


Genetic and ultrasound assessment in recurrent fetal malformations: a case report on TUBA1A gene variation
 0
0 
Abstract
To elucidate the etiology of recurrent fetal brain developmental malformations accompanied by other structural ultrasound anomalies in a Chinese non-consanguineous couple, we performed a comprehensive evaluation of prenatal ultrasound phenotypes across their two consecutive pregnancies. Copy number variation sequencing and exome sequencing were applied to the amniotic fluid sample obtained from the affected pregnancy. One copy number variation and four candidate missense variants in three genes identified in the initial analysis were reassessed and reclassified according to current clinical and genetic evidence. Our results demonstrated that a maternal mosaic c.7G>A(p.Glu3Lys) variant in the tubulin alpha 1a (TUBA1A) gene was the underlying cause of the couple’s recurrent adverse pregnancy outcomes. These findings enrich the database of pathogenic TUBA1A variants, expand the spectrum of associated prenatal ultrasound phenotypes, and provide valuable insights into the underlying pathogenic mechanisms.
Keywords
INTRODUCTION
Tubulinopathies are a diverse group of neurodevelopmental disorders caused by mutations in tubulin genes, which lead to cortical malformations. The tubulin gene tubulin alpha-1A (TUBA1A) was first identified in 2007 in both mice and humans[1]. Cerebral dysplasia is common in individuals with TUBA1A mutations and often results in intellectual disability[2], cognitive deficits, and epilepsy[3,4]. Recent research has linked TUBA1A to extracerebral anomalies, such as a fetal akinesia deformation sequence (FADS)[5], and optic nerve abnormalities[6] associated with TUBA1A mutations, indicating its role in various developmental processes. Herein, we describe a case involving a Chinese couple with a history of recurrent pregnancies in which fetal ultrasound revealed both intracranial and extracranial anomalies. A heterozygous TUBA1A variant (c.7G>A) was detected during the second pregnancy; subsequent validation and reassessment of the variant’s pathogenicity confirmed its association with the observed fetal phenotype.
PATIENT AND METHODS
Patient description
This was a healthy, non-consanguineous G2P0 Chinese couple. The first pregnancy was terminated at approximately 23 weeks of gestation due to structural abnormalities detected by prenatal ultrasound, involving the brain, heart, and craniofacial regions. Unfortunately, no autopsy or genetic testing was conducted on the aborted fetus at the hospital where the procedure was performed. In the current (second) pregnancy, neither nuchal translucency (NT) screening nor noninvasive prenatal testing (NIPT) detected any abnormalities. However, a recent second-trimester ultrasonography at 17+4 weeks of gestation revealed a constellation of fetal structural anomalies highly concordant with those observed in the prior pregnancy. Consequently, prenatal diagnosis was requested, and informed consent was obtained from the couple for further genetic testing, research, and publication. The ethical approval was obtained from the Ethical Review Committee of the Women and Children’s Hospital, School of Medicine, Xiamen University, China (No. KY-2024-105-H01), and written informed consent was obtained from all participants. At 18 weeks of gestation, the pregnant woman underwent amniocentesis at the Xiamen Women and Children’s Hospital. Following the amniocentesis, the couple elected to terminate the pregnancy due to the multiple abnormal ultrasound findings.
Genetic testing and sequencing parameters
The collected amniotic fluid samples (AFS) were sent to BGI Genomics Co., Ltd. (Shenzhen, China) for singleton copy number variation sequencing (CNV-seq) and exome sequencing (ES). For ES, exome capture and library construction were performed using the NEXome XP Panel v1.0 kit (Cat No. 1001872, Nanodigmbio, Nanjing, China) and the NanoPrep® DNA Library Preparation Kit v2 (Cat No. 1002421, Nanodigmbio, Nanjing, China), respectively. The constructed libraries were subsequently sequenced on the MGISEQ-2000 platform (MGI Tech Co., Ltd., Shenzhen, China) to produce 2 × 150 bp paired-end reads with a minimum sequencing depth of 150×. Candidate variants detected by ES in the AFS were verified by Sanger sequencing. CNV-seq was performed via low-depth whole-genome sequencing on the BGISEQ-2000 platform, with an average sequencing depth of 0.4× and a 100 kb threshold for copy number variation (CNV) calling. Confirmatory ES was conducted on maternal peripheral blood samples by Aegicare Biotechnology Co., Ltd. (Shenzhen, China) using the same protocol as described above, with a minimum sequencing depth of 100×.
