
Viral sequencing to inform the global elimination of dog-mediated rabies - a systematic review

 , , ...
, , ... Abstract
Background: Rabies is a fatal zoonotic disease, present in almost 150 countries. The “Zero by 30” initiative aims to eliminate human deaths from dog-mediated rabies globally by 2030. This systematic review investigates how viral sequencing can contribute to achieving the “Zero by 30” goal by improving understanding of viral circulation and the impact of rabies control measures.
Methods: A comprehensive search of bibliographic databases was conducted focusing on research on rabies from regions with endemic dog-mediated rabies published between 2000 and 2023, adhering to Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines. Data were extracted and synthesised to provide recommendations for further research and application to support rabies control.
Results: 220 studies were identified to have documented rabies virus (RABV) sequences from 94 countries, primarily using first-generation technology to produce partial genomes and with sequencing predominantly conducted overseas rather than in-country. Dogs were identified to be the primary RABV reservoir in these regions, although some studies identified more localised wildlife reservoirs. Clade classifications were commonly based on host association or geographical location, however, lack of standardised methods and nomenclature for classifying lineages limited comparison at higher resolution. Cross-species transmission, and both local and long-distance transmission were identified, although quantitative inference was limited. Sequence data was particularly useful for identifying transboundary spread and incursions, investigating host shifts, and tracing sources of human rabies, with endemicity typically characterised by the identification of multiple co-circulating viral lineages.
Conclusion: There is an urgent need for standardised classification methods and phylogeny-based nomenclature for RABVs, and for improved sequencing capacity in regions with endemic dog-mediated rabies, including proficiency in bioinformatics and phylogenetics. Our findings emphasise the critical need to foster international cooperation and coordinate rabies control efforts to reduce transboundary spread, limit reintroductions and maintain progress towards the 2030 target.

Keywords
INTRODUCTION
Rabies virus (RABV) poses a major public health threat, causing around 60,000 deaths annually, almost exclusively in Low- and Middle-Income Countries (LMICs)[1]. The virus is most commonly transmitted through bites from infected hosts in the orders Chiroptera and Carnivora[2]. Domestic dogs are the main source of transmission to humans but, as a multi-host pathogen, wild carnivores also serve as primary RABV hosts with host-associated variants recorded in certain geographies[3]. For example, wildlife such as raccoons, skunks and foxes each maintain different RABV variants in localities across North America[3]. Generally, RABV is referred to according to these host-associated variants (sometimes termed biotype[4], see defined key terms in Table 1).
Definitions of key terms in the context of RABV
| Term | Definition |
| Cross-species transmission | Transmission events from one (host) species to another, that occasionally result in a host shift, whereby a new transmission cycle is established, but more frequently leading to short-lived chains of transmission or dead-end infections with no onward transmission[5,6] |
| Reservoir | One or more epidemiologically connected populations in which the pathogen persists and from which infection is transmitted to a population of concern i.e., a target population[7]. Domestic dogs are considered maintenance hosts in the reservoir for rabies in many regions, while humans, endangered wildlife, and livestock are often considered target populations[8] |
| Variant | A viral population maintained within a particular reservoir host in a geographically defined area that differs from other viral populations due to either a host shift or diversification within a host species or population i.e., host-association[9] (sometimes called a biotype[4]). RABV variants often show host-associations. Here we differentiate dog- vs. wildlife-associated RABV variants[10] |
| Directionality of transmission | The predominant direction of transmission from one host species, population or location to another. Genomic data can be used to identify how infected hosts are linked to each other, and to infer the source of infection[11]. Transmission networks involve mapping transmission routes or pathways to ascertain who infected whom and have been inferred for RABV using parsimony-based approaches and advanced Bayesian frameworks[11] |
| Transboundary spread | pathogen spread across administrative boundaries. We differentiate human-mediated spread, often over long distances e.g., > 50 km, from local dispersal due to host behaviour (rabid dogs typically bite animals within 1 km of their location, but can sometimes run over 20 km)[12]. An incursion (or introduction) is the spread of a pathogen into a new area, either where that pathogen was historically absent, had been previously eliminated (i.e., re-introduction), or where the pathogen is already present. In the latter example, an incursion might be identified from genetic data when a distinct viral lineage is found in an area where other lineages are circulating[13,14] |
| Phylogeny or phylogenetic tree | A branching diagram or tree showing the evolutionary relationships between sequences or species. These generally incorporate nucleotide substitution models, and can include taxonomic and temporal information. The most widely applied methods for tree building are Neighbour Joining, Maximum Parsimony, Maximum Likelihood and Bayesian Inference |
| RABV-GLUE | Is a flexible software system for interpreting sequencing data with functionality for storage and interpretation. The software is freely available and can be directly downloaded for viral sequence analysis or can be used via the web interface[15]: http://rabv-glue.cvr.gla.ac.uk |
| Clade | A monophyletic group with a single common ancestor[16]. Shared ancestry therefore defines the initial pathogen emergence and spread[17]. RABV is designated into clades or subclades (sometimes referred to as major and minor clades), usually associated with specific geographic areas and/or hosts. A lineage (sometimes referred to as a subtype) is a group of related sequences (typically a smaller monophyletic group contained within a larger subclade) defined by statistical support of their phylogenetic placement and genetic differences from their most recent common ancestor (mrca)[18]. Clades and subclades can be classified differently depending on the hosts and geographical location. In this review, publications may have designated clades/subclades/lineages according to the author’s naming system. To allow for comparison between publications, a table of publication-specific names and corresponding RABV-GLUE designations is available in Supplementary Table 1 |
| Phylodynamics | The study of how epidemiological, immunological, and evolutionary processes (inter)act to shape viral phylogenies. Examples include studies of spatial diffusion, sometimes incorporating geographical or population structure i.e., phylogeography[19] |
| Genomic/genetic surveillance | Surveillance involving sequence data to characterise an infectious agent and infer additional information about its dynamics. Genomic refers specifically to WGS i.e., the entire pathogen genome (> 10 kb for RABV), whereas partial genome sequencing is the generation of a sequence of a specified genetic region that can be used to identify the organism. For RABV, 400 bp fragments are used for diagnostics, and many phylogenetic studies sequence the N gene, G gene and/or G-L intergenic regions |
| 1st generation (or Sanger) sequencing | Type of sequencing which produces DNA fragments labelled by chemical modified nucleotides (dideoxynucleotides) during nucleotide elongation. Sanger sequencing sequences one fragment at a time[20] |
| NGS | Parallel approaches that sequence millions of fragments simultaneously, hence have higher throughput than 1st generation approaches. NGS technologies are divided into 2nd generation technologies which analyse clonal representations of the input DNA before sequencing amplified DNA clones, e.g., Illumina MiSeq and Ion Torrent, vs. 3rd generation single molecule sequencing technologies, which can produce longer reads than 2nd generation platforms but typically have higher error rates, e.g., Oxford Nanopore and PacBio[20] |
Phylogenetic analysis enables further classification of RABV diversity into clades, subclades and lineages. The RABV genome is 12 kilobases (kb) in length[21], comprising five genes encoding the nucleoprotein (N), phosphoprotein (P), matrix protein (M), glycoprotein (G) and the large polymerase protein (L)[22]. Like other RNA viruses, RABV exhibits elevated mutation rates because of the absence of proofreading[23]. These mutation rates foster genetic diversity, facilitating tracking of viral spread and enhanced understanding of viral dynamics.
There is no treatment for rabies once clinical signs begin, but post-exposure prophylaxis (PEP) correctly administered shortly after exposure is almost 100% effective in preventing the fatal onset of disease[24]. However, a highly effective canine vaccine is available to prevent disease in the primary reservoir, and therefore prevent transmission to humans. Canine rabies elimination is possible through mass dog vaccination, as demonstrated in Europe, North America, and parts of Asia and Latin America[25]. Several countries where dog-mediated rabies was endemic have now been declared rabies-free or are approaching elimination as a result of sustained dog vaccination[25]. According to the World Health Organisation, to eliminate dog-mediated rabies vaccination campaigns need to achieve coverage of at least 70% and be conducted annually for at least three years[26]. Rabies incidence in Latin America has declined dramatically over recent decades due to coordinated regional dog vaccination programs[25]. In contrast, most LMICs in Asia and Africa have not allocated sufficient budget to control this disease, and access to dog vaccines remains limited. Confounding the lack of vaccines, typically, rabies surveillance has also been poor. Additional challenges to rabies control include lack of understanding of dog ownership patterns, population sizes, and accessibility for vaccination as well as cultural practices including dog meat consumption[27]. To address these challenges, international organisations joined forces under the United Against Rabies collaboration to advocate for the global goal of “Zero by 30”; to end human deaths from dog-mediated rabies by 2030[28].
Surveillance plays a critical role in infectious disease control[29]. Surveillance entails the continuous, systematic collection, analysis, interpretation, and timely dissemination of health-related information[30], serving as the foundation for planning, execution and evaluation of public health strategies. For instance, surveillance provides data on the effectiveness of interventions, supporting decision-making for initiatives like “Zero by 30”[31]. Increasingly, surveillance also involves genetic data, for pathogen diagnosis and risk assessment, as well as to identify the source of outbreaks and to characterise pathogen spread[32]. Linked with locations, pathogen genetic data have uncovered disease movement; from global migration dynamics to local transmission pathways for pathogens such as Influenza virus[33,34], Ebola virus[35], Zika virus[36], Yellow fever virus[37,38], Mpox virus[39-42] and SARS-CoV-2[43]. Sequencing approaches have the potential to enhance rabies surveillance and provide actionable information to inform control programs locally and globally as part of “Zero by 30”. For example, viral sequence data can distinguish undetected local circulation from incursions and potentially identify their sources[44]. More generally, sequencing could provide insights into how rabies circulates within populations and the processes responsible for its maintenance in specific localities[32,45].
Use of pathogen sequence data for surveillance is, however, not yet routine in most LMICs. Constraints include lack of local sequencing capacity, competent personnel and laboratory resources, and these are affected by costs of, and access to, reagents and consumables, as well as power supplies and cold chain[35]. Sequencing technologies have become more affordable and efforts are underway to improve accessibility[46]. Indeed, growth in sequencing capacity during the COVID-19 pandemic provided evidence of the feasibility of scaling up molecular diagnostics, but also highlighted operational challenges. For example, in Nigeria, the number of laboratories capable of molecular identification of SARS-CoV-2 increased from four to 72 in 2020[47]. In this systematic review, our goal was to examine the extent of the application of genetic approaches to RABV surveillance in regions with endemic dog-mediated rabies (much of Africa, Asia, and parts of Latin America) and how, going forward, these approaches can contribute to the “Zero by 30” goal.
