


Drug-based approaches to modulate mitochondrial condition in the case of atherosclerosis: focus on correction of mitochondria dysfunction

Abstract
Mutations in mitochondrial DNA can cause mitochondrial diseases. This review focuses on the main functions of mitochondria and the effect of mutations in mtDNA on the processes of mitophagy, mitodynamics and mitochondrial biogenesis. The main mitochondrial diseases associated with specific mutations in mtDNA are reviewed, with an emphasis on atherosclerosis. It is assumed that mtDNA mutations can provoke pathological changes in the intima of the human aorta and activate a specific immune response, ultimately leading to the development of atherosclerosis. Special attention is paid to the methods of targeted therapy of mitochondrial diseases with the use of antioxidants, mitodynamics modifiers, and phototheranostics.
Keywords
INTRODUCTION
Mitochondria are two-membrane eukaryotic organelles that perform a variety of functions. They appeared 1.5-2 billion years ago as a result of endosymbiosis with alphaproteobacteria[1]. The mitochondria’s main role as the powerhouse of the cell is to provide it with ATP as the energy source, which is formed through the oxidative phosphorylation (OXPHOS) process. In addition, mitochondria are involved in other processes such as signaling, calcium maintenance, beta-oxidation of fatty acids, and regulation of apoptosis and the cell cycle[2].
Due to their endosymbiotic origin, mitochondria have their own ring-shaped DNA (mtDNA). However, the main part of the mitochondrial proteins and RNA genes resides in the nucleus, with a smaller part encoded in mtDNA. Human mtDNA contains 16,569 base pairs, comprising the light (L) and heavy (H) chains[3]. The heavy chain contains a large amount of guanine and encodes 12 subunits of the oxidative phosphorylation system, 2 rRNAs (12S and 16S), and 14 tRNAs. The light chain is rich in cytosine and encodes one subunit of the oxidative phosphorylation system and 8 tRNAs[4]. In total, mtDNA contains genes encoding 2 rRNAs, 13 electron transport chain proteins, and 14 tRNAs. A non-coding regulatory region known as the D-loop (bias loop) is also present in mtDNA. It shifts the reading frame of light chain genes during replication and transcription[5]. Genes in mtDNA are located more condensed than in nuclear DNA. Hence, there is an increased risk of polymerase disruption during replication and, consequently, the accumulation of mutations in mtDNA.
Mutations in mtDNA can negatively affect vital functions and cause inherited diseases. These mutations are categorized into heteroplasmic and homoplasmic, based on the proportion of mitochondria with mutated mtDNA. Heteroplasmic mutations are found in a share of mtDNA copies, and their phenotypic expression is determined by the mutation frequency. In the homoplasmic case, mutations are found in all copies of mtDNA in a cell. mtDNA mutations predominantly negatively affect the functioning of the oxidative phosphorylation system and transcription processes, which leads to the inability of mitochondria to mitophagy, increased oxidative stress due to ROS accumulation, inflammatory processes, impaired anabolism and catabolism[6]. Mitochondrial diseases are primarily transmitted through the maternal line[7]. Aside from hereditary transmission, mtDNA mutations can also be somatic, which are predominantly able to accumulate in the elderly age[8]. The most common diseases associated with mutations in mtDNA are progressive external ophthalmoplegia, MELAS, maternal inherited diabetes mellitus, maternal inherited deafness, Leigh syndrome, Alzheimer’s disease, Parkinson’s disease, cancer, and atherosclerosis[9,10].
In atherosclerosis, mutations in mtDNA can potentially affect inflammatory processes and also cause a specific immune response in endothelial and subendothelial cells[11]. Such pathological processes in artery walls can be caused by mutations in the mtDNA genes encoding rRNA12S, UUR, and CUN recognizing codon tRNA-Leu, as well as subunits of the electron transport chain: 1, 2, 5, 6 NADH Dehydrogenase and cytochrome B[12]. It was also found that the inflammatory response in atherosclerotic arteries is caused by the activation of macrophages by multiply modified low-density lipoproteins (mmLDL)[13].
Currently, one of the most promising pathways for the therapy of mitochondrial diseases is the activation of mitophagy processes in cells containing mutated mtDNA. Mitophagy is a type of autophagy that reduces the number of mitochondria when there is a lack of nutrients, lyses mitochondria with damaged functions, and reduces the frequency of mutational mtDNA[14]. Thus, activation of mitophagy in therapeutic treatment can potentially help to reduce the frequency or severity of diseases associated with mutations in mtDNA.
MITOCHONDRIA (FUNCTIONS AND ROLE IN CELLS)
The functions of mitochondria in the cell are to provide energy, regulate the cell signaling, apoptosis and mitophagy, cell cycle, beta-oxidation, synthesize reactive oxygen species (ROS), activate the response of the endoplasmic reticulum to stress, and maintain the calcium level in the cell. Mutations in mtDNA result in dysfunctions of these processes in mitochondria, as well as an increase in ROS levels, which can lead to mitochondrial diseases[15].
The main function of mitochondria is the synthesis of ATP. Mitochondria make the greatest contribution to cell energy provision due to the processes of oxidative phosphorylation (OXPHOS), which occurs in the electron transport chain (ETC) of mitochondria. ETCs include four respiratory complexes: NADH:ubiquinone oxidoreductase (complex I), succinate:ubiquinone oxidoreductase (complex II), ubiqunol:cytochrome c oxidoreductase (complex III), cytochrome-c oxidase (complex IV), and electron carriers: ubiquinol pool and cytochrome C. During oxidative phosphorylation, a proton gradient is formed, due to which ATP synthase (complex V) can synthesize ATP by transferring protons from the intermembrane space through the Fo region to the mitochondrial matrix, in the F1 region. Conformational change of the catalytic subunits causes an ATP synthesis reaction (ADP + Pi = ATP + H2O)[16]. Reduction equivalents in ETC come from the Krebs cycle, which also takes place in mitochondria. The Krebs cycle not only provides ETC with reducing equivalents (NADH and FADN2), but also participates in the metabolism of amino acids, carbohydrates, and fatty acids, as well as maintaining calcium levels. A by-product of oxidative phosphorylation is reactive oxygen species (ROS), which are mainly produced by complexes I and III in the electron transport chain[17]. In addition to ROS, which include hydrogen peroxide (H2O2), superoxide radical (O2-•), and hydroxyl radical (OH•), the by-products of oxidative phosphorylation are S-nitrosothiols (RSNO) and reactive nitrogen species (RNS), which include the nitroxyl anion (NO-), nitroxyl cation (NO+), nitrate (NO3-). In small amounts, ROS, RSNO, and RNS can be absorbed by the antioxidant system or used to maintain redox balance, and perform signaling functions, activate transcription factors, and participate in apoptosis processes[18,19]. In addition, ROS can both take part in cell proliferation, facilitating cell growth by regulating the activity of kinases (Src, MAPKs, and Akt), transcription factors (NF-kB), and be responsible for inflammatory processes and specific immune responses through phosphorylation and RIG-I mediated dimerization of IRF-3 and subsequent production of IFN-β[20,21].
The consequences of mtDNA mutations are malfunctions of OXPHOS, antioxidant systems, and gene transcription processes, which lead to increased oxidative stress. Higher ROS levels negatively affect the integrity of nearby mtDNA, proteins, and mitochondrial membranes. In cases of critical ROS accumulation, increased oxidative stress can lead to activation of the cell death pathway and the appearance of inflammatory foci in tissues and organs. The occurrence of atherosclerosis, diabetes, dyslipidemia, and hypertension is directly associated with an increased ROS level in the mitochondria[22]. When modeling the processes of oxidative stress in atherosclerotic Apoe -/- mice with superoxide dismutase-2 (SOD2) deficiency, the development of atherosclerosis t was enhanced[23].
The mitochondrial life cycle consists of three main stages: mitochondrial dynamics, which in turn consists of the processes of mitochondrial fusion, fission, mitophagy, and mitochondrial biogenesis. In a mutation process, the life cycle of mitochondria can be disrupted due to abnormal gene transcription and, consequently, mitochondrial life cycle proteins malfunction, which contributes to an even greater accumulation of copies of mutated mtDNA.
As previously mentioned, one of the main processes occurring in the organelle is mitochondrial dynamics. Mitochondrial dynamics (MtDy), also known as mitochondrial turnover, includes fusion stages that occur with the help of Opa1 (Optic Atrophy 1), Mfn-1(mitofusin-1), Mfn-2 (mitofusin-2) proteins, and fission stages that include formation of a daughter mitochondria which involve proteins-1 (Drp-1) (Dynamin-related protein 1) and Fis-1 (mitochondrial fission 1 protein). In this process, ROS act as signal molecules that induce fusion and fission processes. The efficiency of the fusion and fission processes in the mitochondrial life cycle is maintained by the level of mitochondrial population in the cell and their energy function[24]. Mitochondrial dynamics (MtDy) contributes to the mitophagy of dysfunctional or damaged mitochondria. In most cases, after the fission process, one of the mitochondria becomes depolarized and loses the Opa1, Mfn-1, and Mfn-2 proteins, which can activate mitophagy processes. The other mitochondrion becomes hyperpolarized and can then enter the process of mitochondrial turnover[25]. Mitochondrial turnover processes have a great influence on oxidative phosphorylation, apoptosis, and mitophagy[26].
Another important process in mitochondria is mitophagy. Mitophagy is a type of autophagy that controls the number of mitochondria and helps reduce the number of mitochondria with damaged functions or mutated copies of mtDNA. There are several types of mitophagy: basal mitophagy, stress-induced mitophagy, and programmed mitophagy[27]. Mitophagy can also be divided into two types: ubiquitin-dependent and ubiquitin-independent mitophagy. Ubiquitin-dependent mitophagy may follow one of several pathways. PTEN-induced kinase 1 PINK1/parkin pathway operates in mitochondria with impaired membrane potential and is triggered by PTEN-induced kinase 1 (PINK1)[28]. Contrary to this, in intact mitochondria, PINK1 can be degraded by the ubiquitin ligases E3, UBR1, UBR2, and UBR4[29]. When the membrane potential of mitochondria is disrupted, PINK1 phosphorylates S65 in ubiquitin on the outer mitochondrial membrane, which in turn indirectly activates E3 ligase via Parkin to continue mitophagy[30]. Another type of mitophagy, Parkin-independent mitophagy, is divided into three subtypes: receptor-mediated, lipid-mediated, and ubiquitin-mediated mitophagy[31]. Receptor-mediated mitophagy involves BNIP3, BNIP3L, BCL2L13, FUNDC1, FKBP8, and PHB2 interacting in the mitochondrial matrix with LIR, which binds to microtubules-associated 1A/1B LC3S, attaching the phagosome for further mitophagy[32]. In lipid-mediated mitophagy, the inner mitochondrial membrane breaks, after which cardiolipins bind the mitochondria to the phagosome, while ceramide from the mitochondrial matrix enters the cytoplasm, binds to LC3B, and promotes further mitophagy[33]. In ubiquitin-mediated mitophagy, P62, VPS13D, DRP1, and other ubiquitin ligases bind to ubiquitin chains, facilitating the connection of the mitochondria to the phagosome[34].
