

Thick electrode design for lithium-ion batteries from an ion-electron coupled transport perspective: from independent regulation to cooperative design
 0
0 Abstract
Thick electrodes can boost energy density and lower costs in lithium-ion batteries without altering chemistries, but they impede ion and electron transport, causing kinetic degradation. This work reviews multiscale kinetic optimization strategies and clarifies the fundamental limitations of independently enhancing ionic or electronic transport. By introducing the coupled ion-electron transfer theory, it demonstrates that reactions are governed by the spatiotemporal coordination of ion and electron transport, rather than either pathway alone. From a macro-flux perspective, transport mismatch is amplified in thick electrodes, resulting in spatially shifting reaction-inactive regions across the thickness. A reformulated Damköhler number incorporating both transport pathways defines a hybrid ion-electron control zone for uniform reactions. Based on this framework, dual-continuous, spatially decoupled networks and spatially coupled mixed ion-electron conductors are proposed as rational design principles. This work shifts thick-electrode design from empirical structural tuning toward mechanism-based system engineering.
Keywords
INTRODUCTION
To address the stagnation of energy density improvement in conventional lithium-ion batteries, thick electrode design has emerged as a promising device-level strategy[1-3]. By increasing electrode thickness, higher gravimetric and volumetric energy densities can be achieved, accompanied by simplified stacking configurations and reduced manufacturing costs[4-7]. Nevertheless, the transition from thin to thick electrodes fundamentally alters the internal transport environment, introducing severe kinetic penalties. As thickness increases, ion and electron transport pathways become substantially elongated and more tortuous due to the random assembly of multicomponent phases[8,9]. The resulting heterogeneous pore networks and fragile conductive contacts lead to pronounced concentration polarization, internal resistance, and reaction heterogeneity[10]. Under such conditions, electrochemical activity is often confined near the separator or current collector. Consequently, transport kinetics, rather than intrinsic material properties, become the primary bottleneck limiting the practical energy and power performance of thick electrodes.
Among the two charge carriers, sluggish ion transport is widely regarded as the dominant limitation in thick electrodes. To mitigate this issue, extensive efforts have been devoted to engineering fast ion transport pathways across multiple length scales[11,12]. At the particle level, reducing solid-state diffusion lengths through particle downsizing or constructing hierarchical porous architectures has proven effective in enhancing electrolyte accessibility[13]. At the mesoscale, directional pore engineering strategies enable vertically aligned channels that significantly reduce tortuosity and facilitate rapid ion transport[14]. At the electrode scale, gradient architectures in porosity, particle size, or composition are employed to spatially match ionic flux with local reaction demand, alleviating ion depletion and homogenizing reaction distributions[15]. Collectively, these strategies demonstrate that rational structural design can partially compensate for the intrinsic transport limitations imposed by increased thickness.
In parallel, efficient electron transport is equally indispensable, yet it is often more susceptible to mechanical deformation and interfacial degradation during cycling[16]. Conventional electronic networks formed by zero-dimensional conductive additives rely on fragile point-to-point contacts. To address this challenge, conductive network design has evolved toward higher-dimensional and hierarchical architectures. One-dimensional carbon nanotubes and two-dimensional graphene sheets establish long-range conductive bridges and low-resistance interfacial contacts[17]. Synergistic combinations of multidimensional conductive agents further ensure electronic connectivity across particle, mesoscale, and electrode-scale domains. For ultrathick electrodes, three-dimensional porous current collectors and integrated conductive scaffolds provide continuous electronic backbones and mechanical reinforcement, transforming inefficient interparticle hopping into direct backbone conduction[18]. Additional interfacial strategies, including surface conductive coatings and controlled dispersion of conductive additives, further reduce charge-transfer resistance and stabilize electronic pathways[19].
Despite substantial progress, most existing strategies still focus on independently accelerating either ionic or electronic transport. However, electrode performance is governed by the synchronous supply of ions and electrons to electrochemical reaction interfaces. Mismatch between ionic and electronic fluxes inevitably leads to localized polarization, reaction heterogeneity, and premature capacity loss, even when one transport pathway is highly optimized. This limitation underscores the necessity of moving beyond isolated pathway engineering toward a coupled ion-electron transport perspective.
