

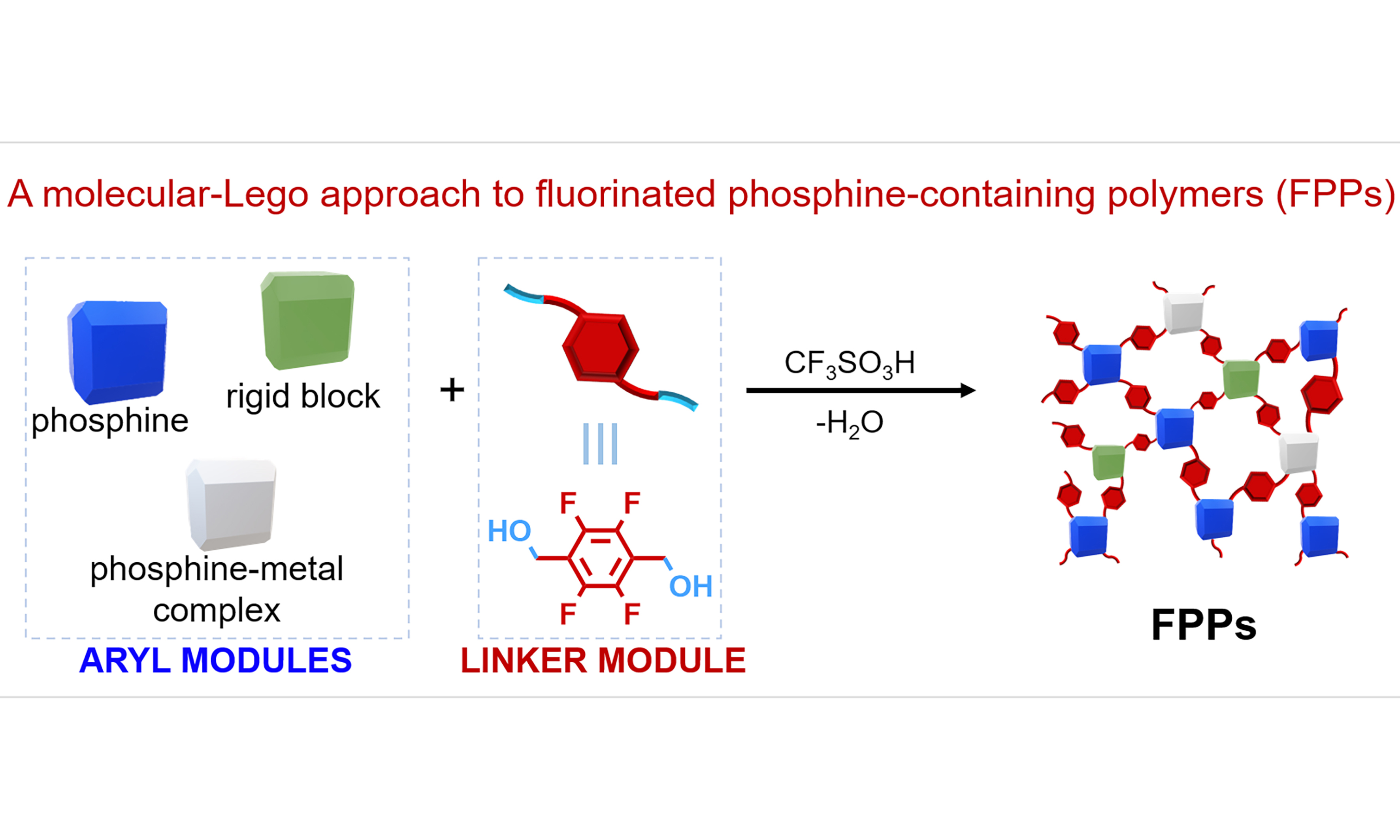
A molecular-Lego approach to fluorinated phosphine-containing polymers enables efficient CO2 transformation
 0
0 Abstract
Phosphine-containing porous organic polymers are candidate materials that could realize heterogenization of the homogeneous phosphine-metal catalysts. However, their succinct synthesis from the commercialized phosphines directly is still a challenge. This work pioneers a Brønsted acid-catalyzed molecular Lego assembly, enabling modular construction of fluorinated phosphine-containing polymers (FPPs) from commercial building blocks - aryl phosphines or their metal complexes - in one step. By simply introducing a third molecular-Lego module with rigid skeleton (e.g., spirobifluorene, triptycene), the porosity of FPPs can be precisely engineered while maintaining good to high yields (60%-98%), demonstrating the plug-and-play versatility of this strategy. This work presents the synthesis of 18 different structures of FPPs with distinct structures. Among them, the Ru complex-derived FPPs-10 shows excellent CO2 uptake capability (1.15 mmol·g-1 at 0 °C) and exhibits exceptional performance in
Keywords
INTRODUCTION
Phosphine ligands constitute a cornerstone of transition-metal catalysis, enabling precise control of steric and electronic environments at metal centers through strategic ligand design[1-3]. These homogeneous systems have demonstrated exceptional activity and selectivity for diverse transformations including cross-
Current synthetic strategies for PPOPs - including coupling polycondensation[24], radical polymerization[25-27], and aldimine condensation[28] - universally require pre-functionalized phosphine monomers [Scheme 1A]. A fundamental challenge arises from the oxygen sensitivity of trivalent phosphorus species, which significantly complicates both monomer synthesis and purification processes. Furthermore, these conventional approaches exhibit additional intrinsic limitations: (i) transition-metal-catalyzed methods[29] inevitably introduce metallic impurities that compromise material purity; and (ii) dynamic covalent chemistry typically yields polymeric networks with unsatisfactory hydrolytic stability[28,30]. Friedel-Crafts polymerization using Lewis acid catalysts (e.g., FeCl3, AlCl3) can directly incorporate non-functionalized aryl phosphines into knitting aryl network polymers (KAPs)[31,32] [Scheme 1B]; however, this method faces two major drawbacks: metal contamination from catalysts and potential phosphine ligand degradation. Solid-state 31P nuclear magnetic resonance (NMR) studies reveal a striking contrast: KAPs-embedded PPh3 shows signals at δ 20-
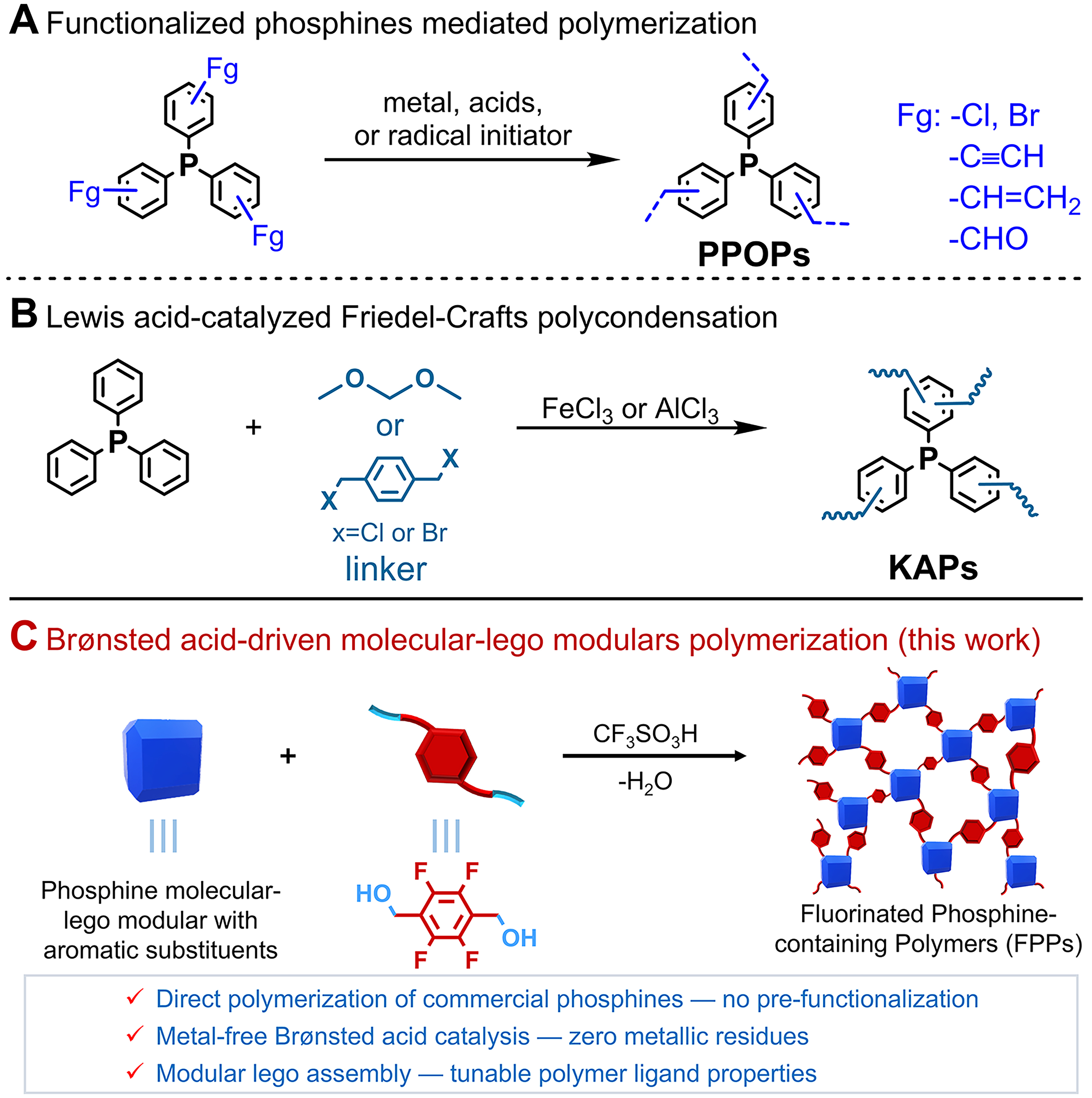
Scheme 1. Synthetic strategies for porous phosphine-containing polymers. PPOPs: Phosphine-containing porous organic polymers; KAPs: knitting aryl network polymers.