Mitochondrial biogenesis is another key process that occurs in the mitochondria. Mitochondrial biogenesis is a process that stimulates the formation and development of new mitochondria. This process is predominantly carried out by SIRT1, AMPK, NRF-2, PGC-1α, and PPARα. In addition to mitochondrial biogenesis, SIRT1, AMPK, and NRF-2 also engage in the activation of mitophagy, autophagy, and inhibit inflammasome formation. AMPK and SIRT1 promote mitochondrial biogenesis by activating PGC-1α. Moreover, AMPK and SIRT1 are involved in mitophagy, autophagy, and metabolic pathways. SIRT1 is indirectly involved in autophagy via activation of Rab7, a G protein that provides autophagosome-lysosome fusion, and also through deacetylation of the FOXO1 transcription factor, enhancing the transcription of genes necessary for autophagy[35]. AMPK also promotes autophagy by phosphorylating ULK1 and AMPK is also capable of inhibiting mTORC1, which in turn has anti-autophagosomal activity[36]. During fasting with amino acids and glucose, inhibition of mTORC1 occurred simultaneously with phosphorylation of ULK1-Ser555 and activation of AMPK. In addition, phosphorylation of ULK1-Ser555 promotes switching from canonical autophagy to mitophagy-specific pathways after AMPK activation[37]. AMPK and SIRT1 activate mitophagy processes by enhancing the transcription of Parkin and Pink1. In addition, AMPK and SIRT1 are involved in metabolic pathways, mutually promoting their activation. PGC-1α stimulates mitochondrial biogenesis by affecting the transcription factors NRF-1, ERRα (estrogen-related receptor alpha), and PPARα[38]. PGC-1α, acting on the transcription factor NRF-1, promotes transcription of Tfam, NRF-2, and TFB1, which in turn stimulate mtDNA replication[39]. When PGC-1α acts on ERRα, it increases the expression of SIRT3, which is responsible for enhancing the transcription of ETC and SOD2 proteins[40]. Through PPARα, PGC-1α increases the expression of CPT1 (carnitine palmitoyltransferase-1) and UCP-2, which reduces the production of superoxide in the mitochondria[41]. Transcription factor NRF-2, in turn, takes part in biogenesis by activating NRF-1.
Proinflammatory processes may also originate in the mitochondria. With the accumulation of mutations in mtDNA, mitophagy may be disrupted, leading to an even greater increase in ROS levels, which causes increased oxidative stress and contributes to cell death and the occurrence of inflammatory processes in tissues. The potential site of ROS formation is complex I and III of the mitochondrial respiratory chain[42]. Thus, mitochondrial mutations in the genes encoding subunits of complexes I and III lead to an increase in oxidative stress. The process of mtDNA release into the cytoplasm as a result of apoptosis depends on the level of ROS and NLRP3 in the inflammasome[43]. As a result of apoptosis, BAK/BAX proteins increase the permeability of the outer membrane for cytochrome c, which causes its release into the cytoplasm and destruction of mitochondrial contents, while BAK/BAX form sufficiently wide pores in the inner mitochondrial membrane[44]. mtDNA is able to enter the cytoplasm through those pores and cause inflammatory processes. Inflammation by macrophages can be induced by lipopolysaccharides (LPS) and ATP under the action of NLRP3 and ROS, where the presence of NLRP3 in a cell will be the limiting factor[43]. The extracellular innate immune response of cGAS-TMEM173 (STING) and IgI synthesis are also mediated by the presence of mtDNA in the cytoplasm as a marker of apoptosis[45]. Thus, mtDNA in the cytoplasm is a powerful proinflammatory factor mediated by high levels of oxidative stress and the presence of NLRP3. Thioredoxin (TRX) and thioredoxin-interacting proteins (TXNIPs) may be involved in inhibiting inflammasome formation. The formation of inflammasome depends on the interaction between TXNIP and NLRP3. If the cell does not experience oxidative stress, then inflammasomes are not formed due to the complexation of TXNIP with TRX[46]. Under oxidative stress, the TXNIP complex with TRX breaks down due to the oxidation of TRX. It should be noted that NRF-2, SIRT1, and AMPK inhibit inflammasome formation. Thus, NRF-2, indirectly through SIRT1 stimulation, increases the expression of TRX and TRX-reducing thioredoxin reductase[47]. In addition, NRF-2-driven gene transcription limits NLRP3 inflammasome activity through downregulation of NF-κB activation, and CASP1, IL1B and IL18 expression[48]. AMPK, in turn, reduces TXNIP transcription and also accelerates the proteasomal degradation of TXNIP[49]. Furthermore, quercetin not only decreases the levels of the NLR family but also exerts an anti-inflammatory effect by promoting mitophagy, which consequently reduces mtROS accumulation and NLRP3 inflammasome activation[50].
DISEASES ASSOCIATED WITH MITOCHONDRIA DYSFUNCTION
The discovery of mutated mtDNA in human cells is a relatively recent development, dating back to 1988[51]. Currently, more than 200 mitochondrial diseases associated with mutations in more than 350 genes are known[52]. Mitochondrial diseases are more common in the elderly than in children and can be caused by the aging process as well as environmental pollution, physical inactivity, smoking, and alcohol consumption. Mitochondrial diseases can be associated with mutations occurring both in mitochondrial and nuclear DNA encoding mitochondrial proteins.
As shown earlier, mtDNA mutations can be homoplasmic or heteroplasmic. The phenotypic manifestation of heteroplasmic mutations is determined by the frequency of occurrence of mutational mtDNA, which is specific to a particular disease and is called the phenotypic threshold effect. Mitochondrial diseases associated with mtDNA can be inherited through the maternal line, as well as be somatic, and accumulate with age.
Diseases connected with mtDNA mutations and dysfunctions of mitochondria
Mutations in mtDNA are associated with high levels of oxidative stress, which is a consequence of improper functioning of oxidative phosphorylation and antioxidant systems. Interestingly, more than half of all mtDNA mutations occur in the gene encoding 22 tRNAs[53]. Kearns-Sayre syndrome (KSS)[54,55], Leber Hereditary optic neuropathy (LHON)[56-59], Leigh Syndrome (LS)[60-63], Myoclonic epilepsy with ragged red fibers (MERRF)[58,64], Mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS syndrome)[62,64-66], Progressive External ophthalmoplegia (CPEO/PEO)[67,68], monoclonal immunoglobulin deposition disorder (MIDD)[69-71], diabetes (NIDDM)[72-76], NARP syndrome[58], Alzheimer’s and Parkinson’s diseases[77-80], cancer[53,81], non-alcoholic liver disease (NAFLD)[82,83], atherosclerosis, cardiomyopathy, encephalomyopathy, and other diseases are associated with mutations in mtDNA[84].
Mitochondrial dysfunction in the context of atherosclerosis pathogenesis
Atherosclerosis is a disease related to a thickening of walls of large enough elastic arteries, eventually leading to almost complete occlusion of the circulation[85]. Atherosclerosis is manifested in the formation of atherosclerotic lesions, which, in addition to many different features, contain so-called foam cells[86]. Multiply modified low-density lipoproteins (mmLDL) are capable of induction of foam cell formation (by macrophages, for example). It should be mentioned that one of the most cited in the literature atherogenic modifications of LDL is oxidation (oxLDL)[87]; however, desialylation of LDL was also shown to be a potential main modification leading to LDL acquisition of atherogenic properties[88]. Interestingly enough, it was recently shown that native LDL and mmLDL have different effects on mitophagy in monocyte-like cells[89].
For a long time, it was unclear whether atherosclerosis is a consequence of mutations or their cause. However, using prone to atherosclerosis C57BL/6J mice "linewith" apolipoprotein (Apo) E (-/-) gene deficiency, it was discovered that a significant number of mtDNA deletions accumulate in mice by the age of 2 months, while phenotypic manifestations of atherosclerosis in the form of plaques occur in mice aged 6 or more months[90]. These results show that mutations of mtDNA contribute to the formation of atherosclerosis.
Thus, atherosclerosis can also be considered a mitochondrial disease that can cause other cardiovascular diseases, such as atherosclerotic heart disease, myocardial infarction, brain stroke, circulatory disorders, and heart failure. For instance, a correlation was found between stroke and myocardial infarction in patients and mutations in the control region (CR) of mtDNA. A study revealed that mtDNA mutations m.16145G>A and m.16311T>C are a potential genetic prerequisite for stroke, while m.72T>C, m.73A>G, and m.16356T>C are a factor for myocardial infarction[91]. Using RT-PCR, it was identified that the following mtDNA mutations are most likely to be connected with cardiovascular system diseases: C3256T (gene MT-TL1), G12315A (gene MT-TL2), G13513A (gene MT-ND5) and G15059A (gene MT-CYB)[92].
Atherosclerosis is supposedly correlated to some mtDNA haplogroups. Thus, haplogroups A and M7a were reliably associated with coronary atherosclerosis, while haplogroup D4a was with myocardial infarction[93]. There are also differences in the prevalence of mitochondrial mutations associated with atherosclerosis in different populations, as shown by Kirichenko et al.[94]. In a study of the association of mtDNA mutations with asymptomatic atherosclerosis in women, it was found that correlations between the heteroplasmy levels of mutations C3256T, G14709A, G12315A, G13513A and G14846A may be associated with haplogroups[95].
One of the markers of mitochondrial functioning is mtDNA copy number (mtDNA-CN), which reflects the number of mitochondrial genomes per mitochondria and the number of mitochondria per cell. It was found that a decreased level of mtDNA-CN in patients leads to an increase in the risk coefficient for coronary heart disease, stroke and CVD, while an increased level of mtDNA-CN lowers this coefficient[96]. According to another research, the level of mtDNA-CN in patients with CHD is significantly lower compared to control[97]. The number of copies of mtDNA depends on both genetic and external factors. Abnormalities in mtDNA-CN levels are associated with increased oxidative stress, activation of the immune response, aging, environmental factors, and bad habits. It was also found that a lowered level of mtDNA-CN can be connected with atherosclerosis development[98]. However, there is also an opinion that the effect of mtDNA-CN on the development of CHD is insignificant[99].
Cells of the immune system other than macrophages (including T- and B-cells) also contribute to manifestations of atherosclerosis[100]. There are certain differences in atherosclerosis development between men and women[101]. Inorganic phosphate may play a role in atherosclerosis[102,103], which is not a big surprise considering the importance of phosphate for living organisms. Polyphosphates, which can be found in many organisms from humans[104] to yeast[105], have many functions and are closely associated with mitochondrial activity, and can be an under-investigated component potentially influencing atherosclerosis development and atherosclerosis-caused complications due to their connection with platelets, coagulation, and inflammation[106].
Pathophysiological changes in endothelium are one of the first manifestations of initial atherosclerosis. Under normal conditions, endothelial cells regulate the lumen of the blood vessel by expanding and narrowing it. However, in the context of atherosclerotic lesions, due to oxidative stress, the activity of endothelial nitric oxide synthase (eNOS) in endothelial cells decreases, weakening the synthesis of NO, which is responsible for preventing the expression of endothelial cell adhesion molecules, chemokines, and inhibiting platelet aggregation[107].
Atherosclerotic lesions also affect vascular smooth muscle cells (VSMC). Dedifferentiation processes occur: synthetic VSMC reduce the expression of contractile proteins, increase proliferation, and reconstruct the extracellular matrix (ECM), leading to wall thickening, functional impairment, development of atherosclerotic plaque, and narrowing of the vascular lumen[108].
Atherosclerotic mutations also correlate with age: mutations G12315A, G14459A, and G15059A were strongly associated with the age of patients, while antiatherogenic mutations (single-nucleotide substitutions A1555G and G14846A) were negatively correlated with age[109]. When studying heteroplasmic mtDNA mutations in groups of patients with coronary heart disease, arterial hypertension, and healthy ones, a pattern was shown in which the individual load of heteroplasmic single nucleotide variants (mtSNVs) increases with age, and the age effect was stronger for low-level heteroplasmy (heteroplasmic fraction, HF, 5%-10%)[110]. Patients with coronary heart disease (CHD) also showed an increased number of heteroplasmic mtDNA variants in the control region (CR), deletions of heteroplasmic mtDNA, and single-nucleotide variants of heteroplasmic mtDNA[111].
Atherosclerosis is closely associated with aging, which in turn is closely associated with the increase in amyloid-caused pathologies, and thus it is quite logical that amyloid deposits can be found in atherosclerotic lesions[112]. It seems to be a reasonable approach to consider the use of the anti-amyloid and anti-prion systems in attempts to prevent or delay atherosclerosis development, at least in some preliminary studies. The similarities between amyloids in yeasts and animals on a molecular level make possible further application of the results of numerous yeast prion and amyloid research on animals and humans[113-115].