ION-ELECTRON COOPERATIVE TRANSPORT MECHANISMS AND PRINCIPLES
Under large overpotentials, reaction currents no longer follow the exponential dependence predicted by classical kinetics. Instead, they exhibit saturation behavior, indicating that interfacial reaction kinetics become rate-limiting even when macroscopic transport pathways appear unobstructed[20]. The conventional Butler-Volmer framework, which treats ionic and electronic transport as independent sequential processes, fails to capture this phenomenon. Bazant et al. pointed out that Butler-Volmer inadequately describes high-current behavior in embedded electrode materials because they neglect ion-electron interactions within the transition state of the reaction[21]. To resolve this discrepancy, the coupled ion-electron transfer (CIET) theory provides a revised microscopic description of interfacial reactions. CIET emphasizes that Li+ intercalation is an intrinsically cooperative process requiring the simultaneous insertion of ions and electrons. Rather than proceeding along a single reaction coordinate, the interfacial reaction follows a minimum-free-energy pathway on a two-dimensional energy landscape defined by ion site availability and electronic reorganization [Figure 1A]. Consequently, reaction kinetics are constrained by the slower of the two processes.
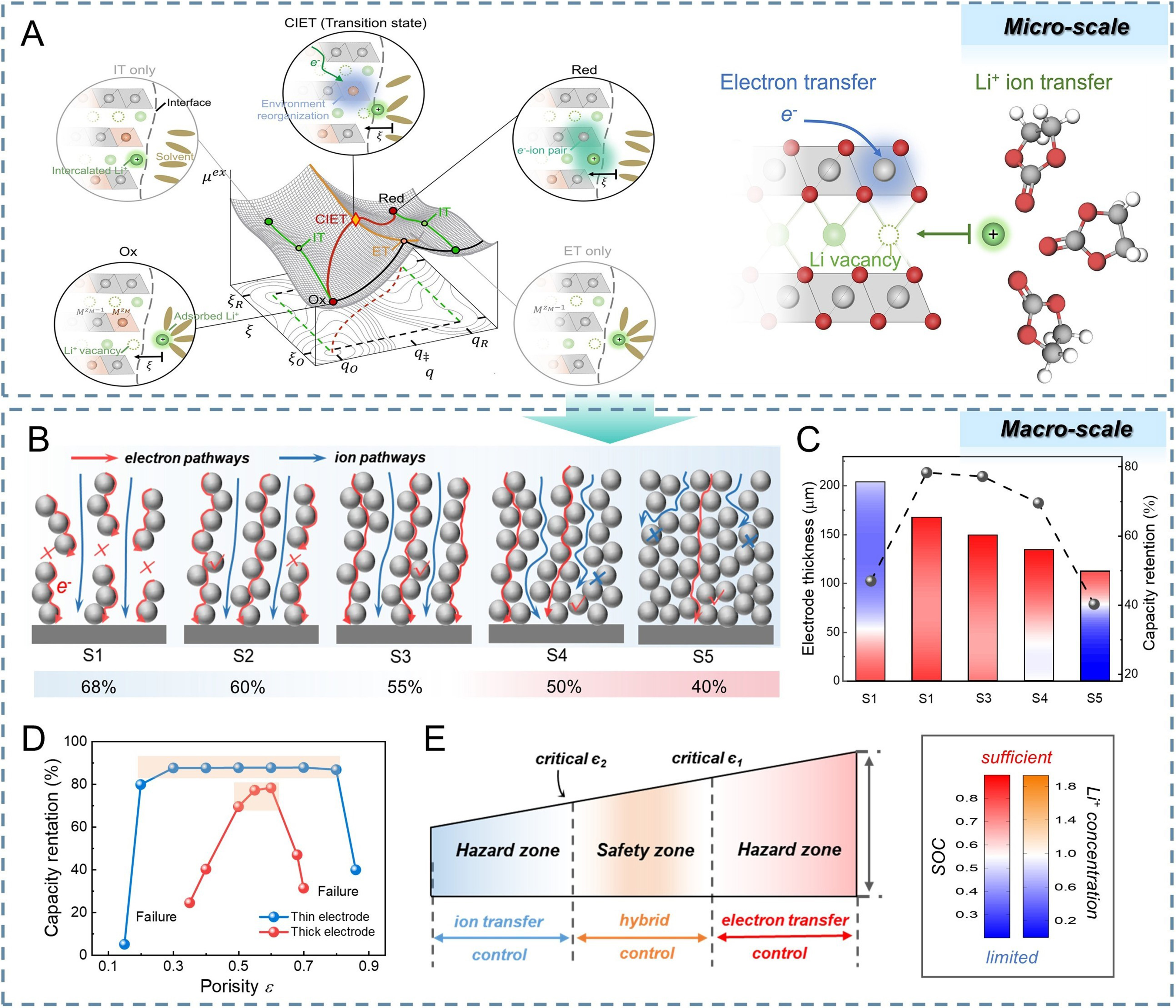
Figure 1. Mechanism of ion-electron coupled transport at different scales. (A) CIET theory at the active material interface on the microscopic scale. This figure is quoted with permission from Ref.[21], Copyright © 2025 American Association for the Advancement of Science; (B) Regulation of ion and electron transport capabilities via a calendering strategy; (C) Heterogeneous lithiumation distribution in electrodes with varying porosities; (D) Capacity retention of thin and thick electrodes at different porosities; (E) The hybrid ion-electron control mechanism ensuring that the electrode remains within the capacity safety zone. (B-E) are quoted with permission from Ref.[22], Copyright © 2024 Wiley. CIET: Coupled ion-electron transfer; IT: interfacial transfer; ET: electron transfer; SOC: state of charge.
