

Discriminating dyslipidemias - a South African perspective
 0
0 
Abstract
Clinical observation and scientific research have established that premature atherosclerotic vascular disease is strongly associated with markedly elevated low-density lipoprotein (LDL) cholesterol levels. Genetic disorders that impair LDL clearance via LDL receptors (LDLRs) are classified into heterozygous and homozygous familial hypercholesterolemia (FH) phenotypes. The heterozygous FH (HeFH) phenotype is characterized by LDL cholesterol levels exceeding 4.9 mmol/L, the presence of tendon xanthomas, and premature heart disease. In contrast, homozygous FH (HoFH) is characterised by LDL cholesterol levels above 13 mmol/L, both cutaneous and tendon xanthomas, and may lead to atherosclerotic vascular disease manifesting in childhood. FH is relatively common and occurs at even higher frequencies in populations founded by small ancestral groups, as documented in South Africa through investigations conducted in tertiary healthcare settings. Despite significant advances in the diagnosis and treatment of lipid and lipoprotein disorders - many of which are part of the differential diagnosis of FH - support for the diagnosis and management of FH has declined. While existing clinical guidelines address most cases of hypercholesterolemia, a subset of individuals with severe dyslipidemias requires more specialized evaluation. Referral criteria are proposed to help identify these patients. Given the high cardiovascular risk associated with severe hypercholesterolemia and the availability of effective treatments, there is an urgent need to strengthen, coordinate, and integrate clinical and laboratory services in South Africa to differentiate among the various causes of these disorders.
Keywords
INTRODUCTION
Since Harbitz and Müller first linked atherosclerosis and xanthomas[1], and familial hypercholesterolemia (FH) was identified as a disorder caused by defects in the low-density lipoprotein (LDL) receptor[2], additional genetic mutations have been discovered that lead to severe hypercholesterolemia and markedly increased risk of atherosclerosis[3]. Distinguishing among the various causes of severe hypercholesterolemia is essential for optimizing risk management , adapting dietary and pharmacologic interventions, and providing accurate genetic counseling. Current guidelines for the management of dyslipidemia appropriately recommend assessing global atherosclerotic vascular disease risk by considering multiple factors to guide treatment decisions. FH and related genetic lipid disorders almost inevitably promote atherosclerosis, which may manifest as early as the first decade of life in individuals with homozygous FH (HoFH), but can be mitigated with pharmacotherapy[4].
In cases of severe hypercholesterolemia accompanied by normal or only modestly elevated triglyceride levels, a thorough clinical assessment by an informed clinician is warranted. The patient’s ancestry may suggest the presence of a founder mutation. Determining whether the inheritance pattern is autosomal dominant or recessive can help identify the underlying cause of the dyslipidemia and facilitate genetic investigation. Secondary causes, such as hypothyroidism or nephrotic syndrome, should also be considered. Several lipid metabolic disorders may present with manifestations affecting the nervous system, skin, or other organs. Physical examination should include documentation of anthropometric measures relevant to cardiovascular risk, clinical signs of dyslipidemia, vascular function, neurological manifestations, and abnormalities of the reticuloendothelial system. Routine laboratory evaluation includes measurement of triglycerides, total cholesterol, high-density lipoprotein (HDL) cholesterol, and LDL cholesterol. Additional assessments such as apolipoprotein B (apoB) and lipoprotein (a) [Lp(a)] levels enhance risk assessment. Although Lp(a) seldom contributes more than 1 mmol/L (38.7mg/dL) of cholesterol, its measurement remains clinically relevant. Polyacrylamide gradient gel electrophoresis of apoB-containing lipoproteins is useful for diagnosing dysbetalipoproteinemia[5] and detecting lipoprotein X (LpX)[6]. Chromatographic techniques can identify abnormal sterol profile. Genetic testing confirms the clinical diagnosis, clarifies inheritance patterns in family screening, and will become increasingly important as gene-editing therapies advance.
