


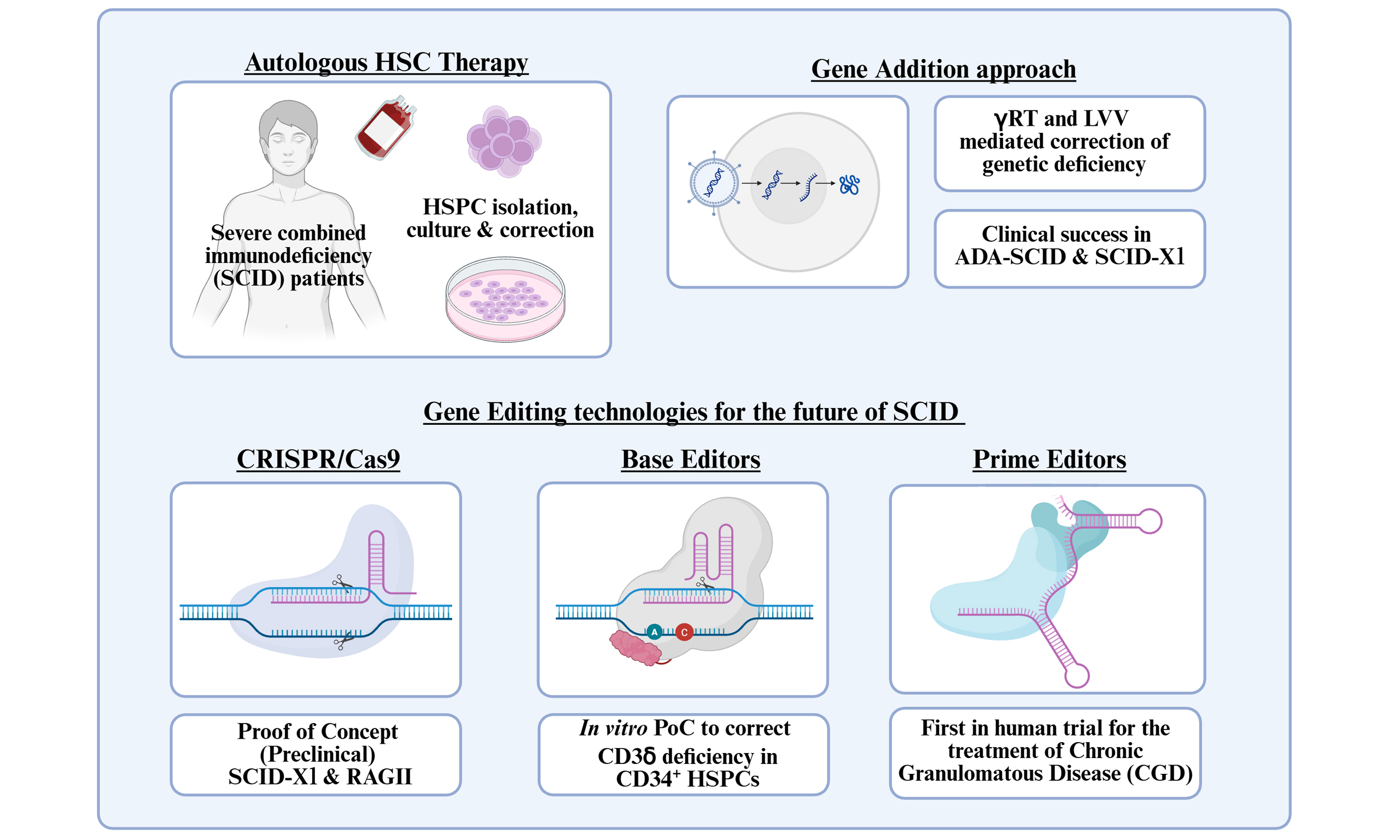
Novel gene-editing technologies: applications of CRISPR-Cas9, base editing, and prime editing in SCID gene therapy
 0
0 Abstract
The use of autologous haematopoietic stem cell gene therapy is increasingly recognised as a promising treatment option for severe combined immunodeficiency diseases (SCID). This approach seeks to correct the underlying genetic cause of SCID conditions, potentially allowing a single treatment to restore a healthy immune system for the lifespan of the patient. To date, such gene therapy has relied on the use of gamma-retroviruses or lentiviruses to deliver genetic material to a patient’s haematopoietic stem cells before reinfusion. This approach has had notable successes in the clinic for conditions including X-linked severe combined immunodeficiency (SCID-X1), Artemis-SCID, and adenosine deaminase-SCID. However, significant hurdles have been met when using viral-mediated gene addition, primarily linked to the potential risk of insertional mutagenesis; however, for certain SCID forms, there are also limitations associated with the regulation and levels of gene expression achievable. This has driven the development of new gene editing technologies for the treatment of SCID conditions. CRISPR (clustered regularly interspaced short palindromic repeats)-Cas9 (CRISPR-associated protein 9), base editing and prime editors are all actively under investigation, mainly in the preclinical stage to understand their potential applications. In this review, we explore gene editing approaches that are in development for the treatment of SCID. While initial results look promising, significant challenges need to be overcome before their clinical use. Such technologies represent an exciting new wave of treatment options for SCID patients.
Keywords
INTRODUCTION
Severe combined immunodeficiency (SCID) is a group of rare, life-threatening genetic disorders characterized by a severely impaired immune system[1-3]. It can be caused by variants in various genes involved in the development and function of T cells, B cells, and natural killer (NK) cells. Due to the critical function of the immune system in maintaining health, children are usually diagnosed early in life (typically 3-6 months of age). Infants fail to thrive, struggle to gain weight and grow, and become susceptible to both opportunistic and recurrent infections. This is primarily due to an inability to mount protective humoral and cellular immune responses, with initial susceptibility normally associated with the loss of the protective effect of maternal immunoglobulins. Early diagnosis and treatment are essential, as SCID patients normally succumb to life-threatening infections within the first year of life. Clinical management of SCID has historically focused on strict isolation to minimise infection risk while providing prophylactic antibiotics and/or immunoglobulin replacement therapy[4]. However, such treatments can only manage symptoms and reduce the risk of infection, and ultimately, a long-term corrective procedure that allows durable reconstitution of immune function is necessary.
Significant advances have now been made in our understanding and characterisation of the different forms of SCID, their genetic basis, inheritance pattern and immunophenotype [Table 1 summarises the SCID forms where autologous haematopoietic stem cell (HSC) gene therapy (HSC-GT) has been used clinically][5-7]. Of the SCID conditions described to date, X-linked SCID (SCID-X1) is the most common, accounting for 30%-40% of patients. It is driven by variants affecting the common g chain (gc). While this protein was first identified as a component of the Interleukin 2 receptor (IL2R), it was later found to be a subunit of numerous cytokine receptors including IL-4, IL-7, IL-9 and IL-15. These cytokines are critical to the maintenance and survival of both mature T and NK cells; therefore, their deficiency leads to severe lymphopenia specifically affecting these cell types while the development of other lineages, such as B cells, is unaffected. Variants in other genes now known to drive SCID conditions include Janus kinase 3 (JAK3), IL7Ra, Recombinase-activating genes (RAG1/2), Cluster of Differentiation 45 (CD45), CD3z, and Zeta-chain-associated protein kinase 70 (ZAP-70). These genes are well characterised and their immunological mechanisms are well understood. However, additional forms of SCID have been identified that are not linked with immunological genes per se. Autosomal deficiency of adenosine deaminase (ADA), a gene involved in the metabolism of adenosine and deoxyadenosine (dAdo), leads to loss of all lymphocytes owing to the toxic build-up of metabolites from the purine salvage pathway[8].
