


RNA-binding proteins in skeletal disorders: insights into molecular mechanisms and possible therapeutic targets
 0
0 Abstract
RNA-binding proteins (RBPs) can form complex regulatory networks by binding to numerous transcripts, thereby exerting precise control over post-transcriptional gene regulation. Defects in their functions contribute to numerous human skeletal disorders by modifying RNA processing and regulation. Advancements in comprehending the molecular mechanisms of RBP functions are facilitating the development of effective therapies. Here, we delineated RBPs involved in bone development and skeletal disorders, highlighting recent advancements in this evolving field, focusing on mechanisms and therapeutic implications.
Keywords
INTRODUCTION
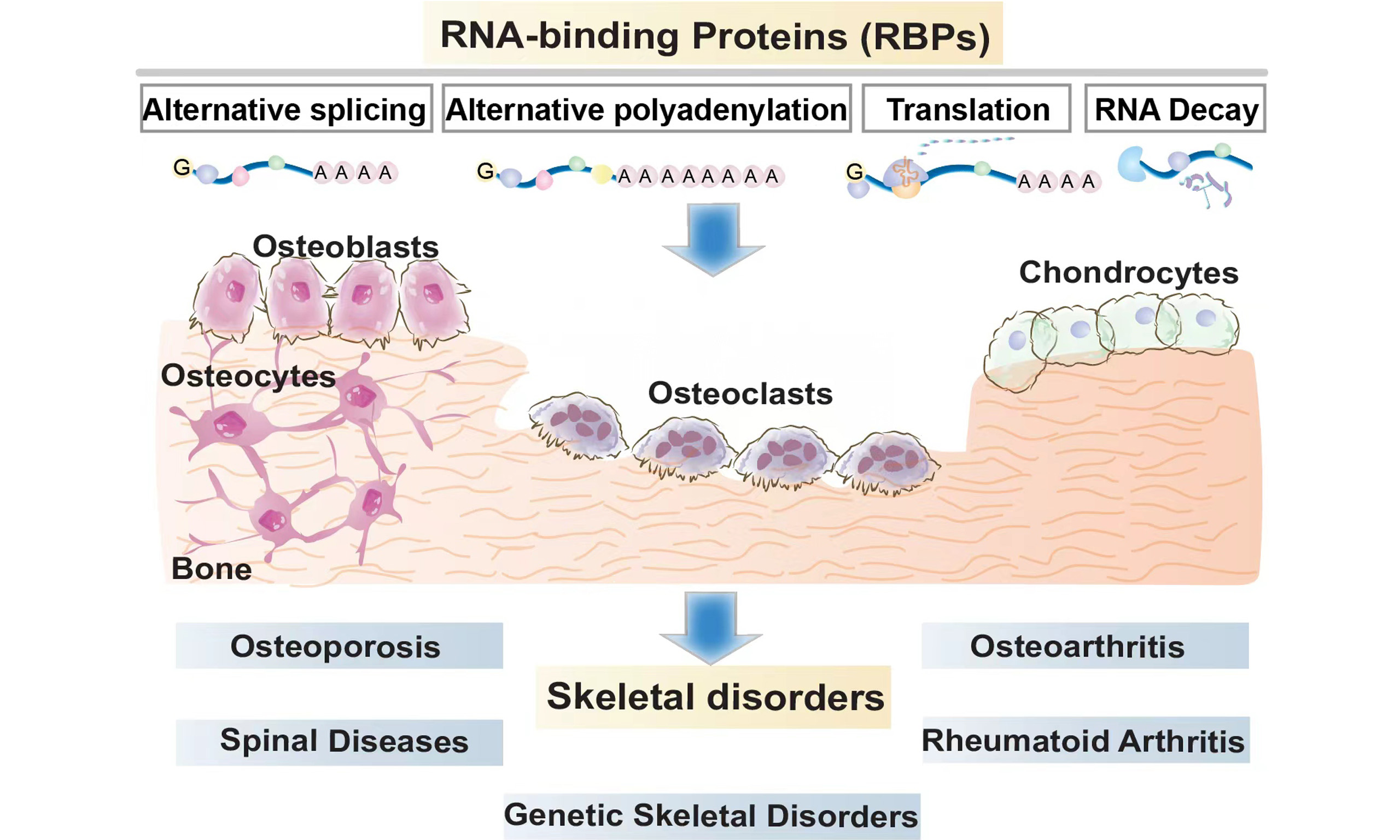
The skeleton is a metabolically active organ composed of cartilage and bone, comprising three principal cell types: osteoblasts (OBs), osteoclasts (OCs), and chondrocytes. OCs are formed through the fusion of mononuclear precursors, resulting in the formation of terminally differentiated mature and giant cells[1]. OBs differentiate directly from mesenchymal precursors or indirectly through a cartilage intermediate[2]. At sites of bone remodeling, OCs secrete regulatory factors that sequentially recruit osteoblast-lineage cells, thereby facilitating bone matrix deposition and mineralization[3,4]. Within this bone homeostasis, osteocytes are terminally differentiated OBs embedded within the bone matrix. They generate paracrine and endocrine factors, detect mechanical forces, regulate mineral metabolism, and sustain intercellular communications[5].
Any imbalance in bone remodeling, caused by inadequate activity of OBs or excessive activation of OCs, leads to diminished bone mass and weakened bone strength, thereby increasing the risk of fractures[6]. Although the role of post-transcriptional regulators, including microRNAs (miRNAs), is well-studied in cellular development, skeletal homeostasis, and disease conditions, the function of regulated RNA-binding proteins (RBPs) remains unclear.
Classic RBPs are modular proteins composed of well-defined RNA-binding domains (RBDs). They create ribonucleoproteins (RNPs) with cellular RNA molecules, affecting their fate from transcription through translation[7]. RBPs are evolutionarily conserved, with approximately 2% being tissue-specific, predominantly associated with mRNA- and non-coding affinities. These are attributed to the presence of one or more RBDs that interact with RNA in a sequence- and structure-dependent fashion, including the common RNA recognition motif (RRM), K-homology, DEAD/DEAH [Asp-Glu-Ala-Asp (referring to the conserved amino acid sequence D-E-A-D in the helicase motif II)/Asp-Glu-Ala-His (referring to the conserved sequence D-E-A-H in the corresponding motif)] helicase, and zinc-finger domains, and approximately 30 other less abundant domains[8].
However, there is another subfamily of RBPs that lack canonical RBDs, termed non-canonical RBPs (ncRBPs). Despite the identification of numerous ncRBPs and the characterization of their RNA interactomes, their roles in bone diseases remain unclear[9]. As “moonlighting” proteins, numerous ncRBPs are established metabolic enzymes, membrane proteins, or signaling molecules[10]. This dual functionality allows them to fulfill critical roles in the bone microenvironment and bone metabolism.
Mutations or expression alterations in RBPs frequently result in tissue-specific deficiencies, affecting skeletal development, mineral density maintenance, and bone integrity. Therefore, this study provides an updated overview of the role of RBPs in skeletal disorders and clarifies the underlying molecular mechanisms, highlighting potential therapeutic applications.
MOLECULAR MECHANISMS OF RBPs IN BONE HOMEOSTASIS
RBPs are a class of cytoplasm-nucleus shuttle proteins that regulate all stages of pre-mRNA processing, including 5’-end capping and splicing to 3’-end cleavage and polyadenylation in the nucleus. Additionally, RBPs closely regulate the encapsulation of mature mRNAs into RNP complexes, thereby facilitating their translocation into the cytoplasm for translation. RBP-mRNA interactions also influence mRNA stability and the timing and mechanisms of mRNA degradation [Figure 1][11,12]. Given the extensively documented studies on disabling RBP functions in skeletal disorders, we review the dysregulation or dysfunction of various RBPs affecting bone homeostasis and emphasize several well-studied RBPs of functional significance [Table 1].
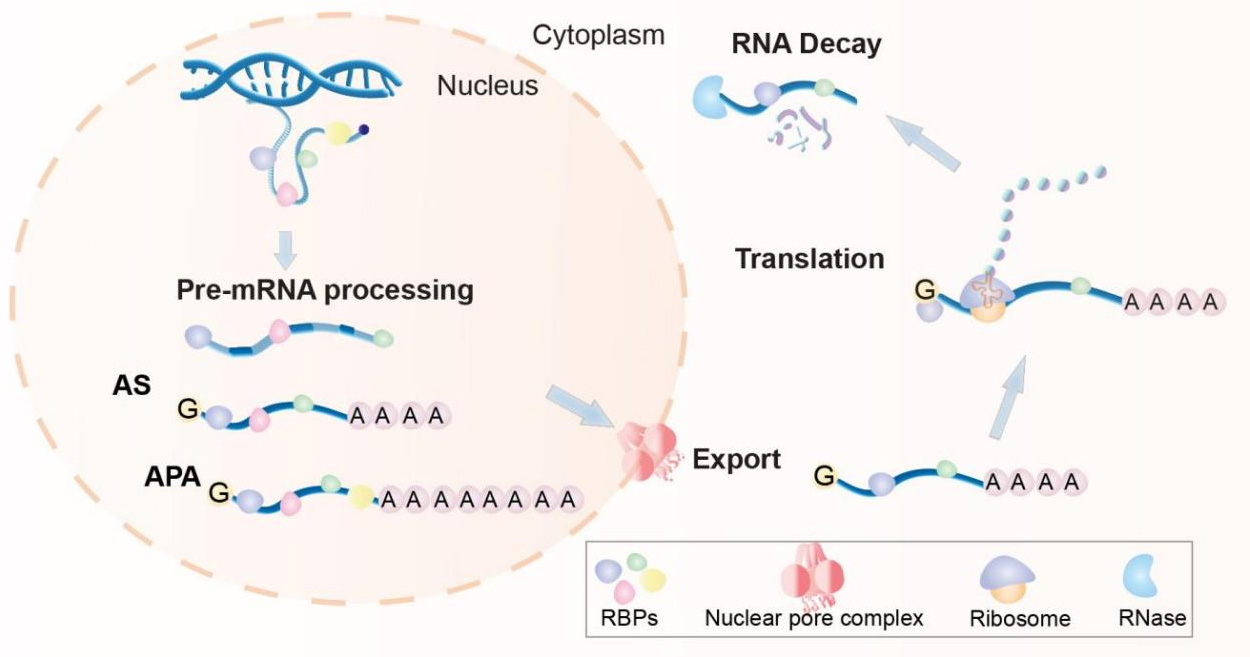
Figure 1. Interactions with RNA-binding proteins regulate RNA metabolism at multiple levels. From birth to decay, RNA fate is controlled by various dynamically changed RNA-binding proteins (RBPs, colored shapes), encompassing processes at the nuclear [alternative splicing (AS), alternative polyadenylation (APA)] and cytoplasmic (transport, translation, and degradation) levels.
