


Connecting academia and industry for innovative drug repurposing in rare diseases: it is worth a try

Abstract
There are different approaches to drug repurposing (DR) depending on the status of the repurposable drug/molecule (approved, investigational, withdrawn, shelved), the context, and the stakeholders involved. The purpose of this perspective paper is to highlight the complexity of academia-industry collaborations in DR for rare diseases and go beyond stereotypes to consider realistic and mutually reinforcing cooperation among various stakeholders, including not only academia and industry but also regulators, legal experts, and payers, leading to benefits for patients with unmet medical needs. Key questions are addressed through the presentation of select DR case studies. Some ongoing and promising European and international initiatives are introduced and some recommendations are proposed.
Keywords
INTRODUCTION
Drug repurposing (DR) is defined as the process of identifying new therapeutic uses for approved or investigational drugs beyond their original medical indications. It is considered to have significant advantages over developing an entirely new drug: fewer risks, lower costs, and shorter timelines. Public-specific programs promote DR initiatives while pharmaceutical companies integrate them as a strategy in the life cycle management of their products. Indeed, DR represents a relevant opportunity to find treatments, especially in the field of rare diseases, where 95% of the 7,000 to 8,000 rare diseases have no approved treatment. A few well-known success stories illustrate very well successful academia-industry DR collaborations, and may be viewed as the tip of the iceberg. There are also bitter and anonymous failures of collaboration.
The purpose of this perspective paper is to explore academia-industry collaborations in DR of medicinal products for rare diseases through their main challenges and successes and through select DR case studies. Key questions are raised to understand why, when, and how to implement a relevant collaboration. Recommendations are also proposed.
DEFINITION OF DRUG REPURPOSING
DR is also known as drug repositioning, reprofiling, retasking, rediscovery, rescue, etc. While reflecting on different types of situations encountered and described in the literature[1], all these terms are synonyms and refer to a similar process that identifies new indications for approved, investigational, failed, or shelved drugs/compounds. At present, the terms are all used interchangeably. For consistency purposes, this article will refer to all terms as “drug repurposing”. As of November 1, 2022, a search on PubMed listed 1,973 results mentioning “drug repurposing” and 861 results mentioning “drug repositioning” for 2022.
DRUG REPURPOSING PROVIDES AN OPPORTUNITY FOR RARE DISEASES
Much has been said about DR over the last two years during the coronavirus (COVID-19) pandemic. An editorial entitled “COVID-19 and the DR Tsunami”, published in July 2020, reflects an unprecedented wave to repurpose existing drugs[2]. The aim is to vastly accelerate the usually long approval process of drugs[3], which is a common goal and an objective also shared with rare disease patients desperately waiting for treatments.
Currently, there are an estimated 350 million people worldwide living with a rare disease. To date, 7,000 to 8,000 rare diseases have been identified, with 250-280 new diseases discovered annually. Only about 5% of rare diseases are estimated to have approved treatments.
Between 2000 and 2021, 245 orphan drugs, so named because of the rare diseases they treat, have been approved in Europe. They cover a total of 148 rare diseases[4]. About 20% of orphan drugs are repurposed drugs[5].
There is an urgent need for treatments, as most of the rare diseases are life-threatening[6]. DR may be a particularly attractive option for the development of treatments for rare diseases. Of note, oncology and infection are together the most common disease areas for DR so far[7].
SELECT EXAMPLES OF SUCCESSFUL DRUG REPURPOSING
The most famous case of DR is probably the development of sildenafil, which is also a good example of both investigational and approved DR[8]. Sildenafil, a phosphodiesterase 5 (PDE5) inhibitor initially explored as a treatment for angina pectoris and hypertension by Pfizer, was tested unsuccessfully in these diseases and eventually demonstrated efficacy in treating erectile dysfunction (ED) (with the trade name Viagra®) and, later on, pulmonary arterial hypertension (PAH) (with the trade name Revatio®)[9].
The clinical trials for angina, which started in 1989, were disappointing, and sildenafil failed in the phase II trial. However, a side effect (induction of penile erection) was serendipitously found during the phase I and II clinical trials and redirected sildenafil to the treatment of ED. This indication was approved in Europe and the United States in 1998. After its serendipitous repurposing for ED, sildenafil was then repurposed (this time intentionally) for the rare disease PAH[10]. This indication was approved in Europe and the United States in 2005[11].
Of note, sildenafil acts on the same target, PDE5, to treat both ED and PAH. PDE5 is a key enzyme involved in the regulation of cyclic guanosine monophosphate (cGMP)-specific signaling pathways in normal physiological processes, such as smooth muscle contraction and relaxation. Angina pectoris is a chest pain associated with coronary heart disease. Since PDE5 hydrolyzes cGMP in the cardiopulmonary vasculature, researchers aimed to establish a new anti-anginal agent using PDE5 inhibitors to prolong cGMP activity and promote vasodilation of the coronary arteries. However, early inconclusive findings indicating minimal PDE5 expression in cardiomyocytes led to the abandonment of this research approach[12]. PDE5 is the predominant PDE in the corpus cavernosum, where its catalytic site degrades cGMP, the key second messenger in the mediation of penile erection. In men with ED, selective inhibition of PDE5 leads to an increase in cGMP in cavernosal tissue and improves erectile function.
Persistent PAH in newborns has been shown to be linked to PDE5 overexpression and overactivation. PAH is characterized by increased pulmonary vascular resistance due to vasoconstriction of the small pulmonary arteries and arterioles. In blood vessels, cGMP relaxes vascular smooth muscles, leading to vasodilation and increased blood flow. By inhibiting PDE5 and raising the intracellular concentration of cGMP, sildenafil is an effective pulmonary vasodilator.
Understanding the mechanisms of action (MOAs) of drugs is critical not only for drug development, but also for DR. The identification of drug MOAs has been primarily based on pharmacological experiments. Identification of MOAs through biological pathways involving a drug and its targets is a more recent alternative approach for diseases not currently treated by a drug, to predict new uses of existing drugs and to repurpose them. Such an approach can be of real interest also for drugs with unknown underlying MOAs.
