

From hype to hope: foundational requirements for NF1 gene therapy success
 0
0 
Abstract
Gene therapy for Neurofibromatosis type 1 (NF1), defined as a collection of approaches that restore the quantity and function of neurofibromin, holds promise for treating various NF-associated clinical manifestations by addressing the root cause of the condition. The Gilbert Family Foundation’s Gene Therapy Initiative (GTI) launched one of the first focused research efforts to support a broad array of gene replacement, gene editing, and other strategies aimed at upregulating neurofibromin levels to mitigate symptoms and advance therapies for NF1. Efforts to discover and evaluate novel gene therapies to treat NF1 are hindered by foundational gaps in the field, the absence of quantitative assays, and a lack of robust preclinical models with reliable functional readouts. This perspective begins by shedding light on those key barriers that continue to hinder progress in NF1 gene therapy, outlines some of the strategies being explored to overcome them and offers pointers to additional strategies that merit further exploration. We then highlight several promising gene therapy strategies now in development. Successfully addressing the challenges described earlier is critical to realizing the full potential of these emerging and novel therapeutic approaches, setting the stage for effective and durable NF1 gene therapy interventions.
Keywords
INTRODUCTION
Neurofibromatosis type 1 (NF1) is a complex genetic disorder caused by mutations in the NF1 gene, which encodes neurofibromin, a tumor suppressor protein critical for regulating cell growth and proliferation. Neurofibromin functions as a GTPase-activating protein (GAP) that reduces Ras signaling by stimulating the hydrolysis of active RAS-GTP to inactive RAS-GDP, classifying NF1 as a RASopathy. NF1 affects approximately 1 in 2,500 to 3,000 individuals worldwide, with an estimated 100,000 affected in the United States. It can be inherited in an autosomal dominant manner or arise from spontaneous germline mutations. The disease exhibits a wide variability in its presentation, even within the same family.
Neurofibromin is ubiquitously expressed, with the highest levels in the central nervous system, particularly in neurons, glial cells, Schwann cells, and peripheral nerve trunks. Haploinsufficiency of neurofibromin or its complete loss impacts multiple cellular processes such as receptor-mediated signaling and cell fate decisions, which contribute to NF1’s broad and variable phenotypes[1]. NF1-associated tumors can develop anywhere in the nervous system and range from benign neurofibromas to malignant peripheral nerve sheath tumors (MPNSTs). Benign cutaneous neurofibromas, often disfiguring, are a hallmark feature of NF1. Plexiform neurofibromas, present in up to 50% of patients, carry the risk of malignant transformation. Additionally, 15%-20% of NF1 patients develop optic pathway gliomas, which can impair vision due to retinal ganglion cell loss. High-grade gliomas, though rare, are a leading cause of NF1-associated mortality. Complications of NF1 can extend to cardiovascular problems, chronic pain, and learning challenges, with approximately 50% of patients experiencing cognitive or academic difficulties[2].
Addressing NF1’s complexity requires innovative approaches to tackle its genetic and molecular underpinnings. The Gilbert Family Foundation’s (GFF) Curing NF initiatives reflect this strategy by supporting the development of cutting-edge technologies and collaborative research in the community of NF1 researchers to address the disorder’s most pressing challenges. Through these efforts, GFF-funded researchers aim to better understand the molecular underpinnings of NF1’s diverse manifestations, improve therapeutic options, and ultimately enhance the quality of life for patients. Launched in 2018, these efforts focus on advancing gene therapy for all NF1 manifestations, vision restoration for patients with
THE GILBERT FAMILY FOUNDATION’S GENE THERAPY INITIATIVE: A THREE-COMPONENT APPROACH
The gene therapy initiative (GTI) research consortium efforts are primarily at the preclinical proof-of-concept stage and are designed to lay a strong foundation for future breakthroughs in NF1 treatment. As illustrated in Figure 1 and elaborated below, the GTI long-term strategy has three interconnected components that synergistically reinforce each other to support: (1) discovery research to fill critical knowledge gaps in NF1-associated biochemical pathways, pathology, and other unmet research needs, (2) enabling technology and infrastructure, such as assays, delivery systems, and preclinical models to advance NF1 research, and (3) advancing gene, cell, and other novel therapeutic approaches to treat NF1.
