


Histological and biochemical methods to assess aminoacyl-tRNA synthetase expression in human post-mortem brain tissue
 0
0 
Abstract
Aminoacyl-tRNA synthetases are essential, non-redundant enzymes that catalyze the charging of tRNAs with their cognate amino acids. This reaction is a prerequisite for protein translation in all cells. Mutations in human aminoacyl-tRNA synthetases are often associated with defects of the peripheral and central nervous system and are the underlying cause of many rare diseases including neuropathies and leukodystrophies. A comprehensive understanding of aminoacyl-tRNA synthetase expression domains is key to understanding these disorders and developing novel targeted treatment strategies. Here, we describe histological and biochemical methods to analyze the expression pattern of the aspartyl-tRNA synthetase AspRS in human post-mortem brain tissue. The same methods can readily be applied to other members of the aminoacyl-tRNA synthetase superfamily or, more generally, to other cytosolic proteins in the human brain.
Keywords
INTRODUCTION
Protein synthesis is an evolutionarily highly conserved process that requires an intimate interplay of specialized ribonucleic acids (RNAs) and proteins. The importance of this process is highlighted by the many diseases resulting from mutations in genes encoding the involved proteins and RNAs[1]. Before transfer RNAs (tRNAs) can transfer amino acids to the nascent polypeptide chain, they need to be charged with their cognate amino acid, a reaction catalyzed by specific aminoacyl-tRNA synthetases (ARSs). A human cell contains 37 cytosolic and mitochondrial synthetase genes, all of which are encoded on nuclear chromosomes. The correct charging of tRNAs is essential for the fidelity of protein synthesis and there is no redundancy amongst ARSs. Consequently, dysfunctional tRNA charging is not tolerated. To date, pathological mutations that cause neurological diseases have been identified in 24 ARS genes[2]. Intriguingly, most of the pathological ARS mutations primarily manifest in deficits of the peripheral and central nervous system, including encephalopathies, Charcot-Marie-Tooth neuropathies, leukodystrophies, or cerebellar ataxia. This indicates a high susceptibility of neural cells to disturbances in protein synthesis[3].
The leukodystrophy Hypomyelination with Brain stem and Spinal cord involvement and Leg spasticity (HBSL) is caused by recessive missense mutations of the DARS1 gene encoding the cytosolic aspartyl-tRNA synthetase (AspRS)[4]. For most patients, the disease manifests from 3-36 months of age. The symptoms include leg spasticity, inability to walk unsupported or articulate, nystagmus, cognitive impairment, epilepsy, and premature death. In addition to infantile-onset patients, the disease spectrum has been broadened by the description of HBSL cases with late adolescent onset and a milder disease course[5]. Interestingly, HBSL patients present with similar symptoms as patients afflicted with Leukoencephalopathy with Brain stem and Spinal cord involvement and elevated Lactate (LBSL), which is caused by missense mutations of the DARS2 gene encoding mitochondrial AspRS[6,7]. Although the two conditions might share a similar underlying pathophysiology, cytosolic and mitochondrial AspRS are not functionally redundant, as genetic deletion of either of the two genes in mice is embryonically lethal and cannot be compensated for by the other[8,9].
The development of accurate HBSL animal models is crucial to advance the understanding of the disease mechanism and to develop targeted and effective treatments. However, the lack of redundancy amongst ARSs and the essential nature of the AspRS enzyme have made disease modelling challenging, as complete knockout of the DARS1 gene resulted in early embryonic lethality[9] and the introduction of patient-specific, disease-causing DARS1 point mutations into the mouse genome has failed to replicate the full HBSL disease phenotype[10,11]. In the absence of precise animal models, detailed knowledge of the AspRS expression pattern can provide valuable insight into etiology and help define therapeutic targets. Our recent gene expression studies using mouse[9] and human[12] brain tissue from neurologically healthy individuals revealed that, despite ubiquitously expressed in all cells, the highest AspRS levels were present in neuronal lineage cells with comparably little immunoreactivity present in oligodendrocytes, astrocytes, and microglia. Anatomically, AspRS expression was highly enriched in the cerebellum, a region responsible for motor control and particularly affected in HBSL patients. Here, we describe in detail the histological and biochemical methods used to analyze the AspRS expression pattern in human post-mortem brain tissue. These protocols can readily be adapted to characterize other members of the ARS protein family or, more broadly, other cytosolic proteins in the human brain.