Herein, we developed a Brønsted acid-catalyzed molecular-Lego polycondensation for constructing fluorinated phosphine-containing polymers (FPPs) [Scheme 1C]. The molecular Lego modules with aryl phosphine ligands or their metal complexes were efficiently assembled with the linker module 2,3,5,6-tetrafluoro-1,4-benzenedimethanol (TF) via Friedel-Crafts polymerization in the presence of trifluoromethanesulfonic acid (CF3SO3H). This methodology demonstrates excellent substrate versatility, with mono-phosphines, flexible/rigid-bridged bis-phosphines, and aryl-substituted metal-phosphine complexes all afforded FPPs in high yields (89%-98%). Remarkably, the introduction of rigid molecular Lego modules (e.g., 9,9’-spirobifluorene, tetraphenylmethane, 1,3,5-triphenylbenzene, triptycene) significantly improved the porosity of FPPs, enhancing their Brunauer-Emmett-Teller (BET) surface areas by several tens-fold. This enhancement is exemplified by comparing FPPs-1 (the polymer derived from PPh3 and TF via CF3SO3H-catalyzed Friedel-Crafts condensation) with a surface area of 14.6 m2·g-1 to FPPs-15 (incorporating 1,3,5-triphenylbenzene as a third component with PPh3 and TF), which achieved a substantially higher surface area of 470.2 m2·g-1. Moreover, the resulting FPPs exhibit excellent catalytic performance in CO2 conversion. Among them, FPPs-10 - polymerized from Ru complex [trans-Ru(dppe)2(Cl)2, 1h] and TF - showed outstanding CO2 adsorption capacity (1.15 mmol·g-1 at 0 °C and 1.01 mmol·g-1 at 25 °C) along with remarkable catalytic activity and stability in the N-formylation of dimethylamine (DMA) with CO2 and H2. Notably, the turnover number (TON) for N,N-dimethylformamide (DMF) production (TONDMF) reached 1.41 × 105 (nDMF/nRU). Additionally, the catalyst demonstrated broad substrate compatibility, efficiently converting various primary and secondary alkylamines into their corresponding formamides. Overall, this work not only establishes a concise and efficient Lego-assembly strategy for synthesizing phosphine-containing porous polymers but also provides a novel approach to heterogenizing metal/phosphine catalysts.
EXPERIMENTAL
Materials
All the commercially available reagents were used without further purification. Among them, 2,3,4,5,6-Pentafluorobenzyl alcohol and 2,2,6,6-tetramethylpiperidine were purchased from Bidepharm. Mesitylene, DMA (2 M in methanol), TF, diethylamine, piperidine, pyrrolidine, morpholine, benzylamine, trifluoromethanesulfonic acid, triphenylphosphine, bis(diphenylphosphino)methane, 1,2-bis(diphenylphosphino)ethane, and Wilkinson’s catalyst [RhCl(PPh3)3] were purchased from Adamas-beta®. H2 and CO2 were purchased from Dalian GuangMing Special Gas Products Co., Ltd. The Ru(PPh3)Cl2 was prepared as reported[46] and Ru complexes (1g and 1h) were prepared from Ru(PPh3)Cl2 following the previous method[47].
Characterizations
Details regarding the characterization instruments and methods can be found in the Characterizations section of the Supplementary Materials.
Synthesis method
Synthesis of FPPs
To a 50 mL Schlenk flask, PPh3 (0.13 g, 0.5 mmol) and TF (0.32 g, 1.5 mmol) and trifluoromethanesulfonic acid (8 mL) were successively added. The flask was placed into liquid nitrogen bath (77 K), evacuated to high vacuum using the Schlenk technique. After sealing, the flask warmed up to room temperature slowly. The above procedure was repeated twice. After ultrasonication for 10 min, the flask was placed into an oil bath and heated to 100 °C for 24 h. The flask was then cooled to 0 °C and quenched with 30 mL degassed water. The brown solid was crushed and washed with degassed water (100 mL × 3). The solid was then treated with a triethylamine solution in ethanol (0.5 mol/L, 20 mL) for 30 min, followed by sequential washing with ethanol (100 mL × 3), THF (100 mL × 3) and CH2Cl2 (100 mL × 3). The brown polymer was obtained after drying in vacuum for 24 h at 50 °C (0.37 g, 98% yield). Other FPPs were prepared following the above procedures.
Synthesis of Ru catalysts through post-synthetic metalation
A suspension of FPPs-1 (0.2 g) in methanol (10 mL) was placed in a 50 mL Schlenk flask, to which RuCl3 (0.01 g) was added. The mixture was heated to 85 °C and stirred for 12 h under N2. After 12 h, the solid was filtered and washed with methanol (10 mL × 6). The solid was dried under vacuum at 50 °C for 12 h and the resultant sample was denoted as Ru/FPPs-1. The Ru/FPPs-5 and Ru/FPPs-6 were prepared from FPPs-5 and FPPs-6 following the above procedures, respectively.
A suspension of FPPs-1 (0.2 g) in methanol (10 mL) was placed in a 50 mL Schlenk flask, to which RuCl3 (0.01 g) was added. The mixture was heated to 85 °C and stirred for 12 h under N2. After 12 h, the solid was filtered and washed with methanol (10 mL × 6). The solid was dried under vacuum at 50 °C for 12 h and the resultant sample was denoted as Ru/FPPs-1. The Ru/FPPs-5 and Ru/FPPs-6 were prepared from FPPs-5 and FPPs-6 following the above procedures, respectively.
Procedure for the N-formylation of dimethylamine
Typically, 10 mL of DMA solution (2 M in methanol, 20 mmol), Ru catalyst (10 mg for Ru/FPPs and 6.8 mg for FPPs-10), and mesitylene (200 μL, internal standard for 1H NMR analysis) are successively added into a 100 mL autoclave with a magnetic stir bar. The autoclave was sealed and the inner air was replaced with CO2 three times. Then CO2 (35 bar) and H2 (45 bar) were charged. The mixture in the autoclave was heated to 140 °C and stirred at 800 rpm for a certain period. Then, the autoclave was cooled to room temperature and the gas was released slowly. The yield of the products was analyzed by 1H NMR spectroscopy with mesitylene as an internal standard.
RESULTS AND DISCUSSION
Although TF has been employed as a linker in porous organic polymer synthesis[48,49], the compatibility of phosphines with this system remains uncertain due to the potential formation of benzyltriphenylphosphonium salts[38,50,51]. To investigate this uncertainty, we conducted a model reaction between PPh3 (1 equiv.) and 2,3,4,5,6-pentafluorobenzyl alcohol (1 equiv.) in the presence of CF3SO3H. The reaction proceeded at 100 °C under vacuum for 24 h, followed by aqueous workup and dichloromethane extraction of organic products. Gas chromatography-mass spectrometry (GC-MS) analysis showed complete consumption of the alcohol substrate and revealed three major peaks attributable to unreacted PPh3, monosubstituted (A), and disubstituted (B) products [Supplementary Figure 1], confirming efficient C–C bond formation[52]. 31P NMR characterization of the crude mixture exhibited a signal at -5.64 ppm that shifted to 30.19 ppm upon H2O2 oxidation [Supplementary Figure 2]. Our comprehensive characterization reveals the reaction pathway excludes phosphonium salt formation [Supplementary Figure 3], establishing the TF linker as an effective platform for the modular, Lego-like assembly of arylphosphine building blocks into well-defined hypercrosslinked polymer networks.