Aging is one of the factors promoting the development of cardiovascular diseases, such as loss of myocardial contractility, elasticity of the left ventricle and coronary arteries, endothelial dysfunction, and arterial stiffness[116]. Aging results in telomere shortening, DNA damage, metabolic disorders, increased oxidative stress, mitochondrial damage, disruption of mitophagy, mitodynamics and mitochondrial biogenesis[117]. Telomere length has functional value in atherosclerosis. Shortened telomeres can be observed in plaque VSMC, macrophages, and endothelial cells, as well as in circulating lymphocytes[118]. It is worth noting that an increased level of oxidative stress can affect telomeres: damaging their integrity[119] and inhibiting the work of telomerase, leading to apoptosis[120]. At the moment, it is still unclear whether telomere shortening is the cause of atherosclerosis or is a sign of plaque progression[118]. A study on G4 mice (with telomerase deficiency and severe telomere dysfunction) by means of quantitative analysis of reverse transcriptase polymerase chain reaction and Western blotting of G4 tissues confirmed a decrease in the expression of PGC-1α, PGC-1β, NRF-1, ERRα, PPARα and TFAM[121]. A direct connection was found between telomere dysfunction and suppression of PGC-dependent processes of biogenesis, mitochondrial functioning, gluconeogenesis, and protection against oxidative stress.
Atherosclerosis, like some other diseases, is characterized by chronic inflammation. Additionally, it can be considered an autoimmune disease[122], and thus, certain approaches for the treatment of autoimmune diseases may also be helpful in the case of atherosclerosis[123]. The inflammatory response due to increased macrophage activity could be potentially caused or modified by mutations in mtDNA. Two homoplasmic mutations were detected in the m.A1811G and m.G9477A regions and three heteroplasmic mutations in the m.G14459A, m.A1555G, and m.G12315A regions, which may be responsible for the activation of the inflammatory response by macrophages[11].
Atherosclerotic manifestations in arterial walls are associated with mutations in the mtDNA genes encoding rRNA12S, UUR, and CUN recognizing codon tRNA-Leu, as well as subunits of the electron transport chain: 1, 2, 5, 6 NADH Dehydrogenase and cytochrome B[12]. Both homoplasmic and heteroplasmic mutations may be connected with atherosclerosis. Recent studies have shown that there are mutations in mtDNA regions del562G, m.1555A>G, m.14459G>A, and m.14846G>A connected with atherosclerosis development[124].
The study also showed that mtDNA mutations A1555G in the MT-RNR1 gene, as well as C3256T in the MT-TL1 gene, G12315A in the MT-TL2 gene, and G15059A in the MT-CYB gene are associated with atherosclerosis, since they were significantly more common in lipofibrous plaques compared to non-atherosclerotic intima[12]. For mutations associated with atherosclerosis in the mtDNA genes MT-RNR1 (rRNA 12S), MT-TL1 (tRNA-Leu, recognizing UUR), MT-ND2, MT-ND5 (NADH dehydrogenase subunits 2 and 5) and MT-CYB (cytochrome b), the average level of heteroplasmy significantly differed in atherosclerotic lesions of the aortic intima compared to intact tissue[125]. There are numerous mtDNA mutations associated with atherosclerotic plaque and found in leukocytes upon atherosclerosis: deletion at 4977 position and missense mutations m.1555A>G, m.3256C>T, m.3336T>C, m.13513G>A, m.15059G>A, m.12315G>A, m.14459G>A, and m.5178C>A, m.9477G>A, m.3243A>G and m.3256C>T[126]. The level of heteroplasmic single-nucleotide mutations C3256T, T3336C, G12315A, G13513A, G14459A, G14846A, and G15059A correlates with the size of carotid atherosclerotic plaques[127]. In the study of normal and atherosclerotic aortic wall tissue, correlations were found between various types of atherosclerotic lesions and mitochondrial mutations: mutations G12315A and G14459A were associated with total and primary atherosclerotic lesions of the aortic intima segments and lipofibrous plaques, mutation C5178A was associated with fibrous plaques and total atherosclerotic lesions. A1555G had an anti-atherosclerotic effect in the primary lesion of lipofibrous cells. The G14846A mutation was antiatherogenic for lipofibrous plaques[90]. Atherosclerotic plaques contain higher amounts of mutated mtDNA compared to unaffected arteries[128]. Next-generation sequencing revealed an increased number of heteroplasmic mtDNA variants in atherosclerotic lesions of the aortic intima compared to non-atherosclerotic aortic intima[129].
Proinflammatory activity in the case of atherosclerosis is enhanced by mmLDL. mmLDL are capable of producing autoantibodies with the further formation of immune complexes. Autoantibodies are a class of G immunoglobulins that form an antibody-antigen complex with the protein particle of LDL. When autoantibodies interact with native LDL, LDL have been shown to acquire the properties of mmLDL[130]. Thus, the formation of immune complexes increases LDL atherogenicity. Moreover, mmLDL can increase the phagocytic activity of macrophages, leading to proinflammatory reactions. Thus, the accumulation of lipids in the cells of the aortic intima leads to an even greater immune response, and also contributes to the formation of atherosclerotic plaque. In atherosclerotic plaque, the production of an inflammatory environment is maintained by cytokines and chemokines[13].
As mentioned above, the inflammatory response is activated by the increased production of ROS, which contributes to the occurrence of oxidative stress in the mitochondria. The inflammatory response is characterized by the release of mtDNA from the damaged mitochondria and the consequent assembly of inflammasomes by NLRP3 as a reaction to it. NLRP3 can form an inflammasome both in response to DAMPs (damage-associated molecular patterns)[131] and directly through the release of mtDNA into the cytoplasm. Consequently, the production of ROS and the outflow of potassium ions both increase[43]. Inflammasomes can be connected with atherosclerosis[132] and many other diseases[133], including cancer[134].
Mitochondria play an important role in maintaining the homeostasis of immune responses in the body through signaling pathways. For example, the mitochondrial signaling pathway derived from PGC-1α (gamma coactivator of the 1-alpha receptor activated by the peroxisome proliferator), which indirectly activates TFAM (mitochondrial transcription factor A) via NRF, provides stability, replication, and transcription of mtDNA, and is also involved in immune responses[19]. During inflammatory processes, in macrophage cells, PGC-1α can activate the pathway responsible for reducing ROS production, and PGC-1β production activates mitochondrial biogenesis and stops the production of cytokines that cause inflammation[135]. Thus, the PGC-1 protein family has antioxidant and anti-inflammatory properties, preventing the formation of inflammasomes and activating mitophagy processes. Signaling pathways can be further used to suppress inflammatory foci in the treatment of many diseases, including atherosclerosis. Currently, cytoplasmic hybrids based on the THP-1 cell line containing mitochondrial mutations associated with atherosclerosis are being developed for atherosclerosis research[136].
MITOCHONDRIA AS A THERAPEUTIC TARGET
Since mutations in mitochondria cause many diseases, mitochondria are the most important targets in therapeutic treatment. The pursuit of targeted therapy for diseases associated with mitochondrial mutations is in its nascent stages. Nonetheless, there are already several scientific researches devoted to the treatment of mitochondrial diseases using targeted therapy. The most promising methods of treating mitochondrial diseases include the usage of antioxidants, mitochondrial dynamics modifiers, and photodynamic therapy.
Antioxidants in mitochondria-targeted therapy are used to reduce the amount of ROS and consequently reduce the impact of oxidative stress. They can also affect the processes of mitodynamics, mitophagy, and inflammatory activity [Figure 1]. Natural antioxidants play an important role in targeted therapies. The most common natural antioxidants include B vitamins, vitamin C, and vitamin E, as well as ferulic acid, berberine, lipoic acid, astaxanthin, glucosamine, melatonin, and coenzyme Q10.

Figure 1. The effect of antioxidants on mitochondria. Antioxidants contribute to the reduction of ROS levels. They also influence the processes of mitophagy, mitochondrial biogenesis, and inflammatory activity. Glucosamine, ferulic acid, melatonin, berberine, astaxanthin, coenzyme Q10, and lipoic acid are natural antioxidants. MitoQ, EUK-8, and EUK-134 are synthetic antioxidants. Glucosamine, ferulic acid, and melatonin activate SIRT1, stimulating the processes of mitophagy. Berberine, affecting AMPK, also activates mitophagy. Astaxanthin is a PPARa agonist that stimulates mitophagy and mitochondrial biogenesis by activating UCP-2 and CPT-1. Coenzyme Q10 inhibits NLRP3, suppressing inflammatory activity and activating biogenesis processes. Lipoic acid and MitoQ activate NRF-2, stimulating the processes of mitophagy and mitochondrial biogenesis. MitoQ, in addition to NRF-2, activates PINK, also stimulating mitophagy processes. EUK-8 and EUK-134 simulate the work of SOD by suppressing inflammatory activity.
Vitamins are organic compounds come with the food necessary for maintaining the optimal functioning of the body. Some vitamins are also involved in maintaining redox balance and reducing ROS levels, as well as helping to activate mitophagy processes. B vitamins, which are essential for maintaining the nervous system, affect the processes of nuclear transcription and metabolism, including energy metabolism, as well as the processes occurring in the mitochondria. Vitamin B1 (Thiamine) helps reduce oxidative stress in mitochondria and also enhances autophagosomal activity, thus increasing the degradation of mutational mitochondria[137]. Thiamine deficiency affects mitochondrial division and mitophagy by activating ER stress markers such as heat shock proteins A (HSP70), 5 (HSPA5), Xbox-binding protein 1 (XBP1), CHOP, transcription activating factor 6 (ATF6), eukaryotic initiation factor 2A (eIF2a), and caspase 12[137]. Derivatives of the vitamin B2 (riboflavin), Flavin mononucleotide, and Flavin adenine dinucleotide in particular, play an important role in the transport of hydrogen and particularly in the oxidation-reduction reactions of ETC complexes I and II[138]. It is assumed that the lowered level of riboflavin in cells can lead to disturbances in the work of respiratory complexes I and II, which contributes to increased oxidative stress. Presently, no direct correlations between mitophagy processes and the level of riboflavin in mitochondria have been found. Vitamin B3 (nicotinamide) helps protect mitochondria from oxidative stress by increasing the activity of mitochondrial nicotinamide nucleotide transhydrogenase (NNT), which transports electrons from NADH to NADP[139]. Nicotinamide can also induce mitophagy by activating the Fis1, Drp1, and Mfn1 proteins involved in mitochondrial fusion and division[140]. The active form of vitamin B6 (pyridoxine), known as pyridoxal, is also able to activate mitophagy processes through the mitochondrial isoform of serine, hydroxymethyl transferase (SHMT2), which plays an important role in serine metabolism[141]. Vitamin B9 (folic acid) is an important antioxidant that promotes mitophagy. It was shown that mitophagy is activated through the PINK1-mediated pathway when methyl-β-cyclodextrin is added to folate, which also enhances LC3 conversion[142]. Vitamin C is one of the most important antioxidants, as it directly affects the reduction of ROS levels, protecting mitochondria from oxidative stress. Vitamin C contributes to the activation of mitophagy processes by activating the PINK1/parkin pathway. Vitamin E has significant antioxidant and anti-inflammatory properties and is successfully used in the treatment of cardiovascular diseases. Its antioxidant properties have also proven themselves in the treatment of atherosclerosis. Vitamin E reduces the level of oxidative stress, as well as the level of oxidized LDL, and also regulates the activity of cyclooxygenase and cytosolic phospholipase A2, thereby reducing the level of leukotrienes and increasing the level of prostacyclin[143].