METHODS
This review was conducted according to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines[48], following a protocol developed a priori to ensure methodological reproducibility and transparency [Supplementary File 1].
Eligibility criteria
Studies must address either dog, human or terrestrial wildlife rabies (i.e., not focus on bat rabies), and use molecular techniques and genetic sequence data, either for diagnosis [diagnostic polymarase chain reaction (PCR) products] or surveillance. We excluded studies reported as literature reviews that did not present genetic sequence data, or focus on rabies, and that were not published in English.
Search strategy
A systematic search was undertaken on PubMed, Web of Science and Google Scholar databases to identify original studies published between 2000 and 2023. Advanced searches with Boolean operators and quotations were performed using the search terms “Rabies AND (genom* OR sequenc* OR molecular OR phylo*) AND (control OR surveillance OR eliminat*)” as illustrated in Supplementary Table 2 with an example of the medical subject headings (MeSH) terms from one of the search engines provided in Supplementary Table 3. Further manual searches were performed for additional relevant studies.
Screening
We reviewed the title and abstracts of all articles that met the inclusion criteria. Where detail was lacking, the entire article was reviewed before its inclusion or exclusion was determined. A first screening phase based on titles and abstracts was conducted independently by three independent reviewers (GJ, CB and MM), and out-of-topic studies excluded. During this phase, studies that were not covering rabies from Asia, Africa or Latin America were also excluded. A second screening phase based on the full texts was conducted by GJ and CB using a standardised eligibility form. Discrepancies observed between reviewers were resolved through discussion. Duplicate studies were removed.
Data extraction
A form designed for this review was used for data extraction [Supplementary Table 4]. The fields extracted included: authors, year of publication, country, study aim, study design, species from which samples were collected, numbers of samples (tested and confirmed for rabies and RABV sequences), sample type, sequence type [Whole Genome Sequencing (WGS) or partial genome, indicating the length and section sequenced], sequencing platform, type of phylogenetic analysis, outcome of the study, and the main study findings, including any recommendations for control measures derived from analysis of sequencing data.
Data synthesis
The main characteristics of the studies were summarised in tabular form (as per the data extraction proforma). Data analysis and visualisation was carried out in R (version 4.0.3)[49].
RESULTS
Study selection
The database search identified 1,558 publications, of which 161 were excluded as duplicates using automation software[50]. Manual searches identified an additional 23 relevant publications. After screening and assessing eligibility, 220 were retained for systematic review [Figure 1].

Figure 1. Flow diagram of the article selection process following PRISMA guidelines. PRISMA: Preferred Reporting Items for Systematic Reviews and Meta-Analyses.
Study Characteristics
The 220 articles generated new RABV sequences from 94 countries [Figure 2], with most from China (n = 54 publications) and Brazil (n = 19) while six undertook large-scale meta-analyses with additional sequencing from multiple countries[51-56]. An average of two studies presenting new RABV sequences were published per year [Figure 2], with most in 2013 (n = 16) and 2015 (n = 17). All studies generated RABV sequences from brain tissue samples, with some on Flinders Technology Associates (FTA) cards[21,57-59] and four including alternative sample types (nuchal biopsy, cerebrospinal fluid and salivary glands). Most publications (n = 188) reported results from partial sequences only, using 1st generation sequencing, mostly the N gene (n = 119). Other studies sequenced the G, P or M genes or the G-L intergenic region, and 54 were multi-gene [Figure 3]. Twenty-nine studies generated WGS, with hotspots in China (n = 10) and Tanzania (n = 5), and nine used multiple platforms (1st and 2nd[15,52,60-63] or 1st and 3rd generation platforms[64,65]). In the last decade, 3rd generation sequencing (Nanopore) increased, as did sequencing output.
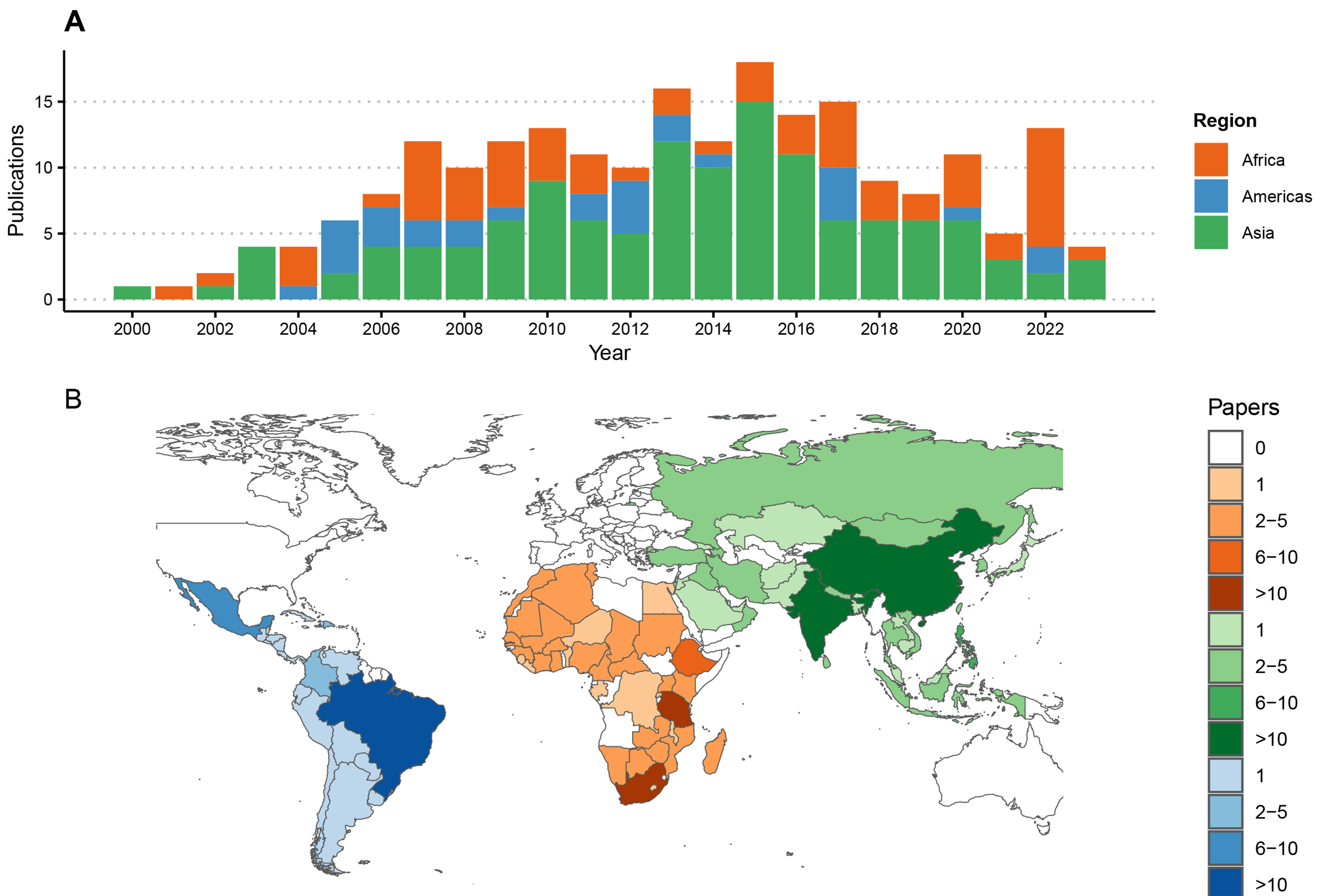
Figure 2. Publications reporting RABV sequences by region from 2000 to 2023. (A) Time series of publications; (B) Numbers of publications reporting sequence data by country, shaded by the number of publications and coloured by geographic region. Each publication was attributed to one or multiple countries based on the origin of the RABV sequences, including five studies reporting travel-associated human cases according to the country of origin. One study describing the global distribution of lyssaviruses[52], was excluded from this figure. RABV: Rabies virus.

Figure 3. Summary of RABV sequencing by country, including the sequencing platform, type of sequence generated, location of sequencing and analyses undertaken. The colour intensity increases with the number of publications per country; note the different colour scale by regions; (A) Americas, (B) Africa, (C) Asia. Only twenty nine publications generated WGS (from 4 countries) and nine conducted phylogeographic analyses. Three publications present data from across multiple countries, with two of these increasing both the number of countries with WGS and phylogeographic analyses[51,52]. An enlarged version of this figure is available as Supplementary Figure 1. RABV: Rabies virus; WGS: Whole Genome Sequencing.
Sequences were generated from 94 of 149 countries in endemic regions (61.7%). Among the endemic regions, countries in Africa conducted the least in-country sequencing (42.8%, 14/27) whereas countries in Asia conducted the most (76.6%, 23/30, Figure 3). In terms of sequencing output, Asia led with 6,715 sequences (381 WGS), followed by Africa with 3,757 sequences (315 WGS) then Latin America with 1,143 sequences (26 WGS). Tree-building methods used for analyses in publications included Neighbour Joining
Circulating RABV
A large proportion of the 220 studies reviewed described the diversity and distribution of circulating RABVs. Broadly, RABV can be classified into dog-related and bat-related viruses, both of which split into major clades, subclades and lineages[15,52], often associated with geographies and hosts. Publications from Latin America typically differentiated dog-related rabies vs. bat-related rabies[56,60,61], and dog-derived and bat-derived vs. skunk variants[56]. In Africa and Asia, RABV diversity was classified predominantly by geographical clustering of dog-related viruses. Generally, there was a lack of standardisation beyond global clade nomenclature, with many studies (n = 92) introducing ad hoc names to refer to diversity within global clade assignments. Figure 4 uses RABV-gene-linked by underlying evolution (GLUE)[15] designations to depict circulating clades and a key indicates alternative names used in publications

Figure 4. The spatial distribution of dog-associated RABV clades reported from Asia, Africa and Latin America. Each country is shaded according to the number of publicly available sequences with at least 90% gene or genome coverage and labelled with a pie chart representing the RABV phylogenetic clades (denoted as Major.Minor clade in the legend e.g., Cosmopolitan. AF1a is major clade Cosmopolitan and minor clade AF1b, while sequences lacking resolution for a minor clade designation are assigned “NA”) within each country and sized according to the number of publications. Note that major and minor clades are annotated according to RABV-GLUE designations[15], but may be annotated differently in primary publications. The correspondence between annotations are detailed in Supplementary Table 1. RABV: Rabies virus; GLUE: gene-linked by underlying evolution.
