

The synergistic effects of Pt-OH and Pt0 enhanced the low-temperature catalytic performance of Pt/CNTs for preferential CO oxidation in a H2 stream
 0
0 
Abstract
In the current era of pursuing low carbon, hydrogen energy emerged as a pivotal zero-carbon energy source, but faces a critical challenge in terms of its practical implementation. Trace CO contaminants (~1% CO) in industrial hydrogen streams derived from hydrocarbon reforming can irreversibly poison Pt electrodes of proton-exchange-membrane fuel cells. The preferential oxidation of CO in H2-rich stream (PROX) is considered as an effective strategy to eliminate CO. In this work, we synthesized the catalyst of 0.3Pt/CNTs-CIW via a colloidal impregnation method, which achieves unprecedented performance in PROX, delivering 100% CO conversion with 50% CO2 selectivity across an exceptionally broad operational temperature window from 20 to 200 °C with a minimal Pt content of 0.3 wt%. Various characterizations reveal that the high efficiency derives from the active structures with the synergies of Pt-OH and Pt0. The adsorption of CO is weakened by the construction of Pt-OH and couples with OH in the form of COOH* which is then oxidized by OH* derived from adsorbed O2 and H2 on Pt0 with a low activation energy, resulting in high efficiency of CO oxidation. Beyond CO-PROX, this bifunction activation paradigm offers transformative potential for diverse catalytic systems involving competitive adsorption and redox coupling, such as low-temperature oxidation of methane and volatile organic compounds abatement.
Keywords
INTRODUCTION
Hydrogen energy, recognized for its zero-carbon and clean properties, is at the forefront of research in the quest for carbon neutrality. Global industrial hydrogen predominantly comes from steam reforming of coal or natural gas. In general, the produced hydrogen contains approximately 0.5 to 2 vol% CO, which necessitates further purification to meet the stringent purity requirements for applications such as fuel cells and processes such as ammonia synthesis[1,2]. Particularly, for proton-exchange-membrane fuel cells - a key application of hydrogen, CO levels must be reduced to below 10 ppm to prevent the poisonous effect of CO on the Pt anode. The preferential oxidation of CO in a rich H2 atmosphere emerges as a crucial technology for achieving the high purity levels essential for hydrogen utilization[3].
For CO-PROX (preferential oxidation of CO in H2-rich stream) reaction, supported Pt catalyst has been intensively studied as it is taken as the most promising candidate for practical applications[4]. However, the strong adsorption of CO on Pt inhibits oxygen adsorption and activation, leading to inferior activity at low temperatures[5]. The popular and effective approaches are to alter the adsorption strength of reactants or products by regulating the particle sizes, the electronic states, or the coordination environments of Pt species[6]. Notable progress has been achieved through innovative catalyst design. A breakthrough development involved the synthesis of FeOx-supported isolated Pt single-atom catalysts, where the formation of high-valent Pt centers with partially vacant 5d orbitals effectively reduced CO adsorption energy, leading to exceptional CO-PROX performance[7]. Complementary studies demonstrate that introducing promoter elements (e.g., K[8], Co[9,10], Cu[11,12], or Ce[13]) could optimize Pt electronic configurations, thereby weakening CO adsorption through electronic effects. Particularly insightful work by Chen et al. revealed that hydroxyl-decorated Pt species on Al2O3 imparted superior activity of CO oxidation than those of metallic Pt and PtOx, attributed to the optimal balance between CO adsorption weakening and oxygen activation enhancement[14].
The other strategies for alleviating CO poisoning problems are to engineer the synergistic sites for the non-competitive adsorptions of CO and O2 via using supports with abundant oxygen vacancies[15], adding structural promoters[16,17] or constructing the metal-oxide interfaces[18]. The excellent activity for CO oxidation and superior sulfur tolerance were obtained on Pt/SnO2 by the strong metal support interaction, which drove the formation of the PtSn or Pt3Sn intermetallic compound shells at the Pt-SnO2 interface[19]. These intermetallic shells strategically restricted CO diffusion and adsorption on Pt particles. The construction of a ZnO shell on ZnPt nanoparticles could trap CO as surface carbonates at low temperatures and release CO2 at a critical temperature, relieving the poison effect of CO on Pt[20]. Notably, carbon supports inherently lack oxygen vacancy-mediated activation and bifunctional metal-oxide interfaces. Interface engineering and bifunctional design have emerged as effective strategies to address the limitations. A representative example involves architecting Pt-CuO nano-islands on mesoporous silica substrates, where the created metal-oxide interfaces significantly enhanced O2 adsorption/activation capabilities while maintaining efficient CO adsorption sites, thereby optimizing the overall reaction kinetics[11]. This interfacial synergy concept has been further developed in Pt-Fe/multi-walled carbon nanotube (MWCNT) systems, where the catalytic surface bifurcated into distinct functional domains: Pt-Fe sites preferentially adsorbed and activated O2, while adjacent Pt domains selectively interact with CO molecules, achieving spatial separation of reactant adsorption processes[21]. The introduced CoO species and Pt species on carbon nanotubes (CNTs) played synergistic effects in CO-PROX reaction, where CoO served as an oxygen supply and Pt provided sites for CO adsorption[10]. A sandwich surface [NiO1-x/Pt/Ni/Pt(111)] could enhance the catalytic activity for CO oxidation where the highly dispersed NiO1-x nanostructures on Pt provided sites for O2 dissociation to alleviate CO poisoning on Pt and the subsurface Ni species could facilitate the reaction of CO with atomic O species[22]. However, it is still a rigorous challenge over the carbon materials-supported single metal of Pt catalysts for achieving complete CO conversion (XCO) at low temperatures and sustaining in a wide temperature window, let alone with low Pt loadings to reduce the cost.
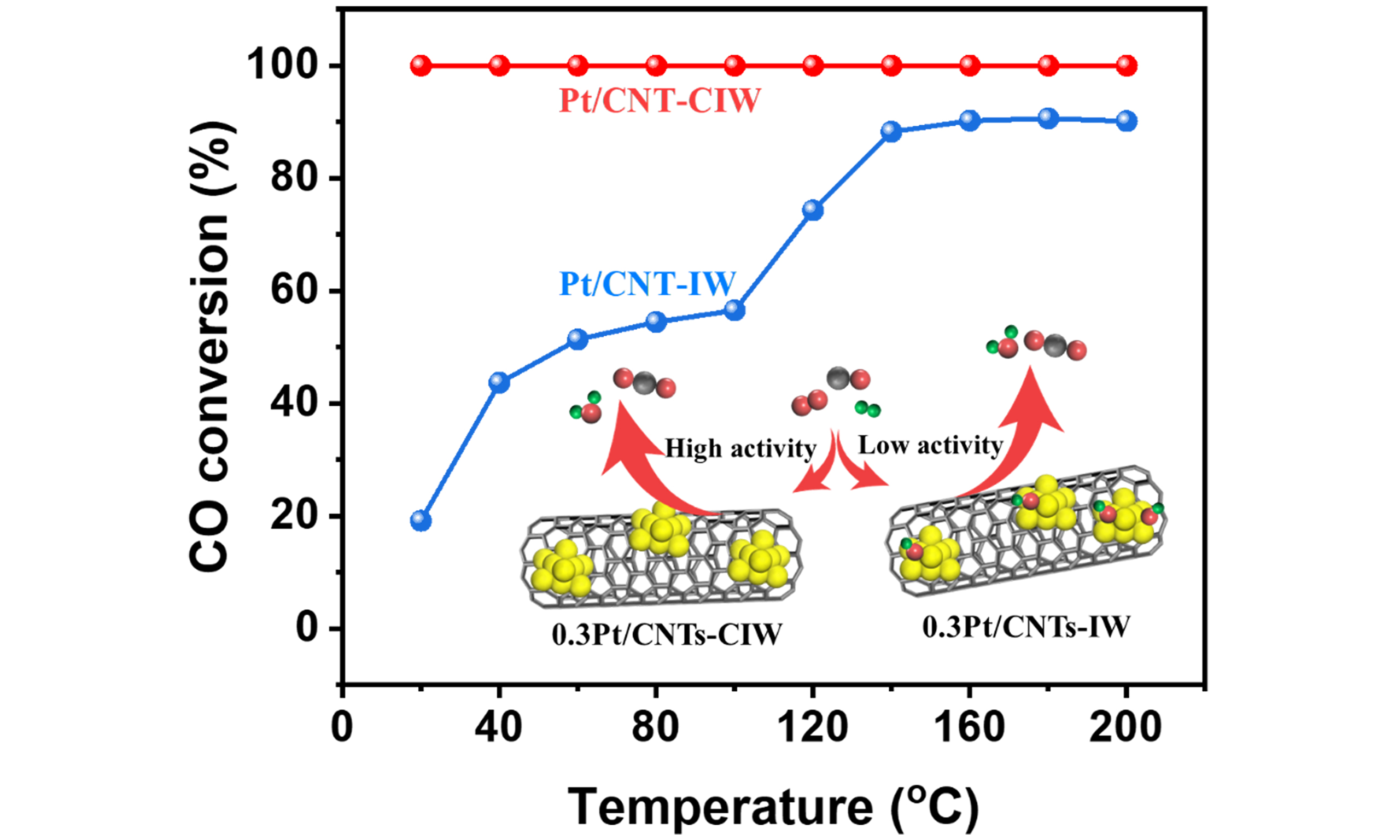
Herein, CNTs were used as support, which possessed high surface area, superior thermal conductivity and abundant surface groups for noble metal anchoring[23,24]. We engineered the Pt clusters bonded with OH groups on the CNTs support (Pt/CNTs-CIW) via a colloidal-impregnation method. The catalyst of Pt/CNTs-IW prepared by the impregnation method was taken as a comparison. Comparative performance evaluation in CO-PROX revealed striking differences: The OH-modified Pt/CNTs-CIW demonstrates complete XCO across an exceptionally broad temperature window (20-200 °C) at ultralow Pt loading
EXPERIMENTAL
Synthesis of catalysts
CNTs were purchased from Jiangsu Xianfeng Nanomaterials Technology Co., Ltd. Ethylene glycol (EG, ≥ 99.5%) and sodium hydroxide (NaOH, ≥ 98.0%) were purchased from Kermel Chemical Reagent Company. Chloroplatinic acid hexahydrate (H2PtCl6·6H2O) was purchased from Huawei Ruike Technology Co., Ltd. All the reagents were used as received and without further purification.