Ferulic acid has anti-inflammatory and antioxidant effects, and these properties have been confirmed in animal and cell culture studies[144]. Ferulic acid can activate SIRT1 (Sirtuin 1), which leads to the activation of mitophagy processes in the cell. In recent clinical trials, the administration of 1,000 mg of ferulic acid to patients with hyperlipidemia had shown a beneficial effect on serum lipid composition, with a decrease in the level of systemic markers of oxidative stress and a reduction in plasma C-reactive protein levels by one-third[145]. There are studies showing the therapeutic effect of ferulic acid in the treatment of Alzheimer’s disease[146], cardiovascular diseases[147], diabetes[148], neurodegenerative disorders[149], and cancer[150]. Berberine is a natural analog of metformin used in the treatment of diabetes. Berberine has a high antioxidant activity and also contributes to the initiation of mitophagy processes by activating AMPK (AMP-activated protein kinase)[151]. Berberine is used as a therapy for type 2 diabetes, cardiovascular diseases, and depression[152]. Lipoic acid is often used in clinical cases as it promotes the activation of NRF-2 transcription[153]. NRF-2, activated by lipoic acid, promotes mitochondrial biogenesis and mitophagy processes. Lipoic acid is clinically prescribed for the treatment of patients with diabetes mellitus[154], cancer[155], gastroenterological diseases[156], and Alzheimer’s disease, Huntington’s, and Parkinson’s syndromes[157]. Astaxanthin, extracted from algae, is also a natural antioxidant used as a natural medicine. Astaxanthin is an agonist of PPARα that is used to protect ETC from ROS[158]. Astaxanthin has proven itself as a drug with anti-inflammatory activity in the treatment of liver diseases, including liver cancer[159], heart failure[160], and eye diseases such as age-related macular degeneration (AMD), cataracts, dry eye disease, and glaucoma[161]. Glucosamine is also used in medicine as an anti-inflammatory agent. Glucosamine promotes O-GlcN protein acylation, increases the activity of SIRT1, and therefore activates mitophagy processes[162,163]. Glucosamine is mainly used in the treatment of osteoarthritis[144]. Melatonin is widely used in medicine. It promotes the activation of mitophagy and mitochondrial biogenesis by initiating transcription of SIRT1 and NRF-2 through activation of the transcription factor BMAL1[164,165]. According to recent data, melatonin shows anti-inflammatory and antioxidant effects in the treatment of neurodegenerative disorders, COVID-19, cardiovascular diseases[166], insomnia, and cancer[167], and it also tackles autism spectrum disorders (ASD)[168]. Currently, ferulic acid, berberine, lipoic acid, astaxanthin, glucosamine, and melatonin have proven themselves as medicinal substances with antioxidant properties used in the treatment of age-related macular degeneration (AMD). Coenzyme Q10 (ubiquinone) has also proven to be one of the best antioxidants in clinical practice. Ubiquinone participates in oxidative phosphorylation, transferring electrons from complex I and II to complex III of ETC. A lack of coenzyme Q10 can lead to excessive ROS accumulation and cause oxidative stress. Treatment of patients with CoQ10 inhibited NLRP3 activation and restored mitochondrial biogenesis[169]. In addition, in patients with atherosclerosis, CoQ10 reduced the inflammatory activity of macrophages, lipid accumulation, and suppressed the development of foam cells in the intima[170]. Coenzyme Q10 is also used in the treatment of other cardiovascular diseases.
In addition to natural antioxidants, synthetic antioxidants are also used in the treatment of mitochondrial diseases. The most well-known synthetic antioxidants are MitoQ, EUK-8, and EUK-134. MitoQ is a more effective analog of coenzyme Q10. MitoQ has proven itself in the treatment of cardiovascular diseases, improving the mitochondria functioning in blood vessels during therapy. When applied to mice with
In addition to antioxidants, substances that can influence the processes of mitochondrial dynamics are used in the treatment of mitochondrial diseases [Figure 2]. Most of the drugs used in mtDNA disease treatment affect PGC-1α, AMPK, SIRT1, and PPARα. Metformin is one of the mitodynamics modifiers. It is mainly used in the treatment of diabetes mellitus and oncology. Metformin affects AMPK (AMP-activated protein kinase), suppressing inflammatory processes[174]. AMPK also stimulates the mitophagy of mitochondria with mutated mtDNA. Metformin application on AMPK inhibits the release of proinflammatory factors controlled by TNF-NF-kB, and endothelial cell atherogenicity consequently decreases[175]. In millimolar concentrations, metformin is able to inhibit complex I in the mitochondrial ETC, which leads to suppression of the activity of the electron transfer chain, resulting in a change in the energy charge of liver adenine nucleotides, a decrease in gluconeogenesis, and activation of AMPK[176]. Fenofibrate, which also changes the dynamics of mitochondria, is used in the treatment of cardiovascular diseases, paraplegia, quadriplegia[177], hyperlipidemia, and cancer[45]. Fenofibrate can change the dynamics of mitochondria by stimulating PPARα[178]. PPARα, by activating UCP-2 (mitochondrial uncoupling protein 2), reduces the production of mitochondrial superoxide and CPT-1 (carnitine palmitoyl transferase I), which activates beta-oxidation of fatty acids[179]. In general, exposure to fenofibrate promotes the activation of mitochondrial biogenesis and the mitophagy of mutational mitochondria. Bezafibrate affects mitochondrial biogenesis through the PPAR-PGC-1α pathway, being a PPAR agonist[180]. Bezafibrate is mainly used in dyslipidemia treatment[181]. Thus, bezafibrate in carnitine palmitoyl transferase 2 (CPT2) deficiency increases the intensity of long-chain fatty acid oxidation and reduces muscle pain[182]. Resveratrol is also a mitodynamics modifier. Resveratrol is used in the treatment of cancer[183], diabetes[184], and cardiovascular diseases[185]. Resveratrol contributes to changes in mitochondrial biogenesis by activating sirtuins, including SIRT1, as well as PGC-1α[186]. SRT2104 has similar SIRT1 activating properties. SRT2104 has only recently been studied for therapeutic use. It was found that SRT2104 can significantly inhibit NF-kB activation and enhance the expression of SIRT1 in microglia, promoting glial cell repair in ischemic or reperfusion brain injuries[187]. Administration of SRT2104 to elderly people increased the efficiency of oxidative phosphorylation in mitochondria, which led to faster recovery of adenosine diphosphate and phosphocreatine after exercise[188]. AICAR (aminoimidazole carboxamide ribonucleoside) is a mitodynamics modulator that promotes the activation of mitophagy processes. AICAR is studied as a potential anti-diabetes substance, and it has already been successfully used to treat hyperinsulinemia[189]. AICAR alters mitodynamics by activating AMPK, which in turn activates PGC-1α[190]. Activation of AMP protein kinase signaling pathways leads to an increase in the intensity of mitochondrial biogenesis, and an increase in ATP production in fibroblasts in patients with a deficiency or malfunction of the respiratory complex I[191]. Epicatechin, which is particularly abundant in dark chocolate, also has a major impact on mitochondrial biogenesis. When epicatechin was applied to mice, the overall level of biogenesis increased due to an increase in the production of ETC proteins, porins, mitophilins, and mitochondrial transcription factor A (TFAM). The number of mitochondria and their cristae also rose. When epicatechin was applied to the coronary arteries of cattle, mitochondrial biogenesis mediated by activation of citrate synthase increased[192]. Currently, studies are being conducted on the applicability of epicatechin in the therapeutic treatment of diabetes mellitus[193], non-alcoholic fatty liver diseases (NAFLD)[194], cardiovascular diseases[195], cisplatin nephropathy[196], and oncological diseases[197]. RTA 408 is a synthetic isoprenoid with mitodynamics modifying activity. RTA 408 enhances biogenesis by activating nuclear respiratory factor 2 (NRF-2)[198]. NRF-2 also promotes mitophagy activation. Recent studies have also shown that RTA-408 suppresses NF-kB signaling by inhibiting TRAF6 attraction to STING[199]. Currently, the effectiveness evaluation of RTA 408 in the treatment of osteoporosis[199], cognitive dysfunction[200], and diabetes[201] is being conducted. Statins are the most commonly used substances in the treatment of cardiovascular diseases, including atherosclerosis. In the treatment of atherosclerosis, statins are most often used together with coenzyme Q10. Statins are inhibitors of the enzyme 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase, an enzyme that controls the rate of the mevalonate pathway. Statins, acting on HMG-CoA reductase, block the synthesis of cholesterol[202]. Statins show numerous positive therapeutic effects: they reduce cholesterol levels, possess antioxidant and anti-inflammatory activity, improve endothelial cell functions, inhibit heart hypertrophy, stop processes of apoptosis in cardiomyocytes, and also work as immunomodulators[203,204]. Statins contribute to the regulation of inflammatory processes by inhibiting NF-κB-dependent NLRP3 expression and IL-1b synthesis, as shown in the human monocytic cell line THP-1[205]. However, serious side effects of statins, caused by the inhibition of HMG-CoA reductase, crucial for several vital metabolic pathways, were observed. The most common side effects in patients using statins are muscle spasms, liver damage, and non-insulin-dependent diabetes[206]. In some cases, statin therapy had to be suspended.

Figure 2. The effect of mitochondrial dynamics modifiers on the processes of mitochondrial biogenesis. Metformin and AICAR act on AMPK by activating PGC-1a, and consequently suppress inflammatory processes, inhibit TNF–NF-Kb, and also stimulate the processes of mitophagy and biogenesis. Fenofibrate and bezafibrate activate PPARa, stimulating the processes of mitophagy and mitochondrial biogenesis, by activating UCP-2 and CPT-1. Epicatechin, acting on TFAM, activates mitochondrial biogenesis. Resveratrol activates SIRT1 and PGC-1a, stimulating the processes of mitophagy and mitochondrial biogenesis. SRT2104, Statins, and RTA-408 suppress inflammatory activity by inactivating NLRP3 through the inhibition of NF-kB. SRT2104 and RTA-408 stimulate the process of mitophagy and mitochondrial biogenesis by activating SIRT1 and NRF-2 correspondingly.
Currently, studies are focused on the applicability of photodynamic therapy to the treatment of mitochondrial diseases [Figure 3]. Photodynamic therapy involves delivering drugs directly to target cells, which are then activated under the influence of radiation to trigger biochemical reactions, thereby exerting a therapeutic effect on cells, tissues, or organs. Photodynamic therapy is based on three main pillars: photosensitizer, light, and oxygen. Currently, the possibility of using photosensitizers in the treatment of cancer is being studied[207]. In addition to cancer, photodynamic therapy is a promising treatment for age-related macular degeneration (AMD)[208]. During photodynamic therapy, a photosensitizer exposed to a certain spectrum of radiation, penetrating the cell, becomes excited. The photosensitizer can transition spontaneously from the excited state to the triplet state, and react with molecular oxygen to produce ROS, which in turn can induce cell death, destruction of the vascular system, or the emergence of an immune response[209]. One of the photosensitizers is the triphenylamine derivative Mito-1. In the cell, Mito-1 produces singlet oxygen acting on mitochondria, increasing the intensity of mitophagy processes[210]. Hypericin is a photosensitizer that potentially proved itself in the treatment of cancer. Hypericin is a hydroxylated phenanthroperylenequinone with a delocalized π system in its aromatic rings[211]. Due to its hydrophobicity, hypericin is mainly accumulated in endoplasmic reticulum membranes, lysosomes, Golgi apparatus, and mitochondria[212]. The antitumor properties of hypericin are caused by its effect on mitochondrial signaling pathways, mitogen-activated protein kinase p38 (MAPK), JNK, PI3K, homologous CCAAT-enhancer-binding protein (CHOP)/TRIB3/Akt/mTOR, TRAIL/TRAIL receptor, c-Met, and Ephrin-Eph[213]. Another promising photosensitizer is protoporphyrin IX (PpIX), which is also used in the treatment of oncological diseases. Protoporphyrin can be used to diagnose porphyria, atherosclerosis, COVID-19, and cancer[214].