Three RABV clades are reported to circulate in Africa; the “Cosmopolitan”, “Africa 2” (AF2) and “Africa 3” (AF3), that subdivide into subclades and lineages[52]. The Cosmopolitan clade was found across 27 African countries and split into AF1a, AF1b, AF1c and AF4 subclades. AF1a was broadly distributed across the continent, and predominant in northern and eastern Africa. AF1b was mainly in eastern and southern Africa, while AF1c and AF4 were found in Madagascar and Egypt respectively[51]. The AF2 clade is found in 14 countries, mainly across West and Central Africa. The AF3 clade, which is associated with viverrids, is found in Southern Africa, with sequences from South Africa and Botswana[52] [Figure 4]. In Asia, RABVs were categorised into four major clades: Cosmopolitan, Arctic (specifically the Arctic-like RABV, which has been found circulating across eastern and southern Asia), Indian Subcontinent, and Asian. The Cosmopolitan clade was widespread in Western[57,63-66], Eastern[67-75], and Northern Asia[76]. Five subclades of the Asian clade (SEA1-SEA5) were found within Eastern and Southeast Asia[77-80]. The Indian Subcontinent clade was prevalent in South Asia[81-84], with a few sequences from Western and Eastern Asia. While the Arctic-like clade was found in parts of Eastern[68,85-89] and South Asia[21,58,81,90-95]. In the Americas clades were categorised into dog-maintained and dog-derived variants, including established wildlife foci in skunks, coyotes, gray foxes and mongoose[56].
Concurrent circulation of divergent clades and subclades within countries, and more locally, was commonly reported across Asia and Africa. This was a less common feature in Latin America, where there are fewer remaining dog-mediated rabies foci and, overall, less dog-related RABV sequence data (almost all partial and just 26 WGS). Six African countries reported co-circulation of the Cosmopolitan and AF2 clades (Cameroon, the Central African Republic, Chad, Ghana, Gabon and Nigeria)[13,14,51,52,96,97]. One study identified co-circulation of Cosmopolitan AF1b subclade and AF3 clade in Botswana, but from different parts of the country; AF1b in the north and AF3 in the south[63]. Similarly six African countries reported co-circulation of Cosmopolitan AF1a and AF1b subclades (Cameroon, Chad, Ghana, Mozambique, Kenya and Uganda)[13,96,98-101]. All of the four major clades found in Asia were seen co-circulating within and between provinces in China[72-75,102-105]. In Southeast Asian countries, only the Asian clade was seen, except in Vietnam where the Cosmopolitan clade was also reported[80]. More differentiated subclades were reported from archipelagic Southeast Asian countries such as Indonesia (Asian SEA1b)[106-108], the Philippines (SEA4), Sri Lanka (Indian-Sub)[82,83], and Taiwan (SEA5)[109-111], with geographically-associated names used. As geographic resolution increased, naming systems became more ad hoc. For example, a study from the Philippines identified nine lineages (GrL1-9) within Group L of SEA4 subclade, named because of circulation within Luzon Island[112].
Many studies used nomenclature only meaningful to that study or research group. For example, sequences from China were variously referred to as groups I-IV[113,114], and lineages A-F[73,115]. The same nomenclature could represent different diversity across studies. For example, group I referred to SEA1b sequences in one study[113], but SEA2a in another[114]. Another study defined “Indian” lineages I & II[81], which were re-classified by RABV-GLUE [Arctic NA (i.e., no minor clade assigned), Arctic AL1a, Asian SEA1a, Indian-sub] revealing much broader diversity and geographic distribution than user-defined terminology implied. In Latin America, most studies (n = 29) grouped RABVs into lineages named by geographical location or species involved, without following a common nomenclature.
Host species
Studies reported RABVs from a broad range of domestic and wildlife hosts and humans. Sequences from domestic animals included dogs, cats, cows, goats, sheep, pigs, camels and horses. RABVs were found in wild carnivores such as foxes, jackals, hyenas, mongoose, African wild dogs, wolves, ferret badgers, and civets as well as occasionally in herbivores (e.g., nilgai and kudu) and more unusual wildlife like monkeys and bears. Eighty-five publications reported sequences from across this species range, 19 from just dogs and livestock, 18 from dogs and wildlife, and 15 from dogs and humans. Cross-species transmission, resulting in spillover, were reported in 63.7% (n = 137) of studies: 13.1% (n = 18/137) reporting transmission between domestic dogs and wildlife, and 86.9% (n = 119) reporting transmission from dogs to both wildlife and livestock. In regions with endemic dog-mediated rabies most livestock outbreaks were attributed to spillover from dogs, except for outbreaks in livestock in Latin America from vampire bat rabies. However, spillover from wildlife to both dogs and to livestock was also observed, as seen in a livestock outbreak linked to foxes in northwest China[72]. Atypical cases, such as rabies in a chicken bitten by a dog, also highlight spillover events in endemic areas[84].
Domestic dogs were predominantly identified as the reservoir of RABVs within the defined scope of this study. Successful control of dog-mediated rabies across Latin America has left bats as the main RABV reservoir, but emergence of independent wildlife-associated cycles were attributed to spillovers from bat, arctic fox and canine rabies[116]. For example, RABVs were associated with skunks in north-central Mexico, with coyotes in west-central Mexico, gray foxes in Colombia, crab-eating foxes in Brazil and mongoose in Caribbean islands[56]. Two publications from Africa reported other reservoirs: horizontal transmission of a canid variant (AF1b) in kudu in Namibia[117], and the distinct AF3 clade circulating in yellow mongoose in Southern Africa[62]. Evolutionary rates inferred from AF3 were outliers from dog-related clades, suggesting host-specific adaptation[52]. In Asia, a unique ferret badger variant was found in Taiwan, distinct from other Asian subclades[118,119].
Studies examined reservoir dynamics, investigating new variants and quantifying transmission within and between species. For example, an Israeli study showed emergence of a dog-related clade that was distinct from fox-associated clades that were predominant before fox-targeted oral rabies vaccination began. The emerging dog-related clade might have indicated a host shift, but sequence similarity to Turkish dog isolates suggested cross-border introduction as the cause of the clade emergence[120]. Bayesian phylogenetic inference from WGS of samples from Turkey spanning from 1999 to 2015 was used to refine the date of a host shift that did occur from dogs to foxes to ~1997[121]. A European red fox-associated variant reported from hyenas in Tanzania[122], was concluded to stem from laboratory contamination as other studies found that sequences from both wildlife and dogs all belonged to typical and closely-related dog-mediated variants belonging to clade AF1b[9]. Further analysis confirmed the central role of domestic dogs in maintaining circulation of AF1b in this region[123]. In the same setting, sequencing an African civet cat case that was presumed to be spillover of dog-mediated rabies, also unexpectedly identified a novel highly divergent lyssavirus[124], and no cases of this variant have been found in dogs. The majority of spillover examples identified were from dogs into wildlife, but a limited number of publications reported spillover from wildlife to dogs or other animals, for example, from Coyote into dogs in Mexico[56].
Human rabies
In total 24 studies (16 from Asia, 6 from Africa, and 2 from Latin America) reported sequences from human rabies cases, with a total of 219 human samples sequenced. Most of these studies (n = 19) identified human cases to be the result of dog-mediated rabies only. Three studies reported wildlife exposure only including 1 raccoon exposure from Nepal[66] and 2 fox exposures, one from India[125] and one from Mexico[126]. The report of raccoon exposure in Nepal may be a misidentification, as raccoons are not native to the country. It’s possible that the biting animal was a red panda, which is indigenous to Nepal. Both species share similarities in head features, dentition, and ringed tails[127]. A longitudinal study from South Africa reported a majority of human cases from dog-mediated rabies as well as three cases spread from mongoose (AF3)[128]. One of the Latin American publications reported a veterinarian who became infected while handling rabid livestock that was determined through genetic analysis to be vampire bat rabies[129].
Several of the human cases occurred in rabies-free countries and were resolved through phylogenetic analysis. Eight were traced to immigrants with exposure histories in countries with endemic dog-mediated rabies where PEP was not received before travel to the (rabies-free) country of diagnosis. Analysis of the patient isolates revealed similarity with cases in postulated countries of origin, including a case in the UK imported from Nigeria[130] and a case in France imported from Mali[131], two unrelated cases in Qatar both imported from Nepal[66], and cases imported from the Philippines to the UK[132], Japan[133] and Finland[134], as well one that likely originated from insectivorous bats involving a Mexican immigrant who was bitten by a fox, and who died in California[126]. Similarly a sequence from a patient in France, with recent travel history to Mali but no known exposure, belonged to AF2 subclade that circulates in West Africa[131].
Human cases with prolonged incubation periods were also identified (normal incubation periods range from 1 week to 8 months). One human case in rabies-free Australia was traced to exposure before relocation. The 10-year-old of Vietnamese origin had stayed in Hong Kong before immigrating to Australia 5 years prior to symptoms onset. Phylogenetic analysis revealed the isolate’s distinctness from Australian Bat Lyssavirus and Vietnamese RABV lineages; instead grouping with a China-associated lineage, suggesting acquisition in Hong Kong and prolonged incubation[135]. Two human cases imported to Japan also had incubation periods exceeding 8 months[133]. Most human cases resulted from bites, with two exceptions in Africa where victims were scratched or licked[136,137]. The first case involved a 26-month old child scratched by a puppy in Gauteng Province, South Africa. Genetic analysis linked this case to an outbreak that spread in dogs in southern Johannesburg following an introduction from KwaZulu-Natal[137]. In the second case, a 6-year-old child from KwaZulu-Natal had no history of being bitten. However, a neighbour’s dog died of unknown causes three days before the child’s death. Although rabies was not suspected or tested for in the dog at the time, the authors speculate that this dog was the child’s most likely source of exposure to rabies, possibly through non-bite contact with saliva (e.g., lick or scratch). Phylogenetic analysis of other cases circulating in the region identified genetic similarity to the child’s sample, further supporting the hypothesis that the child was infected through local exposure[136].
RABV movement
Phylogeographic analysis identified local transmission and long-distance movement of RABVs based on the geographic association or displacement of clades, subclades and lineages. Some studies revealed situations characterised by sustained local circulation, with distinct lineages and closely related sequences confined within specific geographic areas. For instance, in Nigeria, sequences within AF2 (AF2-1 and AF2-2) clustered in Northern and Southern Nigeria respectively[130]. Similar clustering was observed in India[81,94] and major island groups in the Philippines[78,138]. However, local circulation was often not confined by political borders across contiguous landscapes. For example, samples from the border between Brazil (Mato Grosso do Sul state) and Bolivia clustered, reflecting frequent movement across this boundary[139].