Reassessment and reclassification of the variant
Candidate variants are reviewed and reclassified in accordance with the American College of Medical Genetics and Genomics (ACMG)/the Association for Molecular Pathology (AMP) guidelines[7,8], using the Bayesian classification framework from ClinGen Sequence Variant Interpretation (SVI) Working Group[9], and its standardized scoring system, wherein each criterion is assigned an integer point ranging from -8 to +8[10].
RESULTS
Prenatal ultrasound findings
By systematically reviewing the patient’s medical records, we retrieved prenatal ultrasound data from both pregnancies [Table 1]. The ultrasound findings showed strong similarities between the two fetuses. Both exhibited cerebral dysplasia, featuring hydrocephalus, cerebellar dysplasia with vermian agenesis, cystic midline cerebellar lesions, absent corpus callosum, and cavum septum pellucidum [Figure 1A-D]. Non-neurological anomalies were also present in both cases, including cleft palate, ventricular septal defect, unilateral (right) microtia, and mild bilateral renal pyelectasis [Figure 2A-D].
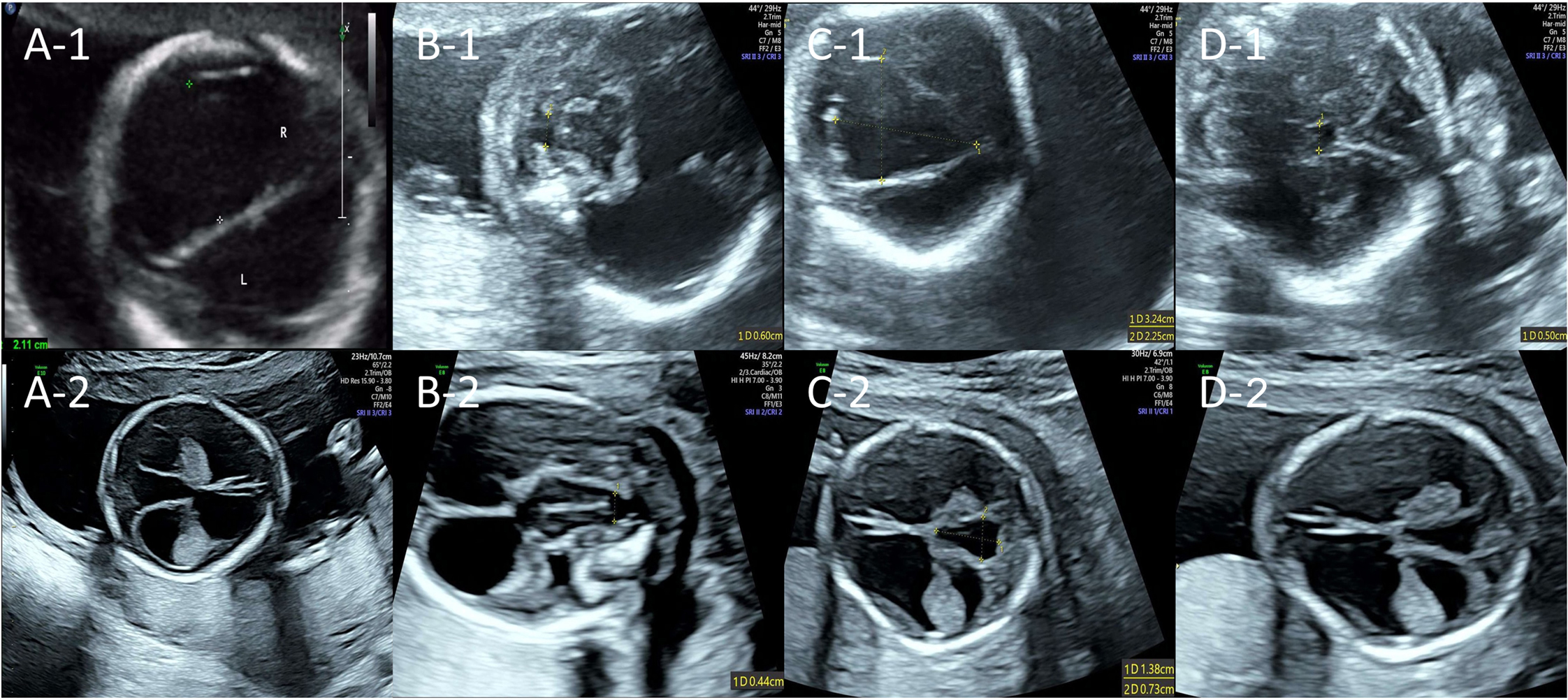
Figure 1. Fetal brain ultrasound findings of two consecutive pregnancies. The upper row shows the prenatal ultrasound from the first pregnancy at 22+4 weeks of gestation; the lower row shows that from the second pregnancy at 17+4 weeks of gestation. (A-1,2) Ultrasound suggested hydrocephalus; (B-1,2) Ultrasound