Encouraged by the positive outcomes from model reaction, we subsequently attempted to construct FPPs using TF as the linker with PPh3 as the Lego building blocks (molar ratio of TF to PPh3 is 3/1) in the presence of CF3SO3H. A monolithic solid was formed after condensation for 24 h at 100 °C under vacuum. After being crushed, the polymer was washed with H2O, EtOH, and CH2Cl2 to afford insoluble solid (denoted as FPPs-1) in a 96% yield [Figure 1A]. The chemical structure of FPPs-1 was fully characterized. To reveal the polymer structure at the molecular level, FPPs-1 was first analyzed by 13C solid-state NMR spectroscopy. As shown in Figure 1B, the signals ranging from about δ 141 to 138 ppm correspond to the fluorinated aryl carbon in the FPPs-1. The peaks at δ 132 to 118 ppm are assigned to the non-fluorinated aromatic carbons. The resonance peaks near δ 28 ppm represent the methylene groups that connected the phosphine ligands and fluorinated aromatic rings. In the solid-state 31P NMR spectrum, FPPs-1 shows a broad peak centered at about δ -3.4 ppm assigned to phosphorus of the embedded PPh3 [Figure 1C]. To further verify the presence of trivalent phosphorus species (P(III)) in the polymer framework, FPPs-1 was reacted with iodomethane (CH3I). Solid-state 31P NMR analysis of the resulting product revealed a distinct chemical shift at δ 21.4 ppm [Supplementary Figure 4], confirming the formation of quaternary phosphonium salts (P(V)) via alkylation. This observation unambiguously demonstrates that the phosphorus centers in FPPs-1 exist as P(III) species within the polymeric backbone. Then, the attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectrum was recorded to identify the characteristic groups on FPPs-1. As shown in Figure 1D, the bands near 1,478 and 1,593 cm-1 are assigned to the skeleton stretching vibration of benzene rings (highlighted in gray)[48]; the bands at around 1,273, 1,033, and 1,001 cm-1 correspond to the stretching vibrations of aromatic C–F bonds (highlighted in blue)[53], and the bands at 1,404 and 2,930 cm-1 could be attributed to aliphatic C–H bond stretching vibration (highlighted in purple)[54]. Notably, the absence of signal near 3,256 cm-1 in FPPs-1 confirms the complete consumption of all –OH groups in TF, demonstrating the system’s high reactivity in forming hyper-crosslinked polymers.
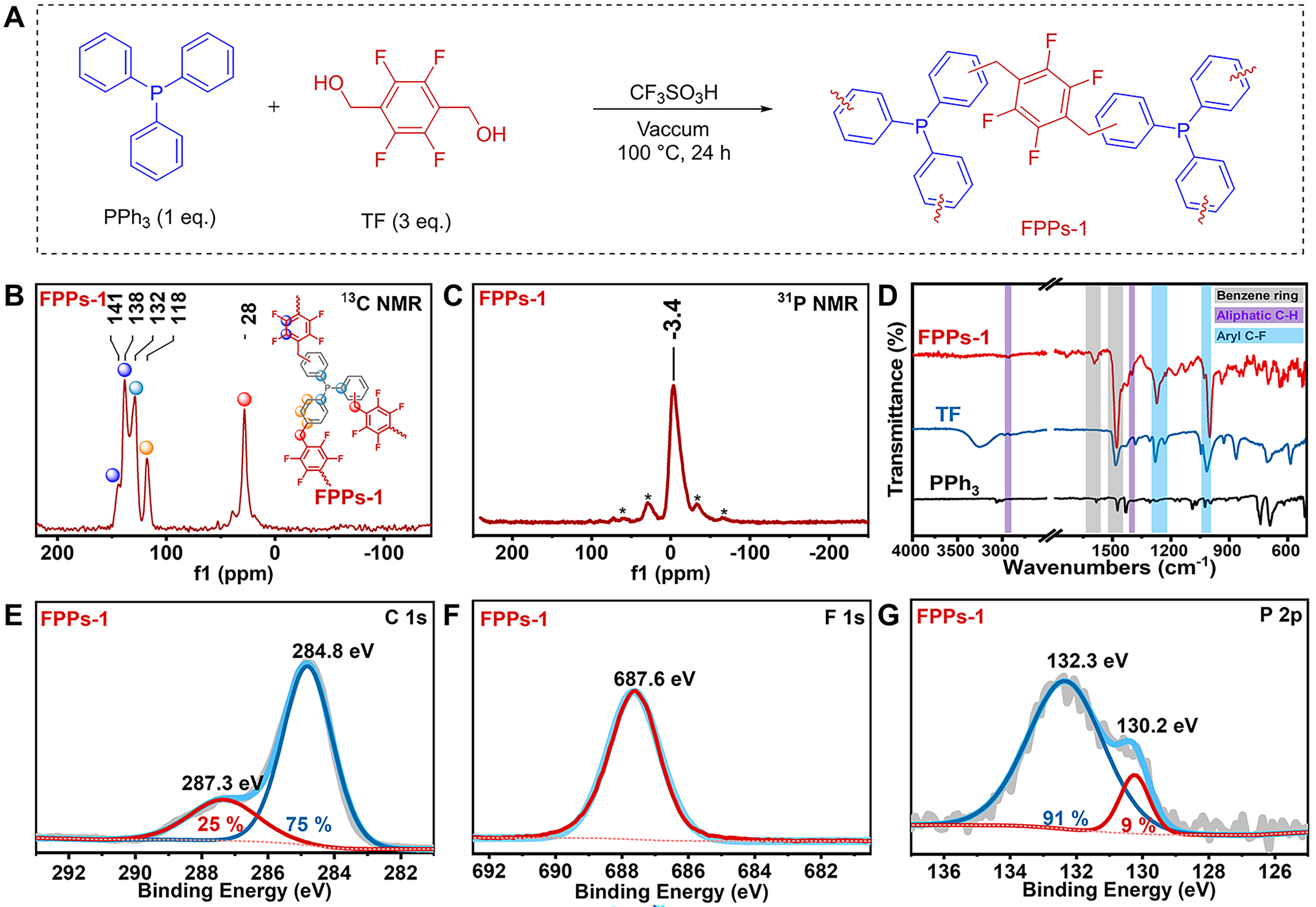
Figure 1. Synthesis and characterization of FPPs-1. (A) Preparation of FPPs-1; (B) Solid-state 13C NMR spectrum; (C) Solid-state 31P NMR spectrum. Spinning sidebands are marked with asterisks (*); (D) ATR-FTIR spectra; (E-G) fine C 1s, F 1s, and P 2p XPS spectra, respectively. FPPs: Fluorinated phosphine-containing polymers; NMR: nuclear magnetic resonance; ATR-FTIR: attenuated total reflectance Fourier transform infrared.
Subsequently, X-ray photoelectron spectroscopy (XPS) analysis was performed on FPPs-1. As shown in Supplementary Figure 5, elemental analysis of FPPs-1 revealed compositions of 28.37% F, 70.09% C, and 1.54% P according to the XPS survey. These values align closely with theoretical atomic percentages modeled from reactant feed ratios, indicating high-fidelity polymerization between the PPh3 and TF. As shown in Figure 1E, the C1s binding energy of fluorinated carbons is around 287 eV, and the binding energy of other carbons is located at 284.8 eV. The atomic ratio of the fluorinated carbons in FPPs-1 is 25%, close to the calculated ratio of 28.6%. As revealed in the F1s XPS spectrum [Figure 1F], the aromatic C–F bonds are in a form of semi-covalent as their binding energy is located at 687.6 eV[55]. The P 2p XPS spectrum of FPPs-1 resolves into two peaks at 130.2 and 132.3 eV, corresponding to phosphorus in PPh3 and oxidized PPh3, respectively [Figure 1G]. Although ~91% of surface phosphine groups undergo oxidation during synthesis, no oxidized PPh3 signal appears in the solid-state 31P NMR spectrum. This demonstrates that the hyper-crosslinked polymer matrix effectively shields encapsulated PPh3 from oxidation. Moreover, the thermal stability of FPPs-1 was investigated by thermogravimetric analysis (TGA) under a nitrogen atmosphere. FPPs-1 exhibits excellent thermal stability, with an initial decomposition temperature of