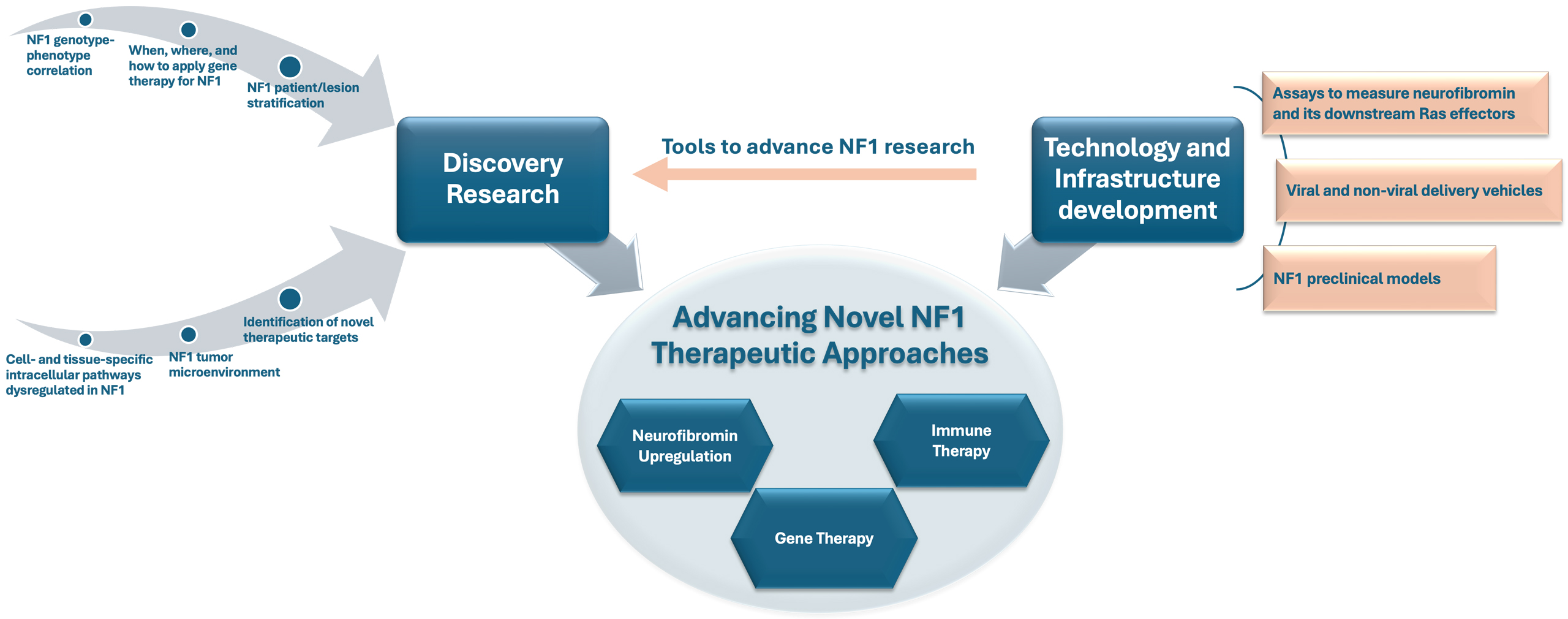
Figure 1. The Gilbert Family Foundation’s Gene Therapy Initiative (GTI) Three-Pronged Strategy for Advancing Novel NF1 Therapeutic Approaches. The GTI framework integrates supporting discovery research with technology and infrastructure development to propel progress in NF1 gene therapy. Discovery research identifies key molecular mechanisms and therapeutic targets within the heterogeneous NF1 disease landscape, while technology and infrastructure development provide essential tools such as assays and preclinical models that support both discovery and the evaluation of novel therapeutic strategies.
Discovery research to fill critical knowledge gaps in NF1 pathways, pathology, and other unmet needs within the NF1 research field
Advancing our understanding of NF1 at both molecular and clinical levels
Advancing our understanding of NF1 at both the molecular and clinical levels is critical for developing therapeutic interventions to prevent or treat various clinical manifestations resulting from altered cell fates and cell states caused by pathological reduction in neurofibromin levels in various cell lineages. This includes enhancing our knowledge of cell- and tissue-specific intracellular pathways dysregulated in NF1, genotype-phenotype correlations, and expanding natural-history studies for all NF1 manifestations to understand disease progression, establish biomarkers, design clinical trials, and inform preclinical translational strategies for testing novel therapies including gene therapy.