Preparation of Pt colloid
Pt colloid was synthesized via an alkaline ethylene glycol (EG) reduction method[25]. Specifically, a solution of H2PtCl6·6H2O in EG with a concentration of 3.8 M and a solution of NaOH in EG with a concentration of 0.25 M were mixed at room temperature. After being stirred at 800 rpm for 20 min to obtain a homogeneous solution, the mixture was transferred to the oil bath at 160 °C and then continuously stirred in the protection of N2 atmosphere. After
Preparation of Pt/CNTs
Firstly, the Pt/CNTs catalysts were prepared via a colloidal impregnation with Pt colloid as the precursor. A certain volume of Pt colloid was taken and diluted with EG, and then the CNTs dried at 110 °C were added dropwise into the solution. The mixture was manually stirred at room temperature for approximately 0.5 h and then left to stand at room temperature overnight. Then, the sample was dried in a vacuum oven at 120 °C for 12 h to remove the solvent. The obtained catalyst was denoted as xPt/CNTs-CIW, where x represents the mass fraction of Pt. For comparison, a sample prepared by the incipient wetness impregnation method using H2PtCl6·6H2O as the precursor was denoted as xPt/CNTs-IW (x represents the mass fraction of Pt).
Characterizations of catalysts
The Pt loading of the catalyst was determined by an Inductively Coupled Plasma-Optical Emission Spectrometer (ICP-OES) (720ES, Agilent, USA). The crystal structures of Pt/CNTs were determined using an X-ray powder diffractometer (XRD) on X’pert PRO, Panalytical, Netherlands, where the Cu Kα radiation (λ = 0.15432 nm, 40 kV, 40 mA) was used. The intensity data were collected in the 2θ range from 10° to 80° with a scanning speed of 5°·min-1. The surface areas and pore size distributions of the samples were calculated by the MultiPoint Bruner-Emmet-Teller (BET) method according to the N2 adsorption-desorption isotherms at -196 °C, the data of which was collected on the instrument of Autosorb-IQ, Quantachrome. To observe the distribution of Pt and the particle size, the reduced catalysts were studied by high-resolution transmission electron microscopy (HRTEM) and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) on the apparatus of Tecnai G2 F30 S-Twin (FEI, USA).
H2-programmed temperature reduction (H2-TPR) experiments were carried out on the chemical absorption analyzer (VDSorb-91i, Vodo, China). First, about 50 mg of the sample was loaded into a U-shaped quartz reactor, and subjected to thermal pre-treatment under Ar flow at 110 °C for 1 h to eliminate physically adsorbed water and surface contaminants. After cooling to room temperature, the catalyst was heated to
Surface electron state analysis was tested through quasi in situ X-ray photoelectron spectroscopy (XPS) using Thermo Scientific Escalab 250Xi with a monochromatic Al Kα X-ray (hν = 1,486.6 eV). Before measurement, the catalysts were pretreated with H2 for 30 min at 300 °C, after which the temperature was lowered to ambient temperature followed by passivation with 1 vol.% O2/Ar for 2 h. Then the reduced samples were immediately sealed in an Ar-filled sample tube and transferred to the analysis chamber as soon as possible. Charge compensation was achieved by referencing adventitious carbon (C1s = 284.8 eV) to account for work function differences between the sample and spectrometer.
CO temperature-programmed reduction (CO-TPR) experiments were conducted on a custom-built temperature-programmed chemisorbed device coupled with mass spectrometric detection. First, approximately 50 mg of the sample was loaded into a U-shaped quartz reactor and subjected to reductive pre-treatment in 5 vol.% H2/Ar for 30 min at 300 °C. Following cooling to room temperature, the system underwent rigorous flushing with inert gas (Ar, 30 mL·min-1) for 30 min to eliminate residual hydrogen and adsorbed water. The catalyst was exposed to a CO/Ar mixture (30 mL·min-1) while executing a controlled temperature ramp (10 °C·min-1) from 25 to 600 °C. Evolved gas analysis was performed via online mass spectrometry (Hyde Analytical, UK), monitoring hydrogen evolution (m/z = 2) and carbon dioxide formation (m/z = 44) as proxies for reduction pathways.
The temperature programming surface reaction (TPSR) experiments were conducted in a self-assembled temperature-programmed chemisorbed device, and the exhaust gas was detected by an online mass spectrometry (Hyde Company, UK). A 50 mg sample was added into a U-shaped quartz tube and pre-reduced by 5 vol% H2/Ar at 300 °C for 30 min and cooled to room temperature in the gas of Ar. The gas of
The hydrogen isotope tracing experiment was also conducted on a self-assembled temperature-programmed chemisorbed device, and the exhaust gas was detected by an online mass spectrometry (Hyde Company, UK). The sample was reduced by 5 vol% H2/Ar at 300 °C for 30 min and cooled to room temperature in the gas of Ar. The gas of 40 vol% D2/4 vol% O2/Ar with a flow rate of 30 mL·min-1 was injected into the reaction tube for adsorption followed by Ar. Afterward, the gas was switched to CO2 gas (30 mL·min-1), and simultaneously, the tube was heated from room temperature to 600 °C. The signals were identified using online mass spectroscopy.
Evolution of catalytic performance
The performances of catalysts for CO-PROX were evaluated in a continuous flow reactor with a fixed bed under atmospheric pressure. A certain amount of catalyst was filled into a U-shaped quartz reaction tube with an inner diameter of approximately 8 mm and a length of 250 mm. The loaded catalyst and quartz wool form a sandwich shape, with a catalyst in the middle. Before measurements, the catalyst was reduced in
XCO was calculated by
The CO2 selectivity (SCO2) was quantitatively determined through oxygen mass balance analysis. The selectivity metric was defined as
Where
As for the kinetic measurements, the samples were diluted with quartz sand and the performance was evaluated at a promoted WHSV to ensure that the XCO was lower than 20%. The CO reaction rate was calculated based on the XCO, as given in
where FCO represents the molar gas flow rate of CO in mol·h-1 and the mPt is the mass of Pt.
The apparent activation energy of the reaction was determined by the Arrhenius equation:
where k represents the reaction rate constant, A is the pre-exponential factor, R represents the gas constant, and T is the temperature.
During the test of CO reaction order αCO, the oxygen partial pressure PO2 remained constant while the partial pressure of CO was tuned. Correspondingly, the CO partial pressure PCO remained constant while the partial pressure of O2 was regulated during the measurement of O2 reaction order αO2. The values of αCO and αO2 were respectively calculated according to
RESULTS AND DISCUSSION
Catalytic performance for CO-PROX
The CO-PROX performances of Pt/CNT catalysts were rigorously evaluated under a simulated reformate gas mixture (1 vol% CO, 1 vol% O2, 40 vol% H2, balance Ar) across a technologically critical temperature window (20-200°C). This range specifically targets the operational requirements of low-temperature water-gas shift (WGS) reactors, where effective CO removal must precede downstream hydrogen utilization in fuel cell systems[26]. As observed in Figure 1, on the pure support of CNTs, no XCO is detected. With the loading of
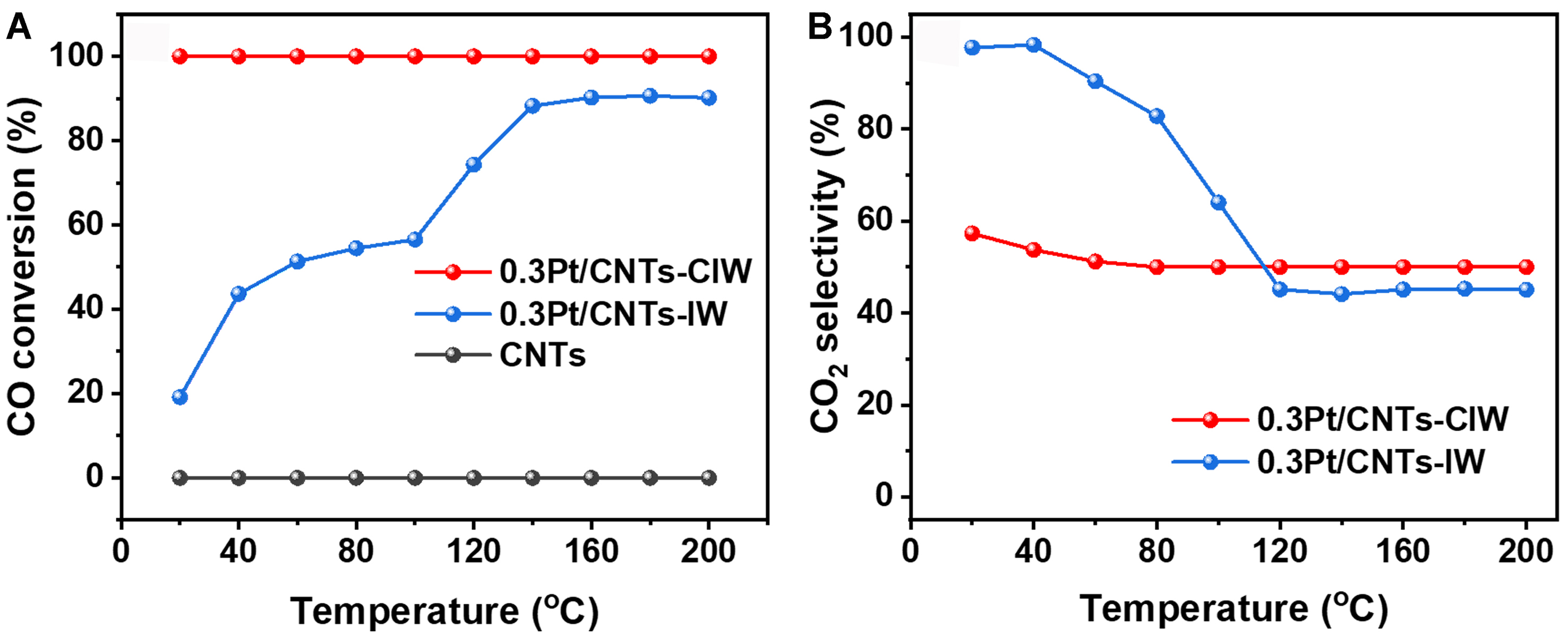
Figure 1. (A) Temperature-dependent CO conversion profiles and (B) CO2 selectivity evolution in CO-PROX over CNTs, 0.3Pt/CNTs-CIW, and 0.3Pt/CNTs-IW catalysts. Reaction conditions: 1 vol% CO + 1 vol% O2 + 40 vol% H2, and Ar balance, WHSV:
Comparisons between Pt/CNTs catalyst and the reported Pt-based catalysts in CO-PROX reaction
| Catalyst | Pt loading (wt%)a | CO: O2: H2 | WHSV (mL·h-1·gcat-1) | T100 (°C)b | CO2 Sel. (%) | Ref. |
| Pt/CNTs-CIW | 0.3 | 1:1:40 | 18,000 | 20-200 | 50 | This work |
| Pt/CNTs-p | 3 | 2:1:97 | 25,000 | 40 | 100 | [27] |
| Pt-Fe/CB | 4 | 1:0.5:50 | 30,000 | 30-45 | 100 | [28] |
| Cu@Pt-Fe/CB | 0.9 | 1:0.5:50 | 30,000 | 30-40 | 100 | [29] |
| 0.75Pt0.2Fe/ND@G | 0.75 | 1:0.5:48 | 45,000 | 30-40 | 100 | [17] |
| Pt-Co/CNTs | 4 | 1:1:50 | 30,000 | 40-200 | 51 | [10] |
| Pt-Fe/CNTs | 4 | 1:1:49 | 120,000 | 40-200 | 50 | [21] |
| Pt-Sn/AC-N | 1 | 1:1:60 | 24,000 | / | 41 | [30] |
| Pt-Ce/AC | 1 | 1:1:60 | 24,000 | 50 | 50 | [31] |
| Pt/Al2O3-R200 | 1 | 1:0.5:40 | 20,000 | 20-100 | 100 | [14] |
| Pt3Co/α-Al2O3 | 0.87 | 1:1:50 | 75,000 | 140-260 | 50 | [9] |
Catalyst structures
The catalyst of Pt/CNT-CIW shows robust catalytic performance in CO-PROX reaction where 100% XCO is obtained in a wide temperature range of 20-200 °C, which outperforms the catalyst of Pt/CNT-IW. As shown in Scheme 1, the superior catalyst of Pt/CNT-CIW was synthesized via a colloid impregnation method with Pt colloid as a precursor, while Pt/CNT-IW was prepared by the wetness impregnation method with H2PtCl6 as a precursor. Variations in synthesis methods can lead to distinct catalyst structures, which in turn have a significant impact on their catalytic performance.
