


Long-term treatment with insulin-like growth factor-1 in Phelan-McDermid syndrome: a case report
 0
0 Abstract
Phelan-McDermid syndrome (PMS) is a chromosomal microdeletion syndrome generally caused by loss-of-function variants or deletions affecting the SHANK3 gene. We report on a case of a 19-year-old woman with a diagnosis of PMS, autism, and developmental disability. She has been under clinical care since the age of 9 and received treatment with subcutaneous IGF-1 from 11 to 15 years of age. The treatment spanned 2 periods, totaling 35 months, interspersed with a 16-month off-treatment interval. Clinically significant improvement was evident during the treatment periods, particularly in the Social Responsiveness Scale, the Aberrant Behavior Checklist, and clinical assessments, contrasted with a clear deterioration during the off-treatment period. Sleep difficulties worsened during the first period, and EKG repolarization abnormalities emerged during the second period, ultimately leading to definitive treatment discontinuation. In conclusion, an experimental long-term on-off-on treatment with IGF-1 in an adolescent with PMS resulted in mixed results, showcasing positive clinical improvements alongside potentially severe adverse events in the long run.

Keywords
INTRODUCTION
Phelan-McDermid syndrome (PMS) is a chromosomal microdeletion syndrome generally caused by loss-of-function variants or deletions in the SHANK3 gene. PMS is a heterogeneous disorder, and the phenotype is associated with neurological and non-neurological features, including hypotonia, global developmental disorder, severe language delay or impairment, and minor dysmorphic features. Frequently, it is associated with autism and cardiological, digestive, and motor diseases or impairments[1].
The SHANK3 gene codes for a protein that plays an important role as a scaffolding protein of postsynaptic glutamatergic receptors. Haploinsufficiency causes synaptic function and plasticity anomalies shown with electrophysiological measures in SHANK3 deficient mice[2]. These mice in turn, show motor deficits, less social sniffing, and reduced vocalizations in comparison with wild-type mice[2,3].
The insulin-like growth factor-1 (IGF-1) is a molecule that crosses the blood-brain barrier and contributes to cell survival, synaptic maturation, and plasticity[4,5]. It is commercially available, with an approved indication for congenital IGF-1 deficiency. Preclinical studies with murine and human neuronal models of SHANK3 deficiency suggested that IGF-1 can reverse synaptic and motor learning deficits. Following this evidence, a pilot 3-month controlled trial of IGF-1 in 9 children with PMS was conducted to assess its effect on social function and repetitive behaviors[6]. A significant improvement in both social and repetitive difficulties was reported, with good tolerance.
Recombinant IGF-1 (rhIGF-1 or mecasermin) has been used in different syndromes associated with low height and in neurodevelopmental disorders[7]. The drug is generally well-tolerated in the short term. IGF-1 and human growth hormone (hGH) have been shown to reverse neurobehavioral deficits in PMS[8]. The majority of side effects of short-term GH replacement therapy are related to sodium and water-retaining properties and reduction in insulin sensitivity. Long-term concerns are mainly related to the potential induction of cell growth and proliferation in response to GH and IGF-1, raising the theoretical possibility of increased risk of tumor recurrence and de novo neoplasia[9].
It was in this context that we initiated the treatment of an 11-year-old PMS girl with concomitant autism and intellectual disability, without a pre-specified time course. Over the course of treatment, we systematically assessed social and other behavioral difficulties together with adverse events. The literature supporting the treatment included in this study is limited, prompting us to adopt a highly personalized approach. We prospectively administered the intervention with meticulous monitoring over a prolonged treatment course, with treatment phases separated by an off-treatment period. We present the procedures for implementation of the experimental treatment, clinical course, adverse events, and decisions about starting and interrupting treatment.