SEVERE PRIMARY (GENETIC) HYPERCHOLESTEROLEMIA
A diagnosis of FH is considered when total cholesterol exceeds 7.0 mmol/L (270 mg/dL) or LDL cholesterol exceeds 4.9 mmol/L (190 mg/dL), and triglycerides are below 5 mmol/L (425 mg/dL), allowing for potential influences from incidental genetic variation and postprandial states.
Differential diagnoses for FH are summarized in Table 1. The phenotype can be affected by other genes, lifestyle factors, and secondary causes. The most common cause of HeFH is a mutation in the LDL receptor (LDLR) gene. Autosomal dominant inheritance is seen in mutations involving the LDLR, apoB, and proprotein convertase subtilisin/kexin type 9 (PCSK9) gain-of-function variants. HeFH typically leads to coronary events in men during their fifth decade of life[7,8]. Polygenic severe hypercholesterolemia has been associated with multiple genes, with the specific variants varying across populations[9]. Congenital analbuminemia[10] is a rare, autosomal recessive disorder. Lysosomal acid lipase deficiency, also rare, can be treated with enzyme replacement therapy using sebelipase alfa[11]. Oligogenic hypercholesterolemia, such as the co-occurrence of LDLR and APOB variants, presents with an intermediate phenotype between HeFH and HoFH. The most common cause of HoFH is the inheritance of biallelic pathogenic LDLR variants, with each parent contributing a mutation that causes HeFH. The HoFH phenotype may also result from recessive LDL receptor adaptor protein 1 deficiency[12] or ABCG5/ABCG8 transporter deficiency, as seen in phytosterolemia[13]. Phytosterolemia can mimic HeFH depending on dietary sterol intake. It responds well to dietary restriction of phytosterols and cholesterol, but poorly to statin therapy. Markedly elevated cholesterol levels can also occur with LpX[14], commonly associated with cholestasis, even in the absence of severe liver dysfunction. As LpX lacks apoB, apoB levels will not increase significantly from the previous state of health. Cerebrotendinous xanthomatosis is a rare autosomal recessive disorder that usually manifests in childhood or adolescence with neurological degeneration and cataracts. However, milder variants may present later with xanthomas and hypercholesterolemia[15]. Dysbetalipoproteinemia can also present with severe hypercholesterolemia in the absence of marked hypertriglyceridemia. This can lead to misdiagnosis as FH, especially in cases involving dominantly inherited apoE variants. Although ApoE2 homozygosity affects about 1% of the population, dysbetalipoproteinemia only develops in the presence of additional metabolic stress[16].
Severe Hypercholesterolemia Phenotypes. Phenotypes to consider when plasma or serum total cholesterol exceeds 7.0 mmol/L, in the absence of severe hypertriglyceridemia
| Phenotype | Lipoproteins | Clinical characteristics | Genes |
| Heterozygous FH (HeFH) | LDL-C 4.9-12 mmol/L | Autosomal dominant. Tendon xanthomas and coronary artery disease after age 25. Tendon xanthomas are common in adults but not always present. Good response to statins; additional therapy may be needed | LDLR, APOB, PCSK9 GOF, APOE del167 |
| Polygenic FH | LDL-C 4.9-9 mmol/L | No clear inheritance pattern. Xanthomas typically absent. Responds to statins. Involves multiple gene variants; 12 originally described. Atherosclerotic risk elevated, though lower than in HeFH | PCSK9, CELSR2, APOB, ABCG8, SLC22A1, HFE, MYLIP, ST3GAL4, NYNRIN, LDLR, APOE |
| Oligogenic FH / double heterozygotes | LDL-C 9-15 mmol/L | Similar to HeFH. Both parents are hypercholesterolemic. Responds to statins | Two different HeFH-related genes |