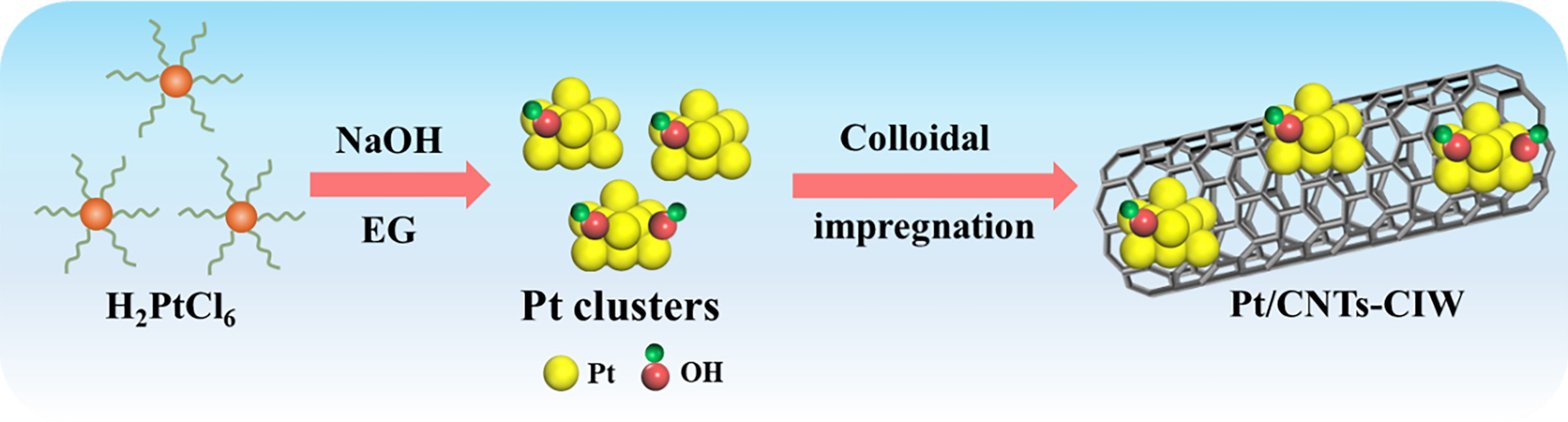
Scheme 1. Schematic diagram of synthesis for Pt/CNTs-CIW catalysts. CIW: Colloidal impregnation-wetting; IW: incipient wetness impregnation; CNTs: carbon nanotubes.
The loadings of Pt on Pt/CNT-CIW and Pt/CNT-IW were determined by ICP. As listed in Supplementary Table 1, the loadings of Pt are 0.30 wt% and 0.27 wt%, respectively, which are similar to the theoretical amount of Pt. The N2 isothermal adsorption curves and the pore size distributions of CNTs, Pt/CNT-CIW and Pt/CNT-IW are displayed in Supplementary Figure 2. Accordingly, the BET surface areas are 247, 207 and 229 m2·g-1, with the pore sizes being centered at 53.7, 44.0 and 48.6 nm [Supplementary Table 1], respectively. The decrease of the BET surface area and pore size can be attributed to the partial occupation of pores by Pt species. The crystal phases of Pt/CNT-CIW and Pt/CNT-IW after reduction were detected by XRD. As shown in Supplementary Figure 3, the typical peaks of CNTs (PDF#41-1487) are observed while the Pt crystal phases (PDF#04-0802) are absent on the catalysts of 0.3Pt/CNT-CIW and 0.3Pt/CNT-IW, indicating the Pt species are in the states of high dispersion[32,33]. In order to observe the Pt particles more intuitively, HAADF-STEM and HRTEM characterizations are performed. The images of Pt colloid were collected to observe the morphologies of Pt particles. As exhibited in Supplementary Figure 4, the Pt species are in a uniform dispersion with a narrow distribution of 1-2 nm. After anchoring the Pt colloid on the support of CNTs, as shown in Figure 2A and B, the size of Pt clusters is revealed with an average size of around 1.5 nm on the reduced 0.3Pt/CNT-CIW according to the statistical results of around 200 particles. A similar average size of Pt at around 1.6 nm is obtained on 0.3Pt/CNT-IW Figure 2C and D. Although the two catalysts possess approximate average size of Pt, particle size distribution is more concentrated on 0.3Pt/CNT-CIW, benefiting from the superiority of the EG reduction method which is versatile for synthesizing size-controlled metal nanoparticles. In addition, the lattice fringes of 0.23 nm are observed in a larger magnification, which can be ascribed to the exposed Pt (111) facets[34]. Based on the distinction of Pt particle size on these two catalysts, it can be deduced that the different performances in CO-PROX are not determined by the particle size, probably from the electronic properties or coordination environment of Pt.
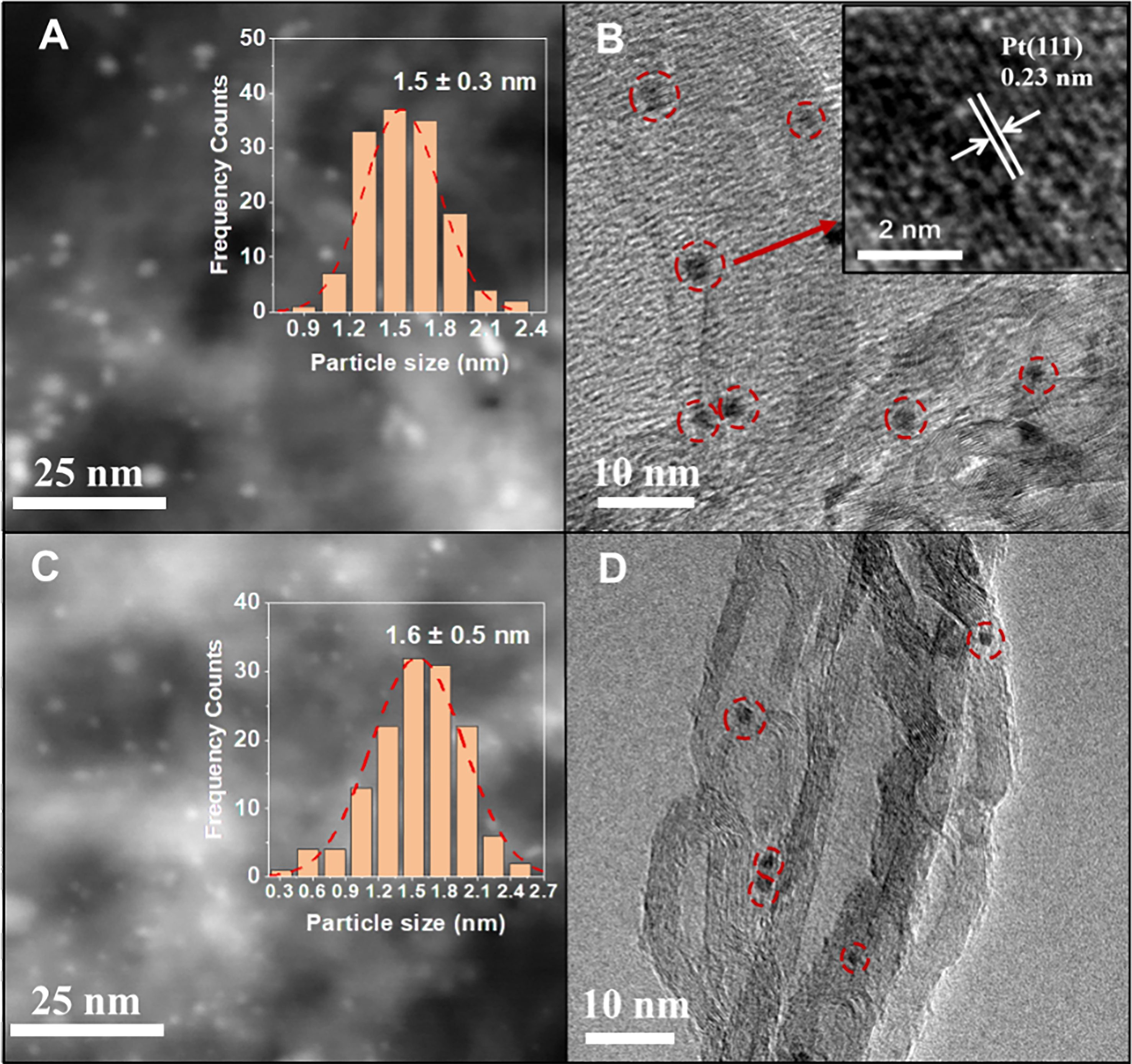
Figure 2. HAADF-STEM and HRTEM images of the reduced 0.3Pt/CNTs-CIW (A and B) and 0.3Pt/CNTs-IW (C and D), the Pt clusters in (B and D) are marked by red cycles. CIW: Colloidal impregnation; CNTs: carbon nanotubes; HAADF-STEM: high-angle annular dark-field scanning transmission electron microscopy; HRTEM: high-resolution transmission electron microscopy; IW: incipient wetness impregnation.