suggested cerebellar dysplasia with vermian agenesis; (C-1,2) Ultrasound suggested a cystic midline cerebellar lesion (suspected arachnoid cyst); (D-1,2) Ultrasound suggested a complete absence of corpus callosum and cavum septum pellucidum.
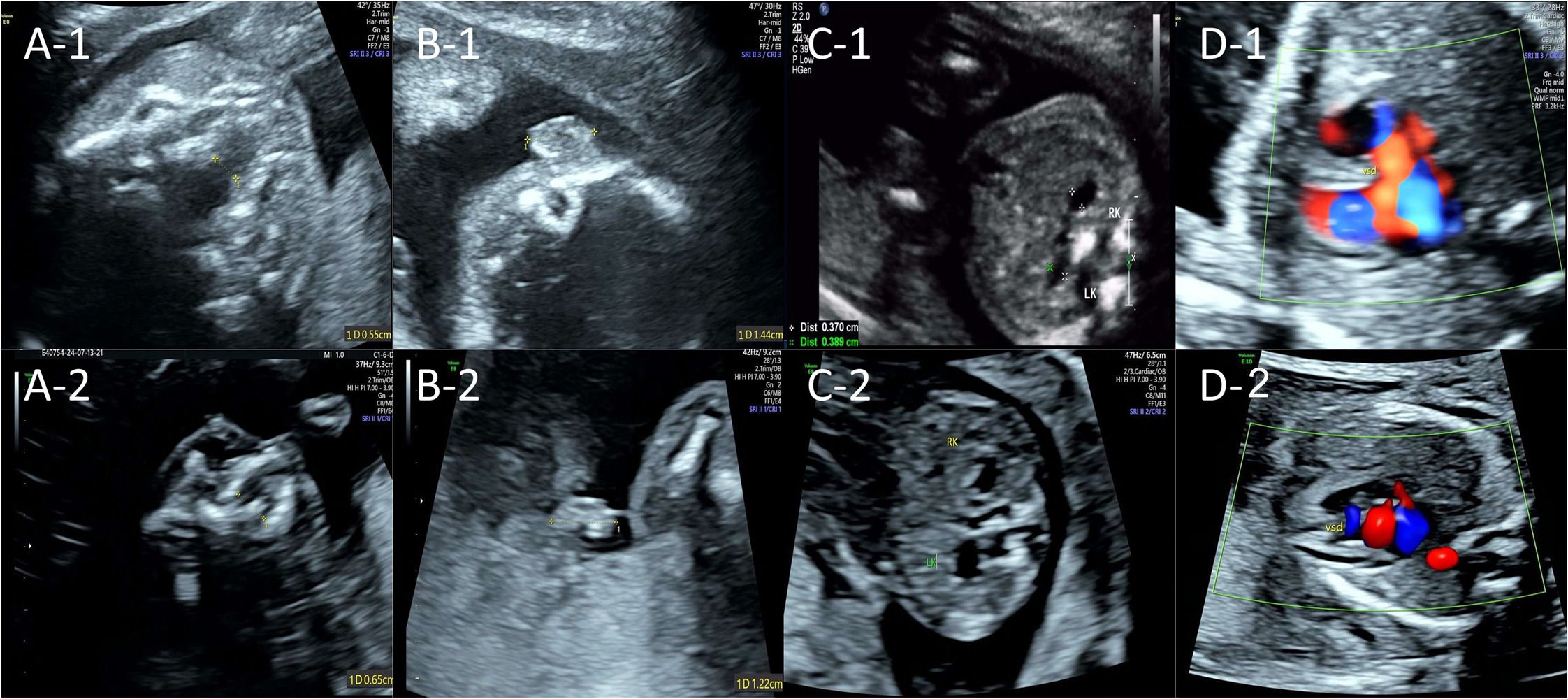
Figure 2. Fetal non-neurological ultrasound findings of two consecutive pregnancies. The upper row shows the prenatal ultrasound from the first pregnancy at 22+4 weeks of gestation; the lower row shows that from the second pregnancy at 17+4 weeks of gestation. (A-1,2) Ultrasound suggested a cleft palate; (B-1,2) Ultrasound suggested unilateral (right) microtia; (C-1,2) Ultrasound suggested mild bilateral renal pyelectasis; (D-1,2) Ultrasound suggested a ventricular septal defect.
Prenatal ultrasound findings of fetuses during two pregnancies
| First pregnancy (GW: 22+4) | Second pregnancy (GW: 17+4) | |
| Prenatal ultrasound findings | Cerebral anomalies: (1) Hydrocephalus (R: 2.11 cm vs. L: 1.97 cm) (2) Cerebellar dysplasia with vermian agenesis (0.6 cm) (3) Cystic midline cerebellar lesion (suspected arachnoid cyst: 3.24 cm * 2.25 cm) (4) Absence of the corpus callosum and cavum septum pellucidum Non-neurological anomalies: (1) Cleft palate (0.55 cm) (2) Ventricular septal defect (3) Unilateral (R) microtia(R: 1.44 cm vs. L: 1.80cm) (4) Mild bilateral renal pyelectasis | Cerebral anomalies: (1) Hydrocephalus (R: 1.46 cm vs. L: 1.45 cm) (2) Cerebellar