In fact, biological pathway analysis based on drug targets (genes and proteins) may reveal new MOAs and also new clinical functions of existing drugs[13]. Alpelisib provides a good example of innovative DR in this respect by illustrating how the analysis of a biological pathway can lead to the treatment of diseases other than the ones initially investigated.
Alpelisib, an inhibitor of the catalytic p110 alpha subunit of the phosphatidylinositol-4,5-bisphosphate
Cancer-associated PIK3CA gene mutations result in the production of an altered p110 alpha subunit. Strongly and permanently activated due to the presence of a gain-of-function mutation, p110 alpha allows the phosphatidylinositol 3-kinase (PI3K) to signal without regulation, promoting oncogenesis and tissue overgrowth[14]. Patients with PIK3CA-mutated, hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative tumors have resistance to chemotherapy and poor outcomes[15].
Developed by Novartis, alpelisib was approved by the United States Food and Drug Administration (FDA) as Piqray® in combination with fulvestrant for advanced or metastatic breast cancer with a PIK3CA mutation. Fulvestrant is a selective estrogen receptor antagonist that works both by downregulating and degrading the estrogen receptor. Piqray® is used with fulvestrant after hormone treatment alone has failed.
Activating PIK3CA mutations are also found in PROS, a group of genetic disorders and various clinical entities that lead to the overgrowth of various body parts. The congenital lipomatous overgrowth, vascular malformations, epidermal nevi, and skeletal anomalies (CLOVES) syndrome, a rare disorder first described as a distinct syndrome, is one subtype of this spectrum. The severity of PROS is highly variable, ranging from localized overgrowth to severe, extensive, and life-threatening overgrowth affecting major vessels
The PI3K/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) pathway is involved in many biological processes, including cell metabolism, proliferation, survival, and growth. Overactivity of the PI3K/AKT/mTOR pathway promotes oncogenesis and tissue overgrowth[16]. It is then relevant to investigate the potential of some anticancer drugs to treat rare overgrowth syndromes such as PROS. As in cancer, PIK3CA mutations in PROS occur as post-zygotic events, but unlike in cancer, these mutations arise during embryonic development[17]. Different components of the PI3K/AKT/mTOR signaling pathway can be specifically targeted to treat PROS. The PIK3CA protein is involved in the first intracellular signal transduction step of the mTOR pathway. Alpelisib acts at the top of the PI3K/AKT/mTOR signaling pathway, whereas sirolimus, a direct inhibitor of mTOR, acts at the end [Figure 1]. Both molecules have been tested in PROS[14].
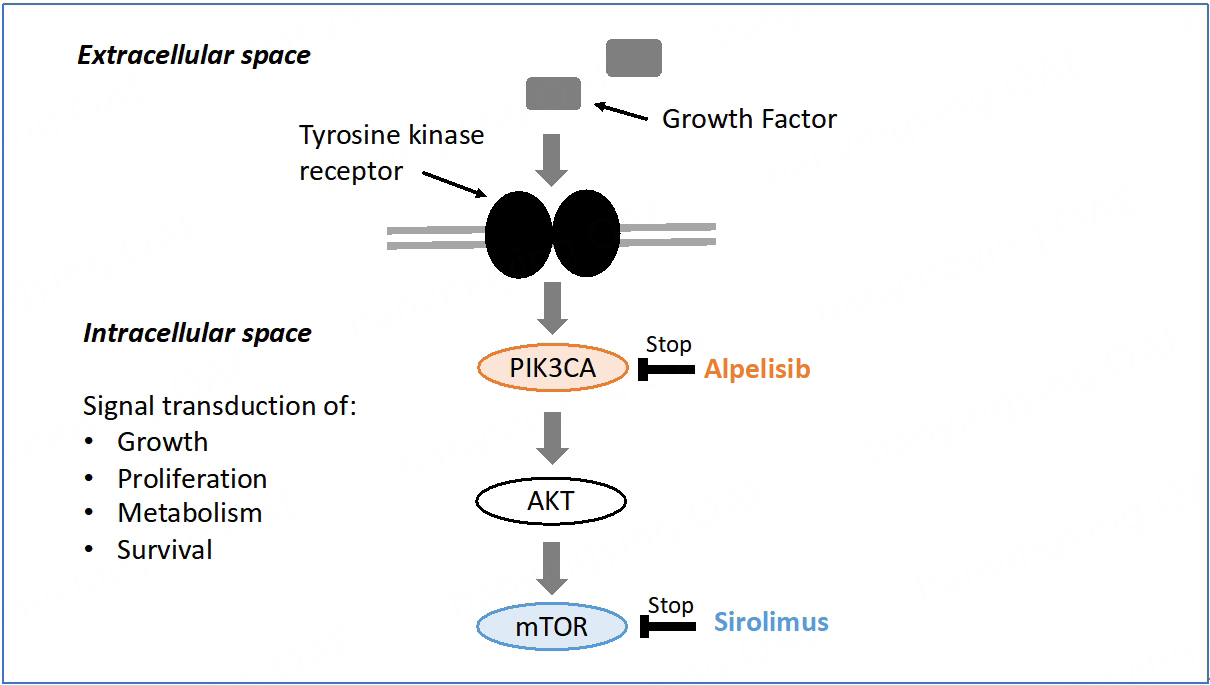
Figure 1. mTOR signal transduction pathway[15]. mTOR: Mammalian target of rapamycin; PIK3CA: phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha; AKT: protein kinase B.
However, since activation of mTOR is responsible for some, but not all, the biological effects caused by PI3K gain-of-function, alpelisib proved to be a more attractive drug candidate in PROS by providing a direct outcome on affected tissues while reducing the risk of off-target effects.
Alpelisib (BYL719) was in a phase III randomized, double-blind, placebo-controlled study (SOLAR-1) conducted in patients with PIK3CA-mutated, HR-positive, HER2-negative advanced breast cancer, demonstrating a tolerable safety profile. Building on these results, Venot et al.[18] decided to explore the therapeutic potential of alpelisib in PROS. After achieving impressive outcomes, first on PROS mouse models, and then on two patients suffering from extremely severe and life-threatening clinical manifestations of PROS/CLOVES syndrome, Venot et al.[18] were authorized to administer BYL719 to 17 additional patients with PROS. Their study was published in 2018, supporting PIK3CA inhibition as a promising therapeutic strategy in patients with PROS[18].