Most publications mentioned human-mediated long-distance viral movement but only about 25% substantiated their claims through phylogeographic analysis. In Tanzania, discrete phylogeographic analysis revealed long-distance movement between regions (> 750 km apart)[140], and sequences from distant ecosystems (Serengeti and Tarangire, > 200 km apart) grouped together[9], with seasonal migration of pastoralists (and their dogs) proposed as an explanation. In Asia, long-distance movement was attributed to waves of human migration from China to Southeast Asia[141]. Indeed, increased trade between and within countries[142], including animal trade[75,104] and the dog meat trade[77,113,143], were identified as playing a role in RABV incursions and expansion.
Three sources of evidence were used to infer transboundary spread: co-circulation of divergent RABVs, clustering of related sequences from adjacent countries/regions and phylogeographic analyses using location data associated with sequences. Studies reported co-circulating divergent viruses in 31 countries, likely caused by introductions that persisted[13,14,96,99,100,144]. For example, AF1a, AF1b and AF2 were found co-circulating in southern Cameroon, with the AF1b and AF2 sequences most closely related to sequences from the Central African Republic[96]. RABVs circulating in neighbouring countries were often closely related, particularly across shared land borders, like Azerbaijan[57] with Georgia, Bangladesh[90] with India, and Tibet[142] with Nepal. There were also instances of emerging subclades within one country closely connected to another. For example, Nepalese isolates identified as a new lineage within the Arctic-like
Several studies used phylogeographic analyses to infer RABV spatiotemporal dynamics[74,79,111,141,145], particularly the direction and speed of dispersal[18,68,146,147]. Faster dispersal was associated with anthropogenic factors. Dog-associated lineages in Brazil dispersed at 30.5 kilometres per year (km/year) in comparison to a lower rate, 9.5 km/year, in crab-eating foxes, which was attributed to human activities driving dispersal of dog-associated lineages[147]. A high velocity of dog-meditated rabies (18.1 km/year), was estimated in Iran, using a novel analytical framework, revealing spread linked to accessible areas associated with high human density[18]. This estimate is similar to the average dispersal rate in Northern Africa (19.5 km/year) where, again, landscape accessibility was an important driving factor[18,32].
The time of introduction and history of rabies spread in various areas was investigated. A publication used historical records and phylogenetic analysis to show that rabies was only present in bats and skunks in the Western Hemisphere, with canine rabies rare or absent among dogs of Native Americans, before the arrival of new dog breeds imported during European colonisation[56]. A more comprehensive recent study combined partial and whole genome sequences to reconstruct movement more precisely, revealing how colonial empires influenced the global spread of RABVs[148].
Human-mediated long-distance movement was identified through phylogeographic analysis as the source of incursions into previously rabies-free areas, posing a challenge to maintaining rabies freedom. Examples identified introductions to historically rabies-free areas such as island provinces in Indonesia[138] and the Philippines[78], and to areas where rabies had been eliminated such as Pemba, (an island off mainland Tanzania[149]), Gauteng Province in South Africa, and N’Djamena, Chad[13].
Geographical features were discussed in fifteen papers, with rivers, lakes and mountain ranges shown to be natural barriers[32,150,151]. For example, sequences within AF1b subclades (AF1b-1 and AF1b-11) were separated by the Zambezi river and Kariba lake in Zambia and Zimbabwe respectively[150]. Transboundary spread between Lesotho and KwaZulu-Natal in South Africa was limited by the Drakensberg and Maloti mountain ranges, which constrain movement of people and animals[151]. In Asia, three lineages from SEA5 found in ferret badgers were segregated by mountain ranges and rivers[111,119]. Transmission corridors that facilitated dissemination included: (1) transportation networks; (2) the presence and size of dog populations (tied to human populations); and (3) anthropogenic factors (trade, agriculture and urbanisation). For example, in Southeast Brazil, urbanisation was reported to play a central role; dog population size was correlated with human populations, meaning higher density regions had more dogs and therefore more RABV diversity[152]. Road networks were often shown to be associated with increased movement, consistent with human-mediated transport of incubating animals. RABV detected from different cities in Mozambique were closely related, despite long distances between them[100]. Sequences from the AF2 clade were proposed to result from an introduction to Bangui, the capital of the Central African Republic, as the closest related sequences were from neighbouring Chad[14]. In Asia, transport routes were correlated with the RABV distribution in Thailand[153], and China[154]. At more local scales in Tanzania phylogeographic analysis revealed that presence of dogs, rather than density, predicted spread[155].
Rabies control
All studies provided recommendations for rabies control and prevention, but most were generic and not specifically inferred from sequences. These studies recommended mass vaccination of dogs, as well as oral vaccination in specific wildlife populations where variants had emerged[156,157]. Other recommendations included dog population management[76,113,158,159], monitoring the health of animals for trade and consumption[75,80,97,129], and raising awareness, particularly in communities identified as “high-risk”[155,158,160,161]. Only 33 publications provided targeted recommendations grounded in genetic evidence. Some reported spillover events, emphasising the necessity for enhanced surveillance (n = 12) for specific wildlife populations[59,72,93,95]. Outbreak investigations pinpointed sources of incursions, and recommended monitoring for animal transport and at borders[78,82,138,162] plus surveillance and control measures in rabies-free areas or areas with low-incidence, where introductions pose risks[89,149]. Likewise, identification of novel variants and genetic diversity prompted suggestions for oral vaccines and baits tailored to hosts, e.g., for ferret badgers[118,143]. There were no instances of vaccine-derived cases/outbreaks in the review, but genomic surveillance would be a crucial tool to identify and monitor such occurrences.
Widespread coexistence of diverse lineages and insights into their evolutionary history, transmission dynamics and dispersal rates from more complex phylogeographic analysis, highlighted the urgency of addressing transboundary transmission, which requires coordinated effort[65,80,140]. Vaccination campaigns focusing solely on urban localised dog populations have demonstrated short-lived success due to rabies circulation across land borders. Examples include Chad and Central African Republic[13,14], India and Bhutan[58], and Peru and Brazil bordering Bolivia[163]. The same was true between islands, for example Pemba, off Tanzania[155], and within and between archipelagic countries in Southeast Asia[78,108,138] and also was identified at the borders of states in India[164]. Therefore, recommendations included scaling up dog vaccination beyond urban centres to encompass surrounding rural areas, along with coordinating transboundary dog vaccination to minimise spread into cities or between neighbouring countries or administrative units such as states or provinces[13,14].
DISCUSSION
In this review, we focused on how genetic data informs understanding of rabies dynamics and its control. Findings from 220 studies demonstrate sequencing as a potentially powerful tool for contributing to “Zero by 30”. However, information from sequences is often not fully or consistently synthesised into specific, actionable recommendations. When used effectively, sequencing has been instrumental in tracing incursions into rabies-free areas and highlighting extensive transboundary transmission that necessitates coordination of control nationally and regionally[78,138,149]. Although RABV diversity differs across these regions with endemic dog-mediated rabies, spillovers and transboundary movement was repeatedly reported, emphasising the importance of coordinated transboundary vaccination efforts and surveillance. As countries approach the “endgame”, i.e., the final stages of an elimination programme where disease is still circulating but at much reduced levels, genetic data is expected to become increasingly useful, providing greater insights for monitoring emerging issues such as spillover and adaptation to alternative hosts and potential re-emergence in dogs.
A major challenge is how classification of phylogenetic diversity and associated nomenclature, beyond the clade level, is not standardised and how varied terminologies (subtypes, subclades, subclasses, clusters etc.) are used. In most publications from Africa and Asia, groupings were designated numbers or letters based on subjectively defined clusters in phylogenies, while Latin American studies often employed antigenic variant classification rather than evolutionary (phylogenetic) relationships. Inconsistent terminology hampered a clear understanding of circulating lineages and their geographic distribution, and hindered their use as reference points for further research[15]. Many studies used nomenclature only meaningful within the context of that particular study and classifications often differed across related studies [Supplementary Table 1]; employing RABV-GLUE[15] to classify minor clades revealed these inconsistencies [Figure 4]. The significance of discerning lineages lies in implications for control, for example, differentiating incursions from undetected local transmission and identifying the scale of circulation. Variations in the portion of the genome sequenced (gene-specific) and sequence length contributed to inconsistencies. Most publications used partial genome sequencing, often targeting the N gene, a relatively conserved region used as a diagnostic marker. But, this limited sampling missed important variation, which may define rare and divergent lineages or improve resolution across narrower spatio-temporal windows. Just 20% of publications used WGS, which can provide deeper understanding.
We found an urgent need to increase in-country sequencing capacity in endemic countries, with relatively few generating sequences in-country. Regionally, Africa lagged behind Latin America and Asia in terms of capacity, but had a higher output than Latin America, likely because few countries in the region remain endemic for dog-mediated rabies. Most sequencing was driven by research, with routine sequencing not yet part of surveillance in endemic regions. Consequently, there is a shortage of sequences, with largest contributions from China and Brazil (upper-middle-income countries with existing networks and resources), whereas several countries had limited representation; some with just one publication. The scarcity of sequences, which poses a challenge to characterising RABV diversity and understanding transmission, could be due to different cultural, intrinsic and socio-economic factors. In the aftermath of the COVID-19 pandemic, sequencing capabilities have expanded, becoming more affordable and accessible. This increased capacity has practical applications for responding to rabies outbreaks[149].
Although dogs were identified as the primary host responsible for most transmission in regions with endemic dog-mediated rabies which were the focus of this study, RABVs broad host range means it is capable of transmission among multiple species. This versatility creates potential for new reservoirs, raising concerns about the effectiveness of control measures, and about these reservoirs being a source of re-emergence[160]. In areas where dog-mediated rabies has largely been eliminated, there were several examples of transmission cycles in wildlife, some of which resulted from spillover from dogs. In Latin America, where canine rabies incidence has reduced dramatically from coordinated regional control, intriguing reservoir dynamics have emerged. For example, three major enzootic cycles, distinct from dog rabies foci in Mexico (skunks, coyotes and gray foxes) highlight a complex maintenance dynamic[165]. Similarly, transmission cycles have established in ferret badgers independently in both Taiwan and China[118,119,166]. These findings underscore the complex reservoir dynamics of RABV and importance of understanding host associations and cross-species transmission. As dog-mediated rabies declines, this understanding will be necessary to enable targeted control, either directing efforts at blocking transmission and spread between source and target populations or controlling infection within new reservoirs. The risk of rabies spillover from wildlife foci to dogs is a serious challenge for “Zero by 30”. When such reservoirs exist, tailored surveillance and control measures are crucial to mitigate the threat of re-emergence in dog populations as a result of spillover. This may eventually require countries to undertake control measures in wildlife reservoirs, but more imminently underscores the need to eliminate rabies from dog populations before wildlife foci can establish.