MATERIALS
Post-mortem brain tissue
Human post-mortem brain tissue was provided by the New South Wales Brain Bank (project no. PID391). All procedures were approved by the UNSW Sydney Human Research Ethics Advisory Panel D (approval number: HC16223). The tissue samples were taken from five male subjects aged between 55 and 57 years who died from cardiovascular disease. Individuals did not suffer from neurological diseases and the brain tissue was free from overt brain pathology. The post-mortem interval, which is the time between the death of the person and the time point at which the tissue was taken, ranged between 12 and 39 h. Brain regions analyzed included motor cortex, hippocampus, cerebellum, and brainstem (pons). Frozen tissue blocks were used for immunoblotting and quantitative real-time PCR (qRT-PCR), and formalin-fixed, paraffin-embedded, 4 µm thin sections were used for immunohistochemistry. Sections from the motor cortex, hippocampus, and cerebellum were cut in the coronal plane; sections from the brainstem were cut transversally.
Antibodies
The following primary antibodies were used for immunoblotting: mouse anti-AspRS (monoclonal; Santa Cruz Biotechnology #sc-393275; 1:500 dilution), which is specific for amino acids 170-467 of the human AspRS protein; rabbit anti-AspRS (polyclonal; Novus Biologicals #NBP1-85937; 1:500 dilution), which is specific for amino acids 1-135 of the human AspRS protein; rabbit anti-GAPDH (Cell Signaling #2118S; 1:5,000 dilution); mouse anti-β-Actin clone C4 (Sigma-Aldrich #mab1501; 1:10,000 dilution). Goat anti-mouse and goat anti-rabbit HRP-conjugated secondary antibodies were obtained from Dianova
For immunohistochemistry, the following primary antibodies were used: mouse anti-AspRS (monoclonal; Santa Cruz Biotechnology #sc-393275; 1:50 dilution) and rabbit anti-NeuN (Cell Signaling #12943S;
TaqMan assays
The following TaqMan assays were used for qRT-PCR: DARS1 (Applied Biosystems #Hs00154683_m1) and β-Actin (Applied Biosystems #Hs01060665_g1).
METHODS
Sample preparation for Western blotting and qRT-PCR
Before protein and RNA lysates can be prepared for Western blotting and qRT-PCR, respectively, the frozen human brain tissue from individual brain regions needs to be homogenized in liquid nitrogen using mortar and pestle until a fine powder is obtained. It is important to keep the tissue in liquid nitrogen at all times during the homogenization process. When homogenization is complete and all liquid nitrogen has evaporated from the mortar, the tissue powder is distributed into two Eppendorf tubes. The tubes are weighed before and after filling to determine the total tissue weight. Homogenized tissue can be kept on dry ice for further downstream applications or stored long-term at -80 °C. One tube is used to prepare a protein lysate for Western blot analysis; the second tube is used for RNA isolation for qRT-PCR.