| Homozygous FH (HoFH) | LDL-C 12-25 mmol/L | Cutaneous and tendon xanthomas. Both parents hypercholesterolemic. Variable response to statins; minimal benefit in cases with null mutations. PCSK9 inhibitors often ineffective | Mostly LDLR |
| Autosomal Recessive Hypercholesterolemia (resembles HoFH) | LDL-C 12-18 mmol/L | Recessive inheritance. Cutaneous and tendon xanthomas. Better statin response than HoFH due to residual LDL receptor function | LDLRAP1 |
| Phytosterolemia (may resemble HeFH or HoFH) | LDL-C 4.9-20 mmol/L | Recessive. Cutaneous and tendon xanthomas. Highly responsive to dietary changes; statins ineffective. Ezetimibe is beneficial | ABCG5 and/or ABCG8 |
| Cerebrotendinous xanthomatosis (may resemble HeFH) | LDL-C 3-10 mmol/L | Recessive. Marked tendon xanthomas. May present with cataracts, ataxia, and dementia, or just xanthomas and hypercholesterolemia. Responds to bile acid sequestrants, ezetimibe, and sometimes statins | CYP27A1 (Cholesterol 27-hydroxylase) |
| Analbuminemia (considered in HeFH) | LDL-C 4.9-10 mmol/L | Recessive. Presents with low blood pressure, elevated ESR, and altered drug metabolism. Responds well to statins | Albumin |
| Lysosomal acid lipase deficiency (considered in HeFH) | LDL-C 4.9-10 mmol/L ± VLDL (TG) | Recessive. Hepatomegaly and enzyme abnormalities. Diminished LAL activity in leucocytes. Responds to statins; enzyme replacement available | LIPA (LAL) |
| Lipoprotein X | TC 7.0 -65 mmol/L; TG may be mildly elevated | Cutaneous xanthomas. Usually secondary to intra- or extrahepatic cholestasis. Jaundice may be mild. Bile acid disorders typically recessive | Bile acid synthetic enzyme defects |
| Dysbetalipoproteinemia | TC > 7.0 mmol/L; TG < 5.0 mmol/L; LDL low/absent, cholesterol in VLDL-IDL | Inherited in a recessive or dominant pattern with delayed penetrance. Cutaneous and tendon xanthomas. May present with severe hypertriglyceridemia. Responds to dietary changes, statins, and fibrates | APOE, LIPC |
FAMILIAL HYPERCHOLESTEROLEMIA IN SOUTH AFRICA
Despite recognition of founder effects in several communities and international calls to action regarding FH[17], severe hypercholesterolemia continues to receive inadequate attention in South Africa. Medical schools, established about a century ago, developed specialized clinics and laboratories - jointly operated by universities and provincial governments at teaching hospitals - to provide tertiary care. Patients with severe or unusual conditions benefited from this concentration of expertise. For example, severe heart failure due to premature ischemic heart disease was managed with cardiac transplantation[18], in at least one patient who possibly had FH.
HoFH was first identified in the (white) Afrikaner community[19], where subsequent investigations uncovered mutations in the LDLR gene[20]. Heritable lipid disorders in Black patients included reports of HoFH[21] and dysbetalipoproteinemia[22]. An Indian patient with HoFH was also reported[23]. Advanced laboratory investigations - including cell culture, ultracentrifugation, and genetic testing - were conducted at the University of Cape Town, Groote Schuur Hospital, and the Red Cross War Memorial Children’s Hospital. In 1988, the Lipid and Atherosclerosis Society of Southern Africa (LASSA) was founded, drawing participation from over 200 medical and scientific professionals, primarily affiliated with medical schools. Referral criteria were established to evaluate severe dyslipidemia and related disorders, as described in Table 2. Considerable expertise was accumulated in diagnosing and managing FH[8]. Plasma exchange was offered for patients with HoFH, who also benefited from statin therapy[24].