CASE REPORT
LD has been attending the Special Care Program for Autism Spectrum Disorders (ASD or autism), AMITEA[10], since she was 9 years old. She had been diagnosed with PMS (carrying a 22q13 deletion of approximately 270 kilobases) with developmental disability, ASD, language delay, hypotonia, squint, kyphosis, and constipation. Other developmental challenges included sleep difficulties, mastication problems, pain hyposensitivity, and occasional heteroaggressive outbursts. Her mental age was approximately 3 years at the time of inclusion in the program and she had a marked attention deficit and hyperactive behaviors. No major dysmorphic features were present.
At intake in our outpatient clinic, she presented with simple-phrase language, mostly consisting of echolalia. She had great difficulties in understanding, low communication intents, poor non-verbal communication, hyperactivity, inattention, emotional dysregulation, marked motor and language stereotypies, and mannerisms. Additionally, she exhibited repetitive behaviors, such as opening and closing doors or hair pulling, restrictive interests (including baby dolls), and a high pain threshold. At 9 years of age, she was prescribed methylphenidate, which improved her attention; she was reported to be playing better and showing improvement in pain sensitivity. However, her sleep deteriorated, and she experienced episodic overexcitement, leading to the discontinuation of treatment. Subsequently, clonidine was prescribed, which effectively improved hyperactivity and attention deficit symptoms and was continued for several years. A course of treatment with oxcarbazepine was attempted to address emotional dysregulation, but its efficacy was unclear, leading to its discontinuation. Other previous pharmacological treatments included risperidone in early infancy (for behavioral difficulties), with very limited effect, and melatonin (for difficulties falling asleep), with no effect. In terms of behavioral treatments, she had undergone intensive early intervention therapy, applied behavior analysis (ABA)-based intervention, speech and language therapy, physiotherapy, sensory integration, and hippotherapy. She had been placed in a Special Education provision since infancy.
She was or had been under the care of specialists in Neurology, Ortho-traumatology, Gastroenterology, Nephrology, and Cardiology to follow up on reflux and musculoskeletal problems, and organic pathologies had been ruled out.
First-degree family history included an insulin-like growth factor-producing pituitary tumor in her father and spondylarthrosis in her mother. Second-degree relatives included an aunt with Patau Syndrome. Third-degree relatives had two cases of developmental delay and one case of autism.
In light of preliminary evidence at the time suggesting a potential positive effect of IGF-1 in PMS, treatment was initiated in October 2015. The family, actively engaged in the establishment and administration of the Spanish Phelan-McDermid Association and closely connected with international experts, actively participated in all the stages leading to the decision to initiate this treatment. They played a pivotal role, from drawing attention to ongoing trials to explicitly consenting to the initiation of the intervention, as well as subsequent decisions regarding stopping and re-starting the treatment.
METHODS AND MATERIALS
Treatment was started and conducted in close collaboration with the department of Endocrinology. After excluding contraindications to the treatment and providing training to the parents for the subcutaneous administration of IFG-1, a progressive dosage, aiming for 0.12 mg per kilogram per day, was initiated. Dose was selected following all available information including the recently published pilot study with 9 patients with PMS[6]. Regular endocrinologist controls following standard guidelines for the monitoring of the use of IGF-1[9] were conducted throughout the course of the treatment, including glycemia monitoring, IGF-1 levels, and echocardiography. Cardiological reviews with electrocardiogram (EKG) and echocardiography were also carried out regularly.
Monthly psychiatric follow-ups were conducted, utilizing various validated instruments in addition to clinical assessments. Instruments used included the clinician-rated Clinical Global Impression-Severity (CGI-S)[11], and parent-rated Social Responsiveness Scale (SRS-2)[12] and the Aberrant Behavior Checklist-Community (ABC-C)[13]. The CGI-S assesses the severity of impairment in global functioning on a 1-7 Likert scale. The SRS-2 is a 65-item measure, with each item rated from 0 to 4, identifying the presence and severity of autistic symptoms. The ABC-C comprises five subscales evaluating irritability/agitation, social withdrawal, stereotypic behavior, hyperactivity, and inappropriate speech. In addition to these monthly measurements, we administered the Vineland Adaptive Behavior Scale-Third edition (VABS-3)[14] at the beginning and end of each treatment period. We used the parent-rated version of the VABS-3 assessment, which occurs via a comprehensive questionnaire that covers different areas of functioning and adaptive behavior.
