


An appraisal of emerging dystrophin restoration therapies in Duchenne muscular dystrophy
 0
0 Abstract
Duchenne muscular dystrophy (DMD) is an X-linked, progressive muscle disorder caused by pathogenic variants in the DMD gene and resulting in a complete loss of dystrophin protein expression. As of now, there is no cure for DMD, and despite improvements in standard of care, there are significant unmet needs for disease modifying treatments. This article provides an overview of emerging therapies aimed at dystrophin restoration, emphasizing exon skipping and gene therapy, within the rapidly evolving landscape for Duchenne muscular dystrophy.
Keywords
INTRODUCTION
Duchenne muscular dystrophy (DMD) is a devastating degenerative muscle disease that affects approximately 1:5,000~10,000 males. DMD prevalence reported in the literature is variable, ranging from 0.9 to 16.8 per 100,000 males. A recent meta-analysis showed a pooled global prevalence of 5.3 cases per 100,000 males[1,2]. The disease is characterized by childhood-onset muscle weakness, which progresses to loss of motor function and premature death due to respiratory and cardiac insufficiency[1]. Data from the Muscular Dystrophy Surveillance, Tracking and Research Network (MD STARnet) showed that over the last 15-20 years, the mean age at diagnosis in the United States has been 4.9 years. While there is no cure for DMD, advances in standard of care, including systemic corticosteroid use, cardioprotective interventions, non-invasive ventilatory support and a multidisciplinary management model, have been shown to slow disease progression and delay the onset of comorbidities[1]. These improvements have led to a median life expectancy of 30 years, with reported variability ranging from 21 to 40 years[3]. The time of diagnosis, access to available treatments, and other socio-economic factors can also affect survival.
Despite recent advancements, significant unmet needs remain in DMD treatment. Early diagnosis is essential, as it allows timely intervention, improves clinical outcomes, and enhances quality of life[4].
MOLECULAR BASIS OF DISEASE
The DMD gene is located on the X chromosome and is the largest known human gene, comprising 79 exons (2.3 Mb) and encoding the protein dystrophin[1]. Most mutations in the DMD gene are large deletions (68%) or duplications (11%), and the remaining 21% are small pathogenic variants. Half of that 21% are nonsense mutations, and small deletions, small insertions and splice site mutations represent 5%, 2% and 3%, respectively[5]. Reports of variations in mutation-type frequency across different cohorts and countries suggest that ethnic background may contribute to mutation diversity[6-9]. Deletions and duplications are concentrated with increased frequency in two hot spots in the DMD gene: exons 45-55 and exons 2-10[10]. These regions have structural characteristics that increase susceptibility to instability and breakage, leading to a higher rate of rearrangements and an increased mutation frequency[11-13].
Dystrophin is a cytoskeletal protein localized to the sarcolemma, connecting the dystrophin-associated protein complex (DAPC) and the intracellular cytoskeleton c-actin. This interaction protects the sarcolemma against damage that can occur due to the forces involved in muscle contraction. Dystrophin contains four functional domains: the actin-binding amino-terminal domain (ABD), a central rod domain, a cysteine-rich domain, and a carboxy-terminal domain. Pathogenic variants in the DMD gene also abolish dystrophin expression in stem cells, which affects cell polarity and mitosis. Stem cells lacking functional dystrophin undergo aberrant asymmetric division and impaired myogenic differentiation[1,10].
The pathophysiological mechanisms involved in DMD secondary to the lack of dystrophin lead to (1) membrane instability, (2) calcium dysregulation within the muscle, (3) inflammation, (4) mitochondrial dysfunction, and (5) increased oxidative stress. With repeated injury and regeneration over time, chronic inflammation leads to muscle fiber fibrosis, reducing contractile function[14].
HISTORICAL BACKGROUND AND NATURAL HISTORY OF THE DISEASE
DMD was the first genetic muscle disorder to be systematically described. Early reports in the first part of the 19th century were made by Meryon and Conte, followed by monographic works by Duchenne and Gowers[15]. The first unequivocal clinical and pathological reports of DMD were by Edward Meryon in his paper, “On granular and fatty degeneration of the voluntary muscles” on November 28th, 1851. He also recognized the X-linked recessive inheritance of the disease[16].
Duchenne examined a larger cohort of patients, refined the clinical observations and also introduced the needle muscle biopsy technique to aid in diagnosis[17].
Over time, several natural history studies have described the clinical milestones of the disease including loss of ambulation, need for ventilatory support, scoliosis, cardiomyopathy and mortality. Factors that can impact the timing of key clinical milestones include the individual genotype and corticosteroid regimen[18].
NEW APPROACHES ON DIAGNOSIS
Newborn screening initiatives
As the landscape for Duchenne muscular dystrophy is quickly evolving, new approaches for early diagnosis are of vital importance.
Newborn screening (NBS) for DMD is fully implemented in China. In the United Kingdom, a long-standing optional pilot program has been performed in Wales. In December 2019, the U.S. Food and Drug Administration (FDA) approved an assay using creatine kinase MM (CK-MM) isoform in dried blood test as a first tier for NBS. However, a second-tier or confirmatory test is necessary[19-24].
As of now in the United States, two states - Minnesota and Ohio - have both approved and implemented NBS. Massachusetts, New York and Arizona have approved NBS but have not yet begun screening, while at least 12 other states have pending legislation[25].
NBS provides significant advantages for both patients and their families. It can provide information to guide future reproductive planning, early access to mutation-targeted therapies (when available), and eligibility for clinical trials. Despite these advantages, the availability of this program in the United States remains restricted, with full implementation in only two states which limits the potential for early diagnosis and treatment.
Long-read sequencing
A variety of molecular techniques have been used to identify causative variants in DMD. Multiplex ligation-dependent probe amplification (MLPA) is the conventional method used to detect deletions and duplications, while next generation sequencing (short-read sequencing) can detect small variants and exon deletions/duplications; collectively, both methods have a diagnostic yield above 95%. Nevertheless, approximately 2%-7% of cases remain unsolved. Many of these undetected variants are deep intronic mutations that can generate pseudoexons, microindels, substitutions, large-scale deletions, and duplications. RNA analysis of muscle tissue might be able to identify some of these mutations; nevertheless, the invasive nature of this test and clinical availability present significant challenges. Lastly, given the presence of rare and complex structural variants, a subgroup of patients remains undiagnosed. The introduction of third generation technologies, long-read sequencing, through several platforms has significant advantages providing precise identification of structural and complex variants in the DMD gene. Long-read sequencing is a promising technology with potential to improve diagnostic yield, particularly in cases involving complex genomic variants[26-29].