Four years later, in 2022, the FDA granted accelerated approval to alpelisib under the brand name Vijoice® to treat severe manifestations of PROS in patients aged two years or older. This indication for Vijoice® is approved based on real-world response rate and duration of response findings in 57 patients in the
ACADEMIA-INDUSTRY COLLABORATION: WHY, WHEN, AND HOW
The two successful DR cases mentioned above - alpelisib in PROS and sildenafil in ED - are good examples of a successful academia-industry DR collaboration. In one case, investigators reported the side effects of sildenafil that became a repurposed indication in ED, and Pfizer drove the repurposing efforts; in the other case, an academic research team attempted independent repurposing of alpelisib in PROS, a new and rare indication further developed by Novartis.
While there are remarkable DR success stories that hit the headlines, there are also bitter and anonymous failures of collaboration. Various reasons may explain the poor cooperation between industry and academia. These reasons need to be properly understood if we want to orient efforts in appropriate directions. Key questions may be addressed: why, when, and how to implement a relevant collaboration. They deserve clear answers through a broader and more structured debate involving all stakeholders. In the meantime, some suggestions can be made, and some existing initiatives at European (for some of them) and international levels can be reported to encourage and stimulate new opportunities.
WHY
Why get academia and industry to work together? There are many reasons to support such collaborations. Even if they have different expectations and constraints, academia and industry may be complementary and bring added value to each other in a highly regulated environment.
Academia as a source of innovation
Pharma’s business model fundamentally depends on product innovation to create value. Patent expiry and generic competition approximately every ten years are real challenges for pharmaceutical companies, which look to academia for valuable knowledge and innovation[20]. The contribution of academia is crucial in terms of basic research, the identification and validation of new targets, and clinical trials to evaluate the efficacy and safety of drug candidates. Academia and industry may be caricatured as the alpha and the omega, as they have complementary enterprises throughout the drug value chain, from innovation to validation. Research for rare diseases starts in academia[21], development is shared among stakeholders, and commercial launch is managed by industry.
Different expectations and constraints, but a shared goal of improving patient health
It has become commonplace to highlight the gap between academia and industry in terms of cultures and practices. Although patenting is key for both, the value of secrecy for pharma continues long after patent filing and grant. Pharmaceutical companies still consider results collected from clinical trials to be confidential information or trade secrets, even after submission to regulatory agencies. Academics, for their part, are under pressure to increase their publication count. The “publish or perish” culture is a well-known practice existing within academic institutions. However, once the patent has been filed, the invention may be published. It is then crucial for a researcher to present his/her work at conferences, exchange with peers, and win grants.
Highly regulated drug development process
Academic researchers have neither the capacity nor the finances to develop a drug candidate until the launch of the finished product. Moreover, they are often unaware of what evidence needs to be submitted as part of an application for marketing authorization (MA) or which complex drug development challenges will have to be addressed. Pharmaceutical companies operate in a very highly regulated area, from key nonclinical studies, phase I-III clinical trials, chemistry, manufacturing and controls (CMC), and formulation activities to regulatory submissions, commercial launch activities, and life-cycle management[7]. Drug development requires a comprehensive and well-thought-out development strategy with a detailed roadmap.
Drug development is a risky and costly process
The time required to develop a new drug is about 10-15 years and the cost is around $2 billion. Despite significant investments of time and money, approval rates for new drugs are about 10%. DR has been proposed as an interesting strategy with fewer risks, lower costs, and shorter timelines. Indeed, repurposed drugs are generally approved sooner (3-12 years), at reduced (50%-60%) cost and lower risk, with approval rates close to 30%[22]. However, in some cases, DR may be an expensive, time-consuming, and risky process[23]. Furthermore, it does not always succeed. Similar to de novo drug development, DR may also fail in late-stage development. That was the case for latrepirdine, originally developed and marketed as an H1 antihistamine for the treatment of skin allergy and allergic rhinitis. Latrepirdine was repurposed as a treatment for Huntington’s disease. Following encouraging phase II trials and preliminary reports showing its neuroprotective functions and ability to enhance cognition in animal models, it failed to show efficacy in phase III trials in Huntington’s disease patients[24].
Some authors have underlined the importance of confirmatory validation studies for successfully translating academic results into industry practice before making larger investments[25]. Such gaps between academia and industry should favor rather than discourage a move toward stronger collaboration. Both have their respective roles in a drug development process; there is complementarity rather than duplication, and there may be mutual support in learning from one another. Moreover, while repurposing previously relied on serendipity, it has more recently evolved thanks to continuous advances in the field of data science and to approaches integrating - among others - computational assistance using Big Data and Artificial Intelligence[26].
WHEN
There is no ideal timing to start a collaboration
There are rather special circumstances leading to potential opportunities or not. For instance, one of the nightmares for a pharmaceutical company is encountering safety issues during pivotal studies close to the approval stage. That was the case for alpelisib when Venot et al. asked for the molecule to be tested in PROS[18]. Although based on an exhaustive literature review and spectacular first results in mice and patients, the alpelisib request was badly timed. The phase III trial of alpelisib in breast cancer was still ongoing. The detection of a potentially serious adverse event during a clinical trial in PROS could have jeopardized the drug development for both indications.
Academia-industry collaboration may start as early as possible within a co-development framework or should be at least anticipated, so as to avoid unexpected issues such as the unpleasant surprise for the industry to learn that one of its drugs is unexpectedly repurposed by an academic team in an out-of-scope indication, or the huge frustration for the academic team who will not be able to develop a promising candidate owned by industry for not being part of its strategy. It must be noted here that large pharmaceutical companies do not have the same expectations as smaller ones. They prefer late-stage development projects (phase II and onwards), where they have relevant experience and can mitigate risks.