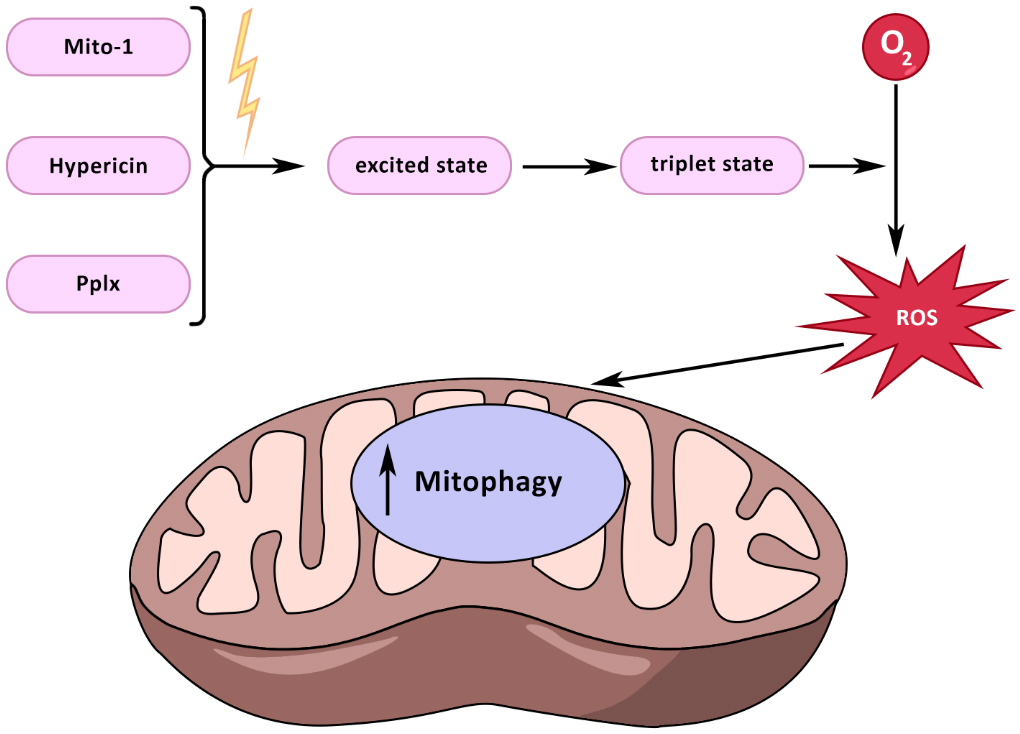
Figure 3. The effect of photodynamic therapy on mitochondria. Photosensitizers such as Mito-1, hypericin, and Pix, under the influence of a certain wavelength, enter the excited state. From the excited state, they can spontaneously enter the triplet state. The triplet photosensitizer, when interacting with O2, turns it into ROS. ROS in mitochondria contribute to the activation of mitophagy processes.
Photodynamic therapy is a promising method for the treatment of mitochondrial diseases, mainly proven in curing oncological diseases. Photodynamic therapy is one of the safest types of cancer treatment.
Therapies for reducing mutations in mtDNA (mitochondrial condition modulation)
mtDNA mutation prevention is one of the most promising methods of their treatment. Such therapy includes genetic engineering methods alongside the use of pharmacological substances.
Although research into gene therapy methods for treating mitochondrial diseases has begun relatively recently, it is actively pursued and evolving. There is a potential for genetic engineering to emerge as the most popular therapeutic method in the future. Genetic engineering techniques are based on removing mutant sites of the DNA and replacing them with fully functional counterparts. The current tools under development for the removal of defective sites include zinc fingers (mtZFN) and effector nucleases such as transcription activators or TALEN[215]. The greatest success of gene therapy has been achieved in the treatment of LHON syndrome. Most patients with LHON have mutations in mtDNA encoding subunit 4 of the NADH dehydrogenase complex (MT-ND4). By using an adenovirus (AAV) that carries the mitochondrial gene and a viral capsid VP2 that promotes fusion with the mitochondrial target sequence, gene insertion occurs and ND4 expression normalizes[216]. Thus, genetic engineering can provide a way to reduce mtDNA heteroplasmy for the treatment of mitochondrial diseases.
Some pharmacological preparations and nutrients also contribute to the reduction of mutational mtDNA levels. Maintaining the function of mtDNA depends, among other things, on the nuclear DNA genes, which ensure the synthesis of mtDNA and maintain the level of nucleotides. Impaired transcription of these genes contributes to the depletion of mtDNA and the appearance of many deletions in it[179]. One of the substances that supports mtDNA synthesis is thymidine kinase 2 (TK2), which is primarily responsible for the restoration of pyrimidine nucleotides. If TK2 is deficient, mtDNA synthesis is disrupted and myopathic mtDNA depletion syndrome occurs[217]. Thus, nucleoside bypass therapy is used to maintain mtDNA synthesis in patients with TK2 deficiency. In a study conducted on mice with a lack of TK2, it was found that deoxythymidine and deoxycytidine promote the restoration of mtDNA synthesis and increase the activity of oxidative phosphorylation[218]. Currently, nucleoside bypass therapy is infrequently used due to the absence of confirmed clinical trials supporting its effectiveness.
CONCLUSION
Mitochondria serve various functions in eukaryotic cells and play a significant role in ontogenesis. That is why defects in mtDNA may negatively affect the organism and cause various diseases. As mentioned in the paper, mutations in mtDNA and mitochondrial dysfunction are associated with and thus can potentially contribute to the development of atherosclerosis. Currently, targeted therapy seems to be the most promising method of treating mitochondrial diseases, as it can directly affect defective mitochondria. Targeted therapy for mitochondrial diseases, including atherosclerosis, is still undergoing thorough studies and has not yet been implemented in clinical practice. The most promising methods of targeted therapy are the use of antioxidants, pharmacological substances that change mitochondrial dynamics, and photodynamic therapy. These types of targeted therapy stimulate mitophagy, the removal of mutated mitochondria from the cell. Another approach to treating mitochondrial diseases is gene therapy. Modern genetic engineering methods have limited applicability so far. It is much more difficult to implement gene therapy in vivo compared to the use of pharmacological substances or nutrients. Currently, the most promising pathway in the treatment of mitochondrial diseases is targeted therapy that affects the mitochondria with antioxidants and modifiers of mitochondrial mitodynamics.
DECLARATIONS
Authors’ contributions
Conceptualization: Bezsonov E
Original draft preparation; figures: Gavrilova D
Editing: Bezsonov E, Gavrilova D, Degtyarevskaya T
Availability of data and materials
Not applicable.
Financial support and sponsorship
The research was supported by the Russian Science Foundation, grant number 22-25-00457.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2023.
REFERENCES
3. Anderson S, Bankier AT, Barrell BG, et al. Sequence and organization of the human mitochondrial genome. Nature 1981;290:457-65.
4. Barshad G, Marom S, Cohen T, Mishmar D. Mitochondrial DNA transcription and its regulation: an evolutionary perspective. Trends Genet 2018;34:682-92.
5. Sbisà E, Tanzariello F, Reyes A, Pesole G, Saccone C. Mammalian mitochondrial D-loop region structural analysis: identification of new conserved sequences and their functional and evolutionary implications. Gene 1997;205:125-40.
6. Berardo A, Musumeci O, Toscano A. Cardiological manifestations of mitochondrial respiratory chain disorders. Acta Myol 2011;30:9-15.
8. Fukui H, Moraes CT. Mechanisms of formation and accumulation of mitochondrial DNA deletions in aging neurons. Hum Mol Genet 2009;18:1028-36.
9. Nesbitt V, Pitceathly RDS, Turnbull DM, et al. The UK MRC mitochondrial disease patient cohort study: clinical phenotypes associated with the m.3243A>G mutation-implications for diagnosis and management. J Neurol Neurosurg Psychiatry 2013;84:936-8.
10. Lake NJ, Compton AG, Rahman S, Thorburn DR. Leigh syndrome: one disorder, more than 75 monogenic causes. Ann Neurol 2016;79:190-203.
11. Orekhov AN, Gerasimova EV, Sukhorukov VN, Poznyak AV, Nikiforov NG. Do mitochondrial DNA mutations play a key role in the chronification of sterile inflammation? Curr Pharm Des 2021;27:276-92.
12. Sobenin IA, Sazonova MA, Postnov AY, Salonen JT, Bobryshev YV, Orekhov AN. Association of mitochondrial genetic variation with carotid atherosclerosis. PLoS One 2013;8:e68070.
13. Gencer S, Evans BR, van der Vorst EPC, Döring Y, Weber C. Inflammatory chemokines in atherosclerosis. Cells 2021;10:226.
15. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev 2014;94:909-50.
16. van der Bliek AM, Sedensky MM, Morgan PG. Cell biology of the mitochondrion. Genetics 2017;207:843-71.
17. Brand MD, Orr AL, Perevoshchikova IV, Quinlan CL. The role of mitochondrial function and cellular bioenergetics in ageing and disease. Br J Dermatol 2013;169:1-8.
18. Lin DS, Huang YW, Ho CS, et al. Oxidative insults and mitochondrial DNA mutation promote enhanced autophagy and mitophagy compromising cell viability in pluripotent cell model of mitochondrial disease. Cells 2019;8:65.
19. Salnikova D, Orekhova V, Grechko A, et al. Mitochondrial dysfunction in vascular wall cells and its role in atherosclerosis. Int J Mol Sci 2021;22:8990.
20. Hayat MA. Autophagy: cancer, other pathologies, inflammation, immunity, infection, and aging. 2015. Available from: https://linkinghub.elsevier.com/retrieve/pii/C2013018837X [Last accessed on 5 Dec 2023].
21. Soucy-Faulkner A, Mukawera E, Fink K, et al. Requirement of NOX2 and reactive oxygen species for efficient RIG-I-mediated antiviral response through regulation of MAVS expression. PLoS Pathog 2010;6:e1000930.
22. Marchio P, Guerra-Ojeda S, Vila JM, Aldasoro M, Victor VM, Mauricio MD. Targeting early atherosclerosis: a focus on oxidative stress and inflammation. Oxid Med Cell Longev 2019;2019:8563845.
23. Sentman ML, Brännström T, Westerlund S, et al. Extracellular superoxide dismutase deficiency and atherosclerosis in mice. Arterioscler Thromb Vasc Biol 2001;21:1477-82.
24. Schrepfer E, Scorrano L. Mitofusins, from mitochondria to metabolism. Molecular Cell 2016;61:683-94.
25. Twig G, Elorza A, Molina AJA, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 2008;27:433-46.
26. Westermann B. Bioenergetic role of mitochondrial fusion and fission. Biochim Biophys Acta 2012;1817:1833-8.
27. Palikaras K, Lionaki E, Tavernarakis N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol 2018;20:1013-22.
28. Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol 2010;191:933-42.
30. Kane LA, Lazarou M, Fogel AI, et al. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol 2014;205:143-53.
31. Villa E, Marchetti S, Ricci JE. No Parkin zone: mitophagy without parkin. Trends Cell Biol 2018;28:882-95.
32. Liu L, Sakakibara K, Chen Q, Okamoto K. Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res 2014;24:787-95.
33. Sentelle RD, Senkal CE, Jiang W, et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat Chem Biol 2012;8:831-8.
34. Ohtake F, Tsuchiya H, Saeki Y, Tanaka K. K63 ubiquitylation triggers proteasomal degradation by seeding branched ubiquitin chains. Proc Natl Acad Sci USA 2018;115:E1401-8.
35. Hariharan N, Maejima Y, Nakae J, Paik J, Depinho RA, Sadoshima J. Deacetylation of FoxO by Sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ Res 2010;107:1470-82.
36. Kim J, Guan KL. Regulation of the autophagy initiating kinase ULK1 by nutrients: roles of mTORC1 and AMPK. Cell Cycle 2011;10:1337-8.