Underreporting and misdiagnosis of human rabies remains a significant issue and can create a false impression of low burden in rabies endemic settings across Africa and Asia. While enhancing diagnostic capacity and overall surveillance is crucial to address underreporting, we also emphasise the pivotal role that genetic data can play in strengthening human rabies surveillance. Rabies-free countries experience imported human cases from exposures in endemic countries[167-169]. Oftentimes, these imported cases were confirmed through epidemiological investigation, but genetic data identified the sources of infections, as well as ambiguous cases, without a clear route of exposure (e.g., no bite history) or origin (e.g., the migrant from Mexico and the veterinarian in Brazil), or with unusually long incubation periods[133,135-137]. These human cases identified from countries that are free from dog-mediated rabies highlight how these technologies could be applied to strengthen human rabies diagnosis and source attribution within endemic countries.
Unlike infections in humans, which are effectively dead-end hosts, movement of infected animals is the predominant factor contributing to RABV establishment in new geographic settings. Publications highlighted both local host movement and long-distance human-mediated movements, however, evidence on transmission links and directionality were sparse, with only a few studies (25%) employing phylogeographic analyses. Most used genetic relatedness of viruses from different locations, based on interpreting phylogenies, without quantitative inference. When carried out, phylogeographic analyses illuminated environmental features as both natural barriers and drivers of spread, with human-related factors driving dispersal towards more populated and accessible areas. These analyses have potential to guide spatial targeting of vaccination, and enhancement of surveillance in at-risk areas[18,89,170]. Frequent reintroductions highlighted epidemiological connectivity of landscapes over which vaccination needs scaling and coordination across political/administrative boundaries[13], with more recent studies showing how sequencing can be used to monitor the impacts of and threats to dog vaccination programmes[13,98,149].
Broader context
Our review has relevance to the broader application of genetic surveillance to pathogens. The recent increase in third-generation sequencing of RABV has potential to further expand given the focus on sequencing capacity for pandemic response. Deployable sequencing has become a key component of outbreak response, with portable lab equipment and sequencing platforms facilitating on-site, real-time, genomic surveillance[33,40,43]. Feasibility and utility of deployable sequencing for rabies surveillance has been demonstrated[46,98,149], with use of Nanopore’s MinIon reducing costs and turnaround times[34,46], and comprehensive protocols, bioinformatic pipelines and open-source user-friendly software and classification tools becoming more available[171-175]. Sequencing for routine surveillance of endemic zoonoses could build and sustain pandemic preparedness at the human-animal-environment interface. While COVID-19 accelerated application of genomic surveillance, it also highlighted stark global disparities in access to sequencing, and bioinformatic expertise remains a bottleneck[176,177]. Since the pandemic investment in LMICs has begun but much more is required[178].
RABV also serves as a model system to understand and manage cross-species transmission and spillover. Instances highlighted in this review, reflect a broader ecological pattern with significant public health and ecological implications. Spillovers can be precursors to larger outbreaks, as evidenced by recent epidemics of Influenza, Ebola, Zika, and COVID-19, while swift sequencing and analysis can inform public health responses and containment[33-35,40,43].
Limitations of the study
While we endeavoured to comprehensively review global regions with endemic canine rabies, it is important to acknowledge the limitations inherent in our study. Genetic studies on rabies demonstrating progress in its elimination, conducted in countries or regions such as Canada, the USA, and Eastern Europe, where dog rabies has been eliminated for some time, were not included. This approach may have limited our ability to discover additional interventions and significant insights from the perspective of these countries. Our review, while focusing on publications from endemic regions may have failed to identify publications documenting importations in rabies-free countries. We supplemented our searches by manually adding relevant instances (~10% of papers), but some studies, particularly those published in non-English journals, may have been overlooked. Despite substantial manual curation efforts in RABV-GLUE to enhance GenBank metadata[179], inconsistencies and gaps persist, potentially hindering data mining efficiency for sequences not associated with a publication. We also only retrospectively and manually identified some studies that did not report the use of sequencing in their abstract. Not all papers transparently report methods, in particular many did not report locations where sequencing was undertaken which we assumed was done in-country. We therefore likely overestimated sequencing capacity for some countries, though we expect our conclusions are robust. We included only studies published after the year 2000, which limited the scope of the study, as it did not capture some of the earliest genomic studies conducted on RABV. However, this also meant most of the methods reported were more comparable and aligned with the 1st, 2nd and 3rd generation sequencing platforms defined in Table 1.
Conclusions and future recommendations
The promise of genetic data for informing rabies control is evident, but its full potential remains untapped as most publications advocate generic measures that lack specificity derived from sequencing. More demanding phylodynamic analysis, integrating geographical, epidemiological, and genetic data to yield more detailed and quantitative understanding, requires greater expertise and computational resources that have not been accessible in LMICs. Future research would benefit from more WGS as well as leveraging the power of existing data through analyses that integrate partial and WGS[148]. Applied insights to be gained by enhancing rabies surveillance with sequencing, lie in the knowledge of what is circulating and how it is spreading while we gear towards elimination. A standardised nomenclature system for categorising RABV diversity, would facilitate clear communication and collaboration among researchers, healthcare professionals and policymakers[175]. We recommend efforts to develop a robust taxonomic classification system under the International Committee on Taxonomy of Viruses (ICTV) that is capable of integrating all existing and newly identified RABV sequences. Scaling up sequencing in endemic countries, with laboratory networking and a more unified terminology for exchange of information and updates on new variants and lineages could enhance risk assessment and control strategies. Genetic results underscore the need for international and regional coordination in controlling transboundary spread, to accelerate progress and maintain gains. Expanding sequencing initiatives and fostering collaborative efforts will support the “Zero by 30” goal, and serve as a prime example of a genomics-informed One Health approach, building capacity for the future[180].
DECLARATIONS
Authors’ contributions
Design: Brunker K, Thumbi SM, Hampson K, Oyugi JO
Literature search: Jaswant G, Bautista CT
Data analysis: Jaswant G, Bautista CT, Hampson K, Brunker K, Ogoti B, Changalucha J, Campbell K, Mutunga M
Manuscript writing: Jaswant G, Bautista CT
Manuscript editing: Hampson K, Brunker K
Availability of data and materials
Data and code to reproduce the analyses and figures are available from our public repository https://github.com/RAGE-toolkit/RABV_geneticSurv_review.
Financial support and sponsorship
This work was supported by Wellcome [207569/Z/17/Z, 224520/Z/21/Z to KH], the UK Medical Research Council [MR/X002047/1 to KB], a Genomics and Modelling for the Control of Viral Pathogens (GeMVi) fellowship to GJ funded by the National Institute for Health Research (NIHR) [176382], a Philippines Department of Science and Technology (DOST) and British Council studentship to CB, a training fellowship in Public Health and Tropical Medicine [110330 to SMT] and Institutional Strategic Support Fund grants at the University of Glasgow [204820 to KB].
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2024.
Supplementary Materials
REFERENCES
1. Hampson K, Coudeville L, Lembo T, et al; Global Alliance for Rabies Control Partners for Rabies Prevention. Estimating the global burden of endemic canine rabies. PLoS Negl Trop Dis 2015;9:e0003709.
2. Worsley-Tonks KEL, Escobar LE, Biek R, et al. Using host traits to predict reservoir host species of rabies virus. PLoS Negl Trop Dis 2020;14:e0008940.
3. Rabies - Bulletin - Europe. Epidemiology of Rabies. Available from: https://www.who-rabies-bulletin.org/site-page/epidemiology-rabies. [Last accessed on 28 May 2024].
4. Sabeta CT, Janse van Rensburg D, Phahladira B, et al. Rabies of canid biotype in wild dog (Lycaon pictus) and spotted hyaena
5. Faria NR, Suchard MA, Rambaut A, Streicker DG, Lemey P. Simultaneously reconstructing viral cross-species transmission history and identifying the underlying constraints. Philos Trans R Soc Lond B Biol Sci 2013;368:20120196.
6. Viana M, Mancy R, Biek R, et al. Assembling evidence for identifying reservoirs of infection. Trends Ecol Evol 2014;29:270-9.
7. Haydon DT, Cleaveland S, Taylor LH, Laurenson MK. Identifying reservoirs of infection: a conceptual and practical challenge. Emerg Infect Dis 2002;8:1468-73.
8. Lembo T, Hampson K, Kaare MT, et al. The feasibility of canine rabies elimination in Africa: dispelling doubts with data. PLoS Negl Trop Dis 2010;4:e626.
9. Lembo T, Haydon DT, Velasco-Villa A, et al. Molecular epidemiology identifies only a single rabies virus variant circulating in complex carnivore communities of the Serengeti. Proc Biol Sci 2007;274:2123-30.
10. Fisher CR, Streicker DG, Schnell MJ. The spread and evolution of rabies virus: conquering new frontiers. Nat Rev Microbiol 2018;16:241-55.
11. Rose R, Hall M, Redd AD, et al. Phylogenetic methods inconsistently predict the direction of HIV transmission among heterosexual pairs in the HPTN 052 cohort. J Infect Dis 2019;220:1406-13.
12. Mancy R, Rajeev M, Lugelo A, et al. Rabies shows how scale of transmission can enable acute infections to persist at low prevalence. Science 2022;376:512-6.
13. Zinsstag J, Lechenne M, Laager M, et al. Vaccination of dogs in an African city interrupts rabies transmission and reduces human exposure. Sci Transl Med 2017;9:eaaf6984.
14. Bourhy H, Nakouné E, Hall M, et al. Revealing the micro-scale signature of endemic zoonotic disease transmission in an African urban setting. PLoS Pathog 2016;12:e1005525.
15. Campbell K, Gifford RJ, Singer J, et al. Making genomic surveillance deliver: a lineage classification and nomenclature system to inform rabies elimination. PLoS Pathog 2022;18:e1010023.
16. Baum D. Reading a phylogenetic tree: the meaning of monophyletic groups. 2008. Available from: https://www.nature.com/scitable/topicpage/reading-a-phylogenetic-tree-the-meaning-of-41956/. [Last accessed on 28 May 2024].
17. Pybus OG, Rambaut A. Evolutionary analysis of the dynamics of viral infectious disease. Nat Rev Genet 2009;10:540-50.
18. Dellicour S, Troupin C, Jahanbakhsh F, et al. Using phylogeographic approaches to analyse the dispersal history, velocity and direction of viral lineages - Application to rabies virus spread in Iran. Mol Ecol 2019;28:4335-50.