Line extension or label expansion is not the right time for a collaboration
DR may be part of an anticipated strategy from the industry in the early drug development process. In contrast, line extension or label expansion is typically planned from the outset of a development program. These activities are part of life cycle management, which aims to maximize the value of existing products with new indications, improved formulation, new packaging, or other modifications, and to allow for patent extensions[27]. Only the MA holder of a drug can currently apply for an extension of its MA. When drugs are repurposed based on on-target effects (as with sildenafil and alpelisib), it means that work may be undertaken by the pharmaceutical company owning the molecule. Company data are not available to academic investigators.
Intellectual property considerations are a critical element when starting a collaboration
Drugs are protected by patents and supplementary protection certificates and may benefit from another form of market exclusivity, as in the case of orphan drugs (seven years in the United States, ten years in Europe).
Patents can cover the existing product, its underlying chemical or biological make-up, as well as new indications, dosage regimes, or mechanisms of delivery. Supplementary protection certificates are intellectual property (IP) rights that serve as an extension to a patent right. They all give the owner the right to prevent others from making, using, or selling the drug without permission. Patent protection is critical for developing a new therapy.
It is challenging to obtain strong patent protection when repurposing compounds. By definition, the structure of a repurposed drug is known, and a novel patent claim to the active pharmaceutical ingredient is not possible. Composition of matter (COM) claims, which protect a drug regardless of its use, are one of the classic and powerful claim types. However, such claims cannot be applied to repurposed drugs in the vast majority of cases. Instead, repurposed drugs are eligible for patent protection through use claims or method of treatment (MOT) claims. MOT patents may be perceived as second-tier to COM patents; however, in some cases, they seem to be a good way of protection and commercial success for some companies[28].
IP position is critical in ensuring the ability to achieve a collaboration based on partnership and dialogue. IP may fall into one of two major categories, active or expired, each leading to a different set of issues.
IP is still in force
Alpelisib is an example of a successful academia-industry DR collaboration in this category leading to a treatment for patients, although the company was not interested in its development at the very beginning. Alpelisib was being evaluated for breast cancer at that time.
If an academic researcher has discovered a new indication for an approved or investigational drug, then early engagement with the potential industry partner is highly recommended, since the company is the patent owner. It will be a crucial step in the process. The objective is twofold: to know if the company is interested in the development, and to obtain access to the drug and potential funding for studies performed under a material transfer agreement (MTA).
Pharmaceutical companies frequently engage in preclinical research collaboration with academics. Complex knowledge transfer processes and substantial challenges in terms of IP may arise. Generating new IP has to be discussed, anticipated, and validated in a consortium agreement in order to organize the rights and obligations of all stakeholders and protect the researchers’ discovery and IP[7]. If the company holding the IP agrees to provide the drug but refuses to provide funding to support preclinical or clinical programs, it will be difficult for the researcher alone to move forward with the drug, unless new patent protection can be obtained. New formulations or drug forms for a known active pharmaceutical ingredient may be developed for a new indication and provide academics with an opportunity for patent protection. Academic investigators may also decide to perform clinical studies without the support of the company and with no guarantee - even if the trial is successful - of a newly approved indication. Repurposing an approved and active pharmaceutical ingredient for a new indication would only benefit the original manufacturer who owns the IP for the COM. Scientific and nonscientific understandable reasons may explain the company’s refusal, such as insufficient remaining patent life, a lack of expertise and interest in the new therapeutic area, or anticipated regulatory risks. A recent study explored innovative mechanisms to fund independent clinical research initiated and led by researchers from academia with repurposed medicines[29]. Focus is put on social impact bonds, crowdfunding, or public-private partnerships to conduct expensive phase III clinical trials with, again, no guarantee of successful achievement.
IP has expired
In this case, the drug is off-patent or generic and widely available at a low price. Propranolol is an example of a successful academia-industry DR collaboration in this category, leading to a new treatment for patients [Figure 2].
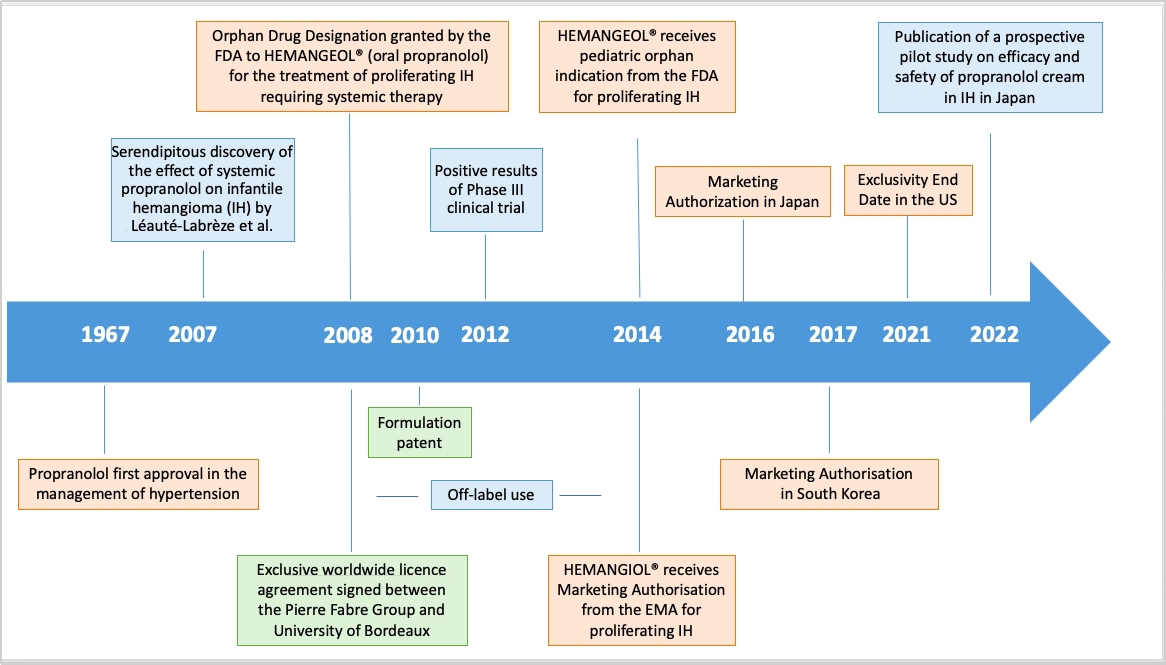
Figure 2. Repurposing propranolol in proliferating infantile hemangioma: key milestones. FDA: Food and Drug Administration.