Targets and dysregulation of RNA-binding proteins associated with bone homeostasis
| RBP | Basic RBP mechanisms | Experimental system | Targets | Bone metabolism | Reference |
| SF3B4 | Alternative splicing | C2C12 cells; ATDC5 cells | BMPR-IA | ↓Osteogenic differentiation ↓Chondrocyte differentiation | [15] |
| p54nrb | Alternative splicing | Transgenic mice | COL2A1 | ↑Chondrocyte differentiation | [16] |
| MSI1 | Translation regulation | MC3T3-E1 cells | MACF1 | ↑Osteoblast differentiation | [21] |
| MSI2 | Translation regulation | Bone marrow mesenchymal stem cells; bone marrow macrophages | Cebpα | ↑Osteoblastogenesis ↑Osteoclastogenesis | [22] |
| SAMD4 | Translation regulation | Mesenchymal progenitors | MIG6 | ↑Osteoblast differentiation ↑Chondrogenesis and chondrocyte differentiation | [24] |
| ZFP36 | mRNA stability | Knockout mice | - | ↓Osteoclast activity | [28] |
| ZFP36L1 | mRNA stability | Knockout mice | PPARγ2; HSPA1A | ↑Osteoblastic differentiation ↑Chondrocyte apoptosis | [29,30] |
| HuR | mRNA stability | Primary mouse osteoblasts; mesenchymal stromal cells | LARP6; HOTAIR | ↑Osteoblast differentiation | [32,33] |
| METTL14 | m6A writer | Bone marrow mesenchymal stem cells; bone marrow macrophages | BECLIN-1; GPX4 | ↑Osteogenic differentiation ↓Osteoclast differentiation | [36,37] |
| METTL3 | m6A writer | MC3T3-E1 cells | BECLIN-1; SMAD7; SMURF1 | ↓Osteogenic differentiation | [38-40] |
| FTO | m6A eraser | Osteoclast precursors | Cyclin A2; CDK2 | ↑Osteoclast formation | [45,46] |
| ALKBH5 | m6A eraser | Mesenchymal stem cells | PRMT6 | ↓Osteogenic differentiation | [47] |
| YTHDF1 | m6A reader | Bone marrow mesenchymal stem cell | ZNF839 | ↑Osteogenic differentiation | [48] |
| IGF2BP2 | m6A reader | MC3T3-E1 cells | SRF | ↑Osteogenic differentiation | [49] |
| YTHDF2 | m6A reader; | Umbilical cord mesenchymal stem cells | FBLN1 | ↓Osteogenic differentiation | [50] |
Alternative splicing
Alternative splicing (AS) is a co-transcriptional mechanism facilitated by the spliceosome - commonly involving exon skipping, intron retention, alternative 5’ splicing sites, alternative 3’ splicing sites, and mutually exclusive exon splicing. In bone homeostasis, specialized RBPs modulate this process by identifying pre-mRNA sequence elements and guiding the splicing machinery to cleavage and ligation sites. This produces multiple mature mRNA isoforms from identical pre-mRNA, affecting mRNA stability and protein diversity[13,14].
For instance, splicing factor 3B subunit 4 (SF3B4) participates in RNA splicing in the nucleus, where it interacts with bone morphogenetic protein receptor-IA (BMPR-IA) to impede osteogenic and chondrocyte differentiation[15]. Similarly, the 54-kDa nuclear RNA-binding protein (p54nrb) regulates chondrogenesis through specific AS. Minigene analysis and mutant p54nrb construction verified that it regulates collagen type II alpha 1 (Col2a1) mRNA splicing through two RRMs. Transgenic expression of mutant p54nrb (with Col2a1-Cre) impairs chondrogenesis and delays endochondral ossification[16]. Although these studies revealed the role of AS in OB and chondrocyte differentiation, its precise role in OC differentiation remains unclear, presenting a potential direction for subsequent studies.
Alternative polyadenylation
Alternative polyadenylation (APA) is a significant post-transcriptional regulation that produces a widespread mechanism that generates transcript isoforms with variable 3’ untranslated regions (3’UTR) length by identifying distinct polyadenylation signals[17]. APA frequently alters solely the 3’UTR of the mRNA, which differs in its interactions with RBPs, consequently influencing mRNA localization, translation, and stability[18]. Although APA is frequently dysregulated in cancer, little information exists regarding APA events in the bone domain. To date, only one study has indicated that APA enhances gene expression during endochondral bone formation. Specifically, APA‐induced Col1a1/2 3’UTRs shortening may evade repression by miR-29a-3p‐mediated gene repression[19].
Translation
RBP-mediated translation control mechanisms frequently focus on the translation initiation stage, where translation efficiencies fluctuate according to the binding affinities of RBPs with the 5’ or 3’ UTR of target mRNAs[20]. Musashi1 and Musashi2 belong to the Musashi (MSI) family, which regulates stem cell fate by modulating translational efficiency. They specifically bind to r(G/A)U1-3AGU sequences (MSI binding elements, MBEs) located at the 3’ UTR of the target mRNA. This binding inhibits the poly-A binding protein from interacting with the extension initiation complex, suppressing translation. Recent studies demonstrated that MSI1 stabilizes microtubule and actin crosslinking factor 1 (Macf1), thereby promoting MC3T3-E1 osteogenic differentiation through the Wnt/β-Catenin pathway[21]. MSI2 may bind to MBEs at the 3’UTR of CCAAT (Cytosine-Cytosine-Adenine-Adenine-Thymine)/enhancer binding protein alpha (mouse Cebpα) mRNA, thereby promoting osteoblastogenesis in bone marrow mesenchymal stem cells (BMSCs)[22]. It is significantly expressed in OCs, facilitating nuclear factor-kappa B (NF-κB) activation, thereby stimulating OC formation and survival from bone marrow macrophages (BMMs)[23]. Furthermore, sterile alpha motif domain containing protein 4 (SAMD4) functions as a repressor of translational mechanisms, and its deficiency in mesenchymal progenitors exhibits impaired OB differentiation, chondrogenesis, and chondrocyte differentiation. This correlates with increased synthesis of mitogen-inducible gene 6 protein, which binds to SAMD4 through stem-loop structures known as “Smaug recognition elements”[24].
Stability
RNA stability determinants encompass terminal structures of eukaryotic mRNAs, specifically the 5’ cap and 3’ poly(A) tail. Loss of either structure is adequate to impede translation and facilitate decay[25]. Specific cis-elements in the target mRNA, including AU-rich, CU-rich, or GU-rich elements, facilitate mRNA decay through interacting with various RBPs[26]. For instance, members of the zinc finger protein 36 families, including ZFP36 and ZFP36L1, are RBPs that facilitate mRNA degradation by binding to AU-rich elements in the 3’UTR[27]. According to Zhang et al., ZFP36 depletion in mice results in severe bone loss, inflammation, and increased OC activity[28]. Moreover, Zfp36l1+/- mice demonstrate cartilage degradation, osteophyte formation, and increased subchondral bone plate thickness. Mechanistically, ZFP36L1 facilitates osteoblastic differentiation by enhancing peroxisome proliferator-activated receptor gamma 2 (Pparγ2) mRNA degradation or inhibiting cartilage destruction by suppressing chondrocyte apoptosis, a process that targets heat shock protein family A member 1A (Hspa1a)[29,30].
Human antigen R (HuR) comprises three RRMs and a hinge region, with its activity partially dependent on its cellular localization. Upon activation, HuR translocates from the nucleus to the cytoplasm and interacts with ARE in the 3’UTRs of target mRNAs, thereby modulating their stability and translation. For instance, its interaction with la-related protein 6 (Larp6) has been demonstrated to stabilize RNA, thereby enhancing osteogenic differentiation[31,32]. This interaction is further stabilized by specific ncRNAs. For instance, circStag1 recruits the HuR protein or HuR-mediated lncRNA HOX transcript antisense RNA (HOTAIR) nucleocytoplasmic translocation into the cytoplasm to facilitate osteogenesis in mesenchymal stem cells (MSCs)[33]. Notably, in co-culture and conditioned-medium systems, HuR-deficient osteocytes inhibit OCs formation by indirectly regulating osteoprotegerin (OPG) expression, rather than by altering OPG mRNA stability[34].
Methylation of N6 nitrogen on adenosine is the primary internal modification observed in eukaryotic messenger RNA[35]. Recent studies suggest that N6-methyladenosine (m6A) methylation may affect RNA stability. Methyltransferase-like 14 (METTL14) is crucial for appropriate skeletal development. As an m6A methyltransferase, it promotes osteogenic differentiation of BMSCs and inhibits OC differentiation of BMMs by stabilizing Beclin-1 mRNA or destabilizing glutathione peroxidase 4 (Gpx4) mRNA[36,37].
Conversely, another m6A methyltransferase, methyltransferase-like 3 (METTL3), functions as a suppressor of osteogenic differentiation of OBs by destabilizing Beclin-1, Sma-and Mad-related protein 7 (Smad7), and SMAD-specific E3 ubiquitin protein ligase 1 (Smurf1) mRNA or by promoting miR‐7212‐5p maturation[38-40]. Additionally, it inhibits the extracellular matrix (ECM) synthesis in endplate chondrocytes and apoptosis and hypertrophic differentiation through m6A-mediated protein degradation[41-43]. METTL3 targets the m6A functional site in circ_0008542, and the upregulation of circ_0008542 enhances the target gene receptor activator of nuclear factor kappa beta (Rank) and initiates OC bone absorption by competing with miR-185-5p[44]. Although both are m6A methyltransferases, they serve distinct roles in bone homeostasis.