Western blotting
For Western blotting, the homogenized tissue powder is resuspended in 10 µL lysis buffer [50 mM Tris-HCL pH 7.4, 1 mM EDTA pH 8.0, 250 mM NaCl, 1% v/v Triton-X, and protease inhibitors (Roche Complete #04693124001)] per 1 mg of tissue weight. Following sonication using a probe sonicator (Branson Digital Sonifier 450) at 10% sonication amplitude for 1 min, the tissue suspension is spun down at full speed in a conventional benchtop centrifuge and the supernatant is transferred to a new tube. The protein concentration of the lysates is determined by Bradford protein assay using Quick Start Bradford 1x Dye Reagent (Bio-Rad #500-0205). 30 µg of protein from each brain region is mixed with five times sample buffer (15 g SDS, 15.6 mL 2M Tris pH 6.8, 57.5 g glycerol, 16.6 mL β-mercaptoethanol) and loaded onto a 10% acrylamide gel (Bio-Rad Mini-PROTEAN gel system). Following separation by SDS-PAGE, proteins are transferred onto a PVDF membrane (Bio-Rad #162-0177). To avoid unspecific antibody binding, the PVDF membrane is incubated in blocking solution (4% milk powder and 0.1% Tween in PBS) for at least one hour at room temperature. Primary antibodies are then applied in blocking solution overnight at 4 °C. Following three 10 min washing steps with PBS-T (0.1% Tween in PBS), the membrane is probed with secondary HRP-coupled antibodies in blocking solution for 1 h at room temperature and washed three times again in PBS-T, each 10 min. Afterwards, the membrane is developed using Clarity Western ECL substrate (Bio-Rad #170-5060) and subsequently imaged employing the Bio-Rad ChemiDoc MP imaging system. Protein bands are quantified using the densitometry function in ImageJ and AspRS protein levels in different brain regions are normalized to the housekeeping proteins β-Actin and GAPDH. A comprehensive AspRS expression study comparing protein levels across different regions of the human brain was conducted following the protocol described here[12]. This study found significantly higher AspRS levels in the cerebellum compared to the other brain regions analyzed.
Quantitative real-time PCR
For qRT-PCR analysis, the tissue powder is resuspended in 350 µL (for up to 20 mg of tissue) or 600 µL (for 20-30 mg of tissue) buffer RLT (from the Qiagen RNeasy MiniKit #74106) plus 1% β-mercaptoethanol and homogenized using a pellet pestle attached to the Kontes pellet pestle motor. Afterwards, the lysate is centrifuged for 3 min at full speed in a conventional benchtop centrifuge and the supernatant is transferred to a fresh tube. RNA extractions are performed using the RNeasy MiniKit (Qiagen #74106) following the manufacturer’s instructions including an on-column DNase digest using the RNase-free DNase kit (Qiagen #79254) to get rid of any genomic DNA. At the end of the extraction procedure, the RNA is eluted from the column in 50 µL RNase free water and the concentration is determined using the NanoDrop microvolume spectrophotometer. An equal amount of RNA from each brain region is used for cDNA synthesis using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems #4368813) in a conventional thermocycler. Afterwards, qRT-PCR can be performed in any suitable real-time PCR machine employing specific TaqMan probes. Data is analyzed using the comparative ΔΔCT method for relative quantification of DARS1 expression normalized to the housekeeping gene β-Actin. Using this method, DARS1 mRNA levels were analyzed across different human brain regions, revealing a significant enrichment in the cerebellum in line with the AspRS protein data[12].
Immunohistochemistry
Formalin-fixed and paraffin-embedded sections were received on glass slides from the New South Wales Brain Bank. The sections were cut at 4 μm thickness to ensure adequate penetration of antibodies. Brain sections included the motor cortex, hippocampus, and cerebellum, which were all cut in the coronal plane, and sections from the brainstem (pons), which were cut in the transverse plane.