Propranolol, a non-selective beta-blocker patented in 1962, was granted FDA approval in 1967 as an antihypertensive drug. In 2008, Léauté-Labrèze et al. from the Bordeaux University Hospital reported its antiangiogenic therapeutic properties in 11 cases of infantile hemangioma (IH), at which time propranolol was already available as a generic drug[30]. IH occurs in 4.5% of infants. Most IHs resolve spontaneously, but 12% are complex and severe forms with functional and even life-threatening complications, such as breathing difficulties, and require systemic treatment[31]. Following this serendipitous discovery, Pierre Fabre Dermatologie Laboratories and the University of Bordeaux formed a partnership in 2008 to co-develop and launch Hemangiol® in Europe and Hemangeol® (a different brand name for the same oral formulation) in the United States in 2014 for the treatment of proliferating IH requiring systemic therapy. For information, the brand name Hemangiol® was changed into Hemangeol® in the United States in accordance with the FDA’s best practices in developing proprietary names guidance, according to which “io” is used as an infix to suggest a high iodine content[32]. In terms of organization and assignment of tasks, resources, and operational skills, the company took charge of the regulatory dossier, including the development of a pediatric formulation and a pivotal study performed on 460 babies between five weeks and five months of age at treatment initiation. The objective of the study was to select the right dosage and confirm the efficacy and tolerance of propranolol in infants with proliferating IH requiring systemic therapy. The clinicians at the Bordeaux University Hospital were involved in the study design and management as well as the drafting and publication of the manuscript[33].
Pharmaceutical companies may be reluctant to initiate collaborations with academia to repurpose off-patent drugs. Once drugs lose patent protection, generics can be allowed for sale, leading to off-label use and commercial risks. Because it was possible to dissolve commercially available propranolol tablets or to use a non-approved preparation of liquid propranolol made up by the hospital pharmacy, off-label use of the drug posed a significant challenge[34]. This was due to risks from inappropriate dosage and from the use of excipients non-recommended for infants, since the therapy was adapted from adult dosage to pediatric populations without an appropriate formulation for young children.
Nevertheless, different actions may be put in place to protect companies and patents are the first step. For propranolol in IH, new patents were granted to academia (second medical use patent in 2008) and industry partners (formulation patent in 2010), offering protection for 20 years until 2028 and 2030, respectively.
In addition to patent protection, regulatory agencies are offering incentives for the development of orphan and pediatric drugs for rare diseases, such as Pediatric-Use Marketing Authorization (PUMA) in Europe and Orphan Drug Designation (ODD) in the United States.
Thanks to PUMA, drugs can benefit from ten years of market protection, including eight years of data exclusivity plus an additional two years of market exclusivity[35].
An ODD is a status delivered by the FDA in the United States or the European Medicines Agency (EMA) in Europe to medicines developed for rare diseases; it may be requested by sponsors for a previously unapproved drug or for a new orphan indication for an already marketed drug. It provides incentives including tax credits for clinical trials, exemption from user fees, and market exclusivity after approval of the drug. In 2008, an ODD was granted by the FDA to Hemangeol®, which could benefit from a seven-year market exclusivity starting in 2014, when the drug was approved. An ODD was not granted in the European Union for Hemangiol® because of “salami slicing”, which denies the possibility of restricting the target population to severe forms only (100% of IHs are considered in the European Union vs. only 12% in the United States).
Other MAs have also been granted to the drug in other countries, such as Japan in 2016 and South Korea and Australia in 2017. However, MA does not mean market access and patient access. Each country has individual requirements for pricing/reimbursement with independent Health Technology Assessments (HTAs). If the submitted data package does not support a positive decision from one country’s HTA, then the product is at risk of partial reimbursement or even no reimbursement in that country. A cost-utility analysis was conducted in Italy in 2015 to assess the costs and clinical benefits of Hemangiol® 3.75 mg/mL (oral solution) in comparison to corticosteroids, considered as the standard of care in the treatment of proliferating IHs. Propranolol was considered cost-effective compared to corticosteroids from the Italian National Health Service perspective[36].
DR relies on a real strategy that involves actors dealing with different constraints but aiming at the same final expectation. Propranolol represents a good use case outlining the success and failure scenarios of DR for all the reasons exposed above. Many lessons can be learned from this case, although it is not always possible to replicate them. An effective IP strategy is an essential tool for identifying risks and opportunities when repurposing medicines and setting out on new projects[37], but it is only part of the puzzle. Market access strategy is critical for the successful launch of a therapy. Landscape assessment is still more relevant in the rare disease ecosystem, where literature and natural history are limited. Close collaboration with clinicians is essential to better understand the burden of the disease and patients’ needs and work together on the clinical development program to optimize pediatric formulations and patient outcomes while keeping in mind the expectations of regulatory agencies and payers.
A paper published in 2022 compares the price of Hemangiol® approved for IH with that of its off-label alternative, not approved for the same indication, in Germany[38]. The study underlines the clinical development and registration program value taken into account by payers in the price obtained by the approved product. The price difference is significant but relatively lower for Hemangiol® compared with two other orphan drugs presented in the paper. Although the results may not be generalized, they may foreshadow a positive return on investment for Hemangiol®. In any case, it is a success story with an approved treatment covering an unmet medical need for babies and infants with severe IH.