20. Kchouk M, Gibrat JF, Elloumi M. Generations of sequencing technologies: from first to next generation. Biol Med 2017;9:3. Available from: https://www.walshmedicalmedia.com/open-access/generations-of-sequencing-technologies-from-first-to-next-generation-0974-8369-1000395.pdf. [Last accessed on 28 May 2024]
21. Pant GR, Lavenir R, Wong FY, et al. Recent emergence and spread of an Arctic-related phylogenetic lineage of rabies virus in Nepal. PLoS Negl Trop Dis 2013;7:e2560.
22. Wunner WH, Larson JK, Dietzschold B, Smith CL. The molecular biology of rabies viruses. Rev Infect Dis 1988;10:S771-84.
23. Voloch CM, Capellão RT, Mello B, Schrago CG. Analysis of adaptive evolution in Lyssavirus genomes reveals pervasive diversifying selection during species diversification. Viruses 2014;6:4465-78.
24. WHO. Weekly Epidemiological Record, 2018, vol. 93, 20. Available from: https://www.who.int/publications-detail-redirect/WER9320. [Last accessed on 28 May 2024].
25. WHO. WHO expert consultation on rabies. 2005. Available from: https://apps.who.int/iris/bitstream/handle/10665/85346/9789241209823_eng.pdf?sequence=1. [Last accessed on 28 May 2024].
26. WHO. WHO Expert consultation on rabies: WHO TRS N°1012. Available from: https://www.who.int/publications-detail-redirect/WHO-TRS-1012. [Last accessed on 28 May 2024].
27. Nguyen AKT, Vu AH, Nguyen TT, et al. Risk factors and protective immunity against rabies in unvaccinated butchers working at dog slaughterhouses in Northern Vietnam. Am J Trop Med Hyg 2021;105:788-93.
28. Minghui R, Stone M, Semedo MH, Nel L. New global strategic plan to eliminate dog-mediated rabies by 2030. Lancet Glob Health 2018;6:e828-9.
29. Cutts FT, Waldman RJ, Zoffman HM. Surveillance for the expanded programme on immunization. Bull World Health Organ 1993;71:633-9.
30. Thacker SB, Berkelman RL. Public health surveillance in the United States. Epidemiol Rev 1988;10:164-90.
31. Wallace RM, Reses H, Franka R, et al. Establishment of a high canine rabies burden in Haiti through the implementation of a novel surveillance program [corrected]. PLoS Negl Trop Dis 2015;9:e0004245.
32. Talbi C, Lemey P, Suchard MA, et al. Phylodynamics and human-mediated dispersal of a zoonotic virus. PLoS Pathog 2010;6:e1001166.
33. Bedford T, Cobey S, Beerli P, Pascual M. Global migration dynamics underlie evolution and persistence of human influenza A (H3N2). PLoS Pathog 2010;6:e1000918.
34. Lemey P, Rambaut A, Bedford T, et al. Unifying viral genetics and human transportation data to predict the global transmission dynamics of human influenza H3N2. PLoS Pathog 2014;10:e1003932.
35. Quick J, Loman NJ, Duraffour S, et al. Real-time, portable genome sequencing for Ebola surveillance. Nature 2016;530:228-32.
36. Quick J, Grubaugh ND, Pullan ST, et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat Protoc 2017;12:1261-76.
37. Giovanetti M, Pinotti F, Zanluca C, et al. Genomic epidemiology unveils the dynamics and spatial corridor behind the Yellow Fever virus outbreak in Southern Brazil. Sci Adv 2023;9:eadg9204.
38. Faria NR, Kraemer MUG, Hill SC, et al. Genomic and epidemiological monitoring of yellow fever virus transmission potential. Science 2018;361:894-9.
39. Roychoudhury P, Sereewit J, Xie H, et al. Genomic analysis of early monkeypox virus outbreak strains, Washington, USA. Emerg Infect Dis 2023;29:644-6.
40. Kugelman JR, Johnston SC, Mulembakani PM, et al. Genomic variability of monkeypox virus among humans, Democratic Republic of the Congo. Emerg Infect Dis 2014;20:232-9.
41. Forni D, Cagliani R, Molteni C, Clerici M, Sironi M. Monkeypox virus: the changing facets of a zoonotic pathogen. Infect Genet Evol 2022;105:105372.
42. Li Y, Hou J, Sun Z, et al. Monkeypox virus 2022, gene heterogeneity and protein polymorphism. Signal Transduct Target Ther 2023;8:278.
43. Faria NR, Mellan TA, Whittaker C, et al. Genomics and epidemiology of the P.1 SARS-CoV-2 lineage in Manaus, Brazil. Science 2021;372:815-21.
44. Trewby H, Nadin-Davis SA, Real LA, Biek R. Processes underlying rabies virus incursions across US-Canada border as revealed by whole-genome phylogeography. Emerg Infect Dis 2017;23:1454-61.
45. Layan M, Dellicour S, Baele G, Cauchemez S, Bourhy H. Mathematical modelling and phylodynamics for the study of dog rabies dynamics and control: a scoping review. PLoS Negl Trop Dis 2021;15:e0009449.
46. Brunker K, Jaswant G, Thumbi SM, et al. Rapid in-country sequencing of whole virus genomes to inform rabies elimination programmes. Wellcome Open Res 2020;5:3.
47. Okeke IN, Ihekweazu C. The importance of molecular diagnostics for infectious diseases in low-resource settings. Nat Rev Microbiol 2021;19:547-8.
48. Page MJ, Moher D, Bossuyt PM, et al. PRISMA 2020 explanation and elaboration: updated guidance and exemplars for reporting systematic reviews. BMJ 2021;372:n160.
49. The R project for statistical computing. Available from: https://www.r-project.org/. [Last accessed on 28 May 2024].
50. Ouzzani M, Hammady H, Fedorowicz Z, Elmagarmid A. Rayyan-a web and mobile app for systematic reviews. Syst Rev 2016;5:210.
51. Talbi C, Holmes EC, de Benedictis P, et al. Evolutionary history and dynamics of dog rabies virus in western and central Africa. J Gen Virol 2009;90:783-91.
52. Troupin C, Dacheux L, Tanguy M, et al. Large-scale phylogenomic analysis reveals the complex evolutionary history of rabies virus in multiple carnivore hosts. PLoS Pathog 2016;12:e1006041.
53. Hayman DT, Fooks AR, Marston DA, Garcia-R JC. The global phylogeography of lyssaviruses - challenging the ‘Out of Africa’ hypothesis. PLoS Negl Trop Dis 2016;10:e0005266.
54. Bourhy H, Reynes JM, Dunham EJ, et al. The origin and phylogeography of dog rabies virus. J Gen Virol 2008;89:2673-81.
55. Oem JK, Kim SH, Kim YH, Lee MH, Lee KK. Reemergence of rabies in the southern Han river region, Korea. J Wildl Dis 2014;50:681-8.
56. Velasco-Villa A, Mauldin MR, Shi M, et al. The history of rabies in the Western Hemisphere. Antiviral Res 2017;146:221-32.
57. Zeynalova S, Shikhiyev M, Aliyeva T, et al. Epidemiological characteristics of human and animal rabies in Azerbaijan. Zoonoses Public Health 2015;62:111-8.
58. Tenzin, Wacharapluesadee S, Denduangboripant J, et al. Rabies virus strains circulating in Bhutan: implications for control. Epidemiol Infect 2011;139:1457-62.
59. Nadin-Davis SA, Turner G, Paul JP, Madhusudana SN, Wandeler AI. Emergence of Arctic-like rabies lineage in India. Emerg Infect Dis 2007;13:111-6.
60. Sato G, Kobayashi Y, Shoji Y, et al. Molecular epidemiology of rabies from Maranhão and surrounding states in the northeastern region of Brazil. Arch Virol 2006;151:2243-51.
61. Carnieli P Jr, de Novaes Oliveira R, de Oliveira Fahl W, et al. Phylogenetic analysis of partial RNA-polymerase blocks II and III of Rabies virus isolated from the main rabies reservoirs in Brazil. Virus Genes 2012;45:76-83.
62. Johnson N, Letshwenyo M, Baipoledi EK, Thobokwe G, Fooks AR. Molecular epidemiology of rabies in Botswana: a comparison between antibody typing and nucleotide sequence phylogeny. Vet Microbiol 2004;101:31-8.
63. Nadin-Davis SA, Simani S, Armstrong J, Fayaz A, Wandeler AI. Molecular and antigenic characterization of rabies viruses from Iran identifies variants with distinct epidemiological origins. Epidemiol Infect 2003;131:777-90.
64. Ismail MZ, Al-Hamdi NK, Al-Amery AN, et al. Quantifying and mapping the burden of human and animal rabies in Iraq. PLoS Negl Trop Dis 2020;14:e0008622.
65. Horton DL, Ismail MZ, Siryan ES, et al. Rabies in Iraq: trends in human cases 2001-2010 and characterisation of animal rabies strains from Baghdad. PLoS Negl Trop Dis 2013;7:e2075.
66. Oude Munnink BB, Farag EABA, GeurtsvanKessel C, et al. First molecular analysis of rabies virus in Qatar and clinical cases imported into Qatar, a case report. Int J Infect Dis 2020;96:323-6.
67. Boldbaatar B, Inoue S, Tuya N, et al. Molecular epidemiology of rabies virus in Mongolia, 2005-2008. Jpn J Infect Dis 2010;63:358-63.
68. Meng S, Sun Y, Wu X, et al. Evolutionary dynamics of rabies viruses highlights the importance of China rabies transmission in Asia. Virology 2011;410:403-9.
69. Tuvshintulga B, Batmagnai E, Bazarragchaa E, Dulam P, Sugar S, Battsetseg B. Detection and molecular characterization of rabies virus in Mongolia during 2008-2010. Int J One Health 2015;1:26-31.
70. Yin JF, Wang JL, Tang Q, et al. Identification of animal rabies in Inner Mongolia and analysis of the etiologic characteristics. Biomed Environ Sci 2014;27:35-44.
71. Zhang Y, Vrancken B, Feng Y, et al. Cross-border spread, lineage displacement and evolutionary rate estimation of rabies virus in Yunnan Province, China. Virol J 2017;14:102.
72. Feng Y, Wang W, Guo J, et al. Disease outbreaks caused by steppe-type rabies viruses in China. Epidemiol Infect 2015;143:1287-91.
73. Tao XY, Tang Q, Rayner S, et al. Molecular phylodynamic analysis indicates lineage displacement occurred in Chinese rabies epidemics between 1949 to 2010. PLoS Negl Trop Dis 2013;7:e2294.