In addition to spontaneous reports of adverse events and regular blood analyses (including IGF-1 plasma levels), the Scale of the Udvalg for Kliniske Uendocndersøgelser (UKU)[15], a comprehensive rating scale designed to assess general side effects of psychotropic drugs, was completed monthly.
PROCEDURE AND OUTCOMES
(a) Courses of treatment: The target dose of IGF-1 was consistently administered for 10 months, starting in October 2015 when LD was 11 years old, and continued until the end of June 2016. The treatment was then interrupted. Considering the positive progression of symptoms while on treatment and their deterioration when IGF-1 was discontinued, treatment was resumed 16 months later in November 2017 when LD was 13 years old and continued until December 2019, totaling 25 months in this second period, when it was definitely discontinued (at 15 years of age). Dose was adjusted based on weight approximately every 6 months, ultimately reaching a final dose of 5.2 mg per day. Both treatment courses were administered under the European Medicines Agency’s “compassionate use” basis, which allows the use of unauthorized medicines under very restrictive and strict conditions. Treatment was authorized and provided by the hospital, following a plea supported by a clinical report and a comprehensive review of the literature and the current state-of-the-art regarding the condition and available treatments.
(b) Adverse events observed included potential sleep deterioration during the initial treatment phase, and hypersensitivity, subcutaneous tissue swelling, and cardiac repolarization abnormalities after 25 months of the second period. These last symptoms (with the possibility of risk of hypertrophic cardiomyopathy) led to the ultimately definitive discontinuation of treatment. The EKG revealed a sinus rhythm at 86 beats per minute, a normal axis, and a narrow QRS at the beginning and end of Period 1. Negative T waves were observed in leads II and III, with normal T waves in all precordials at the start of Period 1 and negative T waves in aVF and V4-V5 before Period 2. One year after discontinuing IGF-1, EKG went back to normal parameters.
(c) Behavioral outcome. Figure 1 illustrates the progression of symptoms over the two treatment periods and the off-treatment interval. During the first treatment period, the SRS-2 showed a progressive improvement of more than 30 points in the total scale, which is considered a clinically significant change[16]. Throughout the off-treatment period, symptoms deteriorated to a slightly worse score than at the beginning of the treatment two years earlier. The reintroduction of treatment resulted in a similar magnitude of symptom improvement as seen in the first period. A parallel trend was observed in four of the five subscales of the ABC-C. Irritability, hyperactivity, social withdrawal, and speech abnormalities improved during the on-treatment periods and deteriorated during the off-treatment interval. No change was observed in VABS-3 age equivalents, which were, at all times, around 3 years of age.