HOW
Implementing a relevant academia-industry collaboration requires a strategy to move forward. There are many opportunities for industry to partner with academia and vice versa, encompassing a wide range of collaboration forms varying in the extent of involvement and risk taken by the stakeholders. Identifying research opportunities, applying to private-public calls for proposals, forming an early-stage partnership, developing new methods for the design of small population clinical trials, understanding the natural history of the disease to improve treatment strategies, or advancing real-world evidence to support payer decisions are just a few examples. How to implement the collaboration is the key question. A case-by-case answer is probably the best strategy, since there are so many different factors to consider for a complete and successful achievement. Such factors are related to the stakeholders themselves, their profiles, skills, interests, willingness, and commitment, but they are also dependent on the candidate repurposing drug, its IP and freedom to operate, its safety and toxicity profile, and the data already accumulated toward gaining regulatory approval[27].
A brief overview of key suggestions and current initiatives in DR is given hereafter.
Educational programs for academia
Academic researchers need to be guided and trained in IP, regulatory procedures, and market access to understand the constraints and expectations of the industry. Technology transfer offices or neutral and trusted third parties (public or private) may support them to this end. First of all, it is extremely important for academia to understand industry needs when dealing with practical problems to which they can apply their knowledge. Academic research may be closely linked to practical implications in an industrial environment[39]. It is also important for academia to be aware of the pharmaceutical development of a drug and for the industry to be sensitized to the key role and importance of publications, grant approvals, and international networking for academia, notably for Ph.D. students and early career researchers. There is a real need for mutual knowledge of expectations and constraints from both academia and industry in order to facilitate interactions and enable fruitful collaboration. Both must learn from each other.
Pilot project from the EMA and the safe and timely access to medicines for patients expert group
Another way of implementing relevant academia-industry collaborations is provided by European initiatives dealing with DR. In October 2021, the EMA and the Heads of Medicines Agencies (HMA) launched a pilot project to support the repurposing of drugs. That project is a follow-up to the European Commission Expert Group on Safe and Timely Access to Medicines for Patients (STAMP) discussions. The objective is to support not-for-profit organizations, including academia, to generate sufficient evidence on the use of a well-established off-patent drug in a new indication. Academic researchers will be connected with regulatory agencies for early scientific advice and with companies for regulatory applications. These investments must also be recognized in pricing and reimbursement policies to make access a reality for all patients[40,41]. Candidate projects were selected in June 2022.
Horizon Europe funding program for research and innovation
Being part of consortia involving teams from industry, academia, subject matter experts, and patient associations may help academic partners build lasting collaborations and networks. A call for proposal “Tackling diseases (2021) (HORIZON-HLTH-2021-DISEASE-04)” was proposed under the Horizon Europe program and closed on September 21, 2021. Thirteen proposals among 253 submitted were related to the topic “Building a European innovation platform for the repurposing of medicinal products”. Among key outcomes, proposals had to address a widened collaboration to set up a sustainable platform and an innovative repurposing model integrating different components such as IP, as well as methodological, scientific, regulatory, and financial aspects. Particular attention was given to supporting academic-driven research[42].
International Rare Diseases Research Consortium task force on drug repurposing
As a brief overview, one could also mention the recent International Rare Diseases Research Consortium (IRDiRC) task force on DR based on the IRDiRC Orphan Drug Development Guidebook[43]. The DR guidebook is aimed at facilitating DR for rare diseases by identifying relevant available tools and practices, defining a list of building blocks (tools, incentives, resources, and initiatives), and creating detailed factsheets for each building block. The factsheets will be based on a systematic literature review and the expertise of the task force members. They will also provide advice and recommendations. A web application and an article are expected by the end of 2023.
DR platforms are examples of tools and resources to be found in this DR guidebook. The DR platform established by the FDA is still available, and can now be located at cure.ncats.io. CURE ID (Infectious Disease) is a collaboration between the FDA and the National Center for Advancing Translational Sciences (NCATS), part of the National Institutes of Health (NIH). The FDA and the NCATS have made critical updates to CURE ID to make it a more effective tool in the COVID-19 public health emergency.
Recommendations from a recent pharmacology and translational research round table
A recent “Pharmacology and translational research” round-table of the Giens Workshops[44] formulated and published a series of recommendations to help overcome sticking points and obstacles to DR[44]. The Giens Workshops are held in Paris every year; they are a think tank whose objective is to advance thinking on current issues in pharmacology and clinical research for therapeutic innovation and health technology assessment. The meetings are organized into round tables, each covering pre-defined themes: translational research; clinical research; health technologies including diagnostics, drugs, and medical devices; health and economics; organizational and regulatory aspects; and topical issues. Each round table aims to bring together experts from various backgrounds. In 2022, one of the round tables was dedicated to DR around the following key points.
Optimizing access to and use of databases
Databases are a basis for DR methods and, therefore, must be designed in a proper manner to achieve relevant results. They can be used to predict drug-target interactions and discover new treatment benefits of the existing drugs. Access must be facilitated to pharmacological databases (chemical and molecule libraries, associated pharmacological data). Although there are open-access databases for searching chemical, biological, and pharmacological data, searching such specific databases is not always straightforward. Another challenge is to gain access to shelved industry compounds and their trial data when companies are reluctant to grant it. It is therefore critical to simplify the procedures for accessing public databases and promote data sharing between public and private structures.
Improving awareness of IP issues
DR can be done on shelved compounds, failed compounds, live patents, and drugs sold on the market, as well as on generic drugs. DR always relies on a previously known drug, but each situation has its own challenges when considering collaborative research and development between pharmaceutical companies and academic researchers. If the repurposing project comes from academic bodies, they will have to discuss and negotiate with the pharmaceutical companies that hold the molecule involved in the project to convince them to agree on the methods for developing and sharing IP.
It is then important to provide all researchers with sufficiently deep training in the legal aspects of the drug development process. A proper DR project requires expert legal counsel to advise on the issues involved in the process, and to identify risks and opportunities when setting out on new projects.
Anticipating preclinical and clinical requirements
The real feasibility of the project, the financial and human resources to be deployed, and the time needed to complete the project should always be analyzed and anticipated. There is a real need for tools or structures capable of providing advice on the model of the scientific advice/protocol assistance of the EMA.