DISEASE MODIFYING TREATMENTS
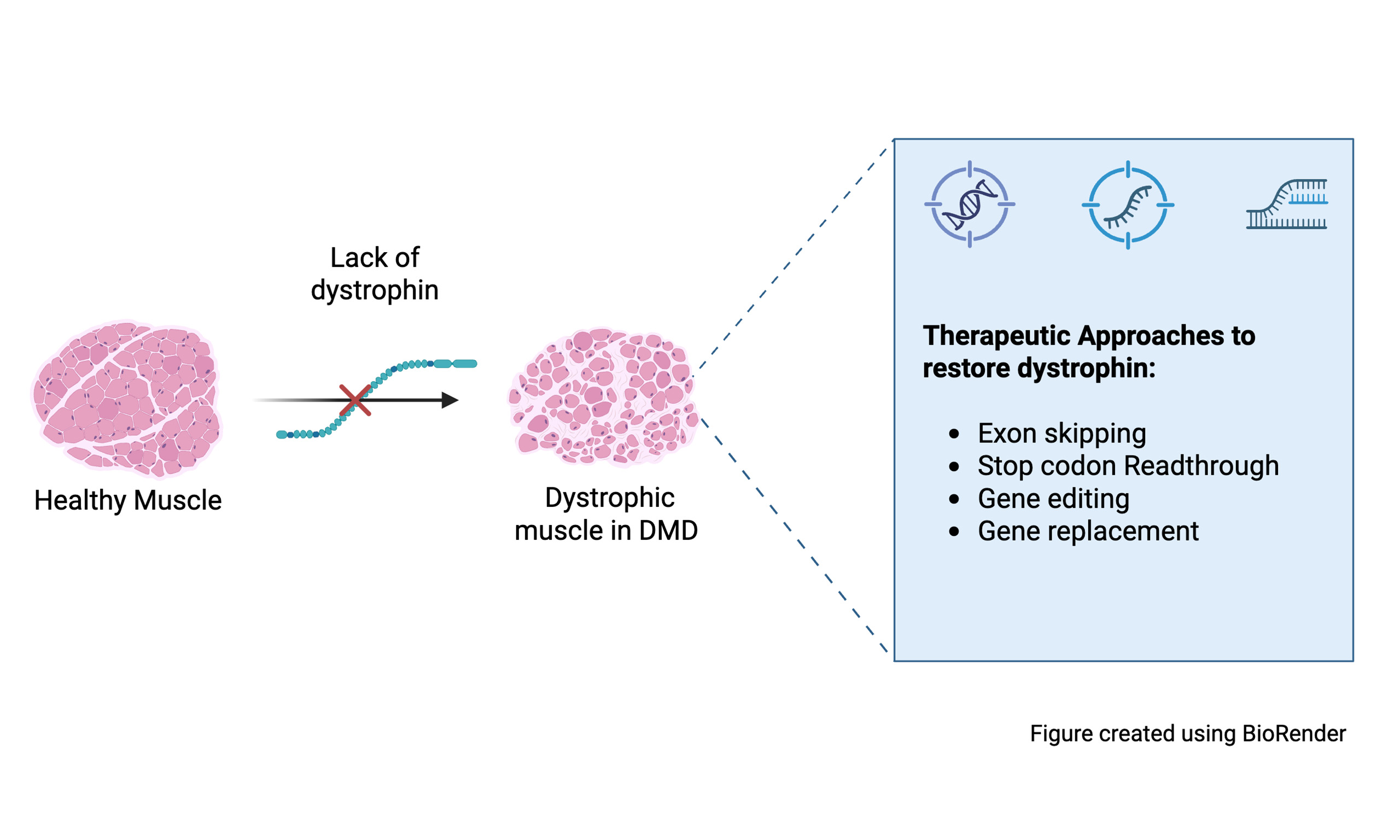
Primary strategies in Duchenne Muscular Dystrophy treatment include: (1) restoring dystrophin at the sarcolemma; (2) upregulation of other genes to replace dystrophin, (homologue utrophin); and (3) managing the downstream effects from the lack of dystrophin which includes inflammation, fibrosis, and oxidative stress[17]. There are several approaches to increase the expression of dystrophin including viral vector mediated gene therapy, exon skipping therapy, read-through of premature stop mutations, and gene editing, and each has advantages and challenges[30] [see Figures 1 and 2].
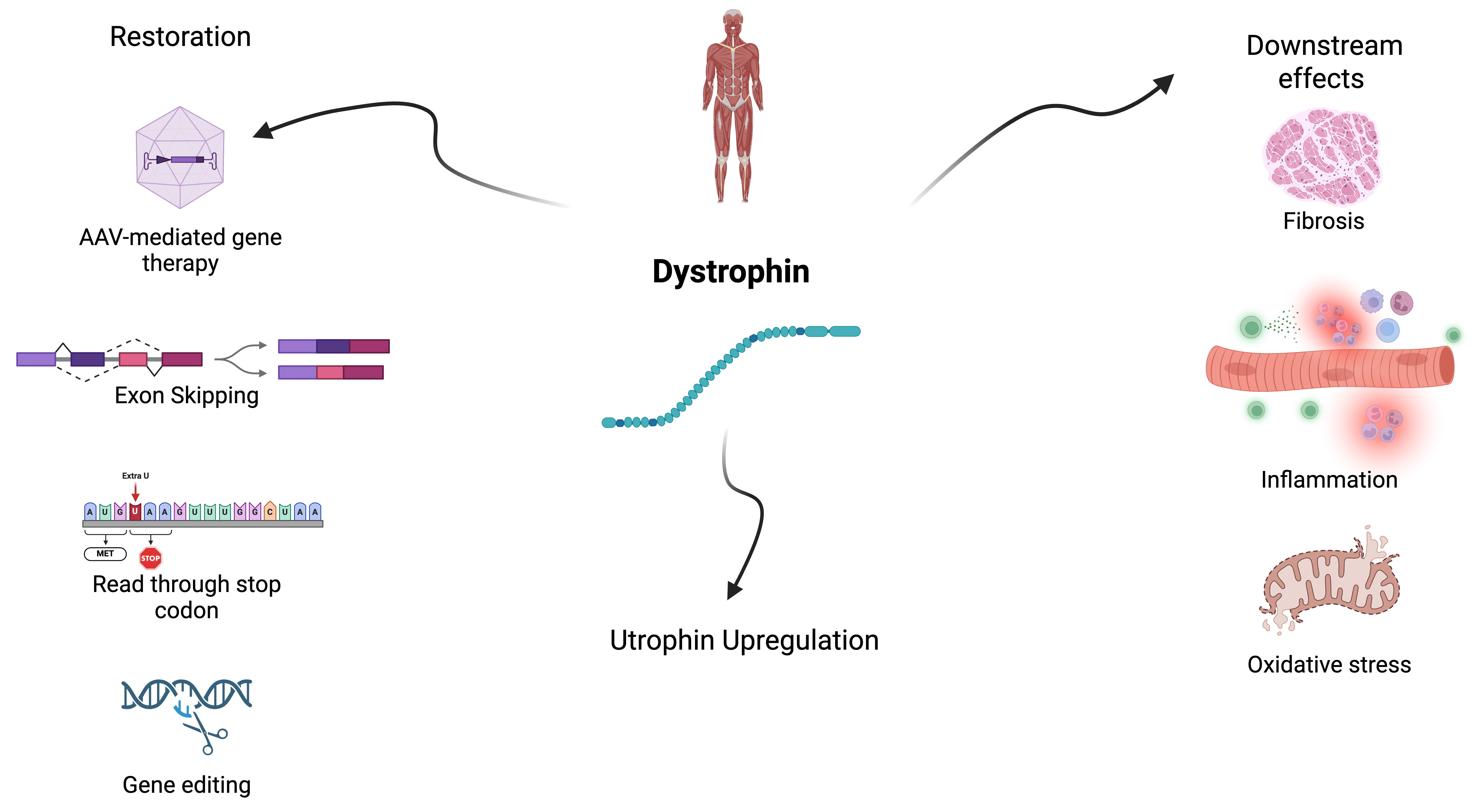
Figure 1. Treatment strategies for Duchenne muscular dystrophy. Created using BioRender (https://BioRender.com/sfg0p18).