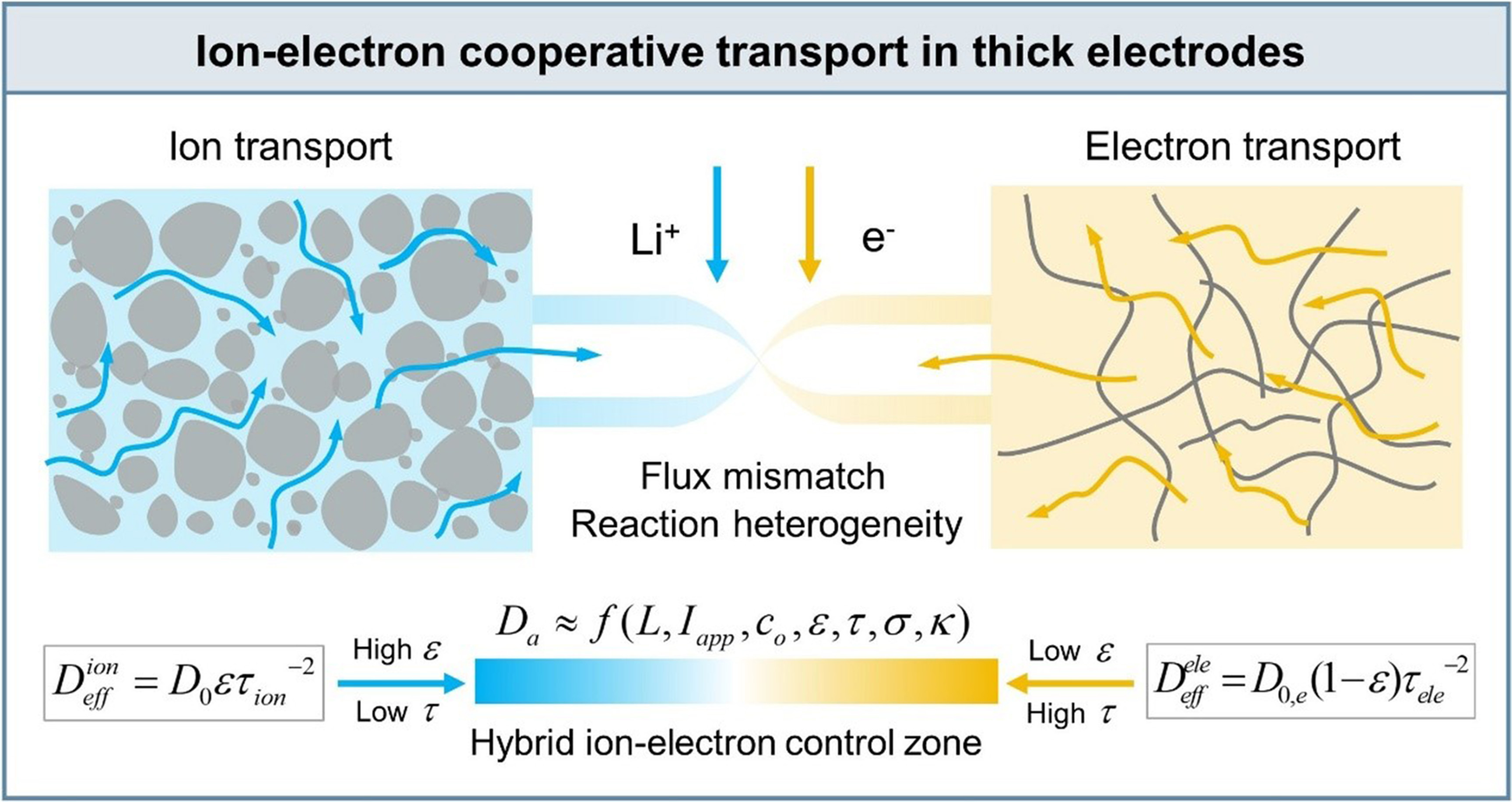
This microscopic coupling inevitably manifests at the macroscopic scale as a matching requirement between ionic and electronic transport fluxes. Fu et al. demonstrated that unbalanced transport properties lead to spatial response heterogeneity [Figure 1B][22]. When porosity is excessively high, ionic resistance is minimized but electronic percolation is weakened, rendering regions near the separator electrochemically inactive. Conversely, overly dense electrodes enhance electronic conduction but impede ion transport [Figure 1C and D]. Uniform reaction distribution is achieved only when ionic and electronic transport capabilities are balanced within the hybrid control zone. Importantly, this balance becomes increasingly sensitive to structural parameters as thickness increases [Figure 1E], indicating that electrode performance is governed not by the independent maximization of either transport pathway, but by their dynamic balance.
To translate ion-electron cooperative transport from qualitative insight into quantitative design, a mechanistic linkage between microstructure and macroscopic kinetics is required. The Damköhler number (Da), defined as the ratio of characteristic diffusion time to characteristic reaction time, is commonly employed to assess transport limitations in thick electrodes. However, conventional models typically assume electronic transport to be non-limiting and thus fail to capture coupled transport effects. Within the CIET framework, the total transport resistance must instead account for both ionic and electronic contributions. Building on this concept, modified Da have been developed to describe reaction current distributions as a function of effective ionic and electronic conductivities[23]. When the depletion time associated with the initial electrolyte concentration (c0) is used as the characteristic reaction timescale, a coupled Da can be derived that explicitly incorporates electrode thickness (L), applied current density (Iapp), and effective transport coefficients (Deff) for both charge carriers[24].
where τcouple denotes the characteristic diffusion time after considering ion-electron coupling, τrxn denotes the characteristic reaction time after considering ion-electron coupling, and F indicates Faraday constant.