Improving regulatory procedures for market access
MA is a regulatory process at the European level and does not necessarily mean market access in all member states of the European Union. Some efforts should be made to achieve better alignment and synergies between regulatory and payers’ requirements. Regulatory agencies assess new treatments’ benefits and harms to the exclusion of economic considerations, whereas payers focus on effectiveness and economic consequences. In France, for instance, the doctrine of the High Authority for Health (HAS) Transparency Committee (TC) needs to evolve both regarding the methodology of potentially admissible trials and the more specific part of the so-called “relevant” clinical comparator. The HAS TC’s main task is to assess medicinal products in order to provide recommendations on reimbursement decisions made by public authorities. Access to reimbursement in France follows MA and requires pharmaceutical companies to submit an application file to the HAS TC.
Creating a public-private partnership support structure on a European scale
The main recommendation made during the round table was the creation of a support structure capable of managing the most complex aspects of repurposing projects. A trusted third party is useful for initiating collaborations and providing the infrastructure and resources for academia and industry to work together. Such a support structure would have dedicated financial resources and expertise at all stages of development until the MA. It could even become the MA holder for the repurposed drug if need be. The Innovative Health Initiative (IHI) is a public-private partnership between the European Union and the European life science industries whose main objective is to translate research and innovation into tangible benefits for patients. Such a structure could be used as a model for DR[45].
CONCLUSION
DR represents a real opportunity and a cheaper and faster way to find and develop new treatments for rare diseases. However, DR is also associated with misconceptions and specific barriers (IP, regulatory, market access, and lack of incentives). It makes no sense to stereotype public-private partnerships and set one community against the other. Without research, there will not be any innovation; without regulatory and commercial development, there will not be any drug on the market. Despite being far away from each other in terms of culture, constraints, and objectives, academia and industry can join forces and build off each other’s strengths to advance research and development of repurposed drugs for patients with rare diseases. This approach has already proved successful in several instances, but its potential is far from being fully exploited. Some fruitful collaborations have resulted in success stories that are widely publicized, but they do not fully reflect reality. The success rates of academia-industry collaboration vary depending on various factors (IP rights of the drug candidate, business strategy, proof of concept robustness, regulatory challenges, commercial aspects, and the type of pharmaceutical company). Academic drug (re)discovery is a vital component, but the lack of knowledge of industry standards can make academics uncomfortable. Educational programs and guidelines could help them better understand industrial constraints and anticipate and adjust accordingly. There is also a need for new and sustainable DR business models involving not only academia and industry but also patient associations, regulators, and payers to equitably address these questions. Some current European and international initiatives such as the pilot project from the EMA and the STAMP expert group, the call for proposals “Building a European innovation platform for the repurposing of medicinal products” within Horizon Europe, and the IRDiRC task force on the DR guidebook should encourage and stimulate new opportunities.
DECLARATIONS
Authors’ contributions
The author contributed solely to the article.
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
The author declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2023.
REFERENCES
1. Murteira S, Ghezaiel Z, Karray S, Lamure M. Drug reformulations and repositioning in pharmaceutical industry and its impact on market access: reassessment of nomenclature. J Mark Access Health Policy. 2013;1:21131.
3. Galindez G, Matschinske J, Rose TD, et al. Lessons from the COVID-19 pandemic for advancing computational drug repurposing strategies. Nat Comput Sci. 2021;1:33-41.
4. European Medicines Agency. Orphan medicinal product designation 2000-2020. Available from: https://www.ema.europa.eu/en/documents/other/orphan-medicines-figures-2000-2020_en.pdf [Last accessed on 7 Apr 2023].
5. European Medicines Agency. Support for development of orphan medicines. Available from: https://www.ema.europa.eu/en/documents/report/report-workshop-support-orphan-medicines-development_en.pdf [Last accessed on 7 Apr 2023].
6. Southall NT, Natarajan M, Lau LPL, et al. IRDiRC Data Mining and Repurposing Task Force. The use or generation of biomedical data and existing medicines to discover and establish new treatments for patients with rare diseases - recommendations of the IRDiRC Data Mining and Repurposing Task Force. Orphanet J Rare Dis. 2019;14:225.
7. Begley CG, Ashton M, Baell J, et al. Drug repurposing: misconceptions, challenges, and opportunities for academic researchers. Sci Transl Med. 2021;13:eabd5524.
8. Shim JS, Liu JO. Recent advances in drug repositioning for the discovery of new anticancer drugs. Int J Biol Sci. 2014;10:654-63.
9. Ghofrani HA, Osterloh IH, Grimminger F. Sildenafil: from angina to erectile dysfunction to pulmonary hypertension and beyond. Nat Rev Drug Discov. 2006;5:689-702.
10. Jourdan JP, Bureau R, Rochais C, Dallemagne P. Drug repositioning: a brief overview. J Pharm Pharmacol. 2020;72:1145-51.
11. Barnett CF, Machado RF. Sildenafil in the treatment of pulmonary hypertension. Vasc Health Risk Manag. 2006;2:411-22.
12. Roy S, Kloner RA, Salloum FN, Jovin IS. Cardiac effects of phosphodiesterase-5 inhibitors: efficacy and safety. Cardiovasc Drugs Ther. ; doi: 10.1007/s10557-021-07275-y. 2021;Online ahead of print.
13. Pan Y, Cheng T, Wang Y, Bryant SH. Pathway analysis for drug repositioning based on public database mining. J Chem Inf Model. 2014;54:407-18.
14. Pagliazzi A, Oranges T, Traficante G, et al. PIK3CA-related overgrowth spectrum from diagnosis to targeted therapy: a case of CLOVES syndrome treated with Alpelisib. Front Pediatr. 2021;9:732836.
15. Scherman D, Fetro C. Drug repositioning for rare diseases: knowledge-based success stories. Therapie. 2020;75:161-7.
16. Chang DY, Ma WL, Lu YS. Role of Alpelisib in the treatment of PIK3CA-mutated breast cancer: patient selection and clinical perspectives. Ther Clin Risk Manag. 2021;17:193-207.
17. Madsen RR, Vanhaesebroeck B, Semple RK. Cancer-associated PIK3CA mutations in overgrowth disorders. Trends Mol Med. 2018;24:856-70.
18. Venot Q, Blanc T, Rabia SH, et al. Targeted therapy in patients with PIK3CA-related overgrowth syndrome. Nature. 2018;558:540-6.