Sections for immunofluorescent labelling [Figure 1] are incubated in a 60 °C oven for 2 h to melt the paraffin surrounding the tissue before placing the slides in xylene for 2 × 10 min to ensure complete solubilization and removal of residual paraffin. The slides are then rehydrated for 2 × 5 min in decreasing concentrations of ethanol in water, starting at 100% ethanol before moving to 96% and 70% ethanol. Finally, the slides are placed in water for a period of 5 min. The deparaffinization and rehydration process is critical to unmask epitopes for immunohistochemical labelling. To further assist the access of antibodies to antigens in the tissue, antigen retrieval can be performed. Different forms of antigen retrieval methods are described in the literature. In our case, citrate buffer antigen retrieval worked best. For antigen retrieval, slides are incubated in 10 mM citrate buffer (1.8 μM citric acid and 8.2 μM sodium citrate tribasic dihydrate in water) and placed in an RHS-1 Microwave Vacuum Histoprocessor (Milestone Medical). The microwave heats the solution to 120 °C and maintains this temperature for an additional 1 min. Slides are then removed from the microwave and allowed to cool down to room temperature. This step is critical to minimize excess background signal. Slides are washed in PBS for 5 min prior to permeabilization with 0.5% TritonX-100 for 3 min. Although permeabilization with TritonX-100 is not critical, our study found that this further assisted in revealing epitopes for antibody staining. Slides are then placed in a Sequenza slide rack (Thermo Scientific), washed 3 times with PBS and blocked in 10% normal goat serum in PBS for 60 min. Primary antibodies are applied in PBS + 10% normal goat serum overnight at 4 °C. Following three washing steps with PBS, fluorophore-coupled secondary antibodies are applied in PBS + 10% normal goat serum for 1 h at room temperature. Slides are washed 3 times with PBS, incubated for 5 min with DAPI and