Summary of SCID conditions & characteristics*
| SCID type | Gene affected | Inheritance Pattern | Immunophenotype (T-/B-/NK-) | Descriptive features |
| X-linked SCID | IL2RG | X-linked recessive | T−/B+/NK− | Most common form; affects common gamma chain (γc) used by multiple cytokine receptors |
| Adenosine deaminase (ADA) deficiency | ADA | Autosomal recessive | T−/B−/NK− | Accumulation of toxic metabolites; affects lymphocyte development |
| RAG1 or RAG2 deficiency | RAG1, RAG2 | Autosomal recessive | T−/B−/NK+ | Critical for V(D)J recombination; causes failure of T and B cell development |
| Artemis deficiency | DCLRE1C | Autosomal recessive | T−/B−/NK+ | Also called radiosensitive SCID; DNA repair defect |
| JAK3 DEFICIency | JAK3 | Autosomal recessive | T−/B+/NK− | JAK3 is downstream of IL2RG; phenotype resembles X-linked SCID |
With clear advances in the early diagnosis of SCID patients and our improved understanding of the molecular basis for these conditions, researchers and physicians have now focused on improved treatment options. Allogeneic haematopoietic stem cell transplant (HSCT), allowing the replacement of genetically defective HSCs with those from healthy donors, has long been viewed as the most promising approach to SCID treatment[9,10]. The first successful allogeneic bone marrow transplant (BMT) for the treatment of SCID was carried out in 1968 using an Human Leukocyte Antigen (HLA)-matched sibling[11]. Successes such as this demonstrated the curative potential of HSC transplant and led to significant advances in the field. However, complications related to graft versus host disease (GvHD), infectious risk linked to the use of cytoreductive myeloablative conditioning (to allow engraftment of allogeneic HSCs) and immunosuppressants (to prevent graft rejection), and the availability of suitable donors have all hindered the use of this approach. Autologous HSC-GT is now being used to overcome these difficulties and has been highly effective in the treatment of SCID. By using haematopoietic stem progenitor cells (HSPCs) from the patient, issues relating to donor mismatch and GvHD are fundamentally avoided. However, unlike allogenic BMT, patient HSPCs must be corrected by genetic modification before transplantation back into the host. As patients themselves act as donors for this procedure, CD34+ HSPCs must first be isolated from the patient. Patients undergo mobilisation [a treatment protocol to encourage HSPC egress from the bone marrow (BM) to the peripheral blood] before apheresis to collect patient cells. This can be achieved by administration of granulocyte colony-stimulating factor (G-CSF) and/or agents such as Plerixafor, a C-X-C motif chemokine receptor 4 (CXCR4) antagonist, which can enhance HSPC release from the BM[12-14]. CD34+ cells are enriched following this procedure before undergoing genetic modification. Once this process is complete, patients undergo a form of conditioning using chemotherapy, such as the alkylating agent busulfan[15], to prepare the BM niche for engraftment. Following conditioning, HSC-GT is performed by infusion of genetically modified HSPCs into the patient [Figure 1][16,17].
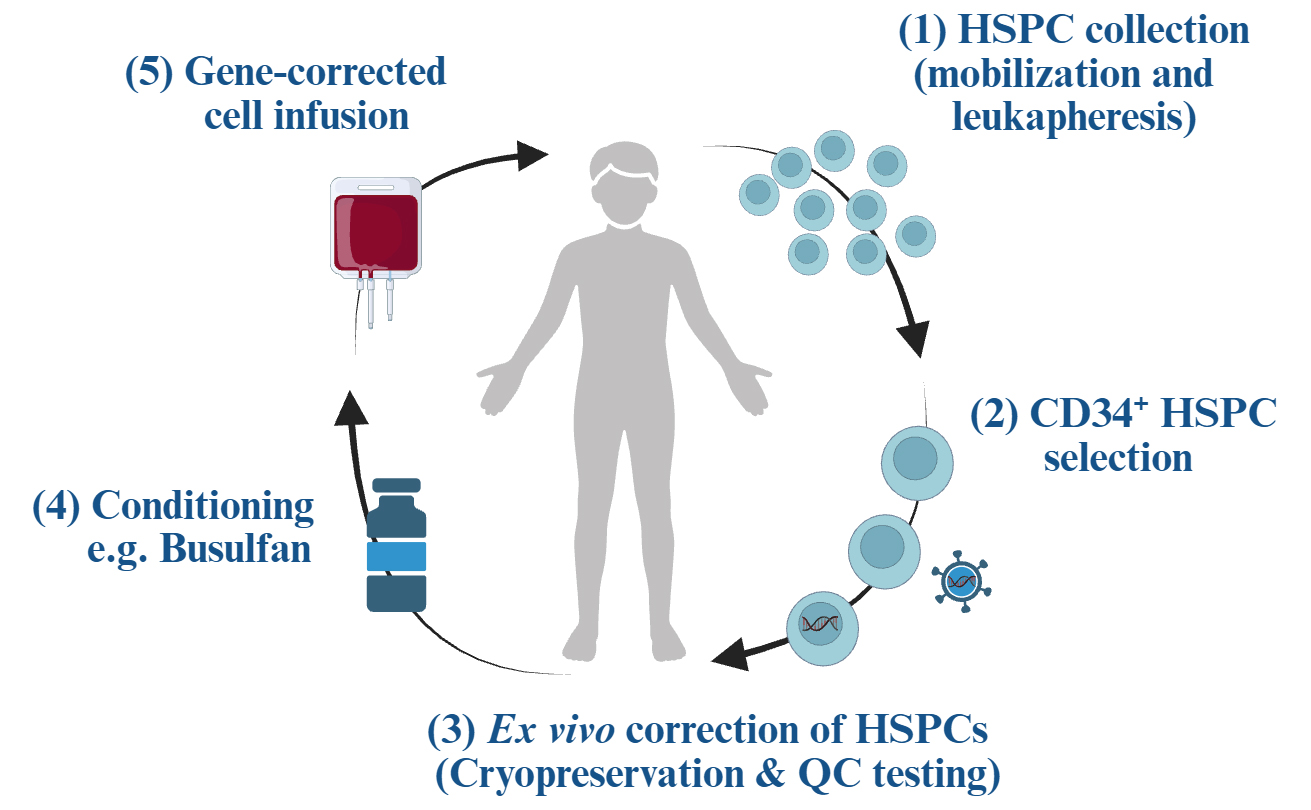
Figure 1. Overview of autologous haematopoietic stem cell transplant. Created in BioRender. Crawford G (2026) https://BioRender.com/3s9v14y. HSPC: Haematopoietic stem progenitor cell; QC: quality check.
The realisation of ex vivo gene-modified autologous HSC gene therapy as a viable treatment option for patients with genetic diseases is highlighted by the successful approval and commercialisation of several HSC-based gene therapy products for immune deficiencies, neurometabolic disorders, and haemoglobinopathies (Table 2 highlights approved ex vivo HSC-GT products). To date, all clinical HSC gene therapy approaches for SCID have utilised ex vivo gene modification using self-inactivating viral vectors, including gamma retroviruses (gRV) and lentiviruses (LVs). While researchers, physicians and private enterprise continue to make significant progress in the development of this approach, several barriers to the widespread clinical application of HSC gene therapies remain. These include challenges relating to: (1) scalability of viral vector production and personalised drug product manufacture; (2) quality control and assurance relating to potency testing and safety assessments; (3) complex regulatory approval processes owing to a lack of standardised expectations. As genetic modification of HSPCs has been shown to be critical for the effective correction of SCID genetic abnormalities, new gene editing technologies are under development to improve the safety and precision of these gene modifications. The use of CRISPR (clustered regularly interspaced short palindromic repeats)-Cas9 (CRISPR-associated protein 9) and base/prime editors in the context of both ex vivo and in vivo HSC gene therapy approaches is under preclinical development for the treatment of SCID, with promising results. Significant challenges still need to be addressed before such strategies can be tested clinically. However, an approved HSC-GT, CasgevyTM, has been successfully tested and approved for the treatment of haemoglobinopathies including sickle cell disease (SCD) and transfusion-dependent thalassaemia (TDT)[18], which utilises CRISPR-Cas9 gene editing of HSCs ex vivo. Such work should provide optimism that these tools can be utilised in the future for the treatment of SCID alongside more established gRV and lentiviral approaches.
