


Audiological findings in an Indian child with Johanson-Blizzard syndrome: a case report

Abstract
Johanson-Blizzard syndrome (JBS) is a rare autosomal recessive disorder characterized by multi-system involvement and facial dysmorphic features. Sensorineural hearing loss is one of the most common manifestations of this pathology. Detailed audiological evaluation in confirmed cases of JBS is essential for the appropriate management of hearing loss. We present the audiological features of a six-year-old Indian girl with Johanson-Blizzard syndrome along with the less emphasized association of café-au-lait spots with JBS.
Keywords
INTRODUCTION
Johanson-Blizzard syndrome (JBS) is a rare autosomal recessive disorder, first described in 1971 by Johanson and Blizzard. It is a very rare multi-system disorder, comprising a wide range of abnormalities including exocrine pancreatic insufficiency, aplasia or hypoplasia of the alae nasi, scalp defects, developmental delay, microcephaly, mental retardation, absence of permanent teeth, sensorineural hearing loss, hypothyroidism, urogenital malformations, imperforate anus, and congenital heart defects[1-3]. The UBR1 gene located on chromosome 15q15.2 is currently the only gene associated with JBS[4]. As per the National Organization of Rare Disorders, JBS affects males and females in equal numbers. Although the exact incidence is unknown, the frequency has been estimated to be 1 in 250,000 births[5] with no reported gender differences. However, no reports on the incidence and prevalence of JBS in the Indian context are available. One of the most common manifestations of this pathology is sensorineural hearing loss (SNHL) of different severity and anomalous development of the inner ear. Sensorineural hearing loss occurs in more than 80% of cases of JBS[6].
There is a dearth of literature reporting audiological findings on JB syndrome. The present case report is one of the first attempts to report the audiological features of an Indian child with JBS with a rare association of café-au-lait spots.
CASE REPORT
The case presented in this report is a six-year-old girl who was referred for detailed audiological evaluation and further management with a genetically confirmed diagnosis of Johanson-Blizzard syndrome. She presented with not hearing and not speaking age appropriately. Her history and medical records revealed she was born out of a consanguineous union and a full-term normal delivery with an immediate birth cry and a birth weight of 2.45 kg. She suffered from secondary apnea requiring resuscitation and neonatal intensive care unit stay for the same. She had no family history of any major medical or genetic disorder. She had clinodactyly of fifth fingers bilaterally and her echocardiogram revealed an atrial-septal defect with persistent ductus arteriosus. She was reported to have normal TSH and a normal 46, XX karyotype. Homozygous mutation of UBR1 gene was detected through molecular genetic testing, thus confirming the diagnosis of Johanson-Blizzard syndrome. She failed the otoacoustic emission screening at the age of 12 days and was reported to have sensorineural hearing loss confirmed by brainstem evoked response audiometry (BERA) at a different medical center.
At the age of three months and six days, she presented to our institute for a detailed audiological evaluation. Her hearing evaluation revealed bilateral severe to profound sensorineural hearing loss confirmed by BERA. Aided behavioral observation audiometry was carried out, and, based on the aided responses and real-ear measurements; the child was fitted with conventional hearing aids bilaterally. The child was recommended for auditory verbal therapy; however, she did not follow up.
The case was lost to follow-up for five years. She reported to our institute again at the age of five years six months to seek admission to the special school for children with hearing impairment on our institute premises and was referred for detailed audiological evaluation as a protocol for school admission. Her mode of communication was mainly by pointing and responding to simple commands by gestures. She had striking facies with a small beak-like nose with hypoplastic nasal alae, absence of permanent teeth, and café-au-lait spots were observed on legs and few spots on hands [Figure 1].