When, where, and how to apply gene therapy effectively in NF1
The successful development of gene therapy for NF1 requires addressing several critical areas of research[1]. A key challenge is determining when, where, and how to apply gene therapy effectively in NF1. First, it is important to determine the cells of origin, their microenvironment, and cell-cell interactions responsible for the development and progression of specific disease manifestations. On this basis, one may determine how restoration of neurofibromin in those cells at various time points of development and progression impacts the associated lesions or manifestations. Leveraging and building upon both seminal and recent single-cell studies in the field has important implications for advancing NF1 gene therapy, offering insights into the early cellular events that govern initiation and progression of NF1-associated lesions, and informing the development of novel therapeutic strategies[3-6].
Emerging experimental studies are beginning to test the therapeutic value of restoring NF1 expression at defined stages of tumor development using relevant model systems. A recent proof-of-concept study has demonstrated that temporally controlled restoration of NF1 expression is sufficient to reverse established neurofibromas[7]. For example, in mouse models harboring NF1 plexiform xenografts with inducible NF1 expression, re-expression or silencing of NF1 after tumor formation led to complete histological normalization of the sciatic nerve in most animals, whereas continued NF1 loss maintained or promoted tumorigenesis[7]. These findings underscore the therapeutic potential of NF1 restoration, even after tumor initiation, and support the feasibility of gene therapy approaches targeting Schwann cells in established disease. Despite the fact that these studies were conducted in a relatively simple and minimally complex NF1 tumor model, they represent an important first step toward broader exploration by developing more platforms that can further investigate the timing and cellular context in which NF1 gene therapy may be most effective, while also helping to define the potential limitations of neurofibromin replacement across diverse clinical manifestations and lesion types.
Additional experiments designed to restore neurofibromin with temporal control in specific cell lineages in genetically engineered mouse models or human induced pluripotent stem cell-derived organoids will be critical. Such experiments may help identify which NF1 manifestations or lesions are most likely to benefit from gene-targeted therapy, ensuring measurable and durable improvements. Additionally, one must determine the quantity of neurofibromin to be restored for therapeutic efficacy, considering factors such as target cell type, NF1 heterozygosity, and mutational landscape. Identifying the specific cell types that must be targeted-such as Schwann cells, stem cell niches, and cells surrounding neurofibromas-and developing strategies for effectively targeting these cells will be essential for successful gene therapy approaches.
Development of enabling technology and infrastructure
Developing assays to measure functional neurofibromin and its downstream effectors
A critical area of focus to enable NF1 research is assay development. Developing more sensitive and reproducible assays with a broader dynamic range to measure functional neurofibromin, Ras activation, and its downstream effector levels is critical for developing successful gene therapy for NF1. Today, most studies still depend on semi-quantitative Western blots that measure neurofibromin itself and, at best, a few downstream Ras effectors. In addition to not necessarily distinguishing between active functional neurofibromin and inactive full-length neurofibromin, this approach suffers from limited sensitivity, narrow dynamic range, and poor reproducibility across laboratories. Baseline neurofibromin levels and the thresholds to reverse or halt specific NF1 manifestations vary and remain unknown; new assays must detect small yet biologically meaningful changes over several orders of magnitude and must do so quickly enough to support high-throughput screening and iterative optimization cycles. Cutting-edge technologies such as single-cell proteomics can provide high-resolution quantitative data[8]. However, these approaches currently face limitations related to cost, instrument accessibility, and throughput, restricting their routine use in large-scale or inter-laboratory applications. A standardized, high-throughput platform that can simultaneously quantify functional neurofibromin and a multiplexed panel of Ras-pathway downstream signaling nodes would allow different labs to directly compare interventions, accelerating preclinical development and providing a quantitative assessment tool for gene- or RNA-based strategies that aim to restore neurofibromin across diverse cell and tissue types. By providing precise, reproducible readouts of neurofibromin levels and Ras signaling across multiple cell and tissue contexts and throughout developmental stages and differentiation states, the same assay will advance NF1 research forward by enabling the characterization of patient-derived organoid models. These demanding performance criteria can be satisfied by a high-sensitivity, multiplexed electrochemiluminescence immunoassay platform, which combines ultrasensitive detection, broad dynamic range, and throughput suitable for screening and
NF1 preclinical model development
While there are several preclinical models currently in use to study various NF1 manifestations, the development, characterization, and optimization of in vivo and in vitro preclinical models that better recapitulate the complexity of NF1 biology and symptoms are necessary to enhance the translational potential of gene therapy. While no single preclinical model may fully capture the full breadth of NF1 manifestations, several established models have yielded valuable insights into specific aspects of the disease. Genetically engineered mouse models of NF1 haploinsufficiency and NF1-associated lesions have contributed significantly to our understanding of the cells of origin of certain tumors and to translational research underpinning the development of MEK inhibitors for treating NF1-associated plexiform neurofibromas[3,9-11]. Nevertheless, significant gaps persist in NF1 preclinical modeling, a view shared by leading investigators in the field, due to challenges such as phenotypic variability, limited reproducibility, and inconsistencies across laboratories regarding available expertise and experimental setups.