To relate this coupled kinetic criterion to electrode microstructure, porous media theory is employed. The effective ionic transport coefficient scales with porosity (ε) and tortuosity (τ). Combining these relations yields a direct structure-performance correlation that quantitatively captures hybrid control zone.
At low porosity, severe pore blockage leads to ion-transport divergence and an ion-limited regime (Da≫1). At high porosity, disruption of the electronic network results in an electron-limited regime, again characterized by Da≫1. In contrast, the hybrid control zone is achieved when ionic and electronic transport timescales are dynamically matched, corresponding to Da ≈ 1. By converting the ion-electron cooperative mechanism into explicit constraints on porosity and tortuosity, this framework establishes a clear theoretical boundary for thick-electrode optimization.
STRATEGIES FOR CONSTRUCTING ION-ELECTRON COOPERATIVE TRANSPORT
Building on microscopic CIET theory and the macroscopic ion-electron hybrid control mechanism, the design of high-performance thick electrodes should prioritize precise matching between ionic and electronic transport networks, rather than isolated enhancement of individual pathways. Table 1 summarizes the respective advantages and disadvantages of the three enhancement strategies: ion transport, electron transport, and coupled transport. The strategies focusing solely on ion transport or electron transport inevitably introduce counteracting limitations: increasing porosity and reducing tortuosity alleviate concentration polarization but often disrupt electronic connectivity, whereas densifying electrodes and increasing conductive additives improve electronic conduction at the cost of ion blockage. In conventional thick electrodes, random packing of multiphase components leads to pore collapse, conductive network degradation, and increased tortuosity during cycling. An ideal thick-electrode architecture should therefore integrate a continuous, low-tortuosity pore network for ion transport with a robust, low-resistance solid conductive skeleton for electron transport. This design ensures that all active sites are simultaneously well-wetted by the electrolyte and efficiently connected to the external circuit [Figure 2A].