19. Canaud G, Gutiérrez JCL, Irvine A, et al. LBA23 EPIK-P1: retrospective chart review study of patients (pts) with PIK3CA-related Overgrowth Spectrum (PROS) who have received alpelisib (ALP) as part of a compassionate use programme. Ann Oncol. 2021;32:S1297.
20. Rosenblatt M. How academia and the pharmaceutical industry can work together: the president’s lecture, annual meeting of the American Thoracic Society, San Francisco, California. Ann Am Thorac Soc. 2013;10:31-8.
21. den Berg S, de Visser S, Leufkens HGM, Hollak CEM. Drug repurposing for rare diseases: a role for academia. Front Pharmacol. 2021;12:746987.
22. Hernandez JJ, Pryszlak M, Smith L, et al. Giving drugs a second chance: overcoming regulatory and financial hurdles in repurposing approved drugs as cancer therapeutics. Front Oncol. 2017;7:273.
23. Fetro C, Scherman D. Drug repurposing in rare diseases: myths and reality. Therapie. 2020;75:157-60.
24. Bharadwaj PR, Bates KA, Porter T, et al. Latrepirdine: molecular mechanisms underlying potential therapeutic roles in Alzheimer’s and other neurodegenerative diseases. Transl Psychiatry. 2013;3:e332.
25. Prinz F, Schlange T, Asadullah K. Believe it or not: how much can we rely on published data on potential drug targets? Nat Rev Drug Discov. 2011;10:712.
26. WHO-EURO. Repurposing of medicines - the underrated champion of sustainable innovation: policy brief. Available from: https://apps.who.int/iris/handle/10665/342567 [Last accessed on 7 Apr 2023].
27. Cha Y, Erez T, Reynolds IJ, et al. Drug repurposing from the perspective of pharmaceutical companies. Br J Pharmacol. 2018;175:168-80.
28. Conour J. Why method of treatment patents for repurposed drugs are worth the investment. Available from: https://www.jdsupra.com/legalnews/why-method-of-treatment-patents-for-92813/ [Last accessed on 7 Apr 2023].
29. Verbaanderd C, Rooman I, Huys I. Exploring new uses for existing drugs: innovative mechanisms to fund independent clinical research. Trials. 2021;22:322.
30. Léauté-Labrèze C, Dumas de la Roque E, Hubiche T, Boralevi F, Thambo JB, Taïeb A. Propranolol for severe hemangiomas of infancy. N Engl J Med. 2008;358:2649-51.
31. Drolet BA, Frommelt PC, Chamlin SL, et al. Initiation and use of propranolol for infantile hemangioma: report of a consensus conference. Pediatrics. 2013;131:128-40.
32. U.S. Department of Health and Human Services; Food and Drug Administration. Best practices in developing proprietary names for human prescription drug products guidance for industry. Available from: https://www.fda.gov/media/88496/download [Last accessed on 7 Apr 2023].
33. Léauté-Labrèze C, Hoeger P, Mazereeuw-Hautier J, et al. A randomized, controlled trial of oral propranolol in infantile hemangioma. N Engl J Med. 2015;372:735-46.
34. Phillips RJ, Penington AJ, Bekhor PS, Crock CM. Use of propranolol for treatment of infantile haemangiomas in an outpatient setting. J Paediatr Child Health. 2012;48:902-6.
35. European Medicines Agency. Paediatric-use marketing authorisations. Available from: https://www.ema.europa.eu/en/human-regulatory/marketing-authorisation/paediatric-medicines/paediatric-use-marketing-authorisations [Last accessed on 7 Apr 2023].
36. Hachem M, Bonamonte D, Diociaiuti A, Mantuano M, Teruzzi C. Cost-utility analysis of propranolol versus corticosteroids in the treatment of proliferating infantile hemangioma in Italy. PharmacoEcon Ital Res Artic. 2015:17.
37. Jadeja N. The role of IP in strategies for repurposing medicines. Available from: http://www.pinsentmasons.com/out-law/analysis/the-role-of-ip-strategies-repurposing-medicines [Last accessed on 7 Apr 2023].
38. Veldman A, Richter E, Hacker C, Fischer D. The use of off-label medications in newborn infants despite an approved alternative being available-results of a national survey. Pharmacy (Basel). 2022;10:19.
39. Ahmed F, Fattani MT, Ali SR, Enam RN. Strengthening the bridge between academic and the industry through the academia-industry collaboration plan design model. Front Psychol. 2022;13:875940.
40. European Commission. Commission Expert Group on safe and timely access to medicines for patients (“STAMP”). Available from: https://health.ec.europa.eu/medicinal-products/pharmaceutical-committee-veterinary-pharmaceutical-committee-and-expert-groups/commission-expert-group-safe-and-timely-access-medicines-patients-stamp_en [Last accessed on 7 Apr 2023].
41. European Medicines Agency. Proposal for a framework to support not-for-profit organisations and academia (institutions and individuals) in drug repurposing. Available from: https://www.ema.europa.eu/en/documents/other/question-answers-repurposing-pilot-project-proposal-framework-support-not-profit-organisations_en.pdf [Last accessed on 7 Apr 2023].
42. European Medicines Agency. Building a European innovation platform for the repurposing of medicinal products. Available from: https://ec.europa.eu/info/funding-tenders/opportunities/portal/screen/opportunities/topic-details/horizon-hlth-2021-disease-04-02 [Last accessed on 7 Apr 2023].
43. Hechtelt Jonker A, Hivert V, Gabaldo M, et al. Boosting delivery of rare disease therapies: the IRDiRC Orphan Drug Development Guidebook. Nat Rev Drug Discov. 2020;19:495-6.
44. Deplanque D, Fetro C, Ferry A, et al. Drug repurposing: from the discovery of a useful pharmacological effect to making the treatment available to the patient. Therapie. 2023;78:10-8.
45. Innovative health initiative. Available from: https://www.ihi.europa.eu/about-ihi [Last accessed on 7 Apr 2023].
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright














Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].