The reduction properties of as-synthesized catalysts were tested by H2-TPR experiments. As shown in Supplementary Figure 5, almost no hydrogen consumption peaks are obtained on the catalyst of 0.3Pt/CNTs-CIW during the whole programming heating procedure from 50 to 600 °C. It is probably due to the fact that the Pt species have been reduced by EG in the synthesis process of Pt colloid and dried under vacuum conditions, which sufficiently protects the Pt from being oxidized[35]. On the catalyst of 0.3Pt/CNTs-IW, two small reduction peaks at 167 and 186 °C can be attributed to the reduction of Pt4+ of H2PtCl6 into low valence. X-ray photoelectron spectra were collected for probing the chemical states of surface Pt species on 0.3Pt/CNTs-CIW and 0.3Pt/CNTs-IW after reduction. As shown in Figure 3A, the spectrum of Pt 4f can be deconvolved into two peaks on both 0.3Pt/CNTs-CIW and 0.3Pt/CNTs-IW. The peak with binding energy of 71.8 eV can be attributed to the metallic Pt species[36]. The other peak located at 72.9 eV belongs to the oxidation state of Pt as the binding energy is exhibited between Pt0 (~71.4 eV) and PtO
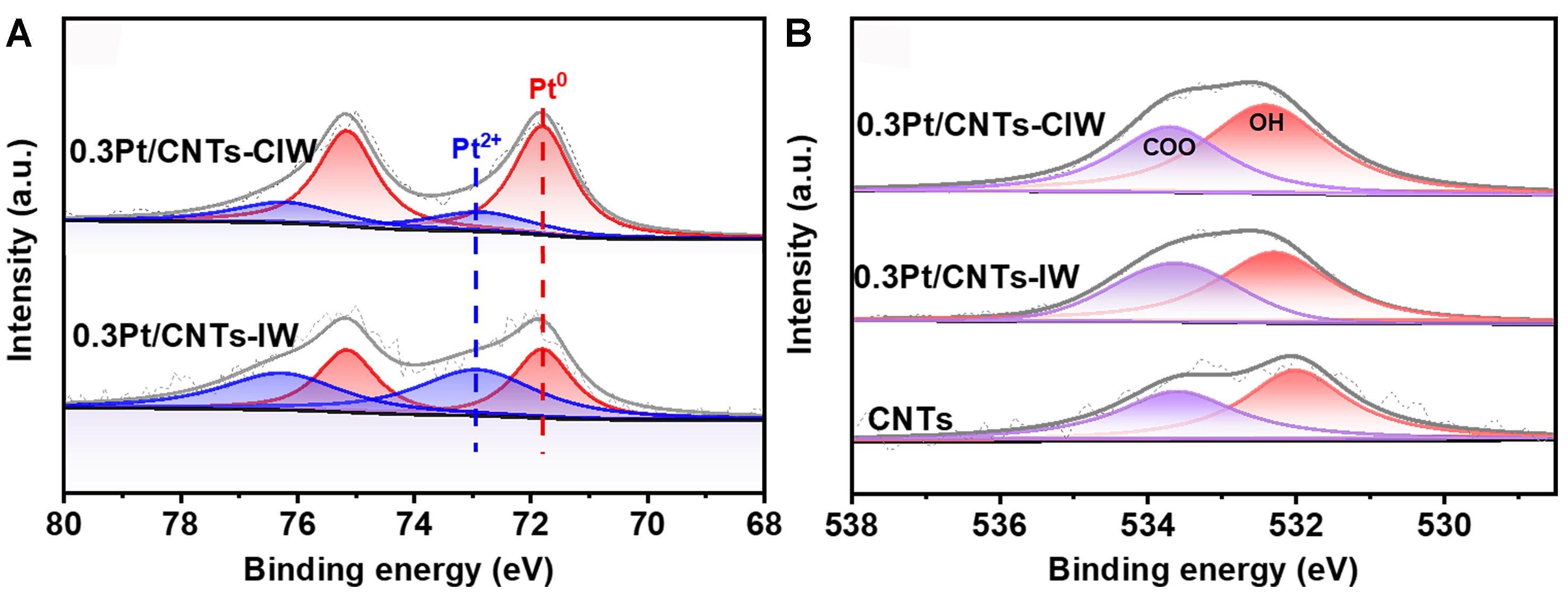
Figure 3. The Pt 4f (A) and O1s (B) XPS spectra of CNTs, 0.3Pt/CNTs-CIW, and 0.3Pt/CNTs-IW. CIW: Colloidal impregnation; CNTs: carbon nanotubes; XPS: X-ray photoelectron spectroscopy.
The surface oxygen species were also investigated by XPS O1s. As displayed in Figure 3B, on the pure CNTs, 0.3Pt/CNTs-IW and 0.3Pt/CNTs-CIW catalysts, both anhydride and esters carboxyl group (-COO-) with binding energy of 533.7 eV and hydroxyls (-OH) with binding energy of 532.4 eV exist[23,39]. As calculated in Supplementary Table 3, the percentages of hydroxyls on the pure CNTs and 0.3Pt/CNTs-IW are similar to the values of 57.8% and 58.9%, respectively, while the value on the catalyst of 0.3Pt/CNTs-CIW increases to 64.3%. The observed hydroxyl (OH) species likely originate from the EG-mediated synthesis of Pt colloids. Previous studies have demonstrated that colloidal nanoparticles synthesized through polyol reduction methods inherently retain surface-bound hydroxyl groups while maintaining absence of organic capping agents, as evidenced by the lack of detectable C-H vibrational modes in surface-sensitive spectroscopy[40]. The Pt species on 0.3Pt/CNTs-CIW with positive valence can be explained by the existence of Pt-OH, which can effectively decrease the adsorption strength of CO[41,42].
To further identify the reducible oxygen species on the surface, the CO-TPR experiments were conducted on the catalysts of 0.3Pt/CNTs-IW and 0.3Pt/CNTs-CIW and the support of CNTs. As shown in Figure 4, the signals of CO2 and H2 are simultaneously detected on the catalyst of 0.3Pt/CNTs-CIW, demonstrating the existence of OH species on Pt species that can react with CO (CO + OH → CO2 + H2)[43]. The presence of OH species on Pt is also verified by the results of O 1s XPS, which exhibited around 64.3% amount on 0.3Pt/CNTs-CIW, much higher than the pure support of CNTs (57.8%). On the catalyst of 0.3Pt/CNTs-IW, there is a little peak at a higher temperature of 440 °C but with no H2 release, which can be attributed to the unreduced PtO by CO. It evidences that almost no OH species exist on the Pt sites as consistent with the result of XPS O1s (58.9% on this sample vs. 57.8% on CNTs). The higher temperature of CO with the unreduced PtO on 0.3Pt/CNTs-IW than the temperature of CO with OH on Pt over 0.3Pt/CNTs-CIW indicates that the hydroxyls decorated on Pt are more active to react with CO than those of the coordinated oxygen species. On the pristine CNTs, there are no peaks of CO and H2, which can be explained by the fact that the OH species without Pt nearby on CNTs are not active for CO oxidation. Therefore, according to the characterizations of XPS and CO-TPR, both Pt-OH and Pt0 species coexist on 0.3Pt/CNTs-CIW surface and Pt-OH tends to be highly active for the process of CO oxidation.
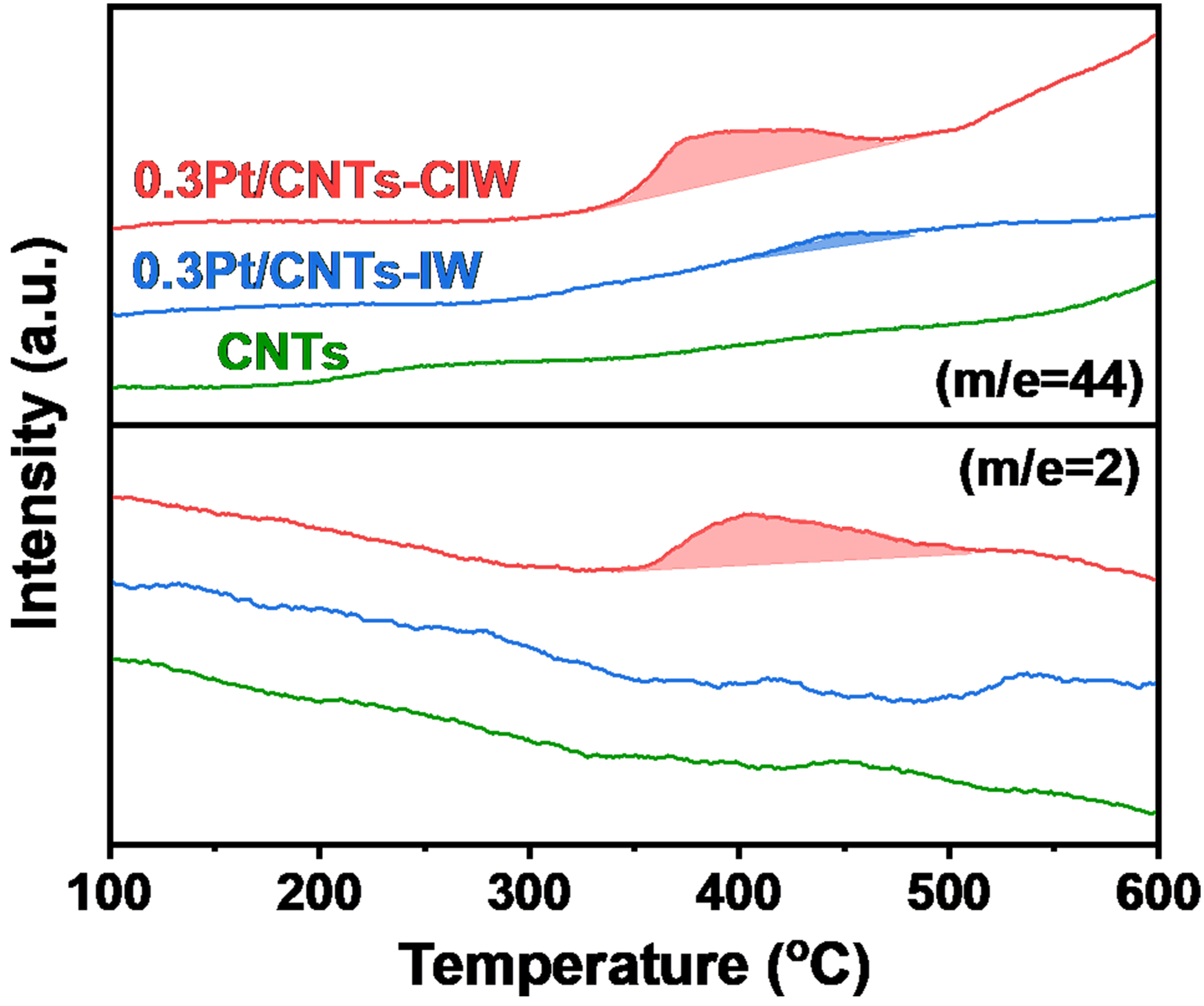
Figure 4. The MS signals of CO2 and H2 during CO-TPR on CNTs, 0.3Pt/CNT-CIW and 0.3Pt/CNT-IW. CIW: Colloidal impregnation-wetting; CNTs: carbon nanotubes; TPR: temperature programmed reduction; MS: mass spectrometry.