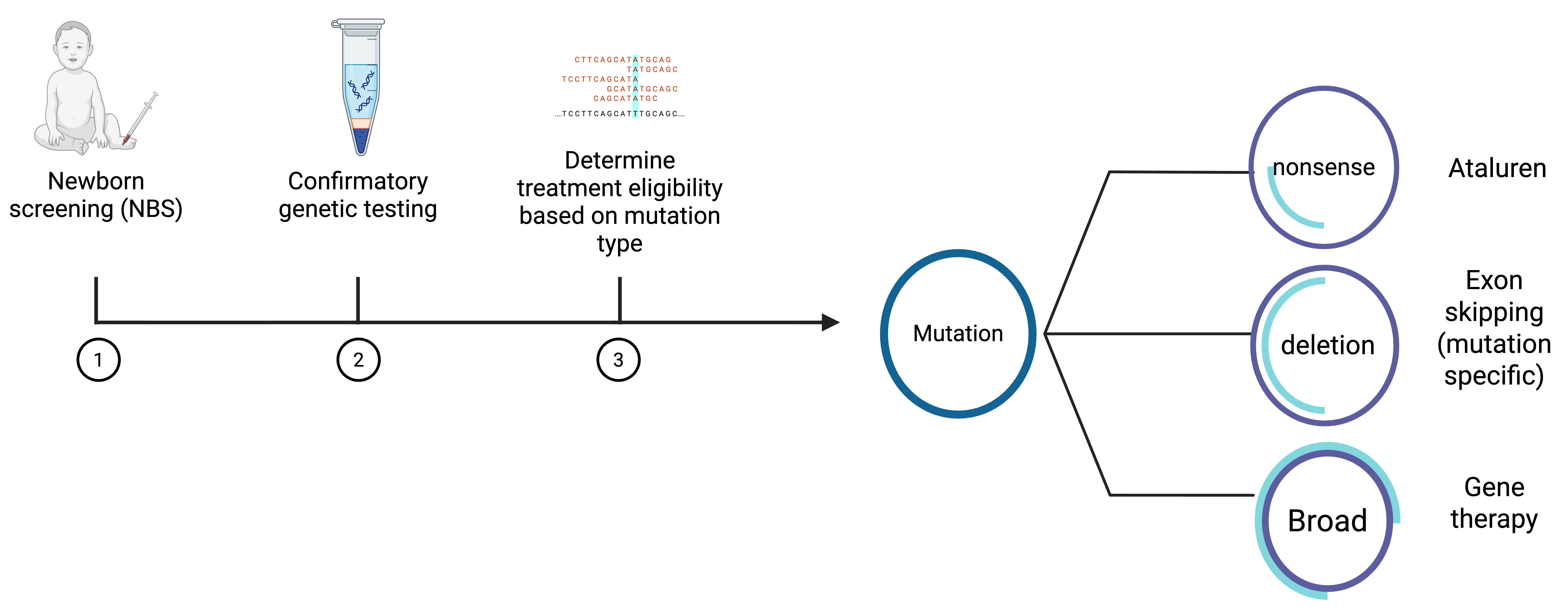
Figure 2. Diagnostic flowchart in Duchenne muscular dystrophy. Created using BioRender (https://BioRender.com/8du86bo).
DNA-mediated therapies
Gene editing
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-mediated gene editing is a promising therapy, theoretically allowing permanent restoration of dystrophin expression. The CRISPR system has two main components: (1) an endonuclease protein (Cas9 is the most commonly used); and (2) a single-guide RNA complementary to the target sequence[31,32]. Myo-editing can occur through homology-directed repair, which, in the presence of a DNA donor template, introduces the desired modification at the target locus; however, this mechanism is active only in proliferating cells. Another way is with nonhomologous end joining that can imprecisely repair the DNA by generating small insertion/deletions at the site of the DNA double strand breaks, which is active in both quiescent and proliferating cells. Myo-editing techniques have shown promising results in preclinical animal models and human cells[33]. Dual-adeno-associated virus (AAV) systems can overcome the packaging limitations of AAV vectors for in vivo CRISPR delivery. However, they require high vector doses, which may limit clinical feasibility. Another major concern of AAV-CRISPR gene therapy is the innate and adaptive immune responses evoked by AAV vectors and Cas proteins[31,33].
Development of editing and delivery methods to reduce viral vector dosing to improve safety and reduce immunogenicity, while maintaining/improving dystrophin restoration efficacy, off-target related safety concerns and durability need to be addressed before clinical implementation[33].
Gene replacement therapy
For monogenic diseases that lead to absent protein expression, the goal of gene replacement therapy is to deliver an intact copy of the mutated gene. In DMD, the transgene is delivered to muscle cells to allow the expression of functional protein and then restore or attenuate the phenotype[15].
The DMD gene (2.3 Mb) and its full-length transcript (14 kb) exceed the carrying capacity of commonly utilized AAV vectors, which is approximately 5 kb. To overcome this limitation, a truncated version of dystrophin (mini/microdystrophin) was developed. The amount of functional microdystrophin required to achieve clinical efficacy varies, and multiple studies in animal models (mice and canines) indicate that approximately 20% of the normal dystrophin level may be sufficient to produce therapeutic effects[1]. Different degrees of disease improvement have been seen in the mouse model with a wide range of dystrophin expression. Functional improvement was seen with as low as 4% dystrophin expression, while 20% levels prevented development of dystrophic symptoms. However, extrapolation of these data to human patients remains uncertain and challenging due to interspecies differences in muscle biology and correlations with clinical outcomes[34]. Another factor to consider is that a truncated dystrophin is likely less effective than the wild-type, and the durability of dystrophin expression is not fully known[35]. Additionally, the accuracy of dystrophin quantification techniques shows variability and sampling error should be considered due to the substantial variation of dystrophin expression within myofibers, between myofibers and even between muscles[30].
