosing of AAV did indeed reduce ataxia in affected animals, however, these animals became blind and developed severe behavioral abnormalities - not an outcome that would benefit a client[59]. Clearly, effective gene therapy of many diffuse neurodegenerative diseases, particularly those where cross-correction does not occur, has not yet been realized in animals with large brains. Improvements in global delivery, immune reactivity, as well as in cost must occur before NPC1 gene therapy could be considered in pets.
Imagining cost-effective, convenient therapies for feline and human NPC disease involves identifying compounds that either (1) allow significant amounts of partially functioning protein to reach its site of activity, or (2) affect downstream targets ideally those that influence disease pathways common to other neurodegenerative diseases such as neuroinflammation and autophagy[63,64]. In the first instance, chaperones may be administered in order to increase the amount of mutant NPC1 protein that escapes endoplasmic reticulum (ER)-degradation and ER-targeted macroautophagy and instead makes it to the lysosomal membrane where even partial activity can affect disease. These proteostatic modulators are currently being discovered and evaluated and could be used to treat several diseases caused by missense mutations[65]. Alternatively, drugs that affect neuroinflammation or autophagy may be useful in treating NPC disease. These compounds include oral rapamycin, capable of restoring microglial homeostasis[66]; oral efavirenz, capable of ameliorating neuronal cholesterol turnover[67]; oral arimoclomol, working presumably through both increasing the pool of trafficked NPC1 protein and by rescuing impaired autophagy[68]; or oral N-acetyl-L-leucine, mitigating dysfunctional lysosome fusion and trafficking[69]. Although these compounds have not been tested in feline NPC disease, the recent FDA approval of both arimoclomol and N-acetyl-L-leucine for human NPC patients may support their use in the treatment of affected cats.
Treating other CNS diseases of cats and dogs
If feline NPC disease is more likely to be managed by the decreased production of affected animals than by therapy, how else might studies in the cat help veterinary patients? It is very likely that the data on calbindin-D as a biomarker and the outcome of AAV-mediated gene therapy studies will impact the therapy of one specific class of neurological disorders seen in veterinary clinics: the hereditary ataxias. Hereditary ataxias are a group of neurodegenerative diseases characterized by cerebellar dysfunction; these diseases have no effective therapy[70]. The NPC cat model has shown that spinal fluid concentrations of calbindin-D is an effective biomarker of Purkinje neuron loss[37] - this diagnostic test could be used to both identify hereditary ataxias with Purkinje cell loss, as well as to monitor the efficacy of therapies. Although not discussed above, other biomarkers including plasma neurofilament light chain are also being evaluated in the feline model that could contribute to markers of neurodegeneration in veterinary patients[71]. Regarding therapy, AAV9 and AAVrh10 vectors have shown a great propensity to transduce Purkinje neurons following intrathecal administration in both the dog and cat[2,3,59,72]. These data provide proof-of-concept for the future therapy of hereditary ataxias in which Purkinje cell dysfunction or death are prominent features. In these ataxias, the failure of gene therapy to transduce all brain neurons is less important since the disease is primarily limited to the cerebellum. Finally, the efficacy of N-acetyl-L-leucine to positively affect the ataxia of NPC disease in human patients should support its evaluation in the treatment of other ataxias.
CONCLUSION
Much work has been done in cats with NPC disease that has helped us to understand the pathogenesis and to develop treatments for this disease. If affected cats could be identified prior to or early in the onset of clinical signs, treatment would be possible if drug/biologic costs were controlled. It is more likely that information acquired on biomarkers of Purkinje cell death and on modulations of pathways common to other neurodegenerative diseases, such as neuroinflammation and autophagy, will help in developing treatment strategies for hereditary ataxias.