The recognition that genetically identical siblings may manifest different disease symptoms and have different disease etiologies underscores the need for more information-rich models that link etiology and symptoms to the underlying molecular pathophysiology. Consequently, there is a need for next-generation models that enable quantitative evaluation of NF1 loss in specific cell lineages and their role in the development of various NF1 manifestations. This could help determine appropriate time points and target cell populations for intervention.
Recent advances in understanding the intracellular developmental programs that drive organ formation from stem cells have enabled the growth of human tissue systems derived from patient biopsies or stem cells[12]. These systems, known as “organoids”, uniquely recapitulate three-dimensional tissue architecture and cell-cell interactions, which are often lost in traditional two-dimensional cultures or some animal models. This feature is particularly valuable for studying NF1-associated cellular heterogeneity, microenvironmental signaling, and the developmental dynamics of NF1-deficient cells. Organoid models are suitable for gene editing, enabling the precise recreation of NF1 gene mutations specific to individual patients. The continued advances in the field of organoids can significantly enhance NF1 gene therapy efforts, as they more accurately replicate the features of human NF1 disease, providing a more faithful system to study NF1 biology. Additionally, organoids or “mini organs” offer a powerful platform for the discovery and preclinical evaluation of gene therapy approaches and other new therapeutic strategies. We regard next-generation organoid systems as additions that can enrich the existing NF1 model repertoire. While it is currently unclear whether organoids derived from patient biopsies will provide the
In September 2023, the Gilbert Family Foundation announced its commitment to build a bricks and mortar research institute focused on finding improved treatments and cures for NF1. The institute, named the “Nick Gilbert Neurofibromatosis Research Institute” or “NGNRI”, will serve as a central forum to unite efforts across the community of NF1 researchers and patient initiatives and to further develop key enabling technologies such as the organoid technologies previously mentioned[13].
Advancing novel NF1 therapeutic approaches
The majority of NF1 gene therapy research aims to develop gene- or RNA-based corrective therapies to address the cellular disruptions caused by the loss of neurofibromin function across multiple body systems. Realizing the potential of NF1 gene therapy requires addressing key gaps: determining optimal timing and targets for therapy, developing robust assays, and improving preclinical models (as outlined in Sections 1 and 2). If delivery and safety challenges are also overcome, gene therapies could offer a one-time or at least a more durable, disease-modifying intervention with broader impact and fewer side effects. MEK inhibitors have validated RAS-MAPK targeting in NF1 but deliver partial and non-curative benefits. While MEK inhibitors represent an important clinical advance, their limitations highlight the pressing need for therapies that address the underlying biology of NF1 rather than primarily mitigating downstream consequences. The FDA-approved MEK inhibitor, selumetinib, has shown effectiveness in a subset of patients; however, there is heterogeneity in patient response, the therapy can cause significant side effects, and plexiform neurofibromas do not completely disappear[14]. Recent clinical trials confirm that MEK inhibitors can shrink plexiform neurofibromas, but the responses are incomplete, variable and come at the cost of chronic toxicity[15-18].
Within this framework, gene therapy strategies are being designed to tackle the root cause of NF1 and halt disease progression by compensating for the loss or dysfunction of neurofibromin through gene replacement via the cellular introduction of genetic material encoding the NF1 gene either in full or in part, mutational targeting by editing NF1 pathogenic variants or by editing, modulating, or overriding pathogenic NF1 mutations at the mRNA level, and enhancing the neurofibromin levels produced by the remaining wild-type NF1 allele in haploinsufficient cells. For a detailed description of the Gilbert Family Foundation’s Gene Therapy Initiative project portfolio, readers can visit the GFF Curing NF website (https://gilbertfamilyfoundation.org/curing-neurofibromatosis/). Next, we highlight some such promising gene therapy tools and approaches that are currently under development.