In general, gene replacement therapies for DMD have focused on the delivery of various micro-dystrophin transgenes. Portions of the DMD coding sequence have been removed to preserve essential protein function while reducing the size to address AAAV vector capacity limits. These constructs differ in terms of the micro-dystrophin structure, tissue-specific promoter and the AAV serotype, such differences may influence the transduction, efficacy and safety. [Table 1] summarize the distinctions between the FDA-approved gene therapy and other investigational drugs currently in clinical trials[35,36].
Summary of different microdystrophin constructs*
| Serotype | Tissue tropism | Promoter | Company | Development progress |
| AAV8 | Skeletal, cardiac | Spc5.12 | Regenxbio | Multicenter, phase I/II/III, open-label study. Entering pivotal phase |
| AAV8 | Skeletal, cardiac | Spc5.12 | Genethon | Completed Phase 1/2. Entering Pivotal phase |
| AAV9 | Skeletal, cardiac, lesser extent CNS | CK8e | Solid Biosciences | Switched to a new generation vector (myo-AAV) |
| AAV9 | Skeletal, cardiac, lesser extent CNS | CK7 | Pfizer | Development was stopped (missed primary point in phase III clinical trial) |
| AAVrh74 | Skeletal, cardiac | MHCK7 | Sarepta/Roche | FDA approved in 2024 (Elevydis). Long-term outcome studies ongoing |
The therapeutic potential of AAV gene therapy depends not only on vector design optimization but also on other strategies, including promoter selection, scaffold choice, and the use of adjunct peptides to enhance transgene expression and minimize toxicity[37,38].
In 2020, Mendell et al. demonstrated in a phase 1/2 study that treatment with SRP-9001 led to improved microdystrophin expression, reduced creatine kinase levels, and increased NorthStar ambulatory assessment (NSAA) scores[39]. In the subsequent phase 3 randomized trial (NCT05096221) with the same molecule, named delandistrogene moxeparvovec (Elevidys), part 1 - change from baseline NSAA score at 52 weeks- did not reach statistical significance. Some of the secondary endpoints showed numeric differences favoring treatment in time to rise (TTR), 10-meter walk/run (10MWR) and 95th percentile stride velocity[39,40].
Based on this data, Elevidys was granted accelerated approval (AA) from the FDA in June 2023 to treat patients with DMD aged four to five years. In June 2024, the label was expanded to a full approval for ambulatory patients and AA for nonambulatory patients over four years of age.
AAV-mediated gene therapy replacement challenges
Durability
DMD is a chronic neuromuscular disease and, as such, requires sustained dystrophin expression. Several studies have shown that AAV vectors can persist in normal tissues, including muscle cells. However, there is some uncertainty regarding the long-term persistence of the transgene in the dystrophic muscle and animal model data might not be fully translatable to patients with DMD[41,42]. Decline in transgene expression can presumably be related to immune response and vector dilution[43]. As the vector genome is thought to remain episomal and not integrate into the host genome, the cell turnover rate in growing muscle, as seen in young patients, and repeated cycles of necrosis and regeneration in dystrophic cells can affect the persistence of the transgene and potentially lead to a dilution effect. It should also be noted that overactivation of the immune system in the microenvironment of dystrophic cells can limit AAV genome persistence[41,44,45].
AAV- immunogenicity
Systemic delivery of high doses of AAV can trigger immune system-mediated toxicity. The complement system is a primary component of innate immunity and in vitro studies suggest that AAV capsid can interact and activate complement components. This activation can also happen through the classic pathway by immune complex formed from anti-AAV capsid antibodies[45-48]. The adaptive immune response to AAV capsid has been reported in gene therapy trials for hemophilia, spinal muscular atrophy (SMA), and other diseases.
To date, two fatal cases of liver failure have been reported following treatment with Elevidys[49]. The pathogenesis of liver injury is not yet fully understood. However, liver injury described in gene therapy clinical trials using AAV vectors could potentially be a result of both innate (possible complement system activation), and adaptive (T-cell mediated) immune response. Additionally, if the transgene is highly immunogenic, it could also trigger immune toxicity[50,51].
Other toxicities related to gene therapy include myocarditis, liver injury and thrombotic microangiopathy (TMA), which can also be related to activation of both innate and adaptive responses against the AAV vector capsid[51].
Some of the strategies under investigation to reduce AAV immunogenicity include inhibition of complement receptor[52], AAV vector coating with a modified polymer to reduce interaction with neutralizing antibodies (Nabs)[53], removal of CpG motifs from the AAV vector[54], and the use of less immunogenic AAV serotypes[55].
Transgene immunogenicity
Dystrophin-specific T cell activation is a major challenge in systemic gene therapy for DMD. Five patients with DMD enrolled in three trials (NCT0428148, NCT04626674, Eudra-CT number, 2020-002093-27), using different microdystrophin transgenes, promoters and AAV products (AAV9, AAV8 and AAVrh74), experienced suspected unexpected serious adverse reactions. These patients presented between 3 and 6 weeks post infusion with symptoms of severe myositis that led to loss of ambulation and weakness of the bulbar and respiratory muscles. The timing of these adverse reactions was consistent with transgene expression and further studies suggested a T-cell-mediated response against specific microdystrophin peptides contained within exons 8 through 11[56,57].
As a result, AAV clinical trials have been modified to exclude patients who have DMD deletions that significantly overlap with those transgene sequences.
Pre-existing immunity
One of the major difficulties in AAV-mediated gene therapy is the presence of pre-existing Nabs against the viral capsid; overall, up to 80% of the human population has antibodies against various serotypes[46].
Pre-existing immunity to AAV vectors is a barrier for treatment eligibility and is primarily due to prior exposure to the wild-type AAV which leads to the formation of Nabs. Seroprevalence differs among serotypes, with studies reporting 32%, 36% and 47% prevalence for AAVrh74, AAV9 and AAV8, respectively. Given the degenerative nature of the disease, it is likely that redosing might be needed in the future. Development of strategies such as pharmacological modulation, removal of circulating antibodies (plasmapheresis), blocking innate immunity (complement antagonist), use of different AAV serotypes and AAV capsid engineering are under investigation[45,58].
RNA-mediated therapies
Exon skipping
Antisense oligonucleotides (ASOs) are short, single-stranded nucleotides that are capable of binding to a specific region of RNA to promote exon-skipping. The main goal is to restore the disrupted reading frame of the dystrophin gene, producing an internally truncated but functional dystrophin protein[1,59].
These therapies are mutation-specific and benefit a subset of patients, approximately 27% of the DMD population. Currently, there are four FDA-conditionally approved ASOs that target exons 45, 51 and 53 [Table 2]. Clinical trials have shown that ASOs restore dystrophin in patients with DMD, albeit at very low levels. The FDA approval was granted based on the surrogate endpoint of dystrophin expression in muscle biopsies of treated patients. However, none of these ASOs has received approval in Europe. Clinical data on Eteplirsen, the first FDA-approved exon skipping therapy, has demonstrated slowing disease progression compared with matched external controls[60]. However, the data remains controversial and it is not yet fully clear if the low levels of dystrophin restoration (even with high intravenous doses) are sufficient and further clinical trials are ongoing to further assess the long-term functional efficacy and safety[61].