DECLARATIONS
Authors’ contributions
Involved with the care of the feline breeding colony and of experimental animals: Vite CH, Gurda BL, Casal ML
Responsible for clinical trials in patients: Porter FD
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
During the preparation of this manuscript, the AI tool ChatGPT (version GPT-5.4 Thinking, OpenAI, released 2026-03-05) was used solely for the preparation of the Graphical Abstract. The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship
Vite CH was supported by R01-NS073661, R01-NS115869, P40-OD010939, Ara Parseghian Medical Research Foundation, Dana’s Angels Research Trust (DART), Race for Adam, National Niemann-Pick Disease Foundation, and Support of Accelerated Research for Niemann-Pick Type C disease (SOAR-NPC). Porter FD was supported by the Intramural Research Program of NICHD, NIH (ZIA HD008988). Gurda BL and Casal ML were supported by P40-OD010939.
Conflicts of interest
Vite CH served on the preclinical scientific advisory board of Vtesse, Sucampo, and Mallinckrodt Pharmaceuticals. Porter FD had a Cooperative Research Agreement with Vtesse, Sucampo, and Mallinckrodt. The authors confirm independence from any sponsors; the content of the article was not influenced by any sponsors.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Gurda BL, Bradbury AM, Vite CH. Canine and feline models of human genetic diseases and their contributions to advancing clinical therapies. Yale J Biol Med. 2017;90:417-31.
2. Bradbury AM, Gurda BL, Casal ML, Ponder KP, Vite CH, Haskins ME. A review of gene therapy in canine and feline models of lysosomal storage disorders. Hum Gene Ther Clin Dev. 2015;26:27-37.
3. Gurda BL, Vite CH. Large animal models contribute to the development of therapies for central and peripheral nervous system dysfunction in patients with lysosomal storage diseases. Hum Mol Genet. 2019;28:R119-31.
4. Platt FM, D’azzo A, Davidson BL, Neufeld EF, Tifft CJ. Lysosomal storage diseases. Nat Rev Dis Primers. 2018;4:27.
5. Ludlaim AM, Waddington SN, Mckay TR. Unifying biology of neurodegeneration in lysosomal storage diseases. J Inherit Metab Dis. 2025;48:e12833.
7. Darios F, Stevanin G. Impairment of lysosome function and autophagy in rare neurodegenerative diseases. J Mol Biol. 2020;432:2714-34.
8. Udayar V, Chen Y, Sidransky E, Jagasia R. Lysosomal dysfunction in neurodegeneration: emerging concepts and methods. Trends Neurosci. 2022;45:184-99.
9. Pentchev PG, Comly ME, Kruth HS, et al. A defect in cholesterol esterification in Niemann-Pick disease (type C) patients. Proc. Natl. Acad. Sci. U.S.A. 1985;82:8247-51.
11. Valle DL, Antonarakis S, Ballabio A, Beaudet AL, Mitchell GA. The online metabolic and molecular bases of inherited disease. McGraw-Hill Education; 2019. Available from: https://ommbid.mhmedical.com/book.aspx?bookID=2709 [Last accessed on 12 Jun 2026].
12. Fraldi A, Annunziata F, Lombardi A, et al. Lysosomal fusion and SNARE function are impaired by cholesterol accumulation in lysosomal storage disorders. EMBO J. 2010;29:3607-20.
13. Walkley S. Pyramidal neurons with ectopic dendrites in storage diseases exhibit increased GM2 ganglioside immunoreactivity. Neuroscience. 1995;68:1027-35.
15. Zervas M, Somers KL, Thrall MA, Walkley SU. Critical role for glycosphingolipids in Niemann-Pick disease type C. Curr Biol. 2001;11:1283-7.
16. Walkley SU, Suzuki K. Consequences of NPC1 and NPC2 loss of function in mammalian neurons. Biochim Biophys Acta. 2004;1685:48-62.
17. Peake KB, Vance JE. Defective cholesterol trafficking in Niemann‐Pick C‐deficient cells. FEBS Lett. 2010;584:2731-9.
18. Vance JE, Peake KB. Function of the Niemann-Pick type C proteins and their bypass by cyclodextrin. Curr Opin Lipidol. 2011;22:204-9.