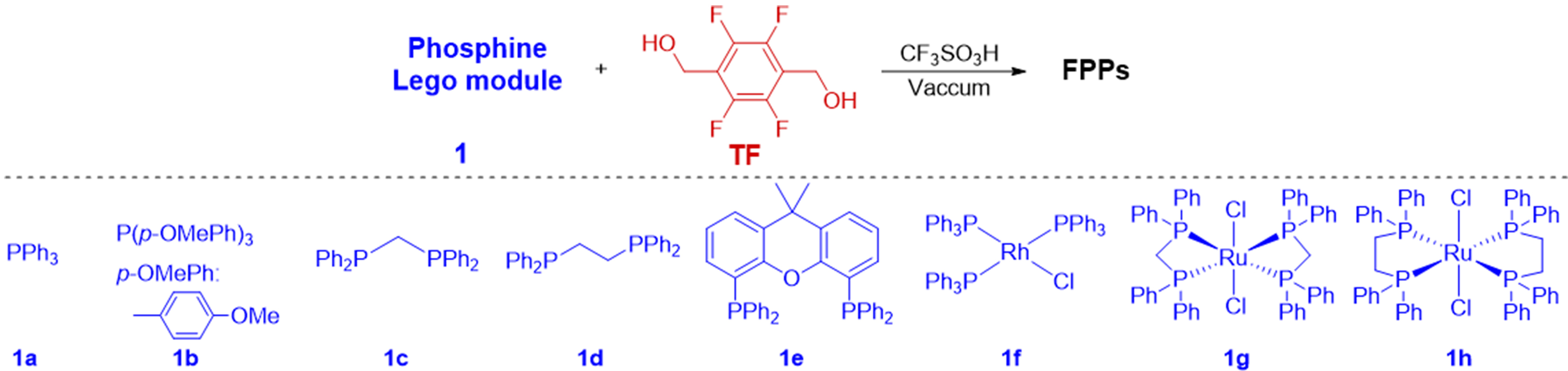
Synthesis of FPPs from TF and different phosphine Lego modules with aromatic substituents via CF3SO3H-catalyzed Friedel-Crafts polymerization: substrate scope of phosphine Lego modulesa
 | |||||||
| Entry | FPPs | 1/TF (mmol) | Surface area (m2∙g-1) | Pore volume (m3∙g-1) | Metal content (%) | 31P NMR (ppm) | Yield (%) |
| 1 | FPPs-1 | 1a/TF = 1/3 | 14.56 | 0.017 | - | -3.4 | 98 |
| 2 | FPPs-2 | 1a/TF = 1/2 | 13.82 | 0.013 | - | - | 98 |
| 3 | FPPs-3 | 1a/TF = 1/1.5 | - | - | - | - | - |
| 4b | FPPs-4 | 1b/TF = 1/2 | 22.41 | 0.024 | - | -13.5* | 98 |
| 5 | FPPs-5 | 1c/TF = 0.5/2 | 13.81 | 0.014 | - | -23.0* | 88 |
| 6 | FPPs-6 | 1d/TF = 0.5/2 | 12.22 | 0.012 | - | -11.3 | 98 |
| 7 | FPPs-7 | 1e/TF = 0.5/3 | 14.82 | 0.015 | - | -18.8* | 98 |
| 8b | FPPs-8 | 1f/TF = 0.25/2.25 | 23.17 | 0.035 | 3.10 | 27.5* | 95 |
| 9b | FPPs-9 | 1g/TF = 0.5/4.0 | 13.19 | 0.010 | 2.70 | 23.2, -24.1 | 98 |
| 10b | FPPs-10 | 1h/TF = 0.5/4.0 | 18.67 | 0.026 | 3.22 | 28.0, -13.9 | 89 |
We attempted to prepare PPh3-containing polymers using alternative Brønsted acid catalysts (HCOOH, CH3COOH, CF3COOH), but none successfully catalyzed the Friedel-Crafts condensation of PPh3 and TF
Based on these results, we systematically evaluated the applicability of various phosphine-based aromatic Lego modules for synthesizing FPPs using TF as the linker and CF3SO3H as the catalyst. The molar ratio between arylphosphine monomers and TF linker was found to be critical for obtaining insoluble hyper-crosslinked FPPs. Using PPh3 (1a) as a representative example, high yields of FPPs were achieved at monomer/TF ratios of 1/3 and 1/2 (Table 1, entries 1 and 2). However, reducing the ratio to 1/1.5 resulted in a gel-like reaction mixture that yielded negligible insoluble polymeric products after THF and DCM solvent washing (Table 1, entry 3). We subsequently investigated substituted monophosphine ligands (1b), alkyl-linked biphosphines (1c and 1d), and rigid-backboned biphosphine ligands (Xantphos, 1e) in polycondensation reactions, all of which successfully afforded FPPs with high yields (about 98%) (Table 1, entries 4-7). The 31P NMR analysis revealed minimal chemical shift variations between polymeric products and free phosphine ligands, suggesting preservation of the phosphine skeleton during Friedel-Crafts polycondensation crosslinking via TF-derived fluorinated groups. Notably, this methodology also demonstrated broad compatibility with metal-coordinated arylphosphine complexes (1f, 1g, and 1h), achieving remarkable yields of 89%-98% (Table 1, entries 8-10). Contrastingly, phosphine-coordinated metal complexes exhibited significant 31P NMR peak shifts upon polymerization, indicating structural reorganization of metal coordination spheres. This phenomenon can be attributed to acid-catalyzed ligand displacement, wherein protonation facilitates the cleavage of metal-phosphine coordination bonds, leading to the formation of protonated phosphorus species[12]. Wilkinson’s catalyst [RhCl(PPh3)3, 1f] exhibits two 31P NMR signals at δ 48.0 and 31.5 ppm[56], corresponding to the two distinct ligated PPh3 groups in different chemical environments. However, these peaks disappear after polymerization, replaced by broad resonances at δ -22.3 and 27.5 ppm with intense spinning sidebands. Solid-state 31P NMR spectra confirmed the emergence of free phosphine signals in FPPs-9 and FPPs-10 (Table 1, entries 9-10), corroborating partial ligand dissociation during polymerization. Elemental analysis revealed substantial metal retention in the FPPs, demonstrating effective metal stabilization through coordination interactions (Table 1, entries 8-10). Despite successful polymerization of both free arylphosphines and arylphosphines-coordinated metal complexes with TF, the resulting materials exhibited limited porosity with BET surface areas below