Referral criteria for tertiary medical services. Based on clinical laboratory results, physical signs, or disease manifestations
| Parameter | Lower limit | Upper limit |
| Triglycerides | Not applicable. (< 0.5mmol/L in abetalipoproteinemia) | > 10 mmol/L - risk of pancreatitis; various causes |
| Total cholesterol | < 2.0 mmol/L - consider hypobetalipoproteinemia, Abetalipoproteinemia | > 7.0 mmol/L - familial hypercholesterolemia (See Table 1) |
| LDL-C | < 1.0 mmol/L (ApoB < 0.5 g/L) - hypobetalipoproteinemia, abetalipoproteinemia | > 4.9 mmol/L (ApoB > 1.2g/L) - familial hypercholesterolemia (See Table 1) |
| HDL-C | < 0.6 mmol/L | > 2.5 mmol/L |
| Clinical indicator | Category 1 | Category 2 |
| Xanthomas | Cutaneous (excluding isolated xanthelasma, which is less reliable); often related to triglyceride elevation or rare sterol disorders (e.g., LpX, hypoalphalipoproteinemias) | Tendinous xanthomas (note prior Achilles rupture may cause thickening); typically due to severe hypercholesterolemia, cerebrotendinous xanthomatosis, or phytosterolemia |
| Premature cardiovascular disease | Metabolic: Homocystinuria, Ehlers-Danlos syndrome, Marfan syndrome | Vascular malformations |
| Congenital Malformations | Early sterol pathway defects: Mevalonate kinase deficiency (hyper-IgD syndrome), CHILD syndrome, Antley-Bixler syndrome, Conradi-Hünermann syndrome, Greenberg dysplasia | Late sterol pathway defects: Dysmorphology, syndactyly, feeding difficulties, cognitive delay (e.g., Smith-Lemli-Opitz syndrome, desmosterolosis, lathosterolosis) |
| Other systems | Neurologic: Adrenoleukodystrophy, Refsum disease, Ichthyosis | Endocrine: Zellweger syndrome, adrenoleukodystrophy |
A grant from the Medical Research Council supported the creation of a dedicated diagnostic laboratory during a time when broader support for tertiary care was being withdrawn. Clinical trials involving new pharmaceutical agents were conducted for patients with HeFH and HoFH, who benefited from specialized expertise and therapeutic advances. Although funding ceased around 2010, it left a legacy of genetically confirmed cases of FH and dysbetalipoproteinemia. Patients with HeFH and clinical evidence of tendon xanthomas were investigated exon-by-exon for mutations in the LDLR and PCSK9 genes, as well as in exons 26 and 29 of APOB. Polymerase chain reaction followed by high-resolution melting analysis identified variants, which were confirmed via restriction enzyme digestion and/or sequencing. Although not all patients underwent full genetic testing, 1,182 were genetically confirmed to have HeFH. Among 87 pathogenic variants in the LDLR gene, seven were novel. The impact of founder effects in the Western Cape was evident, with more than 90% of HeFH cases attributable to 10 specific LDLR variants, listed in descending order based on the mature protein: D206E, V408M, D154N, D200G, del197, G361V, C356Y, R329X, F382S, and E207K. Five of these variants predominated in Afrikaner and mixed-ancestry populations, which also shared several variants. APOB mutations were confined to exon 26 (R3500Q, R3500W, R3531C, H3543Y) and accounted for < 1% of HeFH cases. Similarly, gain-of-function mutations in PCSK9 were rare (< 1%) and included S127R, R469W, R496W, and H553R. The AOPE R145C variant accounted for 20% of dysbetalipoproteinemia cases confirmed by ultracentrifugation or electrophoresis[5], with 75% of patients showing the characteristic apoE2 homozygosity.