Fat mass- and obesity-associated protein (FTO), the first reported m6A demethylase, is essential in maintaining bone mass through its RNA demethylase activity. According to Zhuang et al., FTO enhances osteoclastogenesis by activating NF-κB or stabilizing Cyclin A2 and cyclin-dependent kinase 2 (Cdk2) in a YT521-B homology domain family 2 (YTHDF2)-dependent manner[45,46]. Another RNA demethylase, AlkB homolog 5 (ALKBH5), facilitates m6A methylation of protein arginine methyltransferase 6 (Prmt6) mRNA, consequently inhibiting the osteogenic differentiation potential of MSCs[47]. Current studies indicate that the function of m6A demethylases in bone homeostasis remains controversial.
M6A readers contribute to bone homeostasis by regulating the translation, stability, and degradation of target mRNA. For instance, YT521-B homology domain family 1 (YTHDF1) and insulin-like growth factor 2 mRNA binding protein (IGF2BP2) enhance osteogenic differentiation of BMSCs or MC3T3-E1 by increasing zinc finger protein 839 (Zfp839) mRNA translation efficiency or stabilizing serum response factor (SRF) mRNA[48,49]. Conversely, YTHDF2 mediates fibulin-1 (FBLN1) mRNA degradation, thereby suppressing osteogenic differentiation of umbilical cord MSCs[50]. Collectively, these recent findings highlight the significant role of m6A modification mechanisms in maintaining bone homeostasis. M6A modification machinery may serve as a potential therapeutic target for skeletal disorders. Although the phenotypic effects of these RBPs on bone homeostasis are clear, the precise molecular mechanisms and biophysical interactions driving these processes remain an area requiring further comprehensive investigation.
RBPS AND SKELETAL DISORDERS
Notably, RBPs are crucial in directing the RNA life cycle and function, thereby influencing essential mechanisms for maintaining bone homeostasis. It can shuttle between the nucleus and cytoplasm, participating in nuclear pre-mRNA splicing and polyadenylation, and cytoplasmic mRNA decay and translation, respectively. Considering that RBPs exert precise and responsive control of gene expression, their dysregulation or potential mutations may lead to the emergence of disease phenotypes in complex disorders. Herein, we discuss bone diseases associated with RBP defects and delineate the most representative RBPs for each category, including osteoporosis, spinal diseases, rheumatic diseases, and genetic skeletal disorders, among others [Figure 2].
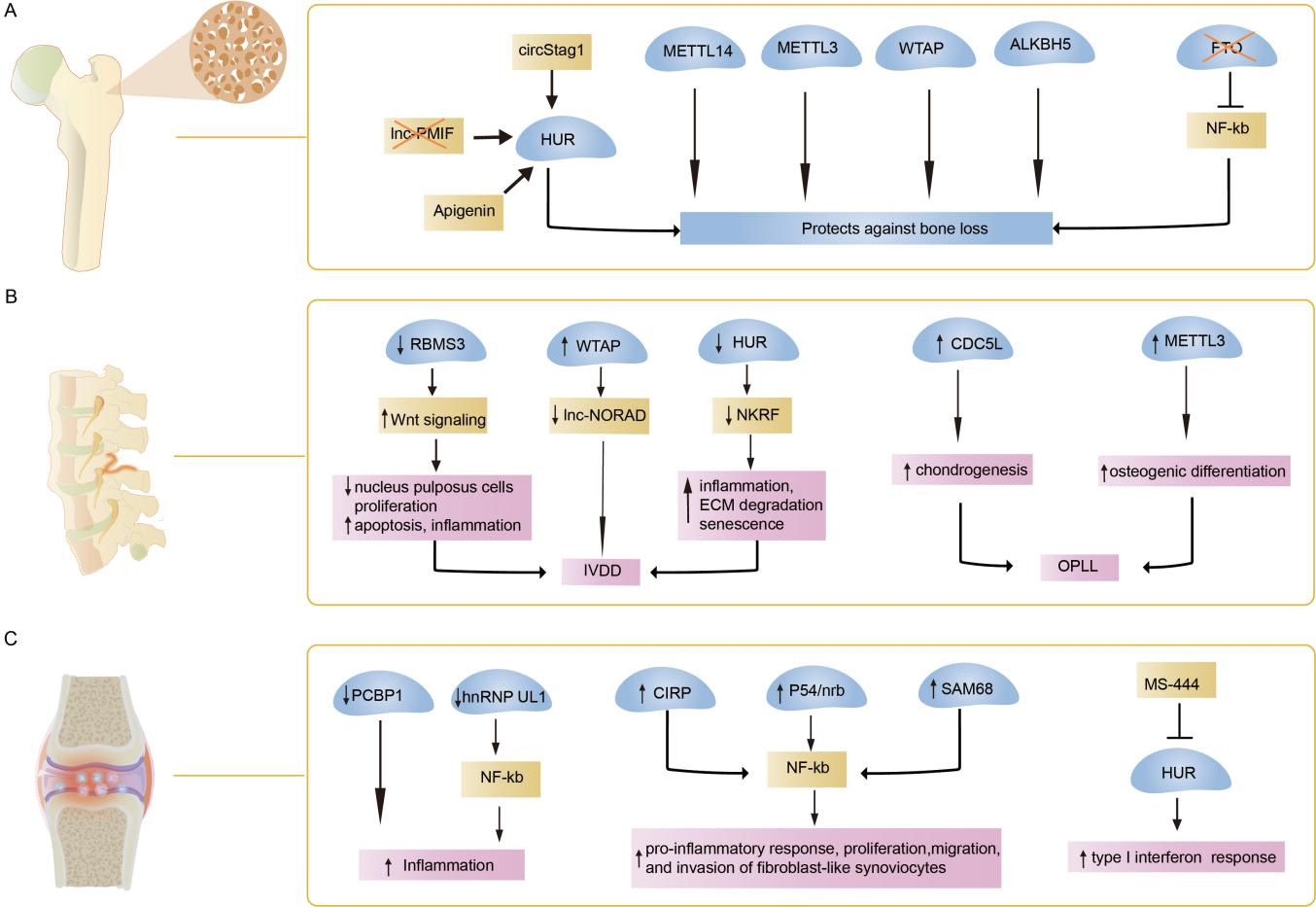
Figure 2. Functions of RNA-binding proteins in bone diseases. The main mechanism of RNA-binding proteins (RBPs) in osteoporosis (A), spinal diseases (B) and rheumatoid arthritis (C) is summarized in this figure. HUR: Human antigen R; lnc-PMIF: long non-coding RNA postulated migration inhibitory factor; circStag1: circular RNA Stag1; METTL14: methyltransferase-like 14; METTL3: methyltransferase-like 3; WTAP: wilms tumor 1-associated protein; ALKBH5: AlkB homologue 5; FTO: fat-mass and obesity-associated protein; NF-κB: nuclear factor-kappa B; IVDD: intervertebral disc degeneration; OPLL: ossification of the posterior longitudinal ligament; RBMS3: RNA binding protein motif single stranded interacting protein 3; lnc-NORAD: long non-coding RNA non-coding RNA activated by DNA damage; NKRF: NF-kappaB repressing factor; ECM: extracellular matrix; CDC5L: cell division cycle 5-like; PCBP1: poly(rC)-binding protein 1; hnRNP UL1: heterogeneous nuclear ribonucleoprotein U-like 1; CIRP: cold-inducible RBP; p54nrb: 54-kDa nuclear RNA-binding protein; SAM68: Src associated during mitosis of 68 kDa.
RBPs and osteoporosis
Osteoporosis, the predominant metabolic bone disease, is characterized by the loss of bone mass, degradation of bone microstructure, and increased bone fragility. As the influences of genetic lesions and environmental factors in osteoporosis are increasingly studied, post-transcriptional regulation mechanisms are becoming prominent in its pathogenesis. Recent studies highlight RBPs as critical players in osteoporosis[51]. For instance, numerous RBP-deficient mice exhibit RNA-binding defects that cause osteoporosis-related phenotypes or exacerbate osteopenia under pathological conditions, including METTL14, YTHDF1, Quaking (QKI), SAMD4, MSI2, iron regulatory protein 2 (IRP2), and METTL3[22,24,36,48,52-54]. Conversely, Src associated during mitosis of 68 kDa (Sam68)-deficient mice retained bone mass during aging, whereas Pumilio2-deficient mice maintained it under ovariectomized (OVX)-induced osteoporosis[55,56]. Given its proven efficacy and safety, targeted therapy aimed at RBP using siRNAs or adeno-associated virus (AAV) has demonstrated potential as an effective and safe approach for treating osteoporosis. The adenovirus-mediated HuR, METTL14, and METTL3 overexpression and the lentivirus-mediated shFTO or Wilms tumor 1-associated protein (WTAP) overexpression effectively mitigate osteoporosis-related symptoms in OVX mice[31,36,45,57]. CircStag1-loaded AAV (circStag1-AAV) and lnc-PMIF siRNA regulate interactions with HuR, effectively enhancing bone formation in OVX rats and aged mice, respectively[33,58]. Apigenin, another HuR activator, mitigates the deterioration of bone mass and trabecular bone micro-architecture in OVX mice and facilitates β‐catenin nuclear translocation, thereby promoting osteogenesis[59]. Exosomes loaded with ALKBH5 injected into the tail vein or exosome-targeted delivery of METTL14 into the marrow cavity of the tibiae ameliorated the osteopenic phenotype in mice[44,60]. Despite some progress in targeted RBP therapy for osteoporosis models, significant advancements are required before these findings can be applied in clinical settings.