EV-based nanocarriers for NF1 and other novel gene delivery technologies
Gene therapies have been identified as a promising avenue for the treatment of NF1[1,19]. However, proper deployment of such therapies remains a significant challenge due to difficulties in delivering large genes like NF1 and targeting cells of interest, such as Schwann cells, where restoration of neurofibromin can have a therapeutic effect in certain manifestations. At the technology development level, advancing innovative and disruptive viral and non-viral gene delivery modalities, along with other gene therapy technologies, would be important. These approaches not only serve as delivery vehicles for NF1 therapeutic payloads but also serve as important tools that can be used in exploring NF1 biology, interrogating the NF1 pathway, and driving the discovery of novel NF1 therapeutic strategies.
Gene delivery using adeno-associated viruses (AAVs) has shown promise in various genetic disorders. However, its application faces several limitations[20]. AAVs have a limited packaging capacity (approximately 4.7 kb), restricting their ability to deliver large genes like NF1. Furthermore, pre-existing immunity against AAVs can reduce transduction efficiency, as individuals may have neutralizing antibodies due to prior exposure. Additionally, long-term gene expression may diminish in dividing cells, necessitating repeated administration, which poses further immunological challenges. These limitations highlight the need for alternative or enhanced delivery systems to effectively target NF1.
Extracellular Vesicles (EVs) are cell-derived natural carriers that play a critical role in mediating intercellular communication by transferring various biomolecules, such as proteins, DNA, and RNA, under both healthy and pathological conditions[21]. Engineered extracellular vesicles (eEVs) represent a promising novel delivery platform that can carry therapeutic payloads[22,23]. Recent studies provide preliminary evidence that EVs can be engineered to carry NF1-relevant therapeutics, including mRNA, plasmid DNA, and protein content, and can effectively deliver these cargos to recipient cells, resulting in the expression of non-mutated NF1[24,25]. Compared to viral vectors, eEVs can potentially overcome key limitations, such as capsid size constraints and redosing challenges, while also improving upon the low transfection efficiencies associated with synthetic carriers. EVs can be readily functionalized with targeting moieties, including peptides, aptamers, and antibodies, to enhance specificity toward target cells and thereby improve therapeutic efficacy. The use of specific ligands to functionalize EVs for targeted delivery to the brain and lung has been demonstrated[23,26]. It is likely that targeted “designer” EVs offer a tractable approach with the potential to overcome key challenges in NF1 gene delivery. Additional studies to refine EV-based nanocarriers, enhancing their tropism to Schwann cells and increasing their relevance for NF1 therapies, are warranted.
Promising novel gene delivery technologies: The development of novel and disruptive gene delivery technologies with inherent biological effects holds significant promise. A systemic lupus erythematosus-associated autoantibody (4H2) can be repurposed as a dual-function therapeutic and gene delivery platform. 4H2 binds endogenous RNA, activates the cytosolic DNA/RNA sensor cGAS[27], and initiates STING-driven inflammatory signaling that is selectively cytotoxic to brain, lung, and breast cancer cells while sparing normal epithelium. In orthotopic glioblastoma models, intravenously delivered 4H2 accumulated in the tumor microenvironment, prolonged survival in a T cell-dependent manner, and enhanced the efficacy of immune-checkpoint blockade. Beyond immunomodulation, 4H2 also served as a non-viral nucleic acid delivery carrier: when complexed with exogenous RNA, it delivered functional transcripts to tumor, brain, and muscle tissues in vivo. These data establish a dual strategy with translational potential for cancer immunotherapy and for gene-replacement approaches[28]. Preliminary data in NF2 highlight the potential of this dual-acting antibody approach, which effectively delivers the NF2 gene into diseased cells while simultaneously eliciting an innate immune response via activation of the cGAS-STING signaling pathway[29]. This dual functionality positions the autoantibody-based immunomodulation/gene delivery strategy as a potentially promising avenue for other genetic diseases such as NF1.
Exploring nonsense suppression as a treatment for NF1
Nonsense mutations, which introduce premature termination codons (PTCs) in the NF1 gene, account for approximately 20% of NF1 cases and lead to truncated, unstable neurofibromin protein. These mutations reduce neurofibromin expression by triggering nonsense-mediated mRNA decay (NMD) or premature translation termination. Readthrough (RT) agents are small molecules capable of inducing ribosomal readthrough of PTCs to restore the expression of full-length functional protein. Such agents have demonstrated efficacy in treating genetic disorders caused by nonsense mutations, including cystic fibrosis and Duchenne muscular dystrophy (DMD)[30].