Kinetic measurements
For exploring the adsorption behaviors of reactants, kinetic experiments were performed which can provide reliable information for the underlying reaction mechanisms and pathways. For CO-PROX reaction, as shown in Figure 5A and B, the reaction orders of CO and O2 are -0.31 and 0.37, respectively, on the catalyst of 0.3Pt/CNT-IW. Correspondingly, the derived rate is given as r = k [CO]-0.31 [O2]0.37. The negative reaction order of CO provides direct kinetic evidence of its poisoning effect on the 0.3Pt/CNT-IW catalyst, stemming from competitive adsorption that preferentially occupies active sites required for O2 activation. The strong Pt-CO interaction originates from the intensive d-π back-donation electronic configuration inherent to Pt0[44]. Besides, although the catalyst was reduced by H2 at 300 °C, the residual chlorine ions persist as Pt-Cl, which can amplify CO chemisorption strength via electronically polarizing the adjacent Pt atoms[45,46]. Therefore, poor catalytic performance of 0.3Pt/CNT-IW catalyst in CO oxidation at low temperatures is obtained, which is attributed to the scarcity of active oxygen species. With the temperature going up to a high level, a significant amount of CO desorbs from the surface of Pt for O2 adsorption and activation, and the reaction is accelerated.
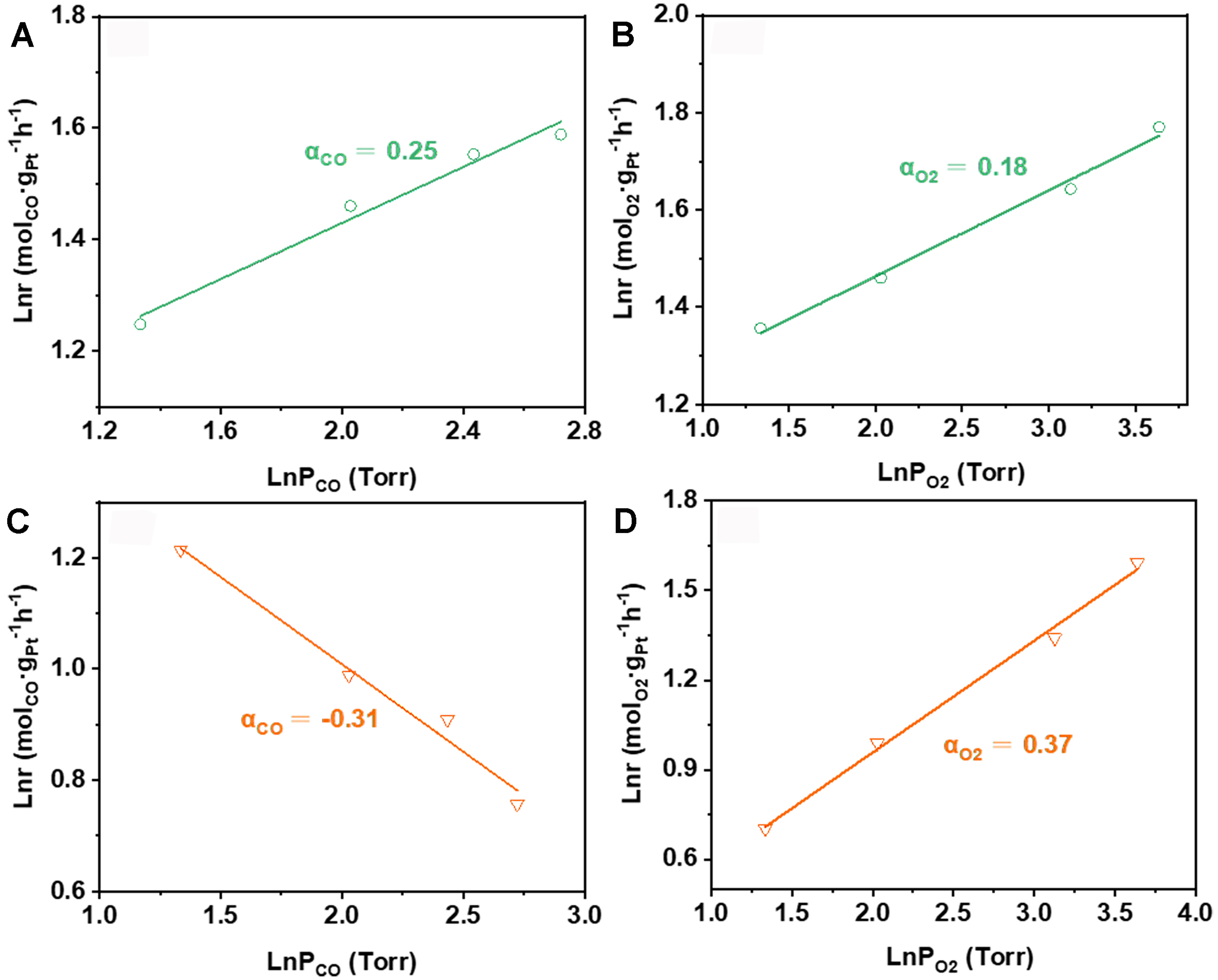
Figure 5. Reaction orders of CO and O2 on (A and C) 0.3Pt/CNTs-CIW and (B and D) 0.3Pt/CNTs-IW at 120 °C. CIW: Colloidal impregnation-wetting; CNTs: carbon nanotubes.
Interestingly, on the catalyst of 0.3Pt/CNT-CIW, the rate is recorded as r = k [CO]0.25 [O2]0.18 based on the calculation of reaction orders Figure 5C and D, where the reaction order of CO flips from negative to positive. This result suggests a hallmark of eliminated CO poisoning effects, benefiting from the presence of hydroxyl groups on Pt clusters[14]. Concurrently, the diminished O2 reaction order (0.18 vs. 0.37 on Pt/CNT-IW) reflects enhanced O2 activation efficiency, enabled by two synergistic mechanisms: Pt0-mediated d-π conjugation effect facilitates O2 dissociation through back-donation of Pt d-electrons to O2 π* orbital; the Pt-OH construction dynamically regulates CO adsorption strength[47,48]. This dual functionality establishes a non-competitive L-H mechanism pathway where spatially separated CO (on Pt-OH) and O2 (on Pt0) activation enable simultaneous reaction processing. The kinetic superiority on 0.3Pt/CNT-CIW also manifests in an ultralow apparent activation energy (Ea = 11.3 kJ·mol-1 [Figure 6A], which is almost half of that on 0.3Pt/CNT-IW (21.7 kJ·mol-1). The higher low-temperature activity of CO oxidation on 0.3Pt/CNT-CIW than that on 0.3Pt/CNT-IW is further ascertained by TPSR profiles [Figure 6B]. Such a dramatic reduction of Ea signifies either rate-determining step alteration (e.g., from O2 dissociation to surface reaction) or pathways bifurcation, consistent with our proposed hydroxyl-assisted mechanism.
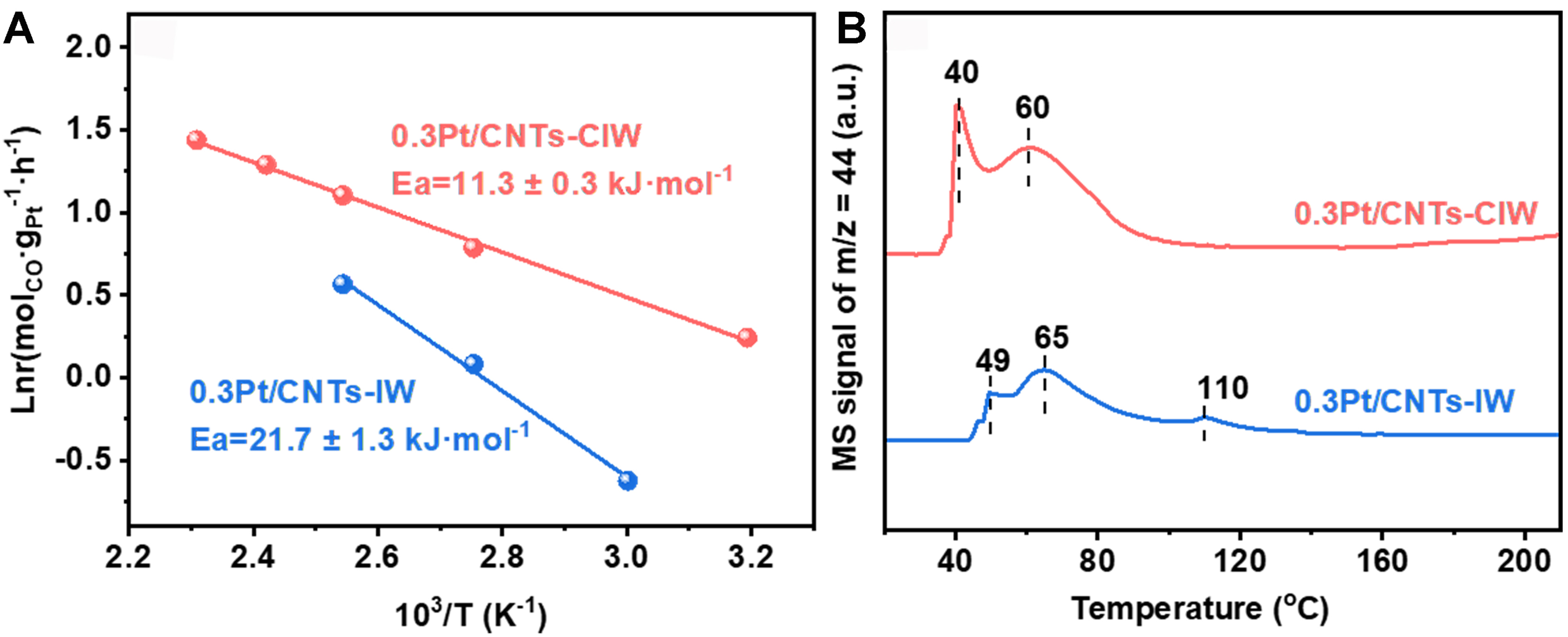
Figure 6. (A) Apparent activation energies and (B) TPSR results of 0.3Pt/CNTs-CIW and 0.3Pt/CNTs-IW. CIW: Colloidal impregnation; CNTs: carbon nanotubes; TPSR: temperature-programmed surface reaction.