Summary of current FDA conditionally approved exon skipping therapies*
| Treatment | Exon | Year of FDA approval | Dose | % DMD patients amenable to treatment | % Dystrophin restoration |
| Eteplirsen (exondys) | 51 | 2016 | 14% | 0.9% after 180 weeks | |
| Golodirsen (vyondis) | 53 | 2019 | 8% | 1% after 48 weeks | |
| Vitolarsen (viltepso) | 53 | 2020 | 8% | 5.9% after 25 weeks | |
| Casimersen (amondys) | 45 | 2021 | 9% | 4.25% after 48 weeks |
ASO treatments have several limitations related to delivery efficiency to the skeletal and cardiac tissues. Approved ASOs are based on phosphorodiamidate morpholino oligomer (PMO) chemistry, which is uncharged and does not bind to serum proteins, thereby limiting tissue distribution and bioavailability due to renal clearance[31]. Delivery to skeletal and cardiac muscle can be improved through pPMO peptide conjugates, transferrin receptor-targeted antibody conjugation, and chemical modifications (see
Summary of ASOs in development*
| ASO | Type of modification | Exon targeted | Company | Current development |
| Vesleteplirsen | conjugate | 51 | Sarepta | Phase 2 multidose ascending dose. Program was discontinued in 2024 due to safety concerns |
| PGN-EDO51 | conjugate | 51 | PepGen | Phase 2 trial was discontinued |
| ENTR-601-044 | conjugate | 44 | Entrada Therapeutics | FDA hold was removed in February 2025 |
| DYNE-251 | Transferrin 1 Antibody associated | 51 | Dyne Therapeutics | Phase 1/2 clinical trial (Active, not recruiting) |
| AOC1044 | Transferrin 1 antibody associated | 44 | Avidity | Phase 1/2 clinical trial (Active, not recruiting) |
| BMN351 | Phosphorothioate chemical modifications | 51 | BioMarin | Phase 1/2 clinical trial (recruiting) |
| WVE-N531 | Inclusion of phosphoryl guanidine (PN), chemical modification | 53 | Wave Life Sciences | Phase 1b/2 open-label study (Active, not recruiting) |
Read-through of premature stop mutations
Nonsense mutations replace an amino acid codon in the messenger RNA (mRNA) by one of the three stop codons - UAA, UGA or UAG, resulting in an inactive, truncated protein. Drugs that induce suppression of these nonsense mutations during translation can increase the read-through of the premature stop signal and produce a full-length protein[65,66].
This therapeutic approach, applicable to all nonsense mutations in DMD, can treat 10%-15% of all DMD patients.
Ataluren is an oral treatment that promotes full-length dystrophin synthesis through ribosomal read-through of an in frame premature stop codon. It is not approved in the United States, and the European Medicines Agency (EMA) is not going to renew its approval in Europe, as clinical trials have failed to prove its effectiveness[67].
Gene replacement surrogates
Utrophin is a cytoskeletal protein that is an autosomal paralogue of dystrophin. Upregulation of utrophin A, the predominant form in skeletal muscle, can potentially compensate for the absence of dystrophin. Several pathways involved in utrophin regulation and expression are currently under investigation[68,69].
Regulatory divergence in treatment approvals
The FDA and the EMA have high concordance in decisions on marketing approvals. However, there are also divergent authorization decisions based on clinical data, efficacy, safety conclusions and regulatory authority differences[70].
Both the FDA and EMA have expedited approval pathways for therapies intended for serious, life threatening and rare diseases. In the United States, the AA program allows authorization based on a surrogate endpoint that is likely to predict a clinical benefit. By contrast, the conditional marketing authorization (CMA) pathway in the European Union requires a higher level of clinical evidence and does not rely on surrogate endpoints. These distinctions are illustrated by the FDA’s approval of exon skipping therapy based on surrogate endpoint and the EMA’s initial approval of Ataluren based on preliminary efficacy evidence and the benefit-risk balance[71].
MEASURES OF MOTOR FUNCTION IN CLINICAL TRIALS AND CLINICAL PRACTICE
Determination of treatment efficacy based on motor function changes must consider many factors. Understanding the natural history and progressive nature of the disease, the effects of standardized administration, and patient behavior or cognition on effort-based motor performance, as well as perception of change and impact on quality of life, are all important when interpreting changes in motor function measures. Clinical outcome assessments (COAs) have been validated to quantitatively measure clinically meaningful functional changes in boys with DMD[72-74]. To better understand true change in response to intervention and interpret clinical trial outcomes, estimates of minimal detectible change (MDC) and anchored minimal clinically important differences (MCIDs) highlighting the relevance of function to patient perception have been established for multiple COAs[73,75-77]. The North Star Ambulatory Assessment (NSAA) incorporates items that may be gained in boys with DMD treated with disease modifying therapies (DMTs) that would typically be lost with typical disease progression. Based on MDC and MCID estimates, score thresholds may be applied to define clinical progression of disease and may aid in distinguishing non-transient functional change using the NSAA. Additionally, timed function tests such as time to climb 4 stairs (4SC) and the Six Minute Walk Distance (6MWD) utilizing continuous data can be sensitive to change. These may be used to convert data to calculate movement velocity and can be interpreted in the context of maturation, using percent-predicted results relative to normative values[78]. The Performance of Upper Limb 2.0 (PUL 2.0) was designed to measure upper extremity function across the range of severity in patients with DMD including those who are nonambulatory[79,80]. The scale measures gross and fine motor function with items that relate to daily function utilizing upper limbs. Results of COAs administered to assess changes in function must incorporate clinical reasoning, knowledge gained from scale validation, and lessons learned from previous clinical trials and clinical care to set realistic expectations for functional prognosis, which may influence clinical decision-making and the recommendations provided to patients and their caregivers.
FROM CLINICAL TRIALS TO REAL-WORLD: LESSONS LEARNED
Extrapolating clinical trial findings to real-world patients is challenging due to multiple influencing factors. The perception of gene therapy benefits can differ among patients, caregivers, and providers. Therefore, it is essential to establish clear definitions and set realistic expectations.
Increases in dystrophin levels after treatment occur gradually over time, and clinical effects might not be apparent until the first year post-treatment. High-dose steroids used in gene therapy can affect short-term functional outcomes. This may create inaccurate perceptions of improvement among patients and families. Discouragement may arise if progress appears to stall during steroid tapering.
A further consideration is the initial expanded label for Elevidys in nonambulatory boys. Families may anticipate outcomes comparable to those observed in younger, ambulatory patients. However, in later disease stages, functional gains are less likely.
This underscores the need for risk stratification for potential cardiac and liver events following gene therapy, particularly in older patients. This should be addressed during the screening process to ensure informed decision-making.
