

Harnessing machine learning for electrochemical CO2 reduction: current progress and future perspectives
 0
0 
MAIN TEXT
Electrochemical carbon dioxide reduction (CO2RR) is regarded as a promising strategy for achieving sustainable carbon utilization, but the complexity and multi-scale characteristics of this reaction process pose significant challenges for the rational design of catalysts. The integration of machine learning (ML) with electrocatalysis can establish cross-scale connections among atomic structure, dynamic interfacial microenvironments, and macroscopic catalytic performance, thereby enabling predictive analysis of catalyst activity, product selectivity, reaction pathways, and electrochemical environment regulation. This outlook integrates recent advances in ML-assisted CO2RR, highlights the key challenges in data quality, feature interpretability, and multiscale integration, and proposes future directions for combining physics-informed modeling with realistic electrochemical reaction environments. With advances in ML, automated experimentation, and multi-source data integration, a self-evolving catalyst design platform is expected to be established.
To achieve sustainable utilization of carbon resources, electrochemical CO2RR is regarded as a key approach to converting carbon dioxide into high-value-added fuels and chemicals[1]. However, CO2RR involves multiple electron-proton cooperative transfers and the generation of various complex intermediates[2]. The CO2RR process is often influenced by multiple factors such as interfacial electric fields, solvents, and surface configurations, resulting in high reaction overpotentials and complex product distributions[3]. Such multiscale coupling makes the rational design of CO₂RR catalysts highly challenging. Traditional trial-and-error experiments and density functional theory (DFT) calculations are often limited by insufficient exploration of chemical spaces and high computational costs, making it difficult to form systematic design principles. With the explosive growth of materials data and computing resources, data-driven catalyst design strategies have created new opportunities for advancing research on CO2RR catalysts[4]. Machine learning (ML) provides a powerful data-driven approach for identifying relationships among catalyst structures, reaction microenvironments, and catalytic performance from large-scale experimental and theoretical datasets [Figure 1][5,6]. It can also help identify key reaction features that are difficult to uncover using traditional empirical methods, thereby supporting the development of performance prediction models with improved generalizability. Moreover, ML offers significant advantages in predicting and optimizing catalyst activity, product selectivity and catalytic reaction pathways, providing key support for high-throughput catalyst screening, mechanism analysis and interpretable catalyst design. As these capabilities continue to develop, ML is gradually becoming a crucial link that connects atomic-scale characteristics, such as electronic structure, with macroscopic catalytic behavior, and is playing an increasingly important role in electrocatalytic CO2RR research[7,8]. In recent years, this technology has demonstrated unique value in four closely related fields: activity prediction, product selectivity prediction, reaction pathway identification, and electrochemical environment modeling.
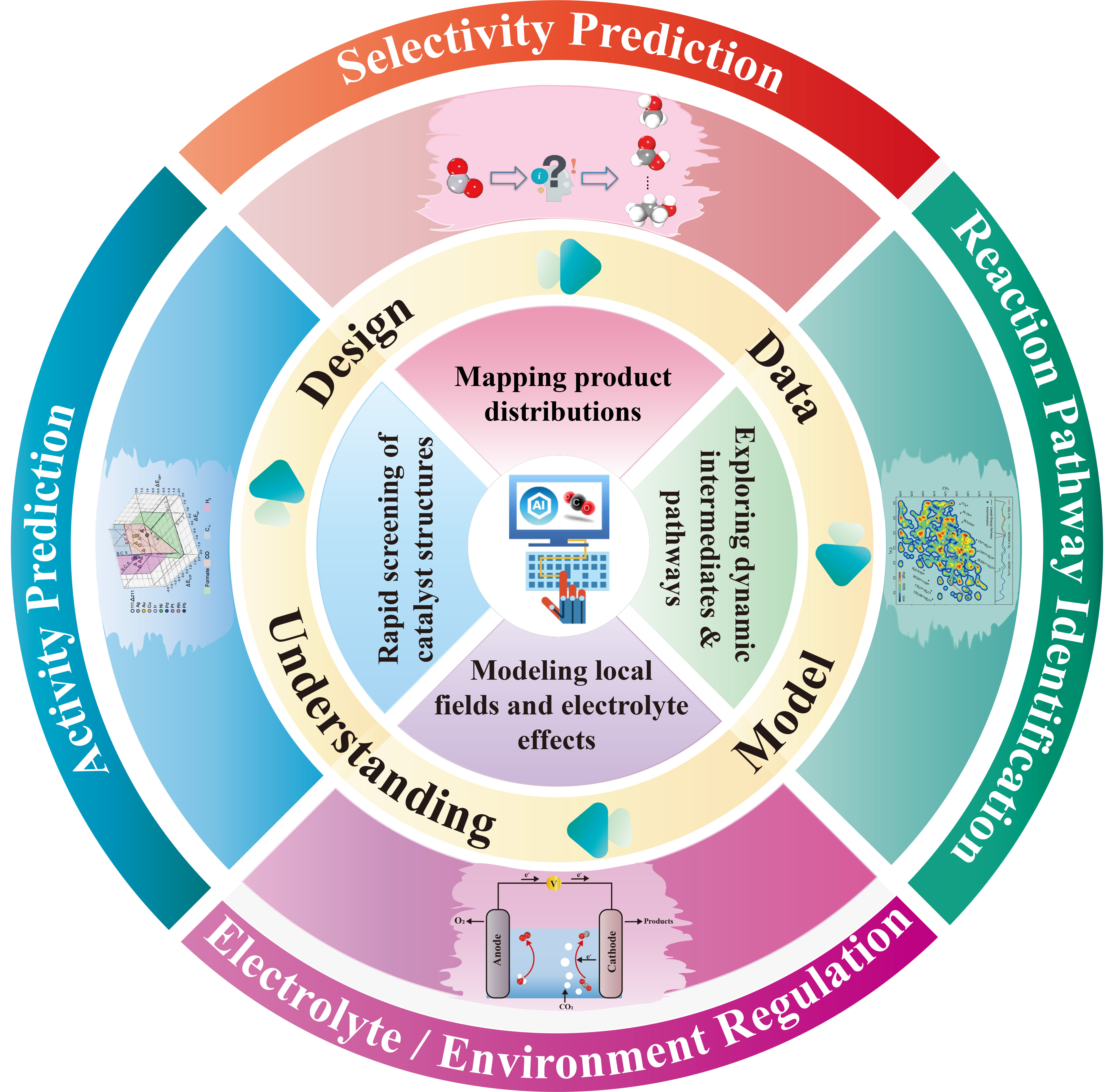
Figure 1. An overview of machine-learning applications in Electrocatalytic CO2RR. ML in four key research directions: catalyst activity prediction (Adapted from Ref.[16] under a Creative Commons Attribution 4.0 International license)[16], product selectivity prediction, reaction pathway identification (Adapted with permission from Ref.[23] Copyright 2022 American Chemical Society)[23], and electrolyte/environment regulation. CO2RR: Carbon dioxide reduction.