Mechanistic elucidation for CO-PROX
The catalyst of 0.3Pt/CNTs-CIW prepared by the colloid impregnation method was found to display impressive activity with complete XCO in a wide temperature window from 20 to 200 °C. Various characterizations such as XPS, CO-TPR and the kinetic results confirm the coexistence of Pt-OH and Pt0 over 0.3Pt/CNTs-CIW and their synergies for CO and O2 activation and oxidation. However, we noted that the role of H2 cannot be neglected. As shown in Supplementary Figure 6, lower conversion of CO with around 58% at 20 °C is displayed during CO oxidation as H2 was removed from the feed gas. In comparison, with the existence of H2, the conversion of CO can be achieved 100%. This result demonstrates that in the process of CO oxidation, H2 plays a promoting role. Furthermore, as shown in Supplementary Figure 7, 0.3Pt/CNTs-CIW exhibits a higher value of specific reaction rate (2.19 molCO·gPt-1·h-1) in CO-PROX that is nearly 3 times higher than the value (0.73 molCO·gPt-1·h-1) in CO oxidation reaction. The presence of H2 could favor the stability of the catalysts by reacting with the activated oxygen species to form hydroxyls[5]. On the catalyst of Ir/Fe(OH)x, the dissociated H on Ir sites could react with the adsorbed O on Fe2+ sites to recover the surface hydroxyls of Fe(OH)x surface, significantly promoting the conversion of CO[47]. Therefore, we speculated that the presence of H2 may participate in the catalytic process of CO oxidation to assist in the maintenance of OH species on Pt in our work.
For studying the participation of H2 in the catalytic cycle of OH, isotope tracing of hydrogen element experiments was carried out. The reduced catalyst of 0.3Pt/CNTs-CIW was exposed to a mixture of O2 and D2 (as a tracer for hydrogen) to a state of adsorption saturation. After Ar purging, CO was introduced followed by which the MS signals of m/z = 44, 18 and 19 (CO2, H2O and HDO, respectively) were recorded during the programmed temperature procedure. As shown in Figure 7A, the introduction of CO resulted in the detection of CO2, along with the signals of both H2O and HDO. It suggests that CO can be oxidized by the surface species that are in the form of OH or OD which are produced from the co-adsorption and activation of O2 and D2 on Pt clusters. To ensure the detected signals of H2O and HDO coming from the CO oxidation process, the programmed temperature procedure was operated under Ar atmosphere. As present in Figure 7B, no H2O and HDO are obtained in the ramping of temperature, indicating that the generated HDO is from the reaction between CO and OD. Besides, it further verified that the active OH groups are not only from the support of CNTs but also from the Pt species that are sufficiently active to oxidize CO, which is identical to the result of CO-TPR. These results indicate the involvement of D2 and provide insights into the reaction pathway and the role of hydrogen in the reaction.
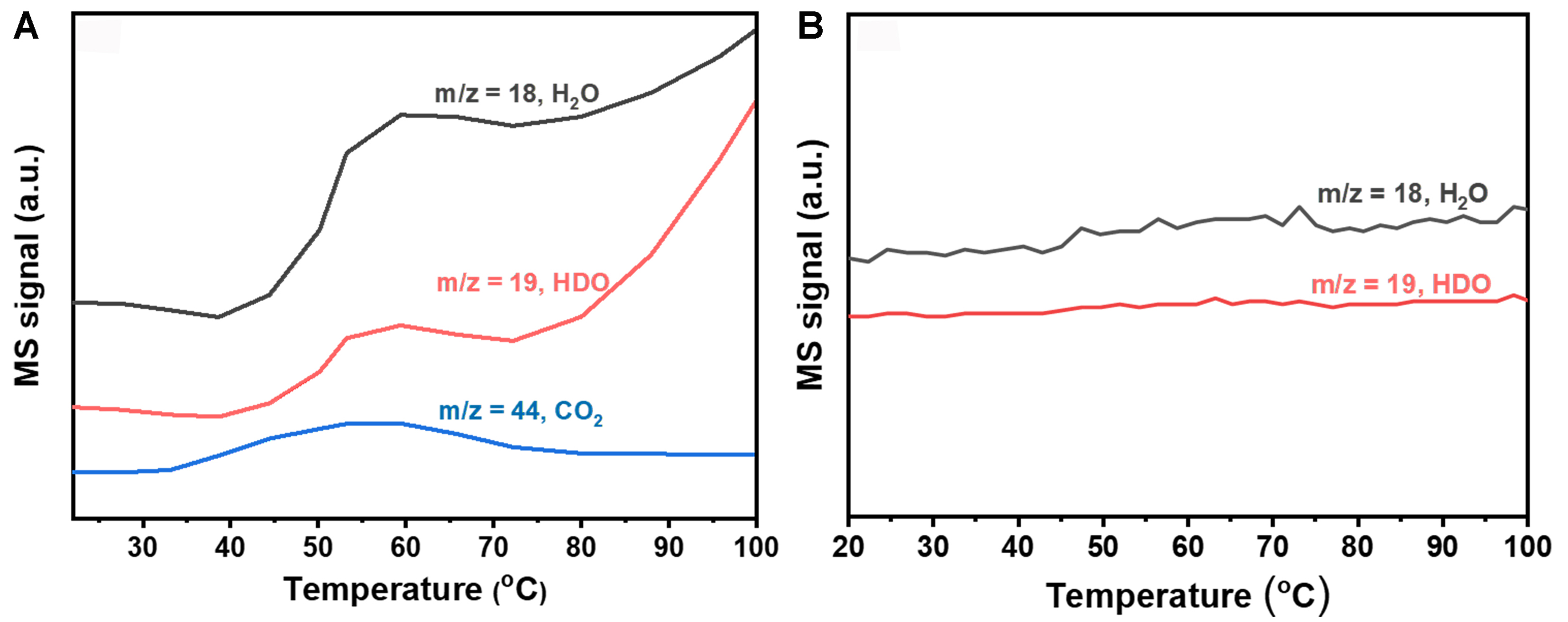
Figure 7. MS signal of CO2, HDO and H2O (m/z = 44, 19, 18) under the atmosphere of CO (A) and Ar (B) over D2 + O2 pre-adsorbed 0.3Pt/CNTs-CIW catalyst. CNTs: Carbon nanotubes; CIW: colloidal impregnation-wetting; HDO: hydrodeoxygenation; m/z: mass-to-charge ratio; MS: mass spectrometry.
For the reaction of CO oxidation, Langmuir-Hinshelwood (L-H) mechanism is considered as a typical reaction mechanism. However, an explicit challenge is that the strong bonding of CO to Pt at low temperatures hindered the adsorption and activation of O2 over Pt species, impeding the catalytic efficiency at low temperatures below 100 °C for CO oxidation[4]. Given the above limitations of the competitive L-H mechanism for Pt-based catalysts, scholars are committed to developing catalysts that could follow the non-competitive L-H mechanism by providing the adsorption and activation sites for O2 on the defect-abundant support or the noble metal-support interfaces or engineering metal-OH structure to weaken CO adsorption[49]. In terms of the reaction pathways with hydroxyls participation for CO oxidation, two general ways are following: one is that the adsorbed CO can be directedly oxidized by the hydroxyls generated from the reaction between the activated oxygen species with the associated H2O or H2[50]; the other one is that the adsorbed CO could couple with surface hydroxyls to form COOH and then oxidized by OH groups with a low activation energy[51].
In this work, we found that the existence of Pt-OH on Pt/CNTs-CIW can effectively weaken the adsorption strength of CO, as evidenced by the transition of CO reaction order from negative to positive in kinetic experiments. This can provide sites for the adsorption and activation of O2 on metallic Pt clusters. CO-TPR experiments reveal that CO oxidation preferentially occurs through reaction with the hydroxyls bonded with Pt rather than those on the support of CNTs. The temperature as high as around 400 °C for CO2 and H2 production during CO-TPR suggests the difficulty of CO being oxidized by hydroxyls. From the result of Supplementary Figure 7, H2 can enhance the catalytic activity of CO-PROX and participate in the process of CO oxidation which is confirmed by the isotopic labeling experiments [Figure 8]. Therefore, the reaction pathway of CO-PROX on Pt/CNTs-CIW is proposed. As shown in Figure 8, the molecules of CO are adsorbed on Pt0 sites and combine with the adjacent Pt-OH to form COOH*. Simultaneously, O2 and H2 undergo dissociative adsorption on metallic Pt clusters to generate active hydroxyl species, OH*, which can react with COOH* to generate CO2 and H2O via a low activation energy. Then the surface hydroxyl regeneration occurs via continuous O2/H2 activation, sustaining the reaction cycle.
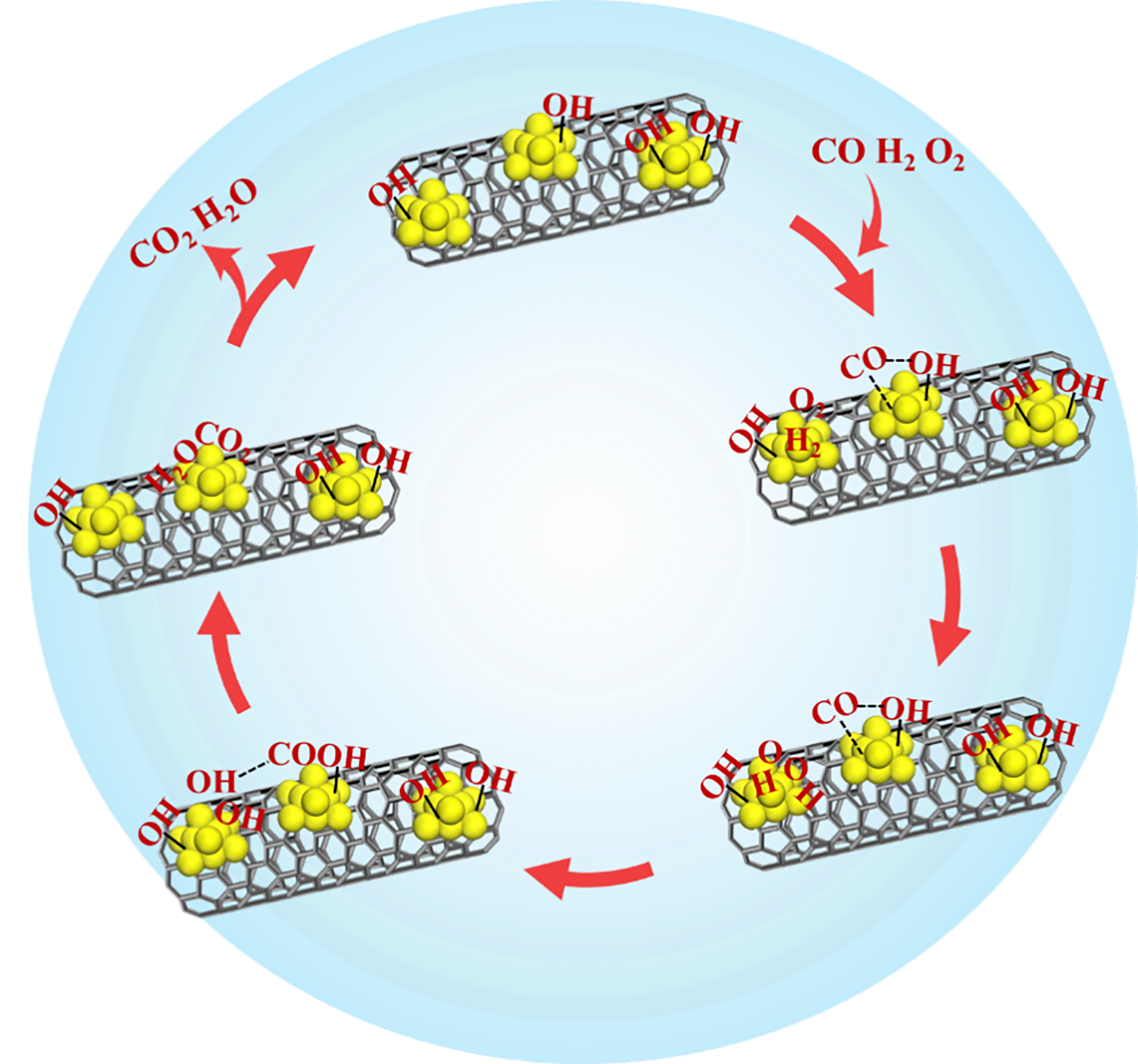
Figure 8. Schematic of proposed reaction pathway over Pt/CNTs-CIW for CO-PROX. CIW: Colloidal impregnation-wetting; CO-PROX: preferential oxidation of CO in H2-rich stream; CNTs: carbon nanotubes.