Ongoing research efforts to accelerate the development and validation of high-throughput screening assays to identify novel RT agents that can suppress PTCs and/or inhibit NMD in NF1 hold promise. In addition, testing the efficacy of RT agents in NF1-specific cell and mouse models carrying distinct PTCs, which mimic the cutaneous and plexiform neurofibromas, as well as the skin hyperpigmentation observed in NF1 patients, is essential. Optimization of the pharmacokinetics and pharmacodynamics of RT agents, with a focus on their impact on neurofibromin expression, Ras signaling, and NF1-associated phenotypes, is also ongoing. Further research is needed to yield critical insights into the molecular mechanisms and therapeutic potential of RT agents for NF1 treatment[31,32].
Exon skipping to treat NF1
For specific NF1 variants, antisense oligonucleotides (ASOs) can be employed to promote exon skipping, restoring functional neurofibromin expression to mitigate tumorigenesis or slow tumor growth. This strategy targets RNA splicing by designing ASOs to exclude specific exons during mRNA processing, effectively “skipping over” pathogenic variants. Exon skipping is a proven therapeutic approach, currently used in clinical treatments for other pediatric rare diseases, including rare and ultra-rare neurological diseases[33,34]. Notably, it has been applied in DMD through the development of exon-skipping therapies aimed at restoring the reading frame of the dystrophin gene. Recent research identified NF1 exons 17 and 52 as viable exon skipping targets, as their exclusion does not compromise neurofibromin function, preserving neurofibromin expression and GAP-related domain (GRD) activity, making them promising therapeutic targets[35,36].
Further research is needed to optimize nonsense suppression and exon-skipping approaches by testing their efficacy in NF1 patient-derived preclinical models to identify the specific NF1 mutations and the clinical manifestation contexts where such therapeutic approaches are likely to be effective.
Rescuing haploinsufficiency by enhancing neurofibromin levels produced by the remaining wild-type NF1 allele
Learning disabilities and behavioral disorders such as attention deficit disorder occur in a significant percentage of children with NF1[37]. These challenges are associated with mutations in one of the two copies of the NF1 gene, leading to reduced levels of neurofibromin[38]. Studies in NF1 mouse models have revealed that these animals develop learning and behavioral problems similar to those observed in humans and thus provide a valuable resource to study the disease and develop new and improved treatment strategies. A novel and innovative approach toward treating learning and behavioral deficits in NF1 relies on boosting levels of the neurofibromin protein within the brain by preventing neurofibromin degradation. Recent work uncovered a new and important mechanism by which neurofibromin levels are regulated within the cell by interacting with FBXW11, an F-box E3 substrate adaptor protein that, notably, also regulates the phosphorylation-dependent ubiquitination and degradation of key factors associated with tumor growth and aggressiveness[39]. Disrupting FBXW11-mediated degradation of NF1 through genetic disruption of FBXW11 can restore neurofibromin protein levels within key brain areas responsible for learning and behavioral disorders associated with NF1[39]. There is potential for this work to provide a strong foundation to advance new gene and small molecule-based therapies toward clinical trials for the treatment of learning disabilities and behavioral disorders in NF1.
CLOSING REMARKS
Advancing gene therapy for NF1 faces numerous complications such as delivery challenges, immunogenicity, and manufacturing hurdles[40,41]. Gene therapy for NF1 holds immense promise, but realizing its full potential demands more than technological innovation. It requires a concerted effort to address persistent foundational gaps in the field. These include deepening our understanding of NF1 at the cellular and molecular levels, including how neurofibromin deficiency alters developmental trajectories, cell fate decisions, and tumor-immune microenvironment interactions, to identify the optimal timing and cellular context for intervention and to improve patient stratification. It also requires developing sensitive and scalable assays to quantify neurofibromin restoration and generating reliable preclinical models that reflect NF1’s heterogeneity and pathophysiology. The Gilbert Family Foundation’s GTI takes a strategic, integrated approach to overcome these obstacles through its three-pronged strategy supporting discovery research, enabling infrastructure, and advancing therapeutic development, including gene therapy, neurofibromin upregulation, and immune-modulatory approaches. These components are not isolated but are mutually reinforcing drivers that generate critical feedback to inform and accelerate progress across the broader therapeutic development ecosystem. By confronting these foundational and translational barriers head-on, the field can transition from proof-of-concept to clinical impact, ultimately transforming NF1 care through durable, disease-modifying gene therapies. Whether the future of NF1 gene therapy is bright or bleak depends on how the field embraces and acts to fulfill these prerequisites.
DECLARATIONS
Authors’ contributions
Conceptualized the perspective and drafted the manuscript: Okaz E, Vinnakota K
Drafting and revising of the manuscript: Venkat P, Muñoz E, Baines I
The manuscript was reviewed and finalized with input from all authors.