Large-scale registries, such as the Strategic Targeting of Registries and International Database of Excellence (STRIDE) Registry and the Cooperative International Neuromuscular Research Group Duchenne Natural History Study (CINRG DNHS), provide robust data to help differentiate between perceived and actual efficacy. For example, in the STRIDE registry, patients receiving ataluren plus standard of care showed a delay in loss of ambulation. On the other hand, data from the CINRG DNHS has shown that glucocorticoid treatment for ≥ 1 year increases median age at loss of ambulation and other functional milestones by
Although gene therapies offer life-changing benefits for patients, the unprecedentedly high cost represents a challenge for reimbursement. Some payers may deny coverage, and others might include several filters before approval which can delay treatment. Various alternative payment models have been proposed and used to pay for these therapies in the United States and internationally. Some of these models address the high budgetary impact of gene therapies by spreading costs over multiple years (amortization) or across multiple patients (risk spreading), to make the cost among the population of patients more predictable. Other models focus on the clinical uncertainty of high-cost therapies by using performance-based agreements. In an outcome-based payment model, compensation is directly linked to the drug’s performance and payment is adjusted based on a predefined outcome, either at the individual level or across the treated population[83]. This model has also been combined with amortization, allowing the benefit of spreading costs over time.
More efforts to implement innovative solutions are needed to ensure patient equitable access to treatment[83].
CONCLUSIONS
Variable results have been reported from multiple clinical trials targeting dystrophin restoration. In general, modest clinical benefits were observed, showing slowed disease progression, but long-term efficacy and durability remain uncertain. Emerging therapies for dystrophin restoration hold promise to address limitations of current treatments, including transfection efficacy, immunotoxicity, alternative immunosuppression regimens, and AAV delivery. Future advancements in dystrophin restoration will require both optimization of current therapeutic strategies and integration of sensitive biomarkers and comprehensive outcome measures to accurately assess clinical response. Additionally, real-world data will be essential to understand the impact across diverse patient populations and guide evidence-based clinical implementation. Nevertheless, multidisciplinary care and steroids remain the mainstay of treatment.
DECLARATIONS
Authors’ contributions
Initial draft, conceptualization, writing and editing: Gonzalez Castillo Z
Writing and editing: Nelson L
Writing, editing and supervision: Batley K, Iannaccone ST
All authors have reviewed and agreed to the published version of the manuscript.
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
Iannaccone ST is an Editor and Editorial Board Member of Journal of Translational Genetics and Genomics. Iannaccone ST was not involved in any steps of editorial processing, notably including reviewers' selection, manuscript handling and decision making, Iannaccone ST has served as a consultant for Sarepta Therapeutics, TRINDS, Merck, BioMarin, Broadstreet and Astellas; Gonzalez Castillo Z has served on advisory boards for ITF Therapeutics and Sarepta Therapeutics; Batley K has received research funding from Catalyst Pharmaceuticals, Sarepta Therapeutics, and NS Pharma. She has served on advisory boards and/or as a speaker for Catalyst Pharmaceuticals, ITF Therapeutics, Sarepta Therapeutics, Pfizer, and Regenxbio; Nelson L declares that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
1. Sun C, Shen L, Zhang Z, Xie X. Therapeutic strategies for duchenne muscular dystrophy: an update. Genes. 2020;11:837.
2. Crisafulli S, Sultana J, Fontana A, Salvo F, Messina S, Trifirò G. Global epidemiology of Duchenne muscular dystrophy: an updated systematic review and meta-analysis. Orphanet J Rare Dis. 2020;15:141.
3. Landfeldt E, Thompson R, Sejersen T, McMillan HJ, Kirschner J, Lochmüller H. Life expectancy at birth in Duchenne muscular dystrophy: a systematic review and meta-analysis. Eur J Epidemiol. 2020;35:643-53.
4. Armstrong N, Apkon S, Berggren KN, et al. The early care (0-3 Years) in duchenne muscular dystrophy meeting report. J Neuromuscul Dis. 2024;11:525-33.
5. Sarker S, Eshaque TB, Soorajkumar A, et al. Mutational spectrum and phenotypic variability of Duchenne muscular dystrophy and related disorders in a Bangladeshi population. Sci Rep. 2023;13:21547.
6. Selvatici R, Rossi R, Fortunato F, et al. Ethnicity-related DMD genotype landscapes in European and non-European countries. Neurol Genet. 2021;7:e536.
7. Amr K, Fahmy N, El-Kamah G. Genomic insights into Duchene muscular dystrophy: analysis of 1250 patients reveals 30% novel genetic patterns and 6 novel variants. J Genet Eng Biotechnol. 2024;22:100436.
8. Zinina E, Bulakh M, Chukhrova A, et al. Specificities of the DMD gene mutation spectrum in Russian patients. Int J Mol Sci. 2022;23:12710.
9. Neri M, Rossi R, Trabanelli C, et al. The genetic landscape of dystrophin mutations in Italy: a nationwide study. Front Genet. 2020;11:131.
10. Bladen CL, Salgado D, Monges S, et al. The TREAT-NMD DMD global database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat. 2015;36:395-402.
11. Esposito G, Tremolaterra MR, Marsocci E, et al. Precise mapping of 17 deletion breakpoints within the central hotspot deletion region (introns 50 and 51) of the DMD gene. J Hum Genet. 2017;62:1057-63.
12. Oshima J, Magner DB, Lee JA, et al. Regional genomic instability predisposes to complex dystrophin gene rearrangements. Hum Genet. 2009;126:411-23.
13. Oudet C, Hanauer A, Clemens P, Caskey T, Mandel JL. Two hot spots of recombination in the DMD gene correlate with the deletion prone regions. Hum Mol Genet. 1992;1:599-603.
14. Bez Batti Angulski A, Hosny N, Cohen H, et al. Duchenne muscular dystrophy: disease mechanism and therapeutic strategies. Front Physiol. 2023;14:1183101.
15. Ricci G, Bello L, Torri F, Schirinzi E, Pegoraro E, Siciliano G. Therapeutic opportunities and clinical outcome measures in Duchenne muscular dystrophy. Neurol Sci. 2022;43:625-33.
16. Tyler KL. Origins and early descriptions of "Duchenne muscular dystrophy". Muscle Nerve. 2003;28:402-22.
17. Fortunato F, Rossi R, Falzarano MS, Ferlini A. Innovative therapeutic approaches for Duchenne muscular dystrophy. J Clin Med. 2021;10:820.
18. Szabo SM, Salhany RM, Deighton A, Harwood M, Mah J, Gooch KL. The clinical course of Duchenne muscular dystrophy in the corticosteroid treatment era: a systematic literature review. Orphanet J Rare Dis. 2021;16:237.
19. Ji C, Kariyawasam DS, Sampaio H, Lorentzos M, Jones KJ, Farrar MA. Newborn screening for Duchenne muscular dystrophy: the perspectives of stakeholders. Lancet Reg Health West Pac. 2024;45:101049.