19. Wojtanik KM, Liscum L. The transport of low density lipoprotein-derived cholesterol to the plasma membrane is defective in NPC1 cells. J Biol Chem. 2003;278:14850-6.
20. Liscum L, Ruggiero RM, Faust JR. The intracellular transport of low density lipoprotein-derived cholesterol is defective in Niemann-Pick type C fibroblasts. J Cell Biol. 1989;108:1625-36.
21. Braak H, Braak E, Goebel HH. Isocortical pathology in type C Niemann-Pick disease: a combined Golgi-Pigmentoarchitectonic study. J Neuropathol Exp Neurol. 1983;42:671-87.
22. Elleder M, Jirásek A, Šmíd F, Ledvinová J, Besley GTN. Niemann-Pick disease type C: study on the nature of the cerebral storage process. Acta Neuropathol. 1985;66:325-36.
23. Lowenthal AC, Cummings JF, Wenger DA, Thrall MA, Wood PA, De Lahunta A. Feline sphingolipidosis resembling Niemann-Pick disease type C. Acta Neuropathol. 1990;81:189-97.
24. Muñana KR, Luttgen PJ, Thrall MA, Mitchell TW, Wenger DA. Neurological Manifestations of Niemann-Pick Disease Type C in Cats. J Vet Intern Med. 1994;8:117-21.
25. Brown DE, Thrall MA, Walkley SU, et al. Feline Niemann-Pick disease type C. Am J Pathol. 1994;144:1412-5.
26. March PA, Thrall MA, Brown DE, Mitchell TW, Lowenthal AC, Walkley SU. GABAergic neuroaxonal dystrophy and other cytopathological alterations in feline Niemann-Pick disease type C. Acta Neuropathol. 1997;94:164-72.
27. Zervas M, Dobrenis K, Walkley SU. Neurons in Niemann-Pick disease type C accumulate gangliosides as well as unesterified cholesterol and undergo dendritic and axonal alterations. J Neuropathol Exp Neurol. 2001;60:49-64.
28. Somers KL, Brown DE, Fulton R, et al. Effects of dietary cholesterol restriction in a feline model of Niemann-Pick type C disease. J Inherit Metab Dis. 2001;24:427-36.
30. Vite CH, Ding W, Bryan C, et al. Clinical, electrophysiological, and serum biochemical measures of progressive neurological and hepatic dysfunction in feline Niemann-Pick Type C disease. Pediatr Res. 2008;64:544-9.
31. Bagel JH, Sikora TU, Prociuk M, et al. Electrodiagnostic testing and histopathologic changes confirm peripheral nervous system myelin abnormalities in the feline model of Niemann-Pick disease type C. J Neuropathol Exp Neurol. 2013;72:256-62.
32. Roszell BR, Tao J, Yu KJ, et al. Pulmonary abnormalities in animal models due to Niemann-Pick type C1 (NPC1) or C2 (NPC2) disease. PLoS ONE. 2013;8:e67084.
33. Gurda BL, Bagel JH, Fisher SJ, et al. LC3 Immunostaining in the inferior olivary nuclei of cats with Niemann-Pick disease type C1 is associated with patterned purkinje cell loss. J Neuropathol Exp Neurol. 2018;77:229-45.
34. Porter FD, Scherrer DE, Lanier MH, et al. Cholesterol oxidation products are sensitive and specific blood-based biomarkers for Niemann-Pick C1 disease. Sci Transl Med. 2010;2:56ra81.
35. Tortelli B, Fujiwara H, Bagel JH, et al. Cholesterol homeostatic responses provide biomarkers for monitoring treatment for the neurodegenerative disease Niemann-Pick C1 (NPC1). Hum Mol Genet. 2014;23:6022-33.
36. Fan M, Sidhu R, Fujiwara H, et al. Identification of Niemann-Pick C1 disease biomarkers through sphingolipid profiling. J Lipid Res. 2013;54:2800-14.