Approved HSC-GT products to date*
| Product | Gene/Modality | Indication | Region(s) approved |
| Strimvelis | ADA gene addition using gamma-retrovirus | Adenosine deaminase deficiency (ADA-SCID) | EU (2016) |
| Zynteglo | bA-T87Q-globin gene addition by lentivirus | β-thalassaemia (TDT) | EU & US (2019 & 2022) |
| Skysona | ABCD1 gene addition by lentivirus | Cerebral adrenoleukodystrophy (CALD) | EU** & US (2021 & 2022) |
| Casgevy (exa-cel) | BCL11A disruption by CRISPR-Cas9 editing (driving fetal haemoglobin production) | Sickle cell disease (SCD) & β-thalassaemia (TDT) | EU & US (2023 & 2024) |
| Lyfgenia (lovo-cel) | bA-T87Q-globin gene addition by lentivirus | Sickle cell disease (SCD) | US (2023) |
| Libmeldy | ARSA gene addition by lentivirus | Metachromatic leukodystrophy (MLD) | EU & US (2020 & 2024) |
SUCCESSES OF HSC-GT FOR SCID
The potential of HSC-GT as a curative treatment for SCID and other primary immunodeficiencies (PIDs) has long been recognised[16,20]. Since the first successful allogenic HSCT for a child with SCID-X1, the field has made significant advancements in the mobilisation and isolation of patient HSPCs, vector design, the process for genetic modification of HSPCs, and our understanding of the need to condition patients prior to HSC-GT. This has allowed autologous HSC-GT to come to the forefront of treatment options for SCID patients. Genetic modification of CD34+ HSPCs has routinely been carried out using a “gene addition” approach utilising viral vectors to integrate target genes[21]. The first approved gene therapy harnessing HSC-GT for clinical use was Strimvelis, for the treatment of ADA-SCID[19]. Strimvelis utilises a modified gRV to correct for the deficiency in the adenosine deaminase (ADA) gene. Migliavacca et al.[22] assessed the efficacy and full safety profile of gRV HSC-GT for ADA-SCID. Of the 43 patients treated, all patients are alive to date with significant improvements in clinical outcomes. Serious adverse events (SAEs) reported in patients were predominantly infectious in nature, and all SAEs were treated and managed clinically, leading to their resolution. However, one patient who received this therapy under compassionate grounds was diagnosed with T-cell acute lymphoblastic leukaemia (T-ALL) in 2020, 4.7 years after receiving Strimvelis treatment[23]. This diagnosis was confirmed to be linked to insertional mutagenesis of the proto-oncogene LIM-domain only protein 2 (LMO2). The significant safety concern between gRVs use, and insertional oncogenic events has been highlighted during several HSC-GT clinical trials. One such trial for the treatment of SCID-X1 initially looked promising with positive outcomes for 9/10 infants treated[24]. However, four of these children developed T-cell leukemias caused by overexpression of LMO2. The risk of such oncogenic events was likely amplified by genetic abnormalities unrelated to vector insertion, including gain-of-function variants in neurogenic locus notch homolog protein 1 (NOTCH1) and deletion of the tumour suppressor gene locus cyclin-dependent kinase 2A (CDKN2A)[25]. However, the link between gRV usage and oncogenesis could not be overlooked. A further clinical trial led by Braun et al.[26] for the treatment of Wiskott-Aldrich syndrome (WAS) again demonstrated the efficacy of HSC-GT. Alarmingly, seven patients of a cohort of ten developed T-cell leukemias, again highlighting the substantial risk of gRV use for ex vivo gene modification. Careful investigation of these cases led to the understanding that the principal risk factor was the viral promoter driving transgene expression. These powerful promoters contain enhancer sequences that have the ability to transactivate neighbouring genes. Given the propensity of gRVs to integrate near transcription start sites (TSS) and also into areas of open chromatin, viral promoters contained in gRVs were able to activate growth-promoting genes or oncogenes, thereby driving clonal expansion[27,28].
Advances in vector technology have led to the use of alternative viruses with significantly improved efficacy and safety profiles. Use of self-inactivating lentiviral vectors (SIN-LVs) is now standard practice as they possess a significantly reduced risk of insertional mutagenesis relative to gRV vectors owing to their integration bias towards TSS and regulatory gene regions[29,30], but most importantly due to the use of internal mammalian promoters that lack transactivation potential. Active clinical trials for the treatment of ADA-SCID using HSC-GT utilizing SIN-LVs to genetically modify cells are ongoing[31]. Such studies were initiated following positive preclinical studies utilising a codon optimised version of ADA under the control of an internal “shortened” human elongation factor 1a (EF-1a) promoter to drive expression[32]. All patients from these studies remain well at the most recent follow-up, with no adverse events or signs of lymphoproliferative complications. Importantly, gene-modified cells expressing the ADA transgene were detectable three months post-transplant, with median ADA enzyme activity measured within or above the normal levels observed in healthy children. Such enzyme activity was linked with adequate detoxification with significantly reduced deoxyadenosine nucleotide levels. Immune reconstitution, including the emergence of CD19+ B cells and CD4+ T cells, was observed three months post-transplant, reaching expected normal levels at 24- and 36-month post-follow-up[31]. Importantly, this restoration resulted in patients being taken off immunoglobulin replacement therapy, confirming their ability to mount normal humoral responses.
The successful use of SIN-LVs has been replicated in further phase I/II clinical trials for other SCID conditions, including SCID-X1. SCID-X1, driven by variants in common gc, results in specific loss of T and NK cells, owing to a lack of survival signals mediated by IL-7 and IL-15 cytokines. Preclinical studies demonstrated the ability of an EF1a-IL2RG construct to fully restore T-cell compartments in genetically deficient animals with normal immunoglobulin (IgM and IgG) also observed in serum samples[33,34]. Importantly, the EF1a-IL2RG vector was assessed for its insertional mutagenesis potential using an in vitro insertional mutagenesis assay, alongside two known vectors with high mutagenic potential[34]. While cells transduced with control vectors displayed a proliferation advantage, observed over numerous assays, EF1a-IL2RG cells showed a significantly lower incidence, in line with mock-transduced controls. Such proliferation phenotypes are normally elicited by the transactivation of neighbouring genes by enhancer sequences within strong viral promoters[29]. No upregulation of these genes was observed in EF1a-IL2RG transduced cells. Such studies provided support for phase I/II studies to confirm the safety and efficacy of this approach in SCID-X1 patients[35].
Current limitations of HSC-GT for the treatment of SCID
In contrast to ADA-SCID and SCID-X1, the use of SIN-LVs and universal promoters has been less successful for the treatment of other SCID conditions including RAG1/2 and Artemis deficiencies. Both RAG1/2 and Artemis proteins are tightly regulated, in a cell-specific and spatial manner during lymphocyte development[36-39]. Immature B cells and developing T cells upregulate RAG1/2 to facilitate the recombination of Variable, Diversity, Joining (VDJ) and T Cell Receptor (TCR) a, b, g and d chain segments to form diverse B Cell Receptor (BCR) and TCR repertoires, respectively[40]. Initial studies to correct RAG1 deficiency in preclinical models using a SIN-LV approach were unsuccessful owing to insufficient RAG expression in vivo (despite the use of the EF1a promoter and high VCNs achieved)[41]. Subsequent studies have refined the SIN-LV approach to enhance RAG1 expression, including: (1) the codon optimisation of the human RAG1 gene; (2) the addition of a woodchuck hepatitis virus posttranscriptional regulatory element (WPRE) sequence in the 3’ Untranslated Region (UTR) of the expression cassette; and (3) the inclusion of a stronger internal viral promoter, myeloproliferative sarcoma virus enhancer, negative control region deleted, dl587rev primer-binding site substituted (MND), to drive gene expression. These refinements were able to achieve robust expression in vivo leading to immune reconstitution of both B and T lymphocytes[42-44]. This therapeutic SIN-LV, designated SIN-MND-co.RAG1, has now entered a phase I/II clinical trial (NCT04797260). However, risks still exist for this approach relating to the use of such a strong viral promoter with enhancer elements and its association with insertional mutagenesis[45,46]. In addition, the use of all these refinements will result in no physiological regulation of the RAG1 gene expressed, with high, universal expression expected throughout the haematopoietic system. This poses a significant concern relating to the genetic instability of cells with high RAG1 under homeostatic conditions. Mechanistically, RAG1 and RAG2 proteins form a complex that can induce specific cleavage of target DNA sequences. As RAG1 expression alone cannot cleave DNA efficiently[47], this SIN-LV strategy is not predicted to drive any genetic instability or pose any risk of malignancies or other pathologies. As part of the safety assessment of this vector, in vitro immortalisation (IVIM) and molecular surrogate assay for genotoxicity assessment (SAGA) assays were used to assess risk of insertional mutagenesis. MND-RAG1 constructs scored negatively in these assays, providing a level of support for their clinical use[48]. However, this will need to be investigated and monitored closely in clinical trials to ensure no safety concerns emerge from this lack of regulation or the potency of the modified SIN-LV. Of note, a recent gene therapy trial for the treatment of cerebral adrenoleukodystrophy (CALD) has reported ≥ 10% of recipients developing haematological malignancies as a consequence of the MND promoter used in this trial[49]. This highlights the importance of vector design and promoter selection when developing HSC gene therapies.