To enhance the porosity of the resulting FPPs, we introduced rigid aryl comonomer modules for copolymerization with arylphosphine and TF linker [Table 2]. Incorporation of 9,9’-spirobifluorene (2a) significantly improved the BET surface area and pore architecture, with the enhancement positively correlated to its feed ratio (Table 2, entries 1-3). Further evaluation extended to tetraphenylmethane (2b), 1,3,5-triphenylbenzene (2c), and triptycene (2d), all of which effectively promoted porous structure development (Table 2, entries 4-6). Notably, 9,9’-spirobifluorene and 1,3,5-triphenylbenzene demonstrated superior performance, generating copolymers FPPs-13 and FPPs-15 with exceptional BET surface areas of 388.54 and 470.23 m2·g-1, respectively. However, the limited solubility of these rigid comonomers in CF3SO3H led to incomplete dissolution even after sonication, resulting in heterogeneous reaction mixtures. This phase-separation phenomenon caused partial monomer exclusion from the polymerization network, consequently reducing product yields (< 90%) compared to systems employing solely arylphosphine ligands.
Synthesis of FPPs from TF, PPh3 and different comonomer modules: substrate scope of comonomer modulea
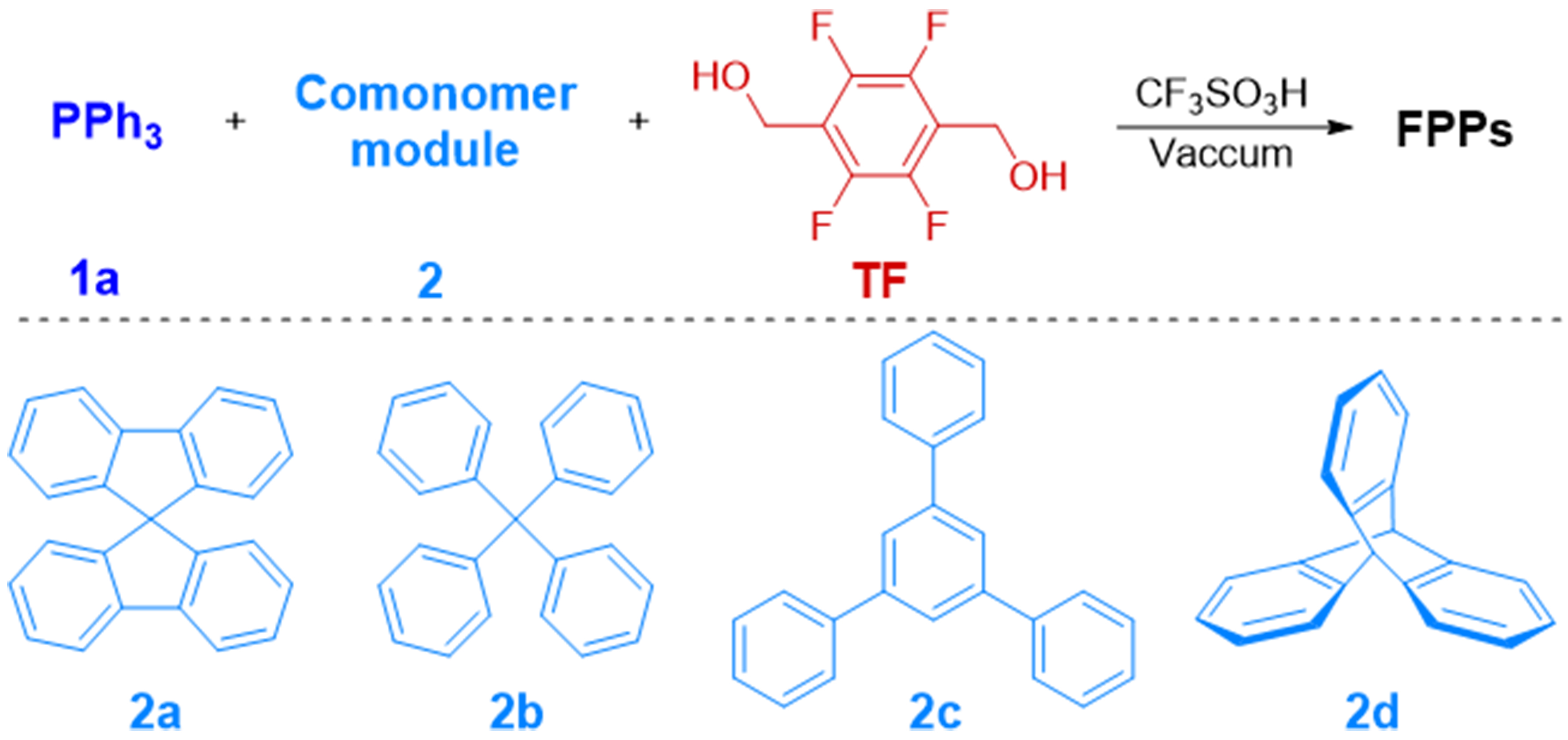 | |||||
| Entry | FPPs | 1/2/TF (mmol) | BET surface area (m2∙g-1) | Pore volume (m3∙g-1) | Yield (%) |
| 1 | FPPs-11 | 1a/2a/TF = 0.5/0.5/2.5 | 61.21 | 0.069 | 88 |
| 2 | FPPs-12 | 1a/2a/TF = 0.5/1/3.5 | 125.60 | 0.108 | 76 |
| 3 | FPPs-13 | 1a/2a/TF = 0.5/1.5/4.5 | 388.54 | 0.299 | 69 |
| 4 | FPPs-14 | 1a/2b/TF = 0.5/1.5/4.5 | 225.01 | 0.23 | 44 |
| 5 | FPPs-15 | 1a/2c/TF = 0.5/1.5/3.75 | 470.23 | 0.389 | 79 |
| 6 | FPPs-16 | 1a/2d/TF = 0.5/1.5/3.75 | 88.55 | 0.124 | 88 |
Building upon the structural advantages of 9,9’-spirobifluorene (2a) as a rigid aryl comonomer, we synthesized a series of FPPs using biphosphine ligand Xantphos (1e), Wilkinson-type
Synthesis of FPPs from TF, spirobifluorene and phosphine Lego modulesa
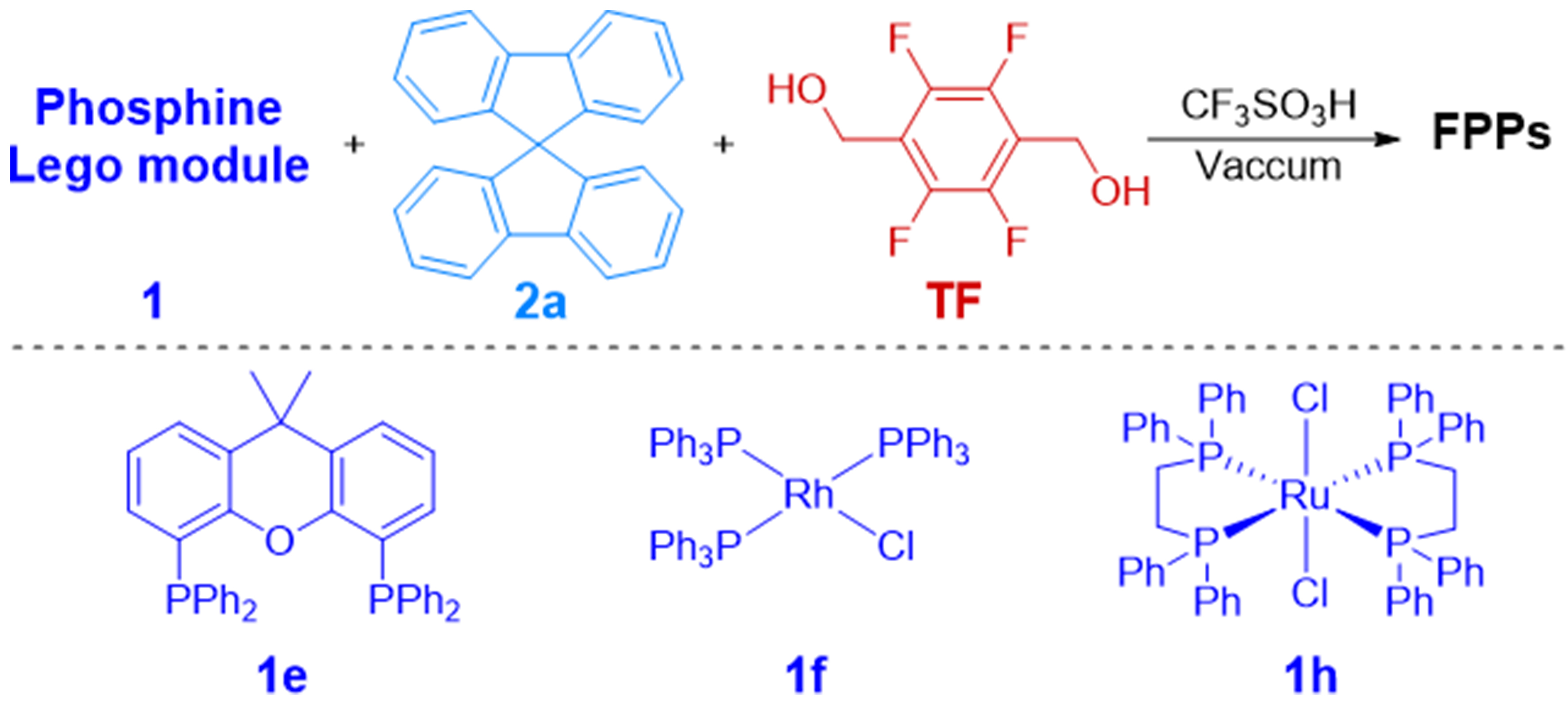 | ||||||
| Entry | FPPs | 1/2/TF (mmol) | BET surface area (m2∙g-1) | Pore volume (m3∙g-1) | Metal content (%) | yield (%) |
| 1 | FPPs-17 | 1e/2a/TF = 0.02/0.1/1.8 | 61.98 | 0.075 | - | 69 |
| 2 | FPPs-18 | 1f/2a/TF = 0.3/0.9/3.15 | 336.77 | 0.286 | 2.74 | 81 |
| 3 | FPPs-19 | 1f/2a/TF = 0.3/1.8/4.95 | 510.95 | 0.397 | 2.11 | 60 |
| 4 | FPPs-20 | 1h/2a/TF = 0.04/0.2/3.6 | 180.67 | 0.168 | 1.35 | 78 |
FPPs exhibit good swelling properties [Supplementary Figure 12], which ensures the mass transfer when they are used as catalyst supports. Subsequently, the post-synthetic metalations were conducted through the reaction of FPPs (FPPs-1, FPPs-5, and FPPs-6 were used) and RuCl3 in methanol under N2 atmosphere and three Ru-containing FPPs (Ru/FPPs-1, Ru/FPPs-5 and Ru/FPPs-6) were obtained. For the CO2-philic nature of the fluorinated polymers[57], these Ru-containing FPPs were tested as a heterogeneous catalyst in the
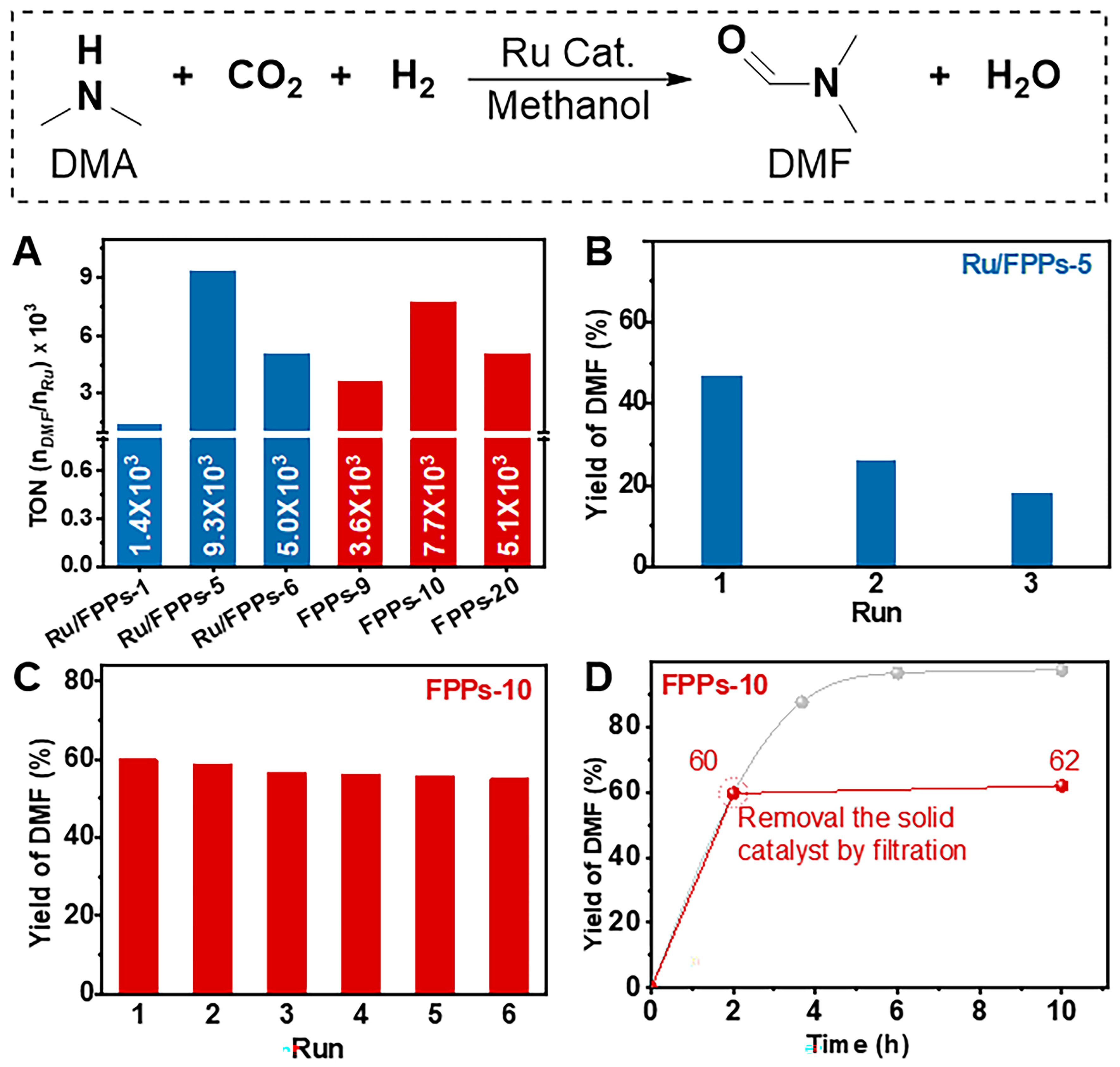
Figure 2. The catalytic performances of various Ru-containing FPPs in the N-formylation of DMA. (A) activity comparison of Ru catalysts in the N-formylation of DMA; (B) recyclability tests of Ru/FPPs-5; (C) recyclability tests of FPPs-10; (D) Filtration test of FPPs-10. FPPs: Fluorinated phosphine-containing polymers; DMA: dimethylamine.