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
1. Staedtke V, Anstett K, Bedwell D, et al. Gene-targeted therapy for neurofibromatosis and schwannomatosis: the path to clinical trials. Clin Trials. 2024;21:51-66.
2. Peduto C, Zanobio M, Nigro V, Perrotta S, Piluso G, Santoro C. Neurofibromatosis type 1: pediatric aspects and review of genotype-phenotype correlations. Cancers. 2023;15:1217.
3. Yang FC, Ingram DA, Chen S, et al.
4. Joseph NM, Mosher JT, Buchstaller J, et al. The loss of Nf1 transiently promotes self-renewal but not tumorigenesis by neural crest stem cells. Cancer Cell. 2008;13:129-40.
5. Le LQ, Liu C, Shipman T, Chen Z, Suter U, Parada LF. Susceptible stages in Schwann cells for NF1-associated plexiform neurofibroma development. Cancer Res. 2011;71:4686-95.
6. Kershner LJ, Choi K, Wu J, et al. Multiple Nf1 Schwann cell populations reprogram the plexiform neurofibroma tumor microenvironment. JCI Insight. 2022:7.
7. Bostanthirige DH, Plante C, Vatasescu JPS, et al. Proof-of-principle of NF1 Gene Therapy in plexiform neurofibroma mice models. bioRxiv 2025;bioRxiv 2025.01.21.634081. Available from: https://www.biorxiv.org/content/10.1101/2025.01.21.634081v1.abstract [accessed 12 August 2025].
8. Goto-Silva L, Junqueira M. Single-cell proteomics: a treasure trove in neurobiology. Biochim Biophys Acta Proteins Proteom. 2021;1869:140658.
9. Wu J, Williams JP, Rizvi TA, et al. Plexiform and dermal neurofibromas and pigmentation are caused by Nf1 loss in desert hedgehog-expressing cells. Cancer Cell. 2008;13:105-16.
10. Jessen WJ, Miller SJ, Jousma E, et al. MEK inhibition exhibits efficacy in human and mouse neurofibromatosis tumors. J Clin Invest. 2013;123:340-7.
11. Zhu Y, Ghosh P, Charnay P, Burns DK, Parada LF. Neurofibromas in NF1: Schwann cell origin and role of tumor environment. Science. 2002;296:920-2.
12. Kim J, Koo BK, Knoblich JA. Human organoids: model systems for human biology and medicine. Nat Rev Mol Cell Biol. 2020;21:571-84.
13. Gilbert Family Foundation Contributes nearly $375 million in partnership with Henry Ford Health to Bring Shirley Ryan AbilityLab to Detroit and create the Nick Gilbert Neurofibromatosis Research Institute. Available from: https://gilbertfamilyfoundation.org/news-and-stories/henry-ford-health-shirley-ryan-ability-lab-nick-gilbert-neurofibromatosis-institute/ [accessed 12 August 2025].
14. Dombi E, Baldwin A, Marcus LJ, et al. Activity of selumetinib in neurofibromatosis type 1-related plexiform neurofibromas. N Engl J Med. 2016;375:2550-60.
15. Gross AM, Dombi E, Wolters PL, et al. Long-term safety and efficacy of selumetinib in children with neurofibromatosis type 1 on a phase 1/2 trial for inoperable plexiform neurofibromas. Neuro Oncol. 2023;25:1883-94.
16. Li L. Disproportionate adverse event signals of selumetinib in neurofibromatosis type I: insights from FAERS. Front Pharmacol. 2024;15:1454418.
17. Chen AP, Coyne GO, Wolters PL, et al; KOMET study investigators. Efficacy and safety of selumetinib in adults with neurofibromatosis type 1 and symptomatic, inoperable plexiform neurofibromas (KOMET): a multicentre, international, randomised, placebo-controlled, parallel, double-blind, phase 3 study. Lancet. 2025;405:2217-30.
18. Moertel CL, Hirbe AC, Shuhaiber HH, et al; for the ReNeu Study Group. ReNeu: a pivotal phase 2b trial of mirdametinib in children and adults with neurofibromatosis type 1 (NF1)-associated symptomatic inoperable plexiform neurofibroma (PN). JCO. 2024;42:3016-3016.
19. Leier A, Bedwell DM, Chen AT, et al. Mutation-directed therapeutics for neurofibromatosis type I. Mol Ther Nucleic Acids. 2020;20:739-53.