Similar concerns were highlighted relating to a lack of physiological gene expression when developing preclinical data for the treatment of Artemis-deficient SCID. High expression of Artemis was shown to be toxic in cell lines including NIH3T3 and 293T[50] when using universal promoters EF1a or phosphoglycerate kinase (PGK). The importance of promoter selection and the consequent expression levels achieved in vivo was investigated in several independent studies. Mostoslavsky et al.[51] demonstrated a significant rescue of lymphocyte development when using SIN-LVs and RAG1-/- animals as a model of SCID (owing to the sensitivity of Artemis knockout animals to conditioning), specifically when using the PGK promoter [Cytomegalovirus (CMV) and EF1a were unable to achieve the same level of restoration]. While B220+ cells and CD4+ T cells were restored to normal levels, cytotoxic CD8+ T cell restoration was incomplete. Supporting these results, SIN-LVs using the PGK promoter were able to rescue lymphocyte development and function using Artemis knockout animals as hosts, in a separate study[52]. However, while these results were very positive, T-cell lymphoma was detected in one animal, raising safety concerns. Nevertheless, these studies confirmed the feasibility of an HSC-GT approach for the treatment of Artemis-SCID and highlighted the need for careful selection of promoters to drive appropriate expression of Artemis protein. The first phase I/II clinical trial of SIN-LV gene therapy for Artemis-SCID included 10 infants treated with genetically modified HSPCs[53]. As preclinical studies had highlighted the need for physiological regulation of Artemis protein, the Artemis transgene was expressed under the control of its own endogenous promoter. All ten children were well at the end of follow-up with immune reconstitution observed 4 weeks post-transplant which was stable for the duration of the study. Four of the six participants who were followed for a 24-month period had successfully received immunisations and mounted protective antibodies highlighting the success of the treatment.
While all past and current clinical trials for the treatment of SCID have utilised gRV and SIN-LVs, they do not come without concerns. The potential for insertional mutagenesis and the lack of physiological regulation of transgenes need to be carefully evaluated before any clinical trial or treatment is initiated. SCID conditions, including SCID-X1, RAG1 deficiency, and Artemis deficiency, all highlight the need for novel gene-editing approaches to restore normal physiological expression of defective genes. Such therapies will result in both improved safety and efficacy for patients undergoing HSC-GT.
NOVEL GENE EDITING TECHNOLOGIES PAVE THE WAY FOR NEW HSC-GT
While the gene addition approach using retroviruses such as gRVs and SIN-LVs has shown much promise and success, concerns relating to insertional mutagenesis and genotoxicity have led to the development of new gene editing approaches. Gene editing allows for genetic abnormalities or mutations to be targeted precisely to preserve the normal physiological expression of a target gene while avoiding the risks posed by viral vectors[54]. In principle, artificial, programmable nucleases, such as zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), or CRISPR/Cas9, induce DNA double-strand breaks (DSBs) that initiate DNA repair processes[55-57]. CRISPR-Cas9 has become the preferred editing tool due to its ease of use, high activity in a wide range of cells, and its notable specificity. Using single guide RNAs (sgRNAs), nucleases are targeted to precise DNA sequences, initiating repair via the non-homologous end joining (NHEJ) pathway or through precise homology-directed repair (HDR). NHEJ repair pathways are undesirable in the context of HSC gene therapy as this process is error-prone, often resulting in insertions or deletions (indels) which are unpredictable in nature[58]. In contrast, the HDR pathway is precise, allowing for the introduction of new sequences (deletions, substitutions or insertions) from an exogenous DNA template possessing homology to the sequences flanking the DSBs. While the CRISPR-Cas9 nuclease system has been recognised for significantly accelerating progress in the gene editing field, it still faces specific challenges in the context of HSC gene therapy. While high specificity of CRISPR-Cas9 mediated editing has been demonstrated in numerous studies, stringent testing, selection and validation of Cas9 variants and sgRNAs was required[59-61]. In addition, DNA repair mechanisms such as HDR are active during S/G2 of the cell cycle, making HDR relatively inefficient in HSPCs, owing to their quiescent nature. Lastly, the need for DSBs to allow for gene editing to occur is not desirable due to the potential for inadvertent recombination effects, the induction of apoptosis[62], and the potential impact of the ‘stemness’ and engraftment potential of HSPCs[63]. Such limitations in CRISPR-Cas9 lead to the development of a further wave of gene editing tools in the form of base and prime editors. Base editing utilises a nickase-only Cas9 (nCas9) fused to a cytidine deaminase or ADA to allow permanent cytosine to thymine (C>T)/guanine to adenine (G>A) base transitions or reverse A>G/T>C base changes, respectively[64,65]. Such editors are highly precise, efficient and require minimal DSB induction. The same team was responsible for combining nCas9 with a reverse transcriptase domain and a multifunctional prime editing guide RNA (pegRNA)[66]. Such technologies are both DSB and HDR independent addressing concerns relating to the induction of DSB [Table 3]. To date, all these new gene editing technologies have been used in early proof-of-concept and feasibility studies to understand their safety and efficacy for the treatment of SCID[20]. Such work has relied on a mixture of in vitro culture systems, in vivo murine modelling, and moving into preclinical studies including the use of larger non-human primates (NHP).
Overview of current gene editing technologies under investigation for clinical application in SCID*
| Feature/technology | CRISPR-Cas9 | Base editors | Prime editing |
| Year developed | 2012[67] | 2016[64] | 2019[66] |
| Main components | Cas9 protein, sgRNA | Cas9 nickase (nCas9) + deaminase + sgRNA | nCas9 + reverse transcriptase + pegRNA |
| Type of DNA break | Double-strand break (DSB) | No DSB (single-strand nick only) | Single-strand nick (no DSB) |
| Cell cycle dependence | Yes - mostly efficient in S/G2 (for HDR) | Less dependent | Less dependent |
| Toxicity | Higher (due to DSB and p53 activation) | Lower than CRISPR-Cas9 | Intermediate to low |
| Editing types | Insertions, deletions, gene knockouts, HDR-based | Transition mutations (C → T, A → G); no indels | Small insertions, deletions, all transition and transversion point mutations |
| Off-target effects | Moderate-high (depends on gRNA specificity) | Lower than CRISPR, but risk of bystander edits | Lowest among the three, but pegRNA design is complex |
| Precision | Medium | High (within narrow editing window) | Very high |
| Efficiency | High (for knockouts), lower for precise edits | High for base changes in suitable sequence contexts | Moderate, but improving |
| Advantages | Simple design; effective knockouts; well-established | Efficient single-base changes; no DSB | High versatility; precise edits without DSB |
| Disadvantages | DSB-related toxicity; low HDR efficiency | Only allows transition mutations; limited edit window | Complex design; larger construct size; lower efficiency |
| Recent research achievements | Successful engraftment of CRISPR/Cas9 edited CD34+ HSPCs in NHP model (Hbf reactivation)[68] | Correction of IL2RG mutation in HSPCs achieved with transplant and engraftment in a murine model of SCID-X1[69] | Patient HSPCs prime-edited to correct SCD allele ex vivo, before successful transplant and lineage maturation in murine models[70] |
| Latest clinical developments | First approved therapy, CasgevyTM, now available for the treatment of SCD and TDT[18] | Phase I/II clinical trial, BEAM-101, for the treatment of severe SCD[71] | First clinical trial, utilizing prime editing technology, PM359, for the treatment of CGD[72] |
APPLICATION OF CRISPR-CAS9 GENE EDITING FOR THE TREATMENT OF SCID-X1
Despite the challenges associated with CRISPR-Cas9 editing for HSCs, numerous labs have begun testing and optimising conditions for its practical application. Efforts have focused on: (1) the target transgenes used; (2) design and validation of sgRNAs to ensure high precision editing (while reducing off-target effects); (3) delivery methods for transfer of CRISPR components to HSPCs to reduce genotoxicity and preserve HSPC function and health and 4) methods to promote HDR pathways to ensure accurate gene editing.
Using a humanised SCID-X1 murine model, Schiroli et al.[73] tested ZFN and CRISPR-Cas9 gene editing approaches to evaluate their efficacy and safety when editing donor HSPCs. This involved the insertion of a fully functional copy of the IL2RG gene into the affected locus before transplantation of gene-modified cells. Engraftment of ≥ 10% corrected HSPCs was sufficient to correct the disease in these models, which is encouraging, as mild conditioning may be compatible with clinical application. The use of adeno-associated virus type 6 (AAV6) as a vehicle for template DNA was also highlighted, resulting in significant improvements in HDR-mediated repair. This approach was further tested and explored in CD34+ HSPCs derived from SCID-X1 patients[74]. CD34+ HSPCs were collected from 6 patients carrying different variants across the IL2RG gene. Corrected cells (median gene correction achieved of 44.5% with a range of 30.1%-47%) were transplanted into NOD SCID gamma (NSG) pups with immune reconstitution monitored and checked 17 weeks after transfer. High levels of engraftment were detected in both BM and spleen of recipients with significant rescue of all immune compartments including the emergence of functional CD3+ T cells. Importantly, the safety of this approach was evaluated in detail during in vivo transplant studies and genotoxicity/off-target analysis. No tumours or abnormalities in haematopoiesis were detected in such studies. Off-target activity of the CRISPR-Cas9 complex was also assessed at 54 predicted genomic sites with insertion/deletion (INDEL) frequencies below the limit of detection. In addition, no evidence was found for karyotypic abnormalities in gene-modified HSPCs. To understand CRISPR-Cas9 competitiveness with LV-integration, Brault et al.[75] carried out a side-by-side study for the correction of the SCID-X1 phenotype in vitro and in vivo[75]. The CRISPR-Cas9 approach targeted exon 1 of the IL2RG locus, for the insertion of a codon-optimised IL2RG complementary DNA (cDNA) donor template delivered by AAV6. In contrast, LV insertion utilised a previously described SIN-LV containing the IL2RG under the EF1a promoter. Viability, haematopoietic potential as measured by colony-forming unit (CFU) assay, and cell cycle analysis were all equivalent between the two approaches. While both approaches rescued T-cell development from CD34+ HSPCs using in vitro differentiation assays, LV-integration was unable to support NK cells. However, there was no selective advantage of CRISPR-Cas9 over LV when gene-modified HSPCs were transplanted into NSG mice. Transplanted animals developed equivalent levels of T and B lymphocytes with serum immunoglobulin levels restored using both approaches. While the efficacy of these approaches may not be in question, the potential for insertional mutagenesis has been a concern with viral vector-mediated approaches. Off-target analysis evaluated 82 potential off-target sites with no evidence of INDELs significantly above background levels. Similar to previous studies using CRISPR-Cas9, no karyotype abnormalities were detected and no differences in overall mortality of recipient NSG animals were observed. Together, these studies provide compelling efficacy and safety data to justify the clinical development of the CRISPR-Cas9-AAV6 platform for the treatment of SCID in the future.