The reaction pathway of catalyzed N-formylation of DMA over FPPs-10 was elucidated by monitoring the reaction mixture via 1H NMR at different reaction periods. As shown in Supplementary Figure 16, the 1H NMR spectrum of the reaction mixture at 0.5 h exhibited strong characteristic peaks assigned to dimethylammonium formate (FA), while the peaks corresponding to DMF were weak. After removing the solid catalyst and heating the mixture at 140 °C for an additional 11.5 h, the FA signals significantly diminished, whereas the DMF signals intensified, indicating thermal dehydration of FA to DMF, consistent with literature reports[58,59]. Thus, the proposed pathway involves: (1) CO2 insertion into the Ru–H bond to generate a Ru-coordinated formate species; (2) dissociation of the formate to form FA in the presence of DMA; and (3) thermal dehydration of FA to DMF [Supplementary Figure 17]. For the efficient catalytic activity of FPPs-10, its CO2 adsorption performance was further evaluated [Supplementary Figure 18]. FPPs-10 demonstrates a CO2 adsorption of 1.15 mmol·g-1 at 0 °C (1.01 mmol·g-1 at 25 °C) with the isosteric heats of adsorption of 10 kJ·mol-1. FPPs-20 give slightly higher CO2 adsorption capability (1.34 mmol·g-1 at 0 °C and 1.17 mmol·g-1 at 25 °C) for its more developed pore structure[60]. The CO2-philic nature of the FPPs may contribute to their high catalytic activity by concentrating CO2 within the catalyst. However, the lower catalytic activity of FPPs-20 compared to FPPs-10 [Figure 2A], despite its superior CO2-philicity, reflects the complex factors influencing N-formylation, which are likely to involve distinct ruthenium active sites, pore architecture, and other effects.
To test the long-term stability of FPPs-10, larger-scale reactions were conducted using dimethylammonium dimethylcarbamate as the amine source. As shown in Supplementary Table 4, the turnover frequency (TOF) was determined to be 1.31 × 103 h-1 after reacting for 60 h. The TOF maintained at 1.17 × 103 h-1 upon prolonging the reaction time to 120 h. Overall, the TOF values of the two reactions are very close, indicating that the catalyst possesses good stability in the long-term reaction time. It is worth noting that the TON reached 1.41 × 105 nDMF/nRU after reacting for 120 h. As shown in Supplementary Table 5, while the FPPs-10 catalyst exhibits lower activity than some Ru catalysts (Ru/PPPOP[58] and POMP 1c[31]) and Ir catalyst (POMP-NHC-Ir 2f[59]) that supported by polymers, its performance is comparable to - or superior to - most reported heterogeneous polymer catalysts, demonstrating the efficacy of this CF3SO3H-catalyzed Lego-like strategy for fabricating advanced heterogeneous catalysts.
The stability of the FPPs-10 was systematically investigated through inductively coupled plasma (ICP), transmission electron microscopy (TEM), scanning transmission electron microscopy (STEM), and XPS characterizations. ICP analysis revealed a slight decrease in ruthenium content from 3.22% in the fresh catalyst to 2.15% after seven reaction cycles [Supplementary Table 6], demonstrating its robust anti-leaching performance. TEM and STEM results confirmed the catalyst’s excellent anti-sintering properties, as no observable ruthenium metal particles were detected in either the fresh or spent catalysts [Supplementary Figure 19]. Energy dispersive X-ray spectrometry (EDS) mapping further verified the uniform distribution of carbon, fluorine, phosphorus, and ruthenium elements throughout the catalyst before and after use, providing additional evidence against metal agglomeration. Comparative XPS analysis of the ruthenium complex (1h) reference sample and FPPs-10 catalyst revealed electronic property variations in C, P, and Ru elements. The C 1s binding energy of spent FPPs-10 remained unchanged, indicating structural stability of the carbon framework, with both fluorinated and non-fluorinated carbon species maintained at comparable ratios [Supplementary Figure 20A]. Polymerization of complex 1h induced a 0.7 eV positive shift in P 2p binding energy, likely due to altered phosphine-ruthenium coordination as previously evidenced by solid-state 31P NMR. Remarkably, the phosphorus electronic environment in FPPs-10 showed minimal changes after multiple cycles, retaining P 2p binding energy at 131.8 eV comparable to the fresh catalyst
Using FPPs-10 as a catalyst, an array of amines was examined in the N-formylation. As shown in Figure 3, the secondary alkyl amines (1A-F) can be converted to the corresponding formamides (2A-F) smoothly in excellent yields. The primary alkyl amines, such as n-butylamine (1G) and benzylamine (1H), can also be easily converted into the corresponding formamides (2G and 2H) with high yields of 98% and 94%, respectively. When it comes to the sterically encumbered amine such as 2,2,6,6-tetramethylpiperidine (1I), only 57% of formamide was found. And the residual 2,2,6,6-tetramethylpiperidine was in the form of ammonium formate for its slow dehydration reactivity. However, both primary and secondary aryl amines
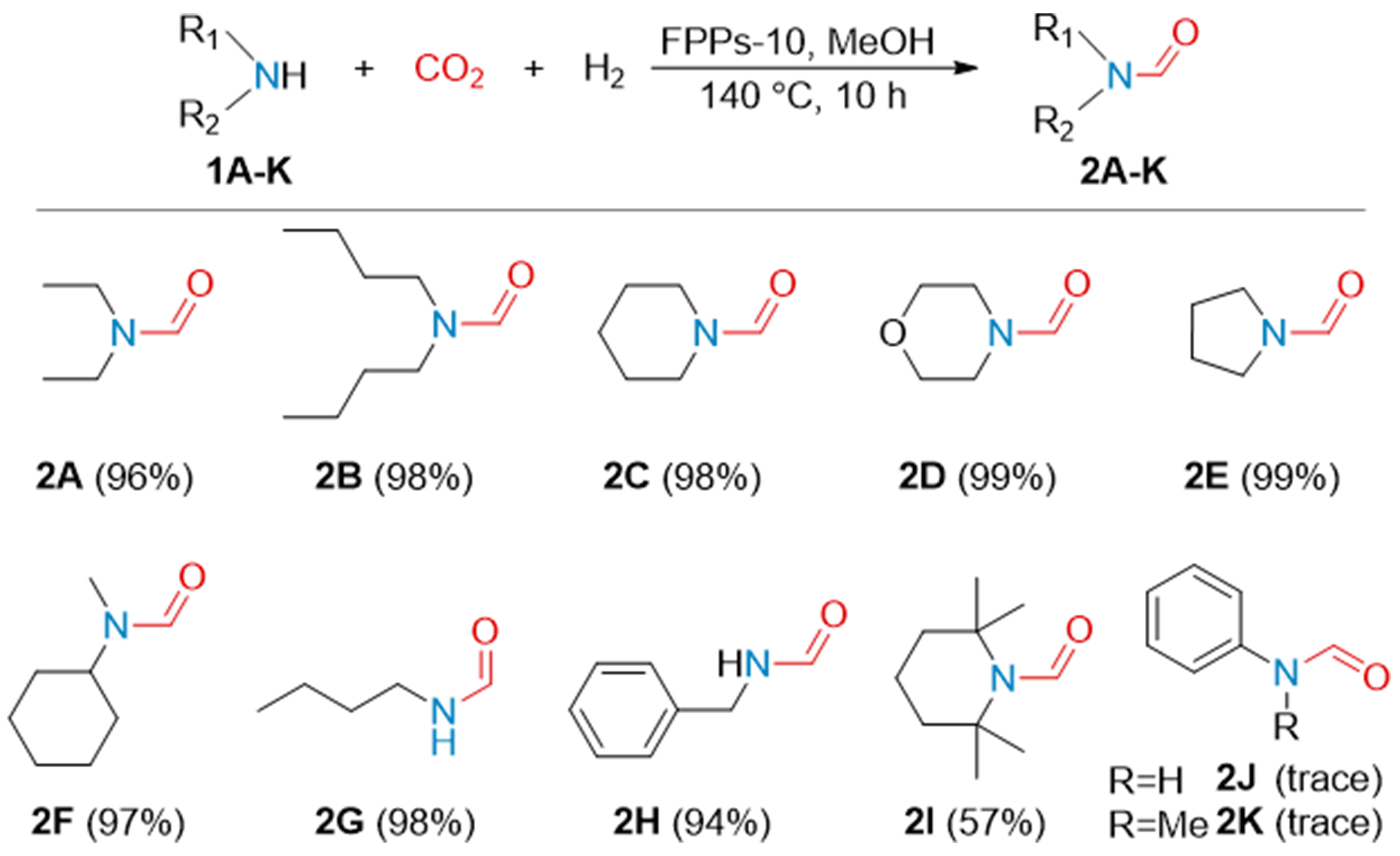
Figure 3. N-formylation of amines with FPPs-10 as catalyst. Reaction conditions: FPPs-10 6.8 mg, amine 5 mmol, MeOH 5 mL, CO2 35 bar, H2 45 bar, 140 °C, 10 h. The yields of formamides were determined by 1H NMR using mesitylene as the internal standard. FPPs: Fluorinated phosphine-containing polymers.