RBPs and spinal diseases
Intervertebral disc degeneration
Intervertebral Disc Degeneration (IVDD) is the primary etiology of chronic lower back pain, which increases in incidence with advancing age. Progressive structural alterations coupled with severe disruptions in metabolic homeostasis facilitate IVDD progression (including abnormal apoptosis, senescence, and pyroptosis of IVD cells; ECM degradation and infiltration of immune cells)[61-63]. Therefore, comprehensively understanding the regulatory mechanisms of IVDD, particularly the primary genetic and epigenetic mechanisms, is essential.
Recent studies have demonstrated that RBPs can regulate degenerative changes in neural progenitor cells (NPCs), thereby influencing the risk of IVDD. Decreased levels of RBP motif single-stranded interacting protein 3 (RBMS3) and HuR have been noted in the nucleus pulposus tissue of patients with IVDD. RBMS3 enhances nucleus pulposus cell proliferation and inhibits their apoptosis and inflammation by inactivating the Wnt/β-catenin signaling pathway, thereby delaying IVDD progression. HuR mitigates inflammation, ECM degradation, and senescence by enhancing NF-kappaB repressing factor (NKRF) mRNA stability and autophagy[64-66]. Conversely, increased protein levels of WTAP enhance lnc-NORAD m6A modification in senescent NPCs, whereas YTHDF2-mediated lnc-NORAD degradation facilitates cellular senescence during IVDD[67].
Ossification of posterior longitudinal ligament
Ossification of Posterior Longitudinal Ligament (OPLL) is an emerging spinal condition among older adults, leading to intractable myelopathy and radiculopathy due to heterotopic ossification of the posterior longitudinal ligament[68]. Recent evidence demonstrated increased levels of cell division cycle 5-like (CDC5L) and METTL3 in patients with OPLL, where they respectively enhance chondrogenesis and osteogenic differentiation[69,70].
Spondylitis
Ankylosing spondylitis is a common, highly heritable spondyloarthropathy that primarily affects the spine joints and pelvis, leading to severe chronic pain[71]. Limited information exists regarding RBP function in spondylitis, with one report indicating that decreased YTHDF2 is a risk factor for new-onset ankylosing spondylitis[72]. Another study found Rio Kinase 3 enriched in peripheral blood mononuclear cells obtained from ankylosing spondylitis, and its suppression reduces osteogenic differentiation in mouse MSC[73].
RBPs and rheumatic diseases
Osteoarthritis
Osteoarthritis (OA) is a leading cause of severe joint pain, creating a significant socioeconomic burden owing to the aging population, with no existing disease-modifying therapies available[74]. This progressive degenerative disease of the cartilage is marked by cartilage degradation, subchondral bone remodeling, and synovial inflammation[75]. Given recent comprehensive reviews regarding aberrant RBP expression and its role in OA regulation, we will concentrate on targeted RBP therapy for OA[76]. Recently, LIN28a was identified in damaged cartilage in humans and mice, where its overexpression enhanced ECM production. In OA mice (with Col2a1-Cre), the loss of Lin28a exacerbates cartilage degradation through high mobility group AT-hook 2 (HMGA2) and Sry HMG box protein 9 (SOX9)[77]. Cre-inducible Lin28 transgenic mice demonstrate increased articular cartilage thickness, which is diminished by the injection of the extracellular regulated protein kinase (ERK) signaling pathway inhibitor U0126[78]. In a surgical destabilization of the medial meniscus model of OA, intra-articular injection of synovium-targeted METTL3 siRNA, adenovirus-mediated shZFP36L1, and lentiviral-mediated pumilio1 (PUM1) overexpression mitigates cartilage damage, preserves cartilage integrity, and inhibits OA pathogenesis[30,79,80]. Notably, ncRBP fascin actin-bundling protein 1 inhibition by NP-G2-044 could mitigate the progression of mechanical overload-induced OA in mice[81]. These data indicate that RBPs and their associated pathway activators or inhibitors may be potential candidates for targeted therapies in OA.
Rheumatoid arthritis
Rheumatoid Arthritis (RA) is a chronic immune-mediated disease characterized by inflammation and destruction of the bone and cartilage in affected joints[82]. M6A regulator level, including ALKBH5, FTO, and YTHDF2, is suppressed in patients with RA, indicating a potential risk factor associated with m6A regulators in RA[83,84]. Recent analyses have proficiently addressed RA-related m6A regulators, including those mentioned above[85].
Poly(rC)-binding protein 1 (PCBP1) and heterogeneous nuclear ribonucleoprotein U-like 1 (hnRNP UL1) - expressed at reduced levels in RA - modulate AS of immune response-related genes and inhibit NF-κB-mediated inflammation, respectively. The conditional deletion of hnRNP UL1 in macrophages through Lyz2-cre enhances the synthesis of inflammatory cytokines[86,87]. Additionally, synovial tissues from patients with RA demonstrate increased levels of cold-inducible RBP (CIRP), p54nrb, and SAM68. They enhance NF-κB-induced pro-inflammatory response, proliferation, migration, and invasion of fibroblast-like synoviocytes in RA[87-89]. MS-444, a potential small-molecule inhibitor of HuR, impeded its dimerization and translocation, thereby diminishing the type I interferon (IFN) response in RA fibroblast-like synoviocytes[90]. Notably, HuR activators mitigate osteoporosis-related phenotypes in mice, whereas HuR inhibitors influence the IFN response in RA. Consequently, the future lies in targeting RBP as a potential therapeutic for skeletal disorders and in developing associated small-molecule compounds as novel therapeutic agents.
RBPs and genetic skeletal disorders
Bone development and homeostasis are disrupted by several genetic diseases. RBP mutations can impair skeletal development, resulting in a spectrum of musculoskeletal abnormalities, predominantly affecting the limbs, hip, and craniofacial skeleton [Table 2].
RNA-binding protein mutations in genetic skeletal disorders with bone phenotypes
| RBP | Disease (OMIM) | RNA metabolism defects | Experimental system | Bone phenotype | Refs |
| RBM10 | TARP syndrome (MIM #311900) | Splicing function | Human case; mouse embryonic pharyngeal arch cell line | Micrognathia; talipes equinovarus | [91,92] |
| SF3B4 | Nager syndrome (MIM #154400) | mRNA splicing | Human case; Knockout mice | Malformations of the craniofacial skeleton and the limbs | [93-96] |
| SF3B2 | Craniofacial microsomia 1 (MIM #164210) | Splicing function | Human case; xenopus; zebrafish; iPSCs | Mandibular hypoplasia | [97-99] |
| EIF4A3 | Robin sequence with cleft mandible and limb anomalies (MIM # 268305) | mRNA stability | Human case; Zebrafish; iPSCs | Mandibular median cleft associated with other craniofacial anomalies and severe limb defects | [100,101] |
| hnRNPA2B1 | Paget disease of bone (MIM #602080) | m6A reader | Human case | Bone deformities, bone pain, and fragility fractures in the pagetic site | [102,103] |
| RBM8A | Thrombocytopenia-Absent Radius Syndrome (MIM #274000) | Splicing function | Human cohort; Zebrafish | A reduction in the number of platelets and the absence of one of the bones in the forearm | [104-106] |
| PUF60 | Verheij syndrome (MIM #615583) | Splicing function | Human case | Craniofacial dysmorphism | [107] |
| EFTUD2 | Mandibulofacial dysostosis with microcephaly (MIM #610536) | Splicing function | Human cohort; Knockout mice | Microcephaly, craniofacial anomalies | [108,109] |
| LARP7 | Alazami syndrome (MIM #615071) | Splicing function | Human case; B-lymphoblastoid cell lines | Microcephalic primordial dwarfism | [110,111] |
| ASCC1 | Spinal muscular atrophy with congenital bone fractures 2 (MIM #616867) | - | Human case; patient-derived fibroblasts | Arthrogryposis multiplex congenita and prenatal fractures of the long bones | [112,113] |
| hnRNPK | Au-Kline Syndrome (MIM #616580) | mRNA stability | Human cohort; Col2a1-Cre mice | Remarkable deformities in the limbs and spine | [114-116] |
| NIPBL | Cornelia de Lange Syndrome 1 (MIM #122470) | Protein translation | Human case; Knockout mice | Limb malformations | [117-121] |
| ERI1 | Spondyloepimetaphyseal dysplasia, Guo-Campeau type(MIM #620663) | rRNA maturation, mRNAs decay | Human case; knockout mice | Severe bone dysplasia affecting the spine and long tubular bones | [122] |
| SH3BP2 | Cherubism (MIM #118400) | - | Human case; knock-in mice | Excessive jaw-bone destruction | [123-125] |
TARP syndrome
TARP syndrome (MIM # 311900) is an X chromosome-linked recessive genetic disorder characterized by Robin sequence (micrognathia, glossoptosis, and cleft palate), congenital heart defects, developmental delay, feeding difficulties, and talipes equinovarus. Mutations of RNA-binding motif protein 10 (RBM10) have been recognized as the causative factor, likely due to the loss of its splicing function for essential developmental regulators, thereby hindering normal palate development[91,92].
Nager syndrome
Nager syndrome (MIM # 154400) exemplifies preaxial acrofacial dysostosis, distinguished by craniofacial skeleton and limb malformations. Rodriguez syndrome has been proposed as a potentially severe variant. SF3B4 haploinsufficiency is confirmed as the primary etiology. Nager syndrome involves mutations that alter SF3B4 synthesis, leading to defective mRNA splicing and decreased expression of target genes in growth plate chondrocytes. The heterozygous mutant mouse (Sf3b4+/-) was initially noted to exhibit diminished body size, homeotic posteriorization of vertebral morphology, and flattened calvaria[93-96].