CRISPR-Cas9 offers a unique opportunity for the treatment of RAG1/2-deficient SCID
The use of CRISPR-Cas9 to allow the insertion of therapeutic RAG1 into the endogenous locus would be desirable allowing for physiological expression to be achieved. Early proof-of-concept studies have successfully demonstrated the restoration of T-cell differentiation in vitro from genetic modified HSPCs[76]. This work utilised two different approaches to restore RAG1 expression by targeting transgene expression in either intron 1 or exon 2 of the Rag1 locus. Intronic insertion failed to restore any immune reconstitution after transplant of HSPCs into NSG animals, possibly due to disruption of gene regulatory components within intron 1. In contrast, exonic insertion produced promising results in vivo with partial rescue of B-cell development. However, while T-cell differentiation could be detected using an in vitro culture system, no development of T cells could be observed in vivo. Nevertheless, this is the first proof-of-concept study to demonstrate RAG1 correction in HSPCs using a gene editing approach. Further investigations to refine this approach will have significant implications for the HSC gene therapy field. While SIN-MND-co.RAG1 constructs were negative using IVIM and SAGA assays during testing, this was not the case for RAG2, which clearly demonstrated a high risk of insertional mutagenesis[48]. Clearly, SIN-LVs have been successful in restoring functional gene expression in several SCID conditions. However, such an approach is highly unlikely to be safe for RAG2-SCID. A significant barrier to early proof-of-concept studies is access to sufficient numbers of CD34+ HSPCs from newly diagnosed PID patients, primarily infants. To address this, Iancu et al.[77] targeted causative SCID genes associated with lymphocyte development, including Artemis and RAG2, for biallelic deletion in healthy donor-derived CD34+ HSPCs to recapitulate SCID phenotypes in vitro. After optimisation of this genetic knockout system, deficient cells were assessed for their lymphocytic differentiation capacity which proved to be successful in reproducing phenotypic deficiencies observed in patients such as blocks in T-cell development. Single allelic gene correction was carried out by gene editing (possible as RAG2-SCID is an autosomal recessive disorder) using AAV6 knock-in template containing functional RAG2 cDNA. Corrected CD34+ HSPCs were able to give rise to T cells in vitro comparable to lymphocyte differentiation achieved by healthy control CD34+ HSPCs. Importantly, deep sequencing analysis of the T cell receptor g (TCRG) loci confirmed that a comparable highly diverse clonogenic population was generated by edited and healthy control progenitors. In addition, the frequency distribution of complementarity determining region 3 (CDR3) lengths was also normal relative to controls, indicating a diverse TCR repertoire had been preserved in these cells. The same group took this approach further, utilizing CRISPR-Cas9-AAV6 to replace the entire RAG2 coding sequence with corrective RAG2 transgene[78]. By doing so, all critical intron sequences that may contribute to the regulation of RAG2 gene expression are included. Such an approach would provide a “one fits all” therapy that could treat all RAG2-SCID patients regardless of the underlying variants. This strategy is important and addresses numerous aspects of current SIN-LV approaches including the lack of physiological regulation (linked to the enhancer/promoter elements used in such vector constructs) and concerns relating to insertional mutagenesis. Future studies will need to confirm the correction of RAG2 phenotypes outside of the T-cell compartment, mainly B lymphocytes and humoral responses. As highlighted in these studies, access to SCID patient samples can be limited, and novel approaches are needed to complete proof-of-concept studies. Chang et al.[79] utilised induced pluripotent stem cells (iPSCs) generated from a JAK3-deficient SCID patient (carrying a C>T nucleotide substitution in exon 14 - C1837T) to test the application of CRISPR-Cas9 gene editing. JAK3 deficiency is characterised by a specific block in T-cell development and a lack of NK cells, owing to the role of JAK3 in cytokine signalling[80]. Using the OP9-DL1/4 in vitro culture system (stromal cells expressing Notch ligands Delta-like-1 or Delta-like-4), T and NK-cell differentiation capacity was confirmed using control iPSCs, while patient-derived JAK3-deficient iPSCs possessed a clear block in T/NK development. Using CRISPR-Cas9 and an AAV6-derived donor template, JAK3 expression was restored in JAK3-deficient iPSCs, confirmed at both RNA (RT-PCR) and protein (Western blot) levels. Using whole genome sequencing, corrected iPSC lines were assessed for potential single nucleotide variants (SNVs) or INDELs at 1,450 potential off-target sites. No variants were introduced in any of these potential off-target sites demonstrating the high degree of specificity provided by the CRISPR-Cas9 platform.
Limitations of CRISPR-Cas9 approach highlighted by preclinical evaluations
These studies have clearly demonstrated early proof-of-concept and the feasibility of CRISPR-Cas9 technology for the genetic manipulation of CD34+ HSPCs ex vivo for the treatment of SCID conditions. However, there are several key limitations that need to be addressed before clinical translation. While high specificity of CRISPR-Cas9 mediated editing has been demonstrated in numerous studies, stringent testing, selection and validation of Cas9 variants and sgRNAs was required[73,77]. In addition, DNA repair mechanisms such as HDR are relatively inefficient in HSPCs, owing to their quiescent nature. Care needs to be taken to ensure culture conditions are optimal to ensure efficient gene repair by HDR, while preserving HSC potency[81-83]. This must include the overall manipulation of CD34+ HSPC cells, ensuring the non-toxic delivery of CRISPR components (Cas9 protein, sgRNAs and donor templates)[84]. To avoid genotoxicity, DNA plasmids are not used routinely as they can trigger type 1 interferon responses leading to disruption of the cell cycle and apoptosis of HSPCs[85]. Instead, donor repair sequences are provided with the use of recombinant adeno-associated viruses (AAVs) including the AAV6 serotype. This specific serotype has been shown to avoid induction of type 1 interferon responses and be able to deliver high levels of repair template to the cell nucleus. The use of electroporation has also been shown to induce DNA damage and cell death in HSPCs, highlighting the need to optimise electroporation conditions[84,86]. All such manipulation of HSPCs may affect their overall stemness, affecting their potency and longevity in vivo. The durability of HSC gene therapy and the persistence of corrected cells are among the hallmark features of these treatments. To date, most preclinical studies to evaluate novel gene editing modalities have focused on murine systems (immunodeficient strains such as NSG) with little work carried out in larger animal models. Studies in NHP models including rhesus macaques has highlighted concerns over the engraftment and long-term contribution of CRISPR-HDR edited cells to haematopoiesis[87,88]. This may be due to functional damage to long-term HSPCs during the HDR process. Such problems were not encountered when using an LV-integration approach.
All these challenges are currently under investigation to improve the overall safety and efficacy of this approach before clinical translation. However, in the context of SCID, gene-modified cells may have a selective advantage following engraftment, which may allow a low frequency of HSPCs corrected by gene editing to achieve positive outcomes[73,89]. Therefore, SCID presents a unique opportunity to successfully demonstrate the potential of CRISPR-Cas9 gene editing and confirm its potential for the future.