37. Nwadike C, Williamson LE, Gallagher LE, Guan JL, Chan EYW. AMPK inhibits ULK1-dependent autophagosome formation and lysosomal acidification via distinct mechanisms. Mol Cell Biol 2018;38:e00023-18.
38. Scarpulla RC. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. BBA Mol Cell Res 2011;1813:1269-78.
39. Gleyzer N, Vercauteren K, Scarpulla RC. Control of mitochondrial transcription specificity factors (TFB1M and TFB2M) by nuclear respiratory factors (NRF-1 and NRF-2) and PGC-1 family coactivators. Mol Cell Biol 2005;25:1354-66.
40. Kong X, Wang R, Xue Y, et al. Sirtuin 3, a new target of PGC-1alpha, plays an important role in the suppression of ROS and mitochondrial biogenesis. PLoS One 2010;5:e11707.
42. Quan Y, Xin Y, Tian G, Zhou J, Liu X. Mitochondrial ROS-modulated mtDNA: a potential target for cardiac aging. Oxid Med Cell Longev 2020;2020:9423593.
43. Nakahira K, Haspel JA, Rathinam VAK, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 2011;12:222-30.
44. Wei MC, Zong WX, Cheng EHY, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 2001;292:727-30.
45. Chen L, Peng J, Wang Y, et al. Fenofibrate-induced mitochondrial dysfunction and metabolic reprogramming reversal: the anti-tumor effects in gastric carcinoma cells mediated by the PPAR pathway. Am J Transl Res 2020;12:428-46.
46. Oslowski CM, Hara T, O’sullivan-murphy B, et al. Thioredoxin-interacting protein mediates ER stress-induced β cell death through initiation of the inflammasome. Cell Metab 2012;16:265-73.
47. Sakurai A, Nishimoto M, Himeno S, et al. Transcriptional regulation of thioredoxin reductase 1 expression by cadmium in vascular endothelial cells: role of NF-E2-related factor-2. J Cell Physiol 2005;203:529-37.
48. Swanson KV, Deng M, Ting JPY. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol 2019;19:477-89.
49. Wu N, Zheng B, Shaywitz A, et al. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol Cell 2013;49:1167-75.
50. Han X, Xu T, Fang Q, et al. Quercetin hinders microglial activation to alleviate neurotoxicity via the interplay between NLRP3 inflammasome and mitophagy. Redox Biol 2021;44:102010.
51. Holt IJ, Cooper JM, Morgan-Hughes JA, Harding AE. Deletions of muscle mitochondrial DNA. Lancet 1988;1:1462.
52. Ng YS, Bindoff LA, Gorman GS, et al. Mitochondrial disease in adults: recent advances and future promise. Lancet Neurol 2021;20:573-84.
54. Mancuso M, Petrozzi L, Filosto M, et al. MERRF syndrome without ragged-red fibers: the need for molecular diagnosis. Biochem Biophys Res Commun 2007;354:1058-60.
55. Zeviani M, Moraes CT, DiMauro S, et al. Deletions of mitochondrial DNA in Kearns-Sayre syndrome. Neurology 1988;38:1339-46.
56. Wallace DC. Mitochondrial defects in neurodegenerative disease. Ment Retard Dev Disabil Res Rev 2001;7:158-66.
57. Rossignol R, Faustin B, Rocher C, Malgat M, Mazat JP, Letellier T. Mitochondrial threshold effects. Biochem J 2003;370:751-62.
58. Schmiedel J, Jackson S, Schäfer J, Reichmann H. Mitochondrial cytopathies. J Neurol 2003;250:267-77.
59. Johns DR, Neufeld MJ, Park RD. An ND-6 mitochondrial DNA mutation associated with Leber hereditary optic neuropathy. Biochem Biophys Res Commun 1992;187:1551-7.
60. Marie SKN, Oba-shinjo SM, Marques-dias MJ, Rosemberg S, Kok F, Reed UC. The prevalence of mitochondrial DNA mutations in Leigh syndrome in a Brazilian series. Med Express 2014;1:239-42. Available from: https://www.scielo.br/j/medical/a/zRN3Vnp8Vj6DDKQBnSHyZkR/?lang=en [Last accessed on 7 Dec 2023]
61. de Vries DD, van Engelen BGM, Gabreëls FJM, Ruitenbeek W, van Oost BA. A second missense mutation in the mitochondrial ATPase 6 gene in Leigh’s syndrome. Ann Neurol 1993;34:410-2.
62. Leng Y, Liu Y, Fang X, et al. The mitochondrial DNA 10197 G >A mutation causes MELAS/leigh overlap syndrome presenting with acute auditory agnosia. Mitochondrial DNA 2015;26:208-12.
63. Shanske S, Coku J, Lu J, et al. The G13513A mutation in the ND5 gene of mitochondrial DNA as a common cause of MELAS or Leigh syndrome: evidence from 12 cases. Arch Neurol 2008;65:368-72.
64. Lorenzoni PJ, Scola RH, Kay CSK, Silvado CES, Werneck LC. When should MERRF (myoclonus epilepsy associated with ragged-red fibers) be the diagnosis? Arq Neuro-Psiquiatr 2014;72:803-11.
65. Wang YX, Le WD. Progress in diagnosing mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes. Chin Med J 2015;128:1820-5.
66. Gil Borlado MC, Moreno Lastres D, Gonzalez Hoyuela M, et al. Impact of the mitochondrial genetic background in complex III deficiency. PLoS One 2010;5:e12801.
67. Yan N, Cai S, Guo B, et al. A novel mitochondrial tRNA(Val) T1658C mutation identified in a CPEO family. Mol Vis 2010;16:1736-42.
68. Elson JL, Swalwell H, Blakely EL, McFarland R, Taylor RW, Turnbull DM. Pathogenic mitochondrial tRNA mutations-which mutations are inherited and why? Hum Mutat 2009;30:E984-92.
69. Zapico SC, Ubelaker DH. mtDNA mutations and their role in aging, diseases and forensic sciences. Aging Dis 2013;4:364-80.
70. Maassen JA, van den Ouweland JM, t Hart LM, Lemkes HH. Maternally inherited diabetes and deafness: a diabetic subtype associated with a mutation in mitochondrial DNA. Horm Metab Res 1997;29:50-5.
71. Chen FL, Liu Y, Song XY, et al. A novel mitochondrial DNA missense mutation at G3421A in a family with maternally inherited diabetes and deafness. Mutat Res 2006;602:26-33.
72. Zhelankin AV, Sazonova MA. [Association of the mutations in the human mitochondrial genome with chronic non-inflammatory diseases: type 2 diabetes, hypertension and different types of cardiomyopathy]. Patol Fiziol Eksp Ter 2012;3:123-8.
73. Wortmann SB, Champion MP, van den Heuvel L, et al. Mitochondrial DNA m.3242G>A mutation, an under diagnosed cause of hypertrophic cardiomyopathy and renal tubular dysfunction? Eur J Med Genet 2012;55:552-6.
74. Ma L, Wang H, Chen J, et al. Mitochondrial gene variation in type 2 diabetes mellitus: detection of a novel mutation associated with maternally inherited diabetes in a Chinese family. Chin Med J 2000;113:111-6.
75. Schaefer AM, Walker M, Turnbull DM, Taylor RW. Endocrine disorders in mitochondrial disease. Mol Cell Endocrinol 2013;379:2-11.
76. Dabravolski SA, Orekhova VA, Baig MS, et al. The role of mitochondrial mutations and chronic inflammation in diabetes. Int J Mol Sci 2021;22:6733.
77. Bender A, Krishnan KJ, Morris CM, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet 2006;38:515-7.
78. Kaukonen J, Juselius JK, Tiranti V, et al. Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science 2000;289:782-5.
79. Spelbrink JN, Li FY, Tiranti V, et al. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat Genet 2001;28:223-31.
80. Van Goethem G, Dermaut B, Löfgren A, Martin JJ, Van Broeckhoven C. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat Genet 2001;28:211-2.
81. Petros JA, Baumann AK, Ruiz-Pesini E, et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc Natl Acad Sci USA 2005;102:719-24.
82. Dabravolski SA, Bezsonov EE, Orekhov AN. The role of mitochondria dysfunction and hepatic senescence in NAFLD development and progression. Biomed Pharmacother 2021;142:112041.
83. Dabravolski SA, Bezsonov EE, Baig MS, Popkova TV, Orekhov AN. Mitochondrial lipid homeostasis at the crossroads of liver and heart diseases. Int J Mol Sci 2021;22:6949.
84. Ding Y, Leng J, Fan F, Xia B, Xu P. The role of mitochondrial DNA mutations in hearing loss. Biochem Genet 2013;51:588-602.
85. Malekmohammad K, Bezsonov EE, Rafieian-Kopaei M. Role of lipid accumulation and inflammation in atherosclerosis: focus on molecular and cellular mechanisms. Front Cardiovasc Med 2021;8:707529.
86. Bezsonov EE, Sobenin IA, Orekhov AN. Immunopathology of atherosclerosis and related diseases: focus on molecular biology. Int J Mol Sci 2021;22:4080.
87. Mushenkova NV, Bezsonov EE, Orekhova VA, Popkova TV, Starodubova AV, Orekhov AN. Recognition of oxidized lipids by macrophages and its role in atherosclerosis development. Biomedicines 2021;9:915.
88. Mezentsev A, Bezsonov E, Kashirskikh D, Baig MS, Eid AH, Orekhov A. Proatherogenic sialidases and desialylated lipoproteins: 35 years of research and current state from bench to bedside. Biomedicines 2021;9:600.
89. Bezsonov E, Borisov E, Vinokurov A, et al. Effects of native and modified low-density lipoproteins on mitophagy. Atherosclerosis 2023;375:98-100.
90. Tian F, Li J, Liu XW, Tong TJ, Zhang ZY. Age-dependent accumulation of mitochondrial DNA deletions in the aortic root of atherosclerosis-prone apolipoprotein E-knockout mice. Arch Gerontol Geriatr 2016;63:72-7.
91. Umbria M, Ramos A, Aluja MP, Santos C. The role of control region mitochondrial DNA mutations in cardiovascular disease: stroke and myocardial infarction. Sci Rep 2020;10:2766.
92. Mitrofanov KY, Zhelankin AV, Shiganova GM, et al. Analysis of mitochondrial DNA heteroplasmic mutations A1555G, C3256T, T3336C, С5178А, G12315A, G13513A, G14459A, G14846А and G15059A in CHD patients with the history of myocardial infarction. Exp Mol Pathol 2016;100:87-91.
93. Sawabe M, Tanaka M, Chida K, et al. Mitochondrial haplogroups A and M7a confer a genetic risk for coronary atherosclerosis in the Japanese elderly: an autopsy study of 1,536 patients. J Atheroscler Thromb 2011;18:166-75.
94. Kirichenko TV, Sobenin IA, Khasanova ZB, et al. Data on association of mitochondrial heteroplasmy and cardiovascular risk factors: comparison of samples from Russian and Mexican populations. Data Brief 2018;18:16-21.
95. Sazonova MA, Sinyov VV, Barinova VA, et al. Mosaicism of mitochondrial genetic variation in atherosclerotic lesions of the human aorta. Biomed Res Int 2015;2015:825468.
96. Ashar FN, Zhang Y, Longchamps RJ, et al. Association of mitochondrial DNA copy number with cardiovascular disease. JAMA Cardiol 2017;2:1247-55.
97. Liu LP, Cheng K, Ning MA, et al. Association between peripheral blood cells mitochondrial DNA content and severity of coronary heart disease. Atherosclerosis 2017;261:105-10.
98. Hu H, Lin Y, Xu X, Lin S, Chen X, Wang S. The alterations of mitochondrial DNA in coronary heart disease. Exp Mol Pathol 2020;114:104412.