Craniofacial microsomia 1
Craniofacial microsomia 1 (MIM #164210) is the second most common congenital facial anomaly, characterized by microtia, mandibular hypoplasia, and preauricular tags. The predominant genetic etiology involves haplo-insufficient variants in splicing factor 3B subunit 2 (SF3B2), present in ~3% of sporadic and ~25% of familial cases. In Xenopus embryos, injecting sf3b2 morpholinos leads to improper formation of cranial neural crest precursors and defective craniofacial cartilage[97,98]. Furthermore, an increase in cell death and a decrease in proliferative capacity have been observed in the differentiation of patient-derived induced pluripotent stem cells (iPSCs) into neural crest cells. Mechanistically, loss-of-function variants in the SF3B2 gene induce extensive mRNA splicing disruption across the transcriptome, especially affecting TP53-dependent cell death, and contribute to these craniofacial features[99].
Robin sequence with cleft mandible and limb anomalies
Robin sequence with cleft mandible and limb anomalies (MIM #268305) (known as Richieri-Costa-Pereira syndrome) is an autosomal-recessive and unique acrofacial dysostosis type caused by mutations in non-coding expansions in the 5’UTR of the eukaryotic translation initiation factor 4A3 (EIF4A3) gene. It features a median cleft in the mandible, accompanied by craniofacial anomalies and considerable limb defects. In zebrafish, loss of eif4a3 leads to underdevelopment of multiple craniofacial cartilage and bone structures. Moreover, impaired neural crest cell development clarifies craniofacial abnormalities identified in patient-derived iPSCs and conditional cre-mediated Eif4a3 mouse models[100,101]. However, the mechanism by which the EIF4A3 mutation affects the specific downstream targets remains unclear.
Paget disease of bone
Paget disease of bone (PDB) (MIM #602080) is a common metabolic bone disorder characterized by increased bone resorption and disorganized bone formation, leading to abnormal focal bone remodeling. Mutations in the m6A reader heterogeneous nuclear ribonucleoprotein A2B1 (hnRNPA2B1) were detected in patients with PDB, who frequently experience bone deformities, pain, and fragility fractures at the pagetic site[102]. Preliminary investigation into degenerative diseases indicates that these mutations may create more potent steric zippers within the prion-like domain of hnRNPA2B1, consequently modifying RNA granule assembly, impairing RNA metabolism, and accelerating fibrillization[103].
Thrombocytopenia-absent radius syndrome
Thrombocytopenia-Absent Radius (TAR) syndrome (MIM # 274000) is a hematological and skeletal disorder characterized by low platelet counts and the absence of one forearm bone. TAR syndrome is generally caused by mutations in RNA-binding motif protein 8A (RBM8A), with haploinsufficiency resulting in severe microcephaly and impaired neurogenesis in mouse models[104,105]. Recent research using the hypomorphic rbm8a zebrafish has demonstrated that the absence of RBM8A inhibits non-canonical Wnt/PCP signaling, resulting in abnormal lateral plate mesoderm patterning and hematopoietic defects[106].
Verheij syndrome
Verheij syndrome (MIM #615583) (8q24.3 microdeletion syndrome) is a rare craniofacial spliceosomopathy characterized by craniofacial dysmorphism, multiple congenital anomalies, and variable neurodevelopmental delay. Pathogenic variants in poly(U) binding splicing factor 60 (PUF60) have been recognized as causative for Verheij syndrome[107]. Although an increasing number of novel PUF60 variants have been identified in the literature, the underlying mechanisms remain a mystery.
Mandibulofacial dysostosis with microcephaly
Haploinsufficiency-inducing deletions or mutations in the spliceosome factor elongation factor Tu GTP binding domain containing 2 (EFTUD2, a GTPase and a core component of the U5 snRNP) were identified in patients with mandibulofacial dysostosis with microcephaly (MFDM, MIM #610536). Affected individuals display microcephaly, craniofacial anomalies, auditory impairment, and dysmorphic features. Conditional knockout models and mechanistic studies have demonstrated that EFTUD2 deficiency impairs cortical histogenesis and drives cortical neuronal attrition. By modulating the splicing patterns of the apoptosis regulators Caspase3 and apoptosis-inducing factor mitochondria-associated 1 (Aifm1), it activates programmed cell death[108,109].
Alazami syndrome
Alazami syndrome (MIM #615071) is an autosomal recessive microcephalic primordial dwarfism characterized by notable facial dysmorphisms and severe intellectual disability. The observed phenotypes may be ascribed to the depletion or loss of functional variants in La-related protein 7 (LARP7), resulting in splicing modification in affected patients, accompanied by diminished 2’-O-methylation of U6 small nuclear RNA[110,111].
Spinal muscular atrophy with congenital bone fractures 2
Spinal muscular atrophy with congenital bone fractures 2 (SMABF2) (MIM #616867) is a rare autosomal recessive neuromuscular disorder resulting from biallelic variants in activating signal co-integrator complex 1 (ASCC1). It is defined by congenital arthrogryposis multiplex and prenatal fractures of the long bones. Multi-omics analyses of patient-derived fibroblasts demonstrate that the downregulation of ASCC1 inhibits the TGF-β/SMAD signaling pathway, consequently suppressing osteoblastogenesis[112,113].
Au-Kline Syndrome
Au-Kline Syndrome (AKS) (MIM #616580) was initially characterized in 2015 as a multiple malformation syndrome distinguished by remarkable deformities in the limbs and spine. De novo missense or intronic variants in heterogeneous nuclear ribonucleoprotein K (HNRNPK) have been associated with AKS[114]. Recent studies demonstrated that depletion of hnRNPK during chondrogenesis using Col2a1-Cre results in dwarfism, marked by impaired survival and premature differentiation of growth plate chondrocytes. Mechanistically, hnRNPK deficiency extends the half-life of Hif1a mRNA, consequently activating the Hif-1 signaling pathway[115]. Recently, the RNA-binding motif protein 42 (RBM42) C-terminal A438T variant disrupted its interaction with hnRNP K, leading to a multisystem disease with musculoskeletal involvement[116].
Cornelia de lange syndrome
Cornelia de Lange Syndrome (CdLS) (MIM #122470) is the most common cohesinopathy, characterized by intellectual disability, growth retardation, limb malformations, and unique facial features. Heterozygous loss-of-function mutations in Nipped-B-Like protein (NIPBL) account for approximately 70% of CdLS cases[117,118]. Surviving Nipbl+/- mice exhibit significant reductions in body size, notable craniofacial changes, and anteriorization of thoracic vertebrae, with left-right asymmetry accompanied by collective changes in osteogenesis-regulatory pathways[119-121].
Spondyloepimetaphyseal dysplasia, Guo-Campeau type
Spondyloepimetaphyseal dysplasia, Guo-Campeau type (SEMDGC) (MIM #620663) is defined by severe bone dysplasia affecting the spine and long tubular bones, leading to substantial short stature with disproportionate extremities. Research has revealed that pathogenic variants in the exoribonuclease 1 (ERI1) gene are responsible for this novel form of bone dysplasia in patients from seven affected families. Mechanistically, missense mutations in the conserved 3’-5’ RNA exonuclease impair enzyme activity, failing to catalyze 5.8S rRNA processing and affecting histone mRNA decay. Furthermore, Eri1 knockout mice exhibit skeletal dysplasia that partially resembles human SEMDGC[122].
Cherubism
Cherubism (MIM #118400) is an autosomal dominant condition associated with severe craniofacial developmental anomalies, notably excessive jaw-bone destruction. Gain-of-function missense variants in c-Abl-SH3 domain binding protein-2 (SH3BP2) have been identified as being responsible for the condition in patients from 12 affected families[123]. In OCs, SH3BP2 phosphorylation seems to activate phospholipase C gamma 2 (PLCG2), resulting in excessive OC activity. Consistently, in the homozygous cherubism knock-in (KI) mouse model (Sh3bp2 KI/+), systemic bone destruction has been observed owing to increased OC formation. Additionally, spleen tyrosine kinase (SYK) inhibitor, entospletinib, abolishes all observed cherubism phenotypes[124,125].
Others
Craniofacial malformations rank among the most common congenital disabilities. Considering the role of RBPs in modifying craniofacial disorders has recently been extensively reviewed[126,127], we presented fundamental research on craniofacial development. A homozygous frameshift variant in hnRNP UL1 was detected in patients exhibiting craniofacial and limb anomalies, consistent with the developmental role of hnRNP UL1 observed in zebrafish[128]. Studies utilizing animal models highlight the essential function of Serine-rich splicing factor 3 (SRSF3), nucleolin, RNA-binding Fox-1 homolog 2 (RBFOX2), and RBMS3 in regulating craniofacial development[129-132].
CONCLUSION AND PERSPECTIVES
This review offers a current and comprehensive overview of RBPs in bone homeostasis and skeletal disorders, elucidating their fundamental mechanisms, potential RBP-targeted therapeutic applications, and prospects. RBPs are recognized as essential regulators of intramembranous and endochondral bone formation. Recently, novel methods for profiling RNA-protein interactions have emerged, significantly expanding our knowledge of the RBP landscape[9,133,134]. However, understanding the roles of various bone homeostasis-specific RBPs and clarifying the complex regulatory network focused on RBPs remains challenging.
Although the roles of RBPs in OBs and OCs have been thoroughly examined, their function in osteocytes has been historically neglected. Osteocytes have long been challenging to study in vitro, partly because these terminally differentiated cells are difficult to isolate and purify, leading to the lack of suitable in vitro cell models. Future studies may employ MLO-Y4 osteocyte-like cells, co-culture systems, and osteocyte-specific conditional knockout models to investigate the in vitro and in vivo function of specific RBPs in osteocytes.
To address the effect of RBP mutations in genetic skeletal disorders, novel cellular models of RBPs using iPSCs synthesized from the fibroblasts of patients offer promising avenues. Mouse models are essential in studying skeletal development, revealing that overexpression or inhibition of RBPs in vivo leads to multiple skeletal defects. Despite significant progress, much remains to be learned regarding how RBPs regulate bone development and homeostasis through mouse genetics. As additional RBPs are individually deleted in bone cells, their distinct and overlapping functions will be clarified.