In terms of activity prediction, researchers have successfully established a quantitative mapping relationship among catalyst structures, electronic properties and adsorption energy through ML models trained with computational and experimental data[9,10]. For instance, previous studies have shown that ML models based on simple physicochemical descriptors such as electronegativity, atomic radius, and d-band center can accurately predict the CO2RR activity of various catalysts, demonstrating strong interpretability and considerable capability for high-throughput catalyst screening[11-13]. These models can not only accurately predict key activity indicators such as overpotential and turnover frequency, significantly improving the screening efficiency of high-performance catalysts, but also identify key descriptors that affect the behavior of active sites (such as coordination geometry and charge distribution), and help researchers deeply understand the intrinsic connection between structure and performance. Product selectivity prediction is another significant breakthrough. The CO2RR process involves multiple competing and complex reaction pathways, which usually lead to the coexistence of various products. By using data-driven machine learning models, researchers can quickly and accurately capture the subtle interdependencies among the morphology, electronic states and reaction conditions of catalysts[14,15], thereby enabling highly accurate prediction of the distribution of target products. By integrating these multidimensional data, researchers have constructed efficient ML-based predictive frameworks, which provide important guidance for improving target-product selectivity and suppressing side reactions[16,17]. In the field of reaction pathway identification, machine learning potentials (MLPs) have become important tools. These models can describe potential energy surfaces with accuracy approaching that of first-principles calculations, while at a substantially reduced computational cost. By combining MLPs with molecular dynamics simulations, researchers can dynamically monitor the surface reconstruction of catalysts, the adsorption of intermediate species, and the evolution of reaction transition states under conditions that more closely resemble real electrochemical environments. For instance, recent simulation studies based on MLPs have successfully revealed how pH modulates the reaction pathway and interfacial reconstruction kinetics over tin-based catalysts, showing good agreement between theoretical prediction and experimental observations[18]. This approach helps elucidate dynamic reaction processes that are difficult to fully capture using conventional static DFT calculations and links microscopic energetics with experimentally observable dynamic behaviors. Moreover, the electrochemical environment should not be regarded as a fixed background, but rather as a dynamic interfacial system jointly governed by local electric fields, electric double-layer structures, ion distributions, and solvent reorganization. These environmental factors can synergistically regulate adsorption configurations, intermediate stability, and reaction barriers, thereby determining catalytic performance and product distribution under operating potentials[19]. By incorporating factors such as electrostatic effects, electric double-layer structure, solvation stability, and pH-dependent surface charge distribution into ML framework, ML can help clarify how local electric fields and ionic species affect catalytic performance and product distribution under the operating potential. Furthermore, when combined with machine learning potentials and molecular dynamics simulations, ML can also be used to investigate solvent dynamics, interfacial reconstruction, and ion-surface interactions under conditions that more closely resemble realistic operating environments[20,21]. Recent studies have further demonstrated that the combination of ML with external electric field simulations can provide