37. Bradbury A, Bagel J, Sampson M, et al. Cerebrospinal fluid calbindin d concentration as a biomarker of cerebellar disease progression in Niemann-Pick type C1 disease. J Pharmacol Exp Ther. 2016;358:254-61.
38. Sidhu R, Kell P, Dietzen DJ, et al. Application of a glycinated bile acid biomarker for diagnosis and assessment of response to treatment in Niemann-Pick disease type C1. Mol Genet Metab. 2020;131:405-17.
39. Mishra S, Kell P, Scherrer D, et al. Accumulation of alkyl-lysophosphatidylcholines in Niemann-Pick disease type C1. J Lipid Res. 2024;65:100600.
40. Stein VM, Crooks A, Ding W, et al. Miglustat improves purkinje cell survival and alters microglial phenotype in feline Niemann-Pick disease type C. J Neuropathol Exp Neurol. 2012;71:434-48.
41. Ward S, O’donnell P, Fernandez S, Vite CH. 2-hydroxypropyl-β-cyclodextrin raises hearing threshold in normal cats and in cats with Niemann-Pick type C disease. Pediatr Res. 2010;68:52-6.
42. Vite CH, Bagel JH, Swain GP, et al. Intracisternal cyclodextrin prevents cerebellar dysfunction and Purkinje cell death in feline Niemann-Pick type C1 disease. Sci Transl Med. 2015;7.
43. Kao ML, Stellar S, Solon E, et al. Pharmacokinetics and distribution of 2‐hydroxypropyl‐β‐cyclodextrin following a single intrathecal dose to cats. J Inherit Metab Dis. 2019;43:618-34.
44. Kurokawa Y, Osaka H, Kouga T, et al. Gene therapy in a mouse model of Niemann-Pick disease type C1. Hum Gene Ther. 2021;32:589-98.
45. Davidson CD, Gibson AL, Gu T, et al. Improved systemic AAV gene therapy with a neurotrophic capsid in Niemann-Pick disease type C1 mice. Life Sci. Alliance. 2021;4:e202101040.
46. Hughes MP, Nelvagal HR, Coombe-tennant O, et al. A novel small NPC1 promoter enhances AAV-mediated gene therapy in mouse models of Niemann-Pick Type C1 disease. Cells. 2023;12:1619.
47. Hughes MP, Smith DA, Morris L, et al. AAV9 intracerebroventricular gene therapy improves lifespan, locomotor function and pathology in a mouse model of Niemann-Pick type C1 disease. Hum Mol Genet. 2018;27:3079-98.
48. Ory DS, Ottinger EA, Farhat NY, et al. Intrathecal 2-hydroxypropyl-β-cyclodextrin decreases neurological disease progression in Niemann-Pick disease, type C1: a non-randomised, open-label, phase 1-2 trial. The Lancet. 2017;390:1758-68.
49. Ottinger EA, Kao ML, Carrillo-Carrasco N, et al. Collaborative development of 2-hydroxypropyl-β-cyclodextrin for the treatment of Niemann-Pick type C1 disease. Curr Top Med Chem. 2014;14:330-9.
50. Mauler D, Gandolfi B, Reinero C, et al. ; and 99 Lives Consortium. Precision medicine in cats: novel Niemann-Pick type C1 diagnosed by whole-genome sequencing. J Vet Intern Med. 2017;31:539-44.
51. Gutić M, Milosavljević MN, Janković SM. Cost-effectiveness of miglustat versus symptomatic therapy of Niemann-Pick disease type C. Int J Clin Pharm. 2022;44:1442-53.
52. Yoon SY, Hunter JE, Chawla S, et al. Global CNS correction in a large brain model of human alpha-mannosidosis by intravascular gene therapy. Brain. 2020;143:2058-72.
53. Yoon SY, Gay-antaki C, Ponde DE, Poptani H, Vite CH, Wolfe JH. Quantitative, noninvasive, in vivo longitudinal monitoring of gene expression in the brain by co-AAV transduction with a PET reporter gene. Mol Ther Methods Clin Dev. 2014;1:14016.