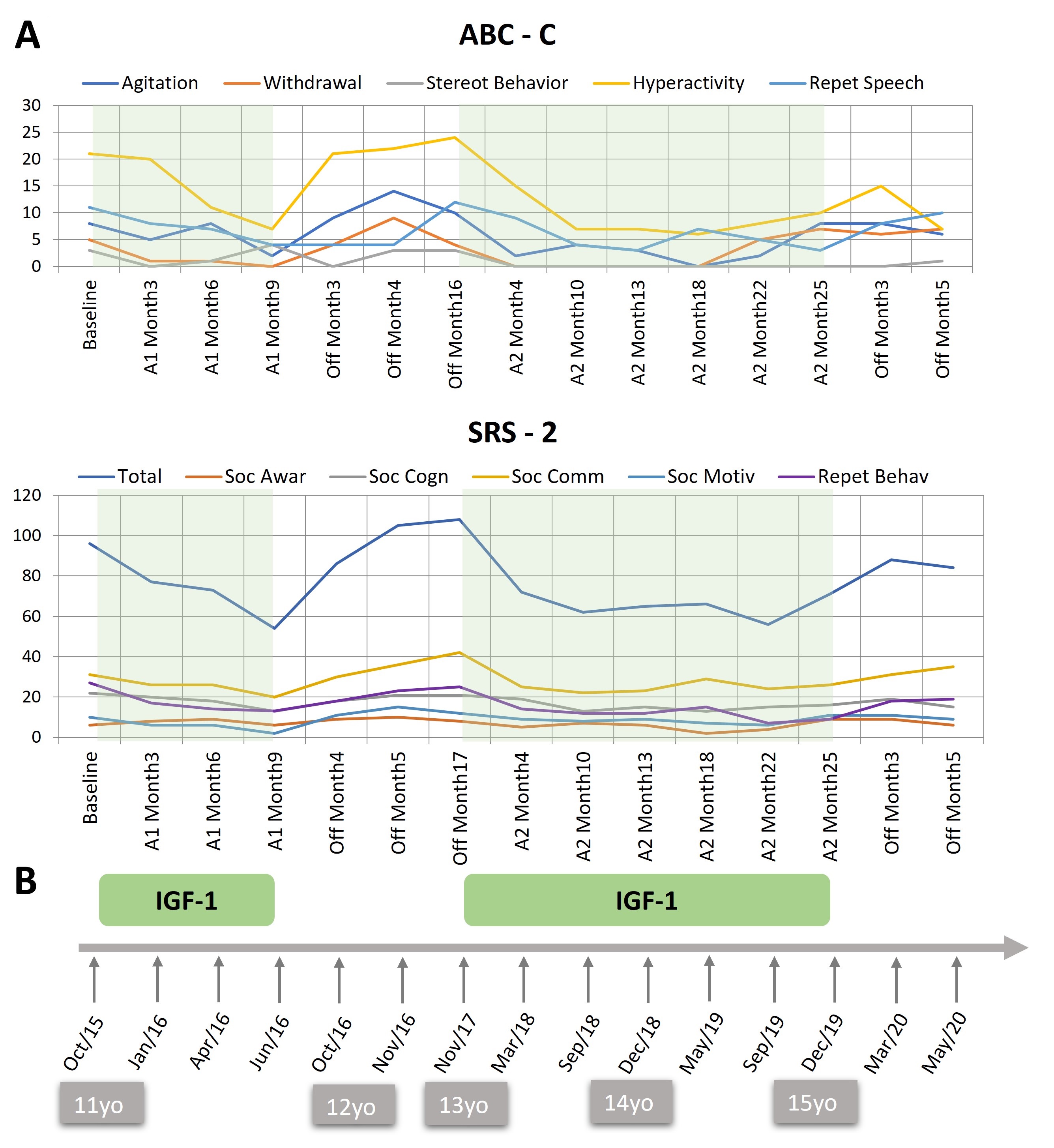
Figure 1. Raw scores on the ABC-C and the SRS-2 during the treatment and off-treatment periods and overview of treatment duration. (A) Treatment periods (A1 and A2) are shaded in green and off-treatment periods are blank. (B) Timeline of treatment periods. Age of the patient is shown in the grey boxes. ABC-C: Aberrant behaviour checklist - community; subscales: Agitation/Irritability, Social withdrawal, Stereotyped behavior, Hyperactivity, Repetitive/Inappropriate speech. SRS-2: Social Responsiveness Scale, subscales: Social awareness, Social cognition, Social communication, Social motivation, Repetitive behavior. Copyright Parellada et al., (2024). This figure is distributed under the terms of the Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License, which enables reusers to copy and distribute the material in any medium or format in unadapted form only, for non-commercial purposes only, and only so long as attribution is given to the creator.