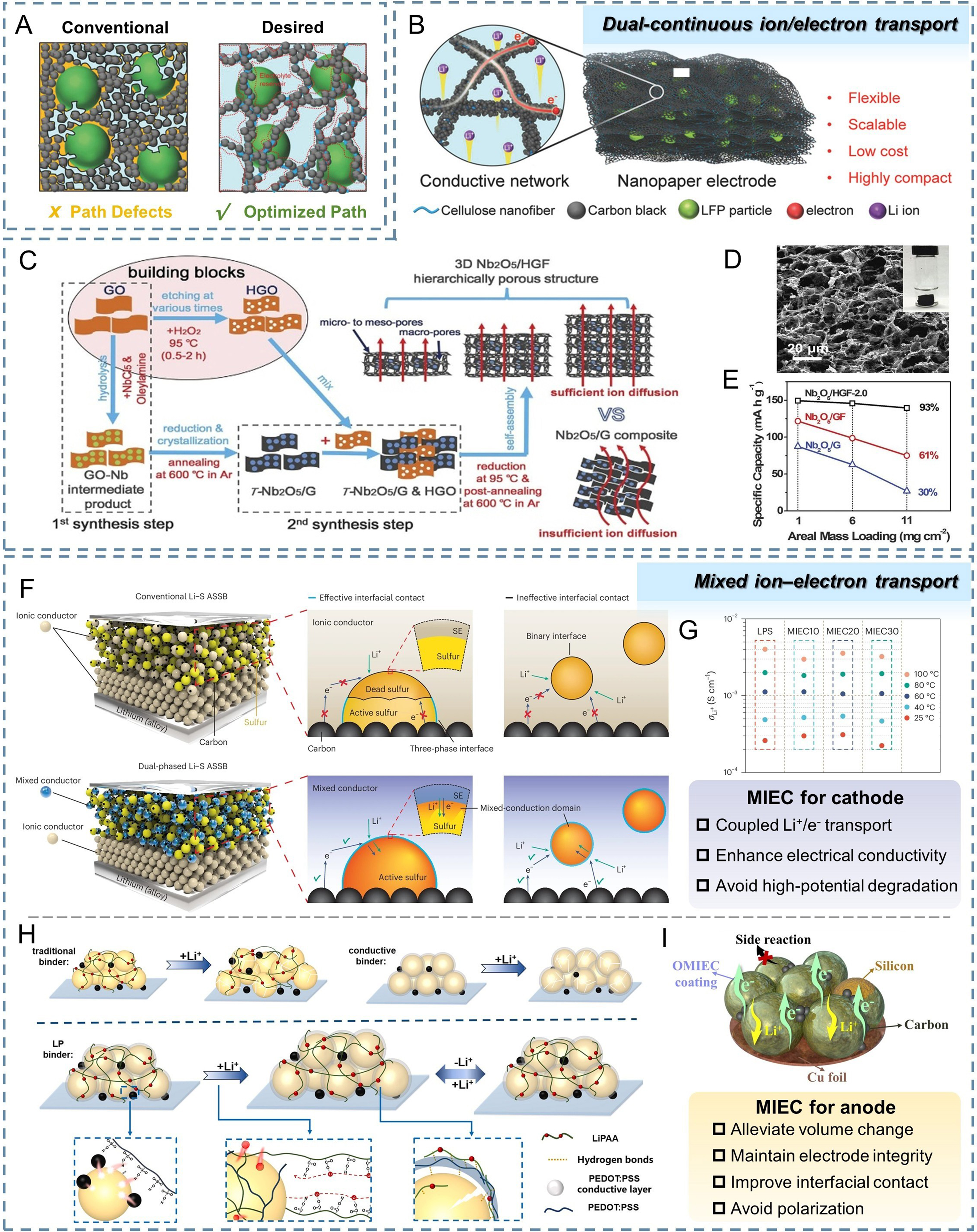
Figure 2. High-performance thick electrodes for constructing ion-electron cooperative transport networks. (A) Schematic of ion-electron transport pathways in conventional electrodes versus desired electrodes; (B) Electrostatic self-assembly enabling spatial decoupling of ion/electron transport pathways. (A and B) is quoted with permission from Ref.[26], Copyright © 2018 Wiley; (C) Two-step synthesis yielding an Nb2O5/HGF composite architecture with independent three-dimensional hierarchical pores; (D) SEM image of the three-dimensional hierarchical porous structure; (E) Comparison of capacity retention rates for electrodes with different loading levels. (C-E) are quoted with permission from Ref.[27], Copyright © 2017 American Association for the Advancement of Science; (F) MIEC introduced a sulfur cathode to establish a spatially coupled ion-electron transport network; (G) The ion conductivity measurements at different temperatures. (F and G) are quoted with permission from Ref.[30], Copyright © 2015 Springer Nature; (H) A trifunctional MIEC serves as a conductive binder for silicon anodes. This figure is quoted with permission from Ref.[31], Copyright © 2024 ELSEVIER; (I) MIEC is coated onto the surface of active particles. This figure is quoted with permission from Ref.[32], Copyright © 2026 ELSEVIER. HGF: Holey graphene framework; SEM: scanning electron microscope; MIEC: mixed ion-electron conductor; LFP: LiFePO4; Go-Nb: graphene oxide Nb2O5; HGO: holey graphene oxide; GF: graphene framework; G: graphene; ASSB: all-solid-state batteries; SE: solid electrolytes; LPS: Li2S·25P2S5; LP: lignin polymer; LiPAA: lithiated polyacrylic acid; PEDOT: poly(3,4-ethylenedioxythiophene); PSS: poly(styrenesulfonate); OMIEC: organic mixed ionic/electronic conductors.