washed again with PBS. Subsequently, slides are washed in 70% ethanol for 5 min and then a few drops of Autofluorescence Eliminator reagent (Millipore #2160) are added directly to the tissue for 3 min to reduce autofluorescence. This step is critical because perfusion of human tissue cannot be performed prior to tissue fixation, potentially resulting in strong autofluorescence signals. Slides are then washed in 70% ethanol to remove residual Autofluorescence Eliminator reagent before being washed in PBS for 5 min. Slides are mounted in ProLong Gold antifade reagent (Thermo Fischer #P101444) as this helps to preserve the fluorescent signal. Fluorescent images can be taken with any suitable confocal microscope (in our study, a Zeiss LSM710 was used). Representative immunofluorescent images depicting AspRS and NeuN (neuronal marker) immunoreactivity are shown in Figure 1. For a detailed analysis of the AspRS expression pattern in human brain tissue, including co-labelling with neuronal, oligodendroglial, astroglial, and microglial markers, please refer to Fröhlich et al., 2018[12].
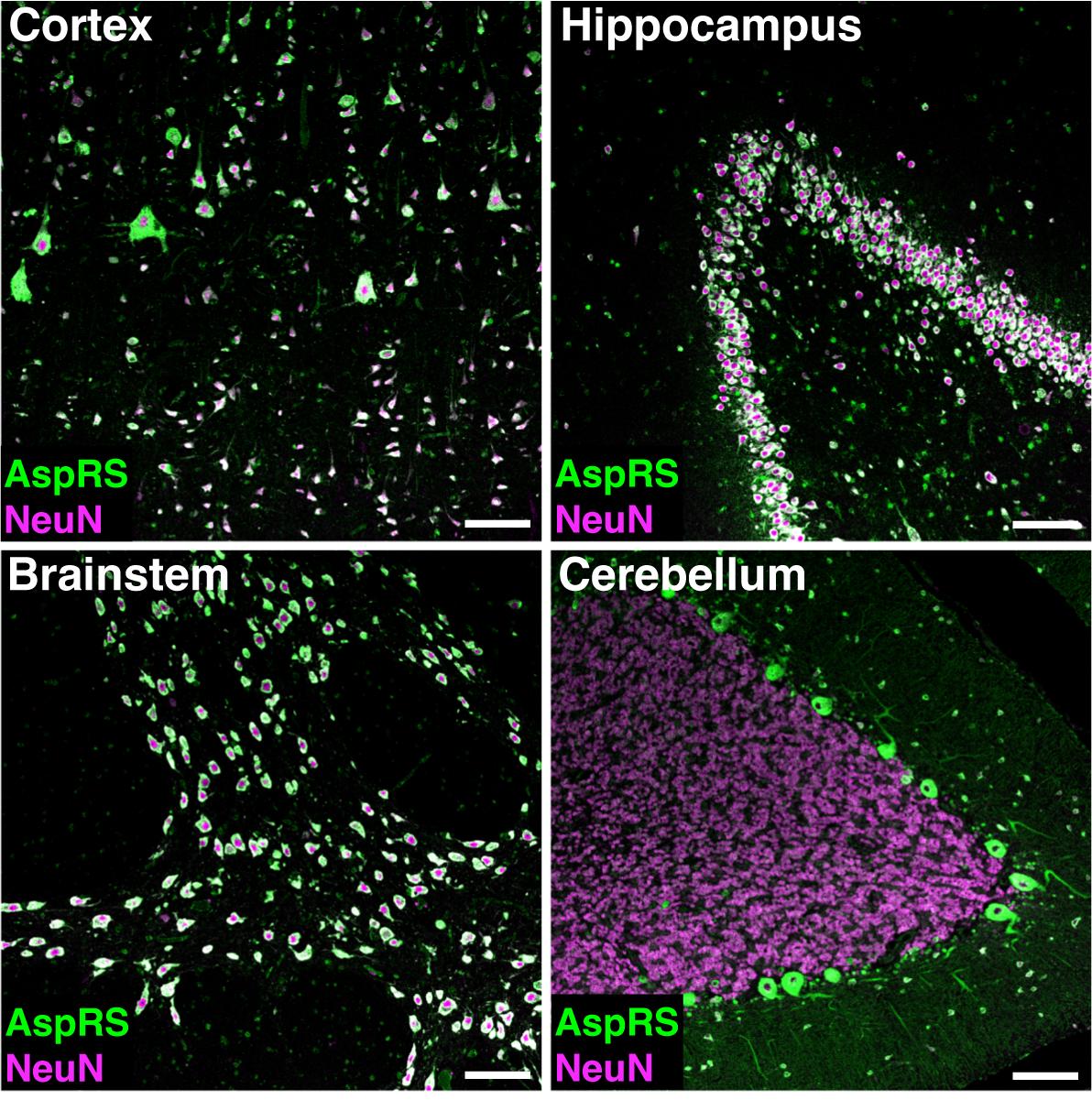
Figure 1. Representative immunofluorescent images showing co-labelling of AspRS (green) and the neuronal cell-type marker NeuN (magenta) in human post-mortem brain sections. Regions displayed include motor cortex, hippocampus, brainstem (pons), and cerebellum. Scale bar: 100 µm.
For immunoperoxidase labelling [Figure 2] with Diaminobenzidine (DAB), the brain sections are dewaxed and rehydrated as described above. After citrate antigen retrieval, sections are incubated with 4%
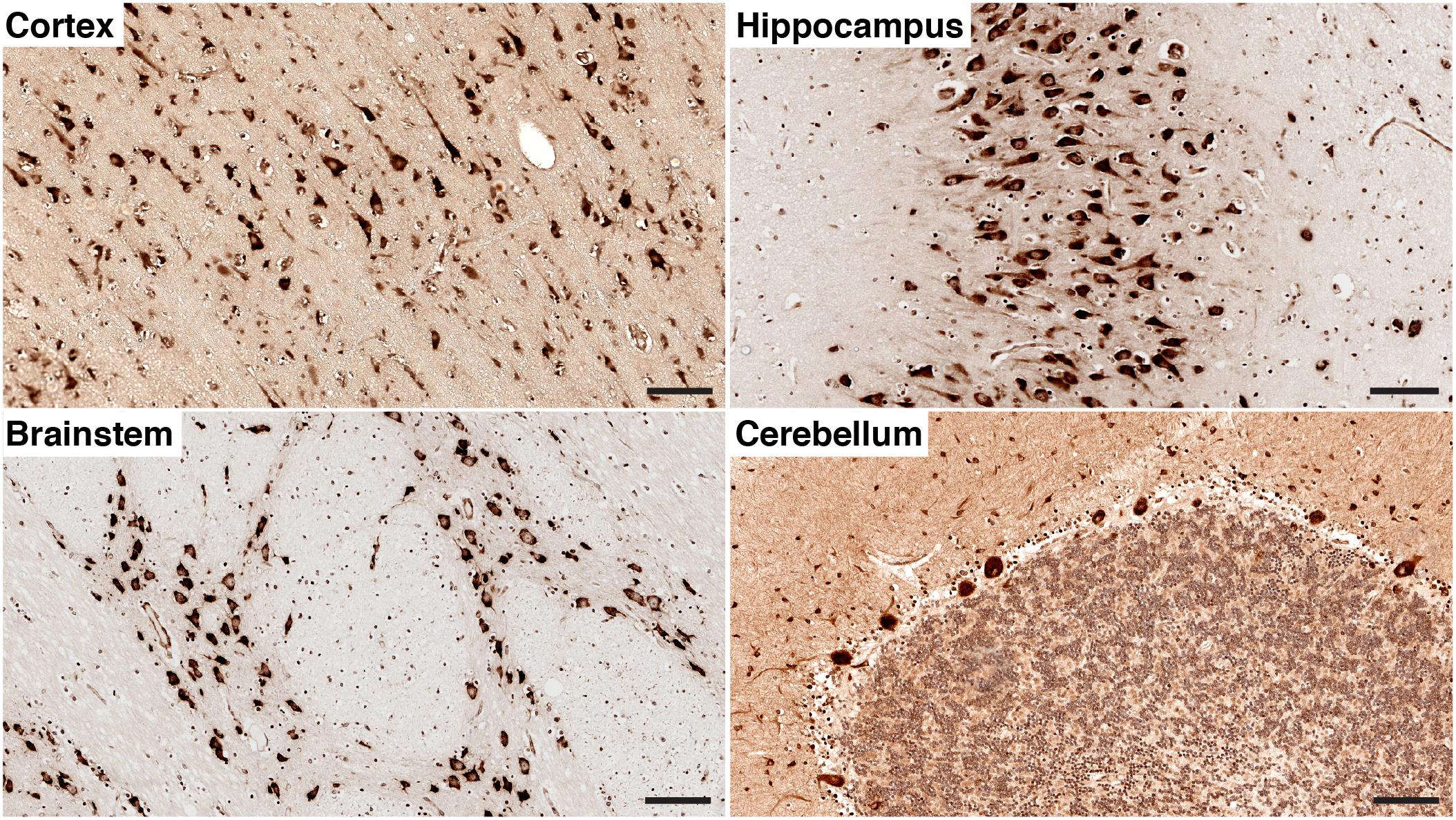
Figure 2. Representative chromogenic detection of AspRS by immunohistochemical labelling with Diaminobenzidine (DAB) in the motor cortex, hippocampus, brainstem (pons), and cerebellum of the human brain. Scale bar: 100 µm.
TECHNICAL NOTES
Western blotting