During the treatment periods, the family spontaneously made remarks such as: “she is more connected”, “she has better attention”, “she is more independent in her leisure time”, “she has been able to enjoy watching a movie together with her cousins for the first time”, “now you can have a conversation with her (said by grandfather)”, “she has said Happy Christmas for the first time and was interested in the Three Wise men Parade” or “she has many more spontaneous contextualized sentences”. Her speech and language therapist reported higher learning speed, going faster across different learning units. At school, teachers observed an increased communication intention, along with increased calmness, attention, and better functional language. During the off-treatment, comments from carers included: “she is less spontaneous”, “participates less”, “she is dysregulated and has tantrums”, and “she is less connected”. Adherence to treatment was excellent and monitored through regular prescription accountability and contact with the Endocrinology Day Hospital, as well as scheduled visits to Psychiatry.
Shortly before the definitive termination of IGF-1 treatment due to adverse events, in November 2019, LD began experiencing occasional and brief episodes of emotional dysregulation and catatonic-like behaviors. These episodes persisted sporadically until a more severe episode began in the summer of 2022, which is described in detail below.
Post-treatment evaluation revealed a progressive worsening of attention deficit and emotional dysregulation to baseline scores. Clonidine and oxcarbazepine were reintroduced but showed a poor response and were ultimately discontinued.
CURRENT SITUATION
In the summer of 2022, 18 months after the final interruption of IGF-1 treatment and aged 17 years, a significant emotional dysregulation period began, characterized by aggressive behavior with new abnormal movements and tremors in the right superior limb and mouth. A neurology specialist reviewed the case, finding no known cause for this deterioration.
In July 2023, LD underwent a brief treatment regimen aimed at addressing her behavioral difficulties. Low doses of aripiprazole and quetiapine were administered, along with the insertion of subcutaneous progestogens to manage dysregulation. Unfortunately, this treatment approach proved ineffective. Moreover, additional symptoms of catatonia, such as lethargy, disconnection from the environment, and facial expressionlessness, emerged. Subsequently, treatment with citalopram was initiated, yielding a positive response characterized by improved mood and regulation.
DISCUSSION
This case shows a long-term treatment (total more than 3 years) of an adolescent with a 22q13 deletion of approximately 270 kilobases (Phelan McDermid Syndrome) with severe developmental disability, ASD, language delay, hypotonia, squint, kyphosis, and constipation. In this case of treatment during adolescence, although there is no improvement in global adaptive function, and mental age remains consistent from the onset of treatment at 11 until its conclusion at 15 (approximately 3 years of age), there are noticeable enhancements in social awareness, participation, effective communication (especially in initiative and contextualization), and behavior. The experimental treatment was administered over two distinct periods, revealing a discernible on-off-on effect corresponding to the treatment intake. On both occasions, once the drug was discontinued, all improved behaviors reverted to baseline scores. Tolerance was generally good throughout most of the treatment duration (over 2 years). However, toward the end, tissue thickening and EKG repolarization abnormalities prompted the decision to discontinue the treatment permanently.
This case prompts the question of whether the years on treatment, marked by enhanced social and communication skills during the challenging adolescent period, may contribute to improved interpersonal relationships and an enhanced overall well-being and quality of life for the patient and her family. These years of treatment, with temporary improvements, can also create false hope for greater or longer improvements, which need to be addressed fully and honestly with the families. In this case, the family felt, both during the treatment course and even now, in retrospect, that the outcome obtained was worth all the effort of the trial. They experienced a better connection with their daughter for a prolonged period of time, which they found highly valuable. The meticulous monitoring of general and molecule-specific adverse events facilitated the early identification of potential severe adverse events, ultimately leading to the final discontinuation of the treatment.
Following the available information at the time of initial treatment mentioned above, IGF-1 and human growth hormone (hGH) have been shown to reverse neurobehavioral deficits in PMS. In terms of clinical studies[8], subsequent investigations included a new short-term pilot study and a randomized controlled trial (RCT) in PMS[17,18], showing improvements in sensory reactivity, hyperactivity and repetitive behaviors, and good tolerability. Tolerability during long-term treatment has not been studied for indications not associated with low height.