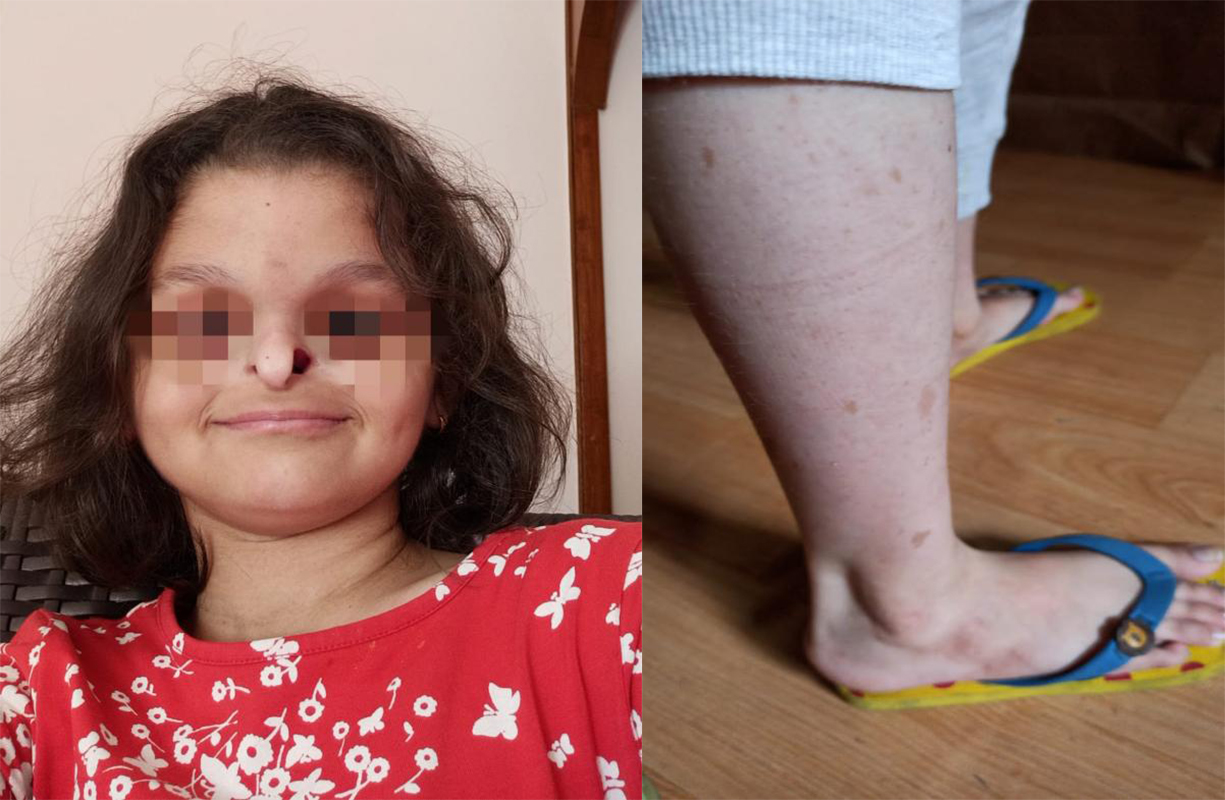
Figure 1. Beak-like nose with hypoplastic nasal alae and café-au-lait spots observed on legs.
A thorough audiological evaluation was carried out. Otoscopic examination revealed the presence of a bilateral cone of light. The middle ear analysis was done using tympanometry, using GSI Tympstar Pro-GSI Grason Stadler with a 226 Hz probe tone, which revealed normal middle ear functioning with bilateral “A” type tympanogram. Reflexometry obtained bilaterally to ipsilateral and contralateral stimulation showed absent acoustic reflexes on ipsilateral and contralateral recording [Figure 2]. Pure tone audiometry (PTA) carried out using a GSI-61 audiometer revealed severe hearing loss in the right ear and profound hearing loss in the left ear. Similarly, there was no response to bone conduction stimulation of the right ear from 250 to 4000 Hz [Figure 3]. Distortion product otoacoustic emissions (DPOAEs) were recorded bilaterally using an ILO V6 instrument with a frequency range of 1000-8000 Hz. DPOAEs were absent bilaterally across all the frequencies, suggestive of outer hair cell dysfunction in both ears.
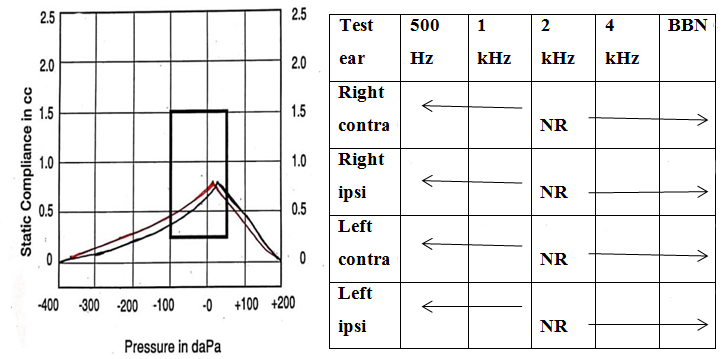
Figure 2. Immittance showing bilateral “A” type of tympanogram with absent acoustic reflexes.