BASE AND PRIME EDITING TECHNOLOGY FOR THE CORRECTION OF SPECIFIC VARIANTS IN SCID
CRISPR-Cas9 gene editing has demonstrated some success in preclinical studies and efforts continue to optimise this platform for genetic modification of CD34+ HSPCs. However, the involvement of DSB, which can be deleterious to these cells, and the low efficiency of the HDR pathway in quiescent HSPCs will continue to be barriers to this approach. Base editing is an alternative approach, allowing for the correction of pathogenic variants without the need for DSBs or DNA templates[64,65]. To date, most base editing studies and successes have been focused on monogenic blood disorders, including β-thalassaemia and SCD[90-92]. Both these genetic disorders are driven by variants in β-globulin (HBB) protein, a crucial component of adult haemoglobin (HbA). By using base editors, many approaches have been tested to correct for the deficiency in b-globulin, including the direct correction of variants in the HBB gene or the reactivation of fetal haemoglobin (HbF) expression that compensates for defects in adult haemoglobin. Such work has resulted in the commencement of phase I/II clinical trials for the treatment for SCD using applicable base editors (NCT05456880, NCT04853576). Beam Therapeutics has enrolled 15 SCD patients, using an adenine base editor (ABE) to introduce specific variants in the promoter regions of HBG1 and HBG2 genes, mimicking naturally occurring variants that drive constitutive production of fetal haemoglobin[71]. This approach utilised ex vivo HSC-GT with busulfan conditioning, similar to the SIN-LV approaches used for the treatment of SCID. In the most recent follow-ups, all patients are well with safety and adverse effects similar to those associated with busulfan conditioning and autologous HSC-GT. Importantly, > 60% induction of HbF has been measured with a significant reduction (< 40%) in sickle haemoglobin (HbS). As such, improvement in numerous clinical outcomes has been observed including normalised or improved markers of haemolysis and oxygen delivery and resolution of anaemia. Such results indicate clear progress in the field and prove that base editing technologies can be successful in the clinic.
First preclinical proof-of-concept studies highlight potential of base editing technology in CD3d-SCID
The use of base and prime editing technologies for the treatment of SCID is still in its early stages. However, variants in common SCID genes including ADA, IL2RG, RAG1/2 and JAK3 have been well characterised, allowing for their correction in principle. An important proof-of-concept study was completed by the Kohn lab utilizing ABEs to correct for a specific variant in the CD3δ gene that drives a rare form of SCID deficiency[93]. CD3d-deficient SCID represents < 1% of all SCID, characterised by a specific loss of T cells. Due to the scarcity of patient samples, development of treatments has been hindered. To overcome this, Jurkat cell lines were generated carrying a specific 202C>T variant in the CD3d gene to mimic the disease phenotype observed in patients. In addition to this cell line, the ABE strategy was tested in healthy CD34+ HSPCs transduced with LV containing the CD3d variant and CD34+ HSPCs collected from a CD3d SCID patient. Transplantation of ABE-edited CD34+ HSPCs into NOD.Cg-KitW-41J Tyr+ Prkdcscid Il2rgtm1Wjl/ThomJ (NBSGW) immunodeficient animals resulted in long-term engraftment and immune reconstitution comparable to untreated and LV-gene-modified CD34+ HSPCs controls. Gene editing was detected in ~80% in all cell types analysed with little to no off-target events. Using artificial thymic organoids (ATOs), McAuley et al.[93] demonstrated a specific block at the double positive (DP) stage of thymic T-cell development associated with a biallelic 202C>T variant using patient-derived CD34+ HSPCs. This deficiency was successfully corrected with the use of ABE resulting in functional T-cells with a diverse TCR repertoire. This important study demonstrated the feasibility of a gene editing approach for SCID treatment that could be applicable to numerous SCID conditions.
The use of prime editors is also under exploration in the context of SCID as we await completion of studies to demonstrate the feasibility, efficacy, and safety of this editing approach. As a parallel milestone, the very first clinical application of a prime-editing gene therapy has begun with the first-in-human treatment of chronic granulomatous disease (CGD) (NCT06559176). Early proof-of-concept and preclinical studies have focused on the use of prime-edited HSC-GT to restore nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity by correcting the delGT variant in the neutrophil cytosolic factor 1 (NCF1) gene, the most prevalent disease-causing variant in the p47phox variant of CGD[72]. Data from the first patient treated with a single dose of prime edited autologous CD34+ stem cells modified ex vivo (PM359) (combined with busulfan conditioning) has been positive, with complete restoration of neutrophil function measured in 58% and 66% of neutrophils analysed at 15 and 30 days post-transplant, respectively. Such studies confirm that prime editing of CD34+ HSPCs can be both effective and safe for patients justifying further studies to fully realise the potential of this gene editing approach.
Challenges for the clinical application of gene editing technologies
While gene editing tools such as base and prime editors address fundamental concerns related to the use of CRISPR-Cas9, much work is needed before each clinical application. The delivery systems used for base and prime editing tools to genetically modify HSPCs are a shared problem with CRISPR-Cas9. Electroporation is commonly used for ribonucleoprotein (RNP) complexes, containing Cas9 protein complexed with guide RNA. While this allows for transient expression and no risk of viral integration, cell viability can be impacted with activation of the tumor suppressor protein (p53)[62]. In addition, the size of payload that can be delivered such as donor DNA repair templates is limited. As highlighted in numerous CRISPR-Cas9 studies, viral vectors can be used to provide donor DNA templates which overcomes this limitation. And while this promotes high efficiency of the HDR pathway, the use of AAV6 can trigger immune responses and effect the long-term engraftment potential of HSPCs. However, as highlighted in this review, the AAV6 serotype has been used across several studies in combination with CRISPR-Cas9 owing to its relative lack of immunogenicity relative to LVs. Lastly, additional delivery methods are currently under development including the use of lipid nanoparticles (LNPs) and nanocarriers. However, their efficiency is relatively low in HSPCs compared to other cell types, making their development for HSPC gene editing less attractive. The main benefit for the use of base editing tools such as cytosine base editors (CBE) and ABEs is the lack of DSBs needed to introduce genetic sequences (in contrast to CRISPR-Cas9 methods). However, detailed analysis has confirmed that this statement is not entirely true as it has been shown that base editing technologies can still induce DSBs, albeit at much lower frequencies. This observation provides scope for more refined approaches to gene editing and the development of novel base editors with higher performance. Clinical application of gene editing technologies also presents unique challenges. The diversity of SCID variants means custom-made strategies will be needed for patients, reducing scalability and increasing costs associated with these treatments. And as the use of such technologies become more widely available, standardisation of regulatory frameworks will be needed to ensure development of therapies and access for patients is not impeded.
CONCLUSIONS AND FUTURE PERSPECTIVES
The potential of HSC-GT for the correction of SCID and other monogenic conditions is beginning to be realised, with new HSC gene therapies being approved and commercialised. However, the genetic modification of CD34+ HSPCs and the correction of underlying genetic abnormalities remain an intense area of research, with continuous advancements being made in the development of novel gene editing tools and the ex vivo HSC gene therapy platform. While almost all HSC gene therapies to date utilise retroviruses, particularly LVs, several platforms are being actively tested in proof-of-concept and preclinical studies. Approval of HSC-GT using CRISPR-Cas9 gene editing for the treatment of haemoglobinopathies, including SCD and TDT, has confirmed the feasibility of this approach for clinical application. This work has been vindicated with the first approval of an ex vivo gene editing product, CasgevyTM, in 2023[18,94]. The outcomes of such trials will be watched closely, with specific attention to the successful engraftment of edited cells, the longevity of therapeutic effects and the overall safety profile of treated patients.
Considering how the research and development field of reprogrammable nucleases has only recently been established, significant progress has been made in understanding their potential usage in SCID, highlighting both advantages and disadvantages compared with currently approved HSC-GT therapies. It is clear that gene addition strategies mediated by LV transfer may have high efficacy and safety in conditions such as ADA-SCID; however, other SCID genotypes will require HSC-GT approaches with more precise gene regulation for optimal outcomes. Evidence is now emerging for the application of both CRISPR-Cas9 and base and prime editing tools in the treatment of numerous SCID conditions, highlighting the versatility of these technologies. Over time, the combination of gene-editing technologies alongside gene addition HSC-GT approaches may become the standard of care, offering patients much-needed treatment options for numerous life-threatening conditions.
DECLARATIONS
Acknowledgements
The Graphical Abstract was created in BioRender. Crawford G (2026) https://BioRender.com/oz4qw7q.
Authors’ contributions
Conceptualisation, writing and editing: Crawford G
Supervision, writing and review: Sagoo P, Gaspar HB
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
All authors declare that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Dvorak CC, Haddad E, Heimall J, et al. The diagnosis of severe combined immunodeficiency (SCID): The Primary Immune Deficiency Treatment Consortium (PIDTC) 2022 definitions. J Allergy Clin Immunol. 2023;151:539-46.
2. Gaspar HB, Gilmour KC, Jones AM. Severe combined immunodeficiency--molecular pathogenesis and diagnosis. Arch Dis Child. 2001;84:169-73.