CLINICAL IMPLICATIONS
The presented case report underscores the importance of incorporating all available sources of evidence when addressing treatments for orphan and rare diseases, especially considering that treatments are often severely limited or even absent in such cases. It also emphasizes the value of exploring alternatives to randomized double-blind placebo-controlled trials to contribute valuable evidence and knowledge beneficial for the management of these disorders. One of these alternative designs is the N-of-1 crossover design, particularly useful in the study and treatment of extreme phenotypes or rare forms of disease, frequently not eligible for standard clinical trials. N-of-1 clinical trials are conducted in one patient at a time, are exploratory, and try to find a personalized and precise treatment for each patient, the right dose, and the right duration of treatment. In the era of multi-omics and precision medicine, multivariate N-of-1 (already considered in Level 1 of evidence)[19] and aggregated N-of-1 trials are ideal vehicles for advancing biomedical and translational science since the insights gained from them can provide insight into how to provide real-time care to each individual[20-22]. These designs, in turn, can overcome the common limitation of meta-analyses, which often yield averaged summary results for heterogeneous groups of patients with very diverse responses to treatments. Proactive and systematic regular and frequent assessment and monitoring of multivariate clinical outcomes and adverse events, all over the course of the treatment, are, as this case illustrates, the way forward to gain a broad understanding of the treatment effect and address potentially harmful effects promptly. N-of-1 clinical trials do not come without drawbacks. There are barriers to generalizing the use of N-of-1 clinical trials. The cost of conducting individualized treatment (preparing the trial, clinical visits, and the cost of some medications) is difficult for institutions or participants to assume. Remote monitoring can help reduce some costs while providing a daily account of response, allowing for quick decisions. Generalizability is threatened. For some conditions, an on-off or randomized on-off design is not appropriate (due to the severity of the conditions, the carry-over effect, and others). Cost-efficacy analyses need to include the advantage of avoiding patients' extensive periods on inefficient medications[23]. Whenever possible, the incorporation of N-of-1 double-blind crossover designs could enhance the standard on-off-on N-of-1 studies. Additionally, it is worth considering intermittent treatments as an alternative approach, as suggested in other conditions[23].
STRENGTHS AND LIMITATIONS
This case exemplifies a stringent experimental treatment regimen characterized by meticulous and standardized monitoring of both outcomes and adverse events. It showcases a highly personalized approach, where the dedication and adherence to treatment by the family, along with institutional support, play pivotal roles. The collection of information from different sources, such as family and clinician-rated instruments, therapists, and school reports, strengthens the interpretation of the results of the trial. The utilization of an on-off-on design and the extended duration of the treatment further contribute to the significance of the findings. In addition to the limitations inherent in all N-of-1 clinical trials, this particular one has further unique constraints. We did not include structured clinician-rated instruments; although we gathered information from multiple sources (parents, teachers, therapists), the placebo effect cannot be ruled out, and we did not incorporate a randomized placebo control design (due to the difficulty and likely ethical concerns associated with sham treatment of IGF-1).
FUTURE DIRECTIONS
The need for expert consensus, continuous learning, networking with international experts, and family involvement in the decision-making process of treatment is the other direct recommendation derived from this case. In the case of PMS, great international efforts are being followed to update knowledge derived from all possible sources of evidence (from anecdotal cases to preclinical work toward meta-analytical reviews) and provide recommendations for management and treatment[24,25]. Through these comprehensive pieces of work, we all gain knowledge about the heterogeneity of responses in different patients and are helped in clinical decision-making, adding to the value of randomized clinical trials and meta-analyses, which summarize overall trends and averages across studies, but do not capture the individual variability and nuances within heterogeneous populations.
CONCLUSION
This clinical case exemplifies a potential approach to treating rare disorders and patients who are challenging to include in standard clinical trials, particularly in the era of personalized medicine. Tailor-made interventions, informed by both preclinical and available clinical evidence, are crucial. Utilizing individualized yet standardized outcome instruments is essential in such cases. Additionally, navigating legal and administrative challenges is imperative. There is a pressing need for scientific innovations to optimize trial designs and interpretation of results in order to maximize the effectiveness of treatments for these conditions.