20. Wang D, Tai PWL, Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov. 2019;18:358-78.
21. Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200:373-83.
22. Tang SN, Salazar-Puerta AI, Heimann MK, et al. Engineered extracellular vesicle-based gene therapy for the treatment of discogenic back pain. Biomaterials. 2024;308:122562.
23. Salazar-Puerta AI, Rincon-Benavides MA, Cuellar-Gaviria TZ, et al. Engineered extracellular vesicles derived from dermal fibroblasts attenuate inflammation in a murine model of acute lung injury. Adv Mater. 2023;35:e2210579.
24. Rincon-Benavides MA, Cuellar-Gaviria T, Alzate-Correa D, et al. Engineered extracellular vesicles as a therapeutic strategy for neurofibromatosis type I. Biomedical Engineering Society (BMES) Annual Meeting; 2023 Oct 11-14; Seattle WA. Available from https://2023bmesannual.eventscribe.net/fsPopup.asp?efp=Sk9RVlVJUUwyMDI3Nw&PresentationID=1315447&rnd=0.4760429&mode=presInfo [accessed 26 August 2025].
25. Rincon-Benavides MA, Cuellar-Gaviria T, Alzate-Correa D, Powell H, Gallego-Pérez D, Higuita-Castro N. Designed extracellular vesicles loaded with NF1 nucleic acid as a novel therapeutic strategy for neurofibromatosis type 1. International Nanomedicine and Drug Delivery Symposium (NanoDDS); 2024 Sep 13-15; Orlando, FL. Available from https://pharmacy.ufl.edu/wordpress/files/2024/09/Final-Program-NanoDDS-2024-1.pdf [accessed 26 August 2025].
26. Ortega-Pineda L, Sunyecz A, Salazar-Puerta AI, et al. Designer extracellular vesicles modulate pro-neuronal cell responses and improve intracranial retention. Adv Healthc Mater. 2022;11:e2100805.
27. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786-91.
28. Chen X, Tang X, Xie Y, et al. A lupus-derived autoantibody that binds to intracellular RNA activates cGAS-mediated tumor immunity and can deliver RNA into cells. Sci Signal. 2025;18:eadk3320.
29. NF2BioSolutions. Available from https://nf2biosolutions.org/non-viral-gene-delivery-zhou-lab-at-yale-university/ [accessed 12 August 2025].
30. Sharma J, Du M, Wong E, et al. A small molecule that induces translational readthrough of CFTR nonsense mutations by eRF1 depletion. Nat Commun. 2021;12:4358.
31. Osum SH, Oribamise EI, Corbière SMAS, et al. Combining nonsense mutation suppression therapy with nonsense-mediated decay inhibition in neurofibromatosis type 1. Mol Ther Nucleic Acids. 2023;33:227-39.
33. Takeda S, Clemens PR, Hoffman EP. Exon-Skipping in Duchenne Muscular Dystrophy. J Neuromuscul Dis. 2021;8:S343-58.
34. McCauley ME, Bennett CF. Antisense drugs for rare and ultra-rare genetic neurological diseases. Neuron. 2023;111:2465-8.
35. Leier A, Moore M, Liu H, et al. Targeted exon skipping of NF1 exon 17 as a therapeutic for neurofibromatosis type I. Mol Ther Nucleic Acids. 2022;28:261-78.
36. Church C. Targeted Exon Skipping of NF1 Exon 52 as a Mutation-Specific Therapeutic for Neurofibromatosis Type 1. 2024 Global NF Conference; 2024 Jun 20-25; Brussels, Belgium. Available from https://www.ctf.org/wp-content/uploads/2024/06/24_NFConferenceAbstractBook_web.pdf [accessed 26 August 2025].
37. Lehtonen A, Howie E, Trump D, Huson SM. Behaviour in children with neurofibromatosis type 1: cognition, executive function, attention, emotion, and social competence. Dev Med Child Neurol. 2013;55:111-25.
38. Botero V, Tomchik SM. Unraveling neuronal and metabolic alterations in neurofibromatosis type 1. J Neurodev Disord. 2024;16:49.
39. Kim TY, Siesser PF, Rossman KL, et al. Substrate trapping proteomics reveals targets of the βTrCP2/FBXW11 ubiquitin ligase. Mol Cell Biol. 2015;35:167-81.
40. Kohn DB, Chen YY, Spencer MJ. Successes and challenges in clinical gene therapy. Gene Ther. 2023;30:738-46.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Issue
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].