Measurements for catalyst stability
Catalyst stability represents a pivotal performance metric, particularly for practical applications requiring sustained operational efficiency. For rigorously assessing the durability of 0.3Pt/CNT-CIW, the accelerated stability was tested under extreme conditions with an elevated space velocity of 22,500 mL·h-1·gcat-1 at 80 °C. As shown in Supplementary Figure 8, the catalyst demonstrates an initial XCO of around 89%, which undergoes rapid attenuation to ~55% in 3 h and remains at this level. Subsequently, with Ar treatment at
Industrial hydrogen streams derived from steam reforming processes typically contain residual CO2
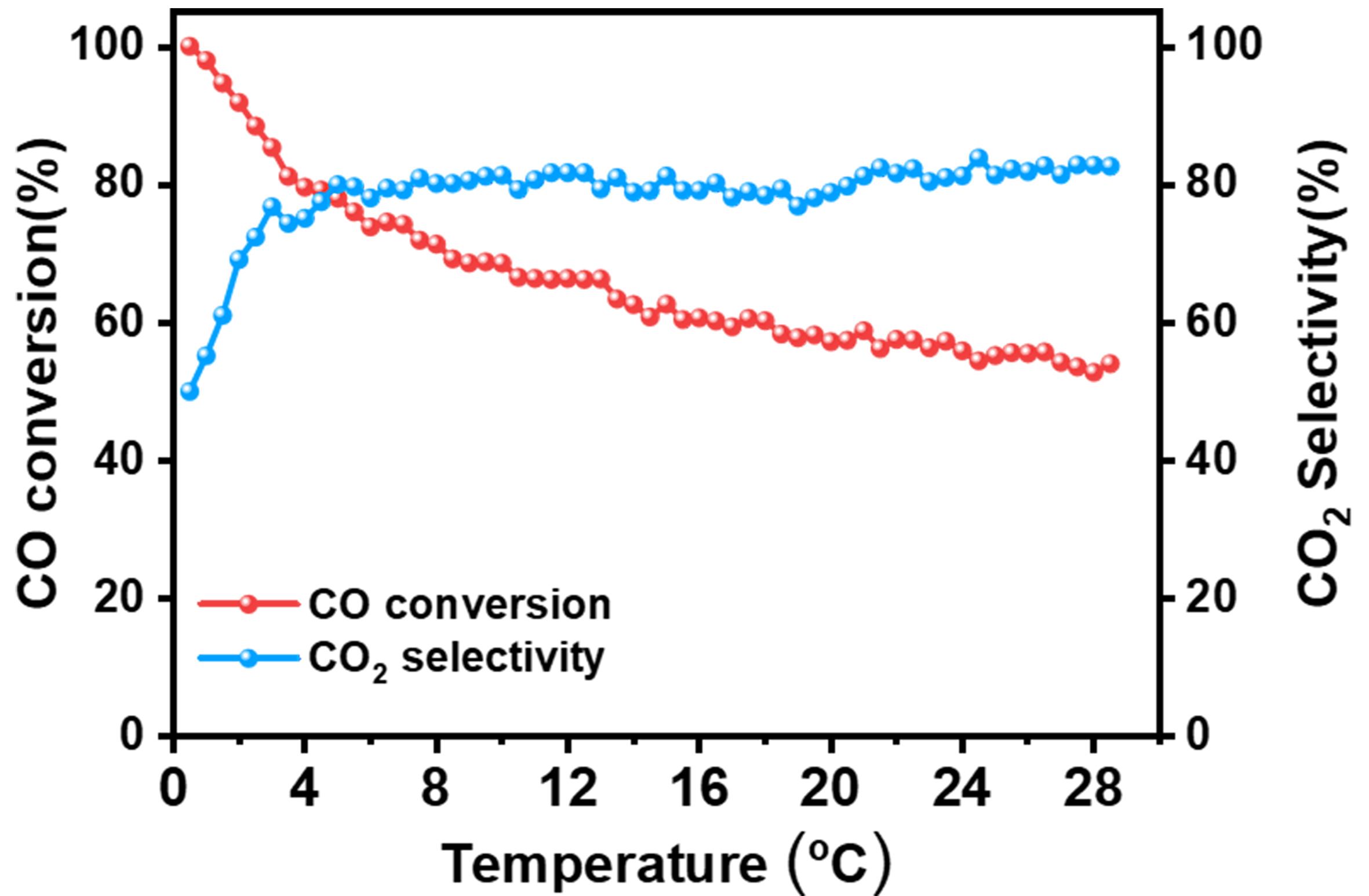
Figure 9. CO conversion and CO2 selectivity profiles with time on stream at 80 °C over 0.3Pt/CNTs-CIW catalyst. Reaction conditions:
CONCLUSION
In summary, the 0.3Pt/CNTs-CIW catalyst, synthesized via a colloidal impregnation strategy, demonstrates exceptional CO-PROX performance compared to its 0.3Pt/CNTs-IW counterpart prepared by conventional incipient wetness impregnation. The 0.3Pt/CNTs-CIW achieves complete XCO (100%) with 50% SCO2 over an unprecedentedly broad operational window (20-200 °C), while maintaining an ultralow Pt loading (0.3 wt%) - a threefold reduction relative to state-of-the-art systems-thereby addressing critical cost barriers for industrial implementation. Mechanistic insights reveal that this superior activity stems from synergistic interplay between adjacent Pt-OH species that electronically weaken CO adsorption and metallic Pt0 sites which facilitate O2 dissociation. The hydrogen isotope tracing experiment proves that the existence of H2 favors the oxidation of CO and participates in the reaction for the cycle of surface hydroxyls. The reaction pathway proceeds through carboxyl intermediates (COOH*) formed by adsorbed CO and hydroxyl groups, which combine with hydroxyls generated from co-activated O2 and H2 on Pt0 sites to yield CO2 and
DECLARATIONS
Authors’ contributions
Designing the experiments, writing the manuscript, performing data analysis and interpretation: Sun, X.
Performing the experiments: Cui, J.; Wang, Y.; Jin, Y.; He, R.
Revising and rewriting some parts of the manuscript: Cheng, Z.; Lan, G.; Qiu, Y.
Providing financial support: Liu, B.; Shi, C.; Sun, X.
Being responsible for the whole work: Li, Y.
Availability of data and materials
The data can be found in the Supplementary Materials. More details are available from the corresponding author upon reasonable request.
AI and AI-assisted tools statement
During the preparation of this manuscript, the AI tool Deepseek (version 3.0, released 2024-12) was used solely for language editing. The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship
This work was supported by the National Natural Science Foundation of China (Nos. 22108248 and 22208305) and Suichang County Key Research and Development Project (SCX2023ZDYF05).
Conflicts of interest
Liu, B. and Shi, C. are affiliated with Zhejiang Micro General New Catalytic Materials Co., Ltd. The other authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
Supplementary Materials
REFERENCES
1. Wei, K.; Wang, X.; Ge, J. Towards bridging thermo/electrocatalytic CO oxidation: from nanoparticles to single atoms. Chem. Soc. Rev. 2024, 53, 8903-48.
2. Park, E. D.; Lee, D.; Lee, H. C. Recent progress in selective CO removal in a H2-rich stream. Catalysis. Today. 2009, 139, 280-90.
3. Liu, K.; Wang, A.; Zhang, T. Recent advances in preferential oxidation of CO reaction over platinum group metal catalysts. ACS. Catal. 2012, 2, 1165-78.
4. Zhang, J.; Shu, M.; Niu, Y.; et al. Advances in CO catalytic oxidation on typical noble metal catalysts: mechanism, performance and optimization. Chem. Eng. J. 2024, 495, 153523.
5. Jing, P.; Gong, X.; Liu, B.; Zhang, J. Recent advances in synergistic effect promoted catalysts for preferential oxidation of carbon monoxide. Catal. Sci. Technol. 2020, 10, 919-34.
6. Cao, J.; Mao, L.; Wang, J.; et al. Stearic acid induced hydrophobization of Pt/Al2O3 catalysts to boost the stability of preferential oxidation of CO in hydrogen. Ind. Eng. Chem. Res. 2024, 63, 14622-30.
7. Qiao, B.; Wang, A.; Yang, X.; et al. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 2011, 3, 634-41.
8. Song, X.; Ke, S.; Ye, Q.; Kang, W.; Guan, Q.; Deng, Z. Innovative charge-tuning for highly dispersed Pt catalysts: achieving deep CO removal in industrial H2 purification for fuel cells. ACS. Appl. Mater. Interfaces. 2024, 16, 52529-38.
9. Shen, J.; Chen, C.; Song, L.; et al. Understanding the impact of hydroxyls on synthesizing the bimetallic alloy Pt3Co on Al2O3 for CO-PROX. Fuel 2025, 379, 133066.
10. Wang, C.; Li, B.; Lin, H.; Yuan, Y. Carbon nanotube-supported Pt-Co bimetallic catalysts for preferential oxidation of CO in a H2-rich stream with CO2 and H2O vapor. J. Power. Sources. 2012, 202, 200-8.