The homogenization method used for protein extraction may vary depending on the kind and amount of tissue. For brain tissue, we found that a combination of sonication and grinding in liquid nitrogen works best. When grinding using mortar and pestle, make sure that the sample remains covered in liquid nitrogen to avoid thawing of the tissue. Before the pulverized tissue is transferred into an Eppendorf tube, ensure that all liquid nitrogen has evaporated from the mortar. Any liquid nitrogen transferred into an enclosed reaction tube can be hazardous as the tube might explode when warming up. During the subsequent sonication step, all cells are lysed by liquid cavitation. An advantage of sonication over other protein extraction methods is that DNA is also sheared during the process and there is no need to treat the sample with DNase. To avoid protein degradation, it is important to keep the samples on ice during sonication.
Choosing the right housekeeping protein is critical for normalization of protein levels, as some housekeepers might be regulated in certain disease conditions or tissues. Normalization against two different housekeeping proteins should yield comparable results and is a good control for the validity of the housekeeper.
Unspecific binding of an antibody - the binding of an antibody to non-target antigens, is easier to identify in Western blotting compared to immunohistochemistry as the molecular weight at which the protein of interest is detected is a strong indication of antibody specificity. Detection of multiple bands on a Western blot can indicate non-specific binding, which needs to be considered if the same antibody were to be used in immunohistochemistry. If uncertain of antibody specificity, peptide blocking should be performed to confirm the validity of the primary antibody. For this, the antibody is pre-absorbed with a blocking peptide corresponding to the epitope of the antibody, which will prevent the binding of the target antigen and consequently reduce signal intensity compared to non-blocked antibodies.