A fundamental inquiry is determining the primary targets of RBPs. High-throughput platforms are utilized efficiently and dependably to evaluate these targets. Current evidence indicates that specific activators and inhibitors of HuR can alleviate bone loss and diminish the IFN response. Furthermore, the SYK inhibitor entospletinib has demonstrated therapeutic potential in the cherubism mouse model, indicating its potential for clinical application. However, further research is required to comprehensively clarify the underlying mechanisms of these effects. Understanding the in vivo specificity of RBPs in drug studies and their role in adult skeletons remains a major challenge.
RBPs interact with the target RNAs to regulate the protein synthesis of the target genes. In the rapidly advancing field of cancer, initial promising RBP therapeutic candidates are currently undergoing clinical trials, including FTO, splicing factor 3B subunit 1 (SF3B1), nucleolin, and RNA-binding motif protein 39 (RBM39). Specifically, small-molecule inhibitors (SMIs) targeting FTO have been formulated as anti-cancer drugs. These SMIs inhibit the catalytic pocket of FTO and impede the binding to m6A-modified targets. Therefore, this evidence endorses the prospective RBP-targeted therapies and aids in the investigation of their potential therapeutic applications in skeletal disorders. Interactions between ncRNAs and RBP are recognized as crucial factors in cancer, and investigating this research direction may reveal novel mechanisms underlying bone diseases. Employing ncRNA-loaded AAV to facilitate interactomes for RBPs may evolve into a simple and efficacious therapeutic strategy in the future. Continuous improvement of RBP-targeted therapies is essential to minimize side effects and enhance efficacy and safety. Addressing these inquiries is essential for comprehensively understanding the functions of RBPs in the skeleton and assessing the benefits and potential drawbacks of RBP-targeted therapies.
DECLARATIONS
Authors’ contributions
Conceptualization, writing - original draft: Xu Y
Supervision, writing - review: Luan X, Wang Y
Validation, writing - editing: Chen S, Xu C
All authors reviewed the manuscript.
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This work was supported by grants from the sub-project of the National Key R&D Program (2022YFC2703702 to Chen S, 2023YFC2705600 to Xu C), National Natural Science Foundation of China (No. 82402000 to Xu Y, 82301935 to Luan X).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Veis DJ, O’brien CA. Osteoclasts, master sculptors of bone. Annu Rev Pathol Mech Dis. 2023;18:257-81.
2. Li ZG, Mathew P, Yang J, et al. Androgen receptor-negative human prostate cancer cells induce osteogenesis in mice through FGF9-mediated mechanisms. J Clin Invest. 2008;118:2697-710.
3. Sims NA, Martin TJ. Osteoclasts provide coupling signals to osteoblast lineage cells through multiple mechanisms. Annu Rev Physiol. 2020;82:507-29.
4. Zhang T, Feng Y, Wang W, et al. Membrane-bound or soluble, mesenchymal or myeloid: which RANKL drives osteoclastogenesis? J Transl Genet Genom. 2026;10:74-88.
5. Wu Y, Gan D, Liu Z, et al. Osteocytes: master orchestrators of skeletal homeostasis, remodeling, and osteoporosis pathogenesis. Front. Cell Dev. Biol. 2025;13:1670716.
6. Cawthray JF, Weekes DM, Sivak O, et al. In vivo study and thermodynamic investigation of two lanthanum complexes, La(dpp)3 and La(XT), for the treatment of bone resorption disorders. Chem. Sci. 2015;6:6439-47.
7. Markmiller S, Soltanieh S, Server KL, et al. Context-dependent and disease-specific diversity in protein interactions within stress granules. Cell. 2018;172:590-604.e13.
8. Gebauer F, Schwarzl T, Valcárcel J, Hentze MW. RNA-binding proteins in human genetic disease. Nat Rev Genet. 2020;22:185-98.
9. Mosca R, Gallardo-dodd CJ, Li Q, et al. RAPseq enables large-scale identification of RBP-RNA interactions and reveals essentials of post-transcriptional gene regulation. Nucleic Acids Res. 2026;54:gkag090.
10. Hentze MW, Sommerkamp P, Ravi V, Gebauer F. Rethinking RNA-binding proteins: riboregulation challenges prevailing views. Cell. 2025;188:4811-27.
11. Seufert L, Benzing T, Ignarski M, Müller R. RNA-binding proteins and their role in kidney disease. Nat Rev Nephrol. 2021;18:153-70.
12. Wang S, Sun Z, Lei Z, Zhang H. RNA-binding proteins and cancer metastasis. Semin Cancer Biol. 2022;86:748-68.
13. Paschalis A, Sharp A, Welti JC, et al. Alternative splicing in prostate cancer. Nat Rev Clin Oncol. 2018;15:663-75.
14. Fu X, Ares M. Context-dependent control of alternative splicing by RNA-binding proteins. Nat Rev Genet. 2014;15:689-701.
15. Watanabe H, Shionyu M, Kimura T, Kimata K, Watanabe H. Splicing factor 3b subunit 4 binds BMPR-IA and inhibits osteochondral cell differentiation. J Biol Chem. 2007;282:20728-38.
16. Hata K, Nishimura R, Muramatsu S, et al. Paraspeckle protein p54nrb links Sox9-mediated transcription with RNA processing during chondrogenesis in mice. J Clin Invest. 2008;118:3098-108.
17. Koscielny G, Texier VL, Gopalakrishnan C, et al. ASTD: The alternative splicing and transcript diversity database. Genomics. 2009;93:213-20.
18. Tian B, Manley JL. Alternative cleavage and polyadenylation: the long and short of it. Trends Biochem Sci. 2013;38:312-20.
19. Khajuria DK, Nowak I, Leung M, et al. Transcript shortening via alternative polyadenylation promotes gene expression during fracture healing. Bone Res. 2023;11:5.
20. Truitt ML, Ruggero D. New frontiers in translational control of the cancer genome. Nat Rev Cancer. 2016;16:288-304.
21. Zou L, Xiang C, Lu M. MSI1 Stabilizes MACF1 to inhibit apoptosis of MC3T3-E1 cells induced by high glucose and promote osteogenic differentiation through Wnt/β-catenin signaling pathway. Mol Biotechnol. 2022;65:1085-95.
22. Suo J, Zou S, Wang J, et al. The RNA-binding protein Musashi2 governs osteoblast-adipocyte lineage commitment by suppressing PPARγ signaling. Bone Res. 2022;10:31.
23. Fujiwara T, Zhou J, Ye S, Zhao H. RNA-binding protein Musashi2 induced by RANKL is critical for osteoclast survival. Cell Death Dis. 2016;7:e2300.
24. Niu N, Xiang J, Yang Q, et al. RNA-binding protein SAMD4 regulates skeleton development through translational inhibition of Mig6 expression. Cell Discov. 2017;3:16050.
25. Roux ME, Rasmussen MW, Palma K, et al. The mRNA decay factor PAT1 functions in a pathway including MAP kinase 4 and immune receptor SUMM2. EMBO J. 2015;34:593-608.
26. Chen XS, Brown CM. Computational identification of new structured cis -regulatory elements in the 3’-untranslated region of human protein coding genes. Nucleic Acids Res. 2012;40:8862-73.
27. Cook ME, Bradstreet TR, Webber AM, et al. The ZFP36 family of RNA binding proteins regulates homeostatic and autoreactive T cell responses. Sci. Immunol. 2022;7:eabo0981.
28. Zhang L, Kwack KH, Thiyagarajan R, et al. Tristetraprolin regulates the skeletal phenotype and osteoclastogenic potential through monocytic myeloid‐derived suppressor cells. The FASEB Journal. 2023;38:e23338.
29. Tseng K, Chen Y, Lin S. Zinc finger protein ZFP36L1 promotes osteoblastic differentiation but represses adipogenic differentiation of mouse multipotent cells. Oncotarget. 2017;8:20588-601.
30. Son Y, Kim H, Choi W, Chun C, Chun J. RNA-binding protein ZFP36L1 regulates osteoarthritis by modulating members of the heat shock protein 70 family. Nat Commun. 2019;10:77.
31. Liu Z, Li B, Hu H, Li X, Zhang X. Potential of RNA-binding protein human antigen R as a driver of osteogenic differentiation in osteoporosis. J Orthop Surg Res. 2022;17:234.
32. Guo Y, Tang CY, Man XF, et al. Insulin‐like growth factor‐1 promotes osteogenic differentiation and collagen I alpha 2 synthesis via induction of mRNA‐binding protein LARP6 expression. Dev Growth Differ. 2017;59:94-103.
33. Chen G, Long C, Wang S, et al. RETRACTED ARTICLE: circular RNA circStag1 promotes bone regeneration by interacting with HuR. Bone Res. 2022;10:32.
34. Fan Z, Kitaura H, Marahleh A, et al. HuR knockdown in MLO-Y4 osteocyte-like cells elevates OPG expression and suppresses osteoclastogenesis in vitro. Int J Mol Sci. 2025;27:430.
35. Doxtader KA, Wang P, Scarborough AM, Seo D, Conrad NK, Nam Y. Structural basis for regulation of METTL16, an S-adenosylmethionine homeostasis factor. Mol Cell. 2018;71:1001-1011.e4.
36. He M, Lei H, He X, et al. METTL14 regulates osteogenesis of bone marrow mesenchymal stem cells via inducing autophagy through m6A/IGF2BPs/beclin-1 signal axis. Stem Cells Translational Medicine. 2022;11:987-1001.
37. Deng M, Luo J, Cao H, Li Y, Chen L, Liu G. METTL14 represses osteoclast formation to ameliorate osteoporosis via enhancing GPX4 mRNA stability. Environ Toxicol. 2023;38:2057-68.
38. Kong Y, Zhang Y, Cai Y, Li D, Yi B, Xu Q. METTL3 mediates osteoblast apoptosis by regulating endoplasmic reticulum stress during LPS-induced inflammation. Cell Signal. 2022;95:110335.
39. Zhang Y, Gu X, Li D, Cai L, Xu Q. METTL3 regulates osteoblast differentiation and inflammatory response via smad signaling and MAPK signaling. Int J Mol Sci. 2019;21:199.