74. Wang L, Wu X, Bao J, Song C, Du J. Phylodynamic and transmission pattern of rabies virus in China and its neighboring countries. Arch Virol 2019;164:2119-29.
75. Liu H, Li L, Yuan X, et al. Rabies viruses in specific wild fur animals in northern China, 2017-2019. Transbound Emerg Dis 2020;67:2307-12.
76. Deviatkin AA, Lukashev AN, Poleshchuk EM, et al. The phylodynamics of the rabies virus in the Russian Federation. PLoS One 2017;12:e0171855.
77. Mey C, Metlin A, Duong V, et al. Evidence of two distinct phylogenetic lineages of dog rabies virus circulating in Cambodia. Infect Genet Evol 2016;38:55-61.
78. Tohma K, Saito M, Demetria CS, et al. Molecular and mathematical modeling analyses of inter-island transmission of rabies into a previously rabies-free island in the Philippines. Infect Genet Evol 2016;38:22-8.
79. Tohma K, Saito M, Kamigaki T, et al. Phylogeographic analysis of rabies viruses in the Philippines. Infect Genet Evol 2014;23:86-94.
80. Yamagata J, Ahmed K, Khawplod P, et al. Molecular epidemiology of rabies in Vietnam. Microbiol Immunol 2007;51:833-40.
81. Reddy RVC, Mohana Subramanian B, Surendra KSNL, et al. Rabies virus isolates of India - simultaneous existence of two distinct evolutionary lineages. Infect Genet Evol 2014;27:163-72.
82. Matsumoto T, Ahmed K, Karunanayake D, et al. Molecular epidemiology of human rabies viruses in Sri Lanka. Infect Genet Evol 2013;18:160-7.
83. Nanayakkara S, Smith JS, Rupprecht CE. Rabies in Sri Lanka: splendid isolation. Emerg Infect Dis 2003;9:368-71.
84. Baby J, Mani RS, Abraham SS, et al. Natural rabies infection in a domestic fowl (Gallus domesticus): a report from India. PLoS Negl Trop Dis 2015;9:e0003942.
85. Boldbaatar B, Inoue S, Tuya N, et al. Molecular epidemiology of rabies virus in Mongolia, 2005-2008. Jpn J Infect Dis 2010;63:358-63.
86. Yang DK, Park YN, Hong GS, et al. Molecular characterization of Korean rabies virus isolates. J Vet Sci 2011;12:57-63.
87. Hyun BH, Lee KK, Kim IJ, et al. Molecular epidemiology of rabies virus isolates from South Korea. Virus Res 2005;114:113-25.
88. Shao XQ, Yan XJ, Luo GL, et al. Genetic evidence for domestic raccoon dog rabies caused by Arctic-like rabies virus in Inner Mongolia, China. Epidemiol Infect 2011;139:629-35.
89. Tao XY, Guo ZY, Li H, et al. Rabies cases in the west of China have two distinct origins. PLoS Negl Trop Dis 2015;9:e0004140.
90. Jamil KM, Ahmed K, Hossain M, et al. Arctic-like rabies virus, Bangladesh. Emerg Infect Dis 2012;18:2021-4.
91. Nagaraja T, Madhusudana S, Desai A. Molecular characterization of the full-length genome of a rabies virus isolate from India. Virus Genes 2008;36:449-59.
92. Cherian S, Singh R, Singh KP, et al. Phylogenetic analysis of Indian rabies virus isolates targeting the complete glycoprotein gene. Infect Genet Evol 2015;36:333-8.
93. Reddy RVC, Mukherjee F, Rana SK, et al. Rabies virus infection in domestic buffaloes and wild animals in India. J Adv Vet Res 2015;5:68-83. Available from: https://advetresearch.com/index.php/AVR/article/view/49. [Last accessed on 28 May 2024]
94. Manjunatha Reddy GB, Krishnappa S, Vinayagamurthy B, et al. Molecular epidemiology of rabies virus circulating in domestic animals in India. Virusdisease 2018;29:362-8.
95. Reddy GBM, Singh R, Singh KP, et al. Molecular epidemiological analysis of wild animal rabies isolates from India. Vet World 2019;12:352-7.
96. Sadeuh-Mba SA, Momo JB, Besong L, Loul S, Njouom R. Molecular characterization and phylogenetic relatedness of dog-derived Rabies Viruses circulating in Cameroon between 2010 and 2016. PLoS Negl Trop Dis 2017;11:e0006041.
97. Kia G, Huang Y, Zhou M, et al. Molecular characterization of a rabies virus isolated from trade dogs in Plateau State, Nigeria. Sokoto J Vet Sc 2018;16:54.
98. Gigante CM, Yale G, Condori RE, et al. Portable rabies virus sequencing in canine rabies endemic countries using the Oxford nanopore MinION. Viruses 2020;12:1255.
99. Hayman DT, Johnson N, Horton DL, et al. Evolutionary history of rabies in Ghana. PLoS Negl Trop Dis 2011;5:e1001.
100. Coetzer A, Anahory I, Dias PT, et al. Enhanced diagnosis of rabies and molecular evidence for the transboundary spread of the disease in Mozambique. J S Afr Vet Assoc 2017;88:e1-9.
101. Hirano S, Itou T, Shibuya H, Kashiwazaki Y, Sakai T. Molecular epidemiology of rabies virus isolates in Uganda. Virus Res 2010;147:135-8.
102. Feng Y, Wang Y, Xu W, et al. Animal rabies surveillance, China, 2004-2018. Emerg Infect Dis 2020;26:2825-34.
103. Tu C, Feng Y, Wang Y. Animal rabies in the People’s Republic of China. Rev Sci Tech 2018;37:519-28.
104. Guo Z, Tao X, Yin C, et al. National borders effectively halt the spread of rabies: the current rabies epidemic in China is dislocated from cases in neighboring countries. PLoS Negl Trop Dis 2013;7:e2039.
105. Feng Y, Wang Y, Hada, et al. Diversity of rabies virus detected in Inner Mongolia, China, 2019-2021. Transbound Emerg Dis 2022;69:249-53.
106. Dibia IN, Sumiarto B, Susetya H, Putra AA, Scott-Orr H, Mahardika GN. Phylogeography of the current rabies viruses in Indonesia. J Vet Sci 2015;16:459-66.
107. Susetya H, Sugiyama M, Inagaki A, Ito N, Mudiarto G, Minamoto N. Molecular epidemiology of rabies in Indonesia. Virus Res 2008;135:144-9.
108. Susilawathi NM, Darwinata AE, Dwija IB, et al. Epidemiological and clinical features of human rabies cases in Bali 2008-2010. BMC Infect Dis 2012;12:81.
109. Tsai KJ, Hsu WC, Chuang WC, et al. Emergence of a sylvatic enzootic formosan ferret badger-associated rabies in Taiwan and the geographical separation of two phylogenetic groups of rabies viruses. Vet Microbiol 2016;182:28-34.
110. Chiou HY, Hsieh CH, Jeng CR, Chan FT, Wang HY, Pang VF. Molecular characterization of cryptically circulating rabies virus from ferret badgers, Taiwan. Emerg Infect Dis 2014;20:790-8.
111. Lin YC, Chu PY, Chang MY, Hsiao KL, Lin JH, Liu HF. Spatial temporal dynamics and molecular evolution of re-emerging rabies virus in Taiwan. Int J Mol Sci 2016;17:392.
112. Saito M, Oshitani H, Orbina JR, et al. Genetic diversity and geographic distribution of genetically distinct rabies viruses in the Philippines. PLoS Negl Trop Dis 2013;7:e2144.
113. Tao XY, Tang Q, Li H, et al. Molecular epidemiology of rabies in Southern People’s Republic of China. Emerg Infect Dis 2009;15:1192-8.
114. Liu Q, Xiong Y, Luo TR, et al. Molecular epidemiology of rabies in Guangxi Province, south of China. J Clin Virol 2007;39:295-303.
115. Zhang YZ, Xiong CL, Zou Y, et al. Molecular characterization of rabies virus isolates in China during 2004. Virus Res 2006;121:179-88.
116. Garcés-Ayala F, Aréchiga-Ceballos N, Ortiz-Alcántara JM, et al. Molecular characterization of atypical antigenic variants of canine rabies virus reveals its reintroduction by wildlife vectors in southeastern Mexico. Arch Virol 2017;162:3629-37.
117. Scott TP, Fischer M, Khaiseb S, et al. Complete genome and molecular epidemiological data infer the maintenance of rabies among kudu (Tragelaphus strepsiceros) in Namibia. PLoS One 2013;8:e58739.
118. Lan YC, Wen TH, Chang CC, et al. Indigenous wildlife rabies in Taiwan: ferret badgers, a long term terrestrial reservoir. Biomed Res Int 2017;2017:5491640.
119. Zhao JH, Zhao LF, Liu F, Jiang HY, Yang JL. Ferret badger rabies in Zhejiang, Jiangxi and Taiwan, China. Arch Virol 2019;164:579-84.
120. David D, Dveres N, Yakobson BA, Davidson I. Emergence of dog rabies in the northern region of Israel. Epidemiol Infect 2009;137:544-8.
121. Marston DA, Horton DL, Nunez J, et al. Genetic analysis of a rabies virus host shift event reveals within-host viral dynamics in a new host. Virus Evol 2017;3:vex038.
122. East ML, Hofer H, Cox JH, Wulle U, Wiik H, Pitra C. Regular exposure to rabies virus and lack of symptomatic disease in Serengeti spotted hyenas. Proc Natl Acad Sci U S A 2001;98:15026-31.
123. Lembo T, Hampson K, Haydon DT, et al. Exploring reservoir dynamics: a case study of rabies in the Serengeti ecosystem. J Appl Ecol 2008;45:1246-57.
124. Marston DA, Horton DL, Ngeleja C, et al. Ikoma lyssavirus, highly divergent novel lyssavirus in an African civet. Emerg Infect Dis 2012;18:664-7.
125. Madhusudana SN, Mani R, Ashwin YB, Desai A. Rabid fox bites and human rabies in a village community in southern India: epidemiological and laboratory investigations, management and follow-up. Vector Borne Zoonotic Dis 2013;13:324-9.
126. Velasco-Villa A, Messenger SL, Orciari LA, et al. New rabies virus variant in Mexican immigrant. Emerg Infect Dis 2008;14:1906-8.
127. Smithsonian’s National Zoo and Conservation Biology Institute. Red panda. Available from: https://nationalzoo.si.edu/animals/red-panda. [Last accessed on 28 May 2024].