The private healthcare sector in South Africa expanded during the 1980s as support for public tertiary care, including lipid clinics, diminished. Both public and private sectors were initially reluctant to adopt cholesterol-lowering therapies for FH, largely due to skepticism regarding the benefits of cholesterol reduction[25]. The shift toward a primary healthcare model led to poorly coordinated care for patients previously followed at specialized lipid clinics[26,27]. As support for tertiary care declined, so did LASSA membership. LASSA eventually joined the South African Heart Association (now SAHeart), which is affiliated with the European Atherosclerosis Society. EAS guidelines for the management of dyslipidemia[28] have been adapted for local use[29,30]. However, public and private healthcare systems maintain separate formularies and have provided limited support for the treatment of severe disorders such as FH. LASSA has not only contributed to national guidelines but also objected to inappropriate clinical management[31-34]. The National Health Laboratory Service (NHLS) Act 37 of 2,000[35] states its intention to provide laboratory services to all South Africans. Nevertheless, it has not expanded testing beyond standard clinical panels. While apolipoproteins A1 and B, Lp(a), and agarose gel electrophoresis are available at some teaching hospitals, additional biochemical and genetic testing for dyslipidemia is not routinely supported. Care for patients with severe lipid disorders has largely deteriorated due to insufficient support, staff attrition, lack of equipment replacement, absence of succession planning, and general failure to sustain the necessary expertise. Medical schools have not succeeded in maintaining lipid clinics or laboratories. LASSA’s attempts to engage the Minister of Health and the Committee of Deans of Health Science Faculties to preserve expertise for the estimated 500,000 people in South Africa with severe lipid and lipoprotein disorders - including at least 200,000 with FH - have been unsuccessful. Once consolidated, South Africa’s expertise could serve as a model for other African countries. Teaching visits to Botswana, Zimbabwe, and Ethiopia have highlighted a clear need for such expertise. The estimated number of individuals with FH in sub-Saharan Africa exceeds 1 million[36].
SOLUTIONS FOR FH AND SEVERE DYSLIPIDEMIA IN SOUTH AFRICA
In modern medical practice, the diagnosis and management of severe disorders are expected to be both timely and cost-effective. Conditions such as HeFH should be recognized and managed at the primary healthcare level, while more complex cases should be referred to specialized clinics. Early identification of FH, along with the initiation of a healthy lifestyle and statin therapy (combined with ezetimibe in some cases), can significantly improve prognosis and reduce the cost of treating complications.
South Africa should consolidate expertise and promote care by establishing at least one (national) center dedicated to the management of the full spectrum of lipid and lipoprotein disorders. Unlike in developed countries, where vascular medicine typically addresses atherogenic conditions, such a South African center should initially manage a broader range of lipid disorders until other specialties gradually assume responsibility for specific clinical services. The laboratory component, however, would likely be best maintained within the national center. This facility would also serve to document the prevalence and characteristics of locally relevant conditions while addressing the country’s healthcare needs.
Undergraduate medical education should emphasize FH and dysbetalipoproteinemia. Although physical signs of dyslipidemia may not always be present, they should be recognized when encountered. These physical manifestations are illustrated in Figure 1. Useful clinical aphorisms include: severe hypertriglyceridemia is associated with cutaneous xanthomas; severe sterol disorders manifest as tendon xanthomas; and mixed hyperlipidemia (e.g., dysbetalipoproteinemia) and extreme sterol disorders (such as HoFH and phytosterolemia) may present with both cutaneous and tendinous xanthomas. Xanthelasma, a planar cholesterol deposit on the eyelids, is less specific and not necessarily indicative of severe dyslipidemia.