DECLARATIONS
Acknowledgments
We hope that this study can help the well-being of individuals with PMS and their families in the future, and for that, we would like to express our gratitude to LD and her family for allowing us to publish this work and for allowing us the very close, and sometimes tedious, monitoring of her progress.
Authors’ contributions
Substantial contributions to the conception or design of the work, or the acquisition, analysis, or interpretation of data for the work: Parellada M, Burdeus-Olavarrieta M, Rodríguez MD
Drafting the work or revising it critically for important intellectual content: Parellada M, Burdeus-Olavarrieta M, San José Cáceres A, Fraguas D
Final approval of the version to be published: Parellada M, Burdeus-Olavarrieta M, San José Cáceres A, Fraguas D, Medrano C, Rodríguez MD
Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved: Parellada M, Burdeus-Olavarrieta M, Fraguas D, San José Cáceres A, Medrano C, Rodríguez MD
Availability of data and materials
The datasets used during the current study are available from the corresponding author on request.
Financial support and sponsorship
This publication has been supported by SERMAS (National Health Service, Spanish Ministry of Health), Spanish Ministry of Science and Innovation, Carlos III Institute of Health (ISCIII) (FI18/00233), CIBERSAM (Mental Health Biomedical Research Network, CB/07/09/0023), Complutense University of Madrid, Ministry of Universities, Phelan-McDermid Association Spain (FIBHGM-CCA033-2016, FIBHGM-CCA027-2020). The results leading to this publication have received funding from the Innovative Health Initiative 2 Joint Undertaking under grant agreement No 777394 for the project AIMS-2-TRIALS. This Joint Undertaking receives support from the European Union’s Horizon 2020 research and innovation programme and EFPIA and AUTISM SPEAKS, Autistica, SFARI. Any views expressed are those of the author(s) and not necessarily those of the funders (IHI-JU2).
Conflicts of interest
Parellada M has served in an advisory or consultancy role for Exeltis and Servier and she is involved in clinical trials promoted by Servier and for F. Hoffmann La Roche Ltd. San José Cáceres A has served in an advisory or consultancy role for F. Hoffmann La Roche Ltd and in clinical trials conducted by Servier. She is currently consulting for Signant Health. Burdeus-Olavarrieta M has nothing to declare. The work presented here is unrelated to the above grants and relationships. Fraguas D has received funding as an advisor or speaker from Angelini, Casen Recordati, Janssen, Lundbeck, Otsuka, and Rovi. Medrano C and Rodríguez MD have nothing to declare.
Ethical approval and consent to participate
According to the policies of the Hospital General Universitario Gregorio Marañón Ethics Committee, publications of individual case reports do not require ethical review.
Consent for publication
Written informed consent for publication of their clinical details was obtained from the parents of the patient.
Copyright
© The Author(s) 2024.
REFERENCES
1. Schön M, Lapunzina P, Nevado J, et al. Definition and clinical variability of SHANK3-related Phelan-McDermid syndrome. Eur J Med Genet. 2023;66:104754.
2. Bozdagi O, Sakurai T, Papapetrou D, et al. Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication. Mol Autism. 2010;1:15.
3. Bozdagi O, Tavassoli T, Buxbaum JD. Insulin-like growth factor-1 rescues synaptic and motor deficits in a mouse model of autism and developmental delay. Mol Autism. 2013;4:9.
4. Hodge RD, D’Ercole AJ, O’Kusky JR. Increased expression of insulin-like growth factor-I (IGF-I) during embryonic development produces neocortical overgrowth with differentially greater effects on specific cytoarchitectonic areas and cortical layers. Brain Res Dev Brain Res. 2005;154:227-37.
5. O’Kusky JR, Ye P, D’Ercole AJ. Insulin-like growth factor-I promotes neurogenesis and synaptogenesis in the hippocampal dentate gyrus during postnatal development. J Neurosci. 2000;20:8435-42.