11. Zhou, X.; Xu, L.; Ma, R.; Chen, Y.; Guan, Q.; Li, W. CuO nano-island anchored Pt catalyst for CO preferential oxidation in H2-rich stream. Chem. Eng. J. 2024, 495, 153295.
12. Li, Y.; Guo, L.; Du, M.; et al. Unraveling distinct effects between CuOx and PtCu alloy sites in Pt-Cu bimetallic catalysts for CO oxidation at different temperatures. Nat. Commun. 2024, 15, 5598.
13. Jardim, E.; Gonçalves, M.; Rico-francés, S.; Sepúlveda-escribano, A.; Silvestre-albero, J. Superior performance of multi-wall carbon nanotubes as support of Pt-based catalysts for the preferential CO oxidation: effect of ceria addition. Appl. Catal. B. Environ. 2012, 113-4, 72-8.
14. Chen, Y.; Lin, J.; Li, L.; Pan, X.; Wang, X.; Zhang, T. Local structure of Pt species dictates remarkable performance on Pt/Al2O3 for preferential oxidation of CO in H2. Appl. Catal. B. Environ. 2021, 282, 119588.
15. Lin, J.; Qiao, B.; Liu, J.; et al. Design of a highly active Ir/Fe(OH)x catalyst: versatile application of Pt-group metals for the preferential oxidation of carbon monoxide. Angew. Chem. Int. Ed. Engl. 2012, 51, 2920-4.
16. Cao, J.; Mao, L.; Wang, J.; et al. Stearic acid induced hydrophobization of Pt/Al2O3 catalysts to boost the stability of preferential oxidation of CO in hydrogen. Ind. Eng. Chem. Res. 2024, 63, 14622-30.
17. Jia, Z.; Qin, X.; Chen, Y.; et al. Fully-exposed Pt-Fe cluster for efficient preferential oxidation of CO towards hydrogen purification. Nat. Commun. 2022, 13, 6798.
18. Gashnikova, D.; Maurer, F.; Sauter, E.; et al. Highly active oxidation catalysts through confining Pd clusters on CeO2 nano-islands. Angew. Chem. Int. Ed. Engl. 2024, 63, e202408511.
19. Wang, S.; Wang, S.; Zong, X.; Wang, S.; Dong, X. CO oxidation with Pt catalysts supported on different supports: a comparison of their sulfur tolerance properties. Appl. Catal. A. Gen. 2023, 654, 119083.
20. Naitabdi, A.; Boucly, A.; Rochet, F.; et al. CO oxidation activity of Pt, Zn and ZnPt nanocatalysts: a comparative study by in situ near-ambient pressure X-ray photoelectron spectroscopy. Nanoscale 2018, 10, 6566-80.
21. Hu, X.; Gao, Y.; Wang, W.; Chen, C. Structural features and catalytic performance in CO preferential oxidation of MWCNT-supported Pt-Fe catalysts. Int. J. Hydrogen. Energy. 2016, 41, 14079-87.
22. Mu, R.; Fu, Q.; Xu, H.; et al. Synergetic effect of surface and subsurface Ni species at Pt-Ni bimetallic catalysts for CO oxidation. J. Am. Chem. Soc. 2011, 133, 1978-86.
23. Gerber, I. C.; Serp, P. A theory/experience description of support effects in carbon-supported catalysts. Chem. Rev. 2020, 120, 1250-349.
24. Tanaka, K. -I, Shou M, Yuan Y. Low temperature PROX reaction of CO catalyzed by dual functional catalysis of the Pt supported on CNT, CNF, graphite, and amorphous-C with Ni-MgO, Fe, and Fe-Al2O3: oxidation of CO via HCOO Intermediate. J. Phys. Chem. C. 2010, 114, 16917-23.
25. Sun, X.; Lin, J.; Chen, Y.; et al. Unravelling platinum nanoclusters as active sites to lower the catalyst loading for formaldehyde oxidation. Commun. Chem. 2019, 2, 129.
26. Sun, X.; Lin, J.; Zhou, Y.; et al. FeOx supported single-atom Pd bifunctional catalyst for water gas shift reaction. AIChE. Journal. 2017, 63, 4022-31.
27. Chen, L.; Bao, Y.; Sun, Y.; Ma, D.; Ye, D.; Huang, B. The graphitic carbon strengthened synergetic effect between Pt and FeNi in CO preferential oxidation in excess hydrogen at low temperature. Catal. Sci. Technol. 2016, 6, 98-106.
28. Xu, H.; Fu, Q.; Yao, Y.; Bao, X. Highly active Pt-Fe bicomponent catalysts for CO oxidation in the presence and absence of H2. Energy. Environ. Sci. 2012, 5, 6313-20.
29. Guo, X.; Fu, Q.; Ning, Y.; et al. Ferrous centers confined on core-shell nanostructures for low-temperature CO oxidation. J. Am. Chem. Soc. 2012, 134, 12350-3.
30. Caglayan, B. S.; Soykal, İ. I.; Aksoylu, A. E. Preferential oxidation of CO over Pt-Sn/AC catalyst: adsorption, performance and DRIFTS studies. Appl. Catal. B. Environ. 2011, 106, 540-9.
31. Şimşek, E.; Özkara, Ş.; Aksoylu, A. E.; Önsan, Z. I. Preferential CO oxidation over activated carbon supported catalysts in H2-rich gas streams containing CO2 and H2O. Appl. Catal. A. Gen. 2007, 316, 169-74.
32. Yang, H.; Wang, C.; Li, B.; Lin, H.; Tanaka, K.; Yuan, Y. Doping effects of Ni-MgO on the structure and performance of carbon nanotube-supported Pt catalysts for preferential oxidation of CO in a H2 stream. Appl. Catal. A. Gen. 2011, 402, 168-75.
33. Chen, L.; Unocic, R. R.; Hoffman, A. S.; et al. Unlocking the catalytic potential of TiO2-supported Pt single atoms for the reverse water-gas shift reaction by altering their chemical environment. JACS. Au. 2021, 1, 977-86.
34. Zhao, Z.; Wang, M.; Ma, P.; et al. Atomically dispersed Pt/CeO2 catalyst with superior CO selectivity in reverse water gas shift reaction. Appl. Catal. B. Environ. 2021, 291, 120101.
35. Quinson, J.; Neumann, S.; Wannmacher, T.; et al. Colloids for catalysts: a concept for the preparation of superior catalysts of industrial relevance. Angew. Chem. Int. Ed. Engl. 2018, 57, 12338-41.
36. Men, Y.; Liu, Y.; Wang, Q.; et al. Highly dispersed Pt-based catalysts for selective CO2 hydrogenation to methanol at atmospheric pressure. Chem. Eng. Sci. 2019, 200, 167-75.
37. Park, J. Y.; Aliaga, C.; Renzas, J. R.; Lee, H.; Somorjai, G. A. The role of organic capping layers of platinum nanoparticles in catalytic activity of CO oxidation. Catal. Lett. 2009, 129, 1-6.
38. Zhang, J.; Gao, M.; Wang, R.; et al. Switching of CO2 hydrogenation selectivity via chlorine poisoning over Ru/TiO2 catalyst. Nano. Res. 2023, 16, 4786-92.
39. Li, Y.; Liu, C.; Su, Y.; Zhao, Y.; Qiao, B. Maximized Ir atom utilization via downsizing active sites to single-atom scale for highly stable dry reforming of methane. Chem. Synth. 2024, 4, 61.
40. Quinson, J.; Inaba, M.; Neumann, S.; et al. Investigating particle size effects in catalysis by applying a size-controlled and surfactant-free synthesis of colloidal nanoparticles in alkaline ethylene glycol: case study of the oxygen reduction reaction on Pt. ACS. Catal. 2018, 8, 6627-35.
41. Chen, Y.; Feng, Y.; Li, L.; et al. Identification of active sites on high-performance Pt/Al2O3 catalyst for cryogenic CO oxidation. ACS. Catal. 2020, 10, 8815-24.
42. DeRita, L.; Resasco, J.; Dai, S.; et al. Structural evolution of atomically dispersed Pt catalysts dictates reactivity. Nat. Mater. 2019, 18, 746-51.
43. Sun, X.; Lin, J.; Wang, Y.; et al. Catalytically active Ir0 species supported on Al2O3 for complete oxidation of formaldehyde at ambient temperature. Appl. Catal. B. Environ. 2020, 268, 118741.
44. Chen, W.; Cao, J.; Fu, W.; et al. Molecular-level insights into the notorious CO poisoning of platinum catalyst. Angew. Chem. Int. Ed. Engl. 2022, 61, e202200190.
45. Chen, L.; Allec, S. I.; Nguyen, M. T.; et al. Dynamic evolution of palladium single atoms on anatase titania support determines the reverse water-gas shift activity. J. Am. Chem. Soc. 2023, 145, 10847-60.
46. Therrien, A. J.; Hensley, A. J. R.; Marcinkowski, M. D.; et al. An atomic-scale view of single-site Pt catalysis for low-temperature CO oxidation. Nat. Catal. 2018, 1, 192-8.
47. Lin, J.; Qiao, B.; Li, L.; et al. Remarkable effects of hydroxyl species on low-temperature CO (preferential) oxidation over Ir/Fe(OH)x catalyst. J. Catal. 2014, 319, 142-9.
48. Sun, X.; Wang, Y.; Cui, J.; Li, Y.; Lin, J. Noble-metal-based catalysts on a scale from nanoparticles to subnanoclusters and single atoms for formaldehyde oxidation at room temperature: a review. ACS. Appl. Nano. Mater. 2024, 7, 3546-63.
49. Chen, Y.; Lin, J. Design of efficient noble metal single-atom and cluster catalysts toward low-temperature preferential oxidation of CO in H2. Int. J. Hydrogen. Energy. 2023, 48, 24788-808.
50. Wang, H. F.; Kavanagh, R.; Guo, Y. L.; Guo, Y.; Lu, G. Z.; Hu, P. Structural origin: water deactivates metal oxides to CO oxidation and promotes low-temperature CO oxidation with metals. Angew. Chem. Int. Ed. Engl. 2012, 51, 6657-61.
51. Zhang, C.; Liu, F.; Zhai, Y.; et al. Alkali-metal-promoted Pt/TiO2 opens a more efficient pathway to formaldehyde oxidation at ambient temperatures. Angew. Chem. Int. Ed. Engl. 2012, 51, 9628-32.
52. Huang, Z.; Hu, S.; Xu, Y.; et al. Water promoting effect in CO oxidation over N-doped Co3O4/Pt nanocatalysts by tailoring metal-support interfaces. Chem. Catalysis. 2023, 3, 100692.
Cite This Article

How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].