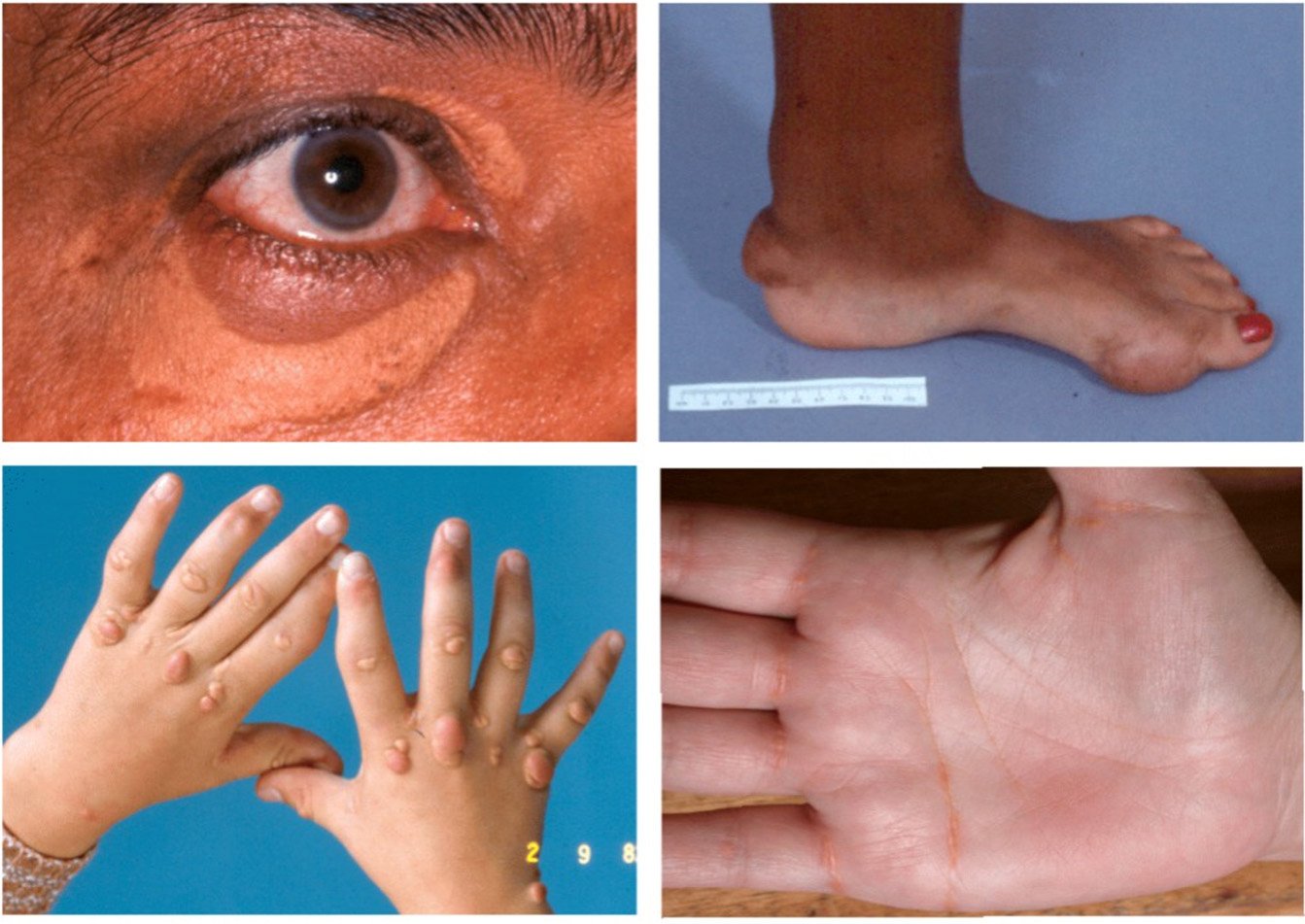
Figure 1. Physical signs of hypercholesterolemia. Top left: Xanthelasma, a planar cutaneous xanthoma located near the orbit, is less specific for FH than arcus cornealis, which appears superiorly and inferiorly at the limbus. Top right: Subcutaneous xanthoma at the ankle and thickened Achilles tendon in a teenager with HoFH. Bottom left: The hands of a child with HoFH showing pathognomonic interdigital web planar xanthomas and cutaneous tuberous xanthomas. Bottom right: Lipid deposits in the creases of the palm and fingers, indicative of dysbetalipoproteinemia.