dysplasia with vermian agenesis (0.44 cm) (3) Cystic midline cerebellar lesion (suspected arachnoid cyst: 1.38 cm * 0.73 cm) (4) Absence of the corpus callosum and cavum septum pellucidum Non-neurological anomalies: (1) Cleft palate (0.65 cm) (2) Ventricular septal defect (0.21 cm) (3) Unilateral (R) microtia(R: 1.22 cm vs. L: 1.35 cm) (4) Mild bilateral renal pyelectasis |
Genetic test results
CNV-seq analysis detected a 106.79 kb deletion {seq[GRCh37]3p14.1p14.1(68316329_68423116)×1} of unknown origin in the fetus and was categorized as a variant of uncertain significance (VUS) in the report. ES analysis of the AFS identified three candidate single-nucleotide variants (SNVs) potentially associated with the fetal phenotype; however, all were classified as VUS [Table 2]. Sanger sequencing confirmed that the EPH receptor B4 (EPHB4): c.2254C>T(p.Leu752Phe) variant in the fetus was paternally inherited, while the two HECT domain and RCC1-like domain 1 (HERC1) missense variants were inherited biparentally [Supplementary Figure 1A]. Owing to the ambiguous peak morphology [Supplementary Figure 1B], initial Sanger sequencing failed to determine the parental origin of the TUBA1A: c.7G>A(p.Glu3Lys) variant. Subsequent ES analysis confirmed low-level mosaicism (~4.43%) at this locus in the maternal sample [Supplementary Figure 1C]; however, this variant was still classified as VUS in the report, given that the mother was unaffected.