quantitative insights into how electric and magnetic fields reshape the electronic structure and reaction energy barriers at electrochemical interfaces. These advances provide new insights into the design of environment-responsive catalysts[22]. This type of modeling technology that incorporates environmental factors helps bridge the gap between atomic-level interactions and macroscopic catalytic performance, thereby providing a more realistic theoretical description of electrochemical operating conditions[23]. To more clearly distinguish different machine learning tasks in CO2RR, Table 1 provides a brief summary of the corresponding typical inputs, prediction targets, and validation strategies. Overall, these advancements indicate that research on CO2RR is shifting from the traditional empirical trial-and-error paradigm toward a research framework that is more precise, efficient, predictive, and mechanism-oriented.
Summary of representative ML Tasks in CO2RR
| ML task | Typical input and target | Prediction target |
| Activity prediction[24,25] | Catalyst composition, structural/electronic descriptors, adsorption-related data | Overpotential, adsorption energy, activity trends |
| Selectivity prediction[25,26] | Catalyst properties, intermediate-binding information, reaction conditions | Product distribution, Faradaic efficiency, selectivity trends |
| Pathway identification[27,28] | Reaction energetics, intermediate structures, dynamic trajectories | Pathway evolution, key intermediates, transition-state-related features |
| Electrochemical environment modeling[29,30] | Interfacial descriptors, electrolyte/potential information, solvent/ion configurations | Interface effects on adsorption, activity, and selectivity |
Although ML has made significant progress in electrocatalytic CO2RR, there are still several obstacles that remain to be addressed. First, the main challenge lies in the availability and quality of the training datasets. Currently, there is an urgent need for high-quality and comprehensive datasets, especially those that systematically cover catalyst structures, reaction energetics as well as electrochemical conditions, such as pH, potential, and electrolyte composition. This deficiency greatly limits the generalizability and transferability of ML models across different catalytic systems and reaction environments. For CO2RR, this challenge is further complicated by several reaction-specific factors, including variability across different experimental platforms, electrolyte-dependent effects, catalyst reconstruction under operating conditions, inconsistent reporting standards, and uncertainties in activity and selectivity measurements. These issues collectively reduce the comparability and reliability of datasets collected from different studies. Additionally, inconsistent in data formats and standards across different sources can lead to inconsistent or ambiguous descriptor definitions. More importantly, most feature values selection still relies on the experience and intuition of researchers rather than unified physical principles. Therefore, establishing a standardized and shareable data platform and developing automated and interpretable feature extraction methods will be the key directions for future development.
Another major challenge lies in model interpretability. Although deep neural networks offer high predictive accuracy, they often operate as a black box, with the opaque decision-making process obscuring the underlying chemical mechanisms. Integrating fundamental physicochemical principles such as thermodynamic constraints, kinetic relationships, and interfacial electric field effects into the ML framework can enhance both the reliability and transparency of the model. The fusion of these physical principles with data-driven learning will help build more intelligent models that can not only provide precise predictions but also help clarify the underlying mechanisms of electrochemical CO2RR.