Public education should highlight the risks of atherosclerosis, particularly premature atherosclerosis resulting from FH. Self-identification of lipid and lipoprotein disorders may be enhanced by displaying photographs of characteristic physical signs at community health centers. Policymakers and healthcare funders - both in the large public sector and the smaller private sector - should be informed about the appropriate management of severe dyslipidemias, particularly FH and dysbetalipoproteinemia. Although clinical guidelines are useful, they have limitations; referral to tertiary care according to the criteria outlined in Table 2 will help ensure accurate diagnosis and optimal management. In the case of FH, statins are now affordable and can safely prevent premature coronary disease, enabling individuals to remain productive and economically active. While not all advanced therapies may be financially viable, expert management can help avoid unnecessary investigations and treatments. Specialized dietary interventions can significantly lower plasma cholesterol (and phytosterol) levels in cases where statins are ineffective[37].
Telemedicine consultations could connect healthcare providers in remote areas with specialists at the national center. Training programs in paediatrics, internal medicine, cardiology, endocrinology, gastroenterology, dermatology, and chemical pathology should include exposure to lipid and lipoprotein metabolism disorders.
In conclusion, distinguishing severe lipid and lipoprotein disorders from moderate dyslipidemias is essential. Accurate diagnosis enables the most effective therapeutic interventions. Unfortunately, lipid and lipoprotein disorders have historically received insufficient attention in South Africa. Improved education for both the public and healthcare professionals will enhance awareness, diagnosis, and treatment of dyslipidemias. The establishment of a dedicated clinical and laboratory center is crucial for identifying severe disorders, advancing local research and experience, and improving care for patients in South Africa and beyond.
DECLARATION
Authors’ contributions
The author contributed solely to the article.
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
The author declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
1. Ose L. Müller-harbitz disease--familial hypercholesterolemia. Tidsskr Nor Laegeforen. 2002;122:924-5.
2. Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232:34-47.
3. Taranto MD, Fortunato G. Genetic heterogeneity of familial hypercholesterolemia: repercussions for molecular diagnosis. Int J Mol Sci. 2023;24:3224.
4. Thompson GR, Blom DJ, Marais AD, Seed M, Pilcher GJ, Raal FJ. Survival in homozygous familial hypercholesterolaemia is determined by the on-treatment level of serum cholesterol. Eur Heart J. 2018;39:1162-8.
5. Blom DJ, Byrnes P, Jones S, Marais AD. Non-denaturing polyacrylamide gradient gel electrophoresis for the diagnosis of dysbetalipoproteinemia. J Lipid Res. 2003;44:212-7.
6. Phatlhane DV, Zemlin AE. Severe hypercholesterolemia mediated by lipoprotein X in a patient with cholestasis. Ann Hepatol. 2015;14:924-8.
7. Slack J. Risks of ischaemic heart-disease in familial hyperlipoproteinaemic states. Lancet. 1969;2:1380-2.
8. Firth JC, Marais AD. Familial hypercholesterolaemia: the Cape Town experience. S Afr Med J. 2008;98:99-104.
9. Talmud PJ, Shah S, Whittall R, et al. Use of low-density lipoprotein cholesterol gene score to distinguish patients with polygenic and monogenic familial hypercholesterolaemia: a case-control study. Lancet. 2013;381:1293-301.
10. Minchiotti L, Caridi G, Campagnoli M, Lugani F, Galliano M, Kragh-Hansen U. Diagnosis, phenotype, and molecular genetics of congenital Analbuminemia. Front Genet. 2019;10:336.
11. Bashir A, Tiwari P, Duseja A. Enzyme replacement therapy in lysosomal acid lipase deficiency (LAL-D): a systematic literature review. Ther Adv Rare Dis. 2021;2:26330040211026928.
12. D’Erasmo L, Di Costanzo A, Arca M. Autosomal recessive hypercholesterolemia: update for 2020. Curr Opin Lipidol. 2020;31:56-61.
13. Windler E, Beil FU, Berthold HK, et al. Phytosterols and cardiovascular risk evaluated against the background of phytosterolemia cases-a German expert panel statement. Nutrients. 2023;15:828.