Quantitative real-time PCR
For RNA work, it is essential to keep the workplace and reagents free of RNases to avoid degradation of the RNA. It is recommended to perform all RNA work in a laminar flow hood.
Similar to protein extraction, tissue homogenization for RNA preparation is performed in two steps. Following liquid nitrogen grinding, cells are lysed using a pellet pestle homogenizer instead of a probe sonicator to avoid shearing of the RNA.
In our study, off-the-shelf TaqMan assays worked best as they provide species-specificity and low unspecific binding. Selection of the appropriate TaqMan assay should take into account that the primers are exon-exon junction spanning to avoid detection of genomic DNA. If analyzing human brain tissue from a carrier of a known ARS mutation, it is essential to select primers and TaqMan probes that anneal to cDNA sequences unaffected by the respective mutation. The specificity of qRT-PCR primer pairs or TaqMan assays should always be validated through sequencing of the amplicon.
Immunohistochemistry
Deparaffinization and rehydration of paraffin-embedded sections are critical to unmask epitopes for immunohistochemical labelling. For some antibodies, it is necessary to perform additional antigen retrieval to further assist the access of the antibody to the antigen. Which antigen retrieval method should be employed depends on the antibody used, and it is worthwhile testing them before the study commences. In our study, citrate antigen retrieval yielded the best results.
To avoid unspecific antibody binding, blocking should be performed in the serum of the host species of the secondary antibody. If this is not possible, we generally have had good experiences using normal goat serum.
For immunofluorescent labelling of antigens, we achieved the best results employing secondary antibodies coupled with Alexa fluorophores.
Human tissue samples inherently give rise to a lot of background signals, as there is no perfusion of tissue prior to fixation, resulting in autofluorescence. There are numerous contributing factors including collagen or elastin, which often illuminate blood vessels in the tissue. Historically, Sudan Black has been used to reduce autofluorescence; however, this also dampens the true fluorescent signal. We have found that the Autofluorescence Eliminator reagent (Millipore #2160) works well on human tissue.
When choosing the most appropriate antibody, it is important to consider the specific advantages and disadvantages of polyclonal versus monoclonal antibodies[13]. Polyclonal antibodies are produced by injecting an animal with the desired antigen to elicit an immune response and produce antibodies against the antigen. Polyclonal antibodies can recognize multiple epitopes of the antigen, which often results in a higher overall affinity and sensitivity to the target protein. The same multi-epitope specificity, however, can result in cross-reactivity with other proteins leading to increased unspecific binding and background staining. In contrast, monoclonal antibodies are produced in vitro by a single B cell clone and recognize only one specific epitope. While this might result in lower overall sensitivity, monoclonal antibodies often show higher specificity with reduced cross-reactivity and, consequently, less background signal. If working with brain tissue from patients affected by a known ARS mutation, a monoclonal antibody that recognizes an epitope outside the mutated amino acid sequence should be used. Alternatively, a polyclonal antibody can be used to ensure multiple epitopes are recognized.
Studies involving antibody-based methods should always include appropriate controls for the specificity of antibodies, such as secondary-only stainings. A comprehensive guideline for the validation of antibodies for use in immunohistochemistry has been developed by Howat et al.[14]. Determination of antibody specificity is crucial in studies on human tissue when genetic manipulations such as overexpression or knockdown via RNA interference of the protein of interest cannot be performed to confirm the validity of an antibody. If positive or negative human control tissue with confirmed up- or downregulated target protein is available, this can be used for establishing antibody specificity. Western blotting can give a first indication if an antibody is specific or not (see above); however, it does not always guarantee antibody specificity in immunohistochemistry applications where the tissue has undergone different processing steps, including fixation and antigen retrieval. A good indication for the specificity of an antibody can also be obtained by ectopically expressing the human protein of interest in a cell culture model such as HEK293 cells with subsequent immunocytochemistry. Finally, peptide blocking, as described above, can be employed to rule out non-specific binding in immunohistochemistry. If the above controls are not possible or yield inconclusive results, orthogonal methods such as in situ hybridization of the corresponding mRNA should be applied to support the antibody-based data.