Comparison of three enhancement strategies
| Strategy | Methods | Advantages | Limitations |
| Ion | Increase porosity, reduce tortuosity, directional channel design, etc. | Reduce concentration polarization | Cause electron breakdown |
| Electron | Reduce porosity, increase conductive agent content, etc. | Reduce ohmic polarization | Cause ion blockage |
| Coupled | Dual-continuous networks, mixed conductive network | Uniform reaction | Structurally complex |
To achieve this goal, two dominant design strategies have emerged. The first relies on spatially decoupled, dual-continuous ion/electron networks, in which ions and electrons are transported through independent yet percolating pathways, each optimized for its own transport requirements[25]. This architectural decoupling mitigates local transport competition and suppresses reaction mismatch. Kuang et al. demonstrated such a strategy by assembling cellulose nanofibers and carbon black into a three-dimensional framework via electrostatic interactions[26]. In this design, interconnected nanopores support ion diffusion while the conductive skeleton independently facilitates electron transport, achieving a volumetric energy density of
The second strategy employs spatially coupled mixed ion-electron conductors (MIECs), which simultaneously transport ions and electrons within a single phase[28]. By bridging ionic and electronic pathways at the molecular or mesoscale level, MIECs eliminate the intrinsic transport bifurcation of conventional electrodes, enabling rapid charge redistribution and reduced polarization. MIECs can function as conductive binders, active frameworks, or artificial interfacial layers, enhancing both ionic and electronic conductivity while stabilizing electrode interfaces[29]. For example, the introduction of MIECs into sulfur cathodes has achieved room-temperature ionic conductivity of 10-4-10-2 S cm-1 and electronic conductivity of 10-3 S·cm-1, resulting in high reversible capacity and excellent cycling stability [Figure 2F and G][30]. Notably, MIEC design priorities differ significantly between cathodes and anodes. Cathodes require high electronic conductivity and oxidative stability, whereas anodes demand balanced transport properties and mechanical robustness to accommodate volume changes and suppress lithium plating. Trifunctional MIECs developed for silicon anodes exemplify this approach, simultaneously providing electronic conduction, ionic transport, and mechanical reinforcement to preserve network integrity [Figure 2H][31]. MIECs can also serve as artificial interfacial coatings on active particles, further mitigating particle fracture and interface degradation [Figure 2I][32]. Moreover, MIECs can reduce polarization in full cells, enabling operation at lower negative-to-positive (N/P) ratios without inducing lithium deposition. Sun et al. significantly improved charge transport kinetics by constructing the MIEC through coating carbon black surfaces with red phosphorus[33]. Even at N/P=1.15, the thick graphite anode effectively suppressed lithium deposition, whereas conventional thick electrodes exhibited pronounced dendrite growth.
Despite their promise, both spatially decoupled architectures and MIEC-based strategies face substantial challenges for industrial implementation. Decoupled designs require precise control over pore orientation, size distribution, and multiphase connectivity, posing challenges for process scalability. MIEC-based approaches depend on complex molecular design and multistep synthesis with finely balanced transport, mechanical, and electrochemical properties, limiting large-scale manufacturability and long-term reliability. Consequently, future thick-electrode development must strike a balance between structural sophistication and manufacturing feasibility, simplifying architectures and material systems while preserving essential ion-electron synergy to bridge the gap between laboratory demonstrations and practical deployment.