99. Willcox JAL, Geiger JT, Morton SU, et al. Neither cardiac mitochondrial DNA variation nor copy number contribute to congenital heart disease risk. Am J Hum Genet 2022;109:961-6.
100. Poznyak AV, Bezsonov EE, Popkova TV, Starodubova AV, Orekhov AN. Immunity in atherosclerosis: focusing on T and B cells. Int J Mol Sci 2021;22:8379.
101. Vakhtangadze T, Singh Tak R, Singh U, Baig MS, Bezsonov E. Gender differences in atherosclerotic vascular disease: from lipids to clinical outcomes. Front Cardiovasc Med 2021;8:707889.
102. Kuro OM. Phosphate as a pathogen of arteriosclerosis and aging. J Atheroscler Thromb 2021;28:203-13.
103. Ellam TJ, Chico TJA. Phosphate: the new cholesterol? The role of the phosphate axis in non-uremic vascular disease. Atherosclerosis 2012;220:310-8.
104. McCarthy L, Downey M. The emerging landscape of eukaryotic polyphosphatases. FEBS Lett 2023;597:1447-61.
105. Kalebina TS, Egorov SN, Arbatskii NP, Bezsonov EE, Gorkovskii AA, Kulaev IS. The role of high-molecular-weight polyphosphates in activation of glucan transferase Bgl2p from Saccharomyces cerevisiae cell wall. Dokl Biochem Biophys 2008;420:142-5.
106. Morrissey JH, Choi SH, Smith SA. Polyphosphate: an ancient molecule that links platelets, coagulation, and inflammation. Blood 2012;119:5972-9.
107. Peng W, Cai G, Xia Y, et al. Mitochondrial dysfunction in atherosclerosis. DNA Cell Biol 2019;38:597-606.
108. Durham AL, Speer MY, Scatena M, Giachelli CM, Shanahan CM. Role of smooth muscle cells in vascular calcification: implications in atherosclerosis and arterial stiffness. Cardiovasc Res 2018;114:590-600.
109. Sazonova MA, Sinyov VV, Barinova VA, et al. Association of mitochondrial mutations with the age of patients having atherosclerotic lesions. Exp Mol Pathol 2015;99:717-9.
110. Calabrese C, Pyle A, Griffin H, et al. Heteroplasmic mitochondrial DNA variants in cardiovascular diseases. PLoS Genet 2022;18:e1010068.
111. Hefti E, Blanco JG. Mitochondrial DNA heteroplasmy in cardiac tissue from individuals with and without coronary artery disease. Mitochondrial DNA A DNA Mapp Seq Anal 2018;29:587-93.
112. Röcken C, Tautenhahn J, Bühling F, et al. Prevalence and pathology of amyloid in atherosclerotic arteries. Arterioscler Thromb Vasc Biol 2006;26:676-7.
113. Wickner RB, Bezsonov EE, Son M, Ducatez M, DeWilde M, Edskes HK. Anti-prion systems in yeast and inositol polyphosphates. Biochemistry 2018;57:1285-92.
114. Bezsonov EE, Groenning M, Galzitskaya OV, et al. Amyloidogenic peptides of yeast cell wall glucantransferase Bgl2p as a model for the investigation of its pH-dependent fibril formation. Prion 2013;7:175-84.
115. Wickner RB, Edskes HK, Bateman DA, et al. Yeast prions: proteins templating conformation and an anti-prion system. PLoS Pathog 2015;11:e1004584.
117. Wu NN, Zhang Y, Ren J. Mitophagy, mitochondrial dynamics, and homeostasis in cardiovascular aging. Oxid Med Cell Longev 2019;2019:1-15.
118. Wang JC, Bennett M. Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ Res 2012;111:245-59.
119. Martinet W, Knaapen MWM, De Meyer GRY, Herman AG, Kockx MM. Elevated levels of oxidative DNA damage and DNA repair enzymes in human atherosclerotic plaques. Circulation 2002;106:927-32.
120. Breitschopf K, Zeiher AM, Dimmeler S. Pro-atherogenic factors induce telomerase inactivation in endothelial cells through an Akt-dependent mechanism. FEBS Lett 2001;493:21-5.
121. Sahin E, Colla S, Liesa M, et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 2011;470:359-65.
122. Gusev E, Sarapultsev A. Atherosclerosis and inflammation: insights from the theory of general pathological processes. Int J Mol Sci 2023;24:7910.
123. Koushki K, Keshavarz Shahbaz S, Keshavarz M, Bezsonov EE, Sathyapalan T, Sahebkar A. Gold nanoparticles: multifaceted roles in the management of autoimmune disorders. Biomolecules 2021;11:1289.
124. Orekhov AN, Poznyak AV, Sobenin IA, Nikifirov NN, Ivanova EA. Mitochondrion as a selective target for the treatment of atherosclerosis: role of mitochondrial DNA mutations and defective mitophagy in the pathogenesis of atherosclerosis and chronic inflammation. Curr Neuropharmacol 2020;18:1064-75.
125. Sobenin IA, Sazonova MA, Postnov AY, Bobryshev YV, Orekhov AN. Mitochondrial mutations are associated with atherosclerotic lesions in the human aorta. Clin Dev Immunol 2012;2012:832464.
126. Zhunina OA, Yabbarov NG, Grechko AV, et al. The role of mitochondrial dysfunction in vascular disease, tumorigenesis, and diabetes. Front Mol Biosci 2021;8:671908.
127. Volobueva A, Grechko A, Yet SF, Sobenin I, Orekhov A. Changes in mitochondrial genome associated with predisposition to atherosclerosis and related disease. Biomolecules 2019;9:377.
128. Yu E, Calvert PA, Mercer JR, et al. Mitochondrial DNA damage can promote atherosclerosis independently of reactive oxygen species through effects on smooth muscle cells and monocytes and correlates with higher-risk plaques in humans. Circulation 2013;128:702-12.
129. Sobenin IA, Zhelankin AV, Khasanova ZB, et al. Heteroplasmic variants of mitochondrial DNA in Atherosclerotic lesions of human aortic intima. Biomolecules 2019;9:455.
130. Freigang S, Hörkkö S, Miller E, Witztum JL, Palinski W. Immunization of LDL receptor-deficient mice with homologous malondialdehyde-modified and native LDL reduces progression of atherosclerosis by mechanisms other than induction of high titers of antibodies to oxidative neoepitopes. Arterioscler Thromb Vasc Biol 1998;18:1972-82.
131. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011;469:221-5.
132. Tall AR, Bornfeldt KE. Inflammasomes and atherosclerosis: a mixed picture. Circ Res 2023;132:1505-20.
133. Bulté D, Rigamonti C, Romano A, Mortellaro A. Inflammasomes: mechanisms of action and involvement in human diseases. Cells 2023;12:1766.
134. Shahbaz S, Koushki K, Ayati SH, Bland AR, Bezsonov EE, Sahebkar A. Inflammasomes and colorectal cancer. Cells 2021;10:2172.
135. Coppi L, Ligorio S, Mitro N, Caruso D, De Fabiani E, Crestani M. PGC1s and beyond: disentangling the complex regulation of mitochondrial and cellular metabolism. Int J Mol Sci 2021;22:6913.
136. Sazonova MA, Sinyov VV, Ryzhkova AI, et al. Creation of cybrid cultures containing mtDNA mutations m.12315G>A and
137. Wang X, Xu M, Frank JA, Ke ZJ, Luo J. Thiamine deficiency induces endoplasmic reticulum stress and oxidative stress in human neurons derived from induced pluripotent stem cells. Toxicol Appl Pharmacol 2017;320:26-31.
138. Colombo B, Saraceno L, Comi G. Riboflavin and migraine: the bridge over troubled mitochondria. Neurol Sci 2014;35 Suppl 1:141-4.
139. Song SB, Jang SY, Kang HT, et al. Modulation of mitochondrial membrane potential and ROS generation by nicotinamide in a manner independent of SIRT1 and mitophagy. Mol Cells 2017;40:503-14.
140. Kang HT, Hwang ES. Nicotinamide enhances mitochondria quality through autophagy activation in human cells. Aging Cell 2009;8:426-38.
141. Gao X, Lee K, Reid MA, et al. Serine availability influences mitochondrial dynamics and function through lipid metabolism. Cell Rep 2018;22:3507-20.
142. Onodera R, Motoyama K, Tanaka N, et al. Involvement of autophagy in antitumor activity of folate-appended methyl-β-cyclodextrin. Sci Rep 2014;4:4417.
144. McCarty MF, Assanga SBI. Ferulic acid may target MyD88-mediated pro-inflammatory signaling - implications for the health protection afforded by whole grains, anthocyanins, and coffee. Med Hypotheses 2018;118:114-20.
145. Bumrungpert A, Lilitchan S, Tuntipopipat S, Tirawanchai N, Komindr S. Ferulic acid supplementation improves lipid profiles, oxidative stress, and inflammatory status in hyperlipidemic subjects: a randomized, double-blind, placebo-controlled clinical trial. Nutrients 2018;10:713.
146. Sgarbossa A, Giacomazza D, di Carlo M. Ferulic acid: a hope for Alzheimer’s disease therapy from plants. Nutrients 2015;7:5764-82.
147. Neto-Neves EM, da Silva Maia Bezerra Filho C, Dejani NN, de Sousa DP. Ferulic acid and cardiovascular health: therapeutic and preventive potential. Mini Rev Med Chem 2021;21:1625-37.
148. Nankar R, Prabhakar PK, Doble M. Hybrid drug combination: combination of ferulic acid and metformin as anti-diabetic therapy. Phytomedicine 2017;37:10-3.
149. Sultana R. Ferulic acid ethyl ester as a potential therapy in neurodegenerative disorders. Biochim Biophys Acta 2012;1822:748-52.
150. Gupta A, Singh AK, Loka M, Pandey AK, Bishayee A. Chapter eight - ferulic acid-mediated modulation of apoptotic signaling pathways in cancer. In: Advances in protein chemistry and structural biology. 2021. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1876162320300912 [Last accessed on 5 Dec 2023].
151. Turner N, Li JY, Gosby A, et al. Berberine and its more biologically available derivative, dihydroberberine, inhibit mitochondrial respiratory complex I: a mechanism for the action of berberine to activate AMP-activated protein kinase and improve insulin action. Diabetes 2008;57:1414-8.
152. Cicero AFG, Baggioni A. Berberine and its role in chronic disease. In: Gupta SC, Prasad S, Aggarwal BB, editors. Anti-inflammatory nutraceuticals and chronic diseases. Cham: Springer International Publishing; 2016. pp. 27-45. Available from: http://link.springer.com/10.1007/978-3-319-41334-1_2 [Last accessed on 5 Dec 2023].
153. Kyung S, Lim JW, Kim H. α-lipoic acid inhibits IL-8 expression by activating Nrf2 signaling in helicobacter pylori-infected gastric epithelial cells. Nutrients 2019;11:2524.
154. Papanas N, Ziegler D. Efficacy of α-lipoic acid in diabetic neuropathy. Expert Opin Pharmacother 2014;15:2721-31.
155. Novotny L, Rauko P, Cojocel C. alpha-Lipoic acid: the potential for use in cancer therapy. Neoplasma 2008;55:81-6.
156. Barut EN, Engin S, Saygın İ, Kaya-Yasar Y, Arici S, Sezen SF. Alpha-lipoic acid: a promising adjuvant for nonsteroidal anti-inflammatory drugs therapy with improved efficacy and gastroprotection. Drug Dev Res 2021;82:844-51.
157. Tóth F, Cseh EK, Vécsei L. Natural molecules and neuroprotection: kynurenic acid, pantethine and α-lipoic acid. Int J Mol Sci 2021;22:403.
158. Krestinina O, Baburina Y, Krestinin R, Odinokova I, Fadeeva I, Sotnikova L. Astaxanthin prevents mitochondrial impairment induced by isoproterenol in isolated rat heart mitochondria. Antioxidants 2020;9:262.