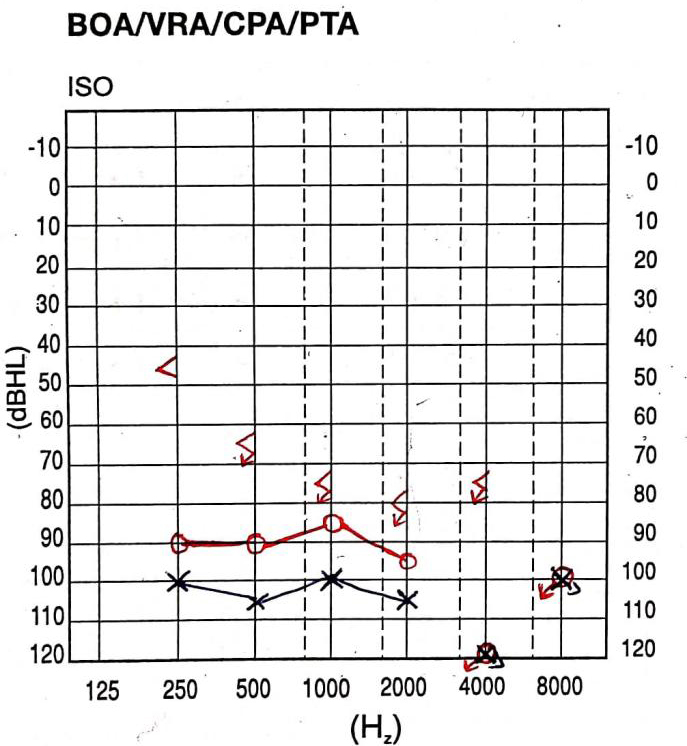
Figure 3. Pure tone audiometry (PTA) showing severe sensorineural hearing loss in the right ear and profound hearing loss in the left ear.
Auditory brainstem responses (ABR) were tracked using clicks, 500 Hz tone burst stimuli using Biologic Navigator Pro version 7.2. Subsequently, bilateral absence of Wave V at 90 dBnHL for rarefaction click and 500 Hz tone burst stimuli was observed. Bone conduction ABR revealed an absence of all ABR waves at 55 dBnHL for alternating click stimuli [Figure 4]. Ear specific aided responses were out of the speech spectrum [Figure 5]. Speech perception assessed through the early speech perception test was fair in audiovisual mode and poor in auditory mode alone, indicating no benefit from conventional hearing aids; hence, she was recommended for cochlear implantation.
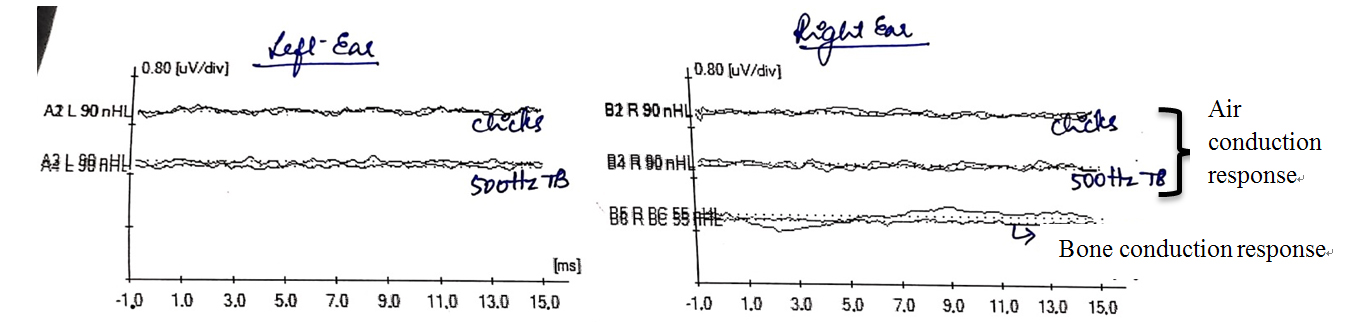
Figure 4. Auditory brainstem response (ABR) showing absence of Waves I, III, and V in response to clicks air conduction and bone conduction stimuli and 500 Hz tone burst stimuli.