Detailed information on the candidate SNV variants identified in the fetus
| Gene | Gene-disease validitya | Transcript | Variant sites and heterozygosity | Origin of variant | Initially reported variant categories (criteria used) | Reclassified variant categories (criteria used) |
| TUBA1A | Definitive | NM_006009.3 | c.7G>A(p.Glu3Lys), het | Maternal (mosaic) | VUS (PM2 + PP2 + PP3) | LP (PM6 + PM2_supporting + PP2 + PP3 + PP4) |
| EPHB4 | Definitive | NM_004444.4 | c.2254C>T(p.Leu752Phe), het | Paternity | VUS (PM2 + PP2 + PP3) | VUS (PM2_supporting + PP2 + PP3) |
| HERC1 | NA | NM_003922.3 | c.14131C>G(p.Pro4711Ala), het | Paternity | VUS (PM2 + PP2 + PP3) | VUS (PM2_supporting + PP2 + PP3) VUS (PM2_supporting + PP2) |
| HERC1 | NA | NM_003922.3 | c.7576A>G(p.Ser2526Glu), het | Maternal | VUS (PM2 + PP2) |
Reclassification of the candidate variants
Given that the etiology of the fetal anomalies remained unidentified in the three initial reports, we performed a comprehensive reanalysis to assess the pathogenicity of the candidate variants. For the copy number loss detected in the fetus, we first mapped the CNV within the TAFA1 gene using the DECIPHER database [Supplementary Figure 2]. We then applied the Pathogenic Very Strong 1 (PVS1) criterion from the SVI guidelines to reclassify the intragenic CNV[11,12], as an alternative to the CNV scoring metric system[13]. Upon querying the ClinGen database (https://www.clinicalgenome.org), we confirmed that the gene-disease association remains uncertain to date. Accordingly, the classification of this CNV was maintained as originally reported.
Subsequently, we focused on four candidate SNVs across three genes. For the variant detected in the EPHB4 gene, although ClinGen has established a gene-disease relationship (EPHB4-related vascular malformation spectrum), the clinical presentation in our patient did not align with the phenotype reported in Online Mendelian Inheritance in Man (OMIM) [Supplementary Table 1]. In addition, the variant was inherited from an unaffected parent, further reducing its likelihood of pathogenicity. Thus, it was retained as a variant of VUS, in accordance with the initial classification. Regarding the candidate variants in HERC1, while the inheritance pattern documented in OMIM is consistent with the familial pattern observed here, the gene-disease association remains ambiguous. Moreover, the phenotypes recorded in OMIM exhibit minimal overlap with the fetal phenotypes in the present case [Supplementary Table 1]. Given these observations, we conclude that there is inadequate evidence to support upgrading the pathogenicity of these two variants.
In comparison to the two candidate genes mentioned above, the phenotypic spectrum associated with the TUBA1A gene demonstrated the strongest concordance with the ultrasonographic phenotypes observed in our cases [Supplementary Table 1]. Consequently, we systematically re-evaluated the original criteria from the report and incorporated additional evidence to reclassify the c.7G>A variant as likely pathogenic [Table 2], thereby identifying this variant as the cause of two adverse pregnancy outcomes in the patient.
DISCUSSION
The role of the TUBA1A gene in brain development
Previous studies indicate that the TUBA1A gene, which encodes the α-tubulin isotype, is essential for microtubule formation and function, and is crucial for neuronal migration, axon guidance, and synapse formation[14]. Mouse models with TUBA1A mutations exhibit impaired neuronal migration and increased branching linked to disrupted microtubule organization and nucleus-centrosome coupling, underscoring its irreplaceable role in neuronal migration and the importance of microtubule flexibility for proper neuronal structure[15]. Similarly, in humans, TUBA1A mutations cause neurodevelopmental disorders known as tubulinopathies - such as lissencephaly and polymicrogyria - highlighting its key role in preserving neuronal integrity and function during brain development[2,16,17].
Ultrasound phenotypes of fetuses with TUBA1A mutation
Compared with postpartum patients, research on the phenotypic characteristics of fetuses with TUBA1A mutations is scarce. From 2007 to 2022, multiple studies reported that such fetuses mostly had cerebral developmental abnormalities, including ventricular dilatation, cerebellar hypoplasia, microcephaly, and gyral abnormalities, all carrying de novo missense mutations in the TUBA1A gene[3,5,18-27]. The current case had multiple cerebral dysplasias; among them, hydrocephalus, vermian agenesis, and absence of the corpus callosum have been previously reported, but cystic midline cerebellar lesions have not been documented before, which broadens the prenatal cerebral phenotypic spectrum of fetuses with TUBA1A mutations.