DECLARATIONS
Acknowledgements
The authors thank the New South Wales Brain Bank for providing the human tissue samples.
Authors’ contributions
Led the project and manuscript preparation: Klugmann M, Fröhlich D
Conducted the research: Klugmann M, Suchowerska AK, Housley GD, Fröhlich D
All authors contributed to and approved the final version of the manuscript.
Availability of data and materials
Not applicable.
Financial support and sponsorship
This work was supported by the European Leukodystrophy Association (ELA 2018-014I2) and the Australian Government Medical Research Future Fund (Leukodystrophy Flagship - Massimo’s Mission; MRFF-ARLKO).
Conflicts of interest
Klugmann M was employed by the company Boehringer Ingelheim Pharma GmbH & Co. KG, while the other authors have declared that they have no conflicts of interest.
Ethical approval and consent to participate
All post-mortem human brain tissue used in this study was provided by the New South Wales Brain Bank (project no. PID391). The study was approved by the UNSW Sydney Human Research Ethics Advisory Panel D (approval number: HC16223). All donors, or their next of kin, provided written informed consent for the use of brain tissue in research, in accordance with the guidelines of the NSW Brain Bank. All tissue samples were de-identified before analysis to protect donor privacy.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2023.
REFERENCES
2. Ognjenović J, Simonović M. Human aminoacyl-tRNA synthetases in diseases of the nervous system. RNA Biol. 2018;15:623-34.
3. Antonellis A, Green ED. The role of aminoacyl-tRNA synthetases in genetic diseases. Annu Rev Genomics Hum Genet. 2008;9:87-107.
4. Taft RJ, Vanderver A, Leventer RJ, et al. Mutations in DARS cause hypomyelination with brain stem and spinal cord involvement and leg spasticity. Am J Hum Genet. 2013;92:774-80.
5. Wolf NI, Toro C, Kister I, et al. DARS-associated leukoencephalopathy can mimic a steroid-responsive neuroinflammatory disorder. Neurology. 2015;84:226-30.
6. Muthiah A, Housley GD, Klugmann M, Fröhlich D. The leukodystrophies HBSL and LBSL-correlates and distinctions. Front Cell Neurosci. 2020;14:626610.
7. Scheper GC, van der Klok T, van Andel RJ, et al. Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat Genet. 2007;39:534-9.
8. Dogan SA, Pujol C, Maiti P, et al. Tissue-specific loss of DARS2 activates stress responses independently of respiratory chain deficiency in the heart. Cell Metab. 2014;19:458-69.
9. Fröhlich D, Suchowerska AK, Spencer ZH, et al. In vivo characterization of the aspartyl-tRNA synthetase DARS: homing in on the leukodystrophy HBSL. Neurobiol Dis. 2017;97:24-35.
10. Fröhlich D, Mendes MI, Kueh AJ, et al. A hypomorphic Dars1D367Y model recapitulates key aspects of the leukodystrophy HBSL. Front Cell Neurosci. 2020;14:625879.
11. Klugmann M, Kalotay E, Delerue F, et al. Developmental delay and late onset HBSL pathology in hypomorphic Dars1M256L mice. Neurochem Res. 2022;47:1972-84.
12. Fröhlich D, Suchowerska AK, Voss C, et al. Expression pattern of the aspartyl-tRNA synthetase DARS in the human brain. Front Mol Neurosci. 2018;11:81.
13. Lipman NS, Jackson LR, Trudel LJ, Weis-Garcia F. Monoclonal versus polyclonal antibodies: distinguishing characteristics, applications, and information resources. ILAR J. 2005;46:258-68.
Cite This Article

How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].