14. Fellin R, Manzato E. Lipoprotein-X fifty years after its original discovery. Nutr Metab Cardiovasc Dis. 2019;29:4-8.
15. Stelten BML, Raal FJ, Marais AD, et al. Cerebrotendinous xanthomatosis without neurological involvement. J Intern Med. 2021;290:1039-47.
16. Marais AD. Apolipoprotein E in lipoprotein metabolism, health and cardiovascular disease. Pathology. 2019;51:165-76.
17. Wilemon KA, Patel J, Aguilar-Salinas C, et al. Reducing the clinical and public health burden of familial hypercholesterolemia: a global call to action. JAMA Cardiol. 2020;5:217-29.
18. Barnard CN. The operation. A human cardiac transplant: an interim report of a successful operation performed at Groote Schuur Hospital, Cape Town. S Afr Med J. 1967;41:1271-4.
19. Seftel HC, Baker SG, Sandler MP, et al. A host of hypercholesterolaemic homozygotes in South Africa. Br Med J. 1980;281:633-6.
20. Gevers W. Three mutations that cause familial hypercholesterolemia in Afrikaners identified-a milestone in South African medicine. S Afr Med J. 1989;76:393-4.
21. Marais AD, Firth JC, Rose AG, Berger GM. Fatal outcome of homozygous familial hypercholesterolaemia in a black patient. A case report. S Afr Med J. 1990;77:588-90.
22. Marais AD, Berger GMB. A diversity of genetic hyperlipoproteinaemias in black patients. Experience at the lipid clinics at Groote Schuur hospital and red cross war memorial children's hospital, Cape Town. S Afr Med J. 1986;70:583-7.
23. Langenhoven E, Warnich L, Thiart R, et al. Two novel point mutations causing receptor-negative familial hypercholesterolemia in a South African Indian homozygote. Atherosclerosis. 1996;125:111-9.
24. Marais AD, Naoumova RP, Firth JC, Penny C, Neuwirth CKY, Thompson GR. Decreased production of low density lipoprotein by atorvastatin after apheresis in homozygous familial hypercholesterolemia. J Lipid Res. 1997;38:2071-8.
25. Davey Smith G, Pekkanen J. Should there be a moratorium on the use of cholesterol lowering drugs? BMJ. 1992;304:431-4.
26. Marais AD, Benatar SR. Planning rational management of chronic diseases-lessons from a lipid clinic. S Afr Med J. 1995;85:340-1.
28. Mach F, Baigent C, Catapano AL, et al. 2019 ESC/EAS guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. 2020;41:111-88.
29. Klug EQ, Raal FJ, Marais AD, et al. South African dyslipidaemia guideline consensus statement: 2018 update A joint statement from the South African Heart Association (SA Heart) and the Lipid and Atherosclerosis Society of Southern Africa (LASSA). S Afr Med J. 2018;108:973-1000.
30. Klug QE, Raal FJ. Heart groups in South Africa advocate for tighter LDL-C control and lipoprotein(a) testing to curb atherosclerotic cardiovascular disease. S Afr Med J. 2024;114:e1973.
32. Marais AD, Blom DJ, Raal FJ. Response to: prescribed minimum benefits complaints: a 5-year retrospective review. S Afr Med J. 2024;114:e2450.
34. Marais AD, Blom DJ, Raal FJ; On Behalf Of The Lipid And The Lipid And Atherosclerosis Society Of Southern Africa. Familial hypercholesterolaemia in South Africa: a reminder. S Afr Med J. 2021;111:700-1.
35. South African Government. National Health Laboratory Service Act 37 of 2000. Republic of South Africa. Available from:https://www.gov.za/documents/national-health-laboratory-service-act [Last accessed on 18 JuL 2025].
36. Marais AD. Familial hypercholesterolaemia and genetic dyslipidaemia: opportunities and needs in management as seen from South Africa. Data presented at the ISA 2012 Satellite Meeting of the International Atherosclerosis Society, Sydney, Australia. 2012.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Issue
Copyright

Data & Comments
Data
0











Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].