3. Heimall J, Cowan MJ. Long term outcomes of severe combined immunodeficiency: therapy implications. Expert Rev Clin Immunol. 2017;13:1029-40.
4. Albin S, Cunningham-Rundles C. An update on the use of immunoglobulin for the treatment of immunodeficiency disorders. Immunotherapy. 2014;6:1113-26.
5. Tasher D, Dalal I. The genetic basis of severe combined immunodeficiency and its variants. Appl Clin Genet. 2012;5:67-80.
6. Aranda CS, Gouveia‐pereira MP, Da Silva CJM, et al. Severe combined immunodeficiency diagnosis and genetic defects. Immunol Rev. 2024;322:138-47.
7. Cirillo E, Giardino G, Gallo V, et al. Severe combined immunodeficiency - an update. Ann N Y Acad Sci. 2015;1356:90-106.
8. Bradford KL, Moretti FA, Carbonaro-sarracino DA, Gaspar HB, Kohn DB. Adenosine Deaminase (ADA)-Deficient Severe Combined Immune Deficiency (SCID): Molecular Pathogenesis and Clinical Manifestations. J Clin Immunol. 2017;37:626-37.
9. Wahlstrom JT, Dvorak CC, Cowan MJ. Hematopoietic stem cell transplantation for severe combined immunodeficiency. Curr Pediatr Rep. 2015;3:1-10.
10. Haddad E, Hoenig M. Hematopoietic stem cell transplantation for severe combined immunodeficiency (SCID). Front. Pediatr. 2019;7:481.
11. Buckley RH. A historical review of bone marrow transplantation for immunodeficiencies. J Allergy Clin Immunol. 2004;113:793-800.
12. Lidonnici MR, Aprile A, Frittoli MC, et al. Plerixafor and G-CSF combination mobilizes hematopoietic stem and progenitors cells with a distinct transcriptional profile and a reducedin vivo homing capacity compared to plerixafor alone. Haematologica. 2017;102:e120-4.
13. Bendall LJ, Bradstock KF. G-CSF: From granulopoietic stimulant to bone marrow stem cell mobilizing agent. Cytokine Growth Factor Rev. 2014;25:355-67.
14. Nakamura N, Jo T, Arai Y, et al. Benefits of plerixafor for mobilization of peripheral blood stem cells prior to autologous transplantation: a dual-center retrospective cohort study. Cytotherapy. 2023;25:773-81.
15. Ciurea SO, Andersson BS. Busulfan in hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2009;15:523-36.
16. John T, Czechowicz A. Clinical hematopoietic stem cell-based gene therapy. Mol Ther. 2025;33:2663-78.
17. Ferrari G, Thrasher AJ, Aiuti A. Gene therapy using haematopoietic stem and progenitor cells. Nat Rev Genet. 2020;22:216-34.
18. Frangoul H, Locatelli F, Sharma A, et al. Exagamglogene autotemcel for severe sickle cell disease. N Engl J Med. 2024;390:1649-62.
19. Schimmer J, Breazzano S. Investor outlook: rising from the ashes; GSK’s European approval of strimvelis for ADA-SCID. Hum Gene Ther Clin Dev. 2016;27:57-61.
20. Zhang Z, Thrasher AJ, Zhang F. Gene therapy and genome editing for primary immunodeficiency diseases. Genes Dis. 2020;7:38-51.
21. Bulcha JT, Wang Y, Ma H, Tai PWL, Gao G. Viral vector platforms within the gene therapy landscape. Sig Transduct Target Ther. 2021;6:53.
22. Migliavacca M, Barzaghi F, Fossati C, et al. Long-term and real-world safety and efficacy of retroviral gene therapy for adenosine deaminase deficiency. Nat Med. 2024;30:488-97.
23. Cesana D, Cicalese MP, Calabria A, et al. A case of T-cell acute lymphoblastic leukemia in retroviral gene therapy for ADA-SCID. Nat Commun. 2024;15:3662.
24. Hacein-bey-abina S, Hauer J, Lim A, et al. Efficacy of gene therapy for X-linked severe combined immunodeficiency. N Engl J Med. 2010;363:355-64.
25. Howe SJ, Mansour MR, Schwarzwaelder K, et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Investig. 2008;118:3143-50.
26. Braun CJ, Boztug K, Paruzynski A, et al. Gene therapy for Wiskott-Aldrich syndrome - long-term efficacy and genotoxicity. Sci Transl Med. 2014;6.
27. Biasco L, Baricordi C, Aiuti A. Retroviral integrations in gene therapy trials. Mol Ther. 2012;20:709-16.
28. Montini E, Cesana D, Schmidt M, et al. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. J Clin Investig. 2009;119:964-75.
29. Modlich U, Navarro S, Zychlinski D, et al. Insertional transformation of hematopoietic cells by self-inactivating lentiviral and gammaretroviral vectors. Mol Ther. 2009;17:1919-28.
30. Bokhoven M, Stephen SL, Knight S, et al. Insertional gene activation by lentiviral and gammaretroviral vectors. J Virol. 2009;83:283-94.
31. Kohn DB, Booth C, Shaw KL, et al. Autologous ex vivo lentiviral gene therapy for adenosine deaminase deficiency. N Engl J Med. 2021;384:2002-13.
32. Carbonaro DA, Zhang L, Jin X, et al. Preclinical demonstration of lentiviral vector-mediated correction of immunological and metabolic abnormalities in models of adenosine deaminase deficiency. Mol Ther. 2014;22:607-22.
33. Zhou S, Mody D, Deravin SS, et al. A self-inactivating lentiviral vector for SCID-X1 gene therapy that does not activate LMO2 expression in human T cells. Blood. 2010;116:900-8.
34. Poletti V, Charrier S, Corre G, et al. Preclinical development of a lentiviral vector for gene therapy of X-linked severe combined immunodeficiency. Mol Ther Methods Clin Dev. 2018;9:257-69.
35. Mamcarz E, Zhou S, Lockey T, et al. Lentiviral gene therapy combined with low-dose busulfan in infants with SCID-X1. N Engl J Med. 2019;380:1525-34.
36. Kuo TC, Schlissel MS. Mechanisms controlling expression of the RAG locus during lymphocyte development. Curr Opin Immunol. 2009;21:173-8.
37. Gilioli G, Lankester AC, De Kivit S, Staal FJ, Ott De Bruin LM. Gene therapy strategies for RAG1 deficiency: challenges and breakthroughs. Immunol Lett. 2024;270:106931.
38. Malu S, De Ioannes P, Kozlov M, et al. Artemis C-terminal region facilitates V(D)J recombination through its interactions with DNA Ligase IV and DNA-PKcs. J Exp Med. 2012;209:955-63.
39. Mansilla-Soto J, Cortes P. VDJ recombination: Artemis and its in vivo role in hairpin opening. J Exp Med. 2003;197:543-7.
40. Nishana M, Raghavan SC. Role of recombination activating genes in the generation of antigen receptor diversity and beyond. Immunology. 2012;137:271-81.
41. Lagresle-peyrou C, Benjelloun F, Hue C, et al. Restoration of human B-cell differentiation into NOD-SCID mice engrafted with gene-corrected CD34+ cells isolated from Artemis or RAG1-deficient patients. Mol Ther. 2008;16:396-403.
42. Garcia-perez L, Van Eggermond M, Van Roon L, et al. Successful preclinical development of gene therapy for recombinase-activating gene-1-deficient SCID. Mol Ther Methods Clin Dev. 2020;17:666-82.
43. Pike-overzet K, Rodijk M, Ng Y, et al. Correction of murine Rag1 deficiency by self-inactivating lentiviral vector-mediated gene transfer. Leukemia. 2011;25:1471-83.
44. Sorel N, Díaz-pascual F, Bessot B, et al. Restoration of T and B cell differentiation after RAG1 gene transfer in human RAG1 defective hematopoietic stem cells. Biomedicines. 2024;12:1495.
45. Montini E, Naldini L, Booth C, Kohn DB, Aiuti A. Balancing efficacy and safety in lentiviral vector-mediated hematopoietic stem cell gene therapy. Mol Ther. 2025;33:6-8.
46. Staal FJ, Pike-overzet K, De Kivit S, et al. Safety and efficacy of gene therapy for RAG1-deficient SCID. Mol Ther. 2025;33:1869-70.
47. Jones JM, Gellert M. The taming of a transposon: V(D)J recombination and the immune system. Immunol Rev. 2004;200:233-48.
48. Schwarzer A, Talbot SR, Selich A, et al. Predicting genotoxicity of viral vectors for stem cell gene therapy using gene expression-based machine learning. Mol Ther. 2021;29:3383-97.