SUMMARY AND OUTLOOK
Thick electrodes offer a chemistry-agnostic pathway to simultaneously enhance energy density and reduce manufacturing costs in lithium-ion batteries by increasing active material loading and minimizing inactive components. However, thickness-induced ion transport limitations and fragile electronic networks often lead to severe capacity loss. Recent multiscale advances highlight ion-electron cooperative transport as a governing mechanism, demonstrating that electrode performance is dictated by the spatiotemporal matching of ionic and electronic fluxes rather than their independent maximization.
The integration of CIET theory with transport architectures based on spatially decoupled dual-continuous networks and spatially coupled mixed conduction pathways provides a mechanistic framework for next-generation thick electrode design. Despite significant progress, further advances are required in three key areas:
(1) Development of CIET-based models beyond conventional pseudo-two-dimensional (P2D) and Butler-Volmer frameworks;
(2) Scalable, cost-effective manufacturing strategies compatible with roll-to-roll processing, such as dry electrode fabrication with mechanically robust binder systems;
(3) Full-cell-level integration emphasizing capacity balancing and kinetic matching between thick cathodes and anodes.
Overall, harnessing coupled ion-electron transport is essential for overcoming the traditional energy-power trade-off and realizing high-energy-density battery systems.
DECLARATIONS
Authors’ contributions
Conceptualization and manuscript design: Fu, K.; Tan, P.
Figure preparation and manuscript writing: Fu, K.
Manuscript discussion: Fu, K.; Li, X.; Sun, K.; Zhai, S.; Yang, H.; Gong, L.; Tan, P.
Manuscript editing and polishing: Tan, P.
Availability of data and materials
No applicable.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
The authors thank the funding support from the National Natural Science Foundation of China (52561145238) and the Chinese Academy of Sciences Project for Young Scientists in Basic Research (YSBR-098).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Ding, F.; Ji, P.; Han, Z.; et al. Tailoring planar strain for robust structural stability in high-entropy layered sodium oxide cathode materials. Nat. Energy. 2024, 9, 1529-39.
2. Lu, X.; Lagnoni, M.; Bertei, A.; et al. Multiscale dynamics of charging and plating in graphite electrodes coupling operando microscopy and phase-field modelling. Nat. Commun. 2023, 14, 5127.
3. Otoyama, M.; Wanibuchi, M.; Takeuchi, T.; et al. LixVSy nanocomposite electrodes for high-energy carbon-additive-free all-solid-state lithium-sulfur batteries. Energy. Mater. 2025, 5, 500126.
4. Yuan, M.; Liu, H.; Ran, F. Fast-charging cathode materials for lithium & sodium ion batteries. Mater. Today. 2023, 63, 360-79.
5. Chen, K.; Namkoong, M. J.; Goel, V.; et al. Efficient fast-charging of lithium-ion batteries enabled by laser-patterned three-dimensional graphite anode architectures. J. Power. Sources. 2020, 471, 228475.
6. Xie, F.; Xu, J.; Liao, Q.; et al. Progress in niobium-based oxides as anode for fast-charging Li-ion batteries. Energy. Rev. 2023, 2, 100027.
7. Li, Y.; Vasileiadis, A.; Zhou, Q.; et al. Origin of fast charging in hard carbon anodes. Nat. Energy. 2024, 9, 134-42.
8. Wang, Y.; Dang, D.; Li, D.; Hu, J.; Zhan, X.; Cheng, Y. Effects of polymeric binders on the cracking behavior of silicon composite electrodes during electrochemical cycling. J. Power. Sources. 2019, 438, 226938.
9. Müller, S.; Pietsch, P.; Brandt, B. E.; et al. Quantification and modeling of mechanical degradation in lithium-ion batteries based on nanoscale imaging. Nat. Commun. 2018, 9, 2340.
10. Ogihara, N.; Itou, Y.; Sasaki, T.; Takeuchi, Y. Impedance spectroscopy characterization of porous electrodes under different electrode thickness using a symmetric cell for high-performance lithium-ion batteries. J. Phys. Chem. C. 2015, 119, 4612-9.
11. Li, X.; Chen, Y.; Lu, Y.; et al. Spatial-dependent coupling of electrochemistry, mass transport, and stress in silicon-graphite composite electrodes for lithium-ion batteries. Adv. Funct. Mater. 2024, 35, 2413560.