CONCLUSIONS
This research breaks through the conventional limitations in synthesizing phosphorus-containing porous polymers by achieving direct molecular Lego-like assembly of phosphine ligands or metal complexes, linker monomer and comonomers via Brønsted acid catalysis. This method demonstrates broad compatibility with commercially available phosphine compounds, including monodentate/bidentate phosphine ligands and metal-phosphine complexes bearing aryl substituents, all of which can be successfully incorporated into FPPs with good yields. Notably, the pore architectures of resulting FPPs can be precisely tailored through modular copolymerization with tunable building blocks. Moreover, the FPPs catalyst demonstrates excellent CO2 adsorption performance and displays high activity, good structural stability and broad alkylamine compatibility in N-formylation. In summary, this “molecular Lego” strategy not only streamlines material preparation but also pioneers a new approach for designing phosphine-based heterogeneous catalysts.
DECLARATIONS
Authors’ contributions
Contributed equally to this work: Wu, M.; Huang, L.; Kang, X.
Conceived the idea: Li, F.; Wu, M.
Designed the experiments: Wu, M.; Huang, L.; Kang, X.
Conducted the experiments: Wu, M.; Huang, L.; Kang, X.; Gao, G.
Analyzed and processed the data: Wu, M.; Huang, L.; Kang, X.; Gao, G.; Huang, Z.
Prepared the manuscript: Wu, M.; Huang, L.; Kang, X.; Sun, P.; Li, F.
Contributed to the revision of the manuscript: Li, F.; Sun, P.
Supervised the project: Sun, P.; Li, F.
Availability of data and materials
More data supporting this work are provided in the Supplementary Materials.
AI and AI-assisted tools statement
During the preparation of this manuscript, the AI tool DeepSeek (DeepSeek-V3, released by Hangzhou DeepSeek Artificial Intelligence Co., Ltd.) was used solely for language editing. The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship
This work was funded by the National Key R&D Program of China (2022YFA1504602), the Natural Science Foundation of China (22472177), and the Light of West China of the Chinese Academy of Sciences.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
Supplementary Materials
REFERENCES
1. King, A. J.; Goicoechea, J. M. Ligand centered reactivity of a transition metal bound geometrically constrained phosphine. Chemistry 2024, 30, e202400624.
2. Coles, N. T.; Sofie, Abels. A.; Leitl, J.; Wolf, R.; Grützmacher, H.; Müller, C. Phosphinine-based ligands: recent developments in coordination chemistry and applications. Coordination. Chemistry. Reviews. 2021, 433, 213729.
3. Kusy, R.; Grela, K. Renaissance in alkyne semihydrogenation: mechanism, selectivity, functional group tolerance, and applications in organic synthesis. Chem. Rev. 2025, 125, 4397-527.
4. Deng, L. F.; Wang, Y.; Xu, S.; et al. Palladium catalysis enables cross-coupling-like SN2-glycosylation of phenols. Science 2023, 382, 928-35.
5. Wu, L.; Ge, C.; Wang, M.; Shi, Z. Phosphorus(III)-compatible asymmetric suzuki-miyaura cross-coupling (P-ASMC) reaction. J. Am. Chem. Soc. 2025, 147, 20657-66.
6. Li, C. C.; Adorján, Á.; Sofiadis, M.; et al. Nitrate reduction for deaminative suzuki-miyaura coupling of anilines. Angew. Chem. Int. Ed. Engl. 2025, 64, e202504012.
7. Gao, S.; Su, L.; Liu, J. Diastereo- and enantioselective pd-catalyzed C-P coupling for axially and P-chiral phosphine oxides via simultaneous dynamic kinetic asymmetric transformation and kinetic resolution. J. Am. Chem. Soc. 2025, 147, 23946-56.
8. Zou, J. Y.; Xing, F. L.; Zi, Q. X.; Du, H. W.; Chen, M.; Shu, W. Asymmetric alkyl-alkyl coupling between electron-deficient and unactivated alkenes to access α-chiral phosphines by Ni catalysis. Sci. Adv. 2025, 11, eadv6571.
9. Guo, S. M.; Huh, S.; Coehlo, M.; Shen, L.; Pieters, G.; Baudoin, O. A C-H activation-based enantioselective synthesis of lower carbo[n]helicenes. Nat. Chem. 2023, 15, 872-80.
10. Mandai, R.; Iwasaki, T.; Nozaki, K. Stable yet strongly lewis-acidic anions enabling cooperative catalysis with cationic transition-metal complexes. Angew. Chem. Int. Ed. Engl. 2025, Epub ahead of print.
11. Gao, S.; Su, L.; Lu, X.; et al. Direct synthesis of poly(adipate)esters via catalytic isomerizing dual hydroesterificative polymerization of 1,3-butadiene. Angew. Chem. Int. Ed. Engl. 2025, 64, e202506438.
12. Wu, M.; Yun, D.; Wang, F.; et al. Sustainable production of emerging diesel additive from butene by palladium-catalyzed alkoxycarbonylation. ACS. Sustainable. Chem. Eng. 2023, 11, 1837-45.
13. Cao, Z.; Wang, Q.; Neumann, H.; Beller, M. Modular and diverse synthesis of acrylamides by palladium-catalyzed hydroaminocarbonylation of acetylene. Angew. Chem. Int. Ed. Engl. 2024, 63, e202410597.
14. Liu, J.; Neumann, H.; Sang, R.; et al. Palladium-catalyzed ethylene methoxycarbonylation using bidentate phosphines: how the stereochemistry of the ligand affects the catalyst’s activity. Chemistry 2025, 31, e202500476.
15. Luo, C.; Wu, C.; Wang, X.; Han, Z.; Wang, Z.; Ding, K. Ruthenium-catalyzed carbocycle-selective hydrogenation of fused heteroarenes. J. Am. Chem. Soc. 2024, 146, 35043-56.
16. Rivera, G.; Ramírez, D.; Martínez, O.; Bernès, S.; Garcia, D.; Yreta, J. Iridium(I) complexes with fluorinated N-heterocyclic carbene and phosphine ligands for the transfer hydrogenation catalysis of ketones and aldehydes. J. Organomet. Chem. 2025, 1036, 123718.
17. Dong, Y.; Yang, P.; Zhao, S.; Li, Y. Reductive cyanation of organic chlorides using CO2 and NH3 via Triphos-Ni(I) species. Nat. Commun. 2020, 11, 4096.
18. Chen, X. W.; Li, C.; Gui, Y. Y.; et al. Atropisomeric carboxylic acids synthesis via nickel-catalyzed enantioconvergent carboxylation of aza-biaryl triflates with CO2. Angew. Chem. Int. Ed. Engl. 2024, 63, e202403401.
19. Li, C.; Chen, X. W.; Liao, L. L.; et al. Nickel-catalyzed atroposelective carbo-carboxylation of alkynes with CO2: en route to axially chiral carboxylic acids. Angew. Chem. Int. Ed. Engl. 2025, 64, e202413305.
20. Liao, H.; Zhang, H.; Wei, X.; et al. Switching of the CO2 hydrogenation selectivity via organic amines over ionic porous polymer-supported Ru single atom catalyst. Chem. Eng. J. 2025, 519, 165038.
21. Kumar, P.; Das, A.; Maji, B. Phosphorus containing porous organic polymers: synthetic techniques and applications in organic synthesis and catalysis. Org. Biomol. Chem. 2021, 19, 4174-92.
22. Kang, X.; Zhang, X.; Yang, Q.; et al. Creation of heterogeneous single-site catalysts for RWGS reaction: ruthenium complex on porous ionic polymers via electrostatic interactions. ACS. Appl. Mater. Interfaces. 2025, 17, 13773-82.
23. Sun, Q.; Dai, Z.; Liu, X.; et al. Highly efficient heterogeneous hydroformylation over rh-metalated porous organic polymers: synergistic effect of high ligand concentration and flexible framework. J. Am. Chem. Soc. 2015, 137, 5204-9.