159. Li J, Guo C, Wu J. Astaxanthin in liver health and disease: a potential therapeutic agent. Drug Des Devel Ther 2020;14:2275-85.
160. Krestinina O, Baburina Y, Krestinin R. Mitochondrion as a target of astaxanthin therapy in heart failure. Int J Mol Sci 2021;22:7964.
161. Giannaccare G, Pellegrini M, Senni C, Bernabei F, Scorcia V, Cicero AFG. Clinical applications of astaxanthin in the treatment of ocular diseases: emerging insights. Mar Drugs 2020;18:239.
162. McCarty MF, O’Keefe JH, DiNicolantonio JJ. Glucosamine for the treatment of osteoarthritis: the time has come for higher-dose trials. J Diet Suppl 2019;16:179-92.
163. Feng KM, Chien WC, Chen JT, et al. The impact of glucosamine on age-related macular degeneration in patients: a nationwide, population-based cohort study. PLoS ONE 2021;16:e0251925.
164. Yu L, Sun Y, Cheng L, et al. Melatonin receptor-mediated protection against myocardial ischemia/reperfusion injury: role of SIRT1. J Pineal Res 2014;57:228-38.
165. Fang J, Yan Y, Teng X, et al. Melatonin prevents senescence of canine adipose-derived mesenchymal stem cells through activating NRF2 and inhibiting ER stress. Aging 2018;10:2954-72.
166. Chitimus DM, Popescu MR, Voiculescu SE, et al. Melatonin’s impact on antioxidative and anti-inflammatory reprogramming in homeostasis and disease. Biomolecules 2020;10:1211.
167. Talib WH, Alsayed AR, Abuawad A, Daoud S, Mahmod AI. Melatonin in cancer treatment: current knowledge and future opportunities. Molecules 2021;26:2506.
168. Lalanne S, Fougerou-Leurent C, Anderson GM, et al. Melatonin: from pharmacokinetics to clinical use in autism spectrum disorder. Int J Mol Sci 2021;22:1490.
169. Chokchaiwong S, Kuo YT, Lin SH, et al. Coenzyme Q10 serves to couple mitochondrial oxidative phosphorylation and fatty acid β-oxidation, and attenuates NLRP3 inflammasome activation. Free Radic Res 2018;52:1445-55.
170. Wang D, Yan X, Xia M, et al. Coenzyme Q10 promotes macrophage cholesterol efflux by regulation of the activator protein-1/
171. Mercer JR, Yu E, Figg N, et al. The mitochondria-targeted antioxidant MitoQ decreases features of the metabolic syndrome in
172. Xiao L, Xu X, Zhang F, et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol 2017;11:297-311.
173. Mahmoudi K, Garvey KL, Bouras A, et al. 5-aminolevulinic acid photodynamic therapy for the treatment of high-grade gliomas. J Neurooncol 2019;141:595-607.
174. Pernicova I, Korbonits M. Metformin-mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol 2014;10:143-56.
175. Hattori Y, Suzuki K, Hattori S, Kasai K. Metformin inhibits cytokine-induced nuclear factor kappaB activation via AMP-activated protein kinase activation in vascular endothelial cells. Hypertension 2006;47:1183-8.
176. LaMoia TE, Shulman GI. Cellular and molecular mechanisms of metformin action. Endocr Rev 2021;42:77-96.
177. La Fountaine MF, Cirnigliaro CM, Hobson JC, et al. Fenofibrate therapy to lower serum triglyceride concentrations in persons with spinal cord injury: a preliminary analysis of its safety profile. J Spinal Cord Med 2020;43:704-9.
178. Pettersen JC, Pruimboom-Brees I, Francone OL, et al. The PPARα agonists fenofibrate and CP-778875 cause increased β-oxidation, leading to oxidative injury in skeletal and cardiac muscle in the rat. Toxicol Pathol 2012;40:435-47.
179. Duncan JG, Fong JL, Medeiros DM, Finck BN, Kelly DP. Insulin-resistant heart exhibits a mitochondrial biogenic response driven by the peroxisome proliferator-activated receptor-α/PGC-1α gene regulatory pathway. Circulation 2007;115:909-17.
180. El-hattab AW, Craigen WJ, Scaglia F. Mitochondrial DNA maintenance defects. BBA Mol Basis Dis 2017;1863:1539-55.
181. De Marco V, Noronha KSM, Casado TC, et al. Therapy of canine hyperlipidemia with bezafibrate. J Vet Intern Med 2017;31:717-22.
182. Bonnefont JP, Bastin J, Laforêt P, et al. Long-term follow-up of bezafibrate treatment in patients with the myopathic form of carnitine palmitoyltransferase 2 deficiency. Clin Pharmacol Ther 2010;88:101-8.
183. Ren B, Kwah MXY, Liu C, et al. Resveratrol for cancer therapy: challenges and future perspectives. Cancer Lett 2021;515:63-72.
184. Huang DD, Shi G, Jiang Y, Yao C, Zhu C. A review on the potential of Resveratrol in prevention and therapy of diabetes and diabetic complications. Biomed Pharmacother 2020;125:109767.
185. Breuss JM, Atanasov AG, Uhrin P. Resveratrol and its effects on the vascular system. Int J Mol Sci 2019;20:1523.
186. Komen JC, Thorburn DR. Turn up the power - pharmacological activation of mitochondrial biogenesis in mouse models. Br J Pharmacol 2014;171:1818-36.
187. Fu CY, Zhong CR, Yang YT, et al. Sirt1 activator SRT2104 protects against oxygen-glucose deprivation/reoxygenation-induced injury via regulating microglia polarization by modulating Sirt1/NF-κB pathway. Brain Res 2021;1753:147236.
188. Libri V, Brown AP, Gambarota G, et al. A pilot randomized, placebo controlled, double blind phase I trial of the novel SIRT1 activator SRT2104 in elderly volunteers. PLoS One 2012;7:e51395.
189. Madhavi YV, Gaikwad N, Yerra VG, Kalvala AK, Nanduri S, Kumar A. Targeting AMPK in diabetes and diabetic complications: energy homeostasis, autophagy and mitochondrial health. Curr Med Chem 2019;26:5207-29.
190. Dombi E, Mortiboys H, Poulton J. Modulating mitophagy in mitochondrial disease. Curr Med Chem 2019;25:5597-612.
191. Golubitzky A, Dan P, Weissman S, Link G, Wikstrom JD, Saada A. Screening for active small molecules in mitochondrial complex I deficient patient’s fibroblasts, reveals AICAR as the most beneficial compound. PLoS One 2011;6:e26883.
192. Moreno-Ulloa A, Cid A, Rubio-Gayosso I, Ceballos G, Villarreal F, Ramirez-Sanchez I. Effects of (-)-epicatechin and derivatives on nitric oxide mediated induction of mitochondrial proteins. Bioorg Med Chem Lett 2013;23:4441-6.
193. Keller A, Hull SE, Elajaili H, et al. (-)-Epicatechin modulates mitochondrial redox in vascular cell models of oxidative stress. Oxid Med Cell Longev 2020;2020:6392629.
194. Chen Q, Wang T, Li J, et al. Effects of natural products on fructose-induced nonalcoholic fatty liver disease (NAFLD). Nutrients 2017;9:96.
195. Chun JH, Henckel MM, Knaub LA, et al. (-)-Epicatechin improves vasoreactivity and mitochondrial respiration in thermoneutral-housed wistar rat vasculature. Nutrients 2022;14:1097.
196. Tanabe K, Tamura Y, Lanaspa MA, et al. Epicatechin limits renal injury by mitochondrial protection in cisplatin nephropathy. Am J Physiol Renal Physiol 2012;303:F1264-74.
197. Chen Q, Li Q, Liang Y, et al. Natural exosome-like nanovesicles from edible tea flowers suppress metastatic breast cancer via ROS generation and microbiota modulation. Acta Pharm Sin B 2022;12:907-23.
198. Rai PK, Russell OM, Lightowlers RN, Turnbull DM. Potential compounds for the treatment of mitochondrial disease. Br Med Bull 2015;116:5-18.
199. Sun X, Xie Z, Hu B, et al. The Nrf2 activator RTA-408 attenuates osteoclastogenesis by inhibiting STING dependent NF-κb signaling. Redox Biol 2020;28:101309.
200. Zhang L, Zhou Q, Zhou CL. RTA-408 protects against propofol-induced cognitive impairment in neonatal mice via the activation of Nrf2 and the inhibition of NF-κB p65 nuclear translocation. Brain Behav 2021;11:e01918.
201. Rabbani PS, Ellison T, Waqas B, et al. Targeted Nrf2 activation therapy with RTA 408 enhances regenerative capacity of diabetic wounds. Diabetes Res Clin Pract 2018;139:11-23.
203. Oesterle A, Laufs U, Liao JK. Pleiotropic effects of statins on the cardiovascular system. Circ Res 2017;120:229-43.
204. Babelova A, Sedding DG, Brandes RP. Anti-atherosclerotic mechanisms of statin therapy. Curr Opin Pharmacol 2013;13:260-4.
205. Kong F, Ye B, Lin L, Cai X, Huang W, Huang Z. Atorvastatin suppresses NLRP3 inflammasome activation via TLR4/MyD88/
206. Apostolopoulou M, Corsini A, Roden M. The role of mitochondria in statin-induced myopathy. Eur J Clin Invest 2015;45:745-54.
207. Yang S, Wu GL, Li N, et al. A mitochondria-targeted molecular phototheranostic platform for NIR-II imaging-guided synergistic photothermal/photodynamic/immune therapy. J Nanobiotechnol 2022;20:475.
208. Wormald RPL, Evans JR, Smeeth LL, Henshaw KS. Photodynamic therapy for neovascular age-related macular degeneration. Chichester, UK: John Wiley & Sons, Ltd; 2003. p. CD002030. Available from: https://doi.wiley.com/10.1002/14651858.CD002030 [Last accessed on 5 Dec 2023].
209. Wen X, Li Y, Hamblin MR. Photodynamic therapy in dermatology beyond non-melanoma cancer: an update. Photodiagnosis Photodyn Ther 2017;19:140-52.
210. Zhang Y, Zhou Q, Bu Y, et al. Real-time imaging mitochondrial viscosity dynamic during mitophagy mediated by photodynamic therapy. Anal Chim Acta 2021;1178:338847.
211. Agostinis P, Vantieghem A, Merlevede W, de Witte PAM. Hypericin in cancer treatment: more light on the way. Int J Biochem Cell Biol 2002;34:221-41.
212. Galanou MC, Theodossiou TA, Tsiourvas D, Sideratou Z, Paleos CM. Interactive transport, subcellular relocation and enhanced phototoxicity of hypericin encapsulated in guanidinylated liposomes via molecular recognition. Photochem Photobiol 2008;84:1073-83.
213. Dong X, Zeng Y, Zhang Z, et al. Hypericin-mediated photodynamic therapy for the treatment of cancer: a review. J Pharm Pharmacol 2021;73:425-36.
214. Courrol LC, de Oliveira Silva FR, Masilamani V. SARS-CoV-2, hemoglobin and protoporphyrin IX: interactions and perspectives. Photodiagnosis Photodyn Ther 2021;34:102324.
215. Gammage PA, Viscomi C, Simard ML, et al. Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo. Nat Med 2018;24:1691-5.
216. El-Hattab AW, Zarante AM, Almannai M, Scaglia F. Therapies for mitochondrial diseases and current clinical trials. Mol Genet Metab 2017;122:1-9.
217. Wang J, El-Hattab AW, Wong LJC. TK2-related mitochondrial DNA maintenance defect, myopathic form. Seattle: University of Washington; 1993. Available from: http://www.ncbi.nlm.nih.gov/books/NBK114628/ [Last accessed on 5 Dec 2023].
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright















Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].