49. Duncan CN, Bledsoe JR, Grzywacz B, et al. Hematologic cancer after gene therapy for cerebral adrenoleukodystrophy. N Engl J Med. 2024;391:1287-301.
50. Multhaup M, Karlen AD, Swanson DL, et al. Cytotoxicity associated with Artemis overexpression after lentiviral vector-mediated gene transfer. Hum Gene Ther. 2010;21:865-75.
51. Mostoslavsky G, Fabian AJ, Rooney S, Alt FW, Mulligan RC. Complete correction of murine Artemis immunodeficiency by lentiviral vector-mediated gene transfer. Proc. Natl. Acad. Sci. U.S.A. 2006;103:16406-11.
52. Benjelloun F, Garrigue A, Demerens-de Chappedelaine C, et al. Stable and functional lymphoid reconstitution in Artemis-deficient mice following lentiviral Artemis gene transfer into hematopoietic stem Cells. Mol Ther. 2008;16:1490-9.
53. Cowan MJ, Yu J, Facchino J, et al. Lentiviral gene therapy for Artemis-deficient SCID. N Engl J Med. 2022;387:2344-55.
54. Liu D, Cao D, Han R. Recent advances in therapeutic gene-editing technologies. Mol Ther. 2025;33:2619-44.
55. Liu X, Li G, Liu Y, Zhou F, Huang X, Li K. Advances in CRISPR/Cas gene therapy for inborn errors of immunity. Front. Immunol. 2023;14:1111777.
56. Xu W, Zhang S, Qin H, Yao K. From bench to bedside: cutting-edge applications of base editing and prime editing in precision medicine. J Transl Med. 2024;22:1133.
57. Kim H, Kim J. A guide to genome engineering with programmable nucleases. Nat Rev Genet. 2014;15:321-34.
58. Allen D, Kalter N, Rosenberg M, Hendel A. Homology-directed-repair-based genome editing in hspcs for the treatment of inborn errors of immunity and blood disorders. Pharmaceutics. 2023;15:1329.
59. Ferrari S, Vavassori V, Canarutto D, et al. Gene editing of hematopoietic stem cells: hopes and hurdles toward clinical translation. Front. Genome Ed. 2021;3:618378.
60. Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 2014;507:62-7.
61. Hsu PD, Scott DA, Weinstein JA, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31:827-32.
62. Dorset SR, Bak RO. The p53 challenge of hematopoietic stem cell gene editing. Mol Ther Methods Clin Dev. 2023;30:83-9.
63. Maganti HB, Bailey AJM, Kirkham AM, Shorr R, Pineault N, Allan DS. Persistence of CRISPR/Cas9 gene edited hematopoietic stem cells following transplantation: a systematic review and meta-analysis of preclinical studies. Stem Cells Transl Med. 2021;10:996-1007.
64. Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420-4.
65. Gaudelli NM, Komor AC, Rees HA, et al. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature. 2017;551:464-71.
66. Anzalone AV, Randolph PB, Davis JR, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149-57.
67. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable Dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816-21.
68. Humbert O, Radtke S, Samuelson C, et al. Therapeutically relevant engraftment of a CRISPR-Cas9-edited HSC-enriched population with HbF reactivation in nonhuman primates. Sci Transl Med. 2019;11:eaaw3768.
69. Zhang L, Li K, Liu Z, et al. Restoring T and B cell generation in X-linked severe combined immunodeficiency mice through hematopoietic stem cells adenine base editing. Mol Ther. 2024;32:1658-71.
70. Everette KA, Newby GA, Levine RM, et al. Ex vivo prime editing of patient haematopoietic stem cells rescues sickle-cell disease phenotypes after engraftment in mice. Nat. Biomed. Eng. 2023;7:616-28.
71. A Phase 1/2 study evaluating the safety and efficacy of a single dose of autologous CD34+ base edited hematopoietic stem cells (BEAM-101) in patients with sickle cell disease and severe vaso-occlusive crises (Beacon Trial). 2022. Available from: https://clinicaltrials.gov/study/NCT05456880 [Last accessed on 20 Mar 2026].
72. Heath JM, Stuart Orenstein J, Tedeschi JG, et al. Prime editing efficiently and precisely corrects causative mutation in chronic granulomatous disease, restoring myeloid function: toward development of a prime edited autologous hematopoietic stem cell therapy. Blood. 2023;142 Suppl:7129.
73. Schiroli G, Ferrari S, Conway A, et al. Preclinical modeling highlights the therapeutic potential of hematopoietic stem cell gene editing for correction of SCID-X1. Sci Transl Med. 2017;9:eaan0820.
74. Pavel-dinu M, Wiebking V, Dejene BT, et al. Gene correction for SCID-X1 in long-term hematopoietic stem cells. Nat Commun. 2019;10:1634.
75. Brault J, Liu T, Liu S, et al. CRISPR-Cas9-AAV versus lentivector transduction for genome modification of X-linked severe combined immunodeficiency hematopoietic stem cells. Front. Immunol. 2023;13:1067417.
76. Castiello MC, Brandas C, Ferrari S, et al. Exonic knockout and knockin gene editing in hematopoietic stem and progenitor cells rescues RAG1 immunodeficiency. Sci Transl Med. 2024;16:eadh8162.
77. Iancu O, Allen D, Knop O, et al. Multiplex HDR for disease and correction modeling of SCID by CRISPR genome editing in human HSPCs. Mol Ther Nucleic Acids. 2023;31:105-21.
78. Allen D, Knop O, Itkowitz B, et al. CRISPR-Cas9 engineering of the RAG2 locus via complete coding sequence replacement for therapeutic applications. Nat Commun. 2023;14:6771.
79. Chang C, Lai Y, Westin E, et al. Modeling human severe combined immunodeficiency and correction by CRISPR/Cas9-enhanced gene targeting. Cell Reports. 2015;12:1668-77.
80. Roberts JL, Lengi A, Brown SM, et al. Janus kinase 3 (JAK3) deficiency: clinical, immunologic, and molecular analyses of 10 patients and outcomes of stem cell transplantation. Blood. 2004;103:2009-18.
81. Rai R, Naseem A, Vetharoy W, et al. An improved medium formulation for efficient ex vivo gene editing, expansion and engraftment of hematopoietic stem and progenitor cells. Mol Ther Methods Clin Dev. 2023;29:58-69.
82. Lomova A, Clark DN, Campo-fernandez B, et al. Improving gene editing outcomes in human hematopoietic stem and progenitor cells by temporal control of DNA repair. Stem Cells. 2019;37:284-94.
83. De Ravin SS, Brault J, Meis RJ, et al. Enhanced homology-directed repair for highly efficient gene editing in hematopoietic stem/progenitor cells. Blood. 2021;137:2598-608.
84. Lattanzi A, Meneghini V, Pavani G, et al. Optimization of CRISPR/Cas9 delivery to human hematopoietic stem and progenitor cells for therapeutic genomic rearrangements. Mol Ther. 2019;27:137-50.
85. Cromer MK, Vaidyanathan S, Ryan DE, et al. Global transcriptional response to CRISPR/Cas9-AAV6-based genome editing in CD34+ hematopoietic stem and progenitor cells. Mol Ther. 2018;26:2431-42.
86. Gundry MC, Brunetti L, Lin A, et al. Highly efficient genome editing of murine and human hematopoietic progenitor cells by CRISPR/Cas9. Cell Reports. 2016;17:1453-61.
87. Aljanahi AA, Lazzarotto CR, Chen S, et al. Prediction and validation of hematopoietic stem and progenitor cell off-target editing in transplanted rhesus macaques. Mol Ther. 2022;30:209-22.
88. Lee B, Gin A, Wu C, et al. Impact of CRISPR/HDR editing versus lentiviral transduction on long-term engraftment and clonal dynamics of HSPCs in rhesus macaques. Cell Stem Cell. 2024;31:455-466.e4.
89. Kume A, Koremoto M, Mizukami H, et al. Selective growth advantage of wild-type lymphocytes in X-linked SCID recipients. Bone Marrow Transplant. 2002;30:113-8.
90. Newby GA, Yen JS, Woodard KJ, et al. Base editing of haematopoietic stem cells rescues sickle cell disease in mice. Nature. 2021;595:295-302.
91. Han W, Qiu H, Sun S, et al. Base editing of the HBG promoter induces potent fetal hemoglobin expression with no detectable off-target mutations in human HSCs. Cell Stem Cell. 2023;30:1624-1639.e8.
92. Liao J, Chen S, Hsiao S, et al. Therapeutic adenine base editing of human hematopoietic stem cells. Nat Commun. 2023;14:207.
93. Mcauley GE, Yiu G, Chang PC, et al. Human T cell generation is restored in CD3δ severe combined immunodeficiency through adenine base editing. Cell. 2023;186:1398-1416.e23.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].