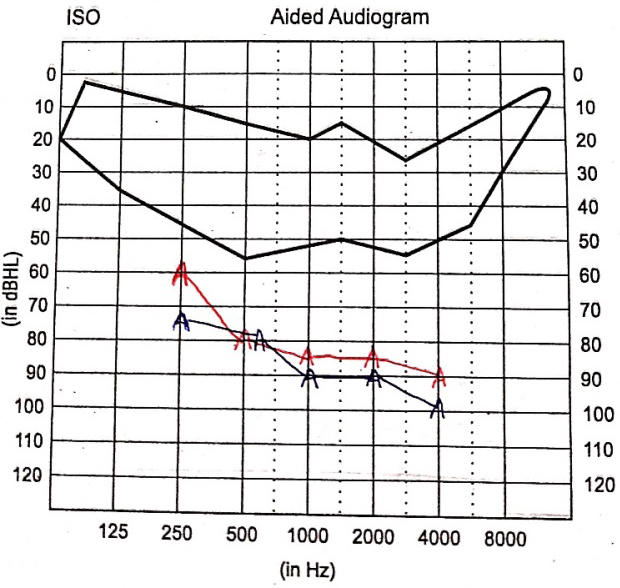
Figure 5. Aided audiogram showing ear specific hearing thresholds with hearing aids bilaterally. Red A: Right ear aided thresholds; Blue A: left ear aided thresholds.
Radiological investigations (CT and MRI brain and temporal bones) were carried out, which revealed a common cystic cavity of the apical and middle turn of bilateral cochlea with normal basal turn and enlarged vestibule typical of Mondini dysplasia.
Detailed speech, language, feeding, and psychological evaluations were carried out. Speech and language evaluation based on Gestural Scale for Hard of Hearing (GSHOH) revealed a delay of one year in reception and three years in expression. Receptive Expressive Emergent Language Skills (REELS) revealed receptive language skills delayed by 4 years and expressive language skills delayed by 3.5 years. Speech and Language Development Chart (SLDC) also was performed, revealing delayed speech and language skills.
Detailed feeding assessment revealed dysphagia in oral phase of swallowing. Psychological evaluation based on Seguin Form Board test (SFB) of intelligence revealed dull-normal level of intellectual functioning (IQ 79) secondary to hearing impairment.
DISCUSSION
The outcomes of this study show the importance of doing a systematic and comprehensive auditory, speech, language, and psychological profiling in children with JBS, which aid in tailoring the management. Facial dysmorphism is one of the constant signs required to make a diagnosis of JBS. A small beak-like nose with hypoplastic nasal alae, absence of permanent teeth, and café-au-lait spots were observed in the present case. The association of café-au-lait spots with JBS is not emphasized in the literature[1,2]. Although Johanson and Blizzard[1] originally reported this in the second case, other authors have not consistently reported this. The presence of a significant number of café-au-lait spots in the present case is an interesting, rare association noted with JBS.
The audiological test battery also revealed that deficits in JBS present themselves in the form of hearing loss. One of the most common manifestations of JBS is sensorineural hearing loss of different severity and anomalous development of the inner ear[7]. The combined results of otoscopy and immittance evaluations rule out any pathology of the outer and middle ear, pointing to inner ear dysfunction. Results from PTA and BERA indicate bilateral sensorineural hearing loss. Our findings are consistent with those of Rosanowoski et al., Ramos et al., and Rawan et al., who reported bilateral severe to profound sensorineural hearing loss as one of the phenotypes of JBS[8-10].
The current study’s findings are consistent with a number of syndromic hearing loss cases involving the inner ear. Otic capsule (inner ear), nasal alae, and mandible are ectodermal in origin[11]. Thus, aplastic nasal alae and Mondini dysplasia along with the presence of severe to profound SNHL can be related manifestations of ectodermal dysplasia.
The use of amplification devices such as hearing aids is recommended to manage speech and language manifestations caused by the disorder in mild to severe SNHL of syndromic origin[8,12]. Despite the use of hearing aids, the aided responses, in this case, were unsatisfactory due to the presence of bilateral Mondini dysplasia.
Several studies have reported severe mental retardation in cases with JBS[13], while only a few studies have reported normal intelligence in cases with JBS[14,15]. However, the child reported in our study had dull-normal intellectual functioning. The observed delay in speech and language development can be attributed to multiple system involvement, delayed global development, and dull-normal intellectual ability, all of which had a detrimental effect on her language development.
In conclusion, the current study is one of the first to report JBS in an Indian child with systematically reported audiological, speech, and language features and café-au-lait spots, a rare and less emphasized association with JBS. The findings of the current study show compromised auditory function in JBS. It also points to the necessity for the involvement of an allied professional team to effectively screen, counsel, and rehabilitate children with JBS before they cross the critical age, which can help to speed up both rehabilitation and audiological outcomes.
DECLARATIONS
Acknowledgment
The authors would like to thank the family for their kind cooperation.