24. Gäumann, P.; Cartagenova, D.; Ranocchiari, M. Phosphine-functionalized porous materials for catalytic organic synthesis. Eur. J. Org. Chem. 2022, 2022, e202201006.
25. Wang, G.; Jiang, M.; Fan, B.; et al. Selective association enforced by the confinement effect to boost the regioselectivity of vinyl acetate hydroformylation. ACS. Catal. 2024, 14, 16014-24.
26. Zhu, Y.; Wang, Z.; Zhao, Y.; Zhou, X.; Zhang, Y.; Yang, Y. Confined trinuclear Ru sites in phosphine-incorporated porous organic polymers for the direct synthesis of alcohols from reductive hydroformylation of alkenes. ACS. Catal. 2024, 14, 4593-600.
27. Wu, M.; Gao, G.; Yang, C.; Sun, P.; Li, F. Highly active Rh catalysts with strong π-acceptor phosphine-containing porous organic polymers for alkene hydroformylation. J. Org. Chem. 2023, 88, 5059-68.
28. Zhao, K.; Wang, H.; Wang, X.; et al. Confinement of atomically dispersed Rh catalysts within porous monophosphine polymers for regioselective hydroformylation of alkenes. J. Catal. 2021, 401, 321-30.
29. Huang, J.; Tarábek, J.; Kulkarni, R.; et al. A π-conjugated, covalent phosphinine framework. Chemistry 2019, 25, 12342-8.
30. Grunenberg, L.; Savasci, G.; Terban, M. W.; et al. Amine-linked covalent organic frameworks as a platform for postsynthetic structure interconversion and pore-wall modification. J. Am. Chem. Soc. 2021, 143, 3430-8.
31. Wen, D.; Chen, J.; Zheng, Q.; Yang, S.; Tu, T. Directly knitted ruthenium pincer complexes with enhanced activity as recyclable single-site catalysts for hydrogenation of CO2 to methanol. CCS. Chem. 2023, 5, 1602-11.
32. Li, B.; Guan, Z.; Wang, W.; et al. Highly dispersed pd catalyst locked in knitting aryl network polymers for Suzuki-Miyaura coupling reactions of aryl chlorides in aqueous media. Adv. Mater. 2012, 24, 3390-5.
33. Jiang, M.; Ding, Y.; Yan, L.; Song, X.; Lin, R. Rh catalysts supported on knitting aryl network polymers for the hydroformylation of higher olefins. J. Catal. 2014, 35, 1456-64.
34. Yang, Y.; Wang, T.; Jing, X.; Zhu, G. Phosphine-based porous aromatic frameworks for gold nanoparticle immobilization with superior catalytic activities. J. Mater. Chem. A. 2019, 7, 10004-9.
35. Yang, Y.; Yang, Y.; Wang, T.; Tian, Y.; Jing, X.; Zhu, G. Highly selective reduction of nitroarenes with gold nano-catalysts immobilized in porous aromatic frameworks. Microporous. Mesoporous. Mater. 2020, 306, 110393.
36. Qiao, S.; Du, Z.; Yang, C.; et al. Phosphine-containing microporous networks: high selectivity toward carbon dioxide to methane. Polymer 2014, 55, 1177-82.
37. Qiao, S.; Huang, W.; Du, Z.; Chen, X.; Shieh, F.; Yang, R. Phosphine oxide-based conjugated microporous polymers with excellent CO2 capture properties. New. J. Chem. 2015, 39, 136-41.
38. Hu, K.; Tang, Y.; Cui, J.; et al. Location matters: cooperativity of catalytic partners in porous organic polymers for enhanced CO2 transformation. Chem. Commun. 2019, 55, 9180-3.
39. Ren, Z.; Lyu, Y.; Song, X.; et al. Dual-ionically bound single-site rhodium on porous ionic polymer rivals commercial methanol carbonylation catalysts. Adv. Mater. 2019, 31, e1904976.
40. Zhang, Q.; Zhang, S.; Li, S. Novel functional organic network containing quaternary phosphonium and tertiary phosphorus. Macromolecules 2012, 45, 2981-8.
41. Hausoul, P. J. C.; Eggenhuisen, T. M.; Nand, D.; et al. Development of a 4,4′-biphenyl/phosphine-based COF for the heterogeneous Pd-catalysed telomerisation of 1,3-butadiene. Catal. Sci. Technol. 2013, 3, 2571.
42. Ji, G.; Li, C.; Xiao, D.; et al. The effect of the position of cross-linkers on the structure and microenvironment of PPh 3 moiety in porous organic polymers. J. Mater. Chem. A. 2021, 9, 9165-74.
43. Jiang, M.; Yan, L.; Ding, Y.; et al. Ultrastable 3V-PPh3 polymers supported single Rh sites for fixed-bed hydroformylation of olefins. J. Mol. Catal. A. Chem. 2015, 404-405, 211-7.
44. Wang, W.; Li, C.; Yan, L.; Wang, Y.; Jiang, M.; Ding, Y. Ionic liquid/Zn-PPh3 integrated porous organic polymers featuring multifunctional sites: highly active heterogeneous catalyst for cooperative conversion of CO2 to cyclic carbonates. ACS. Catal. 2016, 6, 6091-100.
45. Jiang, X.; Zhao, W.; Wang, W.; et al. One-pot approach to Pd-loaded porous polymers with properties tunable by the oxidation state of the phosphorus core. Polym. Chem. 2015, 6, 6351-7.
46. Fox, M. A.; Harris, J. E.; Heider, S.; et al. A simple synthesis of trans-RuCl(C≡CR)(dppe)2 complexes and representative molecular structures. J. Organomet. Chem. 2009, 694, 2350-8.
47. Al-noaimi, M.; Warad, I.; Abdel-rahman, O. S.; Awwadi, F. F.; Haddad, S. F.; Hadda, T. B. Synthesis, structure, spectroscopic properties, electrochemistry, and DFT correlative studies of trans-[Ru(P-P)2Cl2] complexes. Polyhedron 2013, 62, 110-9.
48. Luo, Y.; Yang, Z.; Suo, X.; et al. Robust perfluorinated porous organic networks: succinct synthetic strategy and application in chlorofluorocarbons adsorption. Nano. Res. 2021, 14, 3282-7.
49. Yang, Z.; Guo, W.; Mahurin, S. M.; et al. Surpassing robeson upper limit for CO2/N2 separation with fluorinated carbon molecular sieve membranes. Chem 2020, 6, 631-45.
50. Chen, L. J.; Cai, Z. B.; Li, S. L.; et al. Novel red light-emitting two-photon absorption compounds with large Stokes shifts for living cell imaging. Spectrochim. Acta. A. Mol. Biomol. Spectrosc. 2022, 282, 121660.
51. Chikunova, I. V.; Bakiev, A. N.; Shklyaeva, E. V.; Abashev, G. G. Synthesis and optical and electrochemical properties of new push-pull chromophores containing a 1,2-Di(thiophen-2-yl)ethene fragment. Russ. J. Org. Chem. 2022, 58, 990-6.
52. Vuković, V. D.; Richmond, E.; Wolf, E.; Moran, J. Catalytic friedel-crafts reactions of highly electronically deactivated benzylic alcohols. Angew. Chem. Int. Ed. Engl. 2017, 56, 3085-9.
53. Li, Y.; Wu, Y.; Wang, S.; et al. Fluorinated porous organic frameworks for C2F6/CF4 gases separation. Chem. Synth. 2024, 4, 48.
54. Chiplunkar, P. P.; Pratap, A. P. Utilization of sunflower acid oil for synthesis of alkyd resin. Prog. Org. Coat. 2016, 93, 61-7.
55. Zhao, Y.; Yao, K. X.; Teng, B.; Zhang, T.; Han, Y. A perfluorinated covalent triazine-based framework for highly selective and water-tolerant CO2 capture. Energy. Environ. Sci. 2013, 6, 3684.
56. Brown, T. H.; Green, P. J. Phosphorus-31 and rhodium-103 nuclear magnetic resonance spectra of some rhodium(I) and rhodium(III) phosphine complexes. J. Am. Chem. Soc. 1970, 92, 2359-62.
57. Chen, H.; Yang, Z.; Do-Thanh, C. L.; Dai, S. What fluorine can do in CO2 chemistry: applications from homogeneous to heterogeneous systems. ChemSusChem 2020, 13, 6182-200.
58. Gunasekar, G. H.; Padmanaban, S.; Park, K.; Jung, K. D.; Yoon, S. An efficient and practical system for the synthesis of N,N-Dimethylformamide by CO2 hydrogenation using a heterogeneous Ru catalyst: from batch to continuous flow. ChemSusChem 2020, 13, 1735-9.
59. Shen, Y.; Zheng, Q.; Chen, Z. N.; et al. Highly efficient and selective N-Formylation of amines with CO2 and H2 catalyzed by porous organometallic polymers. Angew. Chem. Int. Ed. Engl. 2021, 60, 4125-32.
60. Ouyang, H.; Peng, M.; Song, K.; Wang, S.; Gao, H.; Tan, B. Synthesis of metal-phenanthroline-modified hypercrosslinked polymer for enhanced CO2 capture and conversion via chemical and photocatalytic methods under ambient conditions. Chem. Synth. 2025, 5, 1.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].