The third major challenge lies in bridging the gap across time and length scales. Electrochemical reactions are fundamentally complex multiscale processes, ranging from atomic-scale charge transfer to mesoscale mass transfer and ultimately to the macroscopic behavior of the entire system. To gain a comprehensive understanding of these processes and achieve closed-loop optimization, it is essential to integrate ML with multiscale simulations and automated experimentation. Notably, with the rapid development of high-throughput robotic experimental systems and self-evolving learning frameworks, this field is undergoing accelerated transformation[31,32]. In the long term, the synergistic integration of physics-based models, multimodal data fusion, and intelligent experiments is expected to establish a new paradigm for catalyst discovery [Figure 2]. Such a closed-loop workflow integrating data, models, and experiments will continuously improve the predictive accuracy of ML models, expand the explorable chemical space, and advance mechanistic understanding of the mechanisms underlying electrochemical CO2 reduction.
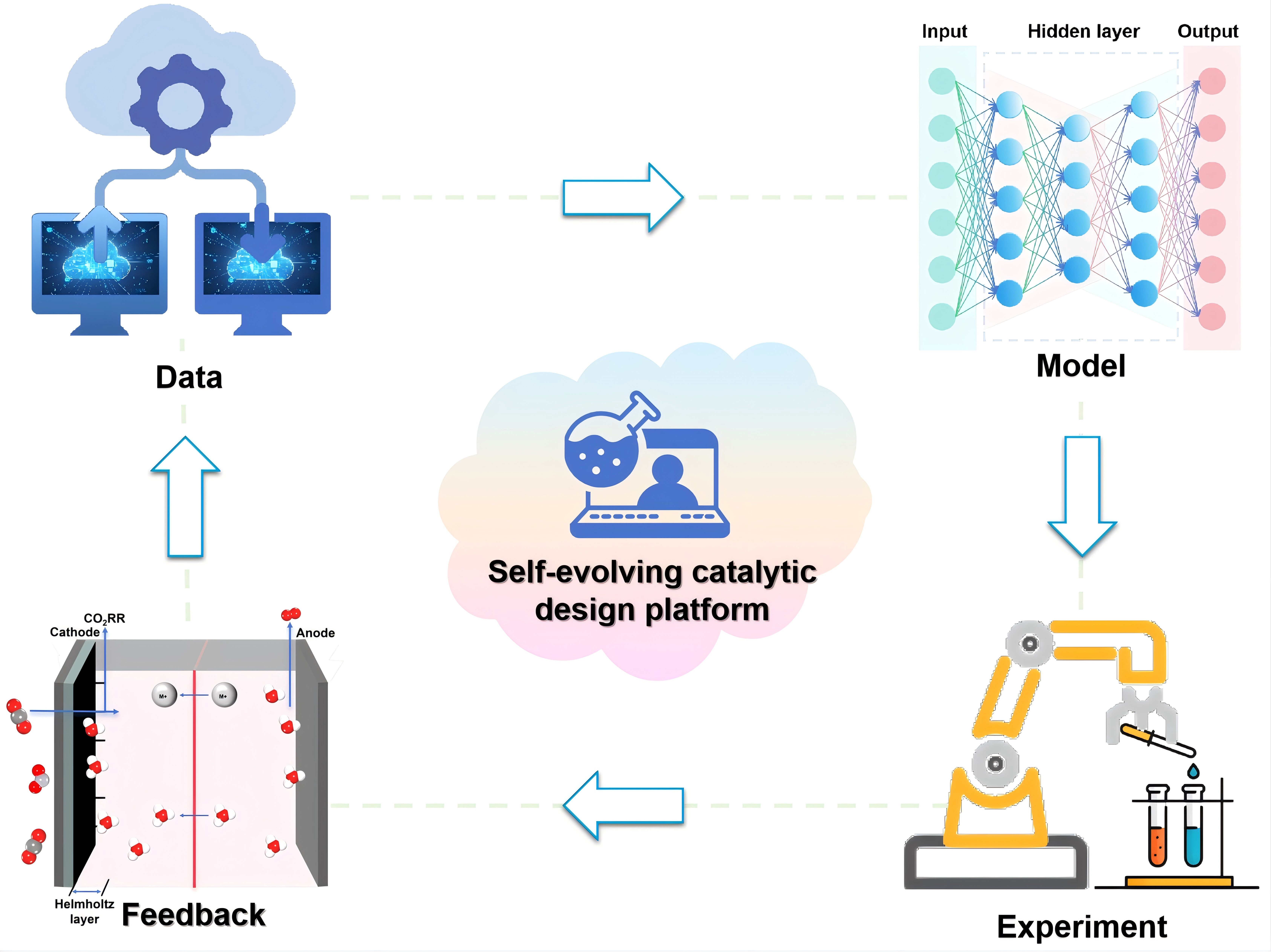
Figure 2. Future roadmap and closed-loop framework for ML-driven design of intelligent CO2RR catalysts. ML: Machine learning; CO2RR: carbon dioxide reduction.