Author’s contributions
Conception or design of the work: Rashmi Bhat, Deena Priya
Data collection: Deena Priya
Data analysis and interpretation: Deena Priya
Drafting the article: Deena Priya, Ghanta Hinduja, Sharanya S
Critical revision of the article: Rashmi Bhat, Deena Priya
Final approval of the version to be published: Deena Priya, Rashmi Bhat, Ghanta Hinduja, Sharanya S
Availability of data and materials
All data supporting the findings of this study are available within the paper.
Financial support and sponsorship
Not applicable.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Ethical approval was waived by The Bangalore Speech and Hearing Research Foundation EthicsCommittee (BSHRF). Case reports and case series involve a small number of human participants, but are notconsidered research subjects, based on the premise that they do not involve researchobjectives and a corresponding protocol, for BSHRF consideration. There being noresearch protocol, case reports are outside the scope of review conducted by BSHRF. Written informed consent to participate from the patients was obtained.
Consent for publication
Consent for publication from the patients was obtained.
Copyright
© The Author(s) 2022.
REFERENCES
1. Johanson A, Blizzard R. A syndrome of congenital aplasia of the alae nasi, deafness, hypothyroidism, dwarfism, absent permanent teeth, and malabsorption. J Pediatr. 1971;79:982-7.
2. Gershoni-Baruch R, Lerner A, Braun J, Katzir Y, Iancu TC, Benderly A. Johanson-Blizzard syndrome: clinical spectrum and further delineation of the syndrome. Am J Med Genet. 1990;35:546-51.
5. NORD. Synonyms of Johanson-Blizzard syndrome. Available from: https://rarediseases.org/rare-diseases/johanson-blizzard-syndrome/ [Last accessed on 24 May 2022].
6. Holcomb MA, Rizk HG, Morris NS, Meyer TA. Bilateral cochlear implantation in a child with Johanson Blizzard syndrome. Int J Pediatr Otorhinolaryngol. 2017;95:69-71.
7. Chugunova TI, Bakhshinian VV, Markova TG, Goikhburg MV, Zherenkova VV. Johanson-Blizzard syndrome: audiological features and results of cochlear implantation. Vestn Otorinolaringol. 2014;(2):90-2.
8. Rosanowski F, Hoppe U, Hies T, Eysholdt U. [Johanson-Blizzard syndrome. A complex dysplasia syndrome with aplasia of the nasal alae and inner ear deafness]. HNO. 1998;46:876-8.
9. Ramos S, Ramos HF, Ramos RF, Peixoto CA, Ramos BF. Johanson-Blizzard syndrome. Braz J Otorhinolaryngol. 2010;76:794.
10. Alwadee RM, Alyousef MY, Yousef EM, Yousef MF. Performance of children with Johanson-Blizzard syndrome after cochlear implantation. Cureus. 2021;13:e19264.
11. Northern JL, Downs MP. Hearing in children. 5th ed. PA: Lippincott Williams & Wilkins; 2002. Available from: https://books.google.com.hk/books?hl=zh-CN&lr=&id=1CUFXsBbBgoC&oi=fnd&pg=PR5&dq=Northern+JL,+Downs+MP.+Hearing+in+children.+5th+ed.+PA:+Lippincott+Williams+%26+Wilkins%3B+2002.&ots=HXI24jmFCf&sig=NbqrT2ITMFZL5FPRENwfBgiUy9M&redir_esc=y#v=onepage&q=Northern%20JL%2C%20Downs%20MP.%20Hearing%20in%20children.%205th%20ed.%20PA%3A%20Lippincott%20Williams%20%26%20Wilkins%3B%202002.&f=false [Last accessed on 24 May 2022].
12. Aubrey S, Crooks B, Rashid M. Pancytopenia from severe cobalamin (vitamin B12) deficiency in Johanson-Blizzard syndrome. Eur J Clin Nutr. 2013;67:1118.
13. Zenker M, Mayerle J, Lerch MM, et al. Deficiency of UBR1, a ubiquitin ligase of the N-end rule pathway, causes pancreatic dysfunction, malformations and mental retardation (Johanson-Blizzard syndrome). Nat Genet. 2005;37:1345-50.
14. Atik T, Karakoyun M, Sukalo M, Zenker M, Ozkinay F, Aydoğdu S. Two novel UBR1 gene mutations ın a patient with Johanson Blizzard Syndrome: a mild phenotype without mental retardation. Gene. 2015;570:153-5.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright














Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].