54. Yoon SY, Bagel JH, O’donnell PA, Vite CH, Wolfe JH. Clinical improvement of alpha-mannosidosis cat following a single cisterna magna infusion of AAV1. Mol Ther. 2016;24:26-33.
55. Hunter JE, Vite CH, Molony CM, O’donnell PA, Wolfe JH. Intracisternal vs intraventricular injection of AAV1 result in comparable, widespread transduction of the dog brain. Gene Ther. 2024;32:184-8.
56. Hunter JE, Molony CM, Panek WK, et al. Effect of disease progression on CSF-directed AAV gene therapy in a large brain animal model of lysosomal storage disease. Mol Ther Methods Clin Dev. 2025;33:101552.
57. Hunter JE, Molony CM, Bagel JH, et al. Widespread correction of brain pathology in feline alpha-mannosidosis by dose escalation of intracisternal AAV vector injection. Mol Ther Methods Clin Dev. 2024;32:101272.
58. Vite CH, Passini MA, Haskins ME, Wolfe JH. Adeno-associated virus vector-mediated transduction in the cat brain. Gene Ther. 2003;10:1874-81.
59. Bradbury AM, Bagel JH, Nguyen D, et al. Krabbe disease successfully treated via monotherapy of intrathecal gene therapy. J Clin Investig. 2020;130:4906-20.
60. Muto Y, Suzuki M, Takei H, et al. Dried blood spot-based newborn screening for bile acid synthesis disorders, Zellweger spectrum disorder, and Niemann-Pick type C1 by detection of bile acid metabolites. Mol Genet Metab. 2023;140:107703.
61. Costa Verdera H, Kuranda K, Mingozzi F. AAV vector immunogenicity in humans: a long journey to successful gene transfer. Mol Ther. 2020;28:723-46.
62. Fratantoni JC, Hall CW, Neufeld EF. Hurler and hunter syndromes: mutual correction of the defect in cultured fibroblasts. Science. 1968;162:570-2.
63. De Marchi F, Munitic I, Vidatic L, et al. Overlapping neuroimmune mechanisms and therapeutic targets in neurodegenerative disorders. Biomedicines. 2023;11:2793.
64. Chu T, Tu X, Yang K, Wu J, Repa JJ, Yan N. Tonic prime-boost of STING signalling mediates Niemann-Pick disease type C. Nature. 2021;596:570-5.
65. Azaria RD, Correia AB, Schache KJ, et al. Mutant induced neurons and humanized mice enable identification of Niemann-Pick type C1 proteostatic therapies. JCI Insight. 2024;9:e179525.
66. Murray CE, Betancourt-trompa DS, Martinez MS, et al. Role of mTORC1 signaling in postnatal microglia activation preceding neurodegeneration in a mouse model for Niemann-Pick disease Type C. PLoS ONE. 2025;20:e0330437.
67. Gascón-bayarri J, Rico I, Sánchez-castañeda C, et al. Efficacy and safety of efavirenz in Niemann-Pick disease type C. Neurotherapeutics. 2025;22:e00706.
68. Shammas H, Kloster Fog C, Klein P, et al. Mechanistic insights into arimoclomol mediated effects on lysosomal function in Niemann-Pick type C disease. Mol Genet Metab. 2025;145:109103.
69. Patterson MC, Ramaswami U, Donald A, et al. Disease-modifying, neuroprotective effect of N-Acetyl-l-leucine in adult and pediatric patients with Niemann-Pick disease type C. Neurology. 2025;105:e213589.
70. Stee K, Van Poucke M, Lowrie M, et al. Phenotypic and genetic aspects of hereditary ataxia in dogs. J Vet Intern Med. 2023;37:1306-22.
71. Casazza K, Cologna SM, Berry‐kravis E, Jarnes J, Porter FD. Biomarker Validation inNPC1: foundations for clinical trials and regulatory alignment. J Inherit Metab Dis. 2025;48:e70075.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].