In addition, previous studies suggested that TUBA1A mutations may be associated with extracranial developmental abnormalities (such as ocular abnormalities), but no relevant abnormalities were detected by prenatal imaging[6,22]. Our study first reported non-neurological ultrasound abnormalities (including cleft palate, ventricular septal defect, etc.) in the prenatal ultrasound phenotypes of such fetuses, which are speculated to be associated with the identified TUBA1A mutation, as no other definitive pathogenic CNVs or SNVs related to these manifestations were detected in the proband.
Reclassification of the VUS association of fetal US findings
In 2020, the ACMG proposed ES for fetuses with structural anomalies if karyotyping and microarray analysis were negative[28], recommending reporting pathogenic (P), likely pathogenic (LP) variants, and phenotype-matched VUS. Recent studies show reanalyzing ES data, especially phenotype-related VUS, can improve diagnostic rates when causes are unclear[29,30].
In the present case, overlapping prenatal ultrasound findings from two pregnancies prompted a genetic investigation; cerebral dysgenesis indicated a potential association between the TUBA1A c.7G>A variant and the disease. Since nearly all previously reported TUBA1A variants in probands were de novo, we initially sought to determine the parental origin of this variant. Sanger sequencing of the mother revealed background peaks at the TUBA1A c.7 locus, suggesting low-level somatic mosaicism, which was subsequently confirmed by ES of maternal peripheral blood. Afterwards, we identified a similar case in which two sisters presented with polymicrogyria due to a TUBA1A c.13A>C mutation, with follow-up analysis confirming the same mosaic mutation in peripheral blood samples from their phenotypically unaffected mother[31]. In accordance with the most recent guidelines issued by the SVI working group[32], germline variants in offspring resulting from parental mosaicism may be classified using *de novo* criteria - such as Pathogenic Strong 2 (PS2) or Pathogenic Moderate 6 (PM6) - provided that parental origin is confirmed and the phenotypic manifestations are specific. In the present case, the PM6 criterion was applied due to insufficient evidence to confirm consanguinity between the proband’s parents.
In addition to the *de novo* criteria, we propose incorporating the Pathogenic Supporting 4 (PP4) criterion to support reclassification of the TUBA1A variant. This criterion applies when a patient’s phenotype or family history is highly specific for a monogenic disorder[7]. Two factors warranted its application in this context: the fetal phenotype exhibited a high specificity for TUBA1A-related tubulinopathies, and the prenatal findings in the couple’s first pregnancy closely mirrored those observed in the second, thereby establishing a pertinent family history. In addition to the newly introduced criteria, the three criteria from the initial ES report were also reassessed. PP2 and PP3 were maintained, whereas Pathogenic Moderate 2 (PM2) was downgraded to a supporting level according to the SVI guideline[33]. Collectively, TUBA1A c.7G>A variant was reclassified using criteria of PM6 (2 points) + PM2_Supporting (1 point) + PP2 (1 point) + PP3 (1 point) + PP4 (1 point), yielding a Bayesian score of six and a “likely pathogenic” interpretation. Based on this conclusion, the couple received comprehensive reproductive guidance.
Limitations of the study
The primary limitation of this study is the lack of in vitro experimental validation to functionally support the pathogenicity of the TUBA1A c.7G>A variant; these will be explored in future research. In addition, as of the submission of this article, the precise efficacy of the fertility guidance provided remains unclear since the couple has not yet achieved a subsequent pregnancy; thus, long-term follow-up is needed for further observation.
CONCLUSION
This study describes a prenatal case with recurrent fetal brain anomalies and concurrent cardiac, facial, and renal abnormalities. By identifying the variant origin and reclassifying its pathogenicity, we confirmed the proband’s phenotype was linked to a maternal mosaic TUBA1A variant. This finding enriches the TUBA1A pathogenic variant database, expands related prenatal ultrasound manifestations, and provides new insights into the gene’s molecular pathways. However, evidence linking TUBA1A variants to extracerebral manifestations is limited, requiring further research to validate our hypothesis.