As ML evolves from a mere predictive tool into an intelligent research partner capable of data-driven reasoning and scientific hypothesis generation, it is expected to reshape the landscape of electrocatalytic CO2 reduction research. Ultimately, it will guide the sustainable development of future carbon-conversion technologies and provide powerful scientific and technological support for achieving the goal of carbon neutrality.
OUTLOOK
Despite the rapid progress achieved in ML-assisted CO2RR research in recent years, several critical issues still need to be addressed before ML can evolve from a high-throughput screening tool into a mechanism-driven catalyst design platform. First, the central challenge lies not merely in the limited amount of data, but in the persistent mismatch among data quality, descriptor consistency, and realistic electrochemical operating conditions. A large portion of existing data is derived either from idealized computational models or from experimental systems operated under substantially different conditions, which greatly limits the transferability of models across different catalyst classes and reaction environments. Second, a common misconception in this field is to equate improved predictive accuracy with deeper mechanistic understanding. In reality, without explicit physical constraints and interfacial environmental information, even highly accurate models may capture only correlations rather than revealing the underlying causal mechanisms. Third, future development should move beyond isolated prediction tasks toward a closed-loop research framework integrating standardized databases, physics-informed and interpretable machine learning, realistic electrochemical interface modeling, and automated experimental validation. In our view, such an integrated strategy represents a key route for transforming machine learning from a data-driven acceleration tool into a more reliable and insightful research platform for catalyst discovery and mechanistic understanding in CO2RR.
DECLARATIONS
Authors’ contributions
Conceived the research: Zhang, Y.; Li, J.
Wrote the manuscript: Zhang, Y.
Supervised the research and revised the manuscript: Zhang, Z.
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This work was supported by the National Key Research and Development Program of China (2023YFA1507904), the National Natural Science Foundation of China (U24B20190, 22375142) and the Innovation Funding Project of Science and Technology, China National Petroleum Corporation (2022DQ02-0408).
Conflicts of interest
Zhang, Z. is a Senior Editorial Board Member of the journal Micro Nano Science. Zhang, Z. was not involved in any steps of the editorial process, including reviewers’ selection, manuscript handling, or decision-making. The other authors declare that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Chang, B.; Pang, H.; Raziq, F.; et al. Electrochemical reduction of carbon dioxide to multicarbon (C2+) products: challenges and perspectives. Energy. Environ. Sci. 2023, 16, 4714-58.
2. Sun, H.; Liu, J. Advancing CO2RR with O-coordinated single-atom nanozymes: a DFT and machine learning exploration. ACS. Catal. 2024, 14, 14021-30.
3. Zhang, Z.; Wang, T.; Cai, Y.; et al. Probing electrolyte effects on cation-enhanced CO2 reduction on copper in acidic media. Nat. Catal. 2024, 7, 807-17.
4. Zhu, Q.; Gu, Y.; Liang, X.; Wang, X.; Ma, J. A machine learning model to predict CO2 reduction reactivity and products transferred from metal-zeolites. ACS. Catal. 2022, 12, 12336-48.
5. Wang, X.; Ye, S.; Hu, W.; et al. Electric dipole descriptor for machine learning prediction of catalyst surface-molecular adsorbate interactions. J. Am. Chem. Soc. 2020, 142, 7737-43.
7. Butler, K. T.; Davies, D. W.; Cartwright, H.; Isayev, O.; Walsh, A. Machine learning for molecular and materials science. Nature 2018, 559, 547-55.
8. Schmidt, J.; Marques, M. R. G.; Botti, S.; Marques, M. A. L. Recent advances and applications of machine learning in solid-state materials science. npj. Comput. Mater. 2019, 5, 83.
9. Back, S.; Yoon, J.; Tian, N.; Zhong, W.; Tran, K.; Ulissi, Z. W. Convolutional neural network of atomic surface structures to predict binding energies for high-throughput screening of catalysts. J. Phys. Chem. Lett. 2019, 10, 4401-8.
10. Zafari, M.; Kumar, D.; Umer, M.; Kim, K. S. Machine learning-based high throughput screening for nitrogen fixation on boron-doped single atom catalysts. J. Mater. Chem. A. 2020, 8, 5209-16.