6. Kolevzon A, Bush L, Wang AT, et al. A pilot controlled trial of insulin-like growth factor-1 in children with Phelan-McDermid syndrome. Mol Autism. 2014;5:54.
7. Réthelyi JM, Vincze K, Schall D, Glennon J, Berkel S. The role of insulin/IGF1 signalling in neurodevelopmental and neuropsychiatric disorders - evidence from human neuronal cell models. Neurosci Biobehav Rev. 2023;153:105330.
8. Moffitt BA, Sarasua SM, Ivankovic D, et al. Stratification of a phelan-McDermid syndrome population based on their response to human growth hormone and insulin-like growth factor. Genes. 2023;14:490.
9. Yuen KCJ, Biller BMK, Radovick S, et al. American association of clinical endocrinologists and American college of endocrinology guidelines for management of growth hormone deficiency in adults and patients transitioning from pediatric to adult care. Endocr Pract. 2019;25:1191-232.
10. Parellada M, Boada L, Moreno C, et al. Specialty care programme for autism spectrum disorders in an urban population: a case-management model for health care delivery in an ASD population. Eur Psychiatry. 2013;28:102-9.
12. Constantino J. Social responsiveness scale (SRS-2). Los Angeles, CA: Western Psychological Services; 2012.
13. Brown EC, Aman MG, Havercamp SM. Factor analysis and norms for parent ratings on the aberrant behavior checklist-community for young people in special education. Res Dev Disabil. 2002;23:45-60.
14. Sparrow SS, Cicchetti DV, Saulnier CA. Vineland adaptive behavior scales, third edition. (Vineland-3)-Complete Kit: PsychCorp; 2016. Available from: https://www.pearsonassessments.com/store/usassessments/en/Store/Professional-Assessments/Behavior/Vineland-Adaptive-Behavior-Scales-%7C-Third-Edition/p/100001622.html [Last accessed on 29 Apr 2024]
15. Lingjaerde O, Ahlfors UG, Bech P, Dencker SJ, Elgen K. The UKU side effect rating scale. A new comprehensive rating scale for psychotropic drugs and a cross-sectional study of side effects in neuroleptic-treated patients. Acta Psychiatr Scand Suppl. 1987;334:1-100.
16. Berven S, Baron M, Deviren V, Glassman S, Bridwell K, Verma K. The assessment of clinically significant differences in treating spinal deformity using the SRS questionnaire: what is the threshold of change that is meaningful to patients? Int J Spine Surg. 2019;13:153-7.
17. Kolevzon A, Breen MS, Siper PM, et al. Clinical trial of insulin-like growth factor-1 in Phelan-McDermid syndrome. Mol Autism. 2022;13:17.
18. Sethuram S, Levy T, Foss-Feig J, et al. A proof-of-concept study of growth hormone in children with Phelan-McDermid syndrome. Mol Autism. 2022;13:6.
19. OCEBM Levels of Evidence Working Group. “The oxford levels of evidence 2”; Oxford centre for evidence-based medicine. 2011. Available from: https://www.cebm.ox.ac.uk/resources/levels-of-evidence/ocebm-levels-of-evidence [Last accessed on 29 Apr 2024].
21. Mirza RD, Guyatt GH. A randomized clinical trial of n-of-1 trials-tribulations of a trial. JAMA Intern Med. 2018;178:1378-9.
22. Schork NJ, Beaulieu-Jones B, Liang WS, Smalley S, Goetz LH. Exploring human biology with N-of-1 clinical trials. Camb Prism Precis Med. 2023;1:e12.
23. Al-Hammadi A, Ruszczak Z, Magariños G, Chu CY, El Dershaby Y, Tarcha N. Intermittent use of biologic agents for the treatment of psoriasis in adults. J Eur Acad Dermatol Venereol. 2021;35:360-7.
24. Srivastava S, Sahin M, Buxbaum JD, et al. Updated consensus guidelines on the management of Phelan-McDermid syndrome. Am J Med Genet A. 2023;191:2015-44.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].