DECLARATIONS
Acknowledgments
We thank the patients and their families for their participation in the study.
Authors’ contributions
Conceptualization, writing - review & editing, funding acquisition: Jiang Y
Investigation, funding acquisition: Wu L
Data curation: Du L, Luo Z, Zhao H
Resources: Lu M
Review & editing: Zhang Y
Availability of data and materials
The data that support the findings of this study are available in the China National Center for Bioinformation at https://bigd.big.ac.cn/gsa-human/browse/HRA012907, reference number HRA012907.
AI and AI-assisted tools tatement
Not applicable.
Financial support and sponsorship
This study was supported by the Healthcare Guidance Project of Xiamen Municipal Bureau of Science and Technology [grant no. F3502Z20244ZD1213] and Xiamen Municipal Special Project for Promoting the Development of Traditional Chinese Medicine [grant no. XWZY-2025-0520].
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Ethical approval was obtained from the Ethical Review Committee of the Women and Children’s Hospital, School of Medicine, Xiamen University (No. KY-2024-105-H01), and written informed consent was obtained from all participants.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
Supplementary Materials
REFERENCES
1. Keays DA, Tian G, Poirier K, et al. Mutations in alpha-tubulin cause abnormal neuronal migration in mice and lissencephaly in humans. Cell. 2007;128:45-57.
2. Aiken J, Buscaglia G, Bates EA, Moore JK. The α-tubulin gene TUBA1A in brain development: a key ingredient in the neuronal isotype blend. J Dev Biol. 2017;5:8.
3. Schröter J, Popp B, Brennenstuhl H, et al. Complementing the phenotypical spectrum of TUBA1A tubulinopathy and its role in early-onset epilepsies. Eur J Hum Genet. 2022;30:298-306.
4. Schröter J, Döring JH, Garbade SF, et al. Cross-sectional quantitative analysis of the natural history of TUBA1A and TUBB2B tubulinopathies. Genet Med. 2021;23:516-23.
5. Brar BK, Thompson MG, Vora NL, et al.; Fetal Sequencing Consortium. Prenatal phenotyping of fetal tubulinopathies: a multicenter retrospective case series. Prenat Diagn. 2022;42:1686-93.
6. Ramirez DA, Anninger WV, Scoles D. Optic nerve hypoplasia and bilateral persistent fetal vasculature due to tuba1a tubulinopathy. Retin Cases Brief Rep. 2025;19:264-6.
7. Richards S, Aziz N, Bale S, et al.; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-24.
8. Wilcox EH, Webb RF, Tshering KC, et al.; ClinGen RASopathy Expert Panel. Updated ACMG/AMP specifications for variant interpretation and gene curations from the ClinGen RASopathy expert panels. Genet Med Open. 2025;3:103430.
9. Tavtigian SV, Greenblatt MS, Harrison SM, et al.; ClinGen Sequence Variant Interpretation Working Group (ClinGen SVI). Modeling the ACMG/AMP variant classification guidelines as a Bayesian classification framework. Genet Med. 2018;20:1054-60.
10. Tavtigian SV, Harrison SM, Boucher KM, Biesecker LG. Fitting a naturally scaled point system to the ACMG/AMP variant classification guidelines. Hum Mutat. 2020;41:1734-7.
11. Abou Tayoun AN, Pesaran T, DiStefano MT, et al.; ClinGen Sequence Variant Interpretation Working Group (ClinGen SVI). Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum Mutat. 2018;39:1517-24.
12. Brandt T, Sack LM, Arjona D, et al. Adapting ACMG/AMP sequence variant classification guidelines for single-gene copy number variants. Genet Med. 2020;22:336-44.
13. Riggs ER, Andersen EF, Cherry AM, et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med. 2020;22:245-57.
14. Belvindrah R, Natarajan K, Shabajee P, et al. Mutation of the α-tubulin Tuba1a leads to straighter microtubules and perturbs neuronal migration. J Cell Biol. 2017;216:2443-61.
15. Leca I, Phillips AW, Ushakova L, Cushion TD, Keays DA. Codon modification of Tuba1a alters mRNA levels and causes a severe neurodevelopmental phenotype in mice. Sci Rep. 2023;13:1215.