128. Weyer J, Szmyd-Potapczuk AV, Blumberg LH, et al. Epidemiology of human rabies in South Africa, 1983-2007. Virus Res 2011;155:283-90.
129. de Brito MG, Chamone TL, da Silva FJ, et al. Antemortem diagnosis of human rabies in a veterinarian infected when handling a herbivore in Minas Gerais, Brazil. Rev Inst Med Trop Sao Paulo 2011;53:39-44.
130. Ogo M, Nel L, Sabeta C. Phylogenetic evidence of the public and veterinary health threat of dog rabies in Nigeria. Nig Vet J 2011;32.
131. Contou D, Dacheux L, Bendib I, et al. Severe ketoalkalosis as initial presentation of imported human rabies in France. J Clin Microbiol 2015;53:1979-82.
132. Smith J, McElhinney L, Parsons G, et al. Case report: rapid ante-mortem diagnosis of a human case of rabies imported into the UK from the Philippines. J Med Virol 2003;69:150-5.
133. Nosaki Y, Maeda K, Watanabe M, et al. Fourth imported rabies case since the eradication of rabies in Japan in 1957. J Travel Med 2021;28:taab151.
134. Rimhanen-Finne R, Järvinen A, Kuusi M, et al. Imported human rabies, the Philippines and Finland, 2007. Emerg Infect Dis 2010;16:1318-9.
135. Johnson N, Fooks A, McColl K. Human rabies case with long incubation, Australia. Emerg Infect Dis 2008;14:1950-1.
136. Coetzee P, Weyer J, Paweska JT, Burt FJ, Markotter W, Nel LH. Use of a molecular epidemiological database to track human rabies case histories in South Africa. Epidemiol Infect 2008;136:1270-6.
137. Sabeta CT, Weyer J, Geertsma P, et al. Emergence of rabies in the Gauteng Province, South Africa: 2010-2011. J S Afr Vet Assoc 2013;84:E1-5.
138. Mahardika GN, Dibia N, Budayanti NS, et al. Phylogenetic analysis and victim contact tracing of rabies virus from humans and dogs in Bali, Indonesia. Epidemiol Infect 2014;142:1146-54.
139. Queiroz LH, Favoretto SR, Cunha EMS, et al. Rabies in southeast Brazil: a change in the epidemiological pattern. Arch Virol 2012;157:93-105.
140. Brunker K, Marston DA, Horton DL, et al. Elucidating the phylodynamics of endemic rabies virus in eastern Africa using whole-genome sequencing. Virus Evol 2015;1:vev011.
141. Gong W, Jiang Y, Za Y, et al. Temporal and spatial dynamics of rabies viruses in China and Southeast Asia. Virus Res 2010;150:111-8.
142. Tao XY, Li ML, Wang Q, et al. The reemergence of human rabies and emergence of an Indian subcontinent lineage in Tibet, China. PLoS Negl Trop Dis 2019;13:e0007036.
143. Zhang HL, Zhang YZ, Yang WH, et al. Molecular epidemiology of reemergent rabies in Yunnan Province, southwestern China. Emerg Infect Dis 2014;20:1433-42.
144. Omodo M, Ar Gouilh M, Mwiine FN, et al. Rabies in Uganda: rabies knowledge, attitude and practice and molecular characterization of circulating virus strains. BMC Infect Dis 2020;20:200.
145. Yu J, Li H, Tang Q, et al. The spatial and temporal dynamics of rabies in China. PLoS Negl Trop Dis 2012;6:e1640.
146. Tian H, Feng Y, Vrancken B, et al. Transmission dynamics of re-emerging rabies in domestic dogs of rural China. PLoS Pathog 2018;14:e1007392.
147. Carnieli P Jr, Ruthner Batista HB, de Novaes Oliveira R, Castilho JG, Vieira LF. Phylogeographic dispersion and diversification of rabies virus lineages associated with dogs and crab-eating foxes (Cerdocyon thous) in Brazil. Arch Virol 2013;158:2307-13.
148. Holtz A, Baele G, Bourhy H, Zhukova A. Integrating full and partial genome sequences to decipher the global spread of canine rabies virus. Nat Commun 2023;14:4247.
149. Lushasi K, Brunker K, Rajeev M, et al. Integrating contact tracing and whole-genome sequencing to track the elimination of dog-mediated rabies: an observational and genomic study. Elife 2023;12:e85262.
150. Muleya W, Chambaro HM, Sasaki M, et al. Genetic diversity of rabies virus in different host species and geographic regions of Zambia and Zimbabwe. Virus Genes 2019;55:713-9.
151. Coetzer A, Coertse J, Makalo MJ, Molomo M, Markotter W, Nel LH. Epidemiology of rabies in Lesotho: the importance of routine surveillance and virus characterization. Trop Med Infect Dis 2017;2:30.
152. Carnieli P Jr, de Novaes Oliveira R, Macedo CI, Castilho JG. Phylogeography of rabies virus isolated from dogs in Brazil between 1985 and 2006. Arch Virol 2011;156:1007-12.
153. Denduangboripant J, Wacharapluesadee S, Lumlertdacha B, et al. Transmission dynamics of rabies virus in Thailand: implications for disease control. BMC Infect Dis 2005;5:52.
154. Ma C, Hao X, Deng H, et al. Re-emerging of rabies in Shaanxi province, China, from 2009 to 2015. J Med Virol 2017;89:1511-9.
155. Brunker K, Lemey P, Marston DA, et al. Landscape attributes governing local transmission of an endemic zoonosis: rabies virus in domestic dogs. Mol Ecol 2018;27:773-88.
156. Carnieli P Jr, Castilho JG, Fahl Wde O, Véras NM, Carrieri ML, Kotait I. Molecular characterization of rabies virus isolates from dogs and crab-eating foxes in Northeastern Brazil. Virus Res 2009;141:81-9.
157. Kobayashi Y, Inoue N, Sato G, et al. Phylogenetic characterization of rabies virus isolates from Carnivora in Brazil. J Vet Med Sci 2007;69:691-6.
158. Ma J, Li S, Yang Y, Wang Q, Huo Y. Epidemiological and phylogenetic analysis of rabies virus isolated from humans in Henan province, China. Arch Virol 2019;164:2811-7.
159. Zhang JY, Zhang B, Zhang SF, et al. Dog-transmitted Rabies in Beijing, China. Biomed Environ Sci 2017;30:526-9.
160. Zhang S, Liu Y, Hou Y, et al. Epidemic and maintenance of rabies in Chinese ferret badgers (Melogale moschata) indicated by epidemiology and the molecular signatures of rabies viruses. Virol Sin 2013;28:146-51.
161. Chao J, Peng Q, Zhao J, et al. Different rabies outbreaks on two beef cattle farms in the same province of China: diagnosis, virus characterization and epidemiological analysis. Transbound Emerg Dis 2021;68:1216-28.
162. Mehta S, Charan P, Dahake R, Mukherjee S, Chowdhary A. Molecular characterization of nucleoprotein gene of rabies virus from Maharashtra, India. J Postgrad Med 2016;62:105-8.
163. Vigilato MA, Clavijo A, Knobl T, et al. Progress towards eliminating canine rabies: policies and perspectives from Latin America and the Caribbean. Philos Trans R Soc Lond B Biol Sci 2013;368:20120143.
164. Gibson AD, Yale G, Corfmat J, et al. Elimination of human rabies in Goa, India through an integrated one health approach. Nat Commun 2022;13:2788.
165. Velasco-Villa A, Orciari LA, Souza V, et al. Molecular epizootiology of rabies associated with terrestrial carnivores in Mexico. Virus Res 2005;111:13-27.
166. Chang JC, Tsai KJ, Hsu WC, et al. Rabies virus infection in ferret badgers (Melogale moschata subaurantiaca) in Taiwan: a retrospective study. J Wildl Dis 2015;51:923-8.
167. Schmiedel S, Panning M, Lohse A, et al. Case report on fatal human rabies infection in Hamburg, Germany, March 2007. Euro Surveill 2007;12:E070531.5.
168. Solomon T, Marston D, Mallewa M, et al. Paralytic rabies after a two week holiday in India. BMJ 2005;331:501-3.
169. European Centre for Disease Prevention and Control. Rabies - Annual epidemiological report for 2019. Available from: https://www.ecdc.europa.eu/en/publications-data/rabies-annual-epidemiological-report-2019. [Last accessed on 28 May 2024].
170. Lumlertdacha B, Wacharapluesadee S, Denduangboripant J, et al. Complex genetic structure of the rabies virus in Bangkok and its surrounding provinces, Thailand: implications for canine rabies control. Trans R Soc Trop Med Hyg 2006;100:276-81.
171. Theys K, Lemey P, Vandamme AM, Baele G. Advances in visualization tools for phylogenomic and phylodynamic studies of viral diseases. Front Public Health 2019;7:208.
172. Hadfield J, Megill C, Bell SM, et al. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics 2018;34:4121-3.
173. O'Toole Á, Scher E, Underwood A, et al. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol 2021;7:veab064.
174. Bautista C, Jaswant G, French H, et al. Whole genome sequencing for rapid characterization of rabies virus using nanopore technology. J Vis Exp 2023.
175. ARTIC network. Available from: https://artic.network/. [Last accessed on 28 May 2024].
176. Brito AF, Semenova E, Dudas G, et al; Bulgarian SARS-CoV-2 sequencing group; Communicable Diseases Genomics Network (Australia and New Zealand); COVID-19 Impact Project; Danish Covid-19 Genome Consortium; Fiocruz COVID-19 Genomic Surveillance Network; GISAID core curation team; Network for Genomic Surveillance in South Africa (NGS-SA); Swiss SARS-CoV-2 Sequencing Consortium. Global disparities in SARS-CoV-2 genomic surveillance. Nat Commun 2022;13:7003.
177. Ling-Hu T, Rios-Guzman E, Lorenzo-Redondo R, Ozer EA, Hultquist JF. Challenges and Opportunities For Global Genomic Surveillance Strategies in the COVID-19 era. Viruses 2022;14:2532.
178. Inzaule SC, Tessema SK, Kebede Y, Ogwell Ouma AE, Nkengasong JN. Genomic-informed pathogen surveillance in Africa: opportunities and challenges. Lancet Infect Dis 2021;21:e281-9.
179. Campbell K. MADDOG: Method for assignment, definition and designation of global lineages. Available from: https://github.com/KathrynCampbell/MADDOG. [Last accessed on 28 May 2024].
Cite This Article
 , ... Kirstyn Brunker
, ... Kirstyn BrunkerHow to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].