12. Yang, Y.; Liu, C.; Su, M.; et al. Kinetic mechanism and structural design of thick electrodes in lithium-ion batteries: challenges and optimization strategies. Small 2025, 21, e06931.
13. Wood, M.; Li, J.; Du, Z.; et al. Impact of secondary particle size and two-layer architectures on the high-rate performance of thick electrodes in lithium-ion battery pouch cells. J. Power. Sources. 2021, 515, 230429.
14. Zhang, X.; Hui, Z.; King, S. T.; et al. Gradient architecture design in scalable porous battery electrodes. Nano. Lett. 2022, 22, 2521-8.
15. Niu, X.; Lu, Y.; Chen, P.; et al. Gradient-matched microstructural engineering for fast-charging, damage-tolerant thick electrodes of lithium-ion batteries. Adv. Energy. Mater. 2025, 15, 2502245.
17. Ju, Z.; Zhang, X.; King, S. T.; et al. Unveiling the dimensionality effect of conductive fillers in thick battery electrodes for high-energy storage systems. Appl. Phys. Rev. 2020, 7, 041405.
18. Pan, S.; Fang, W.; Yan, J.; Zhang, S.; Zhang, H. Multiscale coupled electron-ion transport in semi-solid lithium flow batteries. Energy. Environ. Sci. 2025, 18, 5868-96.
19. Wei, T. S.; Fan, F. Y.; Helal, A.; et al. Biphasic electrode suspensions for li-ion semi-solid flow cells with high energy density, fast charge transport, and low-dissipation flow. Adv. Energy. Mater. 2015, 5, 1500535.
20. Bazant, M. Z. Unified quantum theory of electrochemical kinetics by coupled ion-electron transfer. Faraday. Discuss. 2023, 246, 60-124.
21. Zhang, Y.; Fraggedakis, D.; Gao, T.; et al. Lithium-ion intercalation by coupled ion-electron transfer. Science 2025, 390, eadq2541.
22. Fu, K.; Li, X.; Sun, K.; et al. Rational design of thick electrodes in lithium-ion batteries by re-understanding the relationship between thermodynamics and kinetics. Adv. Funct. Mater. 2024, 34, 2409623.
23. Goel, V.; Chen, K.; Dasgupta, N. P.; Thornton, K. Optimization of laser-patterned electrode architectures for fast charging of Li-ion batteries using simulations parameterized by machine learning. Energy. Storage. Mater. 2023, 57, 44-58.
24. Wang, F.; Tang, M. A quantitative analytical model for predicting and optimizing the rate performance of battery cells. Cell. Rep. Phys. Sci. 2020, 1, 100192.
25. Quilty, C. D.; Wu, D.; Li, W.; et al. Electron and ion transport in lithium and lithium-ion battery negative and positive composite electrodes. Chem. Rev. 2023, 123, 1327-63.
26. Kuang, Y.; Chen, C.; Pastel, G.; et al. Conductive cellulose nanofiber enabled thick electrode for compact and flexible energy storage devices. Adv. Energy. Mater. 2018, 8, 1802398.
27. Sun, H.; Mei, L.; Liang, J.; et al. Three-dimensional holey-graphene/niobia composite architectures for ultrahigh-rate energy storage. Science 2017, 356, 599-604.
28. Xu, Y.; Peng, Y.; Xiong, X.; et al. Electrochemically active materials as critical components for next-generation solid-state electrolytes. Energy. Z. 2025, 1, 100004.
29. Riess, I. Mixed ionic-electronic conductors - material properties and applications. Solid. State. Ion. 2003, 157, 1-17.
30. Wang, D.; Gwalani, B.; Wierzbicki, D.; et al. Overcoming the conversion reaction limitation at three-phase interfaces using mixed conductors towards energy-dense solid-state Li-S batteries. Nat. Mater. 2025, 24, 243-51.
31. Geng, W.; Hu, X.; Zhou, Q.; et al. Rational design of trifunctional conductive binder for high-performance Si anodes in lithium-ion batteries. J. Power. Sources. 2024, 601, 234285.
32. Yu, Y.; Zhu, J.; Zhang, J.; Jiang, M. Bridging conductivity and stability: challenges and progress in organic ionic-electronic conductors for overcoming Si anodes degradation in high-energy lithium-ion batteries. Prog. Mater. Sci. 2026, 156, 101546.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0









Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].