16. Hoff KJ, Aiken JE, Gutierrez MA, Franco SJ, Moore JK. TUBA1A tubulinopathy mutants disrupt neuron morphogenesis and override XMAP215/Stu2 regulation of microtubule dynamics. Elife. 2022;11:e76189.
17. Aiken J, Moore JK, Bates EA. TUBA1A mutations identified in lissencephaly patients dominantly disrupt neuronal migration and impair dynein activity. Hum Mol Genet. 2019;28:1227-43.
18. Cabet S, Karl K, Garel C, et al. Two different prenatal imaging cerebral patterns of tubulinopathy. Ultrasound Obstet Gynecol. 2021;57:493-7.
19. Hebebrand M, Hüffmeier U, Trollmann R, et al. The mutational and phenotypic spectrum of TUBA1A-associated tubulinopathy. Orphanet J Rare Dis. 2019;14:38.
20. Gardner JF, Cushion TD, Niotakis G, et al. Clinical and functional characterization of the recurrent TUBA1A p.(Arg2His) mutation. Brain Sci. 2018;8:145.
21. Yokoi S, Ishihara N, Miya F, et al. TUBA1A mutation can cause a hydranencephaly-like severe form of cortical dysgenesis. Sci Rep. 2015;5:15165.
22. Myers KA, Bello-Espinosa LE, Kherani A, Wei XC, Innes AM. TUBA1A mutation associated with eye abnormalities in addition to brain malformation. Pediatr Neurol. 2015;53:442-4.
23. Shimojima K, Narita A, Maegaki Y, Saito A, Furukawa T, Yamamoto T. Whole-exome sequencing identifies a de novo TUBA1A mutation in a patient with sporadic malformations of cortical development: a case report. BMC Res Notes. 2014;7:465.
24. Okumura A, Hayashi M, Tsurui H, et al. Lissencephaly with marked ventricular dilation, agenesis of corpus callosum, and cerebellar hypoplasia caused by TUBA1A mutation. Brain Dev. 2013;35:274-9.
25. Sohal AP, Montgomery T, Mitra D, Ramesh V. TUBA1A mutation-associated lissencephaly: case report and review of the literature. Pediatr Neurol. 2012;46:127-31.
26. Lecourtois M, Poirier K, Friocourt G, et al. Human lissencephaly with cerebellar hypoplasia due to mutations in TUBA1A: expansion of the foetal neuropathological phenotype. Acta Neuropathol. 2010;119:779-89.
27. Fallet-Bianco C, Loeuillet L, Poirier K, et al. Neuropathological phenotype of a distinct form of lissencephaly associated with mutations in TUBA1A. Brain. 2008;131:2304-20.
28. Monaghan KG, Leach NT, Pekarek D, Prasad P, Rose NC.; ACMG Professional Practice and Guidelines Committee. The use of fetal exome sequencing in prenatal diagnosis: a points to consider document of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2020;22:675-80.
29. Walsh N, Cooper A, Dockery A, O'Byrne JJ. Variant reclassification and clinical implications. J Med Genet. 2024;61:207-11.
30. Li LS, Li DZ. Ongoing reanalysis of prenatal exome sequencing data leads to higher diagnostic yield. Ultrasound Obstet Gynecol. 2022;59:833-4.
31. Jansen AC, Oostra A, Desprechins B, et al. TUBA1A mutations: from isolated lissencephaly to familial polymicrogyria. Neurology. 2011;76:988-92.
32. ClinGen Sequence Variant Interpretation Recommendation for de novo Criteria (PS2/PM6) - Version 1.1. Available from: https://www.clinicalgenome.org/site/assets/files/3461/svi_proposal_for_de_novo_criteria_v1_1.pdf. [Last accessed on 12 Mar 2026].
33. ClinGen Sequence Variant Interpretation Recommendation for PM2 - Version 1.0. Available from: https://www.clinicalgenome.org/site/assets/files/5182/pm2_-_svi_recommendation_-_approved_sept2020.pdf. [Last accessed on 12 Mar 2026].
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].