11. Chen, A.; Zhang, X.; Chen, L.; Yao, S.; Zhou, Z. A machine learning model on simple features for CO2 reduction electrocatalysts. J. Phys. Chem. C. 2020, 124, 22471-8.
12. Del Rio, B. G.; Phan, B.; Ramprasad, R. A deep learning framework to emulate density functional theory. npj. Comput. Mater. 2023, 9, 158.
13. Sun, Z.; Yin, H.; Liu, K.; et al. Machine learning accelerated calculation and design of electrocatalysts for CO2 reduction. SmartMat 2022, 3, 68-83.
14. Bozal-Ginesta, C.; Pablo-García, S.; Choi, C.; Tarancón, A.; Aspuru-Guzik, A. Developing machine learning for heterogeneous catalysis with experimental and computational data. Nat. Rev. Chem. 2025, 9, 601-16.
15. Goldsmith, B. R.; Esterhuizen, J.; Liu, J. X.; Bartel, C. J.; Sutton, C. Machine learning for heterogeneous catalyst design and discovery. AIChE. J. 2018, 64, 2311-23.
16. Mok, D. H.; Li, H.; Zhang, G.; Lee, C.; Jiang, K.; Back, S. Data-driven discovery of electrocatalysts for CO2 reduction using active motifs-based machine learning. Nat. Commun. 2023, 14, 7303.
17. Ren, M.; Guo, X.; Zhang, S.; Huang, S. Design of graphdiyne and holey graphyne-based single atom catalysts for CO2 reduction with interpretable machine learning. Adv. Funct. Mater. 2023, 33, 2213543.
18. Wang, Y.; Wu, Z.; Jiang, Y.; et al. Bridging theory and experiment: machine learning potential-driven insights into pH‐Dependent CO2 Reduction on Sn-based catalysts. Adv. Funct. Mater. 2025, 35, e06314.
19. Zhang, Y.; He, J.; Lai, Z.; Ling, C.; Li, Q.; Wang, J. Machine learning-accelerated kinetic simulations of surface reactions with complex coverage effects. J. Phys. Chem. Lett. 2026, 17, 3307-15.
20. Zhu, J.; Cheng, J. How can machine learning facilitate computational electrochemistry. APL. Comput. Phys. 2026, 2, 020901.
21. Feng, C.; Jiang, B. Machine learning accelerated finite-field simulations for electrochemical interfaces. JACS. Au. 2025, 5, 5939-47.
22. Wang, L.; Zhou, X.; Luo, Z.; et al. Review of external field effects on electrocatalysis: machine learning guided design. Adv. Funct. Mater. 2024, 34, 2408870.
23. Shi, Y.; Kang, P.; Shang, C.; Liu, Z. Methanol synthesis from CO2/CO mixture on Cu-Zn catalysts from microkinetics-guided machine learning pathway search. J. Am. Chem. Soc. 2022, 144, 13401-14.
24. Tran, K.; Ulissi, Z. W. Active learning across intermetallics to guide discovery of electrocatalysts for CO2 reduction and H2 evolution. Nat. Catal. 2018, 1, 696-703.
25. Zhong, M.; Tran, K.; Min, Y.; et al. Accelerated discovery of CO2 electrocatalysts using active machine learning. Nature 2020, 581, 178-83.
26. Nitopi, S.; Bertheussen, E.; Scott, S. B.; et al. Progress and perspectives of electrochemical CO2 reduction on copper in aqueous electrolyte. Chem. Rev. 2019, 119, 7610-72.
27. Unke, O. T.; Chmiela, S.; Sauceda, H. E.; et al. Machine learning force fields. Chem. Rev. 2021, 121, 10142-86.
28. Behler, J.; Parrinello, M. Generalized neural-network representation of high-dimensional potential-energy surfaces. Phys. Rev. Lett. 2007, 98, 146401.
29. Clark, E. L.; Resasco, J.; Landers, A.; et al. Standards and protocols for data acquisition and reporting for studies of the electrochemical reduction of carbon dioxide. ACS. Catal. 2018, 8, 6560-70.
30. Burdyny, T.; Smith, W. A. CO2 reduction on gas-diffusion electrodes and why catalytic performance must be assessed at commercially-relevant conditions. Energy. Environ. Sci. 2019, 12, 1442-53.
31. Raccuglia, P.; Elbert, K. C.; Adler, P. D. F.; et al. Machine-learning-assisted materials discovery using failed experiments. Nature 2016, 533, 73-6.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0









Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].