20. Tavakoli NP, Gruber D, Armstrong N, et al. Newborn screening for Duchenne muscular dystrophy: a two-year pilot study. Ann Clin Transl Neurol. 2023;10:1383-96.
21. Park S, Maloney B, Caggana M, Tavakoli NP. Creatine kinase-MM concentration in dried blood spots from newborns and implications for newborn screening for Duchenne muscular dystrophy. Muscle Nerve. 2022;65:652-8.
22. Chien YH, Lee NC, Weng WC, et al. Duchenne muscular dystrophy newborn screening: the first 50,000 newborns screened in Taiwan. Neurol Sci. 2022;43:4563-6.
23. Jia C, Zhao D, Li Y, et al. Newborn screening and genomic analysis of duchenne muscular dystrophy in Henan, China. Clin Chim Acta. 2023;539:90-6.
24. Moat SJ, Bradley DM, Salmon R, Clarke A, Hartley L. Newborn bloodspot screening for Duchenne muscular dystrophy: 21 years experience in Wales (UK). Eur J Hum Genet. 2013;21:1049-53.
25. Parent Project Muscular Dystrophy. Available from https://www.parentprojectmd.org/advocacy/newborn-screening-action-center/ [Last accessed on 27 Nov 2025].
26. Chen L, Luo X, Wang H, Tian Y, Liu Y. Identifying inversions with breakpoints in the Dystrophin gene through long-read sequencing: report of two cases. BMC Med Genomics. 2024;17:227.
27. Olivucci G, Iovino E, Innella G, Turchetti D, Pippucci T, Magini P. Long read sequencing on its way to the routine diagnostics of genetic diseases. Front Genet. 2024;15:1374860.
28. Owusu R, Savarese M. Long-read sequencing improves diagnostic rate in neuromuscular disorders. Acta Myol. 2023;42:123-8.
29. Ling C, Dai Y, Geng C, et al. Uncovering the true features of dystrophin gene rearrangement and improving the molecular diagnosis of Duchenne and Becker muscular dystrophies. iScience. 2023;26:108365.
30. Wells DJ. What is the level of dystrophin expression required for effective therapy of Duchenne muscular dystrophy? J Muscle Res Cell Motil. 2019;40:141-50.
31. Chen G, Wei T, Yang H, Li G, Li H. CRISPR-based therapeutic gene editing for duchenne muscular dystrophy: advances, challenges and perspectives. Cells. 2022;11:2964.
32. Eslahi A, Alizadeh F, Avan A, et al. New advancements in CRISPR based gene therapy of Duchenne muscular dystrophy. Gene. 2023;867:147358.
33. Chemello F, Olson EN, Bassel-Duby R. CRISPR-editing therapy for duchenne muscular dystrophy. Hum Gene Ther. 2023;34:379-87.
34. Godfrey C, Muses S, McClorey G, et al. How much dystrophin is enough: the physiological consequences of different levels of dystrophin in the mdx mouse. Hum Mol Genet. 2015;24:4225-37.
35. Heslop E, Turner C, Irvin A, Muntoni F, Straub V, Guglieri M; workshop participants. Gene therapy in Duchenne muscular dystrophy: identifying and preparing for the challenges ahead. Neuromuscul Disord. 2021;31:69-78.
36. Roberts TC, Wood MJA, Davies KE. Therapeutic approaches for Duchenne muscular dystrophy. Nat Rev Drug Discov. 2023;22:917-34.
37. Chen SK, Hawley ZCE, Zavodszky MI, et al. Efficacy and safety of a SOD1-targeting artificial miRNA delivered by AAV9 in mice are impacted by miRNA scaffold selection. Mol Ther Nucleic Acids. 2023;34:102057.
38. Xie YL, Wang JY, He Y, et al. The use of melittin to enhance transgene expression mediated by recombinant adeno-associated virus serotype 2 vectors both in vitro and in vivo. J Integr Med. 2023;21:106-15.
39. Mendell JR, Muntoni F, McDonald CM, et al. AAV gene therapy for Duchenne muscular dystrophy: the EMBARK phase 3 randomized trial. Nat Med. 2025;31:332-41.
40. Oskoui M, Caller TA, Parsons JA, et al. Delandistrogene Moxeparvovec gene therapy in individuals with duchenne muscular dystrophy: evidence in focus: report of the AAN guidelines subcommittee. Neurology. 2025;104:e213604.
41. Manini A, Abati E, Nuredini A, Corti S, Comi GP. Adeno-associated virus (AAV)-mediated gene therapy for duchenne muscular dystrophy: the issue of transgene persistence. Front Neurol. 2021;12:814174.
42. Wasala NB, Yue Y, Hu B, et al. Lifelong outcomes of systemic adeno-associated virus micro-dystrophin gene therapy in a murine duchenne muscular dystrophy model. Hum Gene Ther. 2023;34:449-58.
43. Shen W, Liu S, Ou L. rAAV immunogenicity, toxicity, and durability in 255 clinical trials: a meta-analysis. Front Immunol. 2022;13:1001263.
44. Mollard A, Peccate C, Forand A, et al. Muscle regeneration affects adeno associated Virus 1 mediated transgene transcription. Sci Rep. 2022;12:9674.
45. Duan D. Systemic AAV micro-dystrophin gene therapy for duchenne muscular dystrophy. Mol Ther. 2018;26:2337-56.
46. Kumar SRP, Duan D, Herzog RW. Immune responses to muscle-directed adeno-associated viral gene transfer in clinical studies. Hum Gene Ther. 2023;34:365-71.
47. Zaiss AK, Cotter MJ, White LR, et al. Complement is an essential component of the immune response to adeno-associated virus vectors. J Virol. 2008;82:2727-40.
48. Martino AT, Suzuki M, Markusic DM, et al. The genome of self-complementary adeno-associated viral vectors increases Toll-like receptor 9-dependent innate immune responses in the liver. Blood. 2011;117:6459-68.
49. U.S Food and Drug Administration. FDA investigating deaths due to acute liver failure in non-ambulatory duchenne muscular dystrophy patients following ELEVIDYS. Available from https://www.fda.gov/vaccines-blood-biologics/safety-availability-biologics/fda-investigating-deaths-due-acute-liver-failure-non-ambulatory-duchenne-muscular-dystrophy-patients [Last accessed on 19 Nov 2025].
50. Bengtsson NE, Tasfaout H, Chamberlain JS. The road toward AAV-mediated gene therapy of Duchenne muscular dystrophy. Mol Ther. 2025;33:2035-51.
51. Larrey D, Delire B, Meunier L, Zahhaf A, De Martin E, Horsmans Y. Drug-induced liver injury related to gene therapy: a new challenge to be managed. Liver Int. 2024;44:3121-37.