40. Mi B, Xiong Y, Yan C, et al. Methyltransferase‐like 3‐mediated N6‐methyladenosine modification of miR‐7212‐5p drives osteoblast differentiation and fracture healing. J Cell Mol Med. 2020;24:6385-96.
41. He Y, Wang W, Xu X, et al. Mettl3 inhibits the apoptosis and autophagy of chondrocytes in inflammation through mediating Bcl2 stability via Ythdf1-mediated m6A modification. Bone. 2022;154:116182.
42. He Y, Wang W, Luo P, et al. Mettl3 regulates hypertrophic differentiation of chondrocytes through modulating Dmp1 mRNA via Ythdf1-mediated m6A modification. Bone. 2022;164:116522.
43. Xiao L, Hu B, Ding B, et al. N(6)-methyladenosine RNA methyltransferase like 3 inhibits extracellular matrix synthesis of endplate chondrocytes by downregulating sex-determining region Y-Box transcription factor 9 expression under tension. Osteoarthritis Cartilage. 2022;30:613-25.
44. Wang W, Qiao S, Wu X, et al. Circ_0008542 in osteoblast exosomes promotes osteoclast-induced bone resorption through m6A methylation. Cell Death Dis. 2021;12:628.
45. Zhuang J, Ning H, Wang M, et al. Downregulated fat mass and obesity-associated protein inhibits bone resorption and osteoclastogenesis by nuclear factor-kappa B inactivation. Cell Signal. 2021;87:110137.
46. He J, Zhao Y, Zhang Y, Zhang Z, Li D, Xu Q. FTO regulates osteoclast development by modulating the proliferation and apoptosis of osteoclast precursors in inflammatory conditions. Cell Signal. 2024;117:111098.
47. Li Z, Wang P, Li J, et al. The N6-methyladenosine demethylase ALKBH5 negatively regulates the osteogenic differentiation of mesenchymal stem cells through PRMT6. Cell Death Dis. 2021;12:578.
48. Liu T, Zheng X, Wang C, et al. The m6A “reader” YTHDF1 promotes osteogenesis of bone marrow mesenchymal stem cells through translational control of ZNF839. Cell Death Dis. 2021;12:1078.
49. Zhou Z, Chen S, Wu T, et al. IGF2BP2, an RNA‐binding protein regulates cell proliferation and osteogenic differentiation by stabilizing SRF mRNA. J Cell Physiol. 2022;238:195-209.
50. Yang H, Wang W, Liu H, et al. miR615‐3p inhibited FBLN1 and osteogenic differentiation of umbilical cord mesenchymal stem cells by associated with YTHDF2 in a m6A‐miRNA interaction manner. Cell Prolif. 2024;57:e13607.
51. Wu J, Niu L, Yang K, et al. The role and mechanism of RNA-binding proteins in bone metabolism and osteoporosis. Ageing Res Rev. 2024;96:102234.
52. Du T, Yan Z, Zhu S, et al. QKI deficiency leads to osteoporosis by promoting RANKL-induced osteoclastogenesis and disrupting bone metabolism. Cell Death Dis. 2020;11:330.
53. Zhou Y, Yang Y, Liu Y, et al. Irp2 knockout causes osteoporosis by inhibition of bone remodeling. Calcif Tissue Int. 2018;104:70-8.
54. Yan G, Yuan Y, He M, et al. m6A methylation of precursor-miR-320/RUNX2 controls osteogenic potential of bone marrow-derived mesenchymal stem cells. Molecular Therapy - Nucleic Acids. 2020;19:421-36.
55. Richard S, Torabi N, Franco GV, et al. Ablation of the Sam68 RNA binding protein protects mice from age-related bone loss. PLoS Genet. 2005;1:e74.
56. Yoon DS, Choi Y, Lee K, et al. Downregulation of the RNA-binding protein PUM2 facilitates MSC-driven bone regeneration and prevents OVX-induced bone loss. J Biomed Sci. 2023;30:26.
57. Liu J, You Y, Sun Z, et al. WTAP-mediated m6A RNA methylation regulates the differentiation of bone marrow mesenchymal stem cells via the miR-29b-3p/HDAC4 Axis. Stem Cells Translational Medicine. 2023;12:307-21.
58. Li D, Liu J, Yang C, et al. Targeting long noncoding RNA PMIF facilitates osteoprogenitor cells migrating to bone formation surface to promote bone formation during aging. Theranostics. 2021;11:5585-604.
59. Huai Y, Wang X, Mao W, et al. HuR‐positive stress granules: potential targets for age‐related osteoporosis. Aging Cell. 2024;23:e14053.
60. Yang J, Sun B, Wang Z, et al. Exosome-targeted delivery of METTL14 regulates NFATc1 m6A methylation levels to correct osteoclast-induced bone resorption. Cell Death Dis. 2023;14:738.
61. Li G, Zhang W, Liang H, Yang C. Epigenetic regulation in intervertebral disc degeneration. Trends Mol Med. 2022;28:803-5.
62. Sun K, Jiang J, Wang Y, et al. The role of nerve fibers and their neurotransmitters in regulating intervertebral disc degeneration. Ageing Res Rev. 2022;81:101733.
63. Francisco V, Pino J, González-gay MÁ, et al. A new immunometabolic perspective of intervertebral disc degeneration. Nat Rev Rheumatol. 2021;18:47-60.
64. Wang JJ, Liu XY, Du W, Liu JQ, Sun B, Zheng YP. RBMS3 delays disc degeneration by inhibiting Wnt/β-catenin signaling pathway. Eur Rev Med Pharmacol Sci. 2020;24:499-507.
65. Shao Z, Tu Z, Shi Y, et al. RNA-Binding Protein HuR Suppresses Inflammation and Promotes Extracellular Matrix Homeostasis via NKRF in Intervertebral Disc Degeneration. Front. Cell Dev. Biol. 2020;8:611234.
66. Shao Z, Ni L, Hu S, et al. RNA‐binding protein HuR suppresses senescence through Atg7 mediated autophagy activation in diabetic intervertebral disc degeneration. Cell Prolif. 2020;54:e12975.
67. Li G, Ma L, He S, et al. WTAP-mediated m6A modification of lncRNA NORAD promotes intervertebral disc degeneration. Nat Commun. 2022;13:1469.
68. Xu C, Zhang Z, Liu N, et al. Small extracellular vesicle-mediated miR-320e transmission promotes osteogenesis in OPLL by targeting TAK1. Nat Commun. 2022;13:2467.
69. Jokoji G, Maeda S, Oishi K, et al. CDC5L promotes early chondrocyte differentiation and proliferation by modulating pre-mRNA splicing of SOX9, COL2A1, and WEE1. J Biol Chem. 2021;297:100994.
70. Yuan X, Shi L, Guo Y, et al. METTL3 regulates ossification of the posterior longitudinal ligament via the lncRNA XIST/miR-302a-3p/USP8 axis. Front. Cell Dev. Biol. 2021;9:629895.
71. Brown MA, Kenna T, Wordsworth BP. Genetics of ankylosing spondylitis-insights into pathogenesis. Nat Rev Rheumatol. 2015;12:81-91.
72. Luo Q, Guo Y, Xiao Q, et al. Expression and clinical significance of the m6A RNA-binding proteins YTHDF2 in peripheral blood mononuclear cells from new-onset ankylosing spondylitis. Front. Med. 2022;9:922219.
73. Zong H, Liu Y, Wang X, et al. RIOK3 potentially regulates osteogenesis-related pathways in ankylosing spondylitis and the differentiation of bone marrow mesenchymal stem cells. Genomics. 2023;115:110730.
74. Sellam J, Berenbaum F. The role of synovitis in pathophysiology and clinical symptoms of osteoarthritis. Nat Rev Rheumatol. 2010;6:625-35.
75. Kapoor M, Martel-pelletier J, Lajeunesse D, Pelletier J, Fahmi H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat Rev Rheumatol. 2010;7:33-42.
76. Yi Q, Deng Z, Yue J, et al. RNA binding proteins in osteoarthritis. Front. Cell Dev. Biol. 2022;10:954376.
77. Jouan Y, Bouchemla Z, Bardèche-trystram B, et al. Lin28a induces SOX9 and chondrocyte reprogramming via HMGA2 and blunts cartilage loss in mice. Sci. Adv. 2022;8:eabn3106.
78. Kobayashi T, Kozlova A. Lin28a overexpression reveals the role of Erk signaling in articular cartilage development. Development. 2018;145:dev162594.
79. Chen X, Gong W, Shao X, et al. METTL3-mediated m6A modification of ATG7 regulates autophagy-GATA4 axis to promote cellular senescence and osteoarthritis progression. Ann Rheum Dis. 2022;81:87-99.
80. Yoon DS, Lee K, Choi Y, et al. TLR4 downregulation by the RNA-binding protein PUM1 alleviates cellular aging and osteoarthritis. Cell Death Differ. 2022;29:1364-78.
81. Xiao Y, Chen L, Chen D, et al. FSCN1 induces subchondral bone sclerosis in osteoarthritis via modulating actin cytoskeleton dynamics and YAP signaling. Arthritis Res Ther. 2026;28:33.
82. Nygaard G, Firestein GS. Restoring synovial homeostasis in rheumatoid arthritis by targeting fibroblast-like synoviocytes. Nat Rev Rheumatol. 2020;16:316-33.
83. Luo Q, Gao Y, Zhang L, et al. Decreased ALKBH5, FTO, and YTHDF2 in Peripheral blood are as risk factors for rheumatoid arthritis. Biomed Res Int. 2020;2020:5735279.
84. Yao F, Xu C, Gao Y, et al. Expression and clinical significance of the m6A reader YTHDF2 in peripheral blood mononuclear cells from rheumatoid arthritis patients. J Immunotoxicol. 2022;19:53-60.
85. Wu S, Li X, Wu Y, Yin S, Huang C, Li J. N6-methyladenosine and rheumatoid arthritis: a comprehensive review. Front. Immunol. 2021;12:731842.
86. Cao X, Li P, Song X, et al. PCBP1 is associated with rheumatoid arthritis by affecting RNA products of genes involved in immune response in Th1 cells. Sci Rep. 2022;12:8398.
87. Ma Z, Zhou Y, Wang Y, et al. RNA-binding protein hnRNP UL1 binds κB sites to attenuate NF-κB-mediated inflammation. J Autoimmun. 2022;129:102828.
88. Li Q, Guo H, Li H, Zhu Q, Liu Y. P54/nrb prompts rheumatoid arthritis progression mainly by transcriptionally activating NF-κB signaling. Pharmazie. 2017;72:260-4.
89. Sun W, Qin R, Wang R, et al. Sam68 promotes invasion, migration, and proliferation of fibroblast-like synoviocytes by enhancing the NF-κB/P65 pathway in rheumatoid arthritis. Inflammation. 2018;41:1661-70.
90. Herdy B, Karonitsch T, Vladimer GI, et al. The RNA‐binding protein HuR/ELAVL1 regulates IFN‐β mRNA abundance and the type I IFN response. Eur J Immunol. 2015;45:1500-11.
91. Johnston JJ, Teer JK, Cherukuri PF, et al. Massively parallel sequencing of exons on the X chromosome identifies RBM10 as the gene that causes a syndromic form of cleft palate. Am J Hum Genet. 2010;86:743-8.
92. Rodor J, Fitzpatrick DR, Eyras E, Cáceres JF. The RNA-binding landscape of RBM10 and its role in alternative splicing regulation in models of mouse early development. RNA Biol. 2016;14:45-57.
93. Bernier FP, Caluseriu O, Ng S, et al. Haploinsufficiency of SF3B4, a component of the Pre-mRNA spliceosomal complex, causes nager syndrome. Am J Hum Genet. 2012;90:925-33.
94. Mcpherson E, Zaleski C, Ye Z, Lin S. Rodriguez syndrome with SF3B4 mutation: a severe form of nager syndrome? Am J Med Genet A. 2014;164:1841-5.
95. Marques F, Tenney J, Duran I, et al. Altered mRNA splicing, chondrocyte gene expression and abnormal skeletal development due to SF3B4 mutations in rodriguez acrofacial dysostosis. PLoS Genet. 2016;12:e1006307.
96. Yamada T, Takechi M, Yokoyama N, et al. Heterozygous mutation of the splicing factor Sf3b4 affects development of the axial skeleton and forebrain in mouse. Dev Dyn. 2020;249:622-35.
97. Heike C, Hing A, Aspinall C, et al. Clinical care in craniofacial microsomia: a review of current management recommendations and opportunities to advance research. Am J Med Genet C Semin Med Genet. 2013;163:271-82.
98. Timberlake AT, Griffin C, Heike CL, et al. ; University of Washington Center for Mendelian Genomics. Haploinsufficiency of SF3B2 causes craniofacial microsomia. Nat Commun. 2021;12:4680.
99. Rao S, Watt K, Maili L, et al. Splicing defects and cell death cause SF3B2-linked craniofacial microsomia. J Dent Res. 2025;104:1116-26.
100. Favaro FP, Alvizi L, Zechi-ceide RM, et al. A noncoding expansion in EIF4A3 causes richieri-costa-pereira syndrome, a craniofacial disorder associated with limb defects. Am J Hum Genet. 2014;94:120-8.
101. Miller EE, Kobayashi GS, Musso CM, et al. EIF4A3 deficient human iPSCs and mouse models demonstrate neural crest defects that underlie Richieri-Costa-Pereira syndrome. Hum Mol Genet. 2017;26:2177-91.
102. Qi X, Pang Q, Wang J, et al. Familial early-onset paget’s disease of bone associated with a novel hnRNPA2B1 mutation. Calcif Tissue Int. 2017;101:159-69.
103. Kim HJ, Kim NC, Wang Y, et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013;495:467-73.
104. Albers CA, Paul DS, Schulze H, et al. Compound inheritance of a low-frequency regulatory SNP and a rare null mutation in exon-junction complex subunit RBM8A causes TAR syndrome. Nat Genet. 2012;44:435-9.
105. Mao H, Pilaz L, Mcmahon JJ, et al. Rbm8a haploinsufficiency disrupts embryonic cortical development resulting in microcephaly. J. Neurosci. 2015;35:7003-18.
106. Kocere A, Chiavacci E, Soneson C, et al. Rbm8a deficiency causes hematopoietic defects by modulating Wnt/PCP signaling. Dev Biol. 2025;528:34-56.
107. El Chehadeh S, Kerstjens-frederikse WS, Thevenon J, et al. Dominant variants in the splicing factor PUF60 cause a recognizable syndrome with intellectual disability, heart defects and short stature. Eur J Hum Genet. 2016;25:43-51.
108. Lines MA, Huang L, Schwartzentruber J, et al. Haploinsufficiency of a spliceosomal GTPase encoded by EFTUD2 causes mandibulofacial dysostosis with microcephaly. Am J Hum Genet. 2012;90:369-77.
109. Chen L, Li Y, Yu Y, et al. EFTUD2 regulates cortical morphogenesis via modulation of Caspase‐3 and Aifm1 splicing pathways. Adv Sci. 2025;12:e04200.
110. Alazami AM, Al-owain M, Alzahrani F, et al. Loss of function mutation in LARP7, chaperone of 7SK ncRNA, causes a syndrome of facial dysmorphism, intellectual disability, and primordial dwarfism. Hum Mutat. 2012;33:1429-34.
111. Hasler D, Meduri R, Bąk M, et al. The alazami syndrome-associated protein LARP7 guides U6 small nuclear RNA modification and contributes to splicing robustness. Mol Cell. 2020;77:1014-1031.e13.
112. Voraberger B, Mayr JA, Fratzl-zelman N, et al. Investigating the role of ASCC1 in the causation of bone fragility. Front. Endocrinol. 2023;14:1137573.
113. Knierim E, Hirata H, Wolf NI, et al. Mutations in subunits of the activating signal cointegrator 1 complex are associated with prenatal spinal muscular atrophy and congenital bone fractures. Am J Hum Genet. 2016;98:473-89.
114. Choufani S, Mcniven V, Cytrynbaum C, et al. An HNRNPK-specific DNA methylation signature makes sense of missense variants and expands the phenotypic spectrum of Au-Kline syndrome. Am J Hum Genet. 2022;109:1867-84.
115. Chen Y, Wu J, Zhang S, et al. Hnrnpk maintains chondrocytes survival and function during growth plate development via regulating Hif1α-glycolysis axis. Cell Death Dis. 2022;13:803.
116. Chen Y, Yang B, Zhang XM, et al. Biallelic variants in RBM42 cause a multisystem disorder with neurological, facial, cardiac, and musculoskeletal involvement. Protein Cell. 2024;15:52-68.
117. Alonso-gil D, Losada A. NIPBL and cohesin: new take on a classic tale. Trends Cell Biol. 2023;33:860-71.
118. Krantz ID, Mccallum J, Descipio C, et al. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of drosophila melanogaster nipped-B. Nat Genet. 2004;36:631-5.
119. Gu W, Wang L, Gu R, et al. Defects of cohesin loader lead to bone dysplasia associated with transcriptional disturbance. J Cell Physiol. 2021;236:8208-25.
120. Kawauchi S, Calof AL, Santos R, et al. Multiple organ system defects and transcriptional dysregulation in the Nipbl+/- mouse, a model of cornelia de lange syndrome. PLoS Genet. 2009;5:e1000650.
121. Chea S, Kreger J, Lopez-burks ME, Maclean AL, Lander AD, Calof AL. Gastrulation-stage gene expression in Nipbl+/- mouse embryos foreshadows the development of syndromic birth defects. Sci. Adv. 2024;10:eadl4239.
122. Guo L, Salian S, Xue J, et al. Null and missense mutations of ERI1 cause a recessive phenotypic dichotomy in humans. Am J Hum Genet. 2023;110:1068-85.
123. Ueki Y, Tiziani V, Santanna C, et al. Mutations in the gene encoding c-Abl-binding protein SH3BP2 cause cherubism. Nat Genet. 2001;28:125-6.
124. Chester JG, Carcamo B, Gudis DA, et al. PLCG2 variants in cherubism. J Allergy Clin Immunol. 2024;154:1554-8.
125. Yoshimoto T, Hayashi T, Kondo T, Kittaka M, Reichenberger EJ, Ueki Y. Second-generation SYK inhibitor entospletinib ameliorates fully established inflammation and bone destruction in the cherubism mouse model. J Bone Miner Res. 2018;33:1513-9.
126. Forman TE, Dennison BJC, Fantauzzo KA. The role of RNA-binding proteins in vertebrate neural crest and craniofacial development. J Dev Biol. 2021;9:34.
127. Lehalle D, Wieczorek D, Zechi‐ceide R, et al. A review of craniofacial disorders caused by spliceosomal defects. Clin Genet. 2015;88:405-15.
128. Blackwell DL, Fraser SD, Caluseriu O, et al. Hnrnpul1 controls transcription, splicing, and modulates skeletal and limb development in vivo. G3 (Bethesda). 2022;12:jkac067.
129. Dennison BJC, Larson ED, Fu R, Mo J, Fantauzzo KA. Srsf3 mediates alternative RNA splicing downstream of PDGFRα signaling in the facial mesenchyme. Development. 2021;148:dev199448.
130. Dash S, Trainor PA. Nucleolin loss of function leads to aberrant fibroblast growth factor signaling and craniofacial anomalies. Development. 2022;149:dev200349.
131. Cibi DM, Mia MM, Guna Shekeran S, et al. Neural crest-specific deletion of Rbfox2 in mice leads to craniofacial abnormalities including cleft palate. eLife. 2019;8:e45418.
132. Jayasena CS, Bronner ME. Rbms3 functions in craniofacial development by posttranscriptionally modulating TGF-β signaling. J Cell Biol. 2012;199:453-66.
133. Hawkins S, Mondaini A, Namboori SC, et al. ePRINT: exonuclease assisted mapping of protein-RNA interactions. Genome Biol. 2024;25:140.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].