52. Spathis R, Kuriplach DR, Narvesen S, et al. Enhancing AAV-microdystrophin gene therapy after repeat dosing by blocking phagocytosis. Front Immunol. 2025;16:1527840.
53. Pinto MS, Martí-Melero L, Fernandez-Alarcon J, et al. Polymer-based coating of adeno-associated viral particles as a new strategy to evade immune response for DMD treatment. J Control Release. 2025;384:113896.
54. Bertolini TB, Shirley JL, Zolotukhin I, et al. Effect of CpG depletion of vector genome on CD8+ T cell responses in AAV gene therapy. Front Immunol. 2021;12:672449.
55. Li C, Samulski RJ. Engineering adeno-associated virus vectors for gene therapy. Nat Rev Genet. 2020;21:255-72.
56. Bönnemann CG, Belluscio BA, Braun S, Morris C, Singh T, Muntoni F. Dystrophin immunity after gene therapy for duchenne's muscular dystrophy. N Engl J Med. 2023;388:2294-6.
57. Potter RA, Moeller IH, Khan S, et al. Immunologic investigations into transgene directed immune-mediated myositis following delandistrogene moxeparvovec gene therapy. Sci Rep. 2025;15:4.
58. Verma S, Nwosu SN, Razdan R, et al. Seroprevalence of adeno-associated virus neutralizing antibodies in males with duchenne muscular dystrophy. Hum Gene Ther. 2023;34:430-8.
59. Leckie J, Zia A, Yokota T. An updated analysis of exon-skipping applicability for Duchenne muscular dystrophy using the UMD-DMD database. Genes. 2024;15:1489.
60. Iff J, Done N, Tuttle E, et al. Survival among patients receiving eteplirsen for up to 8 years for the treatment of Duchenne muscular dystrophy and contextualization with natural history controls. Muscle Nerve. 2024;70:60-70.
61. Patterson G, Conner H, Groneman M, Blavo C, Parmar MS. Duchenne muscular dystrophy: current treatment and emerging exon skipping and gene therapy approach. Eur J Pharmacol. 2023;947:175675.
62. Aartsma-Rus A. The future of exon skipping for Duchenne muscular dystrophy. Hum Gene Ther. 2023;34:372-8.
63. Matsuo M. 30 years since the proposal of exon skipping therapy for Duchenne muscular dystrophy and the future of pseudoexon skipping. Int J Mol Sci. 2025;26:1303.
64. Chwalenia K, Wood MJA, Roberts TC. Progress and prospects in antisense oligonucleotide-mediated exon skipping therapies for Duchenne muscular dystrophy. J Muscle Res Cell Motil. 2025.
65. Politano L. Read-through approach for stop mutations in Duchenne muscular dystrophy. An update. Acta Myol. 2021;40:43-50.
66. Ng MY, Li H, Ghelfi MD, Goldman YE, Cooperman BS. Ataluren and aminoglycosides stimulate read-through of nonsense codons by orthogonal mechanisms. Proc Natl Acad Sci USA. 2021;118:e2020599118.
67. Mercuri E, Osorio AN, Muntoni F, et al. Safety and effectiveness of ataluren in patients with nonsense mutation DMD in the STRIDE Registry compared with the CINRG Duchenne Natural History Study (2015-2022): 2022 interim analysis. J Neurol. 2023;270:3896-913.
68. Péladeau C, Jasmin BJ. Identifying FDA-approved drugs that upregulate utrophin a as a therapeutic strategy for duchenne muscular dystrophy. In: Maruyama R, Yokota T, editors. Muscular dystrophy therapeutics. New York: Springer; 2023. pp. 495-510.
69. Soblechero-Martín P, López-Martínez A, de la Puente-Ovejero L, Vallejo-Illarramendi A, Arechavala-Gomeza V. Utrophin modulator drugs as potential therapies for Duchenne and Becker muscular dystrophies. Neuropathol Appl Neurobiol. 2021;47:711-23.
70. Kashoki M, Hanaizi Z, Yordanova S, et al. A comparison of EMA and FDA decisions for new drug marketing applications 2014-2016: concordance, discordance, and why. Clin Pharmacol Ther. 2020;107:195-202.
71. Vokinger KN, Hwang TJ, Glaus CEG, Kesselheim AS. Therapeutic value assessments of novel medicines in the US and Europe, 2018-2019. JAMA Netw Open. 2022;5:e226479.
72. Mazzone E, Martinelli D, Berardinelli A, et al. North Star Ambulatory assessment, 6-minute walk test and timed items in ambulant boys with Duchenne muscular dystrophy. Neuromuscul Disord. 2010;20:712-6.
73. Mayhew AG, Cano SJ, Scott E, et al. Detecting meaningful change using the North Star Ambulatory assessment in Duchenne muscular dystrophy. Dev Med Child Neurol. 2013;55:1046-52.
74. McDonald CM, Henricson EK, Abresch RT, et al. The 6-minute walk test and other clinical endpoints in duchenne muscular dystrophy: reliability, concurrent validity, and minimal clinically important differences from a multicenter study. Muscle Nerve. 2013;48:357-68.
75. Henricson E, Abresch R, Han JJ, et al. The 6-minute walk test and person-reported outcomes in boys with Duchenne muscular dystrophy and typically developing controls: longitudinal comparisons and clinically-meaningful changes over one year. PLoS Curr. 2013;5:ecurrents.
76. Ayyar Gupta V, Pitchforth JM, Domingos J, et al. Determining minimal clinically important differences in the North Star Ambulatory Assessment (NSAA) for patients with Duchenne muscular dystrophy. PLoS One. 2023;18:e0283669.
77. Muntoni F, Signorovitch J, Sajeev G, et al. Meaningful changes in motor function in Duchenne muscular dystrophy (DMD): a multi-center study. PLoS One. 2024;19:e0304984.
78. Henricson E, Abresch R, Han JJ, et al. Percent-predicted 6-minute walk distance in duchenne muscular dystrophy to account for maturational influences. PLoS Curr. 2012;4:RRN1297.
79. Mayhew A, Mazzone ES, Eagle M, et al. Development of the performance of the upper limb module for duchenne muscular dystrophy. Dev Med Child Neurol. 2013;55:1038-45.
80. Mayhew AG, Coratti G, Mazzone ES, et al. Performance of upper limb module for duchenne muscular dystrophy. Dev Med Child Neurol. 2020;62:633-9.
81. Bushby K, Finkel R, Wong B, et al. Safety and effectiveness of ataluren in patients with nonsense mutation Duchenne muscular dystrophy: 2022 interim analysis from the STRIDE Registry compared with the CINRG Duchenne Natural History Study. J Neurol. 2023;270:2885-95.
82. Henricson EK, Abresch RT, Cnaan A, et al. The cooperative international neuromuscular research group Duchenne natural history study: Glucocorticoid treatment preserves clinically meaningful functional milestones and reduces rate of disease progression as measured by manual muscle testing and other commonly used clinical trial outcome measures. Muscle Nerve. 2013;48:55-67.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].